
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiple neighboring active sites of an atomically precise copper nanocluster catalyst for efficient bond-forming reactions†
Atanu
Ghosh‡
a,
Arunachalam
Sagadevan‡
 a,
Kathiravan
Murugesan
a,
Stefan Adrian F.
Nastase
a,
Bholanath
Maity
a,
Mohammad
Bodiuzzaman
a,
Aleksander
Shkurenko
b,
Mohamed Nejib
Hedhili
c,
Jun
Yin
d,
Omar F.
Mohammed
b,
Mohamed
Eddaoudi
b,
Luigi
Cavallo
a,
Magnus
Rueping
*a and
Osman M.
Bakr
*a
a,
Kathiravan
Murugesan
a,
Stefan Adrian F.
Nastase
a,
Bholanath
Maity
a,
Mohammad
Bodiuzzaman
a,
Aleksander
Shkurenko
b,
Mohamed Nejib
Hedhili
c,
Jun
Yin
d,
Omar F.
Mohammed
b,
Mohamed
Eddaoudi
b,
Luigi
Cavallo
a,
Magnus
Rueping
*a and
Osman M.
Bakr
*a
aKAUST Catalysis Center (KCC), Division of Physical Sciences and Engineering, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: osman.bakr@kaust.edu.sa; magnus.rueping@kaust.edu.sa
bAdvanced Membranes and Porous Materials Center (AMPMC), Physical Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia
cCore Labs, King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Kingdom of Saudi Arabia
dDepartment of Applied Physics, The Hong Kong Polytechnic University, Hung Hom, Kowloon, 999077, Hong Kong, China
First published on 6th March 2024
Abstract
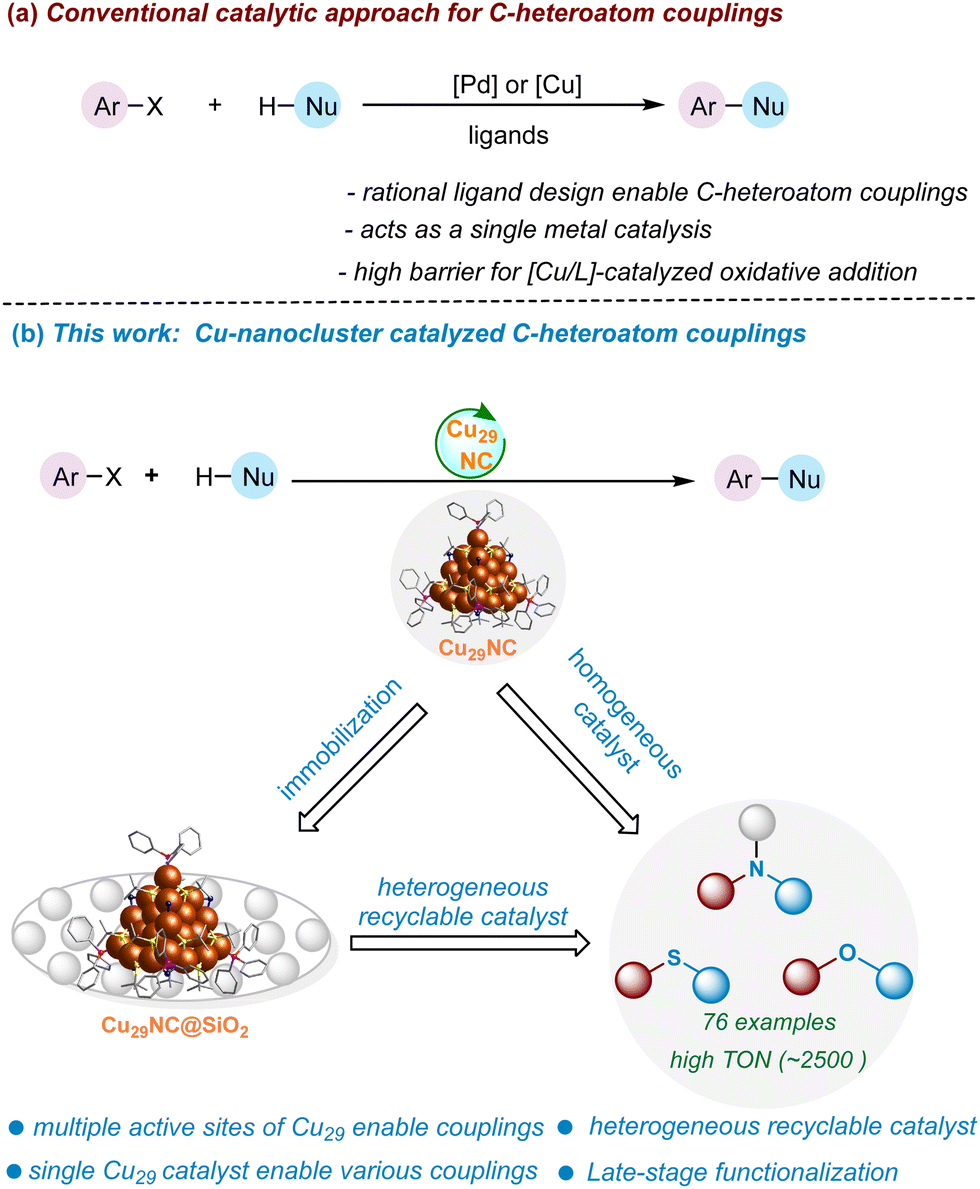
Atomically precise copper nanoclusters (NCs) are an emerging class of nanomaterials for catalysis. Their versatile core–shell architecture opens the possibility of tailoring their catalytically active sites. Here, we introduce a core–shell copper nanocluster (CuNC), [Cu29(StBu)13Cl5(PPh3)4H10]tBuSO3 (StBu: tert-butylthiol; PPh3: triphenylphosphine), Cu29NC, with multiple accessible active sites on its shell. We show that this nanocluster is a versatile catalyst for C-heteroatom bond formation (C–O, C–N, and C–S) with several advantages over previous Cu systems. When supported, the cluster can also be reused as a heterogeneous catalyst without losing its efficiency, making it a hybrid homogeneous and heterogeneous catalyst. We elucidated the atomic-level mechanism of the catalysis using density functional theory (DFT) calculations based on the single crystal structure. We found that the cooperative action of multiple neighboring active sites is essential for the catalyst's efficiency. The calculations also revealed that oxidative addition is the rate-limiting step that is facilitated by the neighboring active sites of the Cu29NC, which highlights a unique advantage of nanoclusters over traditional copper catalysts. Our results demonstrate the potential of nanoclusters for enabling the rational atomically precise design and investigation of multi-site catalysts.
New conceptsIn the realm of modern synthetic chemistry, the focus is on developing efficient catalysts with well-defined active sites. Nanostructured materials, like metal nanoparticles, show promise, but their ill-defined structure hinders mechanistic understanding. Atomically precise nanoclusters (NCs), especially copper nanoclusters (CuNCs), with defined structures, offer a solution. Earth-abundant and cost-effective, CuNCs are emerging materials for sustainable catalysis applications. This work introduces a novel core–shell copper nanocluster, Cu29NC, with a unique shell structure, demonstrating its efficiency in diverse C-heteroatom (C–O, C–N & C–S) bond-forming reactions. Unlike previous CuNCs, Cu29NC serves as a recyclable and external ligand-free catalyst for a broad range of reactions, including late-stage functionalization of complex organic molecules. Experimental and theoretical investigations provide insights into its multi-site catalytic mechanism. We anticipate that the new insights gained from this study will be instrumental in the design, synthesis, and applications of more effective nanocluster catalysts for challenging bonding-forming reactions. |
Introduction
Carbon-heteroatom (C-heteroatom) bond-forming reactions have played a pivotal role in organic transformations for the synthesis of pharmaceutical compounds, natural products, and organic materials.1–7 Pd-catalyzed Buchwald–Hartwig reactions are among the most efficient methods for C-heteroatom cross-couplings.8–11 However, the high cost of the metal as well as ligands hampers their widespread catalytic applications.8,12 Accordingly, the copper-mediated Ullmann–Goldberg protocol13 was devised as a potential cost-effective alternative but often suffered from the usage of stoichiometric amounts of copper and high reaction temperatures (>150 °C).14,15 More recently, the groups of Buchwald,16,17 Hartwig,18 and Ma19–22 have pioneered the use of copper catalysts with several ligand scaffolds (including neutral to anionic) to facilitate the cross-coupling reaction efficiently with a broader substrate scope.23–26 However, as these Cu-catalyzed cross-coupling reactions require ligands – often in excess amounts26–28 – they generate undesired ligand-related waste and may complicate the isolation process. Therefore, there is a pressing need to develop a new catalyst that can demonstrate efficient C-heteroatom coupling reactions without exogenous ligands.A central objective in modern synthetic chemistry is the development of new and efficient catalysts for challenging bond-forming reactions – catalysts which should also have well-defined multiple active sites and are reusable.29–31 Owing to their higher surface area, multiple active sites, and heterogenous nature, nanostructured materials (e.g., metal nanoparticles, MNPs) have shown great potential for catalytic performance as compared to traditional homogenous catalysts.32–38 However, due to their ill-defined structure it is difficult to delineate the role of active sites in the reaction mechanism and, hence, a catalysis-by-design approach cannot be practically implemented in such systems.39 On the other hand, atomically precise nanoclusters (NCs, <3 nm), featuring well-defined positions of metal and ligands while possessing diverse polyhedral structures,39–41 could serve as an ideal catalytic platform to enable the bond-forming reactions with mechanistic understanding owing to their structurally defined multiple active sites and tolerability for recycling. In particular, the catalytic properties of NCs highly depends on the nature of the protecting ligand and motif structure.42–45
To date, the structure–property relationships of nanocluster catalysts have mainly focused on precious metal nanoclusters such as Au and its alloys with Pd, Pt, and Ag for various chemical reactions.39,40,43,46 Due to their earth abundance, cost-effectiveness, and versatile ligand architecture, copper nanoclusters have recently emerged for sustainable catalysis of chemical reactions.37,47–53 The challenge in the development of new core–shell copper nanoclusters (CuNCs) that facilitate unique bond-forming reactions lies not only in their synthesis, but also in the unravelling of the reaction mechanism through atomically precise structure–property relationships of the CuNCs.
Herein we have designed and synthesized a novel core–shell copper nanocluster, [Cu29(StBu)13Cl5(PPh3)4H10]tBuSO3, (Cu29NC) with a high percentage of chloride ligands on the surface. It encompasses a centred cubooctahedron core (Cu13), shielded by a unique tetrahedral shell, Cu16S13Cl5P4. This shell is characterized by three distinct motifs, each exhibiting crown-like structures. The incorporation of chloride ligands in the shell serves to alleviate crowding around the active sites, making it a viable catalyst in organic reactions. We demonstrate that Cu29NC serves as an efficient catalyst to enable the broad range of cross-coupling reactions (C–O, C–N, and C–S) with a high functional group tolerance (Fig. 1). In contrast to previous Cu NCs, such as Cu61NC and Cu28NC, which have proven effective as photocatalysts for certain cross-coupling reactions,52,54 this Cu29NC serve as a recyclable external ligand-free catalyst for a broad range of C-heteroatom coupling reactions, including late-stage functionalization of complex organic molecules. A combined experimental investigation (with various controls) with theoretical DFT modeling (based on the single crystal structure) indicate the participation of multiple neighboring metal atoms in all fundamental steps of the reaction. This work opens new avenues for copper nanocluster-based catalysis and sheds light on their catalytic mechanism by DFT calculation based on the single crystal structure.
 | ||
| Fig. 1 Catalytic methods for C-heteroatom bond-forming reactions. | ||
Results and discussion
Synthesis and characterizations of Cu29 nanocluster
The Cu29 cluster was synthesized by a two-step procedure. In brief, the reduction of Cu(I)-StBu-PPh3 complex (chloroform solution) by ethanolic solution of sodium borohydride (NaBH4) produced the Cu29NC. The synthesis procedure is detailed in the experimental section of the ESI.†A single crystal of appropriate size was used for single-crystal X-ray diffraction (SCXRD). The cluster crystallizes in a triclinic crystal system with P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group (Table S1, ESI†). The total structure of the Cu29 clusters derived from the SCXRD is presented in Fig. 2a. A combined study of SCXRD and mass spectrometry confirms the total molecular formula of the cluster, [Cu29(StBu)13Cl5(PPh3)4H10]tBuSO3 (vide infra). The cluster contains a centered-cubooctahedron core (Cu13, Fig. 2b and Fig. S1, ESI†) which is protected by a unique tetrahedral shell, Cu16S13Cl5P4 (Fig. 2c). The structure contains only one C1 symmetry axis.
space group (Table S1, ESI†). The total structure of the Cu29 clusters derived from the SCXRD is presented in Fig. 2a. A combined study of SCXRD and mass spectrometry confirms the total molecular formula of the cluster, [Cu29(StBu)13Cl5(PPh3)4H10]tBuSO3 (vide infra). The cluster contains a centered-cubooctahedron core (Cu13, Fig. 2b and Fig. S1, ESI†) which is protected by a unique tetrahedral shell, Cu16S13Cl5P4 (Fig. 2c). The structure contains only one C1 symmetry axis.
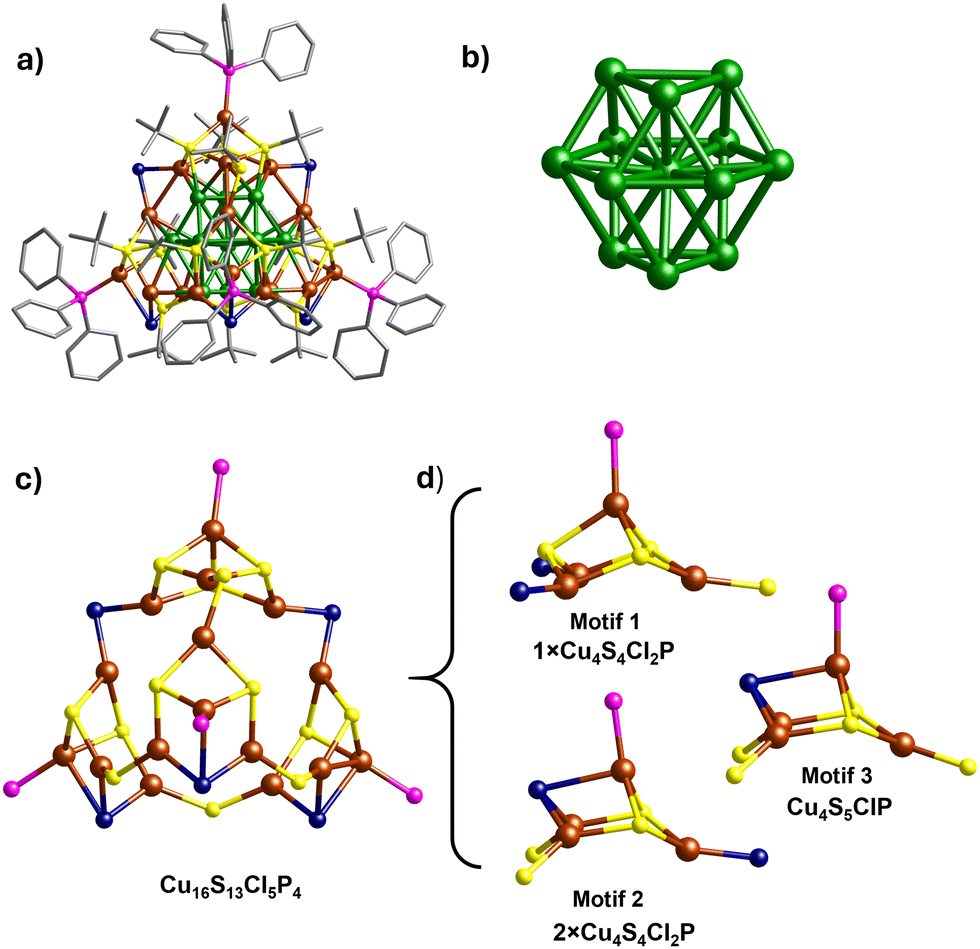 | ||
| Fig. 2 (a) Molecular structure of [Cu29(StBu)13Cl5(PPh3)4H10]+ Cu29NC. (b) Cubo octahedron Cu13 core. (c) Structure of tetrahedral shell, Cu16(StBu)13Cl5(PPh3)4. Carbon and hydrogen atoms are omitted for clarity (d) Structures of three distinct types of motifs. Color legend: green, and brown, copper atoms of the Cu13 core, and shell, respectively; yellow, sulfur; pink, phosphine; dark blue, chlorine; and gray, carbon. | ||
The shell, Cu16S13Cl5P4, is composed of three different types of motifs, motif 1 (Cu4S4Cl2P), motif 2 (Cu4S4Cl2P), and motif 3 (Cu4S5Cl1P) (Fig. 2d). All the motifs have crown-like structures. Motif 1 is connected with two units of motif 2 through two bridging μ2-Cl whereas motif 3 is connected to motif 1 through bridging thiolate ligands. On the other hand, two units of motif 2 and one unit of motif 3 are interconnected through three bridging thiolate ligands. The interactions of motifs 1, 2 and 3 with the Cu13 core are quite different. The surface of the Cu13 core is composed of six distorted square and eight triangle faces (Fig. S2, ESI†). Motif 1 occupies one of the triangular faces whereas motifs 2 and 3 occupy the three-square faces of the Cu13 core (Fig. S3, ESI†). Overall, the Cu29NC occupies a tetrahedral shape where the corners of the tetrahedron are occupied by the triphenylphosphine ligands. Three diverse types of thiolate ligand groups are observed in the Cu29NC (Fig. S4, ESI†). Like the thiolate ligands, two types of bridging modes are also observed for the chloride ligands. Two chloride ligands that connect motif 1 with the two units of motif 2 have μ2 bridging mode (μ2-Cl) and the remaining three chloride ligands have μ3 bridging mode (μ3-Cl). It is worth mentioning that such kind of chloride rich (high chloride to thiolate ratio) shell structure is rare in the core–shell nanocluster family. The presence of a higher percentage of chloride ions reduces the crowding of surface copper atoms and, hence, heightens their probability of being involved in catalysis.
Generally, copper nanoclusters contain hydrides.47,52,55–59 However, it is difficult to locate them using SCXRD for such a larger cluster system. Thus, ESI MS was used to determine the exact composition of the clusters. Details of the sample preparation and instrumental conditions are described in the experimental section of the ESI.† Positive mode ESI MS of the chloroform solution of nanoclusters produced a major peak at m/z 4237.8 in the mass range of m/z 20–10000 (Fig. 3). Upon expansion, the peak shows a characteristic separation of Δm/z = 1 (Fig. S5, ESI†), which confirms the presence of singly charged (1+) species. The assigned composition for the above-mentioned peak is [Cu29(StBu)13Cl5(PPh3)4H10]+. The exact matching of isotopic distributions of experimental (black trace) and simulated (red trace) spectra confirm the assigned composition (Fig. 3a). Furthermore, to verify the number of hydrides, ESI MS of deuterated analogue of Cu29NC, Cu29D, was performed. In case of Cu29D, the peak position is shifted by Δm/z = 10 due to the replacement of ten hydrogen atoms (m/z 10) by deuterium atoms (m/z 20) which further supports the assigned compositions (Fig. 3b). Similar to the reported copper nanoclusters,52,60 we performed geometry optimizations by DFT calculations to locate the ten hydrides (Fig. S6A, ESI†). Details of the calculations are presented in the experimental section of the ESI.† In order to substantiate the existence of the hydrogen atoms within the NC, a 2H nuclear magnetic resonance (NMR) analysis was conducted on Cu29D in 950 MHz spectrometer in CHCl3, as illustrated in Fig. S6B (ESI†). These yielded signals originating exclusively from deuterium (D) atoms within the sample and added CDCl3 for calibration. The resultant NMR spectrum exhibited discernible peaks distributed across a range of chemical shift values, ranging from low to high regions. This observation provides compelling evidence for the presence of deuterium atoms within the NC, indicative of their occupancy in diverse chemical environments.
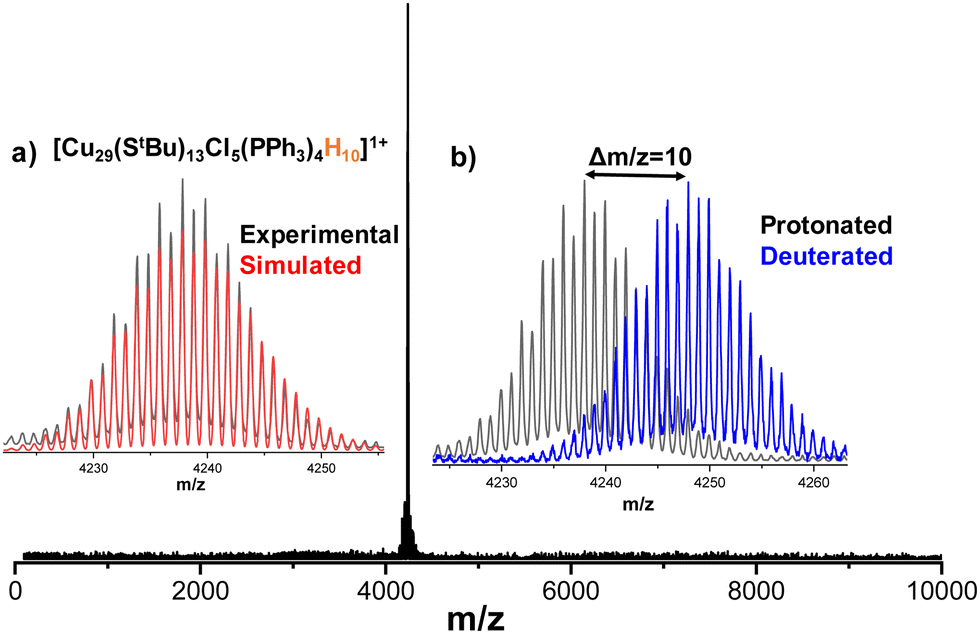 | ||
| Fig. 3 Positive mode ESI MS spectrum of [Cu29(StBu)13Cl5(PPh3)4H10]+ nanocluster. The peak at m/z 4237.8 corresponds to the molecular ion peak. Insets: (a) Exact matching of experimental (black trace) and simulated (red trace) isotopic distributions confirm the assigned compositions. (b) Comparison of protonated, [Cu29(StBu)13Cl5(PPh3)4H10]+, and deuterated, [Cu29(StBu)13Cl5(PPh3)4D10]+, molecular ion peaks. | ||
The unique shell structure along with the easily obtainable clean mass spectrum allowed us to study the cluster further using mass spectrometry (Fig. S7 and S8, ESI†). Furthermore, to identify the counter ion, we performed the ESI MS measurement in the negative mode and observed a peak at m/z 137 (Fig. S9, ESI†). The exact matching of experimental and simulated isotopic distributions confirms the presence of tert-butyl sulfonate (tBuSO3−) as the counter ion, which is in good agreement with SCXRD data (inset, Fig. S9, ESI†).
The oxidation state of copper atoms holds a fundamental significance in catalysis, which can influence the reactivity and selectivity. Thus, we performed X-ray photoelectron spectroscopy (XPS) measurements of the solid powder sample to identify the oxidation states of the copper atoms. XPS survey spectrum presented in Fig. S10a (ESI†) confirms the presence of all the expected elements (Cu, S, Cl, C, and P). High-resolution XPS spectra of Cu 2p, S 2p, C 1s, and Cl 2p are presented in Fig. S10b–e (ESI†). Generally, it is difficult to distinguish Cu(0) and Cu(I) as their 2p binding energy are almost similar. Hence, we gathered Cu LMM Auger spectra of Cu(I)–StBu thiolate and Cu29 cluster (Fig. S11, ESI†). The presence of a sharp peak at 916.3 eV confirms that all the copper atoms in Cu29 nanocluster are in +1 oxidation state which is in good agreement with SCXRD and ESI MS analysis.61 Furthermore, a comparison of Cu LMM Auger spectra Cu(I)-StBu thiolate and Cu29 cluster is presented in Fig. S11 (ESI†) which also supports the presence of only Cu(I) oxidation state in Cu29 nanocluster.
Reaction development
We commenced our investigations with the newly synthesized Cu29NC and tested the cluster in the C–O cross-coupling of p-bromobenzonitrile and p-cresol. Use of 0.05 mol% of catalyst provided 86% of the desired C–O coupled product (Fig. 4 and Table S2, entry 1, ESI†). Most striking, the reaction can be effectively performed with a catalyst loading of 0.025 mol% still providing the coupled product in 88% yield (Fig. 4, and Table S2, Entry 2, ESI†). The optimal temperature was found to be 90 °C while lower temperature resulted in lower yield (Table S2, entries 3 and 4, ESI†). Further we examined various conditions with respect to catalyst, bases, and solvents to understand and improve the catalytic performance of Cu29NC on the cross-coupling reactions (Table S2, entries 5–12, ESI†). Solvent optimizations indicate that CH3CN provided more yield in the case of a C–O coupling reaction while DMSO is better for other cross-couplings (C–N and C–S bond formations, see details in Table S3 and S4, ESI†). In order to better understand the catalyst efficacy, optimization studies were performed in the presence of two different electrophiles (p-bromobenzonitrile and p-bromoanisole) with p-cresol as the nucleophile under standard reaction (Fig. 4). As direct comparison, CuCl was investigated as a catalyst instead of Cu29NC (Fig. 4), leading to lower efficiency (∼22% yield), which is in line with the previous work.17,18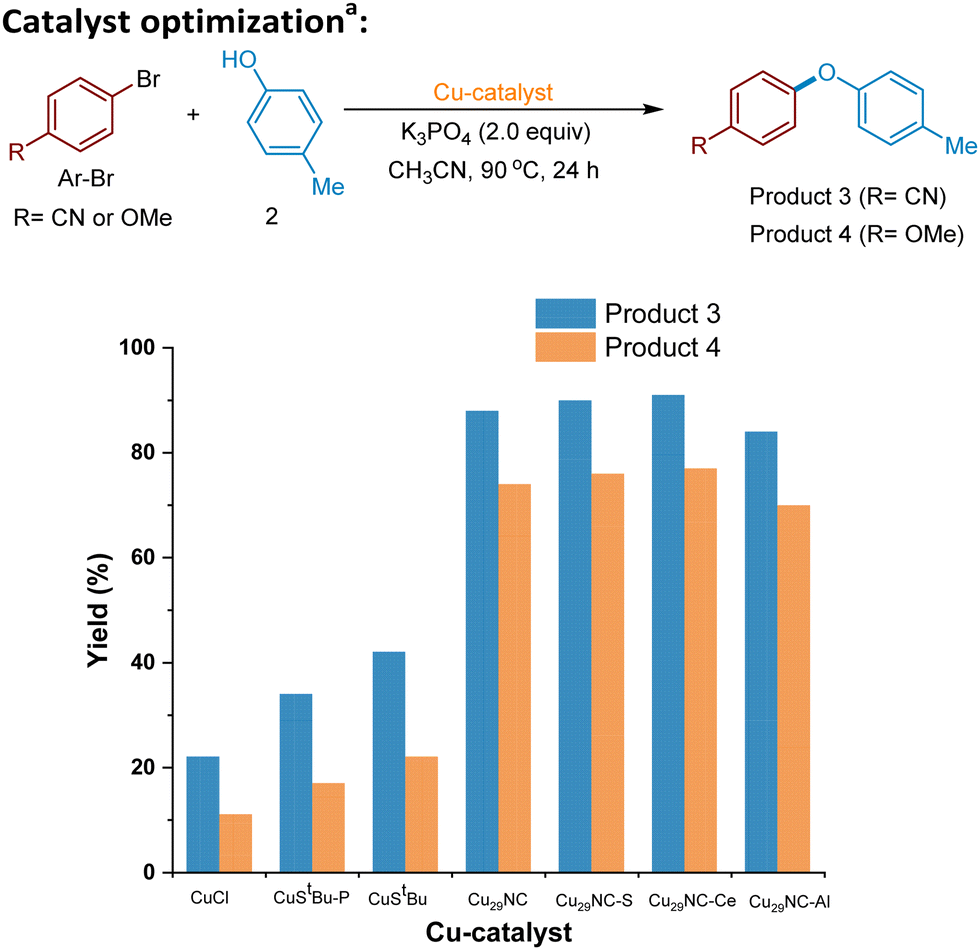 | ||
| Fig. 4 Catalyst optimization. aConditions: Ar–Br (0.3 mmol), 2 (0.375 mmol), Cu-catalyst used such as 5 mol% of CuCl or CuStBu-PPh3, CuStBu or 0.025 mol% of Cu29NC or Cu29-Celite S or Cu29NC-CeO2 or Cu29NC-Al2O3), K3PO4 (2.0 eq.), and acetonitrile (CH3CN) (2 mL). The mixture was stirred at 90 °C for 24 h. Yield refers to GC yield using 1,3,5-trimethylbenzene (mesitylene) as the internal standard. | ||
Control experiments demonstrated that the absence of the Cu29NC catalyst or a base resulted in no reaction or trace amounts observed (Table S2, entries 13 and 17, ESI†). Notably, the current Cu29NC catalyst can be supported on different surfaces (Celite S, Celite 545, alumina (γ), CeO2, and carbon powder) and tested for the C–O coupling reaction. These findings indicate that 0.025 mol% of Cu29-Celite S is an effective C–O cross-coupling catalyst, with similar efficiencies to the unsupported cluster (Fig. 4 and details in ESI†), while also providing higher recyclability than other supports. Furthermore, Cu-complexes, namely Cu(I) StBu-PPh3 and Cu(I) StBu (precursors for the preparation of Cu29NC), were used as catalysts. However, the desired C–O coupling products (3 and 4) were obtained in lower yields with both, activated electrophiles (<42% yield) and unactivated electrophiles (<22% yield) compared to Cu29NC (which yields 88%) (See, Fig. 4). Previously, we used Cu61NC and Cu28NC for photocatalytic C(sp2)− Nhet Ullmann–Goldberg coupling54 and Sonogashira C–C cross-coupling reaction,52 respectively. In this regard, we performed control experiments with Cu61NC and Cu28NC as catalysts instead of Cu29NC for the current C–O cross-coupling reactions, however, they produced low yields, 58% and 63%, respectively (Table S2, entries 8 and 9, ESI†). Thus, we hypothesize that the differently oriented chloride rich motif structures of Cu29NC plays a key role in this reaction's efficiency.
Recyclable Cu29NC catalyst development
In order to convert the Cu29NC nanocluster into a heterogeneous recyclable catalyst, we aimed to attach the catalyst to a support. Thus, we prepared five different supported clusters (Cu29-Celite S, Cu29-Celite 545, Cu29-alumina (γ), Cu29-CeO2, and Cu29-carbon powder) and compared the three-reaction parameters, catalyst loading, performance, and leaching, to find the best heterogeneous catalyst. Details of their synthesis are described in the experimental section of the ESI.† The catalyst systems Cu29-Celite S, Cu29-CeO2, and Cu29-alumina (γ) showed better loading and performance than the remaining two systems (Fig. S12 and S13, ESI†). Interestingly, Cu29–CeO2 and Cu29-alumina (γ) showed a higher amount of leaching after the 1st cycle, whereas a negligible amount of leaching was observed for Cu29-Celite S (Fig. S14, ESI†). Thus, recyclability was further studied with the Cu29-Celite S system. The diffuse reflectance optical spectrum of the pure Cu29 cluster showed two step-like features at 504 and 463 nm (black trace, Fig. S15a, ESI†). These features were also observed for the Cu29-Celite S system (red trace, Fig. S15a, ESI†). A comparison of XPS results confirmed that the oxidation state of Cu atoms remained unaffected during the synthesis of the supported cluster, Cu29-Celite S (Fig. S15b, ESI†). Furthermore, Z-contrast scanning transmission electron microscopy (STEM) analysis of Cu29-Celite S showed that the size of the pure Cu29 cluster also remained (Fig. S15C, ESI†). All these results showed that the Cu29 nanoclusters loaded on Celite S support did not decompose or change shape (Fig. S15, ESI†). It is worth mentioning that the efficiency of this supported catalyst was similar to that of the unsupported form (Fig. 4). Furthermore, we demonstrated that the heterogeneous catalyst could be easily recycled and reused for five cycles with similar activity (Fig. 5, see details in ESI,† Fig. S15). There might be a possibility that the cluster decomposes and releases copper atoms/copper thiolates during the reaction (2nd cycle onwards). However, we detected negligible amounts of the copper in the solution which shows that there is no considerable cluster decomposition during the catalytic reactions (Fig. 5, black trace). In contrast, previous reports on gold clusters have shown that the catalytic efficiency of free clusters is extremely low compared to supported clusters.62,63 Preheat treatment at around 150 °C43,64,65 and partial or complete ligand removal are also required to achieve better efficiency in these reports.43,66 However, our study found that preactivation of the Cu29NC cluster at elevated temperatures is unnecessary, as the performance of both free and supported clusters remains almost the same. Thus, our cluster system provides a synergistic effect between homogeneous and heterogeneous catalysts, setting it apart from other catalytic systems.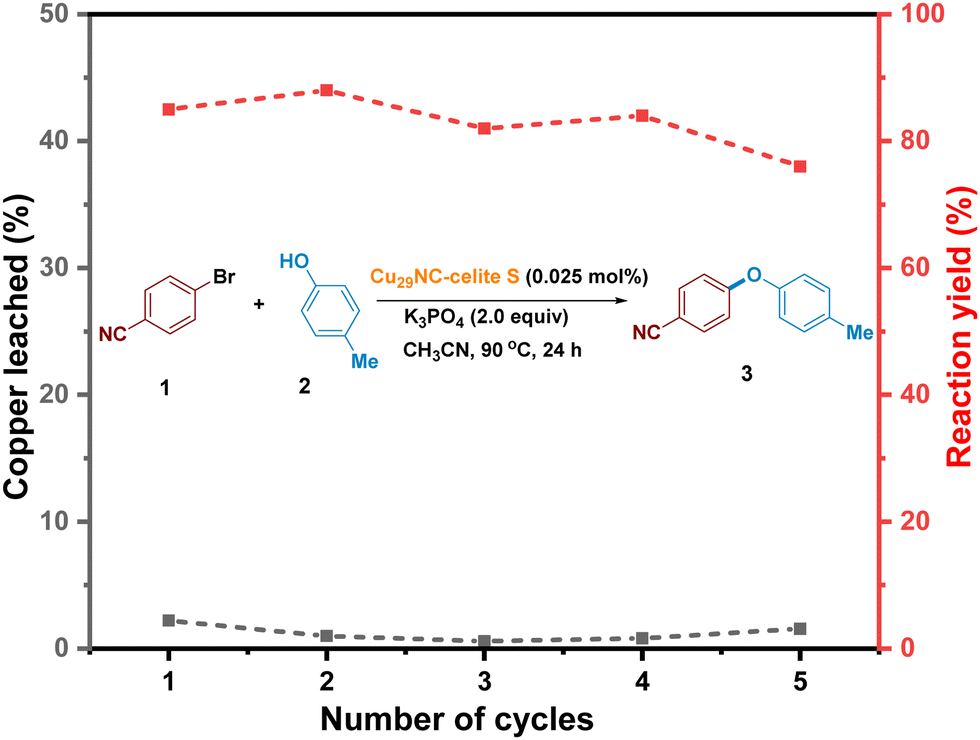 | ||
| Fig. 5 Recycling of Cu29-Celite S catalyst. Percentage of yield and leaching are denoted with red and black traces, respectively. The amount of leached copper is negligible for each cycle. | ||
Substrates scope
With the optimized conditions and recyclability of the catalyst in hand (Fig. 4, Table S2, ESI†), we set out to explore the scope of the C–O cross-coupling using various aryl/heteroaryl bromides (Fig. 6, heterogeneous Cu29-Celite S (Cu29-S) catalyst is used for those mentioned in bracket). In summary, a wide variety of electron-neutral, electron-donating, and electron-withdrawing aryl bromides react well in the C–O coupling reaction, providing the corresponding C–O arylated products in good to excellent yields (69–93% yields). Typically, electron-rich aryl bromides are poor coupling partners due to a higher energy barrier for the oxidative addition step.22 Notably, the Cu29NC performs as an efficient catalyst for electron-rich to neutral aryl bromides (4–7, 76–81% yields). In addition, diverse meta- and ortho-substituted aryl bromides are also amenable to the transformation (9 & 10). Particularly, sterically more hindered 1-bromonaphthalene performed well in this Cu29NC-catalyst process, demonstrating another advancement over previous copper-catalyzed processes.18 In general, heteroaryl bromides are not good coupling partners in copper catalysis, due to their strong coordination with the metal-catalyst, which may hinder the catalytic activity.67–69 Remarkably, the current Cu-cluster catalyzed reaction is viable for a series of heteroaryl bromides (12 and 13 and 25 and 26, 77–85% yields). Next, a combination of various phenols and aryl bromides was employed in and the products were obtained in good to excellent yield (15–26). Notably, phenols bearing chloride and bromide at the para position were also well tolerated and gave the corresponding product without affecting the bromine atom (18 and 19, 71–77%), potentially allowing for the subsequent orthogonal functionalization.70–74 Electron-deficient and heteroaryl phenols are also suitable nucleophiles in this cross-coupling transformation (21 and 26). In addition, phenols containing the unactivated olefin (Eugenol) selectively underwent this reaction and generated their desired C–O coupled product in excellent yield (24 and 25, 84% yields) allowing further functionalization on the olefin bond (i.e; Heck-type reactions of eugenol derivatives).75–78 Moreover, benzylic and aliphatic alcohols were also found to be good coupling nucleophiles in this transformation (27–35). Furthermore, use of ethanol afforded the C–O coupled products in good yields (27, 73% yield). In particular, primary alcohols containing benzylic, CF3, cyclopentyl, n-octanol, and pyridine-substituted alcohols were also good coupling partners with aryl(hetero)bromides (28–35, 69–89% yields). Interestingly, a terminal olefin substituted linear alcohol is also tolerated in this C–O coupling reaction without affecting the C–C double bond (32, 69% yield), allowing follow-up coupling reactions.75,76,78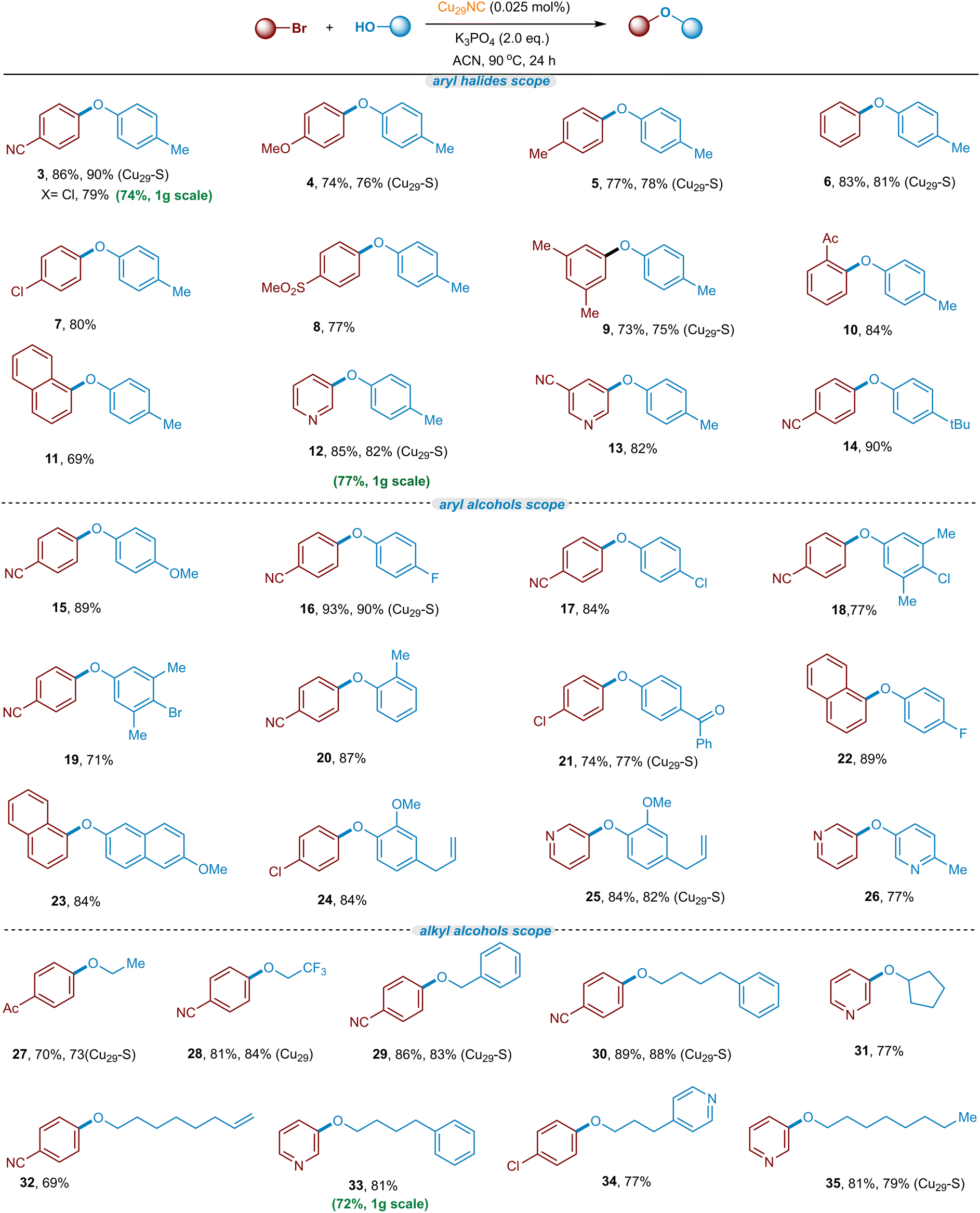 | ||
| Fig. 6 Substrate scope of aryl halides and alcohols. Standard reaction conditions. Yield of isolated product after purification by column chromatography on a silica gel. (Cu29-S) indicates heterogeneous Cu29-Celite S. | ||
Mechanistic studies
To define the reaction mechanism, control experiments (Schemes S4 and S5, ESI†) and quantum mechanical calculations were performed. First, a radical trapping experiment was conducted to check for the presence of radical species in the reaction pool. No radical was identified. Further, a radical clock experiment using aryl bromides 36 and p-cresol 2 under standard conditions results in the non-cyclized product 37 predominating over the cyclized product 38 (Scheme S4, ESI†). These control experiments indicate that the reaction does not proceed through radical formation. The above results also suggest that the oxidative addition of Ar–X (X = Br, Cl is operated by a concerted mechanism instead of a single electron transfer (SET). To further discern between concerted oxidative addition and SET pathways we carried out competition experiments. For example, the reaction with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 4-chlorobenzonitrile (1-Cl) and 1-bromonaphthalene (39) with p-cresol as nucleophile, under standard conditions provides a product ratio of 1:14.2 (Scheme S5a, ESI†). Despite its lower reduction potential of 1-Cl, 39 reacts faster, which can be related to the weaker C–Br bond compared to C–Cl. Consistently, a 1:1 mixture of 1-Cl and 4-iodoanisole (40) leads to a product ratio of 1:12.75 (Scheme S5b, ESI†). Overall, these results indicate that conventional two-electron oxidative addition is operative instead of a radical pathway via SET.54,79
:1 mixture of 4-chlorobenzonitrile (1-Cl) and 1-bromonaphthalene (39) with p-cresol as nucleophile, under standard conditions provides a product ratio of 1:14.2 (Scheme S5a, ESI†). Despite its lower reduction potential of 1-Cl, 39 reacts faster, which can be related to the weaker C–Br bond compared to C–Cl. Consistently, a 1:1 mixture of 1-Cl and 4-iodoanisole (40) leads to a product ratio of 1:12.75 (Scheme S5b, ESI†). Overall, these results indicate that conventional two-electron oxidative addition is operative instead of a radical pathway via SET.54,79
Electronic structure modeling of all possible intermediates along the reaction pathway were performed using a DFT approach80,81 (refer to ESI† for computational methods). In the DFT study we selected two different electrophiles, 4-bromoanisole and 4-chloroanisole, combined with the nucleophilic partner p-cresol. We started by identifying the most active site and elucidating an ensemble effect, which is the involvement of more than one Cu atom in Cu29NC. To achieve this goal, we have relied on the thermodynamic feasibility of the first step, which is (Cu)29–OAr formation via deprotonation by K3PO4. Different (Cu)29–OAr intermediates were modeled (Fig. 7), by considering all the structurally different surface Cu-atoms on motifs 1–3 (Fig. 2). In addition to nine intermediates showing the nucleophile –OAr interacting with one single Cu atom (structures A1–A6 and A8–A10 in Fig. 7), we also identified one intermediate with the –OAr fragment bridging two adjacent Cu atoms, one of which is terminal (structure A7 in Fig. 7). Intermediate A7 is identified as the most stable one, suggesting for an ensemble effect favoring the initial activation of Cu29NC. Other attempts to locate low energy structures with the –OAr fragment bridging two adjacent Cu atoms failed. Overall, the higher stability of structures A4–A7 indicate that motif 2 has more active Cu atoms compared to motifs 1 and 3.
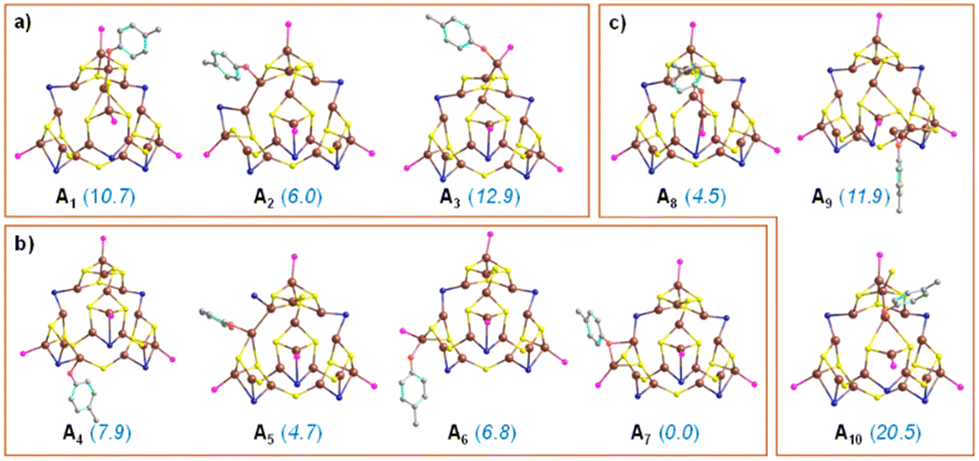 | ||
| Fig. 7 DFT optimized structures and relative energies (in kcal mol−1) of (Cu)29–OAr complexes varying the active Cu-atom in three different regions: (a) motif 1, (b) motif 2, and (c) motif 3. Only surface Cu atoms are projected, and H atoms are omitted for clarity. The energy values are at the PBE-D3 level of theory using implicit solvation model in acetonitrile. | ||
All the intermediates along the reaction pathway and their relative energies are displayed in Fig. 8 involving 4-bromoanisole, and in Fig. S17 (refer to ESI†) for chloro analogue. As discussed above, the reaction starts with formation of the Cu–Nu bond in the presence of a strong base, K3PO4, which is exergonic by 138.1 kcal mol−1. The resulting intermediate A7 undergoes oxidative addition of 4-bromoanisole, since the alternative SET step is endergonic by 104.7 kcal mol−1 and can thus be excluded (Fig. S18, ESI†), consistently with the experimental evidence.
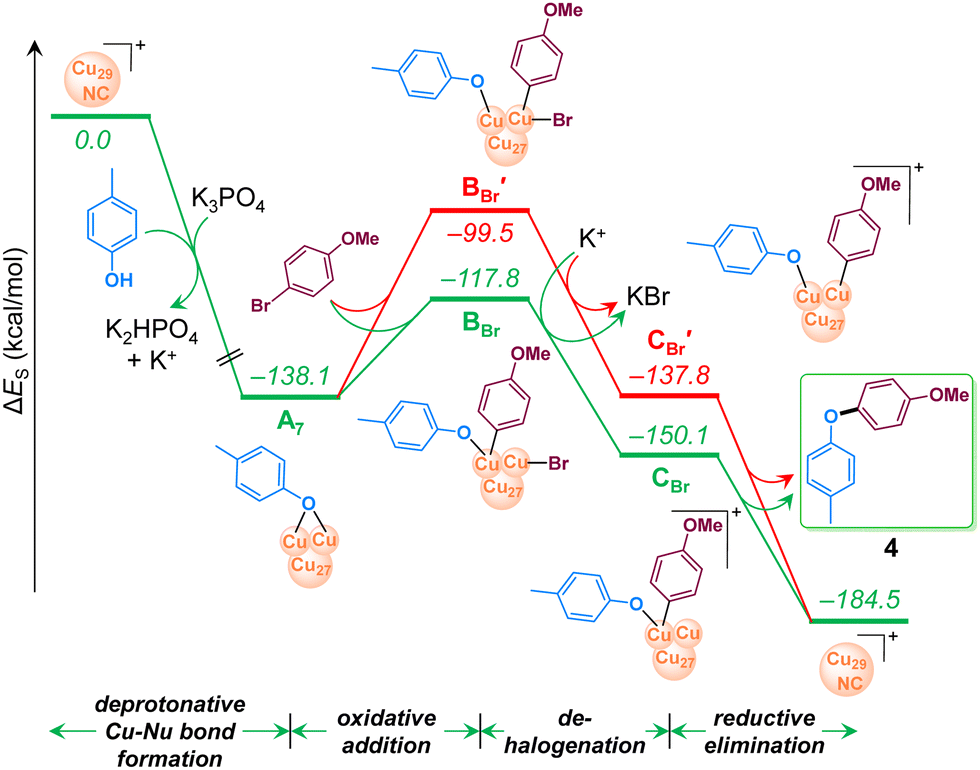 | ||
| Fig. 8 Computed energy profile of Cu29-NC catalyzed C–O coupling reaction of p-cresol using 4-bromoanisole. For energy convention, refer to Fig. 7. | ||
Within the conventional 2e oxidative addition scheme, two possible pathways (green vs. red paths in Fig. 8 were considered depending on the mechanism of Ar–Br activation. The first we considered involves a bimetallic mode of activation, with the Ar and Br moieties bound to two vicinal Cu atoms (green line in Fig. 8), is endergonic by 20.3 kcal mol−1 and leads to intermediate BBr. The next step, debromination from BBr results in the cationic intermediate CBr with an energy gain of 32.8 kcal mol−1. Finally, highly exergonic reductive elimination from CBr results in the desired product and regenerates the starting Cu29NC which is ready to start another catalytic cycle.
The second pathway we considered involves a standard monometallic oxidative addition of Ar–Br at a single Cu-atom (red line in Fig. 8), leading to the intermediate BBr′, which is 18.3 kcal mol−1 more unstable than the bimetallic intermediate BBr. Consistently, the following debrominated intermediate, CBr′, is higher in energy than CBr by 12.3 kcal mol−1. Overall, the above results indicate that the pathway with bimetallic oxidative addition (green line) is more viable than the corresponding monometallic one (red line). Considering the initial reaction of Cu29NC with the HO–Ar nucleophile, also favored by the presence of two nearby Cu atoms, our calculations indicate the role of an ensemble effect in favoring the whole reaction pathway. Consistently, the computed energy profile for 4-chloroanisole (Fig. S17 in ESI†) resembles that of 4-bromoanisole (Fig. 8). To further acquire more insight into the kinetics involved, a linear one-dimensional scan analysis of the potential energy surface (PES) was conducted on the reaction rate-limiting step, oxidative addition of ArBr to A7 (Fig. S19 in ESI†). By creating a series of “images” along the bond (C–Br) breaking during the reaction with a specific increment of the distance between the C and Br with all atoms relaxed, an energy profile was described from which the energy barrier was predicted to be lower towards the formation of BBr compared to that of BBr′. The nudged elastic band and dimer methods were tested and deemed unfeasible due to the large size of the system.
Finally, based on our control experiments and computational results, we propose the catalytic cycle of the Cu29NC catalyzed C-heteroatom coupling reaction, shown in Scheme 1. The mechanism includes four fundamental steps: deprotonative Cu–Nu bond formation, oxidative addition, dehalogenation, and reductive elimination to liberate the product. Notably, though the reaction occurs at the terminal Cu of motif 2, the neighboring Cu also plays a significant role during the reaction pathway. As a result, the ensemble effect is visible in the DFT calculations, which clearly distinguishes the catalytic behavior of a nanocluster from single metal catalysis.
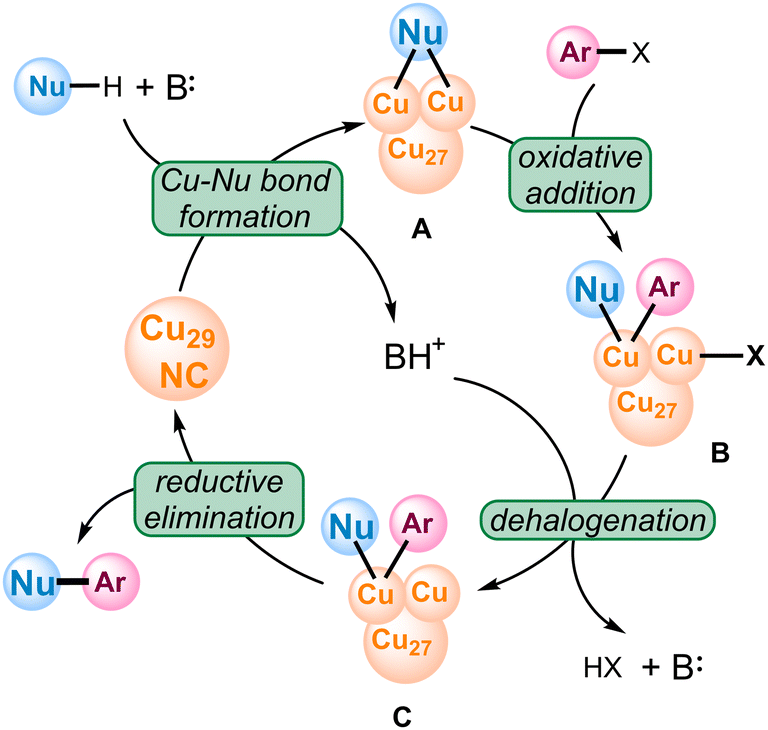 | ||
| Scheme 1 Proposed catalytic cycle. | ||
Extended substrates scope
After successfully demonstrating the C–O cross-coupling reaction, we aimed to explore the C–N and C–S cross-coupling reaction of N & S-nucleophiles with aryl bromides (Fig. 9 and optimization condition see Tables S3 and S4, ESI†). To summarize, secondary aliphatic amines such as pyrrolidine, piperidine, and morpholine readily undergo the desired C–N cross-coupling with various aryl bromides including pyridinyl aryl bromides (41–47, 72–90% yields). This Cu-cluster catalysis is also applicable to C–N cross-couplings of N-heterocycles with various aryl bromide electrophiles. For example, carbazole, indole, pyrazole, and imidazole are found to be competent nucleophiles in this cross-coupling reaction, providing desired C–N arylated products in excellent yield (48–53). Other N-nucleophiles such as N-alkylaniline, sulfonyl amines, benzamide, and oxazolidin-2-one, also reacted well (54–57, 68–76% yields). Besides, we set out to explore the S-nucleophiles with aryl bromides under the optimized Cu29NC catalytic conditions. It is worth noting that, electron-rich and electron-poor thiophenol derivatives readily underwent C–S coupling and the products were isolated in good to excellent yields (58–71, 70–89% yields). Heteroaryl bromides were found to be well-suitable coupling electrophiles in combination with thiophenol nucleophiles (61–65). An aliphatic thioalcohol was also successfully applied (70 and 71).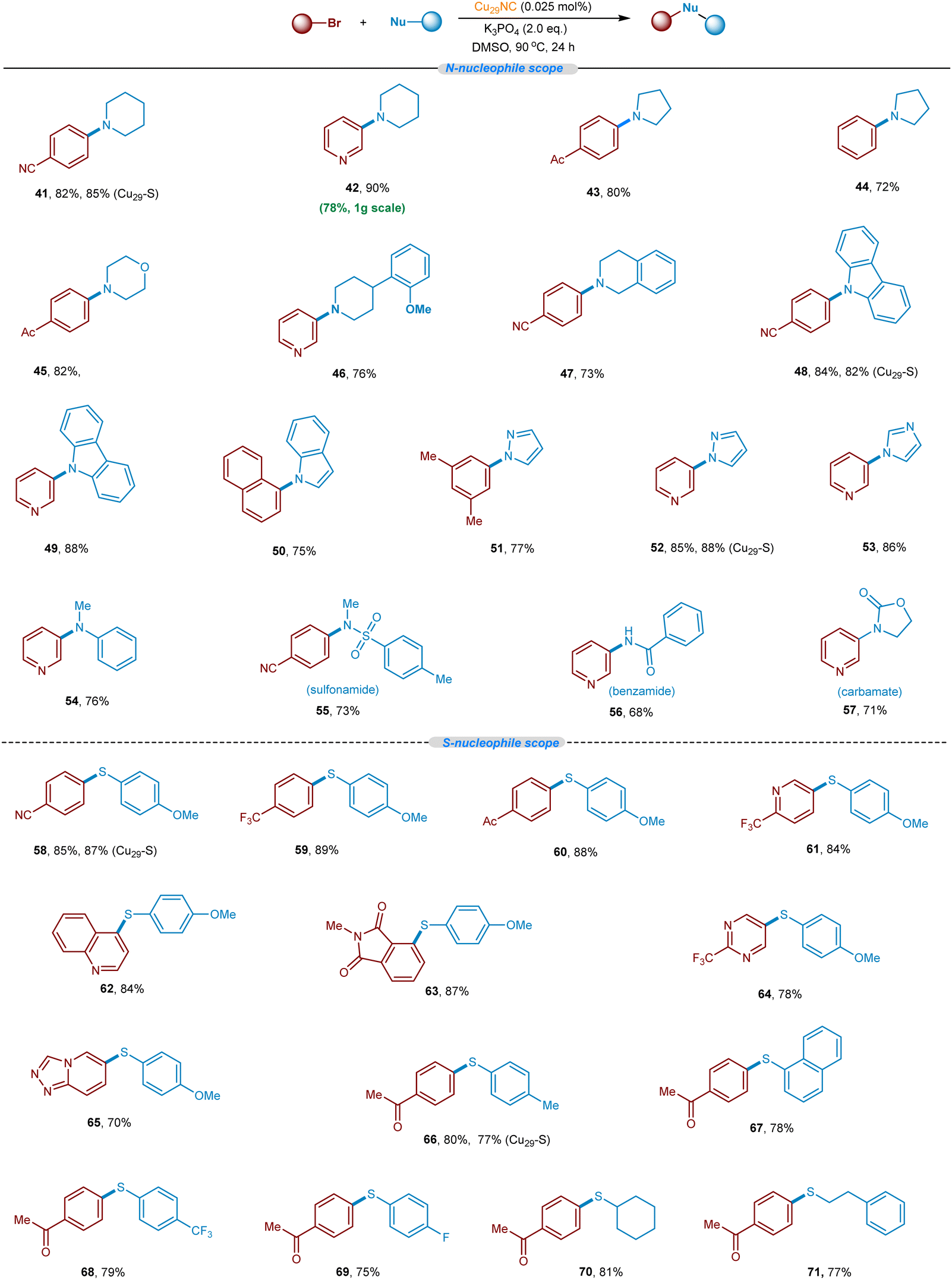 | ||
| Fig. 9 Substrate scope of N-nucleophiles and S-nucleophiles with aryl halides. Standard reaction conditions. Yield of isolated product after purification by column chromatography on a silica gel. (Cu29-S) indicates heterogeneous Cu29-Celite S. | ||
To further highlight the synthetic utility and generality of this new Cu29NC catalytic system, late-stage functionalization of structurally complex organic molecules or drug-like molecules via C–O, C–N, and C–S bond-forming reactions was demonstrated. As summarized in Fig. 10 natural products containing O-nucleophiles such as estrone, tocopherol, are selectively coupled with aryl(hetero)-bromides without affecting other functionalities. The C–O, C–N, & C–S couplings was also accomplished on 1.0 to 2.0 mmole scale (72–82, 65–85% yields). Fenofibrate was reacted with p-cresol, piperidine, and pyrazole, and the corresponding coupled products isolated in very good yield (75 and 77–78, 65–85% yields). Importantly, N-heterocylce containing desloratadine is smoothly coupled with 3-bromo-pyridine to afford the C–N coupled product (79, 74% yield). Furthermore, S-nucleophile-containing drugs/complex structures can be late-stage functionalized with aryl bromides, delivering the corresponding products in good to excellent yields (80–82). Collectively, the current Cu29NC catalysis is viable for the efficient C-heteroatom bond-forming reactions of pharmaceutical molecules bearing multiple functional groups and is thus attractive for pharmaceuticals synthesis and late-stage drug modification or functionalization.
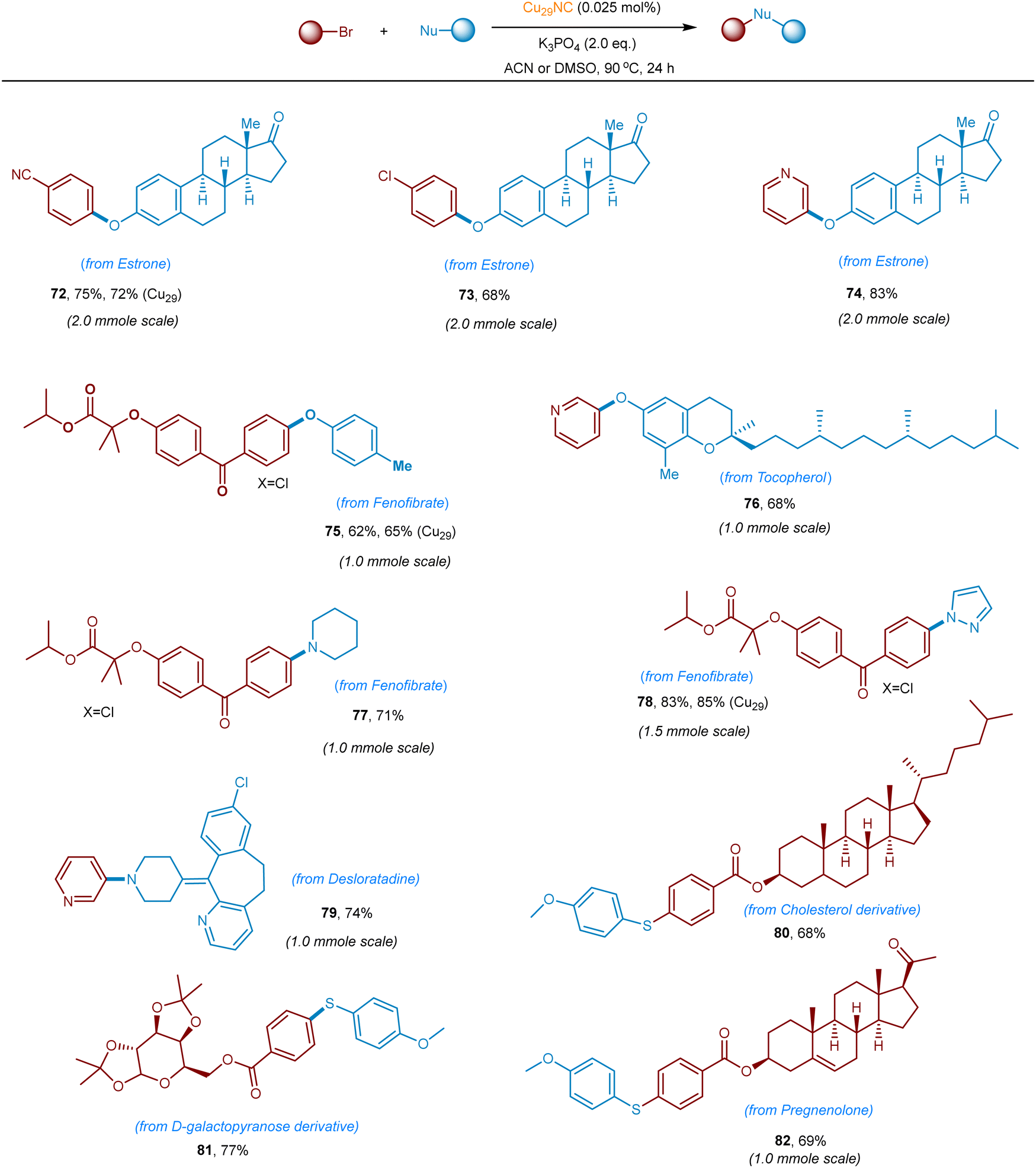 | ||
| Fig. 10 Late-stage functionalization of complex organic and drug molecules. Standard reaction conditions. Yield of isolated product after purification by column chromatography on a silica gel. (Cu29-S) indicates heterogeneous Cu29-Celite S. | ||
Conclusions
In summary, a core–shell nanocluster, [Cu29(StBu)13Cl5(PPh3)4H10]tBuSO3 with a unique shell structure has been synthesized. The shell, [Cu16(StBu)13Cl5(PPh3)4], is composed of three different types of motifs; motif 1 (Cu4S4Cl2P), motif 2 (Cu4S4Cl2P), and motif 3 (Cu4S5Cl1P). The cluster has been utilized as a versatile catalyst in various C-heteroatom coupling reactions of structurally diverse aryl(hetero) halides with a broad range of nucleophiles (alcohols, amines, sulphur, and amides), including, late-stage functionalization of complex organic molecules or pharmaceuticals bearing multiple functional groups. The DFT-based mechanistic study along with control experiments provided insight into the reaction mechanism. Our calculations suggest that the active sites of motif 2 show better catalytic activity than motifs 1 and 3. Further studies showed that the participation of multiple neighboring active sites at the different stages of the reactions play a crucial role for catalytic performance of the Cu29NC. The involvement of multiple active sites of Cu29NC substantially lowered the energy barrier of oxidative addition of aryl halide which was found to be the rate-limiting step. Furthermore, the supported Cu29NC system was shown to exhibit recyclability without significant loss in the catalytic activity. We anticipate that the new insights gained from this study will be instrumental in the design, synthesis, and applications of more effective nanocluster catalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by King Abdullah University of Science and Technology (KAUST).Notes and references
- N. Schneider, D. M. Lowe, R. A. Sayle, M. A. Tarselli and G. A. Landrum, J. Med. Chem., 2016, 59, 4385–4402 CrossRef CAS PubMed.
- I. Ghosh, N. Shlapakov, T. A. Karl, J. Düker, M. Nikitin, J. V. Burykina, V. P. Ananikov and B. König, Nature, 2023, 619, 87–93 CAS.
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- G. Evano, J. Wang and A. Nitelet, Org. Chem. Front., 2017, 4, 2480–2499 RSC.
- T. Qin, B. Liu, K. Zhu, Z. Luo, Y. Huang, C. Pan and L. Wang, TrAC, Trends Anal. Chem., 2018, 102, 259–271 CrossRef CAS.
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- C. H. Burgos, T. E. Barder, X. Huang and S. L. Buchwald, Angew. Chem., Int. Ed., 2006, 45, 4321–4326 CrossRef CAS PubMed.
- G. Mann, C. Incarvito, A. L. Rheingold and J. F. Hartwig, J. Am. Chem. Soc., 1999, 121, 3224–3225 CrossRef CAS.
- S. D. McCann, E. C. Reichert, P. L. Arrechea and S. L. Buchwald, J. Am. Chem. Soc., 2020, 142, 15027–15037 CrossRef CAS PubMed.
- J. D. Laffoon, V. S. Chan, M. G. Fickes, B. Kotecki, A. R. Ickes, J. Henle, J. G. Napolitano, T. S. Franczyk, T. B. Dunn, D. M. Barnes, A. R. Haight, R. F. Henry and S. Shekhar, ACS Catal., 2019, 9, 11691–11708 CrossRef CAS.
- F. Ullmann and P. Sponagel, Ber. Dtsch. Chem. Ges., 1905, 38, 2211–2212 CrossRef CAS.
- G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054–3131 CrossRef CAS PubMed.
- F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954–6971 CrossRef CAS PubMed.
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2010, 1, 13–31 RSC.
- S.-T. Kim, M. J. Strauss, A. Cabré and S. L. Buchwald, J. Am. Chem. Soc., 2023, 145, 6966–6975 CrossRef CAS PubMed.
- R. Ray and J. F. Hartwig, Angew. Chem., Int. Ed., 2021, 60, 8203–8211 CrossRef CAS PubMed.
- S. Bhunia, G. G. Pawar, S. V. Kumar, Y. Jiang and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 16136–16179 CrossRef CAS PubMed.
- Z. Chen, Y. Jiang, L. Zhang, Y. Guo and D. Ma, J. Am. Chem. Soc., 2019, 141, 3541–3549 CrossRef CAS PubMed.
- S. Bhunia, S. De and D. Ma, Org. Lett., 2022, 24, 1253–1257 CrossRef CAS PubMed.
- Q. Yang, Y. Zhao and D. Ma, Org. Process Res. Dev., 2022, 26, 1690–1750 CrossRef CAS.
- C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC.
- S. Daly, M. F. Haddow, A. G. Orpen, G. T. A. Rolls, D. F. Wass and R. L. Wingad, Organometallics, 2008, 27, 3196–3202 CrossRef CAS.
- H.-J. Cristau, P. P. Cellier, S. Hamada, J.-F. Spindler and M. Taillefer, Org. Lett., 2004, 6, 913–916 CrossRef CAS PubMed.
- N. Xia and M. Taillefer, Chem. – Eur. J., 2008, 14, 6037–6039 CrossRef CAS PubMed.
- A. Shafir, P. A. Lichtor and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 3490–3491 CrossRef CAS PubMed.
- K. L. Jones, A. Porzelle, A. Hall, M. D. Woodrow and N. C. O. Tomkinson, Org. Lett., 2008, 10, 797–800 CrossRef CAS PubMed.
- G. V. Smith and F. Notheisz, in Heterogeneous Catalysis in Organic Chemistry, ed. G. V. Smith and F. Notheisz, Academic Press, San Diego, 1999, pp. 1–28 DOI:10.1016/B978-012651645-6/50001-9.
- B. M. Trost, Transition Met. Org. Synth., 2004, 2–14, DOI:10.1002/9783527619405.ch1a.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88–119 CrossRef CAS PubMed.
- L. Rout, S. Jammi and T. Punniyamurthy, Org. Lett., 2007, 9, 3397–3399 CrossRef CAS PubMed.
- Y. S. Panova, A. S. Kashin, M. G. Vorobev, E. S. Degtyareva and V. P. Ananikov, ACS Catal., 2016, 6, 3637–3643 CrossRef CAS.
- S. Jammi, S. Sakthivel, L. Rout, T. Mukherjee, S. Mandal, R. Mitra, P. Saha and T. Punniyamurthy, J. Org. Chem., 2009, 74, 1971–1976 CrossRef CAS PubMed.
- M. B. Gawande, A. Goswami, F.-X. Felpin, T. Asefa, X. Huang, R. Silva, X. Zou, R. Zboril and R. S. Varma, Chem. Rev., 2016, 116, 3722–3811 CrossRef CAS PubMed.
- J. Oliver-Messeguer, L. Liu, S. García-García, C. Canós-Giménez, I. Domínguez, R. Gavara, A. Doménech-Carbó, P. Concepción, A. Leyva-Pérez and A. Corma, J. Am. Chem. Soc., 2015, 137, 3894–3900 CrossRef PubMed.
- B. Mondal, K. Basu, R. Jana, P. Mondal, B. Hansda, A. Datta and A. Banerjee, ACS Appl. Nano Mater., 2022, 5, 7932–7943 CrossRef CAS.
- L. L. Chng, N. Erathodiyil and J. Y. Ying, Acc. Chem. Res., 2013, 46, 1825–1837 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- G. Li, H. Abroshan, C. Liu, S. Zhuo, Z. Li, Y. Xie, H. J. Kim, N. L. Rosi and R. Jin, ACS Nano, 2016, 10, 7998–8005 CrossRef CAS PubMed.
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2021, 121, 567–648 CrossRef CAS PubMed.
- S. Wang, L. Tang, B. Cai, Z. Yin, Y. Li, L. Xiong, X. Kang, J. Xuan, Y. Pei and M. Zhu, J. Am. Chem. Soc., 2022, 144, 3787–3792 CrossRef CAS PubMed.
- W. Zhu, S. Zhang, W. Fan, Y. Yang, H. Zhao, W. Fei, H. Bi, J. He, M.-B. Li and Z. Wu, Precis. Chem., 2023, 1, 175–182 CrossRef CAS.
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS PubMed.
- S. Lee, M. S. Bootharaju, G. Deng, S. Malola, W. Baek, H. Häkkinen, N. Zheng and T. Hyeon, J. Am. Chem. Soc., 2020, 142, 13974–13981 CrossRef CAS PubMed.
- C. Sun, N. Mammen, S. Kaappa, P. Yuan, G. Deng, C. Zhao, J. Yan, S. Malola, K. Honkala, H. Häkkinen, B. K. Teo and N. Zheng, ACS Nano, 2019, 13, 5975–5986 CrossRef CAS PubMed.
- A. W. Cook, Z. R. Jones, G. Wu, S. L. Scott and T. W. Hayton, J. Am. Chem. Soc., 2018, 140, 394–400 CrossRef CAS PubMed.
- C. Zhang, Z. Wang, W.-D. Si, L. Wang, J.-M. Dou, Z.-Y. Gao, C.-H. Tung and D. Sun, ACS Nano, 2022, 16, 9598–9607 CrossRef CAS PubMed.
- A. Sagadevan, A. Ghosh, P. Maity, O. F. Mohammed, O. M. Bakr and M. Rueping, J. Am. Chem. Soc., 2022, 144, 12052–12061 CrossRef CAS PubMed.
- S. Nematulloev, A. Sagadevan, B. Alamer, A. Shkurenko, R. Huang, J. Yin, C. Dong, P. Yuan, K. E. Yorov, A. A. Karluk, W. J. Mir, B. E. Hasanov, M. Nejib Hedhili, N. M. Halappa, M. Eddaoudi, O. F. Mohammed, M. Rueping and O. M. Bakr, Angew. Chem., Int. Ed., 2023, 62, e202303572 CrossRef CAS PubMed.
- C. Dong, R.-W. Huang, A. Sagadevan, P. Yuan, L. Gutiérrez-Arzaluz, A. Ghosh, S. Nematulloev, B. Alamer, O. F. Mohammed, I. Hussain, M. Rueping and O. M. Bakr, Angew. Chem., Int. Ed., 2023, 62, e202307140 CrossRef CAS PubMed.
- A. Sagadevan, A. Ghosh, P. Maity, O. F. Mohammed, O. M. Bakr and M. Rueping, J. Am. Chem. Soc., 2022, 144, 12052–12061 CrossRef CAS PubMed.
- A. Ghosh, R.-W. Huang, B. Alamer, E. Abou-Hamad, M. N. Hedhili, O. F. Mohammed and O. M. Bakr, ACS Mater. Lett., 2019, 1, 297–302 CrossRef CAS.
- C. Dong, R.-W. Huang, C. Chen, J. Chen, S. Nematulloev, X. Guo, A. Ghosh, B. Alamer, M. N. Hedhili, T. T. Isimjan, Y. Han, O. F. Mohammed and O. M. Bakr, J. Am. Chem. Soc., 2021, 143, 11026–11035 CrossRef CAS PubMed.
- Y. Bao, X. Wu, B. Yin, X. Kang, Z. Lin, H. Deng, H. Yu, S. Jin, S. Chen and M. Zhu, Chem. Sci., 2022, 13, 14357–14365 RSC.
- S. Biswas, S. Hossian, T. Kosaka, J. Sakai, D. Arima, Y. Niihori, M. Mitsui, D.-E. Jiang, S. Das, S. Wang and Y. Negishi, Chem. Commun., 2023, 59, 9336–9339 RSC.
- J. O. Tellechea, C. Lima, D. Choi, D. Salazar, M. Samiei, A. Keoseyan, E. Vinogradova, F. Yan, D. Wherritt, M. M. Alvarez and G. Baghdasarian, AAAFM Energy Mater., 2020, 1, 16–26 CrossRef.
- S. Wang, Z. Wu, S. Dai and D.-E. Jiang, Angew. Chem., Int. Ed., 2021, 60, 12289–12292 CrossRef CAS PubMed.
- R.-W. Huang, J. Yin, C. Dong, A. Ghosh, M. J. Alhilaly, X. Dong, M. N. Hedhili, E. Abou-Hamad, B. Alamer, S. Nematulloev, Y. Han, O. F. Mohammed and O. M. Bakr, J. Am. Chem. Soc., 2020, 142, 8696–8705 CrossRef PubMed.
- H. Shen, Q. Wu, S. Malola, Y.-Z. Han, Z. Xu, R. Qin, X. Tang, Y.-B. Chen, B. K. Teo, H. Häkkinen and N. Zheng, J. Am. Chem. Soc., 2022, 144, 10844–10853 CrossRef CAS PubMed.
- G. Li, C. Zeng and R. Jin, J. Am. Chem. Soc., 2014, 136, 3673–3679 CrossRef CAS PubMed.
- G. Li and R. Jin, J. Am. Chem. Soc., 2014, 136, 11347–11354 CrossRef CAS PubMed.
- G. Li, D.-e Jiang, C. Liu, C. Yu and R. Jin, J. Catal., 2013, 306, 177–183 CrossRef CAS.
- K. Sakamoto, S. Masuda, S. Takano and T. Tsukuda, ACS Catal., 2023, 13, 3263–3271 CrossRef CAS.
- X. A. F. Cook, A. de Gombert, J. McKnight, L. R. E. Pantaine and M. C. Willis, Angew. Chem., Int. Ed., 2021, 60, 11068–11091 CrossRef CAS PubMed.
- M. N. Lavagnino, T. Liang and D. W. C. MacMillan, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 21058–21064 CrossRef CAS PubMed.
- A. C. Sather and T. A. Martinot, Org. Process Res. Dev., 2019, 23, 1725–1739 CrossRef CAS.
- R. Chinchilla and C. Nájera, Chem. Soc. Rev., 2011, 40, 5084–5121 RSC.
- A. Hossain, A. Bhattacharyya and O. Reiser, Science, 2019, 364, eaav9713 CrossRef PubMed.
- A. Sagadevan and K. C. Hwang, Adv. Synth. Catal., 2012, 354, 3421–3427 CrossRef CAS.
- D. Roy and Y. Uozumi, Adv. Synth. Catal., 2018, 360, 602–625 CrossRef CAS.
- A. Sagadevan, A. Charitou, F. Wang, M. Ivanova, M. Vuagnat and M. F. Greaney, Chem. Sci., 2020, 11, 4439–4443 RSC.
- A. Deb, S. Bag, R. Kancherla and D. Maiti, J. Am. Chem. Soc., 2014, 136, 13602–13605 CrossRef CAS PubMed.
- S.-S. Wang and G.-Y. Yang, Catal. Sci. Technol., 2016, 6, 2862–2876 RSC.
- S. Reddy Chidipudi, M. D. Wieczysty, I. Khan and H. W. Lam, Org. Lett., 2013, 15, 570–573 CrossRef CAS PubMed.
- P.-Z. Wang, Y. Gao, J. Chen, X.-D. Huan, W.-J. Xiao and J.-R. Chen, Nat. Commun., 2021, 12, 1815 CrossRef CAS PubMed.
- J. W. Tye, Z. Weng, A. M. Johns, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 9971–9983 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2292096. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4mh00098f |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2024 |