
The electronic structure of diatomic nickel oxide†
Nickolas A.
Joyner
,
João Gabriel Farias
Romeu
 ,
Brian
Kent
and
David A.
Dixon
*
,
Brian
Kent
and
David A.
Dixon
*
The University of Alabama, Department of Chemistry and Biochemistry, Shelby Hall, Tuscaloosa AL, 35487-0336, USA. E-mail: Dixondadixon@ua.edu
First published on 17th June 2024
Abstract
The nature of the Ni–O bond is relevant to catalytic and environmental applications. The vibrational frequency and electronic structure of NiO were calculated using CASSCF, icMRCI+Q, CCSD(T), and DFT. CASSCF predicted a quintet state (5Σ−) ground state for the equilibrium bond distance with a state crossing at 1.65 Å, where the triplet (3Σ−) state becomes of lower energy. These states arise from the 3d8(3F)4s2 (3F) and 3d9(2D)4s1 (3D) configurations of Ni. The icMRCI+Q method predicts a triplet (3Σ−) ground state and does not predict a state crossing with the quintet. This state has significant ionic character with the 2pz of O bonding with the 4s/3dz2 of the Ni to form a σ bond. The NiO frequency at the icMRCI+Q level of 835.0 cm−1 is in excellent agreement with experiment; the value of re is 1.5992 Å at this computational level. CCSD(T) predicts ωe = 888.80 cm−1 when extrapolated to the complete basis set limit. Frequencies predicted using CCSD(T) deviate from experiment consistent with the calculations showing large multireference character. A wide array of density functionals were benchmarked. Of the 43 functionals tested, the ones that gave the best prediction of the frequency are ωB97XD, CAM-B3LYP, and τ-HCTH with respective values of 831.8, 838.3, and 837.4 cm−1 respectively. The bond dissociation energy (BDE) of NiO is predicted to be 352.4 kJ mol−1 at the Feller–Peterson–Dixon (FPD) level in good agreement with one of the experimental values. The calculated BDEs at the DFT level are sensitive to the choice of functional and atomic asymptote. Sixteen functionals predicted the BDE within 20 kJ mol−1 of the FPD value.
Introduction
Solid oxide electrocatalysts (SOEC) have been a focus in recent years due to their applications in renewable energy conversion and storage,1 as SOEC catalysts can selectively reduce H2O and CO2 to achieve carbon neutral H2 generation.2–7 However, with such common Ni-based catalysts, CO2 reduction is challenging.8 To increase the efficiency and success of these catalysts, Ni supported on yttria stabilized zirconia (YSZ) is being developed as an SOEC catalyst that can reduce CO2 with minimal challenges. To fine tune the Ni cathode, a wide array of Ni alloys and NiO materials have been proposed, which require further exploration to understand what role these complexes play in CO2 reduction.9–11 It has been established that Ni clusters on YSZ can interact with the oxygens leading to the formation of Ni–O bonds in the solid state. The current goal is to provide insights into the nature of the Ni–O bond by examining diatomic NiO.There have been several computational and experimental studies of diatomic NiO. However, different values have been obtained for the vibrational frequencies as shown in Table 1. Ram and Bernath used Fourier transform infrared emission spectroscopy of the A 3Π–X 3Σ− band of NiO in the near-infrared region and determined the spectroscopic constants for the ground (X 3Σ−) and excited (A 3Π) states by analyzing the vibration–rotation bands.12 For the ground state, they chose the values of ωe and ωexe obtained by Srdanov and Harris in a laser-induced fluorescence (LIF) study including a rotational analysis of NiO.13 Green et al.14 measured the values of ωe and ωexe for NiO in an Ar matrix at 14 K and found similar values to the gas phase spectroscopic values.13 Anion photoelectron spectroscopy experiments on NiO− by Wu et al.15 provided the adiabatic electron affinity (AEA) and harmonic vibrational frequency for the ground state of NiO. The ionization energy (IE) and other thermochemical data of NiO were reported by Watson et al.16 in their high-temperature mass spectrometry experiments. Other experimental work on NiO includes the microwave spectrum of pure rotational transitions of the υ = 0 and υ = 1 vibrational states,17 photoelectron spectroscopy of NiO and NiO−,18 and chemiluminescent reactions of nickel with ozone and measured by mass spectrometry.19 Farber and Srivastava studied the vaporization of NiO(s) and the thermodynamics of reactions of nickel metal with oxygen vapor using effusion-mass spectrometric to obtain a NiO(g) dissociation energy.20 A lower limit to the dissociation energy of 250.9 ± 19.3 kJ mol−1 was reported by Fisher and Armentrout based on reactions of nickel ions with cyclopropane and ethylene oxide using guided ion beam mass spectrometry.21
| Method | r e (Å) | ω e (cm−1) | D 0 (kJ mol−1) | Ref. |
|---|---|---|---|---|
| Experiment | 1.62712 | 12 | ||
| Experiment | 800(50) | 15 | ||
| Experiment | 1.627 | 839.1 ± 0.5 (ωexe = 5.4 ± 0.5) | 13 | |
| Experiment | 837.61 ± 1.1 (ωexe = 5.92 ± 0.6) | 14 | ||
| Experiment | 349.4 ± 6.3 | 20 | ||
| Experiment | 373 ± 3366 ± 30 | 16 | ||
| GVB-CI | 1.60 | 841 | 376 | 22 |
| CISD | 1.608 | 906 | 149 | 23 |
| CISD+Q | 1.591 | 848 | 254 | 23 |
| MRCI | 1.70 | 700 | 24 | |
| MRCI+Q | 1.67 | 690 | 24 | |
| ICACPF | 1.626 | 850 | 362 | 26 |
| DFT/CAM-B3LYP | 1.647 | 838.3 | 214.2 (BDE(1)) | Current |
| DFT/τ-HCTH | 1.621 | 837.4 | 477.6 (BDE(1)) | Current |
| DFT/ωB97XD | 1.652 | 831.8 | 238.7 (BDE(1)) | Current |
| CCSD(T)/CBS | 1.6238 | 872.1 | 352.4 | Current |
| icMRCI+Q/CBS | 1.5992 | 835.0 | 387.7 | Current |
Computational studies of NiO range from density functional theory (DFT), Hartree–Fock, configuration interaction singles and doubles (CISD), and generalized valence bond (GVB) methods. Walch and Goddard used the GVB method to describe the Ni–O bonds in NiO, Ni2O, Ni3O, Ni4O, Ni4O+, Ni5O, and Ni5O+.22 Dolg et al. calculated some of the spectroscopic parameters and binding energies of NiO using ab initio methodologies (SCF, CISD and CISD+Q) with single-electron fit (SEFIT) pseudopotentials and an active space comprising the 4s and 3d Ni atomic orbitals and 2s and 2p O orbitals.23 The predicted values of the bond distance and ωe were highly dependent on the computational methodology. Similar behavior was found by Bauschlicher et al.24 who compared the results from SCF, CISD, CISD+Q, MRCI, and MRCI+Q calculations. These authors noted that the large differences between the CISD and CISD+Q results show that the reference space must be larger, so that multireference CI (MRCI) calculations are necessary. They also disagreed with the description of the bonding by Walch and Goddard III, who predicted that NiO has a covalent bond.19,21 DFT/B3LYP calculations of NiO have also been reported.25 Bauschlicher and Maitre26 have surveyed computational studies of all of the first-row transition metal oxides. They note that it is difficult to calculate the properties of NiO from single reference starting points due to localization of π* singly occupied orbitals on the Ni or O. Their best values were obtained at the CASSCF/ICACPF (internally contracted average coupled pair functional) level with an extended polarized double-ζ basis set on Ni and the aug-cc-pVTZ basis set on the O without the diffuse f.
Computational methods
The equilibrium geometry (re) of NiO was initially optimized using DFT with the hybrid B3LYP exchange correlation functional27–29 the correlation consistent aug-cc-PVDZ basis set for O,30,31 and the aug-cc-pVDZ-PP basis set with an effective core potential32 for Ni.33,34 The B3LYP geometries were used as starting points for calculations at the CCSD(T) level (coupled cluster theory with single and double excitations with perturbative triples).35–41 All coupled cluster calculations were performed with the R/UCCSD(T) approach, where the restricted open-shell Hartree–Fock calculation is performed followed by a relaxation of the spin constraint at the coupled cluster level. The CCSD(T) calculations utilized the third-order Douglas–Kroll–Hess Hamiltonian (DKH3), with the aug-cc-pVNZ-DK42 basis set for O and Ni. This combination of basis sets will be further denoted as aN-DK. Harmonic and anharmonic frequencies (ωe, ωexe) were obtained using a Dunham expansion at the CCSD(T) level.43The state-averaged complete active space self-consistent field (SA-CASSCF)44–48 approach was performed to account for non-dynamical correlation effects and describe the lowest spin-free states, ΛS. To account for the two quasi-degenerate 3Fg and 3Dg atomic states of Ni coupling with the O(3Pg) and to improve the convergence near the equilibrium bond distance, 36 ΛS singlet, 37 ΛS triplet, and 39 ΛS quintet states were optimized in the SA-CASSCF calculations. The atomic aug-cc-pVQZ-DK42 basis set for O and aug-cc-pwCVQZ-DK49 basis set for Ni were used, and this combination will be denoted as awQ-DK. These calculations were carried out in the highest Abelian point group available, C2v. Expectation values of Lz2 were calculated to ensure that both degenerate components of each Λ state were correctly accounted for. The active space of NiO includes 14 electrons in nine orbitals (4 × a1, 2 × b1, 2 × b2, 1 × a2 in C2v symmetry), which have dominant 2p of O and 4s and 3d of Ni.
Dynamic correlation effects were accounted for by using the internally contracted multireference configuration interaction (icMRCI) method taking the SA-CASSCF wavefunctions as a reference, as implemented in MOLPRO.50–52 The relaxed Davidson correction (+Q)53 was included as an estimate of missing quadruple excitations. Only the ground state (3Σ−) was optimized and a potential energy curve (PEC) was calculated around the bond distance but keeping the nine A2 ΛS triplet states as a reference. The same approach was done using second-order perturbation theory (CASPT2)54,55 in order to recover dynamical correlation effects; however, some points on the ground state PEC did not fit on the curve well enough to calculate the vibrational properties. The harmonic, and anharmonic frequencies (ωe, ωexe) were obtained using a Durham expansion43 in the same interval used in the UCCSD(T)/aD-DK calculation, but at the icMRCI+Q/awQ-DK level. Certain points did not fall on the curves and were not included in the calculation of the frequencies. Inclusion of these points resulted in a large sum of squares of residuals in the fitting procedure, a large deviation in the fitted minimum energy, and yielded a value of ωexe > 15 cm−1. For the icMRCI+Q calculation, even small errors in the energies used in the fit had a significant impact on the predicted ωexe for the ground state. Thus, only ωe was calculated from the fit for the icMRCI+Q. Spectroscopic properties produced by the CASSCF, icMRCI+Q, and CCSD(T) calculations were then extrapolated to the complete basis set limit (CBS) using awCnZ-DK (where n = D, T and Q) using the eqn (1) for the energies.
En = ECBS + A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp[−(n − 1)] + Bexp[−(n − 1)2] exp[−(n − 1)] + Bexp[−(n − 1)2] | (1) |
For the DFT benchmark study, the 43 selected functionals are given in Table 2 and consist of a mix of local spin density approximation (LSDA), general gradient approximations (GGA), meta GGA (mGGA), hybrid GGA (HGGA), and hybrid meta GGA (HmGGA) functionals. The optimization and frequency analysis conducted with the given functionals started with the B3LYP optimized geometry. The DFT calculations were done with the aug-cc-pVDZ basis set on O and the aug-cc-pVDZ-PP basis set on Ni (10 electron pseudopotential on Ni). The DFT calculations were performed at the unrestricted spin level.
| Method | Exchange | Correlation | Type |
|---|---|---|---|
| B1B9560 | Becke 96 | Becke 95 | HmGGA |
| B1LYP30,61 | Becke 96 | Lee–Yang–Parr | HGGA |
| B3LYP27,28 | Becke 93 | Lee–Yang–Parr | HGGA |
| B3P8628,65 | Becke 93 | Perdew 86 | HGGA |
| B3PW9128 | Becke 93 | Perdew–Wang 91 | HGGA |
| B97164 | Handy–Tozer's modified B97 | Handy–Tozer's modified B97 | HGGA |
| B97265 | Wilson–Bradley–Tozer's modified B97 | Wilson–Bradley–Tozer's modified B97 | HGGA |
| B9866 | Becke 98 | Becke 98 | HGGA |
| BP8662,67 | Becke 88 | Perdew 86 | GGA |
| BMK68 | Boese–Martin | Perdew 86 | HGGA |
| CAM-B3LYP69 | Becke 88 | Lee–Yang–Par | HGGA |
| HSE0670–74 | Heyd–Scuseria–Ernzerhof | Perdew–Burke–Ernzerhof | HGGA |
| HS0670–74 | Heyd–Scuseria–Ernzerhof | Perdew–Burke–Ernzerhof | HGGA |
| HSE0370–74 | Heyd–Scuseria–Ernzerhof | Perdew–Burke–Ernzerhof | HGGA |
| LC-ωPBE75 | Perdew–Burke–Ernzerhof | Perdew–Burke–Ernzerhof | HGGA |
| M052X76 | Minnesota 05 | Minnesota 05 | HmGGA |
| M0577 | Minnesota 05 | Minnesota 05 | HmGGA |
| M062X78 | Minnesota 062x | Minnesota 062X | HmGGA |
| M0678 | Minnesota 06 | Minnesota 06 | HmGGA |
| M06HF79,80 | Minnesota 06HF | Minnesota 06HF | HmGGA |
| M08HX81 | Minnesota 08HX | Minnesota 08HX | HmGGA |
| M1182 | Minnesota 11 | Minnesota 11 | HmGGA |
| MN12SX83 | Minnesota 12SX | Minnesota 12SX′ | HmGGA |
| MN1584 | Minnesota 15 | Minnesota 15 | HmGGA |
| mPW1LYP27,85 | Barone's modified PW91 | Lee–Yang–Par | HGGA |
| mPW1PBE85–87 | Barone's modified PW91 | Perdew–Burke–Ernzerhof | HGGA |
| mPW1PW9163,85 | Barone's modified PW91 | Perdew–Wang 91 | HGGA |
| mPW3PBE85–87 | Barone's modified PW91 | Perdew–Burke–Ernzerhof | HGGA |
| N12SX83 | N12SX | N12SX′ | HmGGA |
| O3LYP27,88 | Handy's OPTX | Lee–Yang–Par | HGGA |
| PBE1PBE86,89 | Perdew–Burke–Ernzerhof | Perdew–Burke–Ernzerhof | HGGA |
| PBEh1PBE86,90 | 98 Revised PBE | Perdew–Burke–Ernzerhof | HGGA |
| PBE86 | Perdew–Burke–Ernzerhof | Perdew–Burke–Ernzerhof | GGA |
| PBE089 | Perdew–Burke–Ernzerhof | Perdew–Burke–Ernzerhof | HGGA |
| PW9163,91 | Perdew–Wang 1991 | Perdew–Burke–Ernzerhof | GGA |
| SOGGA11X92 | SOGGA11X | SOGGA11X | HmGGA |
| SVWN593,94 | Slater | VWN Functional V | LSDA |
| τ-HCTH95 | τ-Dependent of HCTH | τ-Dependent of HCTH | mGGA |
| TPSSh96 | Tao–Perdew–Staroverov–Scuseria | Tao–Perdew–Staroverov–Scuseria | HmGGA |
| ωB9797 | Becke 97 | Becke 97 | HGGA |
| ωB97X97 | Becke 97 | Becke 97 | HGGA |
| ωB97XD98,99 | Becke 97 | Becke 97 | HGGA |
| X3LYP27,100 | Becke 97 | Becke 97 | HGGA |
A bonding analysis of NiO was performed based on the natural population analysis (NPA) results based on the Natural Bond Orbitals (NBOs)101,102 using NBO7103,104 with the MOLPRO program package at the awQ-DK level.
The DFT calculations were done using the Gaussian 16 program.105 All CCSD(T), CASSCF, CASPT2, and MRCI+Q calculations were performed in the MOLPRO 202150–52 computational chemistry program on computers at The University of Alabama, and the Alabama Supercomputing Authority (Dense Memory Cluster).
Results and discussion
Vibrational frequency
In the multiconfiguration and multireference calculations, the two lowest atomic states arising from the 3d8(3F)4s2 (3F) and 3d9(2D)4s (3D) configurations of Ni were correlated because their associated spin–orbit states are very close in energy. This resulted in a high density of molecular electronic states around the minimum bond distance of the ground state up to 20000 cm−1 relative to the ground state for the SA-CASSCF calculations. A quintet state (5Σ−) was predicted as the ground state, followed by a triplet (3Σ−) state close in energy, with bond distances of about 1.77 Å and 1.69 Å, respectively. Fig. 1 shows this behavior at the SA-CASSCF level.
 | ||
| Fig. 1 Potential energy curves of the low-lying ΛS singlet, triplet, and quintet SA-CASSCF states. | ||
The inclusion of dynamic correlation effects by icMRCI+Q dramatically changed the description of electronic molecular states of NiO. Spin–orbit effects were also included to check whether possible avoided crossings could change the description of the potential energy curves. However, this did not happen for the Ω states resulting from the ΛS 5Σ− (Ω = 0, 1, 2) and 3Σ− (Ω = 0, 1) states. The spin–orbit splitting was 107 cm−1 for the ground state 3Σ− (Ω = 0) and the first excited state 3Σ− (Ω = 1). Table 3 presents the composition of the spin–orbit states that correlate to the 5Σ− and 3Σ− states. Fig. 2 shows the spin–orbit icMRCI+Q/awQ-DK potential energy curves up to 8000 cm−1. The ground state at the icMRCI+Q level was predicted to be the 3Σ−0 and lies more than 4000 cm−1 below the 5Σ−2 state; thus, there is no crossing between the triplet and quintet states as was found at the SA-CASSCF level. There is no significant change in the shape of the PECs and the energy splitting due to the spin–orbit effects is small for NiO, so the following analysis will focus on the ΛS states. Table 4 presents the spectroscopic parameters calculated for the ground state (3Σ−) of NiO using the awT-DK and awQ-DK basis sets, and the extrapolation to the complete basis set (CBS) limit. The basis set has very little effect on the bond distance, which shows a small shortening as more functions are included in the basis set, as well as a small decrease in the ωe and ωexe. The inclusion of spin–orbit coupling lowers the energy of the ground state by 111 cm−1 relative to the value without spin orbit and has no significant impact on the bond distance. In the spin–orbit calculation, a small change in the shape of the PECs around the bond distance decreases the ωe and ωexe for the Ω states associated with the ΛS 3Σ− state.
| State | Ω | ΔE (eV) | ΛS composition |
|---|---|---|---|
| 3Σ−0 | 0 | 0.000 | 98% 3Σ− + 2% 3Π |
| 3Σ−1 | 1 | 0.006 | 99% 3Σ− + 1% 3Π |
| 5Σ−2 | 2 | 0.560 | 95% 5Σ− + 4% 3Π |
| 5Σ−1 | 1 | 0.560 | 99% 5Σ− + 1% 3Π |
| 5Σ−0 | 0 | 0.560 | 100% 5Σ− |
 | ||
| Fig. 2 Potential energy curves of the low-lying Ω states using the spin–orbit icMRCI+Q energies. | ||
| Method | Basis set | r e/Å | ω e/cm−1 | ω e x e | T 1 |
|---|---|---|---|---|---|
| a ω e x e was not calculated. See Computational section for discussion. | |||||
| icMRCI+Q | awT-DK | 1.6023 | 838.5 | ||
| icMRCI+Q | awQ-DK | 1.6003 | 836.1 | ||
| icMRCI+Q | CBS | 1.5992 | 835.0 | ||
| CCSD(T) | aD-DK | 1.63628 | 865.6 | 1.87 | 0.129 |
| CCSD(T) | aT-DK | 1.62481 | 889.08 | 2.08 | 0.126 |
| CCSD(T) | aQ-DK | 1.62276 | 889.40 | 1.93 | 0.126 |
| CCSD(T) | CBS | 1.62177 | 888.80 | 1.82 | |
| CCSD(T) | awT-DK | 1.62496 | 876.09 | 2.29 | 0.1029 |
| CCSD(T) | awQ-DK | 1.62425 | 873.54 | 1.94 | 0.1029 |
| CCSD(T) | CBS | 1.62384 | 872.07 | 1.74 | |
Coupled cluster theory at the CCSD(T) level was used as the benchmark of the single reference methods as shown in Table 4. There is a modest basis set effect from aD to aT with the overall bond distance decreasing by 0.01 Å, and the ωe increasing by 20 cm−1. However, there is little effect of increasing the basis set from aT to aQ on the bond distance and frequency. When correlating the outer core electrons in the system, the bond distance is very similar to the calculations correlating only the valence electrons. The effect on the ωe is modest with the frequencies being roughly 10 cm−1 less than valence only calculations. There are large T1 values for the CCSD(T) calculations showing substantial multi-reference character which arises from the 3d8(3F)4s2 (3F) and 3d9(2D)4s (3D) configurations as described above. The quintet state is 149 kJ mol−1 above the triplet at the awcTZ-DK level of theory.
The potential energy curve calculated at the CCSD(T)/awCQ-DK level is given in Fig. 3. Potential energy curves calculated at the CCSD(T) level were obtained at intervals of 0.01Å as the frequency of NiO is highly sensitive to Re, and to appropriately model the 5Σ− (Ω = 0, 1, 2) and 3Σ− (Ω = 0) state crossing that occurs at approximately at 1.62 Å in the multi-reference calculations. When compared to the results from the multireference and multiconfigurational calculations, icMRCI+Q and CASSCF, respectively, CCSD(T) consistently overestimates ωe. Fig. 3 shows an overlay of the potential energy curves from the CCSD(T), MRCI, MRCI+Q, and CASSCF calculations. Therefore, as a benchmark, methods other than CCSD(T) should be used due to the highly multireference character of the Ni–O bond.
 | ||
| Fig. 3 Potential energy curves of the ground 3Σ− state calculated at the CCSD(T)/awQ-DK level (light blue), the potential energy curve of the 3Σ− state calculated at the icMRCI/awQ-DK level (black), the potential energy curve of the 3Σ− state calculated at the icMRCI+Q/awQ-DK level (green), the potential energy curve of the 5Σ− state calculated at the CASSCF/awQ-DK level (red), and the potential energy curve of the 3Σ− state calculated at the CASSCF/awQ-DK level (purple). | ||
Thermochemistry
The bond dissociation energies (BDEs) of NiO calculated at CCSD(T)/awn-DK and icMRCI+Q/awn-DK levels are given in Table 5. Spin–orbit (SO) constants were calculated at the icMRCI+Q/awn-DK level and these numbers were used to estimate the final BDE values in both cases. Due to the excessive number of singlet, triplet, and quintet electronic molecular states correlating to the two lowest dissociation channels, Ni(3Fg) + O(3Pg) and Ni(3Dg) + O(3Pg), the icMRCI+Q did not converge properly at the dissociation limit. Therefore, only the NiO triplet states (9 A1, 9 B1, 9 B2, 9 A2) were optimized in the SA-CASSCF, and these states were taken as a reference for the ground state optimization in the icMRCI+Q. The Lz2 numbers confirmed the correct convergence in the asymptotic limit at r(Ni–O) = 6 Å.| Calculation | awT-DK | awQ-DK | CBS (awn-DK) |
|---|---|---|---|
| CCSD(T) Quintet | 238.0 | 235.6 | 234.2 |
| icMRCI+Q | 388.8 | 398.7 | 404.3 |
| SO constant (Ni) | 12.0 | 12.0 | 12.0 |
| SO constant (O) | 0.9 | 1.0 | 1.0 |
| SO constant (NiO 3Σ−) | 1.3 | 1.3 | 1.3 |
| SO constant (NiO 5Σ−) | 7.2 | 3.3 | 1.0 |
| ZPE correction 3Σ− | 5.0 | 5.0 | 5.0 |
| ZPE correction 5Σ− | 3.2 | 3.3 | 3.3 |
| FPD/CCSD(T) 5Σ− | 229.1 | 222.6 | 218.9 |
| FPD/CCSD(T) 3Σ− | 362.6 | 356.1 | 352.4 |
| Final icMRCI+Q 3Σ− | 372.2 | 382.1 | 387.7 |
| Watson et al.16 | 373.2 | ||
| Farber and Srivastava20 | 349.4 ± 6.3 |
For the CCSD(T) calculations, the BDE was calculated relative to separate atomic energies, instead of using the supermolecule approximation used for icMRCI+Q. Due to the multi-reference nature of the triplet ground state, the BDE at the FPD/CCSD(T) level was first calculated relative to the excited quintet state where the T1 values are much smaller showing less multi-reference character. The BDE of the ground triplet state was then obtained by using the icMRCI+Q adiabatic energy difference between the triplet ground state and the excited quintet state.
We have previously found that the CCSD(T) energies can be improved by using orbitals from density functional theory.106–108 Thus, another approach to the prediction of the BDE of NiO is to use orbitals generated at the DFT level using the PW91 functional for the triplet state. This approach led to reasonable values of the T1 diagnostic109 on the order of 0.015. The BDE for the triplet state was substantially improved over that using the HF orbitals for the CCSD calculations and is within 10 kJ mol−1 of the value from the quintet state.
The calculated FPD/CCSD(T) and icMRCI+Q BDEs of the triplet ground state of NiO are not in particularly good agreement with each other, differing by 37 kJ mol−1. The experimental results Farber and Srivastava20 and Watson et al.16 differ by 24 kJ mol−1 so they cannot be used to help in distinguishing between the two calculated values. The FPD value is consistent with the lower value of 349.4 ± 6.3 kJ mol−120 within the error bars. The icMRCI+Q value is 14 kJ mol−1 greater than the larger experimental value of 372.2 kJ mol−1.16
Electronic structure
The orbital populations and the charges (q) from NBO analysis for NiO are shown in Table 6. The NBO analysis in Table 7 shows that NiO has significant ionic interactions with a highly polarized σ bond that is predominantly composed of the 2pz on O and the 4s on Ni. Two α unpaired electrons associated with the dπ non-bonding orbitals on nickel result in the 3Σ− ground state. Fig. 4 also shows the SA-CASSCF natural orbitals at the awQ-DK level associated with the nine electrons correlated in the 14 orbitals of the active space. The icMRCI+Q wavefunction for the ground state indicates a strong multireference character. The corresponding major contributions are given in eqn (2)| 0.71 |1σ21δ42σ21π42π2〉 − 0.39 |1σ21δ42σ13σ11π42π2〉 | (2) |
| Property | NiO (3Σ−) |
|---|---|
| q(Ni) | 1.405 |
| q(O) | −1.405 |
| 4s (4sα/4sβ) | 0.49 (0.24/0.24) |
| 3d (3dα/3dβ) | 8.07 (4.96/3.11) |
| O 2p (2pα/2pβ) | 5.39 (2.76/2.63) |
| NBO | Symmetry | occ | Ni%q | Ni%s | Ni%d | O%q | O%s | O%p |
|---|---|---|---|---|---|---|---|---|
| Ni–O | σ | 2.000 | 23.7 | 96.3 | 3.3 | 76.3 | 6.1 | 93.6 |
| LP(Ni) | δ | 4.000 | 100 | 0.0 | 100 | |||
| LP(Ni) | σ | 1.998 | 100 | 3.2 | 96.8 | |||
| LP(Ni) α | π | 1.994 | 100 | 0.0 | 99.9 | |||
| LP(O) | σ | 1.998 | 100 | 94.1 | 5.9 | |||
| LP(O) | π | 3.982 | 100 | 0.0 | 100 |
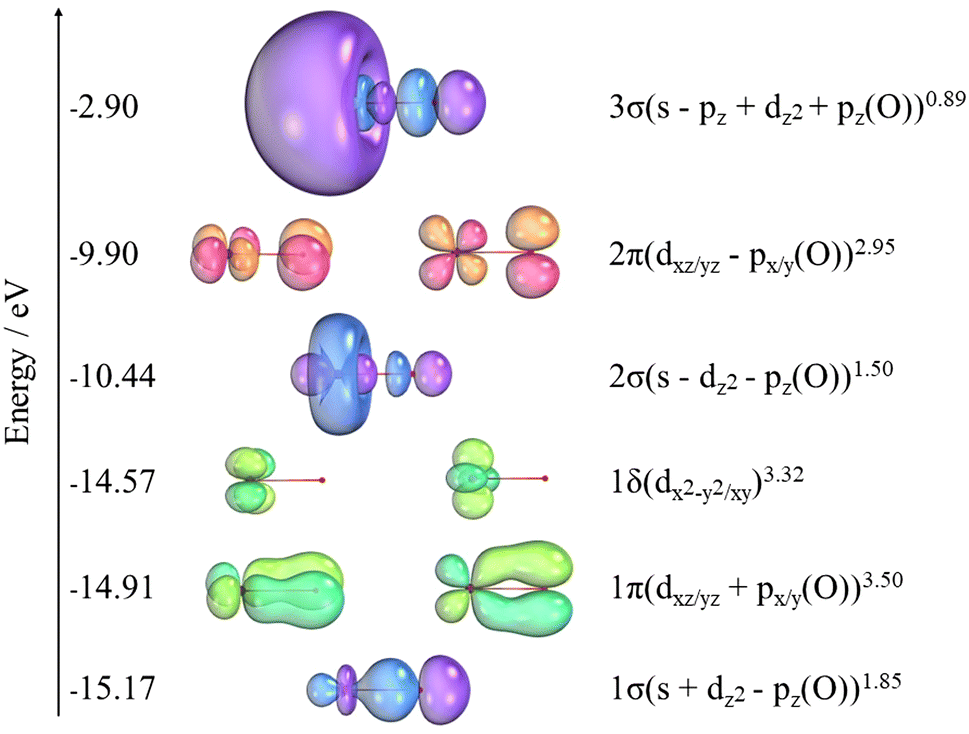 | ||
| Fig. 4 Natural orbitals (SA-CASSCF/awQ-DK) energies and composition for the 14 electrons correlated in the active space. Unlabeled orbitals are on Ni. Ni on the left and O on the right. 80% of the surfaces were accounted for. | ||
There are 10 additional configurations whose coefficients range from 0.10 to 0.20. The triplet state is comprised of two α unpaired electrons associated with the dπ non-bonding orbitals on Ni. Further NBO analysis shows that NiO has significant ionic interactions and a σ bond that is mostly a pz of oxygen combined with the 4s of nickel.
Benchmarking DFT functionals: frequency
Table 8 shows ωe and re calculated using 43 DFT functionals benchmarked against the best multireference value and experiment. Of the functionals tested, 3 functionals predicted ωe in reasonable agreement with experiment and the icMRCI+Q calculations. The three best functionals were CAM-B3LYP, τ-HCTH, and ωB97xd, which predicted ωe within 5 cm−1 of experiment and of the icMRCI+Q calculations. CAM-B3LYP is a long-range corrected form of B3LYP utilizing the Coulomb-attenuating method (CAM). CAM-B3LYP overestimates the Ni–O bond length by approximately 0.02 Å from the experimental value12 of 1.62712 Å and 0.04 Å from the icMRCI+Q value. The CAM-B3LYP result differs from experiment13 by 7.3 cm−1 and from icMRCI+Q calculations by 3.3 cm−1. The ωB97XD functional which also includes long range interactions differs from experiment by 0.8 cm−1 and by 3.2 cm−1 from the best calculated value. The τ-HCTH meta GGA functional differs by 1.7 cm−1 from experiment and 2.4 cm−1 from icMRCI+Q. In comparison to the best functionals, commonly used functionals such as B3LYP, PBE, and M06 predict ωe values that are not in agreement with experiment. The B3LYP value is ∼ 50 cm−1 larger than experiment and similar to the CCSD(T) values. Furthermore, PBE and M06 are 70 cm−1 and 80 cm−1 larger than experiment, respectively.| Functional | r e (Å) | ω e (cm−1) | ΔExpt (cm−1) | ΔicMRCI+Q (cm−1) |
|---|---|---|---|---|
| B1B95 | 1.605 | 900.8 | 61.7 | 65.8 |
| B1LYP | 1.612 | 892.2 | 53.1 | 57.2 |
| B3LYP | 1.611 | 888.5 | 49.4 | 53.5 |
| B3P86 | 1.601 | 908.8 | 69.7 | 73.8 |
| B3PW91 | 1.605 | 901.5 | 62.4 | 66.5 |
| B971 | 1.651 | 743.1 | 96.0 | 91.9 |
| B972 | 1.608 | 884.4 | 45.3 | 49.4 |
| B98 | 1.650 | 743.1 | 96.0 | 91.9 |
| BP86 | 1.616 | 866.6 | 27.5 | 31.6 |
| BMK | 1.603 | 945.6 | 106.5 | 110.6 |
| CAM-B3LYP | 1.647 | 838.3 | 0.8 | 3.3 |
| HSE06 | 1.602 | 910.4 | 71.3 | 75.4 |
| HS06 | 1.602 | 910.2 | 71.1 | 75.2 |
| HSE03 | 1.603 | 911.3 | 72.2 | 76.3 |
| LC-ωPBE | 1.590 | 959.2 | 120.1 | 124.2 |
| M052X | 1.621 | 911.3 | 72.2 | 76.3 |
| M05 | 1.623 | 887.0 | 47.9 | 52.0 |
| M062X | 1.625 | 875.6 | 36.5 | 40.6 |
| M06 | 1.599 | 913.4 | 74.3 | 78.4 |
| M06HF | 1.673 | 797.2 | 41.9 | 37.8 |
| M08HX | 1.632 | 884.0 | 44.9 | 49.0 |
| M11 | 1.618 | 919.8 | 80.7 | 84.8 |
| MN12SX | 1.595 | 938.3 | 99.2 | 103.3 |
| MN15 | 1.624 | 873.8 | 34.7 | 38.8 |
| mPW1LYP | 1.611 | 893.2 | 54.1 | 58.2 |
| mPW1PBE | 1.605 | 911.9 | 72.8 | 76.9 |
| mPW1PW91 | 1.602 | 910.5 | 71.4 | 75.5 |
| mPW3PBE | 1.603 | 903.9 | 64.8 | 68.9 |
| N12SX | 1.645 | 801.9 | 37.2 | 33.1 |
| O3LYP | 1.616 | 858.9 | 19.8 | 23.9 |
| PBE1PBE | 1.602 | 910.0 | 70.9 | 75.0 |
| PBEh1PBE | 1.602 | 912.0 | 72.9 | 77.0 |
| PBE | 1.617 | 862.4 | 23.3 | 27.4 |
| PPE0 | 1.600 | 917.0 | 77.9 | 82.0 |
| PW91 | 1.617 | 866.4 | 27.3 | 31.4 |
| SOGGA11X | 1.663 | 758.4 | 80.7 | 76.6 |
| SVWN5 | 1.586 | 925.9 | 86.8 | 90.9 |
| τ-HCTH | 1.621 | 837.4 | 1.7 | 2.4 |
| TPSSh | 1.611 | 889.3 | 50.2 | 54.3 |
| ωB97 | 1.596 | 947.6 | 108.5 | 112.6 |
| ωB97X | 1.644 | 853.9 | 14.8 | 18.9 |
| ωB97XD | 1.652 | 831.8 | 7.3 | 3.2 |
| X3LYP | 1.610 | 892.0 | 52.9 | 57.0 |
Benchmarking DFT functionals: Ni–O BDE
The Ni–O BDEs with different functionals are given in Table 9. We derive BDE(1) and BDE(2) from eqn (3) and (4) respectively,| NiO → 3Ni + 3O | (3) |
| NiO → 1Ni + 3O | (4) |
521.347 cm−1. The closed shell 1S state corresponding to the 3d10 configuration is at 14728.840 cm−1.
| Functional | BDE(1) | ΔBDE(1) | Ni config BDE(1)a | BDE(2) | ΔBDE(2) | Ni config BDE(2)a |
|---|---|---|---|---|---|---|
| a Ni atom configuration from the NBO analysis. | ||||||
| B1B95 | 347.5 | 4.9 | [Ar]s1d9 | 372.4 | −20.0 | [Ar]d10 |
| B1LYP | 334.9 | 17.5 | [Ar]s1d9 | 455.6 | −103.2 | [Ar]s1d9 |
| B3LYP | 367.0 | −14.6 | [Ar]s1d9 | 475.1 | −122.7 | [Ar]s1d9 |
| B3P86 | 383.7 | −31.3 | [Ar]s1d9 | 551.1 | −198.7 | [Ar]s1d9 |
| B3PW91 | 363.3 | −10.9 | [Ar]s1d9 | 500.5 | −148.1 | [Ar]d10 |
| B971 | 309.7 | 42.7 | [Ar]s2d8 | 457.0 | −104.6 | [Ar]s1d9 |
| B972 | 379.3 | −26.9 | [Ar]s1d9 | 466.2 | −113.8 | [Ar]s1d9 |
| B98 | 304.0 | 48.4 | [Ar]s1d9 | 376.9 | −24.5 | [Ar]d10 |
| BP86 | 463.0 | −110.6 | [Ar]s1d9 | 545.1 | −192.7 | [Ar]s1d9 |
| BMK | 389.5 | −37.1 | [Ar]s2d8 | 565.9 | −213.5 | [Ar]d10 |
| CAM-B3LYP | 214.2 | 138.2 | [Ar]s1d9 | 380.2 | −27.8 | [Ar]s1d9 |
| HSE06 | 191.3 | 161.1 | [Ar]s1d9 | 323.7 | 28.7 | [Ar]s1d9 |
| HS06 | 348.5 | 3.9 | [Ar]s1d9 | 480.0 | −128.5 | [Ar]s1d9 |
| HSE03 | 349.8 | 2.6 | [Ar]s1d9 | 482.6 | −130.2 | [Ar]s1d9 |
| LC-ωPBE | 368.2 | −15.8 | [Ar]s1d9 | 507.2 | −154.8 | [Ar]s1d9 |
| M052X | 307.2 | 45.2 | [Ar]s1d9 | 337.6 | 14.8 | [Ar]d10 |
| M05 | 374.0 | −21.6 | [Ar]s1d9 | 328.0 | 24.4 | [Ar]d10 |
| M062X | 262.4 | 90.0 | [Ar]s1d9 | 294.0 | 58.4 | [Ar]d10 |
| M06 | 364.6 | −12.2 | [Ar]s1d9 | 451.5 | −99.1 | [Ar]s1d9 |
| M06HF | 189.5 | 162.9 | [Ar]s1d9 | 307.4 | 45.0 | [Ar]d10 |
| M08HX | 291.6 | 60.8 | [Ar]s1d9 | 459.5 | −107.1 | [Ar]d10 |
| M11 | 326.1 | 26.3 | [Ar]s2d8 | 438.8 | −86.4 | [Ar]s1d9 |
| MN12SX | 251.9 | 100.5 | [Ar]s2d8 | 384.9 | −32.5 | [Ar]s2d8 |
| MN15 | 353.7 | −1.3 | [Ar]s1d9 | 288.5 | 63.9 | [Ar]d10 |
| mPW1LYP | 342.7 | 9.7 | [Ar]s1d9 | 461.3 | −108.9 | [Ar]s1d9 |
| mPW1PBE | 342.3 | 10.1 | [Ar]s1d9 | 393.8 | −41.4 | [Ar]d10 |
| mPW1PW91 | 338.7 | 13.7 | [Ar]s1d9 | 390.3 | −37.9 | [Ar]d10 |
| mPW3PBE | 371.5 | −19.1 | [Ar]s1d9 | 489.6 | −137.2 | [Ar]s1d9 |
| N12SX | 264.4 | 88.0 | [Ar]s2d8 | 444.7 | −92.3 | [Ar]s1d9 |
| O3LYP | 385.3 | −32.9 | [Ar]s1d9 | 466.7 | −114.3 | [Ar]s1d9 |
| PBE1PBE | 349.4 | 3.0 | [Ar]s1d9 | 479.8 | −127.4 | [Ar]s1d9 |
| PBEh1PBE | 347.8 | 4.6 | [Ar]s1d9 | 479.9 | −127.5 | [Ar]s1d9 |
| PBE | 469.5 | −117.1 | [Ar]s1d9 | 546.7 | −194.3 | [Ar]s1d9 |
| PPE0 | 245.9 | 106.5 | [Ar]s1d9 | 352.2 | 0.2 | [Ar]d10 |
| PW91 | 469.8 | −117.4 | [Ar]s1d9 | 549.4 | −197.0 | [Ar]s1d9 |
| SOGGA11X | 464.6 | −112.2 | [Ar]s1d9 | 512.8 | −160.4 | [Ar]d10 |
| SVWN5 | 587.6 | −235.2 | [Ar]s1d9 | 636.1 | −283.7 | [Ar]s1d9 |
| τ-HCTH | 477.6 | −125.2 | [Ar]s1d9 | 358.9 | −6.5 | [Ar]s2d8 |
| TPSSh | 396.0 | −43.6 | [Ar]s1d9 | 499.3 | −146.9 | [Ar]s1d9 |
| ωB97 | 374.5 | −22.1 | [Ar]s1d9 | 620.5 | −268.1 | [Ar]s1d9 |
| ωB97X | 240.4 | 112.0 | [Ar]s1d9 | 358.6 | −6.2 | [Ar]d10 |
| ωB97XD | 238.7 | 113.7 | [Ar]s1d9 | 283.7 | 68.7 | [Ar]d10 |
| X3LYP | 362.4 | −10.0 | [Ar]s1d9 | 473.9 | −121.5 | [Ar]s1d9 |
In contrast to the issues with the frequency calculations, 16 DFT functionals predicted BDE(1) value within 20 kJ mol−1 of the FPD BDE, with 4 functionals, HS06, HSE03, MN15, and PBE1PBE, predicting the BDE within 4 kJ mol−1 of the FPD value. A majority of the functionals predicted the 3D excited state of Ni as the ground state instead of the 3F state, although this corresponds to only a small energy correction of 2.5 kJ mol−1. The top three performing functionals for the frequency calculations, CAM-B3LYP, τ-HCTH, and ωB97XD all performed poorly, with ΔBDE(1) values of 138.2, −125.2, and 113.7 kJ mol−1, respectively. Calculating the Ni–O BDE with respect to a singlet Ni and correcting the energy with the singlet–triplet splitting of Ni giving BDE(2) does improve many of the poorly performing functionals including CAM-B3LYP and τ-HCTH. The PBE0 functional is in essentially exact agreement with the FPD value. A limitation on the BDE(2) values is the state of the Ni as taken from the NBO analysis as most of the atomic singlet Ni DFT calculations go to an open shell s1d9 singlet rather than the closed shell d10 configuration which introduces additional errors.
Conclusions
The vibrational frequency, bond dissociation energy, and electronic structure of NiO were calculated using a range of methodologies including CASSCF, icMRCI+Q, CCSD(T), and DFT. CASSCF predicted a quintet state (5Σ−) ground state for the equilibrium bond distance with a state crossing occurs at 1.65 Å, where the triplet (3Σ−) state becomes lower energy. icMRCI+Q predicts a triplet (3Σ−) state as the ground state and does not predict a state crossing. The ground state results from the ionic interaction of the O 2pz bonding with the 4s/3dz2 of Ni to form a σ bond. The inclusion of dynamic correlation effects at the icMRCI+Q level altered the electronic structure predicted by CASSCF. At the icMRCI+Q level, the ground state is 3Σ− in agreement with experiment.12,13 The frequency calculated at the icMRCI+Q/CBS level is within 4.1 cm−1 of experiment,13 and the bond distance is within 0.028 Å of experiment.12 The calculated values show little effect of increasing basis set size, with frequencies deviating from the complete basis set level by 3 cm−1 for the triple zeta basis set, and 1 cm−1 for the quadruple basis set.To benchmark the accuracy of non-multireference methods for the frequency, CCSD(T) and DFT were used. CCSD(T) predicts vibrational frequencies that are ∼ 50 cm−1 too high due to the multireference character of the ground triplet state. Correlation of the outer core electrons of Ni improves the calculated frequency but the value still differs by ∼40 cm−1 from experiment. An array of 43 DFT functionals waas benchmarked. The 3 best functionals out of the 43 tested are ωB97XD, CAM-B3LYP, and τ-HCTH which gave frequencies within <10 cm−1 of experiment.
The BDE of NiO was calculated at the FPD/CCSD(T) and icMRCI+Q levels as well as with the 43 DFT functionals. The available experimental data16,20 are not in good enough agreement to distinguish between the FPD/CCSD(T) and icMRCI+Q BDE values for the NiO BDE. The FPD/CCSD(T) value of 352 kJ mol−1 is consistent with the experimental20 BDE of 349.4 ± 6.3 kJ mol−1 whereas the icMRCI+Q BDE of 388 kJ mol−1 is larger than the largest experimental16 value by 14 kJ mol−1. The results show that further experimental investigation of the thermodynamics of the NiO bond is needed. The DFT values for 16 functionals are in agreement within 20 kJ mol−1 of the FPD/CCSD(T) value using the BDE(1) expression but these do not include the 3 best functionals for the frequency predictions. The 3 best frequency functionals have errors ranging from −125 to 138 kJ mol−1 using the BDE(1) expression. Use of the state splitting corrected BDE(2) expression substantially improved the BDE values for the CAM-B3LYP and τ-HCTH functionals, especially the latter value. Note that the use of the BDE(2) expression resulted in worse agreement for most functionals. It is important to calculate the electronic state of the Ni atom used in the BDE equation with each functional so an appropriate experimental correction for the excited state-ground state splitting can be applied.
Comparison of the multireference and single reference results shows that the MRCI approach gives a bond distance for the 3Σ− ground state that is a bit too short but has the right curvature; in contrast, the single reference based CCSD(T) method with a large T1 value demonstrating multireference character yields a good bond distance, but the predicted curvature is too tight. The CCSD(T) method predicts a good bond energy if the energy of the excited 5Σ− state which is dominated by a single configuration. The BDE for the ground 3Σ− state can then be obtained by the energy difference between the 3Σ− and 5Σ− states. The MRCI approach predicts a BDE that is likely to be too large and this can be attributed to the difficulty in calculating the energy at large R. Selected DFT functionals can predict the bond distance and vibrational frequency with some reliability but fail in predicting the BDE unless a carefully selected set of atomic states is chosen.
The current work demonstrates that the vibrational frequency and BDE of NiO can be reliably calculated using advanced correlated molecular orbital theory approaches. If one starts with a single reference-based method such as CCSD(T), the T1 values should be carefully checked for the presence of multi-reference character. When using DFT, it is important to be careful about the choice of the functional as commonly used functionals such as PBE and B3LYP perform poorly when calculating both the spectroscopic properties and BDE of NiO. In addition, care must be taken to examine the electronic state of the bare metal atom as it may not be the ground state. For more complex NiO based systems, it will be important to carefully choose the functional and compromise may need to be made for getting the best functional in terms of vibrational spectroscopy predictions and reaction energies.
Data availability
The data supporting this article have been included as part of the Supplementary Information.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences by UNCAGE-ME, an Energy Frontier Research Center funded by the United States Department of Energy, Office of Science, Basic Energy Sciences under Award DE-SC0012577. D. A. D. thanks the Robert Ramsay Fund at The University of Alabama.References
- Y. Lingting and K. Xie, High-Temperature Electrocatalysis and Key Materials in Solid Oxide Electrolysis Cells, J. Eng. Chem, 2021, 54, 736–745 Search PubMed.
- C. Graves, S. D. Ebbesen and M. Mogensen, Co-Electrolysis of CO2 and H2O in Solid Oxide Cells: Performance and Durability, Solid State Ionics, 2011, 192, 398–403 CrossRef CAS.
- Y. Tao, S. D. Ebbesen and M. B. Mogensen, Carbon Deposition in Solid Oxide Cells during Co-Electrolysis of H2O and CO2, J. Electrochem. Soc., 2014, 161, F337–F343 CrossRef CAS.
- S. D. Ebbesen, X. Sun and M. B. Mogensen, Understanding the Processes Governing Performance and Durability of Solid Oxide Electrolysis Cells, Faraday Disc., 2015, 182, 393–422 RSC.
- S. D. Ebbesen, C. Graves and M. Mogensen, Production of Synthetic Fuels by Co-Electrolysis of Steam and Carbon Dioxide, Int. J. Green Energy, 2009, 6, 646–660 CrossRef CAS.
- Y. Zheng, J. Wang, B. Yu, W. Zhang, J. Chen, J. Qiao and J. Zhang, A Review of High Temperature Co-Electrolysis of H2O and CO2 to Produce Sustainable Fuels Using Solid Oxide Electrolysis Cells (SOECs): Advanced Materials and Technology, Chem. Soc. Rev., 2017, 46, 1427–1463 RSC.
- Y. Li, P. Li, B. Hu, C. Xia and A. Nanostructured, Ceramic Fuel Electrode for Efficient CO2/H2O Electrolysis Without Safe Gas, J. Mat. Chem. A, 2016, 4, 9236–9243 RSC.
- S. Ebbesen, R. Knibbe and M. Mogensen, Co-Electrolysis of Steam and Carbon Dioxide in Solid Oxide Cells, J. Electrochem. Soc., 2012, 159, F482–F489 CrossRef CAS.
- J. Carneiro and E. Nikolla, Nanoengineering of Solid Oxide Electrochemical Cell Technologies: An Outlook. Nano, Research, 2019, 12, 2081–2092 CAS.
- M. Laguna-Bercero, Recent Advances in High Temperature Electrolysis Using Solid Oxide Fuel Cells: A Review, J. Power Sources, 2012, 203, 4–16 CrossRef CAS.
- X.-K. Gu and E. Nikolla, Fundamental Insights into High-Temperature Water Electrolysis Using Ni-Based Electrocatalysts, J. Phys. Chem. C, 2015, 119, 26980–26988 CrossRef CAS.
- R. S. Ram and P. F. Bernath, Fourier Transform Infrared Emission Spectroscopy of a New A 3Πi−X 3Σ− System of NiO, J. Mol. Spectrosc., 1992, 155, 315–325 CrossRef CAS.
- V. I. Srdanov and D. O. Harris, Laser Spectroscopy of NiO: The 3Σ− Ground State, J. Chem. Phys, 1988, 89, 2748–2753 CrossRef CAS.
- D. W. Green, G. T. Reedy and J. G. Kay, Matrix Isolated FeO. NiO, and CoO: Ground State Vibratioonal Frequencies, J. Mol. Spectrscop., 1979, 78, 257–266 CrossRef CAS.
- Hongbin Wu and L.-S. Wang, A Study of Nickel Monoxide (NiO), Nickel Dioxide (OniO), and Ni(O2) Complex by Anion Photoelectron Spectroscopy, J. Chem. Phys., 1997, 107, 16–21 CrossRef CAS.
- L. R. Watson, T. L. Thiem, R. A. Dressler, R. H. Salter and E. Murad, High Temperature Mass Spectrometric Studies of the Bond Energies of Gas-Phase ZnO, NiO, and CuO, J. Phys. Chem., 1993, 97, 5577–5580 CrossRef CAS.
- K. Namiki and S. Saito, Microwave Spectrum of the NiO Radical in the X 3Σ− State, Chem. Phys. Lett., 1996, 252, 343–347 CrossRef.
- T. M. Ramond, G. E. Davico, F. Hellberg, F. Svedberg, P. Salén, P. Söderqvist and W. C. Lineberger, Photoelectron Spectroscopy of Nickel, Palladium, and Platinum Oxide Anions, J. Mol. Spectrosc., 2002, 216, 1–14 CrossRef CAS.
- D. A. Burgard, J. Abraham, A. Allen, J. Craft, W. Foley, J. Robinson, B. Wells, C. Xu and D. H. Stedman, Chemiluminescent Reactions of Nickel, Iron, and Cobalt Carbonyls with Ozone, Appl. Spectrosc., 2006, 60, 99–102 CrossRef CAS PubMed.
- M. Farber and R. D. Srivastava, The Dissociation Energy of NiO and Vaporization Sublimation Enthalpies of Ni, Anal. Calorimetry, 1974, 3, 731–741 CAS.
- E. R. Fisher and P. B. Armentrout, Reactions of Co+, Ni+, and Cu+ with Cyclopropane and Ethylene Oxide. Metal-Methylidene Ion Bond Energies, J. Phys. Chem., 1990, 94, 1674–1683 CrossRef CAS.
- S. P. Walch and W. A. Goddard III, Electronic States of the Nickel Oxide Molecule, J. Am. Chem. Soc., 1978, 100, 1338–1348 CrossRef CAS.
- M. Dolg, W. H. Stoll and H. Preuss, Ab initio Pseudopotential Study of the First Row Transition Metal Monoxides and Iron Monohydride, J. Chem. Phys, 1987, 86, 2123–2131 CrossRef CAS.
- C. W. Bauschlicher, C. J. Nelin and P. S. Bagus, Transition Metal Oxides: CrO, MoO, NiO, PdO, AgO, J. Chem. Phys, 1985, 82, 3265–3276 CrossRef CAS.
- M. A. Abdulsattar and M. T. Matrood, Spin Multiplicity Effects on Electronic and Spectroscopic Properties of NiO Diatomic Molecule: A DFT Study, AIP Conf. Proc., 2023, 2845, 070029 CrossRef CAS.
- C. W. Bauschlicher and P. Maitre, Theoretical Study of First Transition Row Oxides and Sulfides, Theor. Chim. Acta, 1995, 90, 189–203 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, Density-Functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- W. Kohn, A. D. Becke and R. G. Parr, Density Functional Theory of Electronic Structure, J. Phys. Chem., 1996, 100, 12974–12980 CrossRef CAS.
- T. H. Dunning, Gaussian Basis Set for Use in Correlated Molecular Calculations. I. The Atoms Boron Through Neon and Hydrogen, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- R. A. Kendall, T. H. Dunning, Jr. and R. J. Harrison, Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- M. Dolg, Improved Relativistic Energy-Consistent Pseudopotentials for 3d-Transition Metals, Theor. Chem. Acc., 2005, 114, 297–304 Search PubMed.
- S. Li, J. M. Hennigan, D. A. Dixon and K. A. Peterson, Accurate Thermochemistry for Transition Metal Oxide Clusters, J. Phys. Chem. A, 2009, 113, 7861–7877 CrossRef CAS PubMed.
- The basis sets for Ni are available from Prof. Kirk Peterson, Washington State University. https://https:1/chem.wsu.edu/people/faculty/wsu-profile/kipeters/(Accessed September 1, 2023).
- G. D. Purvis and R. J. Bartlett, A Full Coupled-Cluster Singles and Doubles Model: The Inclusion of Disconnected Triples, J. Chem. Phys., 1982, 76, 1910–1918 CrossRef CAS.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, A Fifth-order Perturbation Comparison of Electron Correlation Theories, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS.
- J. D. Watts, J. Gauss and R. J. Bartlett, Coupled-Cluster Methods with Non-iterative Triple Excitations for Restricted Open-Shell Hartree–Fock and Other General Single-Determinant Reference Functions. Energies and Analytical Gradients, J. Chem. Phys., 1993, 98, 8718–8733 CrossRef CAS.
- R. J. Bartlett and M. Musial, Coupled-Cluster Theory in Quantum Chemistry, Rev. Mod. Phys., 2007, 79, 291–352 CrossRef CAS.
- M. J. O. Deegan and P. J. Knowles, Perturbative Corrections to Account for Triple Excitations in Closed and Open Shell Coupled Cluster Theories, Chem. Phys. Lett., 1994, 227, 321–326 CrossRef CAS.
- M. Rittby and R. J. Bartlett, An Open-Shell Spin-Restricted Coupled Cluster Method: Application to Ionization Potentials in N2, J. Phys. Chem., 1988, 92, 3033–3036 CrossRef CAS.
- P. J. Knowles, C. Hampel and H.-J. Werner, Coupled Cluster Theory for High Spin, Open Shell Reference Wave Functions, J. Chem. Phys., 1993, 99, 5219–5228 CrossRef CAS.
- W. A. de Jong, R. J. Harrison and D. A. Dixon, Parallel Douglas-Kroll Energy and Gradients in NWChem: Estimating Scalar Relativistic Effects Using Douglas-Kroll Contracted Basis Sets, J. Chem. Phys., 2001, 114, 48–53 CrossRef CAS.
- J. L. Dunham, The Energy Levels of a Rotating Vibrator, Phys. Rev., 1932, 41, 721–731 CrossRef CAS.
- B. O. Roos, P. R. Taylor and P. E. Siegbahn, A Complete Active Space SCF Method (CASSCF) Using Density-matrix Formulated Super-CI Approach, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- P. E. M. Siegbahn, J. Almlöf, A. Heiberg and B. O. Roos, The Complete Active Space SCF (CASSCF) Method in a Newton-Raphson Formulation with Application to the HNO Molecule, J. Chem. Phys., 1981, 74, 2384–2396 CrossRef.
- H.-J. Werner, Matrix-Formulated Direct Multiconfiguration Self-Consistent Field and Multiconfiguration Reference Configuration-Interaction Methods, Adv. Chem. Phys., 1987, 1 CAS.
- H.-J. Werner and W. Meyer, A Quadratically Convergent Multiconfiguration-Self-Consistent Field Method with Simultaneous Optimization of Orbitals and CI Coefficients, J. Chem. Phys., 1980, 73, 2342–2356 CrossRef CAS.
- H.-J. Werner and W. Meyer, A Quadratically Convergent MCSCF Method for the Simultaneous Optimization of Several States, J. Chem. Phys., 1981, 74, 5794–5801 CrossRef CAS.
- N. B. Balabanov and K. A. Peterson, Systematically Convergent Basis Sets for Transition Metals. I. All-Electron Correlation Consistent Basis Sets for the 3d Elements Sc-Zn, J. Chem. Phys., 2005, 123, 064107 CrossRef PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, Molpro: A General-Purpose Quantum Chemistry Program Package, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CAS.
- H.-J. Werner, P. J. Knowles, F. R. Manby, J. A. Black, K. Doll, A. Heßelmann, D. Kats, A. Köhn, T. Korona, D. A. Kreplin, Q. Ma, T. F. Miller III, A. Mitrushchenkov, K. A. Peterson, I. Polyak, G. Rauhut and M. Sibaev, The Molpro Quantum Chemistry Package, J. Chem. Phys., 2020, 152, 144107 CrossRef CAS PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, W. Györffy, D. Kats, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, S. J. Bennie, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, S. J. R. Lee, Y. Liu, A. W. Lloyd, Q. Ma, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, T. F. Miller III, M. E. Mura, A. Nicklass, D. P. O’Neill, P. Palmieri, D. Peng, T. Petrenko, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and M. Welborn, MOLPRO, version 2020.1, a package of ab initio programs, see https://www.molpro.net. [Accessed July 1, 2022].
- E. R. Davidson and D. W. Silver, Size Consistency in the Dilute Helium Gas Electronic Structure, Chem. Phys. Lett., 1977, 52, 403–406 CrossRef CAS.
- K. Andersson, P. A. Malmqvist, B. O. Roos, A. J. Sadlej and K. Wolinski, Second-Order Perturbation Theory with a CASSCF Reference Function, J. Phys. Chem., 1990, 94, 5483–5488 CrossRef CAS.
- K. Andersson, P. A. Malmqvist, B. O. Roos, A. J. Sadlej and K. Wolinski, Second-Order Perturbation Theory with a Complete Active Space Self-Consistent Field Reference Function, J. Chem. Phys, 1992, 96, 1218–1226 CrossRef CAS.
- D. A. Dixon, D. Feller and K. A. Peterson, A Practical Guide to Reliable First Principles Computational Thermochemistry Predictions Across the Periodic Table, in Annual Reports in Computational Chemistry, ed R. A. Wheeler, Elsevier, Amsterdam, 2012, vol. 8, ch. 1, pp. 1–28 Search PubMed.
- D. Feller, K. A. Peterson and D. A. Dixon, Further Benchmarks of a Composite, Convergent, Statistically-Calibrated Coupled Cluster-Based Approach for Thermochemical and Spectroscopic Studies, Mol. Phys., 2012, 110, 2381–2399 CrossRef CAS.
- K. A. Peterson, D. Feller and D. A. Dixon, Chemical Accuracy in Ab Initio Thermochemistry and Spectroscopy: Current Strategies and Future Challenges, Theor. Chem. Acc., 2012, 131, 1079 Search PubMed.
- D. Feller, K. A. Peterson and D. A. Dixon, The Impact of Larger Basis Sets and Explicitly Correlated Coupled Cluster Theory on the Feller-Peterson-Dixon Composite Method. in Annual Reports in Computational Chemistry, ed. D. A. Dixon, Elsevier, Amsterdam, 2016, vol. 12, pp. 47–78 Search PubMed.
- A. D. Becke, Density-Functional Thermochemistry. IV. A new Dynamical Correlation Functional and Implications for Exact-Exchange Mixing, J. Chem. Phys., 1996, 104, 1040–1046 CrossRef CAS.
- C. Adamo and V. Barone, Toward Reliable Adiabatic Connection Models Free from Adjustable Parameters, Chem. Phys. Lett., 1997, 274, 242–250 CrossRef CAS.
- J. P. Perdew, Density-Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas, Phys. Rev. B, 1986, 33, 8822–8824 CrossRef PubMed.
- J. P. Perdew and Y. Wang, Accurate and Simple Analytic Representation of the Electron Gas Correlation Energy, Phys. Rev. B, 1992, 45, 13244–13249 CrossRef PubMed.
- A. D. Boese and N. C. Handy, New Exchange–Correlation Density Functionals: The Role of the Kinetic-Energy Density, J. Chem. Phys., 2002, 116, 9559–9969 CrossRef CAS.
- P. J. Wilson, T. J. Bradley and D. J. Tozer, Hybrid Exchange–Correlation Functional Determined from Thermochemical Data and Ab Initio Potentials, J. Chem. Phys., 2001, 115, 9233–9242 CrossRef CAS.
- H. L. Schmider and A. D. Becke, Optimized Density Functionals from the Extended G2 Test Set, J. Chem. Phys, 1998, 108, 9624–9631 CrossRef CAS.
- A. D. Becke, Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- A. D. Boese and J. M. L. Martin, Development of Density Functionals for Thermochemical Kinetics, J. Chem. Phys., 2004, 121, 3405–3416 CrossRef CAS PubMed.
- T. Yanai, D. Tew and N. Handy, A New Hybrid Exchange–Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, Hybrid Functionals Based on a Screened Coulomb Potential, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof. Erratum: Hybrid functionals based on a screened Coulomb potential, J. Chem. Phys. 118, 8207, 2003 Search PubMed.
- J. Heyd and G. E. Scuseria, Assessment and Validation of a Screened Coulomb Hybrid Density Functional, J. Chem. Phys., 2004, 120, 7274–7280 CrossRef CAS PubMed.
- J. Heyd, J. E. Peralta, G. E. Scuseria and R. L. Martin, Energy Band Gaps and Lattice Parameters Evaluated with the Heyd–Scuseria–Ernzerhof Screened Hybrid Functional, J. Chem. Phys., 2005, 123, 174101 CrossRef PubMed.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, Influence of the Exchange Screening Parameter on the Performance of Screened Hybrid Functionals, J. Chem. Phys, 2006, 125, 224106 CrossRef PubMed.
- O. A. Vydrov and G. E. Scuseria, Assessment of a Long Range Corrected Hybrid Functional, J. Chem. Phys, 2006, 125, 234109 CrossRef PubMed.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions, J. Chem. Theory Comput., 2006, 2, 364–382 CrossRef PubMed.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, Exchange–Correlation Functional with Broad Accuracy for Metallic and Nonmetallic Compounds, Kinetics, and Noncovalent Interactions, J. Chem. Phys., 2005, 123, 161103 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, The M06 suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-class Functionals and 12 Other Functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Y. Zhao and D. G. Truhlar, Comparative DFT Study of van der Waals Complexes: Rare-Gas Dimers, Alkaline-Earth Dimers, Zinc Dimer, and Zinc-Rare-Gas Dimers, J. Phys. Chem. A, 2006, 110, 5121–5129 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Density Functional for Spectroscopy: No Long-Range Self-Interaction Error, Good Performance for Rydberg and Charge-Transfer States, and Better Performance on Average than B3LYP for Ground States, J. Phys. Chem. A, 2006, 110, 13126–13130 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Exploring the Limit of Accuracy of the Global Hybrid Meta Density Functional for Main-Group Thermochemistry, Kinetics, and Noncovalent Interactions, J. Chem. Theory Comput., 2008, 4, 1849–1868 CrossRef CAS PubMed.
- R. Peverati and D. G. Truhlar, Improving the Accuracy of Hybrid Meta-GGA Density Functionals by Range Separation, J. Phys. Chem. Lett., 2011, 2, 2810–2817 CrossRef CAS.
- R. Peverati and D. G. Truhlar, Screened-Exchange Density Functionals with Broad Accuracy for Chemistry and Solid State Physics, Phys. Chem. Chem. Phys., 2012, 14, 16187–16191 RSC.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, MN15: A Kohn–Sham Global-Hybrid Exchange–Correlation Density Functional with Broad Accuracy for Multi-Reference and Single-Reference Systems and Noncovalent Interactions, Chem. Sci., 2016, 7, 5032–5051 RSC.
- C. Adamo and V. Barone, Exchange Functionals with Improved Long-Range Behavior and Adiabatic Connection Methods without Adjustable Parameters: The mPW and mPW1PW models, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Erratum: Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- A. J. Cohen and N. C. Handy, Dynamic Correlation, Mol. Phys., 2001, 99, 607–615 CrossRef CAS.
- C. Adamo and V. Barone, Toward Reliable Density Functional Methods Without Adjustable Parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158–6169 CrossRef CAS.
- M. Ernzerhof and J. P. Perdew, Generalized Gradient Approximation to the Angle- and Aystem-Averaged Exchange Hole, J. Chem. Phys., 1998, 109, 3313–3320 CrossRef CAS.
- K. Burke, J. P. Perdew and Y. Wang, Derivation of a Generalized Gradient Approximation: The PW91 Density Functional, in Electronic Density Functional Theory: Recent Progress and New Directions, ed. J. F. Dobson, G. Vignale and M. P. Das, Springer, NY, 1998, pp. 81–111 Search PubMed.
- R. Peverati and D. G. Truhlar, A Global Hybrid Generalized Gradient Approximation to the Exchange–Correlation Functional That Satisfies the Second-Order Density-Gradient Constraint and Has Broad Applicability in Chemistry, J. Chem. Phys., 2011, 135, 191102 CrossRef PubMed.
- J. C. Slater, Quantum Theory of Molecules and Solids, McGraw-Hill, New York, 1974, vol. 4 Search PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- A. D. Boese and N. C. Handy, New Exchange–Correlation Density Functionals: The Role of the Kinetic-Energy Density, J. Chem. Phys., 2002, 116, 9559–9969 CrossRef CAS.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, Comparative Assessment of a New Nonempirical Density Functional: Molecules and Hydrogen-Bonded Complexes, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Systematic Optimization of Long-Range Corrected Hybrid Density Functionals, J. Chem. Phys., 2008, 128, 084106 CrossRef PubMed.
- J.-D. Chai and M. Head-Gordon, Long-range Corrected Hybrid Density Functionals with Damped Atom-atom Dispersion Corrections, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- J.-D. Chai and M. Head-Gordon, Systematic Optimization of Long-range Corrected Hybrid Density Functionals, J. Chem. Phys, 2008, 128, 084106 CrossRef PubMed.
- X. Xu and W. A. Goddard, The X3LYP Extended Density Functional for Accurate Descriptions of Nonbond Interactions, Spin States, and Thermochemical Properties, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 2673–2677 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Intermolecular Interactions from a Natural Bond Orbital, Donor–Acceptor Viewpoint, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- F. Weinhold and C. R. Landis, Valency and Bonding: A Natural Bond Orbital Donor–Acceptor Perspective, University Press, Cambridge, U.K., 2005 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, Natural Bond Order 7.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2018.
- E. D. Glendening, C. R. Landis and F. Weinhold, NBO 7.0: New Vistas in Localized and Delocalized Chemical Bonding Theory, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Z. Fang, J. Both, S. Li, S. Yue, E. Aprà, M. Keçeli, A. F. Wagner and D. A. Dixon, Benchmark Calculations of Energetic Properties of Groups 4 and 6 Transition Metal Oxide Nanoclusters Including Comparison to Density Functional Theory, J. Chem. Theor. Comput., 2016, 12, 3689–3710 CrossRef CAS PubMed.
- Z. Fang, Z. Lee, K. A. Peterson and D. A. Dixon, Use of Improved Orbitals for CCSD(T) Calculations for Predicting Heats of Formation of Group IV and Group VI Metal Oxide Monomers and Dimers and UCl6, J. Chem. Theor. Comput., 2016, 12, 3583–3592 CrossRef CAS PubMed.
- Z. Fang, M. Vasiliu, K. A. Peterson and D. A. Dixon, Prediction of Bond Dissociation Energies/Heats of Formation for Diatomic Transition Metal Compounds: CCSD(T) Works, J. Chem. Theor. Comput., 2017, 13, 1057–1066 CrossRef CAS PubMed.
- T. J. Lee and P. R. Taylor, A Diagnostic for Determining the Quality of Single-Reference Electron Correlation Methods, Int. J. Quantum Chem. Symp., 1989, 23, 199–207 CAS.
- A. Kramida, Yu Ralchenko and J. Reader, NIST ASD Team (2023). NIST Atomic Spectra Database (ver. 5.11), [Online]. Available: https://physics.nist.gov/asd [2024, April 22]. National Institute of Standards and Technology, Gaithersburg, MD DOI:10.18434/T4W30F (Accessed April 1, 2024).
- U. Litzén, J. W. Brault and A. P. Thorn, Spectrum and Term System of Neutral Nickel, Ni I, Phys. Scr., 1993, 47, 628–673 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Total energies in a.u. See DOI: https://doi.org/10.1039/d4cp01796j |
| This journal is © the Owner Societies 2024 |