
Anti-plasticizing effect of water on prilocaine and lidocaine – the role of the hydrogen bonding pattern†
Xiaoyue
Xu
 ,
Holger
Grohganz
* and
Thomas
Rades
,
Holger
Grohganz
* and
Thomas
Rades
Department of Pharmacy, University of Copenhagen, Universitetsparken 2, 2100 Copenhagen, Denmark. E-mail: holger.grohganz@sund.ku.dk
First published on 7th May 2024
Abstract
It is generally accepted that water, as an effective plasticizer, decreases the glass transition temperatures (Tgs) of amorphous drugs, potentially resulting in physical instabilities. However, recent studies suggest that water can also increase the Tgs of the amorphous forms of the drugs prilocaine (PRL) and lidocaine (LID), thus acting as an anti-plasticizer. To further understand the nature of the anti-plasticizing effect of water, interactions with different solvents and the resulting structural features of PRL and LID were investigated by Fourier transform infrared spectroscopy (FTIR) and quantum chemical simulations. Heavy water (deuterium oxides) was chosen as a solvent, as the deuterium and hydrogen atoms are electronically identical. It was found that substituting hydrogen with deuterium showed a minimal impact on the anti-plasticization of water on PRL. Ethanol and ethylene glycol were chosen as solvents to compare the hydrogen bonding patterns occurring between the hydroxyl groups of the solvents and PRL and LID. Comparison of the various Tgs showed a weaker anti-plasticizing potential of these two solvents on PRL and LID. The frequency shifts of the amide C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups of PRL and LID due to the interactions with water, heavy water, ethanol, and ethylene glycol as observed in the FTIR spectra showed a correlation with the binding energies calculated by quantum chemical simulations. Overall, this study showed that the combination of weak hydrogen bonding and strong electrostatic contributions in hydrated PRL and LID could play an important role in inducing the anti-plasticizing effect of water on those drugs.
O groups of PRL and LID due to the interactions with water, heavy water, ethanol, and ethylene glycol as observed in the FTIR spectra showed a correlation with the binding energies calculated by quantum chemical simulations. Overall, this study showed that the combination of weak hydrogen bonding and strong electrostatic contributions in hydrated PRL and LID could play an important role in inducing the anti-plasticizing effect of water on those drugs.
1. Introduction
Poor aqueous solubility of active pharmaceutical ingredients (APIs) is recognized as a major challenge in the development of orally administrated drugs.1,2 Converting crystalline drugs into their high-energy amorphous counterparts exhibits promising potential to enhance the solubility and bioavailability of poorly water-soluble APIs.3,4 However, the absence of a crystal lattice also renders the APIs thermodynamically unstable, eventually resulting in crystallization, and thus loss of the solubility advantage.5,6 This creates challenges in amorphous drug development during manufacturing and storage.Adding to the intrinsic thermodynamic instability of amorphous drugs is the fact that adsorbed water further plasticizes amorphous drugs, thus decreasing their glass transition temperatures (Tgs), resulting in additional physical instability.7,8 The plasticizing effect of water is believed to arise from the propensity of water to interact with various functional groups of amorphous drugs through hydrogen bonding.9 Water molecules disrupt the intermolecular forces in the amorphous solids, thus increasing the molecular mobility of amorphous APIs, resulting in the reduced Tgs of such drugs.10,11 However, recent studies observed that water can also have an anti-plasticizing effect on some APIs, including prilocaine (PRL) and lidocaine (LID), resulting in higher Tgs of hydrated amorphous PRL and LID compared with the respective anhydrous amorphous forms of these drugs.12,13 Useful insights into the anti-plasticizing nature of water on amorphous PRL were gained from the work of Ruiz et al. who investigated the molecular interactions between PRL and water by Fourier transform infrared spectroscopy (FTIR).13 The anti-plasticizing effect of water on PRL was ascribed to a dimeric structure of PRL held together by a water bridge, evidenced by the red shift of the amide CO group of amorphous PRL upon hydration.13
In this work, to further investigate the nature of the anti-plasticizing effect of water on the amorphous drugs PRL and LID, interactions and resulting structural properties of PRL and LID were investigated in the presence of different solvents. Heavy water (deuterium oxides) was chosen as a solvent because the deuterium and hydrogen atoms are electronically identical. It was assumed in this study that the substitution of water with heavy water would not affect the anti-plasticizing effect of water for PRL. Ethanol and ethylene glycol were chosen as solvents based on their varying abilities to form hydrogen bonds with PRL and LID. Ethylene glycol was expected to be a potential anti-plasticizer for PRL, because the two hydroxyl groups of this molecule could facilitate the formation of a dimeric structure of PRL bridged by ethylene glycol.13 In contrast, ethanol was assumed to be a plasticizer for PRL and LID due to the molecule only having a single hydroxyl group. The interactions of the solvents with PRL and LID were experimentally investigated by FTIR and theoretically calculated based on density functional theory (DFT). To characterize the quantitative nature of the interactions with different solvents, symmetry-adapted perturbation theory (SAPT) was employed. Overall, our findings indicate that the previous hypothesis, attributing the anti-plasticizing effect of water to a dimer formation through a water bridge, cannot sufficiently account for the observed changes in the Tgs of PRL and LID with water, ethanol, and ethylene glycol.13 Further analysis suggested that the combination of weak hydrogen bonding and strong electrostatic contributions could be an indicator of the anti-plasticization of water in hydrated PRL and LID.
2. Materials and methods
2.1. Materials
Prilocaine (PRL, MW = 220.31 g mol−1) was purchased from Fluorochem Ltd (Hadfield, U.K.). Lidocaine (LID, MW = 234.34 g mol−1) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Water (18.2 MΩ) was freshly prepared using a Milli-Q water system from ELGA LabWater (High Wycombe, U.K.). Absolute ethanol was purchased from VWR (Randnor, PA, USA). Ethylene glycol and heavy water (deuterium oxides) were purchased from Sigma-Aldrich (St. Louis, MO, USA).2.2. Methods
For solvated amorphous PRL and co-amorphous PRL-LID samples, a droplet of heavy water or ethanol was added to the crystalline drug or crystalline mixtures of PRL and LID with mole fractions of LID from 0 to 0.7, with the samples subsequently undergoing solvent evaporation monitored on a microbalance. The evaporation process allowed precise control of the amount of added solvent to reach the desired total sample mass.12 This pathway could not be used for ethylene glycol, due its non-volatile nature. Therefore, a specific amount of ethylene glycol was added directly to the crystalline PRL. The DSC pans were subsequently hermetically sealed and subjected to melt-quenching in a DSC.
For amorphous PRL samples combined with heavy water, solvent-to-drug molar ratios from 10% to 100% were used, corresponding to an earlier study on water.14 For amorphous PRL and co-amorphous PRL-LID samples with ethanol and ethylene glycol, a solvent-to-drug molar ratio of 50% was used.
These unsolvated and solvated amorphous PRL and co-amorphous PRL-LID samples were subsequently used for measuring the Tgs and conducting FTIR analysis.
2.2.2.1. Differential scanning calorimetry (DSC). The Tgs of unsolvated and solvated amorphous PRL and co-amorphous PRL-LID samples were measured using a temperature modulated DSC (TA instruments, New Castle, DE, USA) under a nitrogen gas flow of 50 mL min−1. The samples were heated to 353 K at a rate of 10 K min−1, and equilibrated at 193 K, followed by quench-cooling. The Tg was determined by reheating the sample at 2 K min−1 with a modulation amplitude of 0.2120 K and a period of 40 s. The Tg was taken at the midpoint of the change in heat capacity (ΔCp). All Tg values were determined by three independent samples and are reported as mean ± standard deviation.
2.2.2.2. Calculation of the Tgs of unsolvated and solvated amorphous LID based on co-amorphous PRL-LID systems. It is impossible to obtain pure unsolvated and solvated amorphous LID as drug recrystallization is observed during cooling from the drug melt.14 In our previous study, an approach has been established to calculate the Tgs of unsolvated LID and hydrated LID at a water-to-drug molar ratio of 50%, based on the experimental Tgs of amorphous PRL and co-amorphous PRL-LID systems with mole fractions of LID from 0 to 0.7.12 In that study, the Tgs of unsolvated and hydrated LID at a water-to-drug molar ratio of 50% were determined to be 209.8 ± 0.5 K and 210.7 ± 0.7 K, respectively.12 The Tg of LID solvated with ethanol was calculated using that previously reported approach, based on the experimental Tgs of amorphous PRL and co-amorphous PRL-LID systems.12 As shown in Table S1 (ESI†), at a solvent-to-drug molar ratio of 50%, the calculated Tgs of amorphous LID and co-amorphous PRL-LID systems with ethanol were consistent with the experimental Tgs. Overall, the Tg of LID with ethanol at a solvent-to-drug molar ratio of 50% was found to be 188.9 ± 0.5 K.
2.2.2.3. Calculations of the theoretical Tgs of solvated amorphous PRL and LID. The theoretical Tgs of PRL and LID with ethanol and ethylene glycol were calculated by using the Gordon–Taylor equation:15
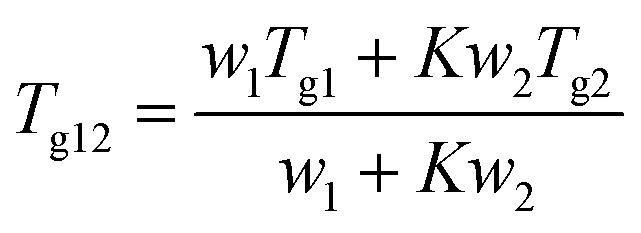 | (1) |
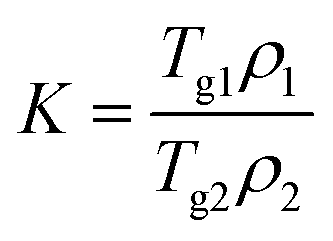 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) glycol = 1.114 g cm−3).20
glycol = 1.114 g cm−3).20
Previous studies have examined the FTIR spectra for unsolvated and hydrated amorphous PRL, as well as for co-amorphous PRL-LID systems.13,22 In this study, the FTIR spectra of amorphous unsolvated and solvated LID were investigated based on the co-amorphous system of PRL and LID with a mole fraction of LID of 0.7,22 due to fast crystallization of pure amorphous LID.14 PRL and LID are compounds featuring amino-amide structures, including the amide CO and amide N–H groups within both compounds, a secondary amine group specific to PRL, and a tertiary amine group specific to LID. Furthermore, the amide CO (1682 cm−1) and N–H (3296 cm−1) groups of amorphous PRL and LID, the amide N–H (1519 cm−1) group of amorphous PRL, and the amide N–H (1492 cm−1) group of amorphous LID, were chosen to analyze molecular interaction with the hydroxyl groups of the solvents.
2.2.4.1. Optimized structural models. One hundred starting structural models of hydrated PRL, PRL with ethanol, PRL with ethylene glycol, hydrated LID, and LID with ethanol were initially generated by the genmer tool under Molclus software.23 These structural models were optimized at the PM6-DH+ level using the MOPAC program to get ten stable structural models with low energies.24 Subsequently, the obtained ten structural models were optimized at the B3LYP-D3(BJ)/def2-TZVPP level using Gaussian 09 software to find the lowest energy model.25 For the structural model of PRL with heavy water, the hydrogen atoms of water in the structural model of hydrated PRL were substituted by deuterium atoms, as the equilibrium geometries are unaffected by isotopic substitution.26
2.2.4.2. Calculation of binding energy and FTIR spectra. For the optimized structural models of solvated drugs, the total electronic binding energies, were evaluated by the single-point energies at the B3LYP-D3(BJ)/jul-cc-pVTZ level. The basis set superposition error (BSSE) was corrected by the Boys and Bernardi's counterpoise (CP) technique.27 The vibrational frequencies of structural models were calculated at the B3LYP-D3(BJ)/def2-TZVPP level. A scaling factor of 0.959 was applied on the calculated vibrations in order to match the experimental FTIR spectra.28 All the calculated vibrational spectra were plotted in the Lorentzian line shape with a full width at half-maximum (FWHM) of 20 cm−1 using the Multiwfn program.29
2.2.4.3. Energy decomposition analysis. Symmetry-adapted perturbation theory (SAPT) was applied at the sSAPT0/jun-ccpVDZ level on the optimized molecular structural models of solvated drugs using the PSI4 program.30,31 The total electronic binding energies, dominating by the interactions between the drugs and solvents, were computed and decomposed into electrostatic, induction, exchange, and dispersion contributions.30,32 The electrostatic contribution describes the Coulombic interactions between the static charge distributions of molecules. The induction contribution arises from the polarization of one molecule by the electric field of another. The exchange effect is strongly repulsive, caused by the overlap of electron wavefunctions from different atoms. The dispersion contribution arises from fluctuations in the electron distribution within an atom or molecule.
3. Results and discussion
The Tgs of PRL and LID upon hydration have been investigated previously with regard to the anti-plasticizing effect of water.12,13 Taking account of previous studies, to maximize the anti-plasticizing effect of water, a water-to-drug molar ratio of 50% was chosen to compare the Tgs of anhydrous and hydrated PRL and LID.12,13 The Tg of hydrated PRL was increased by 4.1 K compared with that of anhydrous PRL, and the Tg of hydrated LID was increased by 0.9 K compared with that of anhydrous LID investigated in a previous study.12 Overall, the Tgs of PRL and LID increase upon hydration, indicating an anti-plasticizing effect of water on PRL and LID. The extent of the Tg increase of PRL and LID upon hydration indicates that the anti-plasticizing potentials of water can be ranked as hydrated PRL > hydrated LID.The interactions between water and PRL have previously been investigated by Ruiz et al.13 It has been shown that water did only bind to the amide CO group of PRL, which is visible in the FTIR spectra of anhydrous versus hydrated amorphous PRL by the red shift of the amide CO group of PRL.13 In our previous study, we also found that only the amide CO groups of amorphous PRL and LID were involved in the molecular interactions between water and co-amorphous systems of PRL and LID at mole fractions of LID from 0.1 to 0.7, according to spectroscopic investigations and quantum chemical simulations.22 Overall, it has been shown that water only interacted with the amide CO groups of PRL and LID.
Given that water only interacted with the amide CO group of PRL, Ruiz et al. pointed out that the addition of water led to a preferential formation of PRL dimers held together by two hydrogen bonds between one water molecule and the amide CO groups of two PRL molecules.13 This hypothesis was rationalized by the observations that the maximum hydration ratio for liquid PRL was at a water-to-drug molar ratio of 50%,33 corresponding to one water molecule binding to two PRL molecules.13 Since LID is structurally related to PRL, and showed similar interactions between water and the amide CO group of LID to those of PRL,22 as well as showing maximum hydration for liquid LID at a water-to-drug molar ratio of 50%,34 a dimeric structure of LID with water could also have been formed between water and the amide CO groups of LID.
3.1. Effect of heavy water on PRL
In order to closer investigate the influence of the hydrogen bonding pattern on the observed and assumed anti-plasticizing effect, water was firstly exchanged with heavy water. The differences of deuterium and hydrogen atoms are associated with their masses, resulting in changes of the vibrational motions of the atoms.35 However, given that the deuterium and hydrogen atoms are electronically identical, it was assumed in this study that the interactions between PRL and water would remain consistent upon substitution with heavy water.26,36 Thus, it was expected that heavy water would exhibit an identical anti-plasticizing impact on PRL as observed with water.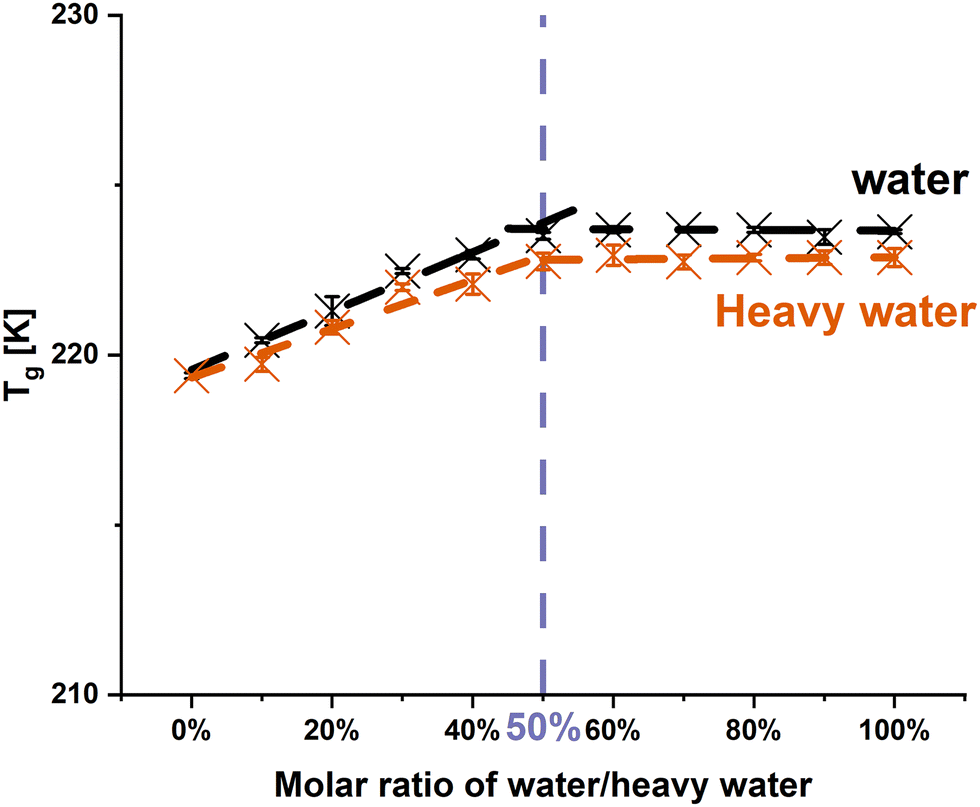 | ||
| Fig. 1 Changes in the Tgs of PRL with the addition of water and heavy water. The solvent-to-drug molar ratios range from 0% to 100%. | ||
O group vibration of PRL was chosen to investigate the molecular interaction of PRL with water and heavy water, as the previous investigation by Ruiz et al. indicated that water only interacted with the amide CO group of PRL.13 The experimental FTIR spectra show that the amide CO groups of PRL were red shifted with the addition of water and heavy water at solvent-to-drug molar ratios of 10%, 30%, and 50% (Fig. 2). A red shift of 2 cm−1 in the amide CO group of PRL was observed when comparing the interactions of heavy water versus water at different solvent-to-drug molar ratios, potentially due to the isotopic substitution with the heavier deuterium atom.35 This further suggests that heavy water has identical binding sites to water, given that the difference in the wavenumbers remained constant at 2 cm−1 at different solvent-to-drug molar ratios.
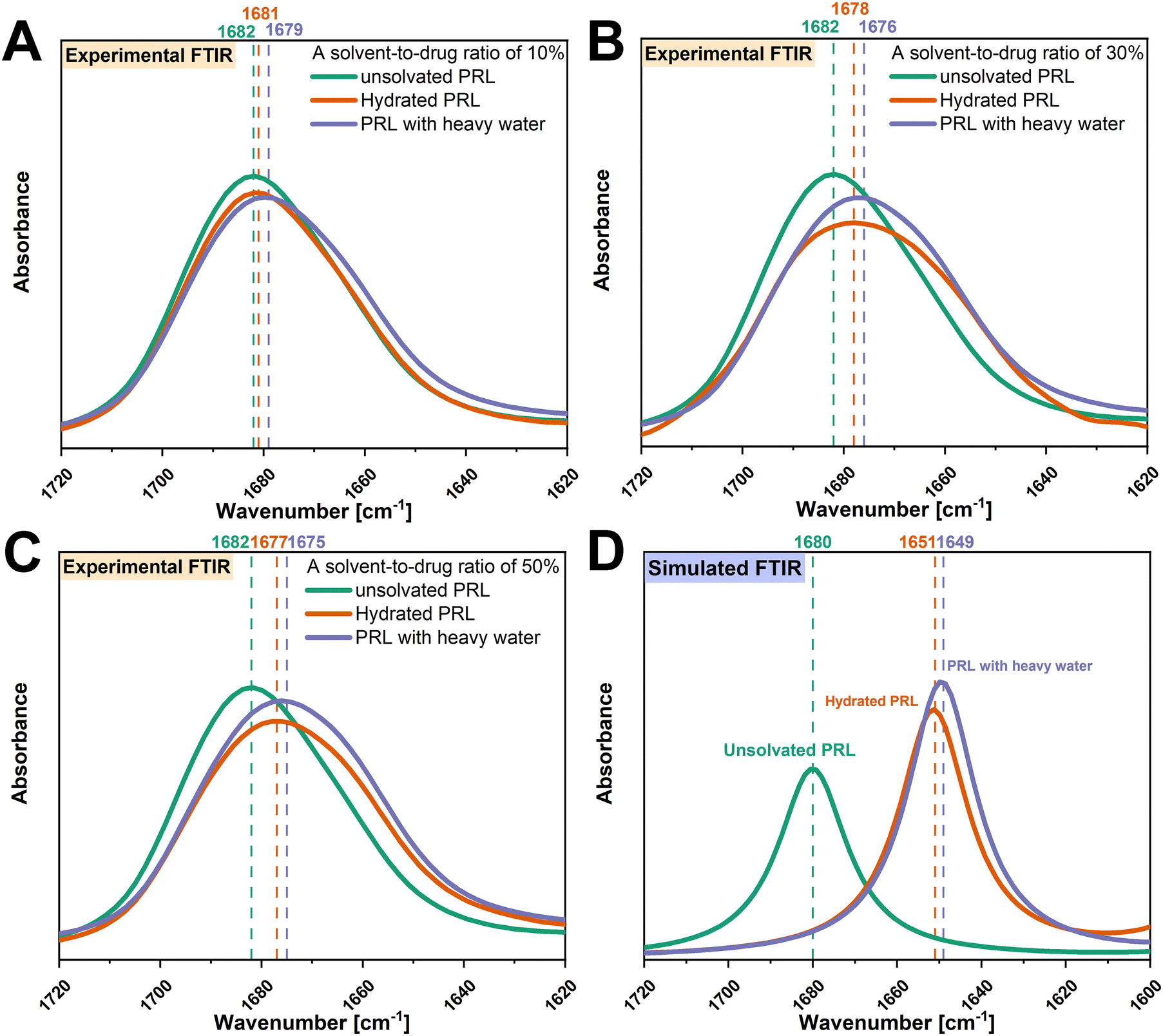 | ||
| Fig. 2 Experimental FTIR spectra of the amide CO groups of unsolvated and solvated PRL with a solvent-to-drug molar ratio of 10% (A), 30% (B), and 50% (C). Simulated FTIR spectra of amide CO groups of unsolvated and solvated PRL with a solvent-to-drug molar ratio of 100% (D). FTIR spectra of unsolvated PRL are denoted in green, hydrated PRL in orange, and PRL with heavy water in blue. The wavenumbers corresponding to the amide CO groups of unsolvated and solvated PRL and LID are denoted in the respective colors. | ||
To further investigate the isotopic effect on the interactions of PRL with water, DFT calculations were performed to calculate the vibrational frequencies of the amide CO groups of PRL with water and heavy water. Since only the interactions of PRL with water and heavy water were of interest, a solvent-to-drug molar ratio of 100% was used to construct the structural models. It was assumed that the isotopic substitution had a negligible effect on the molecular structures of hydrated PRL, because heavy water and water have identical electronic structures.26,36 Thus, the structural model of PRL-heavy water was constructed by the substitution of hydrogen with deuterium atoms, without configurational changes of the structural model of hydrated PRL. In this regard, different interaction of PRL with water and heavy water would result in a variation of the shift between the two solvents, in contrast, a constant shift can be seen as indication of similar hydrogen bonding pattern. The binding sites of water and heavy water were restricted to the amide CO groups of PRL and LID. The simulated structural models of PRL with water and heavy water are shown in Fig. S2 (ESI†). Despite some differences in the wavenumbers between the experimental and simulated FTIR spectra, both experimental and theoretical analysis indicated that the interactions of PRL with water and heavy water resulted in red shifts of the amide CO groups of PRL (Fig. 2). A red shift of 2 cm−1 of the amide CO group of PRL with heavy water versus water was observed in the simulated FTIR spectra, consistent with those observed in the experimental FTIR spectra. Considering that the simulated configuration of PRL remained unchanged when interacting with water and heavy water, the difference of 2 cm−1 in the wavenumbers observed in the simulated FTIR spectra of PRL with water and heavy water, was attributed to the differences in the vibrational motions of the hydrogen and deuterium atoms. These findings rationalized the observations in the experimental FTIR spectra that the red shift of 2 cm−1 in the amide CO group of PRL with heavy water versus water was due to the isotopic substitution at the same binding sites of PRL.
O group of PRL.13
3.2. Effect of ethanol and ethylene glycol on the Tgs of PRL and LID and on the molecular interactions with those drugs
Based on a previous study, the anti-plasticizing effect of water was ascribed to a dimeric structure of PRL held together by a water bridge.13 The possible hydrated structure of PRL could be formed by two hydrogen bonds between the two amide CO groups of two PRL molecules and the two hydroxyl groups of one water molecule.13 From this consideration, the two hydroxyl groups in one water molecule could play an important role in the anti-plasticizing effect, “binding together” two drug molecules. Ethylene glycol thus was chosen as it contains two hydroxyl groups, potentially also facilitating the formation of ethylene glycol-bridged structures with PRL. Ethylene glycol was thus expected to be a possible anti-plasticizer for PRL. In contrast, ethanol was chosen because it only has one hydroxyl group and was thus assumed to be a plasticizer for PRL and LID.
| Experimental Tg (K) | Theoretical Tg (K) | ΔTg (K) | |
|---|---|---|---|
| a The experimental Tgs of unsolvated LID and LID with ethanol were calculated based on the co-amorphous PRL-LID systems, using a previously reported approach.12 | |||
| PRL | 219.4 ± 0.1 | — | — |
| PRL with ethanol | 204.1 ± 0.3 | 190.5 | 13.6 |
| PRL with ethylene glycol | 215.4 ± 1.0 | 209.4 | 6.0 |
| LID | 209.8 ± 0.5a | — | — |
| LID with ethanol | 188.9 ± 0.5a | 185.9 | 3.0 |
O, amide N–H, and amine groups of PRL and LID with ethanol and ethylene glycol were investigated first. As shown in Fig. 3, with the addition of ethanol and ethylene glycol, red shifts of the amide CO groups (1682 cm−1) of PRL and LID were observed. For both solvated drugs, the shifts of the amide N–H groups of PRL (1519 cm−1) and LID (1492 cm−1) were less profoundly changed compared with those of the amide CO groups. No shifts of the N–H groups (3296 cm−1), attributed to the amide N–H groups of PRL and LID and the secondary amine group of PRL, were observed for the solvated amorphous PRL and LID samples. The peak shifts of the amide CO, amide N–H, and amine groups of PRL and LID with ethanol and ethylene glycol exhibited a similar trend as the changes reported in previous studies for hydrated amorphous PRL and LID.13,22 Zeglinski et al. observed a pronounced shift of the amide CO group in the FTIR spectra of N-methylacetamide (NMA), in comparison to the amide N–H group, indicating the formation of hydrogen bonds at the amide CO group of NMA with water.38 Thus, the amide CO groups of PRL and LID exhibited a higher potential to interact with ethanol and ethylene glycol compared to the amide N–H and amine groups, similarly to their interactions with water.
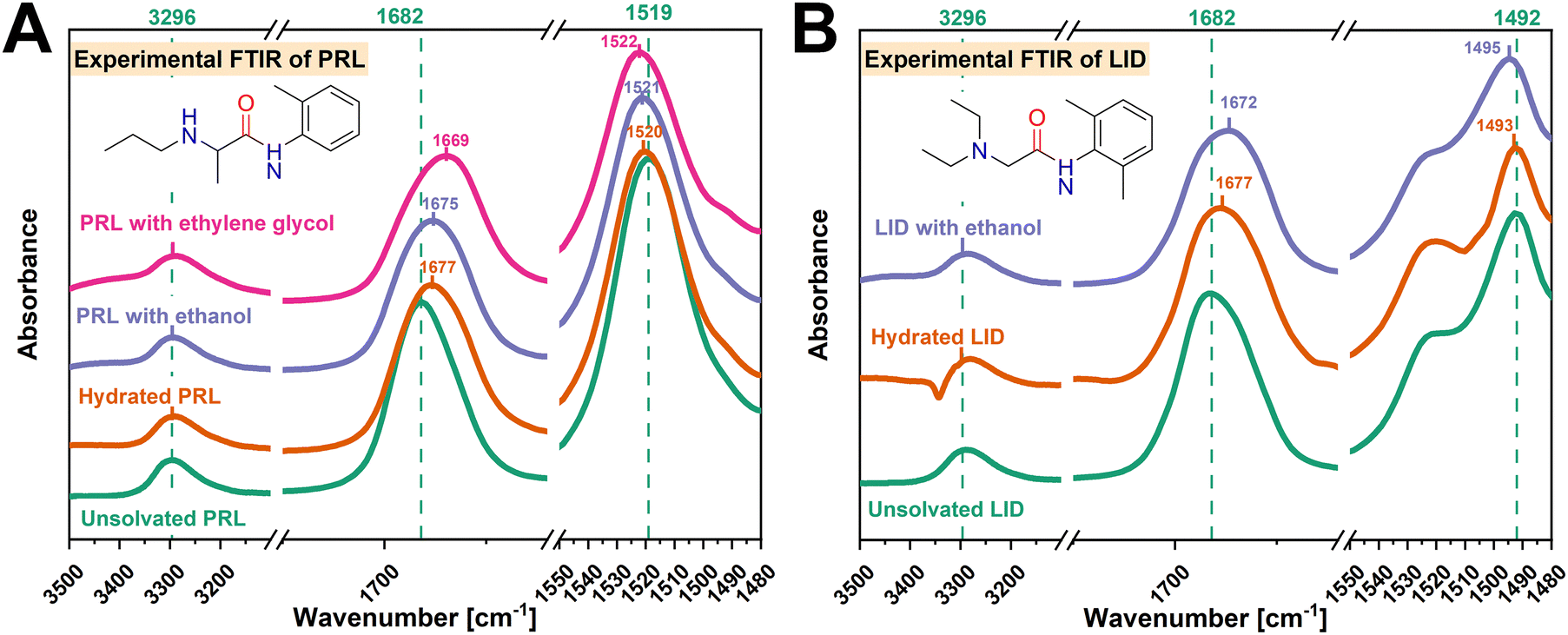 | ||
| Fig. 3 Comparison of experimental FTIR spectra between unsolvated and solvated PRL (A) and LID (B) with a drug-to-solvent molar ratio of 50%. The FTIR spectra of unsolvated PRL and LID are denoted in green, hydrated PRL and LID in yellow, PRL and LID with ethanol in blue, and PRL with ethylene glycol in pink. The wavenumbers corresponding to the amide CO, amide N–H, and amine groups of unsolvated and solvated PRL and LID are denoted in the respective colors. | ||
O groups of PRL and LID exhibited a high potential to interact with ethanol and ethylene glycol. The vibrational frequencies of the amide CO groups of the structural models of PRL and LID with ethanol and ethylene glycol at a solvent-to-drug molar ratio of 100% were calculated by DFT. The simulated structural models of PRL and LID with ethanol and ethylene glycol are shown in Fig. S2 (ESI†). In Fig. 4, the experimental vibrational frequencies of the amide CO groups of solvated PRL and LID are compared with those of the simulated frequencies. This comparative analysis involved referencing against the vibrational frequencies of hydrated PRL and LID. Although there are some differences in the wavenumbers between the experimental and simulated FTIR spectra, the shifts of the amide CO groups of PRL and LID with water, ethanol and ethylene glycol exhibited a similar trend. In both simulated and experimental FTIR spectra, the amide CO groups of hydrated PRL and LID exhibited the smallest shifts, compared to those with ethanol and ethylene glycol.
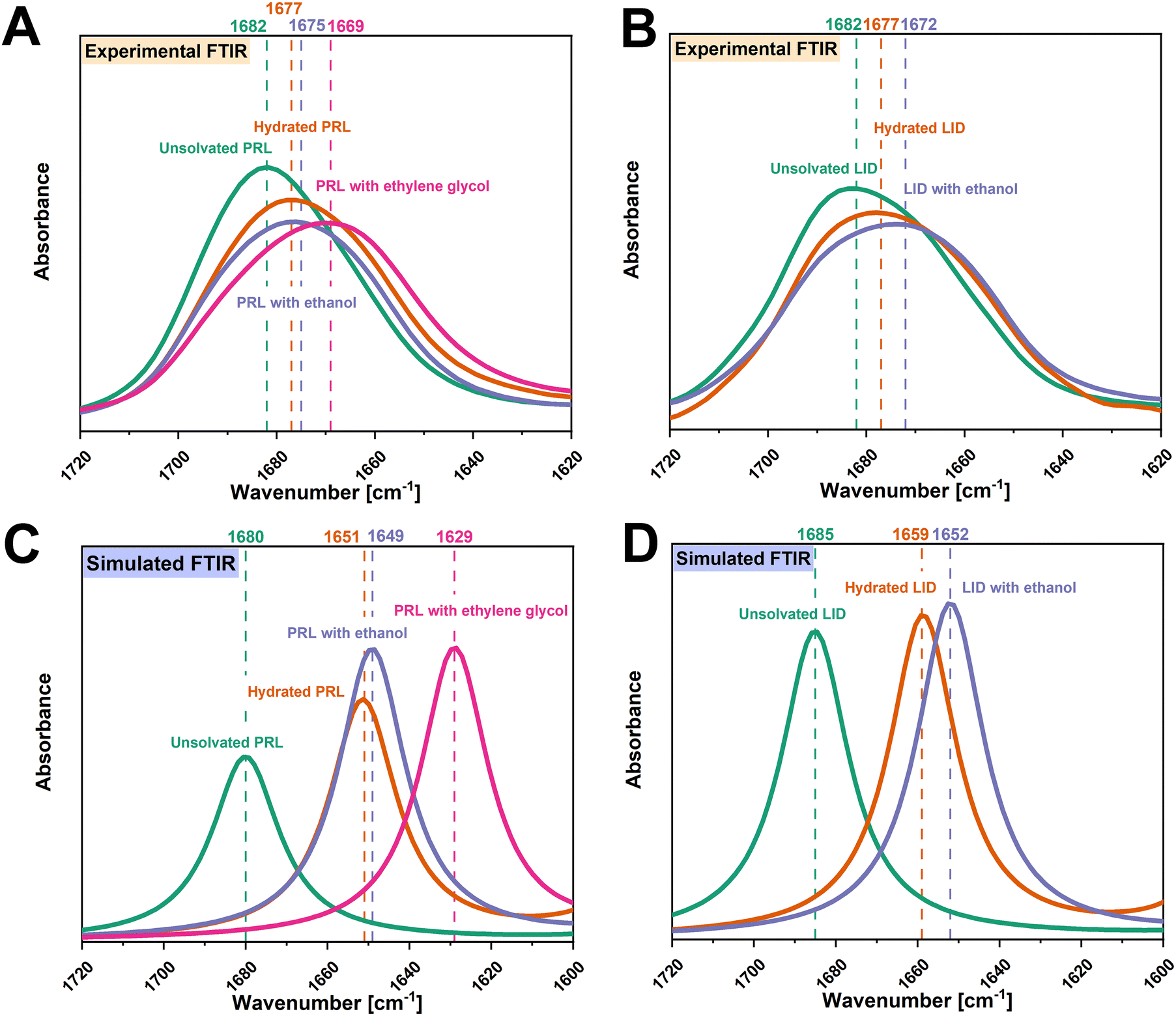 | ||
| Fig. 4 Experimental FTIR spectra of the amide CO groups of unsolvated and solvated PRL (A) and LID (B) with a solvent-to-drug molar ratio of 50%. Simulated FTIR spectra of amide CO groups of unsolvated and solvated PRL (C) and LID (D) with a solvent-to-drug molar ratio of 100%. The FTIR spectra of unsolvated PRL and LID are denoted in green, hydrated PRL and LID in yellow, PRL and LID with ethanol in blue, and PRL with ethylene glycol in pink. The wavenumbers corresponding to the amide CO groups of unsolvated and solvated PRL and LID are denoted in the respective colors. | ||
O, amide N–H, and amine groups of PRL and LID with ethanol and ethylene glycol followed a similar trend as the shifts observed in hydrated PRL and LID. According to the hypothesis of “dimer formation”, the two hydroxyl groups of one ethylene glycol molecule could interact with the two amide CO groups of two PRL molecules.13 These interactions of PRL with ethylene glycol would then be expected to be similar to those observed with water, leading to an anti-plasticizing effect of ethylene glycol on PRL.13 In contrast, ethanol could not act as an anti-plasticizer for PRL and LID, because one ethanol molecule could not interact with the two amide CO groups of PRL and LID.13 However, it was found that although the addition of ethanol and ethylene glycol resulted in a reduction of the Tgs of PRL and LID, the experimental Tgs of those solvated drugs were higher than the theoretical Tgs calculated using the Gordon–Taylor equation. These findings indicate that although ethanol and ethylene glycol overall acted as plasticizers for PRL and LID, they also exhibited “anti-plasticizing potential” due to the observed molecular interactions. In addition, the “anti-plasticizing potential” of ethanol on PRL was more pronounced compared to that of ethylene glycol, assessed by comparing the experimental and theoretical Tgs of PRL with ethanol and ethylene glycol. Thus, the observations in this study do not support the hypothesis that the anti-plasticizing effect of water is related to a dimer formation with a solvent bridge.13
3.3. Simulated structural models of PRL and LID at a solvent-to-drug molar ratio of 50%
In order to visualize the molecular interactions of PRL and LID with water, ethanol, and ethylene glycol at a solvent-to-drug molar ratio of 50%, quantum chemical simulations were performed. The structural models of solvated drugs were generated randomly, by changing the orientations and distances among the components, using two drug molecules of PRL or LID, and one solvent molecule of water, ethanol, or ethylene glycol. The random orientations of molecules in an amorphous phase result in numerous stationary conformational states on the potential energy surface.39 The model with the lowest binding energy was assumed to be the most reasonable model on the potential energy surface for analyzing the structural features, compared with the other constructed model.40 The simulated structural models are shown in Fig. 5. The binding energies of the investigated structural models are shown in Table S2 (ESI†). Hydrogen bonding in hydrated PRL, PRL with ethanol, PRL with ethylene glycol, and hydrated LID, was formed via the acceptors from the amide CO groups of PRL and LID. The structural models thus gave an indication that the amide CO groups of PRL and LID were involved into the interactions with water, ethanol, and ethylene glycol, respectively, which is consistent with the observations in the experimental FTIR spectra. However, for hydrated LID, the model indicated the formation of another hydrogen bond between the amide N–H group of LID and the hydroxyl group of water. In addition, for LID with ethanol, the hydrogen bonding was formed in a different way compared with the other structural models and was based on the amide N–H groups from two LID molecules as donors and the hydroxyl group from one ethanol as an acceptor. The possible structural models of solvated LID indicated that for pure amorphous LID, the amide N–H groups of LID might be involved in the molecular interactions with water and ethanol. However, in the experimental FTIR spectra of solvated LID, the interactions involving the amide N–H groups of LID with water and ethanol were not observed, which were predicted from the experimental FTIR spectra of co-amorphous systems of PRL and LID. Solvent-bridged structures of PRL and LID, via two hydrogen bonds with the amide CO groups from two drug molecules of PRL and LID, were not observed in any of simulated solvated structural models. Overall, these observations do not support the hypothesis that the anti-plasticizing effect of water was due to the interactions of one water molecule with the two amide CO groups of two drug molecules of PRL and LID.13
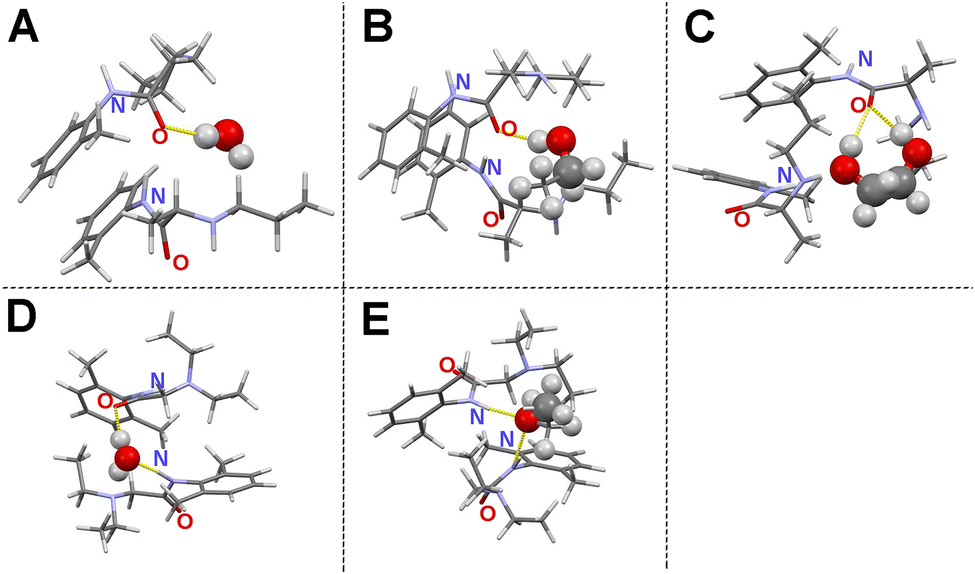 | ||
| Fig. 5 Simulated structural models of hydrated PRL (A), PRL with ethanol (B), PRL with ethylene glycol (C), hydrated LID (D), and LID with ethanol (E) at a solvent-to-drug molar ratio of 50%. Carbon atoms are denoted in dark grey, hydrogen in light grey, oxygen in red, and nitrogen in light purple. The hydrogen bonding with the amide groups of PRL and LID is marked in yellow. | ||
It is known for crystalline structures that dimer formation affects the stability of crystalline solids, specifically due to the long-range order existing in the crystal.41 Dimer structures in the unit cell are repeated over many orders of magnitude in a crystal, thus affecting its physicochemical properties.42 However, for amorphous PRL and LID, the order of the motifs such as the dimeric structure of drugs, is not replicated due to the lack of long-range order. Thus, the anti-plasticizing effect of water on amorphous PRL and LID may not be solely correlated with the individual dimeric structures of PRL and LID bridged by water.
3.4. Binding energy consideration on the anti-plasticizing effect of water
The plasticizing effect of water on amorphous drugs could effectively raise the potential energy minima of the conformational states through the intermolecular interactions.43 In a previous study the plasticizing abilities of additives on proteins according to the differences in their binding energies were investigated based on a computer-aided molecular design platform.44 The results of that study indicated that a low binding energy was indicative of a strong binding affinity between a plasticizer and a protein, thus leading to a strong plasticizing effect.44 The strong interactions between plasticizers and amorphous drugs can break the original intra- and inter-molecular network of the anhydrous drugs, leading to an increase in molecular mobility and a subsequent decrease in the Tgs of the drugs.45 However, it has been shown that intermolecular interactions with water can also reduce the potential energy barrier of some macromolecules more effectively than sterically hindered intramolecular interactions,46,47 and thus may show an anti-plasticizing effect of water. In addition, Wang et al. have reported an anti-plasticizing effect of urea/water mixtures on starch molecules.48 The anti-plasticizing effect of urea/water mixtures benefited from weak intermolecular interactions between water and starch.48 These previous findings highlight the important role of molecular interactions and the corresponding binding energies in influencing the effect of water on amorphous drugs.As discussed above, in both simulated and experimental FTIR spectra, the amide CO groups of hydrated PRL and LID exhibited the smallest shifts, compared to those with ethanol and ethylene glycol. In Fig. 6, the experimental and simulated frequency shifts of PRL and LID solvated with water, ethanol, and ethylene glycol are shown. The frequency shifts of the amide CO groups observed in the FTIR spectra showed a linear correlation with the binding energies of the structural models for both PRL and LID. However, an exception was noted in the experimental frequency shift of LID with ethanol. This deviation showed a potential unique interaction between LID and ethanol, distinct from the other solvated drugs, as shown in Fig. 5. Overall, both the experimental and theoretical analyses indicated that increased binding energies of solvated drugs led to smaller shifts in the amide CO groups of PRL and LID, consistent with previous findings.49,50 The binding energies of hydrated PRL and LID were higher than those with ethanol and ethylene glycol. The higher binding energies of hydrated PRL and hydrated LID indicated that the hydrogen bonds formed in hydrated PRL and LID were weaker compared to those formed with ethanol and ethylene glycol. In addition, the binding energy of hydrated PRL, determined based on the electronic structure calculations, remained constant with the deuterium substitution, when replacing water with heavy water. However, the increased mass of deuterium in the harmonic oscillator results in a shallower potential energy well for interactions with heavy water, compared with hydrogen.51 Furthermore, the small red shifts observed in the amide CO group of PRL when comparing heavy water with water, indicate that the binding strength with heavy water was slightly stronger than with water.52 These findings are consistent with the similar Tg patterns observed for PRL with water and heavy water, but the slightly lower Tg values of PRL with heavy water than with water.
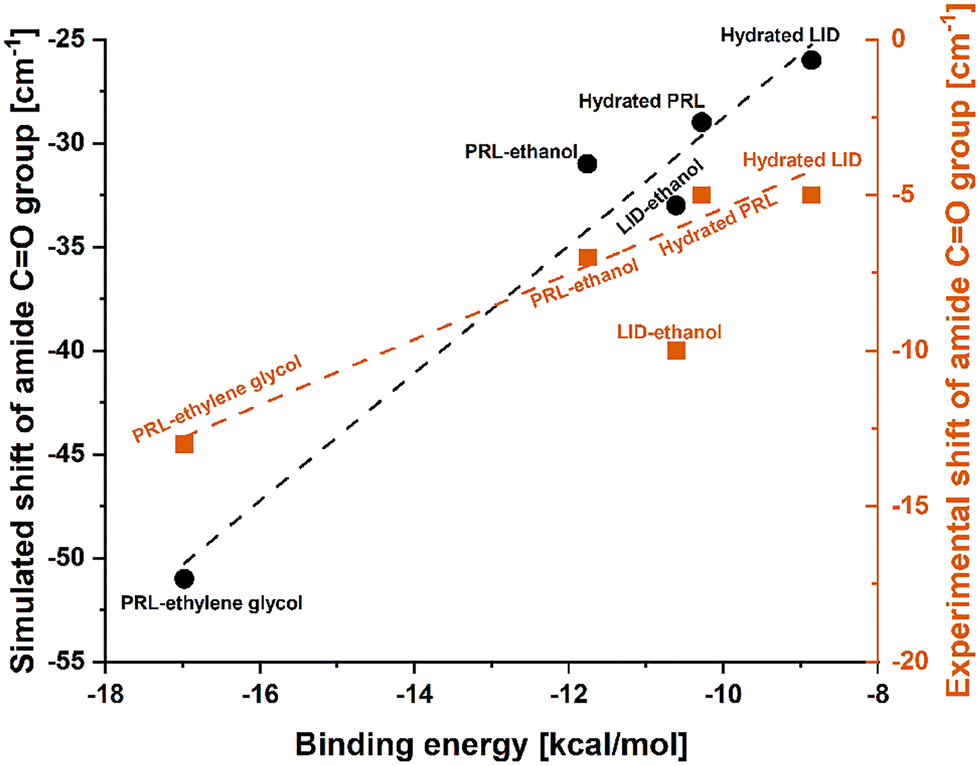 | ||
| Fig. 6 Linear regression fitted to the experimental shifts (R2 = 0.970) and simulated shifts (R2 = 0.955) of the amide CO groups of PRL and LID with water, ethanol, and ethylene glycol, as a function of the corresponding calculated binding energies. The experimental shift of LID with ethanol did not follow the linear relationship with the calculated binding energy. | ||
Overall, the weak interactions of PRL and LID with water could thus be favorable to induce an anti-plasticizing effect of water.
A binding energy decomposition analysis was performed to further explore the nature of the interactions between PRL and LID with water, ethanol and ethylene glycol. Using the SAPT approach, the binding energy was decomposed into electrostatic, induction, exchange, and dispersion contributions. Within the formation of hydrogen bonding, the electrostatic, induction, and dispersion effects contribute as attractive forces, and the exchange effect contributes repulsively. The percentage of each contribution to the total attractive force was calculated as:53
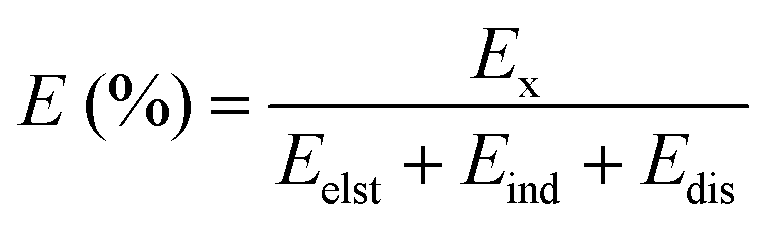 | (3) |
The binding energies and their decompositions obtained with SAPT analysis are summarized in Table S3 (ESI†). The contribution percentages to the total attractive forces are shown in Fig. 7. The electrostatic effect was dominant in the molecular interactions of PRL and LID for the selected solvents, contributing 64.3% for hydrated PRL, 62.1% for hydrated LID, 59.0% for PRL with ethanol, 58.5% for PRL with ethylene glycol, and 54.0% for LID with ethanol. The electrostatic contributions decreased in the order of hydrated PRL > hydrated LID > PRL with ethanol > PRL with ethylene glycol > LID with ethanol. This was matched by the order of anti-plasticizing potentials of hydrated PRL > hydrated LID > PRL with ethanol > PRL with ethylene glycol > LID with ethanol, as discussed previously. Overall, the binding energy decomposition analysis indicated that the electrostatic contributions of hydrated PRL and LID were higher compared with the other solvated drugs with ethanol and ethylene glycol.
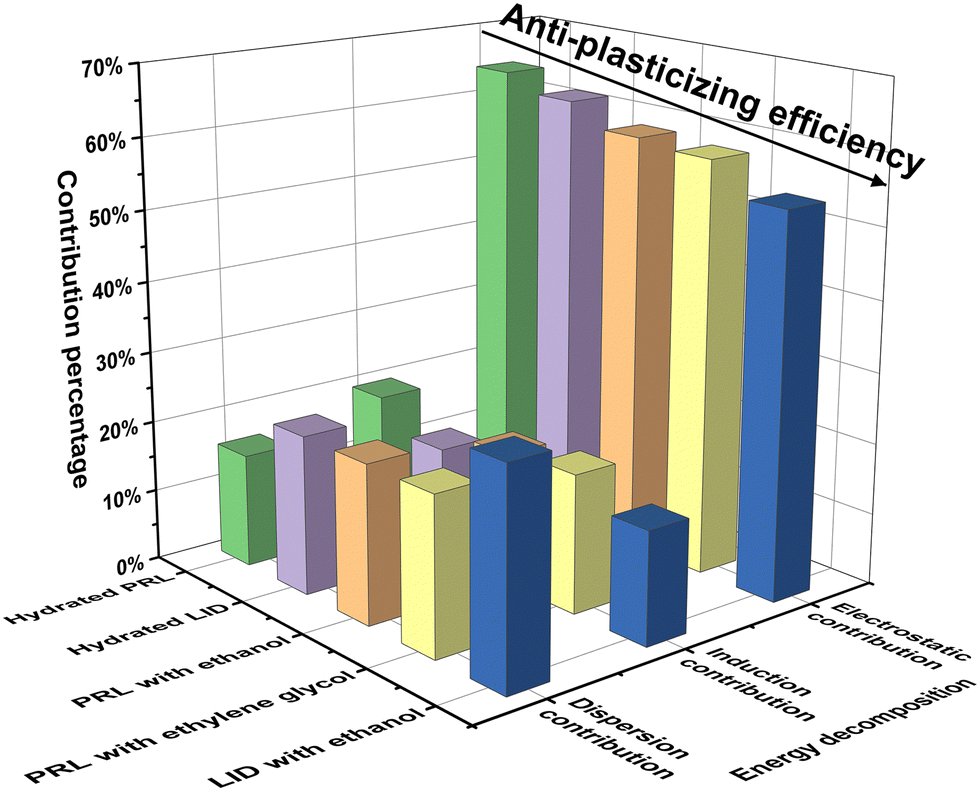 | ||
| Fig. 7 Contribution percentages of the electrostatic, induction, and dispersion effects to the total attractive forces for the structural models of hydrated PRL, hydrated LID, PRL with ethanol, PRL with ethylene glycol, and LID with ethanol, with a solvent-to-drug molar ratio of 100% computed with SAPT. The binding sites of the investigated solvents were restricted at the amide CO groups of PRL and LID. | ||
The percentage contributions of electrostatic forces of hydrated PRL and LID were larger than those of the other structural models. Some studies have already indicated a potential correlation between high electrostatic contributions and an increased Tg of amorphous compounds. Wojnarowska et al. suggested that electrostatic interactions, existing between the hydrochloride salts of PRL and LID could induce an increased Tg of the co-amorphous systems of PRL-HCl and LID-HCl compared with co-amorphous systems of PRL and LID.54,55 Chuang et al. found that the specific electrostatic interactions between potato starch and calcium chloride could stabilize the polymeric matrix and increase the Tg of the potato starch.56 In contrast, it was also found that bulky internal plasticizers effectively plasticized (meth)acrylate polymers by weakening the electrostatic interactions with the polymer.57 Thus, the relative contribution from electrostatic interactions observed in the solvated drugs could be a favorable indicator for assessing the anti-plasticizing potentials of water, ethanol, and ethylene glycol with respect to PRL and LID.
4. Conclusions
In this study, the nature of the anti-plasticizing effect of several solvents on PRL and LID was investigated. Water was earlier shown to increase the Tgs of PRL and LID, and was ascribed for PRL to a dimeric structure of drugs held together by a water bridge. Heavy water was selected as a solvent, because the deuterium and hydrogen atoms are electronically identical. The binding sites of PRL with heavy water were identical to those with water. Ethanol and ethylene glycol were chosen as solvents based on their varying abilities to form hydrogen bonds with the amide CO groups of PRL and LID. Although both solvents showed a decrease in Tg, this decrease was less than expected, thus the solvents showed a certain anti-plasticizing potential. Experimental spectroscopic analysis and quantum chemistry simulations showed that the combination of weak hydrogen bonding and strong electrostatic forces contribute to an anti-plasticizing effect of water on PRL and LID.
Author contributions
Xiaoyue Xu: conceptualization, formal analysis, investigation, writing – original draft. Holger Grohganz: conceptualization, formal analysis, project administration, writing – review & editing, supervision. Thomas Rades: conceptualization, formal analysis, project administration, writing – review & editing, supervision.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Xiaoyue Xu acknowledges the China Scholarship Council (Grant 202008420212) for financial support.References
- L. Di, P. V. Fish and T. Mano, Drug. Discovery Today, 2012, 17, 486–495 CrossRef CAS PubMed.
- Q. Shi, S. M. Moinuddin and T. Cai, Acta Pharm. Sin. B, 2019, 9, 19–35 CrossRef PubMed.
- B. C. Hancock and G. Zografi, J. Pharm. Sci., 1997, 86, 1–12 CrossRef CAS PubMed.
- Y. Kawabata, K. Wada, M. Nakatani, S. Yamada and S. Onoue, Int. J. Pharm., 2011, 420, 1–10 CrossRef CAS PubMed.
- G. Kasten, K. Löbmann, H. Grohganz and T. Rades, Int. J. Pharm., 2019, 557, 366–373 CrossRef CAS PubMed.
- H. Grohganz, K. Löbmann, P. Priemel, K. Tarp Jensen, K. Graeser, C. Strachan and T. Rades, J. Drug Delivery Sci. Technol., 2013, 23, 403–408 CrossRef CAS.
- B. C. Hancock and G. Zografi, Pharm. Res., 1994, 11, 471–477 CrossRef CAS PubMed.
- A. Saleki-Gerhardt and G. Zografi, Pharm. Res., 1994, 11, 1166–1173 CrossRef CAS PubMed.
- A. Newman and G. Zografi, J. Pharm. Sci., 2019, 108, 1061–1080 CrossRef CAS PubMed.
- Y. I. Matveev, V. Y. Grinberg and V. B. Tolstoguzov, Food Hydrocol., 2000, 14, 425–437 CrossRef.
- S. R. Byrn, G. Zografi and X. Chen, Solid-State Properties of Pharmaceutical Materials, John Wiley & Sons, New York, 2017, ch. 15, pp. 213–230 Search PubMed.
- X. Xu, H. Grohganz and T. Rades, Mol. Pharm., 2022, 19, 3199–3205 CrossRef CAS PubMed.
- G. N. Ruiz, M. A.-O. X. Romanini, A. Hauptmann, T. Loerting, E. Shalaev, J. A.-O. Tamarit, L. A.-O. Pardo and R. A.-O. Macovez, Sci. Rep., 2017, 7, 7470 CrossRef PubMed.
- L. I. Blaabjerg, E. Lindenberg, K. Löbmann, H. Grohganz and T. Rades, Mol. Pharm., 2016, 13, 3318–3325 CrossRef CAS PubMed.
- M. Gordon and J. S. Taylor, J. Appl. Chem., 1952, 2, 493–500 CrossRef CAS.
- B. Kabtoul and M. A. Ramos, Phys. Status Solidi A, 2011, 208, 2249–2253 CrossRef CAS.
- C. A. Angell, J. M. Sare and E. J. Sare, J. Phys. Chem., 1978, 82, 2622–2629 CrossRef CAS.
- A. Nagoe and M. Oguni, AIP Conf. Proc., 2008, 982, 185–188 CrossRef CAS.
- R. Simha and R. F. Boyer, J. Chem. Phys., 1962, 37, 1003–1007 CrossRef CAS.
- SciFinder, https://scifinder-n.cas.org/? referrer = scifinder.cas.org, (accessed Feburary 2024).
- A. Kalra, P. Tishmack, J. W. Lubach, E. J. Munson, L. S. Taylor, S. R. Byrn and T. Li, Mol. Pharm., 2017, 14, 2126–2137 CrossRef CAS PubMed.
- X. Xu, T. Rades and H. Grohganz, Int. J. Pharm., 2024, 651, 123807 CrossRef CAS PubMed.
- T. Lu, Molclus program (Version 1.10), https://www.keinsci.com/research/molclus.html (accessed 8, 1, 2023) Search PubMed.
- J. J. P. Stewart. MOPAC2009, Stewart Computational Chemistry, Colorado Springs, CO, USA, 2008 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- S. Scheiner and M. Čuma, J. Am. Chem. Soc., 1996, 118, 1511–1521 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- Database of Frequency Scale Factors for Electronic Model Chemistries, https://comp.chem.umn.edu/freqscale/version3b2.htm, (accessed February, 2024).
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- T. M. Parker, L. A. Burns, R. M. Parrish, A. G. Ryno and C. D. Sherrill, J. Chem. Phys., 2014, 140, 094106 CrossRef PubMed.
- R. M. Parrish, L. A.-O. Burns, D. G. A. Smith, A. A.-O. Simmonett, A. E. DePrince, 3rd, E. A.-O. Hohenstein, U. Bozkaya, A. Y. Sokolov, R. A.-O. Di Remigio, R. M. Richard, J. F. Gonthier, A. M. James, H. R. McAlexander, A. Kumar, M. Saitow, X. Wang, B. P. Pritchard, P. Verma, H. F. r A.-O. Schaefer, K. Patkowski, R. A.-O. King, E. A.-O. Valeev, F. A. Evangelista, J. M. Turney, T. A.-O. Crawford and C. A.-O. Sherrill, J. Chem. Theory Comput., 2017, 13, 3185–3197 CrossRef CAS PubMed.
- I. K. Petrushenko, N. I. Tikhonov and K. B. Petrushenko, Diamond Relat. Mater., 2020, 107, 107905 CrossRef CAS.
- I. B. Rietveld, M. A. Perrin, S. Toscani, M. Barrio, B. Nicolai, J. L. Tamarit and R. Ceolin, Mol. Pharm., 2013, 10, 1332–1339 CrossRef CAS PubMed.
- R. Ceolin, M. Barrio, J. L. Tamarit, N. Veglio, M. A. Perrin and P. Espeau, J. Pharm. Sci., 2010, 99, 2756–2765 CrossRef CAS PubMed.
- J. S. Mugridge, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2012, 134, 2057–2066 CrossRef CAS PubMed.
- D. Wade, Chem. – Biol. Interact., 1999, 117, 191–217 CrossRef CAS PubMed.
- X. Xu, T. Rades and H. Grohganz, Eur. J. Pharm. Biopharm., 2023, 186, 1–6 CrossRef CAS PubMed.
- N. S. Myshakina, Z. Ahmed and S. A. Asher, J. Phys. Chem. B, 2008, 112, 11873–11877 CrossRef CAS PubMed.
- M. T. Ruggiero, M. Krynski, E. O. Kissi, J. Sibik, D. Markl, N. Y. Tan, D. Arslanov, W. van der Zande, B. Redlich, T. M. Korter, H. Grohganz, K. Löbmann, T. Rades, S. R. Elliott and J. A. Zeitler, Phys. Chem. Chem. Phys., 2017, 19, 30039–30047 RSC.
- J. F. C. Silva, M. T. S. Rosado and M. E. S. Eusébio, J. Mol. Struct., 2021, 1242, 1230709 CrossRef.
- M. Yamasaki, W. Li, D. J. D. Johnson and J. A. Huntington, Nature, 2008, 455, 1255–1258 CrossRef CAS PubMed.
- H. Tong, P. Tan and N. Xu, Sci. Rep., 2015, 5, 15378 CrossRef CAS PubMed.
- J. Gupta, C. Nunes and S. Jonnalagadda, Mol. Pharm., 2013, 10, 4136–4145 CrossRef CAS PubMed.
- F. Abookleesh, F. E. S. Mosa, K. Barakat and A. Ullah, Polymers, 2022, 14, 3690 CrossRef CAS PubMed.
- G. Zografi and A. Newman, J. Pharm. Sci., 2017, 106, 5–27 CrossRef CAS PubMed.
- T. A. Shmool, M. Batens, J. Massant, G. Van den Mooter and J. A. Zeitler, Eur. J. Pharm. Biopharm., 2019, 144, 244–251 CrossRef CAS PubMed.
- J. Kölbel, M. L. Anuschek, I. Stelzl, S. Santitewagun, W. Friess and J. A. Zeitler, J. Phys. Chem. Lett., 2024, 15, 3581–3590 CrossRef PubMed.
- J. L. Wang, F. Cheng and P. X. Zhu, Carbohydr. Polym., 2014, 101, 1109–1115 CrossRef CAS PubMed.
- D. Khamar, J. Zeglinski, D. Mealey and C. Rasmuson, J. Am. Chem. Soc., 2014, 136, 11664–11673 CrossRef CAS PubMed.
- J. Zeglinski, M. Kuhs, K. R. Devi, D. Khamar, A. C. Hegarty, D. Thompson and Å. C. Rasmuson, Cryst. Growth Des., 2019, 19, 2037–2049 CrossRef CAS.
- Y. Bai, B. W. J. Chen, G. Peng and M. Mavrikakis, Catal. Sci. Technol., 2018, 8, 3321–3335 RSC.
- A. D. Stephens, J. Kölbel, R. Moons, C. W. Chung, M. T. Ruggiero, N. Mahmoudi, T. A. Shmool, T. M. McCoy, D. Nietlispach, A. F. Routh, F. Sobott, J. A. Zeitler and G. S. Kaminski Schierle, Angew. Chem., 2023, 135, e202212063 CrossRef PubMed.
- S. Emamian, T. Lu, H. Kruse and H. Emamian, J. Comput. Chem., 2019, 40, 2868–2881 CrossRef CAS PubMed.
- Z. Wojnarowska, J. Zotowa, J. Knapik-Kowalczuk, L. Tajber and M. Paluch, Eur. J. Pharm. Sci., 2019, 134, 93–101 CrossRef CAS PubMed.
- Z. Wojnarowska, W. Smolka, J. Zotova, J. Knapik-Kowalczuk, A. Sherif, L. Tajber and M. Paluch, Phys. Chem. Chem. Phys., 2018, 20, 27361–27367 RSC.
- L. Chuang, N. Panyoyai, L. Katopo, R. Shanks and S. Kasapis, Food Chem., 2016, 199, 791–798 CrossRef CAS PubMed.
- M. Klähn, R. Krishnan, J. M. Phang, F. C. H. Lim, A. M. van Herk and S. Jana, Polymer, 2019, 179, 121635 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00995a |
| This journal is © the Owner Societies 2024 |