
Elucidating the degradation mechanism of the nerve agent A-234 using various detergents: a theoretical investigation†
Rongxin
Shi
a,
Lin
Zhang
 a,
Denghui
Ma
ab and
Zexing
Cao
*a
a,
Denghui
Ma
ab and
Zexing
Cao
*a
aState Key Laboratory of Physical Chemistry of Solid Surfaces and Fujian Provincial Key Laboratory of Theoretical and Computational Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, China. E-mail: zxcao@xmu.edu.cn; Tel: +86-592-2186081
bSchool of New Energy, Ningbo University of Technology, Ningbo, 315336, China
First published on 6th May 2024
Abstract
A-234 (ethyl N-[1-(diethylamino)ethylidene]phosphoramidofluoridate) is one of the highly toxic Novichok nerve agents, and its efficient degradation is of significant importance. The possible degradation mechanisms of A-234 by H2O, H2O2, NH3, and their combinations have been extensively investigated by using density functional theory (DFT) calculations. According to the initial intermolecular interaction and the proton transfer patterns between the detergent and the substrate A-234, the A-234 degradation reaction is classified into three categories, denoted as A, B, and C. In modes A and B, the degradation of A-234 by H2O2, H2O, and NH3 is initiated by the nucleophilic attack of the O or N atom of the detergent on the P atom of A-234, coupled with the proton transfer from the detergent to the O or N atom of A-234, whereas in mode C, the direct interaction of H2N–H with the F–P bond of A-234 triggers ammonolysis through a one-step mechanism with the formation of H–F and N–P bonds. Perhydrolysis and hydrolysis of A-234 can be remarkably promoted by introducing the auxiliary NH3, and the timely formed hydrogen bond network among detergent, auxiliary, and substrate molecules is responsible for the enhancement of degradation efficiency.
1. Introduction
A-series nerve agents (NAs), also known as Novichok agents, represent a subclass of extremely poisonous organophosphorus compounds, structurally resembling the well-known G-series and V-series nerve agents.1–4 Notable for their potency, these NAs have attracted widespread international attention, particularly due to their involvement in high-profile incidents in 2018.5 Similar to the G-series and V-series NAs, A-series NAs exert the poisoning effect by inhibiting the activity of acetylcholinesterase (AchE), the enzyme responsible for breaking down acetylcholine. As a result, the accumulation of acetylcholine in synapses occurs, impairing the physiological function of living organisms.6 The prolonged presence of NAs in the environment raises apprehensions about long-term consequences, and a comprehensive understanding of their degradation mechanisms is highly required to develop rapid, convenient and efficient degradation strategies.Great efforts have been made to decontaminate NAs effectively, both experimentally and theoretically.7–27 Metal–organic frameworks (MOFs), leveraging their catalytic properties, have demonstrated that they can facilitate the degradation of NAs.7–13 Additionally, various metal oxides, such as MgO, Al2O3, TiO2, Cu2O, CuO, and ZnO, have considerable potential for adsorbing and decomposing NAs.14–21 The biodegradation of NAs by enzymes under mild conditions, such as phosphotriesterase (PTE) successfully used in degrading organophosphorus pesticides, has also received considerable interest.22–27
In addition, active detergents were used for detoxification of NAs in the past few decades. For example, hydrogen peroxide (H2O2), recognized for its noncorrosive and nonhazardous nature, has shown environmentally friendly behavior in degradation of organophosphorus NAs.28–34 H2O2 in combination with appropriate activators (i.e., bicarbonate, citrate, and molybdate) may decontaminate nerve agents VX (O-ethyl S-[2-(diisopropylamino)ethyl]methylphosphonothioate) and GD (pinacolyl methylphosphonofluoridate) through perhydrolysis, resulting in the formation of their non-toxic phosphonates.30 The microporous activated carbons may adsorb these chemical warfare agents, and the adsorbed nerve agents VX, sarin (O-isopropyl methylphosphonofluoridate), and HD (bis(2-chloroethyl)sulfide) can be efficiently decomposed by using the hydrogen peroxide solution under ambient conditions, which provides a potentially environmentally friendly approach for the detoxification of adsorbed NAs.35
NAs are highly toxic organophosphates (OPs) and they can be degraded through hydrolysis, and H2O as a green detergent might be used for decontamination of NAs. However, previous studies showed that the nerve agent did not react with H2O, and the hydrolysis of VX depends on the pH and temperature.36 In particular, the hydrolytic product of VX from the P–O bond cleavage is almost as toxic as VX in neutral to weakly basic solutions and further detoxification treatment is urgently required. Theoretically, Ramasami and co-workers studied the hydrolysis mechanisms of A-234 using DFT calculations, and the hydrolysis at the acetamidine center is predicted to be more favorable thermodynamically than that at the phosphinate center, leading to the hydrolysis product with the presence of the P–F bond.37 Alternatively, ammonolysis is another potential strategy to degrade NAs.38 Das and co-workers investigated the ammonolysis mechanisms of a nerve agent simulant, O,S-dimethyl methylphosphonothiolate (DMPT), using DFT calculations and molecular dynamics simulations, the stepwise degradation pathway is predicted to be more favorable than the concerted one, and NH3 as the catalyst can effectively reduce the activation energy of the stepwise reaction.39 Besides, Mandal compared the decontamination of nerve agent tabun ([dimethylamino(ethoxy)phosphoryl]formonitrile) by using NH3 and H2O, based on DFT and MP2 calculations, and the former is a more efficient degradation reagent.40
Despite such considerable efforts to deal with the G- and V-series NAs, the knowledge of Novichok agents is still quite limited even now.12,37,41 The existing literature data of A-series NAs primarily focus on their structural and spectroscopic properties from theoretical calculations and mass spectrometric studies.42–45 Note that the perhydrolysis mechanism of A-series NAs by H2O2 and the ammonolysis mechanism of A-series NAs by NH3 are still not well-understood to the best of our knowledge. Herein, the degradation of Novichok agent A-234, one of the A-series NAs as shown in Fig. 1, has been explored by extensive DFT calculations, and the differences in the degradation mechanism and efficiency by H2O, H2O2, and NH3 were discussed. Furthermore, the detoxification performances of different detergent combinations (such as H2O2/H2O, H2O2/NH3, H2O/H2O, and H2O/NH3, where the former serves as the nucleophile and the latter serves as a proton donor in these combinations) toward A-234 have been compared.
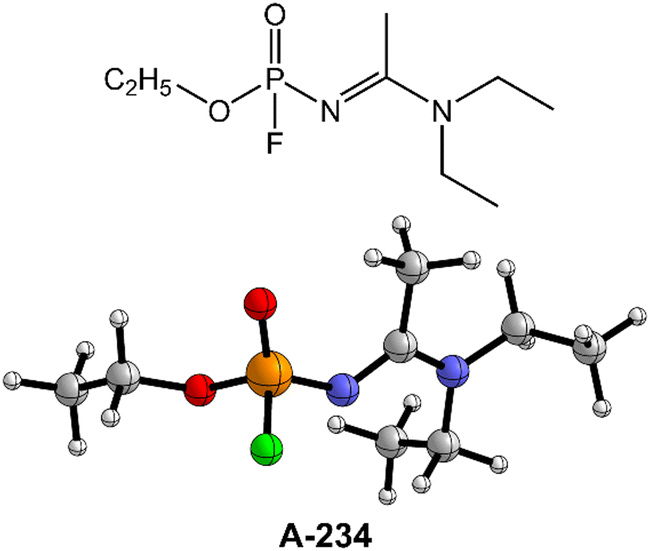 | ||
| Fig. 1 The molecular structure of A-234. | ||
2. Computational details
All calculations were carried out using the Gaussian 16 program.46 Geometric optimizations were performed at the M06-2X47/6-311++G(d,p)48–52 level of theory. The vibrational analysis at the same level was used to confirm the optimized local minima without an imaginary frequency and the transition-state structures with only one imaginary frequency. Furthermore, all transition states were confirmed to connect corresponding reactants and products by the intrinsic reaction coordinate (IRC) calculations.53 Single-point energy calculations with the implicit solvation model based on density (SMD)54 for the solvent water were carried out at the M06-2X/6-311++G(d,p)/SMD level of theory. Different DFT functionals (e.g., M06-2X-D3,55 B3LYP,56 PBE0,57 MN15,58 and ωB97X-D59) and basis sets (e.g., 6-311++G(2d,2p)48–52 and def2-TZVP60) were considered, but no qualitatively different results were found (see Tables S1 and S2 in the ESI†). Unless otherwise stated, here all the thermochemical properties were discussed based on the predicted results at the M06-2X/6-311++G(d,p)/SMD//M06-2X/6-311++G(d,p) level of theory under the standard conditions (298.15 K and 1 atm). Fig. S1–S3 (ESI†) show the predicted relative free energy profiles for the degradation of A-234 by H2O2, H2O, and NH3, based on the gas-phase optimized structures and vibrational frequencies combined with the single-point energy calculation including the solvent effect, showing good agreement with those obtained through M06-2X/6-311++G(d,p)/SMD geometry optimizations and vibrational frequency analyses, and similarly, their corresponding equilibrium geometries of the species involved in the reaction are of good consistency. All the geometric structures are visualized by using CYLview.613. Results and discussion
3.1 A-234 degradation using different detergents
The degradation mechanism of A-234 by H2O2, H2O, and NH3 was examined first. At the initial stage of the reaction, the nucleophilic attack of O atoms for H2O and H2O2 or the N atom for NH3 on the P center of A-234 was considered. During this process, protons from the detergents may be transferred to different atomic sites in A-234, resulting in various degradation products. Accordingly, we categorize the degradation process triggered by the nucleophilic attack of the detergent on A-234 into three reaction modes A, B, and C (see Scheme 1), where protons were transferred to the O, N, and F atoms of A-234, respectively.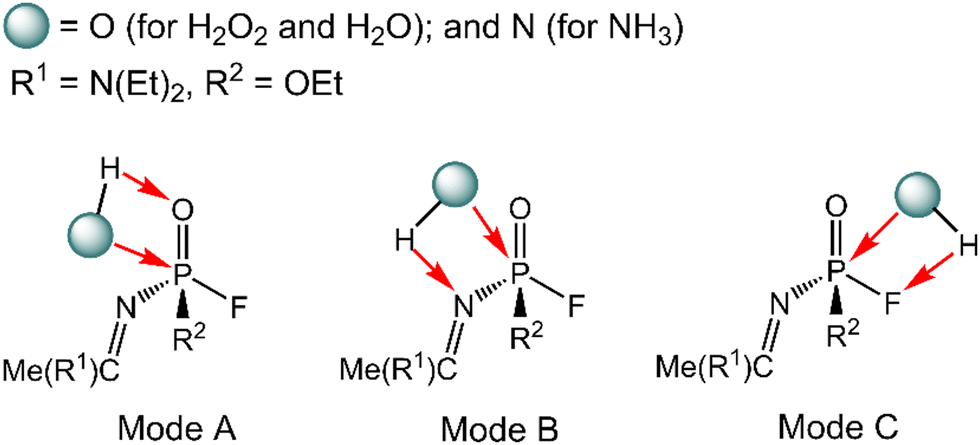 | ||
| Scheme 1 Plausible modes for the nucleophilic attack of H2O2, H2O, and NH3 on A-234. | ||
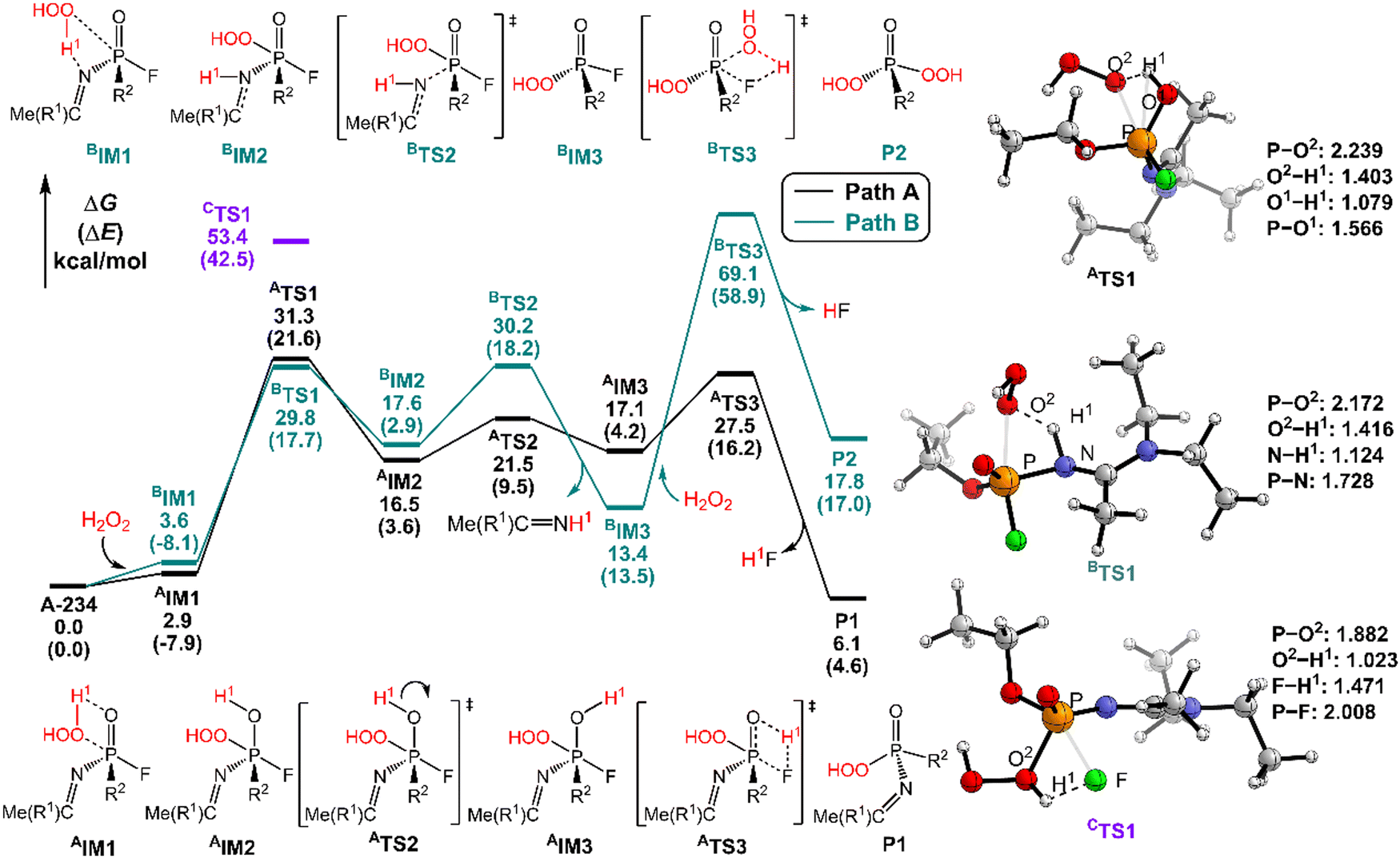 | ||
| Fig. 2 Predicted relative free energy profiles (ΔG in kcal mol−1) and electronic energies (ΔE in parentheses) of the species involved in the perhydrolysis of A-234 by H2O2. The main transition state structures and the selected key bond lengths (in Å) are given. | ||
The perhydrolysis of A-234 by H2O2 through path A begins with the formation of the molecular complex AIM1, where the HOO–H1⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P hydrogen bond interaction stabilizes AIM1. The nucleophilic attack of the O atom in H2O2 on the P atom of A-234, coupled with the proton transfer (H1) from H2O2 to the O atom of A-234, leads to a metastable intermediate AIM2 through the transition state ATS1 with a free energy barrier of 28.4 kcal mol−1. Subsequently, AIM2 evolves into AIM3 through rotation of the P–OH1 moiety with a low free energy barrier of 5.0 kcal mol−1, in which H1 and F atoms are in the same side of P–O. Followed by the coupling of H1 and F atoms in AIM3 and the release of HF, the degradation product P1 is formed through a four-membered ring transition state ATS3 with a free energy barrier of 10.4 kcal mol−1. The degradation path A is an endergonic process with a Gibbs free energy of the reaction ΔG of 6.1 kcal mol−1.
P hydrogen bond interaction stabilizes AIM1. The nucleophilic attack of the O atom in H2O2 on the P atom of A-234, coupled with the proton transfer (H1) from H2O2 to the O atom of A-234, leads to a metastable intermediate AIM2 through the transition state ATS1 with a free energy barrier of 28.4 kcal mol−1. Subsequently, AIM2 evolves into AIM3 through rotation of the P–OH1 moiety with a low free energy barrier of 5.0 kcal mol−1, in which H1 and F atoms are in the same side of P–O. Followed by the coupling of H1 and F atoms in AIM3 and the release of HF, the degradation product P1 is formed through a four-membered ring transition state ATS3 with a free energy barrier of 10.4 kcal mol−1. The degradation path A is an endergonic process with a Gibbs free energy of the reaction ΔG of 6.1 kcal mol−1.
Similar to the path A above, here path B also initiates from a molecular complex BIM1, where the HOO–H1⋯N-(A-234) hydrogen bond interaction is involved in BIM1, differing from AIM1. Afterwards, the H1 is transferred to the N atom coupled with the nucleophilic attack of the O atom of H2O2 on the P center of A-234 yielding a metastable BIM2 through a four-membered ring transition state BTS1 with a free energy barrier of 26.2 kcal mol−1. The cleavage of the P–N bond in BIM2, coupled with the release of Me(R1)CNH, produces an intermediate BIM3viaBTS2 with a free energy barrier of 12.4 kcal mol−1. Note that BIM3 may be toxic owing to the presence of P–F, and thus it needs to be further detoxicated by an additional H2O2 molecule. However, the cleavage of the P–F bond in BIM3 experiences a substantially high free energy barrier of 55.7 kcal mol−1, suggesting that BIM3 could be the end-product of path B. Path B from A-234 to BIM3 is also a thermodynamically endergonic process with a Gibbs free energy of the reaction ΔG of 13.4 kcal mol−1. Although both paths A and B may compete with each other at the initial stage, overall, path A is more favorable for the perhydrolysis of A-234 by H2O2, both kinetically and thermodynamically, compared to path B.
The path C for the perhydrolysis of A-234 by H2O2 involves the four-membered ring transition state CTS1, where the H1 of H2O2 is transferred to the F site coupled with the nucleophilic attack of the O atom of H2O2 on the P atom of A-234, with a substantially high free energy barrier of 53.4 kcal mol−1. Therefore, such a direct activation mode of the P–F bond is considered unfavorable. Clearly, path A is inevitably responsible for the perhydrolysis of A-234 during its degradation by H2O2.
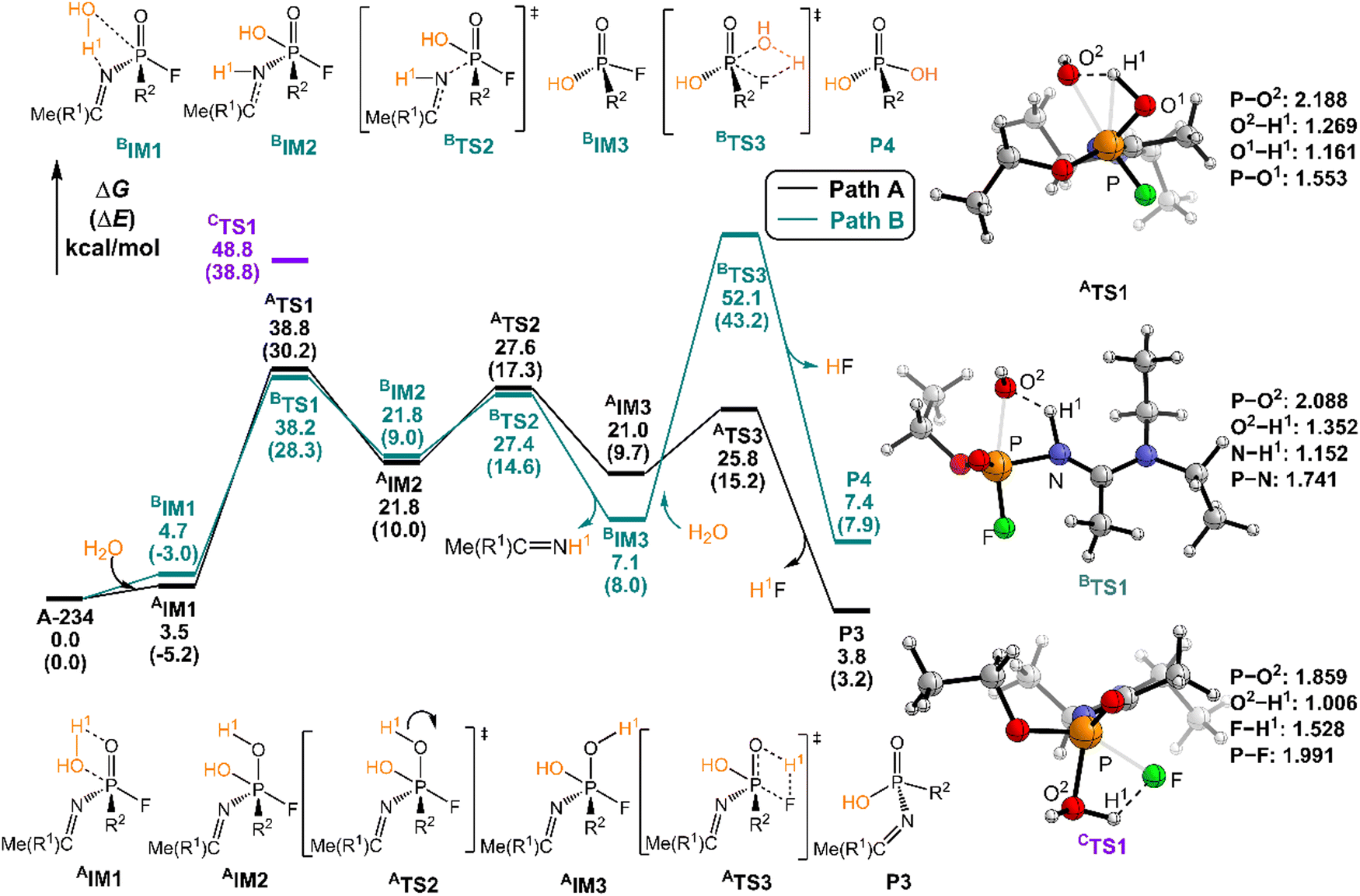 | ||
| Fig. 3 Predicted relative free energy profiles (ΔG in kcal mol−1) and electronic energies (ΔE in parentheses) of the species involved in the hydrolysis of A-234 by H2O. The main transition state structures and the selected key bond lengths (in Å) are given. | ||
In path A, the hydrolysis of A-234 by H2O starts from the generation of a molecular complex AIM1 derived from a hydrogen bond interaction between the H1 atom of H2O and the PO moiety of A-234. Then, the proton H1 shifts to the O atom of the PO moiety, coupled with the nucleophilic attack of the newly generated HO− on the P atom, leading to a metastable intermediate AIM2viaATS1 with a free energy barrier of 35.2 kcal mol−1. Afterwards, the rotation of the –P–OH1 bond in AIM2 leads to the formation of AIM3 through ATS2 with a low free energy barrier of 5.8 kcal mol−1. This isomerization process reduces the distance between H1 and F atoms and boosts the elimination of the F atom in the form of HF. Followed by the formation of the H1F molecule through a four-membered ring transition state ATS3 with a free energy barrier of 4.8 kcal mol−1, the degradation product P3 is obtained. The path A for the A-234 hydrolysis by H2O has a Gibbs free energy of the reaction ΔG of 3.8 kcal mol−1.
In path B, the molecular complex BIM1 with H2O with A-234 is formed first through the HO–H⋯N–(A-234) hydrogen bond interaction. Subsequently, the nucleophilic attack of the O atom of H2O on the P atom, coupled with the proton H1 shift to the N atom, yields a metastable intermediate BIM2 through BTS1 with a free energy barrier of 33.5 kcal mol−1. Note that the P–N bond in BIM2 is remarkably stretched and weakened. With the leaving of the Me(R1)CNH species, an intermediate BIM3 is formed through BTS2 with a free energy barrier of 5.6 kcal mol−1. The hydrolytic breakage of the P–F bond in BIM3 by additional H2O, coupled with the release of HF, generates the degraded product P4 through the four-membered transition state BTS3 with a relatively high free energy barrier of 45.0 kcal mol−1. Similar to the perhydrolysis of A-234 in Fig. 2, the hydrolysis of A-234 through path B is also an unfavorable process, both kinetically and thermodynamically, compared to path A.
In the hydrolysis path C of A-234 by H2O, H2O approaches A-234 from the direction of the P–F bond first. The subsequent degradation experiences CTS1 with a substantially high free energy barrier of 48.8 kcal mol−1, and the proton H1 transfer from H2O to the F atom site of A-234, along with the nucleophilic attack of the O atom of H2O on the P atom of A-234, is involved in the hydrolysis process. Clearly, both paths B and C are less possible for the A-234 hydrolysis.
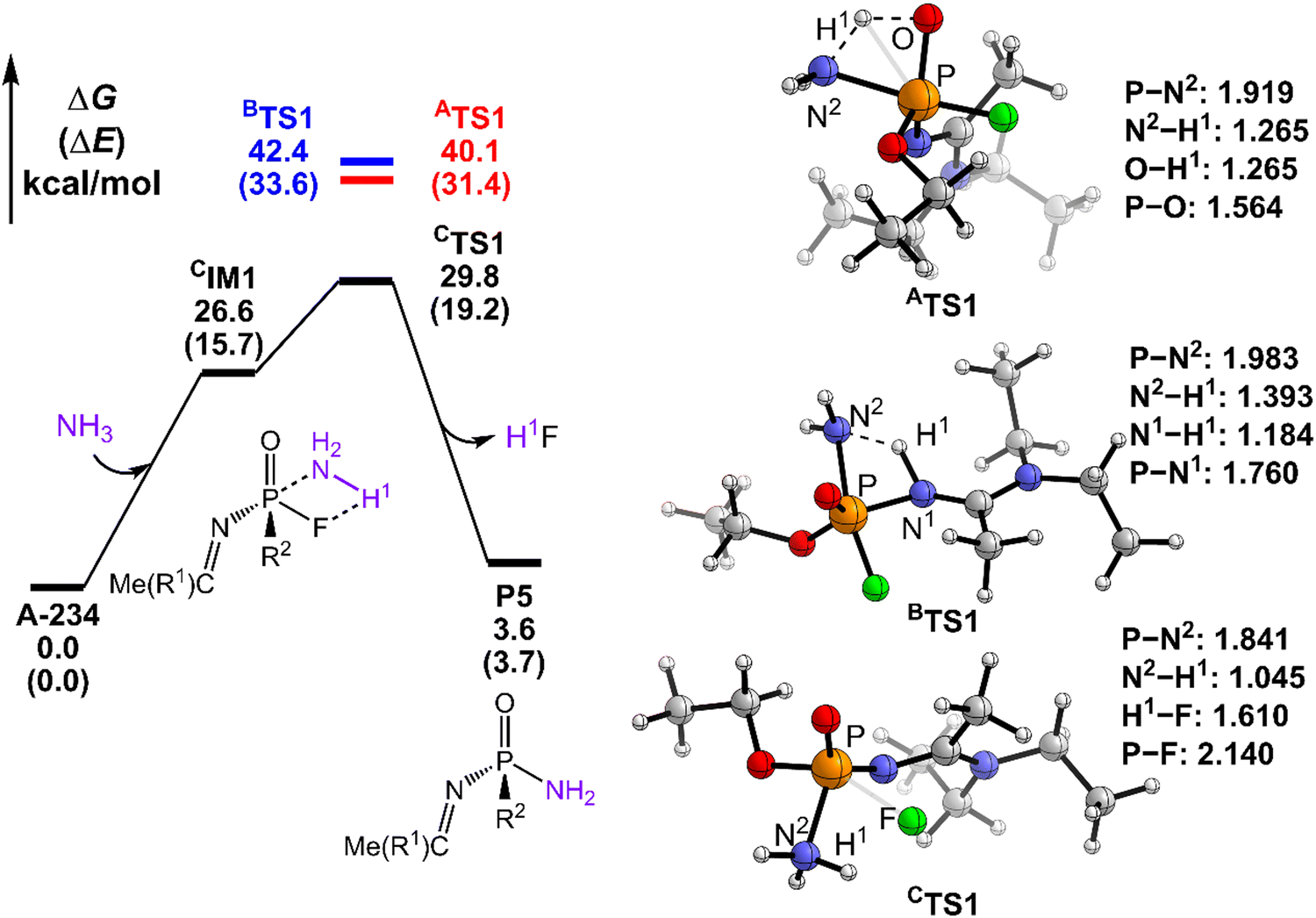 | ||
| Fig. 4 Predicted relative free energy profiles (ΔG in kcal mol−1) and electronic energies (ΔE in parentheses) of the species involved in the ammonolysis of A-234 by NH3, where for paths A and B, only the key transition states are shown in red and blue, respectively. The main transition state structures and the selected key bond lengths (in Å) are given. | ||
Here, the path preference for A-234 degradation by NH3 can be ascribed to the relatively stronger electron-donating ability of NH3 as a weak proton donor than that of H2O2 or H2O, resulting in that the nucleophilic attack of NH3 on the P center of A-234 (path C) is favored. Such superiority of path C is also supported by further IRC analyses related to the key transition states (ATS1, BTS1, and CTS1) in the perhydrolysis and ammonolysis of A-234. For ATS1 and BTS1 in paths A and B, their energy demands are mainly caused by the proton transfer process from the detergents (H2O2 and NH3) to A-234, suggesting that both paths A and B are dominated by the proton transfer step (see Fig. S4a and b and d and e in the ESI†). However, for CTS1 in path C (Fig. S4c and f, ESI†), its energy requirement primarily arises from the cleavage of the P–F bond, along with the proton transfer from the detergent to the F atom of A-234, which can be facilitated by the nucleophilic attack of NH3 on the P center of A-234. Overall, the relatively strong proton-donating ability of H2O2 (or H2O), than that of NH3, may facilitate the perhydrolysis (or hydrolysis) of A-234 through paths A and B, while the ammonolysis of A-234 by NH3 primarily proceeds via path C owing to the stronger nucleophilicity of NH3 than those of H2O2 and H2O.
Overall, the perhydrolysis and hydrolysis of A-234 generally follow path A with relatively low free energy spans and thermodynamically comparative advantages, although path B is competitive at the initial stage of the A-234 degradation. However, the ammonolysis of A-234 is dominated by path C. Notably, the direct degradation of A-234 respectively by using H2O2, H2O, or NH3 experiences relatively high free energy barriers, and thus its efficient degradation requires harsh reaction conditions and the optimal combination of detergents or catalysts.
3.2 A-234 degradation by different combinations of detergents
It was found that the mixture of H2O2 vapor with NH3 gas could degrade the nerve agent VX on solid surfaces at ambient temperature, indicating that the notable combination effect of H2O2 with NH3 toward the decontamination of VX.62 Speculatively, the hydrogen bonding networks formed by the detergents themselves or with solvent molecules exist in the admixture, which may play a crucial role in promoting the degradation of NAs. Therefore, further computational efforts are made to elucidate the combination effects of different detergents (e.g., H2O2/H2O, H2O2/NH3, H2O/H2O, and H2O/NH3) on the degradation efficiency of A-234. In these combinations, the former serves as the nucleophilic reagent and the latter as an auxiliary agent to donate the proton.The predicted degradation mechanisms and relative energies of A-234 by detergent combinations of H2O2/NH3 and H2O2/H2O are shown in Fig. 5. The degradation of A-234 by H2O2/NH3 through path A commences with the formation of the molecular complex 1IM1, where a novel hydrogen bond network is involved in 1IM1. The hydrogen bond interaction plays a crucial role in facilitating the subsequent proton transfer. The proton transfers from H2O2 to the N atom of NH3 (H1) and simultaneously from NH3 to the O atom in the PO moiety (H2) mediated by the hydrogen bond network, coupled with the nucleophilic attack of the resulting HOO− anion to the P atom of A-234, lead to the formation of the metastable 1IM2via a six-membered ring 1TS1 with a free energy barrier of 17.4 kcal mol−1. 1IM2 evolves into the isoenergetic isomer 1IM3 through the rotation of the –P–OH2 bond with a low free energy barrier of 3.7 kcal mol−1, where the hydrogen bond network may facilitate the coupling of H1 and F atoms. Finally, the rebound of H2 to the NH2 moiety to regenerate NH3, coupled with the formation and release of HF, yields the nontoxic P1 through a six-membered ring 1TS3 with a free energy barrier of 5.6 kcal mol−1. Fig. 2 and 5 show that the free energy barrier of the nucleophilic attack in the degradation of A-234 remarkably decreases from 31.3 kcal mol−1 by H2O2 (or 31.0 kcal mol−1 by H2O2/H2O) to 20.5 kcal mol−1 by H2O2/NH3. Here, the presence of NH3 constitutes a hydrogen bonding network to mediate the hydrogen transfer, which enhances the degradation efficiency of H2O2 toward A-234.
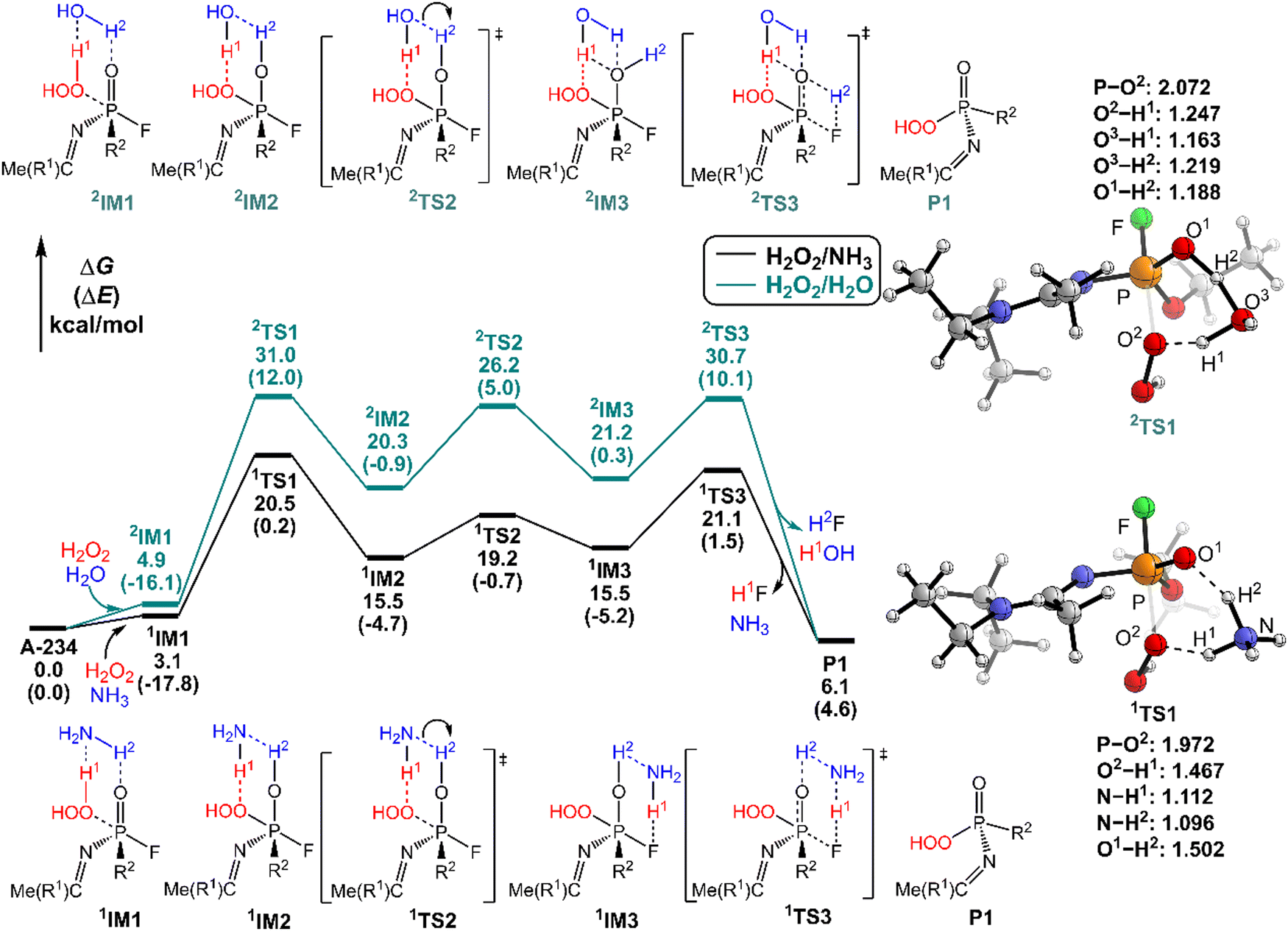 | ||
| Fig. 5 Predicted relative free energy profiles (ΔG in kcal mol−1) and electronic energies (ΔE in parentheses) of the species involved in the perhydrolysis of A-234 by H2O2/NH3 and H2O2/H2O. The main transition state structures and the selected key bond lengths (in Å) are given. | ||
The degradation of A-234 by H2O2/H2O with the H2O as the auxiliary agent was also investigated, and the corresponding results are shown in Fig. 5. Note that the degradation of A-234 by H2O2/H2O takes place on a higher free-energy surface than that by H2O2/NH3, suggesting that NH3 should be a better auxiliary agent than H2O.
As shown in Fig. 6, the degradation mechanisms of A-234 by H2O/NH3 and H2O/H2O were investigated. Compared to the hydrolysis of A-234 using a single molecule of H2O, the presence of NH3 or additional H2O significantly increases the degradation efficiency of H2O toward A-234. Particularly, NH3 as the prodegradant is superior to H2O, and the free energy barrier of the rate-determining step in the degradation of A-234 is remarkably reduced from 38.8 kcal mol−1 by H2O to 29.1 kcal mol−1 by H2O/NH3.
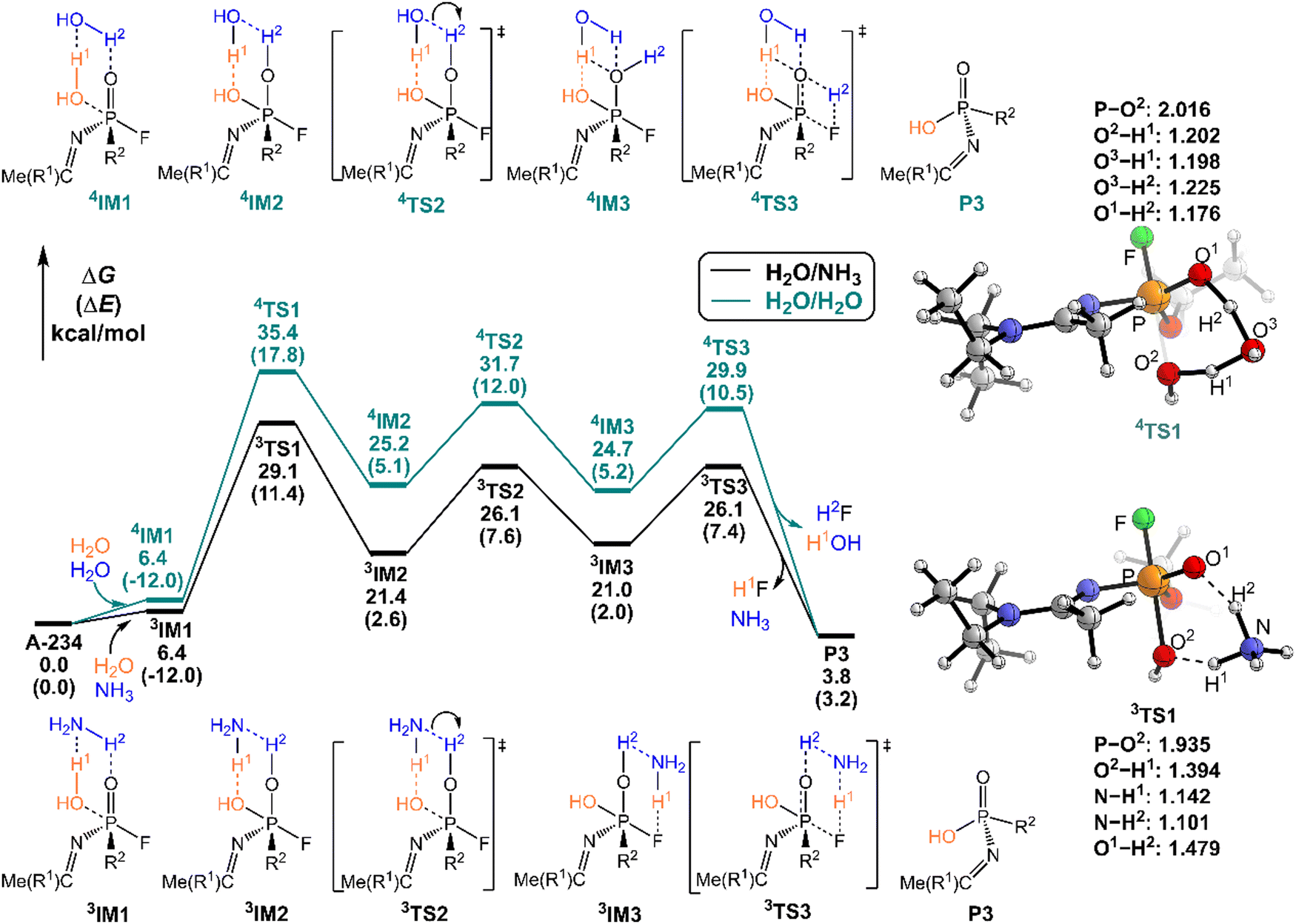 | ||
| Fig. 6 Predicted relative free energy profiles (ΔG in kcal mol−1) and electronic energies (ΔE in parentheses) of the species involved in the hydrolysis of A-234 by H2O/NH3 and H2O/H2O. The main transition state structures and the selected key bond lengths (in Å) are given. | ||
In order to gain an insight into the difference in detoxification efficiency for H2O2 and H2O, as well as the effects of auxiliary agents NH3 and H2O on the degradation of A-234, IRC analysis has been performed on the key transition states, including 1TS1, 2TS1, 3TS1 and 4TS1. As shown in Fig. 7a and c, the hydrogen transfers from O2 to N (H1) and from N to O1 (H2) in both 1TS1 and 3TS1 are concerted but not synchronous, which may be ascribed to different hydrogen bonds O–H⋯N and N–H⋯O that are involved in the concerted proton transfer of perhydrolysis and hydrolysis assisted by NH3. The presence of NH3 facilitates the proton transfer and the formation of strong nucleophiles HOO− and HO−, thereby promoting the degradation of A-234 by H2O2 and H2O. In contrast, during the degradation process assisted by H2O, the proton transfers in 2TS1 and 4TS1 are concerted and synchronous, owing to the involvement of same hydrogen-bond networks. Additionally, the higher basicity of NH3, than that of H2O,63 renders it more receptive to the proton from H2O2 or H2O, further facilitating the formation of strong nucleophiles and thereby promoting the degradation process.
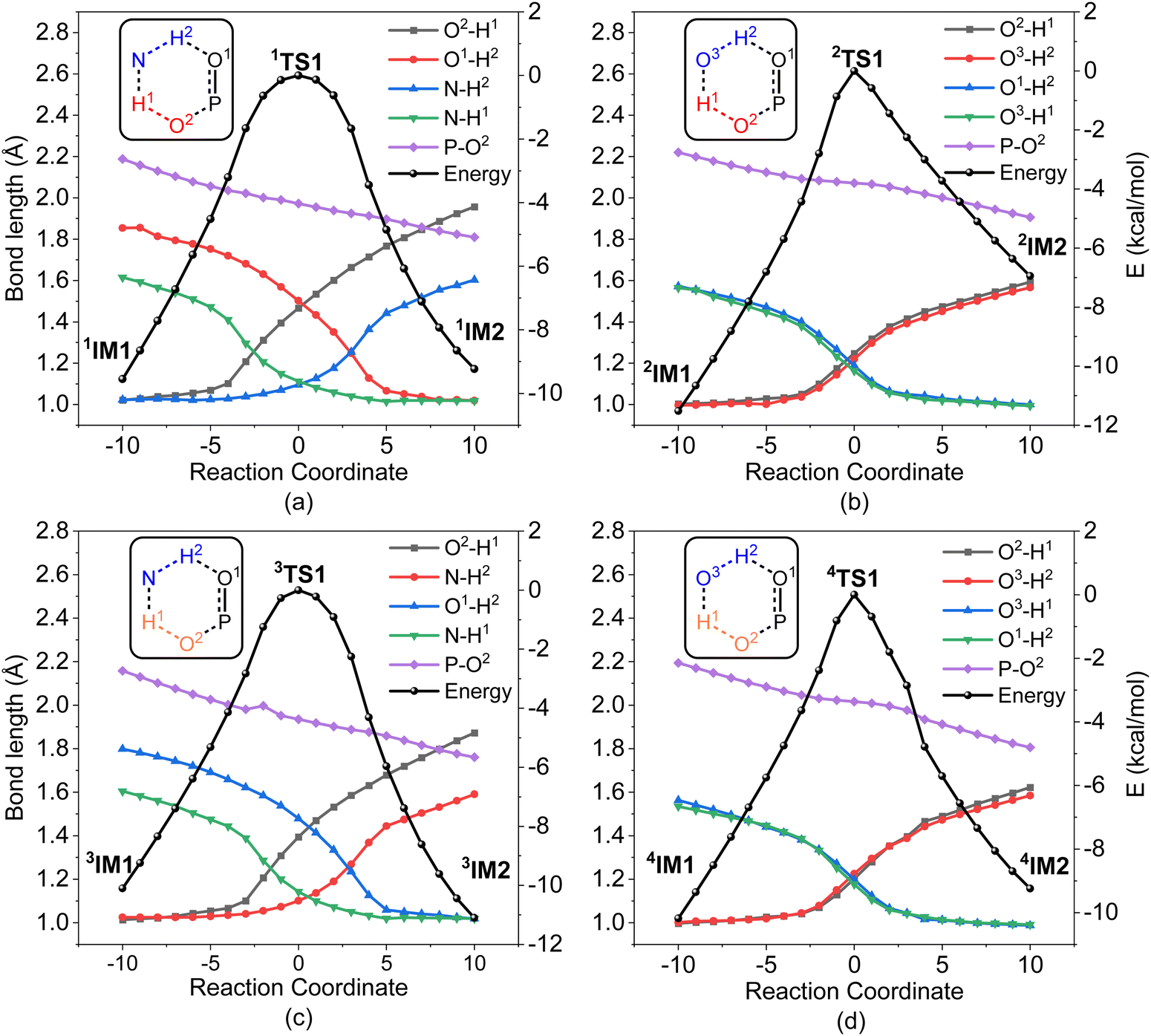 | ||
| Fig. 7 IRC analysis for the key transition states: (a) 1TS1 for H2O2/NH3, (b) 2TS1 for H2O2/H2O, (c) 3TS1 for H2O/NH3, and (d) 4TS1 for H2O/H2O. | ||
Furthermore, regardless of the presence of an auxiliary agent, the degradation efficiency of H2O2 toward A-234 is higher than that of H2O, which is attributed to the superior proton-donating capacity of H2O2, than that of H2O, as evidenced by their respective pKa values (11.6 for H2O2 and 14.0 for H2O).63
4. Conclusions
Here, the plausible degradation mechanisms of A-234 by H2O2, H2O, NH3 and their combinations were investigated by using DFT calculations. The calculated results revealed that the degradation of A-234 by H2O2 and H2O is triggered by the nucleophilic attack on the P center of A-234 coupling with the proton transfer to the O atom of the PO moiety via path A, while H2N–H and F–P-(A-234) tend to interact directly in the ammonolysis of A-234 by NH3via path C. Both H2O2 and NH3 have relatively high degradation efficiency toward A-234 than H2O. More importantly, various combinations of detergents (nucleophile/auxiliary: H2O2/H2O, H2O2/NH3, H2O/H2O, and H2O/NH3) may remarkably promote the degradation of A-234, since the hydrogen bond networks among the detergent, auxiliary, and substrate molecules facilitate the initial proton transfers and the formation of strong nucleophiles. In particular, the perhydrolysis and hydrolysis efficiency of A-234 by H2O2 and H2O can be notably improved with the help of NH3, suggesting that NH3 is a better auxiliary agent than H2O, and this is attributed to the higher basicity of NH3 to promote the proton transfer than that of H2O. Furthermore, the degradation of A-234 and the rate-determining step depend on the auxiliary agent. The present findings provide an in-depth understanding of the degradation of A-234 using small-molecule detergents, which is beneficial to the development of effective decontaminants toward the detoxification of A-series nerve agents.
Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21933009 and 22373078).References
- S. Costanzi, J.-H. Machado and M. Mitchell, ACS Chem. Neurosci., 2018, 9, 873–885 CrossRef CAS PubMed.
- S. Mukherjee and R. D. Gupta, J. Toxicol., 2020, 2020, 3007984 CrossRef PubMed.
- F. R. Sidell and J. Borak, Ann. Emerg. Med., 1992, 21, 865–871 CrossRef CAS PubMed.
- S. W. Wiener and R. S. Hoffman, J. Intensive Care Med., 2004, 19, 22–37 CrossRef PubMed.
- R. Stone, Science, 2018, 359, 1314–1315 CrossRef CAS PubMed.
- B. Sanson, F. Nachon, J.-P. Colletier, M.-T. Froment, L. Toker, H. M. Greenblatt, J. L. Sussman, Y. Ashani, P. Masson, I. Silman and M. Weik, J. Med. Chem., 2009, 52, 7593–7603 CrossRef CAS PubMed.
- Y. Liu, S.-Y. Moon, J. T. Hupp and O. K. Farha, ACS Nano, 2015, 9, 12358–12364 CrossRef CAS PubMed.
- J. E. Mondloch, M. J. Katz, W. C. Isley Iii, P. Ghosh, P. Liao, W. Bury, G. W. Wagner, M. G. Hall, J. B. DeCoste and G. W. Peterson, Nat. Mater., 2015, 14, 512–516 CrossRef CAS PubMed.
- P. Li, S.-Y. Moon, M. A. Guelta, L. Lin, D. A. Gómez-Gualdrón, R. Q. Snurr, S. P. Harvey, J. T. Hupp and O. K. Farha, ACS Nano, 2016, 10, 9174–9182 CrossRef CAS PubMed.
- N. S. Bobbitt, M. L. Mendonca, A. J. Howarth, T. Islamoglu, J. T. Hupp, O. K. Farha and R. Q. Snurr, Chem. Soc. Rev., 2017, 46, 3357–3385 RSC.
- M. L. Mendonca and R. Q. Snurr, ACS Catal., 2020, 10, 1310–1323 CrossRef CAS.
- M. C. de Koning, C. Vieira Soares, M. van Grol, R. P. T. Bross and G. Maurin, ACS Appl. Mater. Interfaces, 2022, 14, 9222–9230 CrossRef CAS PubMed.
- D. Ma and Z. Cao, J. Phys. Chem. C, 2022, 126, 19159–19168 CrossRef CAS.
- A. Michalkova, L. Gorb, M. Ilchenko, O. A. Zhikol, O. V. Shishkin and J. Leszczynski, J. Phys. Chem. B, 2004, 108, 1918–1930 CrossRef CAS.
- A. Michalkova, M. Ilchenko, L. Gorb and J. Leszczynski, J. Phys. Chem. B, 2004, 108, 5294–5303 CrossRef CAS.
- A. Michalkova, J. Martinez, O. A. Zhikol, L. Gorb, O. V. Shishkin, D. Leszczynska and J. Leszczynski, J. Phys. Chem. B, 2006, 110, 21175–21183 CrossRef CAS PubMed.
- V. M. Bermudez, J. Phys. Chem. C, 2007, 111, 3719–3728 CrossRef CAS.
- V. M. Bermudez, J. Phys. Chem. C, 2009, 113, 1917–1930 CrossRef CAS.
- N. Q. Le, C. E. Ekuma, B. I. Dunlap and D. Gunlycke, J. Phys. Chem. C, 2018, 122, 2832–2839 CrossRef CAS.
- R. Tsyshevsky, S. Holdren, B. W. Eichhorn, M. R. Zachariah and M. M. Kuklja, J. Phys. Chem. C, 2019, 123, 26432–26441 CrossRef CAS.
- D. Ma and Z. Cao, J. Phys. Chem. C, 2021, 125, 24396–24405 CrossRef CAS.
- V. K. Rastogi, J. J. Defrank, T.-C. Cheng and J. R. Wild, Biochem. Biophys. Res. Commun., 1997, 241, 294–296 CrossRef CAS PubMed.
- K. E. LeJeune, J. R. Wild and A. J. Russell, Nature, 1998, 395, 27–28 CrossRef CAS PubMed.
- P.-C. Tsai, A. Bigley, Y. Li, E. Ghanem, C. L. Cadieux, S. A. Kasten, T. E. Reeves, D. M. Cerasoli and F. M. Raushel, Biochemistry, 2010, 49, 7978–7987 CrossRef CAS PubMed.
- A. N. Bigley and F. M. Raushel, Chem.-Biol. Interact., 2019, 308, 80–88 CrossRef CAS PubMed.
- F. Fan, Y. Zheng, Y. Zhang, H. Zheng, J. Zhong and Z. Cao, ACS Catal., 2019, 9, 7038–7051 CrossRef CAS.
- J. Yu, Y. Fu and Z. Cao, J. Phys. Chem. B, 2023, 127, 7462–7471 CrossRef CAS PubMed.
- A. M. McAnoy, J. Williams, M. R. L. Paine, M. L. Rogers and S. J. Blanksby, J. Org. Chem., 2009, 74, 9319–9327 CrossRef CAS PubMed.
- E. V. Patterson and C. J. Cramer, J. Phys. Org. Chem., 1998, 11, 232–240 CrossRef CAS.
- G. W. Wagner, Main Group Chem., 2010, 9, 257–263 CAS.
- G. W. Wagner, L. R. Procell, D. C. Sorrick, G. E. Lawson, C. M. Wells, C. M. Reynolds, D. B. Ringelberg, K. L. Foley, G. J. Lumetta and D. L. Blanchard, Jr., Ind. Eng. Chem. Res., 2010, 49, 3099–3105 CrossRef CAS.
- G. W. Wagner and Y.-C. Yang, Ind. Eng. Chem. Res., 2002, 41, 1925–1928 CrossRef CAS.
- Y.-C. Yang, F. J. Berg, L. L. Szafraniec, W. T. Beaudry, C. A. Bunton and A. Kumar, J. Chem. Soc., Perkin Trans. 2, 1997, 607–614 RSC.
- Y. C. Yang, L. L. Szafraniec, W. T. Beaudry and C. A. Bunton, J. Org. Chem., 1993, 58, 6964–6965 CrossRef CAS.
- R. Osovsky, D. Kaplan, I. Nir, H. Rotter, S. Elisha and I. Columbus, Environ. Sci. Technol., 2014, 48, 10912–10918 CrossRef CAS PubMed.
- Y.-C. Yang, Acc. Chem. Res., 1999, 32, 109–115 CrossRef CAS.
- Y. A. Imrit, H. Bhakhoa, T. Sergeieva, S. Danés, N. Savoo, M. I. Elzagheid, L. Rhyman, D. M. Andrada and P. Ramasami, RSC Adv., 2020, 10, 27884–27893 RSC.
- C. J. Leverant, C. W. Priest, J. A. Greathouse, M. K. Kinnan and S. B. Rempe, Int. J. Mol. Sci., 2021, 22, 8653 CrossRef CAS PubMed.
- D. Mandal, K. Sen and A. K. Das, J. Phys. Chem. A, 2012, 116, 8382–8396 CrossRef CAS PubMed.
- D. Mandal, Theor. Chem. Acc., 2020, 139, 169 Search PubMed.
- I. Lyagin and E. Efremenko, Catal. Commun., 2019, 120, 91–94 CrossRef CAS.
- H. Bhakhoa, L. Rhyman and P. Ramasami, R. Soc. Open Sci., 2019, 6, 181831 CrossRef PubMed.
- S. E. Hosseini, H. Saeidian, A. Amozadeh, M. T. Naseri and M. Babri, Rapid Commun. Mass Spectrom., 2016, 30, 2585–2593 CrossRef CAS PubMed.
- K. Jeong and J. Choi, R. Soc. Open Sci., 2019, 6, 190414 CrossRef CAS PubMed.
- L. A. Vieira, J. S. F. D. Almeida, T. C. C. França and I. Borges, Comput. Theor. Chem., 2021, 1202, 113321 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- G. W. Spitznagel, T. Clark, P. von Ragué Schleyer and W. J. Hehre, J. Comput. Chem., 1987, 8, 1109–1116 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 2008, 72, 650–654 CrossRef.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 2008, 72, 5639–5648 CrossRef.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Y. Haoyu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- C. Legault, CYLview20, 2020, https://www.cylview.org Search PubMed.
- S. Gon Ryu and H. Wan Lee, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2015, 50, 1417–1427 CrossRef PubMed.
- W. M. Haynes, CRC handbook of chemistry and physics, CRC Press, Boca Raton, 2014 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional computational details; predicted relative free energy profiles for A-234 degradation by selected detergents at the M06-2X/6-311++G(d,p)/SMD level of theory; estimated reaction rate constants for selected rate-limiting steps; optimized coordinates of all the discussed species. See DOI: https://doi.org/10.1039/d4cp00881b |
| This journal is © the Owner Societies 2024 |