
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne touch is all it takes: the supramolecular interaction between ubiquitin and lanthanide complexes revisited by paramagnetic NMR and molecular dynamics†
Karen
Dos Santos‡
 a,
Alessio
Bartocci‡
bcd,
Natacha
Gillet
e,
Sandrine
Denis-Quanquin
e,
Amandine
Roux
ef,
Eugene
Lin
a,
Zeren
Xu
a,
Raphael
Finizola
e,
Pauline
Chedozeau
a,
Xi
Chen
a,
Cédric
Caradeuc
a,
Mathieu
Baudin
ag,
Gildas
Bertho
a,
François
Riobé
h,
Olivier
Maury
c,
Elise
Dumont
*ij and
Nicolas
Giraud
*a
a,
Alessio
Bartocci‡
bcd,
Natacha
Gillet
e,
Sandrine
Denis-Quanquin
e,
Amandine
Roux
ef,
Eugene
Lin
a,
Zeren
Xu
a,
Raphael
Finizola
e,
Pauline
Chedozeau
a,
Xi
Chen
a,
Cédric
Caradeuc
a,
Mathieu
Baudin
ag,
Gildas
Bertho
a,
François
Riobé
h,
Olivier
Maury
c,
Elise
Dumont
*ij and
Nicolas
Giraud
*a
aUniversité Paris Cité, Laboratoire de Chimie et Biochimie Pharmacologiques et Toxicologiques, UMR CNRS 8601, Paris, France. E-mail: nicolas.giraud@u-paris.fr
bDepartment of Physics, University of Trento, Via Sommarive 14, I-38123 Trento, Italy
cINFN-TIFPA, Trento Institute for Fundamental Physics and Applications, Via Sommarive 14, I-38123 Trento, Italy
dInstitut de Chimie de Strasbourg, UMR 7177, CNRS, Université de Strasbourg, Strasbourg Cedex 67083, France
eUniv. Lyon, ENS de Lyon, CNRS UMR 5182, Université Claude Bernard Lyon 1, Laboratoire de Chimie, F69342, Lyon, France
fPolyvalan SAS, Lyon, France
gLaboratoire des Biomolécules, LBM, Département de chimie, École Normale Supérieure, PSL Université, Sorbonne Université 45 Rue d’Ulm, 75005 Paris, France
hUniv. Bordeaux CNRS, Bordeaux INP, ICMCB UMR 5026, F-33600 Pessac, France
iUniversité Côte d’Azur, CNRS, Institut de Chimie de Nice, UMR 7272, 06108 Nice, France. E-mail: elise.dumont@univ-cotedazur.fr
jInstitut Universitaire de France, 5 rue Descartes, 75005 Paris, France
First published on 1st May 2024
Abstract
The supramolecular interaction between lanthanide complexes and proteins is at the heart of numerous chemical and biological studies. Some of these complexes have demonstrated remarkable interaction properties with proteins or peptides in solution and in the crystalline state. Here we have used the paramagnetism of lanthanide ions to characterize the affinity of two lanthanide complexes for ubiquitin. As the interaction process is dynamic, the acquired NMR data only reflect the time average of the different steps. We have used molecular dynamics (MD) simulations to get a deeper insight into the detailed interaction scenario at the microsecond scale. This NMR/MD approach enabled us to establish that the tris-dipicolinate complex interacts specifically with arginines and lysines, while the crystallophore explores the protein surface through weak interactions with carboxylates. These observations shed new light on the dynamic interaction properties of these complexes, which will ultimately enable us to propose a crystallization mechanism.
Introduction
The versatility of lanthanide ions and complexes has made them powerful tools for investigating biomolecules such as proteins, peptides, lipids, and DNA. Over the last few decades, lanthanide complexes have been successfully used in several applications ranging from binding tags for supramolecular recognition1,2 to chiral sensing3 and even immunoassays.4,5 More recently, they have also been proposed as additives for protein co-crystallization, not only because of their nucleating properties6 but also because of the phasing ability of the lanthanide center for X-ray crystallography.7–9 In addition, lanthanide complexes can act as “molecular glues”,10–12 which can contribute to consolidating protein–protein interfaces. However, the precise mechanism of the interaction between lanthanide complexes and biomolecules in solution remains unclear, and hence there is a need to develop a comprehensive understanding of the dynamic process of non-covalent binding. To this end, one of the most effective experimental techniques that rely on the electronic properties of lanthanides is paramagnetic nuclear magnetic resonance (paramagnetic NMR), which has become a powerful tool for addressing structural features of proteins.13–15 Several labelling strategies of proteins with lanthanide ions (Ln3+) have been reported over the last few decades, either using covalent or non-covalent tags, which were shown to yield remarkable long-range structural details with exceptional clarity.16In this paper, we report a comprehensive structural analysis of the dynamic association between a lanthanide complex and a protein. Capitalizing on our recent work demonstrating the combined use of paramagnetic NMR and molecular dynamics (MD) to study interactions with intrinsically disordered peptides,17 we extend our approach to get insight into another example of a weakly interacting system involving a more rigid protein, ubiquitin, and two complexes of interest, namely tris-dipicolinate and crystallophore (Fig. 1). First, ubiquitin is a small protein that plays a critical role in regulating various cellular processes, including protein degradation,18 DNA repair,19,20 and signal transduction,21,22 which makes it a key component in maintaining cellular homeostasis. Second, the tris-dipicolinate lanthanide complex [Ln(DPA)3]3−, which exists as a racemic mixture of two enantiomers that readily interconvert in water at room temperature,23 has been shown to develop supramolecular interactions with proteins.24–28
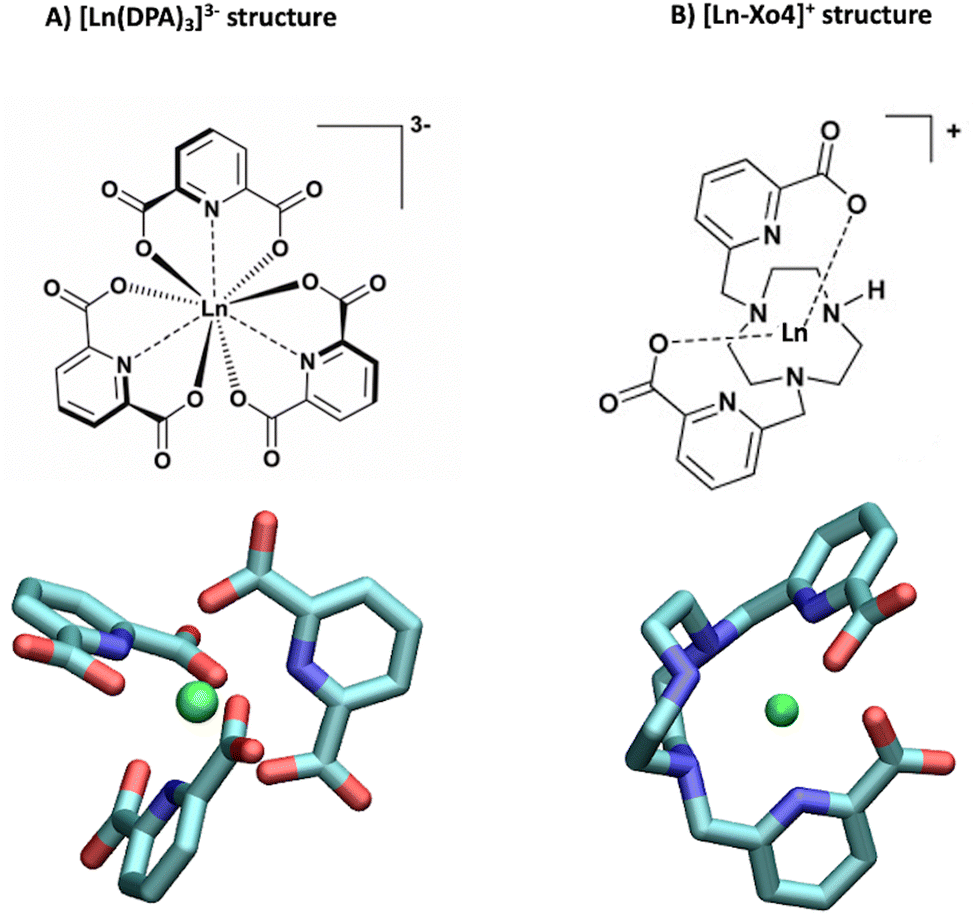 | ||
| Fig. 1 Structures of (A) the tris-dipicolinate lanthanide complex, [Ln(DPA)3]3− and (B) the crystallophore [Ln–Xo4]+.6 | ||
It presents several advantages for probing protein structure and interactions: (i) there is no need to modify the protein chemically because [Ln(DPA)3]3− binds non-covalently to it, (ii) the recovery of the protein can be achieved easily via dialysis and (iii) in many cases, the interaction process results in a fast exchange with respect to chemical shifts between the bound and the free states, which allows for a simple monitoring using, for instance, a NMR titration protocol.28 The interactions between ubiquitin and a series of derivatives of [Ln(DPA)3]3− were successfully described by Wei et al. to demonstrate the potential of using lanthanide complexes as promising non-covalent tags for obtaining angle and distance restraints of proteins in structural biology applications.27 Wei et al. have notably observed that all derivatives of [Ln(DPA)3]3− interact with the first region including residues K6, I44, and H68. They have also reported that positively charged residues R72 and R74 from the C-terminal region are also prone to interact with the complexes; however, they have proposed that under the experimental conditions of their analysis this interaction is a transient one due to the flexibility of the C-terminal tail and did not identify it as a potential secondary interaction site with ubiquitin. Third, the crystallophore (Xo4) lanthanide complex [Ln–Xo4]+, which consists of a cationic Ln3+ with a macrocyclic triazacyclononane bis-picolinate ligand (Fig. 1), has been shown to be a powerful agent for protein crystallography, with optimized properties for nucleation, phasing in X-ray crystallography, and luminescence.6,29–32
One fundamental issue that is raised in this study is the weakness of the interactions that are transiently created between the lanthanide complex and the protein of interest, which often leads to an overall low affinity between the two partners. This supramolecular process yields in general small and sometimes undetectable perturbations of standard NMR spectra recorded for such samples. To overcome this limitation and gain a better insight into this interaction mechanism, we have implemented paramagnetic NMR experiments, using suitably chosen lanthanide metals to generate paramagnetic relaxation enhancements (PREs) or pseudo-contact shifts (PCSs) that are tailored to highlight the regions of the protein involved in the interaction with the lanthanide complex. Furthermore, we have computed all-atom molecular dynamics simulations spanning microseconds, to interpret our NMR data in light of the dynamic picture yielded by the resulting MD trajectories.
Results
Paramagnetic NMR study
Two lanthanide ions were used to investigate the interaction between ubiquitin and the lanthanide complex by NMR: gadolinium (Gd3+) to access PREs and ytterbium (Yb3+) for PCS measurements. Synthetic details are given in the ESI.†Observation of PREs induced by [Gd(DPA)3]3−
We have acquired a series of 2D 1H–1H TOCSY and 1H–15N HSQC spectra of a ubiquitin sample to which we have added 0.2 to 0.8 equivalents of [Gd(DPA)3]3− to measure relaxation enhancements induced by the Gd3+ center.33 The resulting HSQC 2D maps are shown for 0 and 0.8 eq. in Fig. 2. Overall, for several residues we observed a broadening of their correlation upon addition of [Gd(DPA)3]3−, as reflected by the blue correlations that are visible among the 2D maps. The correlation peaks corresponding to the residues L8, K11, Q40, F45, A46, G47, K48, Q49, T66, H68, L71, R72, L73, and R74 undergo a signal extinction in the presence of [Gd(DPA)3]3−, which suggests that these amino-acids are close enough to the Gd3+ center to undergo significant PREs.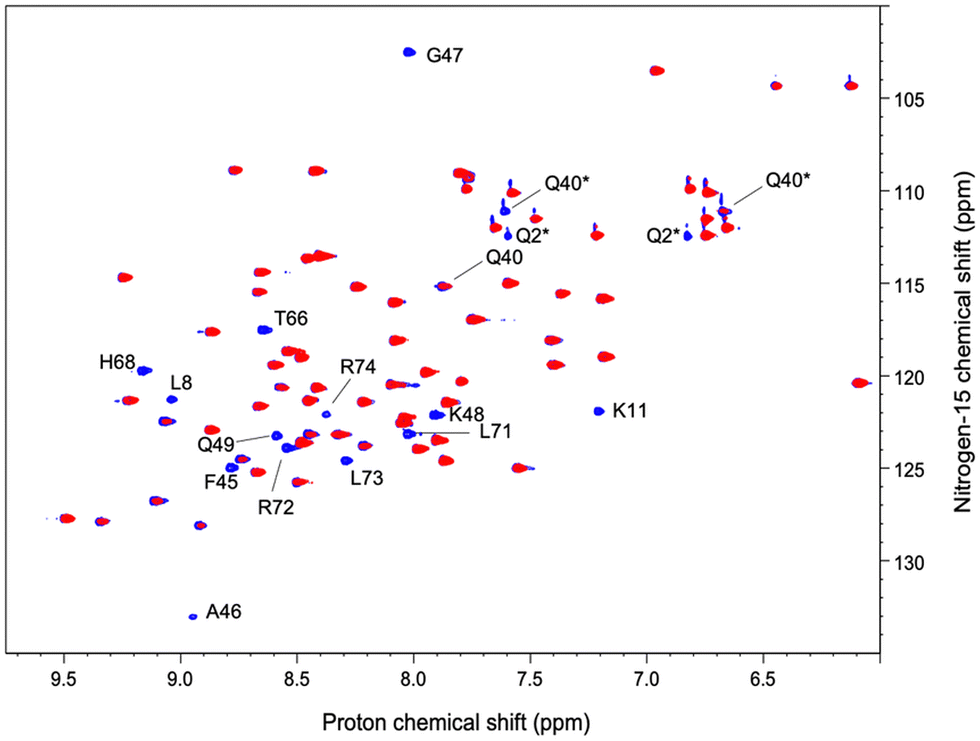 | ||
| Fig. 2 Superposition of 1H–15N HSQC spectra recorded on a ubiquitin sample without (blue) and with (red) 0.8 eq. of [Gd(DPA)3]3−. Signals marked with * correspond to N–Hε correlations from the side-chain of the residue Q40. | ||
To build a more accurate picture of the regions of the protein that are at the interface with the lanthanide complex upon interaction, we have calculated for each N–H correlation of ubiquitin the peak intensity ratio Ipara/Idia, where Ipara and Idia are the intensities of a given correlation in the protein with and without the paramagnetic species, respectively. Fig. 3A shows the evolution of the resulting intensity ratio profile for increasing amounts of [Gd(DPA)3]3−.
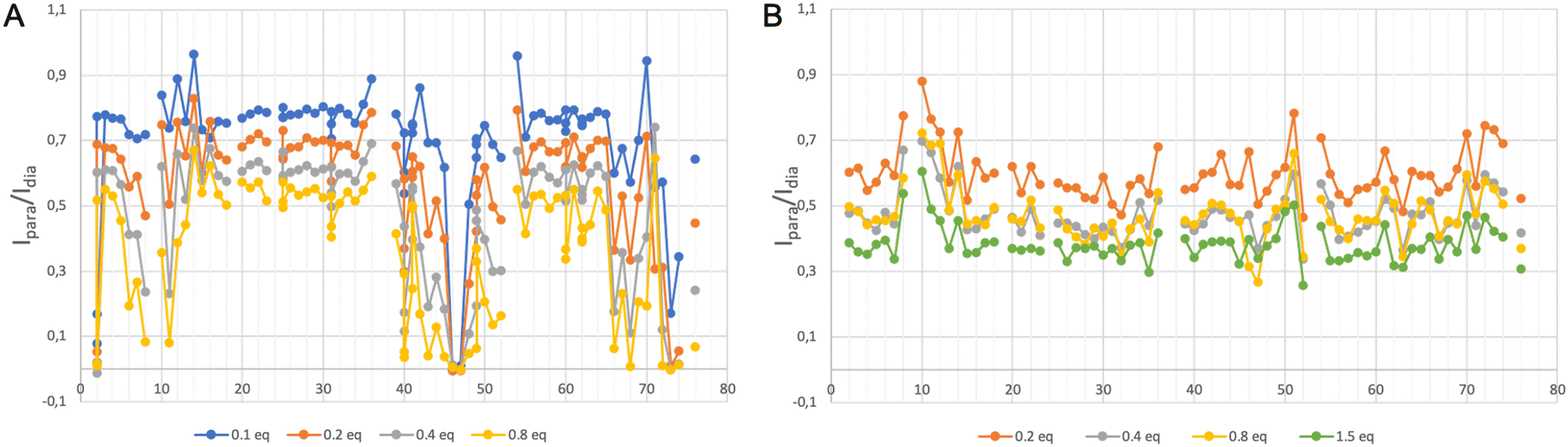 | ||
| Fig. 3 Distribution of the intensity ratio Ipara/Idia determined from the 1H–15N HSQC spectrum of a solution of ubiquitin upon the addition of different molar ratios of (A) [Gd(DPA)3]3− and (B) [GdXo4]+. | ||
Three different regions can be highlighted, which show a lower Ipara/Idia ratio upon addition of [Gd(DPA)3]3−: (i) K6 to T12, (ii) Q40, R42, F45, A46, G47, K48 and Q49, and (iii) T66, H68, L71, R72, L73, R74 and G76. This observation suggests (i) that the paramagnetic complex comes close to these residues during the interaction process and (ii) that there is more than one interaction site in the ubiquitin protein under our experimental conditions.
Measurement of PCS induced by [Yb(DPA)3]3−
We have also acquired a series of 2D 1H–1H TOCSY and 1H–15N HSQC spectra of the ubiquitin sample to which we have added the ytterbium complex, with [Yb(DPA)3]3−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ubiquitin molar ratios ranging from 0 to 10. The choice of the ytterbium center was made based on the tradeoff between the strength of the PCS that we want to measure to highlight the interaction regions on the one hand and the moderate PRE induced by the dipole–dipole interaction with the unpaired electron from this lanthanide ion, which could contribute to making some proton signals undetectable on the other hand.16 A first inspection of the resulting TOCSY maps allows for identifying a set of residues undergoing a significant shift of their N–H correlation upon addition of [Yb(DPA)3]3−: F4, K6, L15, Q41, I44, F45, G47, K48, Q49, L50, D58, I61, Q62, S65, T66, L67, L69, V70, L73, R74 and G76 (the 2D TOCSY and HSQC maps can be found in the ESI.† See Tables S1, S2 and Fig. S1, S2, ESI†). It is worth noting that most of these residues belong to the regions also identified using PREs induced by the Gd complex.
:ubiquitin molar ratios ranging from 0 to 10. The choice of the ytterbium center was made based on the tradeoff between the strength of the PCS that we want to measure to highlight the interaction regions on the one hand and the moderate PRE induced by the dipole–dipole interaction with the unpaired electron from this lanthanide ion, which could contribute to making some proton signals undetectable on the other hand.16 A first inspection of the resulting TOCSY maps allows for identifying a set of residues undergoing a significant shift of their N–H correlation upon addition of [Yb(DPA)3]3−: F4, K6, L15, Q41, I44, F45, G47, K48, Q49, L50, D58, I61, Q62, S65, T66, L67, L69, V70, L73, R74 and G76 (the 2D TOCSY and HSQC maps can be found in the ESI.† See Tables S1, S2 and Fig. S1, S2, ESI†). It is worth noting that most of these residues belong to the regions also identified using PREs induced by the Gd complex.
Fig. 4 shows the shift variation induced on amide protons by the addition of 10 equivalents of [Yb(DPA)3]3− to the ubiquitin sample. Two regions with significant chemical shift variations can be identified, including residues (i) G47–K48–Q49 and (ii) T66–L67, while other residues around these regions exhibit smaller variations. It should be reminded here that on the one hand the shifted signals arise from a fast exchange process of the protein between a free and a “bound” state. It is also possible that this “bound” state can be described as a dynamic association process that would also contribute to partially averaging the observed chemical shift variations by modulating the dipole–dipole interaction between the unpaired electron from the Yb center and the amino-acid N–H group in the protein.34
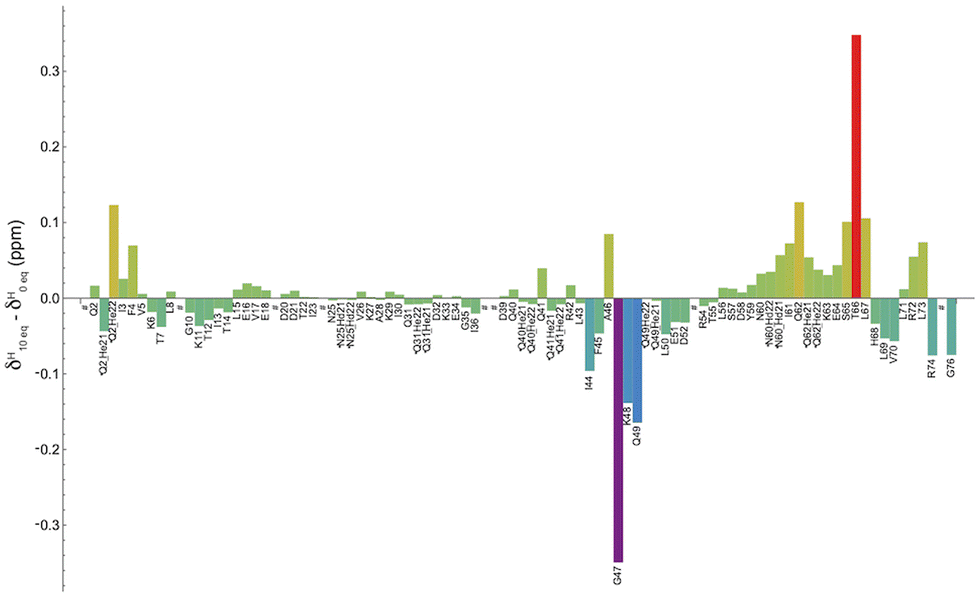 | ||
| Fig. 4 Distribution of the chemical shift variation δH10eq.–δH0eq. of each amide proton in ubiquitin, measured on 2D 1H–15N HSQC spectra recorded for 0 eq. and 10 eq. of [Yb(DPA)3]3−, respectively. Residues marked with * represent the correlation of Hε or Hδ with a nitrogen of the side chain. Residues M1, T9, P19, E24, P37, P38 and G75 presented either weak signals or could not be detected and represented by the # symbol. | ||
On the other hand, the sign and the amplitude of those variations, which reflect the anisotropy of the magnetic susceptibility tensor of the lanthanide ion, depend on the geometry of each interaction site in the protein. A thorough analysis of these variations is out of the scope of this paper, but we can however conclude that the use of this paramagnetic lanthanide ion allows for mapping rather accurately the main regions involved in the weak interaction with the tris-dipicolinate complex.
In conclusion for this part, under the experimental conditions of our analysis, the combination of PRE and PCS analyses reveals three main regions in ubiquitin that are involved in an interaction process with [Ln(DPA)3]3−.
NMR signature for [Ln–Xo4]+ interaction with ubiquitin
We have followed the same protocol as described above to investigate the interaction between ubiquitin and [Ln–Xo4]+. Fig. 3B shows the evolution of the peak ratio Ipara/Idia profile along the ubiquitin sequence as a function of the amount of added [Gd–Xo4]+ complex. These data show an average relaxation enhancement effect that seems to be shared by most of the residues and increases with the amount of [Gd–Xo4]+. Furthermore, a thorough analysis of this peak ratio profile does not allow for observing specific signal extinctions, as it was the case with [Gd(DPA)3]3−, even for the addition of up to 1.5 equivalents of [Gd–Xo4]+. Moreover, no significant chemical shift variation was observed after the addition of [Yb–Xo4]+ neither on the resulting HSQC nor TOCSY maps (see Fig. S2 and S4, ESI†).Given the amount of paramagnetic species that are present in the solution, the amplitude of the observed relaxation effect suggests a very weak though real interaction between [Gd–Xo4]+ and ubiquitin, hence the average relaxation enhancement that is even slightly greater than the one observed with [Gd(DPA)3]3−, all other factors being equal. However, no specific interaction site can be identified through a noticeable perturbation of its spectral signature, which can be explained by two factors: (i) overall, [Gd–Xo4]+ has a net charge of +I compared to the −III charge of [Gd(DPA)3]3−, which can lead to a weaker and thus more diffuse electrostatic interaction mechanism over the surface of the protein; and (ii) although [Gd(DPA)3]3− is known to interact dominantly with cationic residues such as lysine or arginine, [Gd–Xo4]+ is expected to interact with anionic chemical moieties such as carboxylate groups.29
At this stage, another viewpoint is needed to complement NMR analysis with a dynamic picture of the association process. In a previous paper,17 we explored the potential of molecular dynamics for proposing an interaction scenario that can bridge the gap between the transient supramolecular interactions involving a mobile partner, and the average picture yielded by NMR. In the following, we perform MD simulations to decipher the genesis of the binding between the two lanthanide complexes under investigation and ubiquitin.
Dynamics of the interaction of ubiquitin with [Ln(DPA)3]3− and [Ln–Xo4]+ probed by MD simulations
The interaction footprinting of [Ln(DPA)3]3− and [Ln–Xo4]+ was probed along all-atom, explicit solvent molecular MD for an overall simulation time of 3.8 μs (the initial system is shown in Fig. S5, ESI†). Such interactions do not excessively influence the structural stability of the protein (RMSD ∼2 Å), while the flexible loop 72–76 is more involved in fluctuations (see Fig. S13, ESI†). Two main binding sites and a third weaker one can be seen on the 3D density plots shown in Fig. 5, which unambiguously illustrate that [Ln(DPA)3]3− and [Ln–Xo4]+ target distinct binding sites of ubiquitin.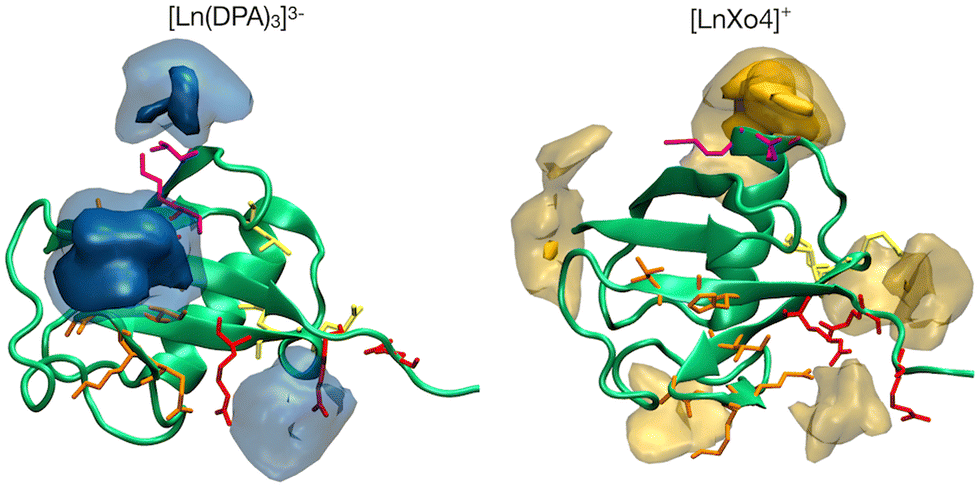 | ||
| Fig. 5 3D densities (in points Å−3) of [Ln(DPA)3]3− (left panel) and [LnXo4]+ (LnXo4, right panel) molecules around the protein. Ubiquitin protein is represented as a green cartoon, while H68 and R42–R74 binding site residues as orange and red lines, respectively. A third binding site is reported for both complexes as magenta lines, corresponding to residues K6, K11 and T12 for Ubi–[Ln(DPA)3]3− and K33 and E34 for Ubi–[LnXo4]+ respectively. Darker colors represent a higher density value (0.5 and 0.15 points Å−3 isosurfaces for [Ln(DPA)3]3− and [LnXo4]+ complexes or molecules, respectively), while lighter ones represent isodensity values of 0.27 and 0.07 points Å−3 for [Ln(DPA)3]3− and [Ln–Xo4]+, respectively. | ||
The different behaviors of the two lanthanide complexes are also evident from the per-residue normalized contact probabilities given in Fig. S8 (ESI†). For [Ln(DPA)3]3−, the relative population of the ligand–protein complex is almost equally shared between two main interaction sites, located around residues: H68–G47 and R42–R72–R74 (Fig. 6). A third, less populated binding site corresponds to the interaction with the surface, solvent-exposed lysine K6. The cluster analysis performed on the concatenated 3.8 μs (see ESI†) allowed us to extract the most representative structures of the ubiquitin–lanthanide complex system in line with the contact probability. The latter are shown in Fig. 6, which depicts the two major binding sites (see also Fig. S9 and S10, ESI†). It is worth noting that these sites do not correspond to a single residue, but to several that cooperatively help to bind the lanthanide tris-dipicolinate complex.
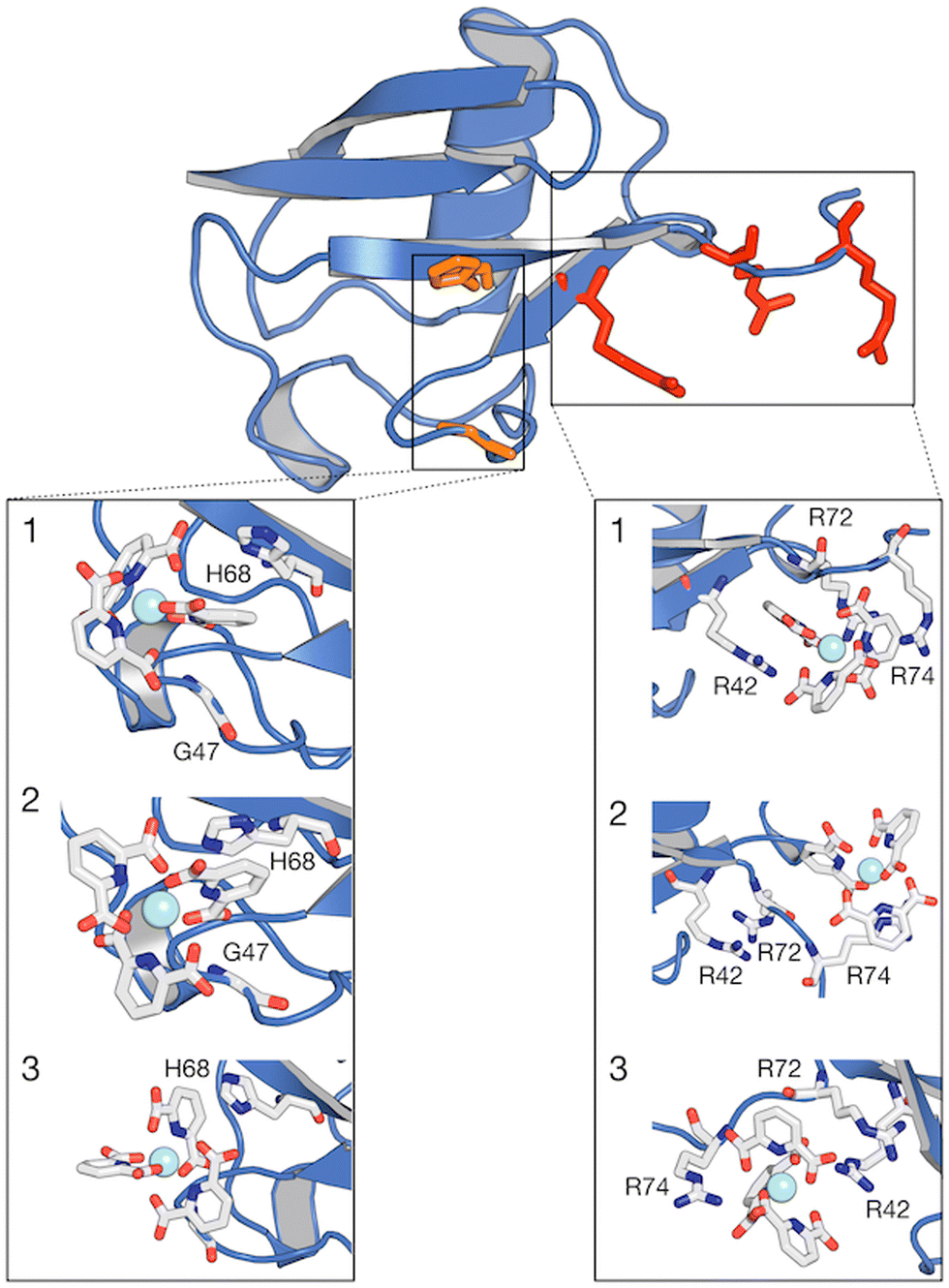 | ||
| Fig. 6 Structure of ubiquitin with regions of higher contact probability with the [Ln(DPA)3]3− highlighted in red colour. Expanded regions showing the 3 most representative structures (1, 2 and 3) of the interaction between ubiquitin and [Ln(DPA)3]3− obtained from cluster analysis. Within the same protein structure, different complexes are captured interacting with the protein. The relative population percentages are 27%, 27% and 25% for structures 1, 2 and 3, respectively. Binding sites H68 and R42–R74 are reported as orange and red green lines and residues are listed in Table S3 (ESI†). | ||
The normalized contact probability (see Fig. S8, ESI†) corroborates that most of the interactions rely on positively-charged (such as K6, K11, K33, R42, H68, R72 and R74) and neutral (such as Q40, G47 and Q49) residues, while no interaction is likely to occur between [Ln(DPA)3]3− and negatively charged residues. In addition, these results suggest that the ubiquitin:[Ln(DPA)3]3− interaction is neither rigid nor gives rise to a single well-defined adduct, instead demonstrating a highly dynamic association between the lanthanide complex and the protein.
Our MD simulations also support that from 2 to 4 [Ln(DPA)3]3− interact with one ubiquitin (see Fig. S11 and S12, ESI†). Conversely, for [Ln–Xo4]+ we observe more than 50% of the relative population of complexes interacting with ubiquitin in one representative structure (Fig. 7), with the most probable stoichiometry values ranging between 1 and 2, reflecting a lower affinity. As expected, [Ln–Xo4]+ molecules are mainly found to interact with negatively charged sidechains, (i) glutamate (E18, E34, and E64) and (ii) aspartate (D32, D51, D58). Few contacts with other sidechains (K33 and Q40) and even hydrophobic residues (G35 and L73) are however also observed. Furthermore, the analysis of the contact probabilities (Fig. S8, ESI†) yields a more accurate picture of the latter interaction process. Indeed, it should be noted that the average contact probability is significantly lower for [Ln–Xo4]+ than that for [Ln(DPA)3]3−, which suggests that [Ln–Xo4]+ explores a broader surface of ubiquitin, with a significantly shorter residence time for each interaction site, highlighting the different behavior of this complex.
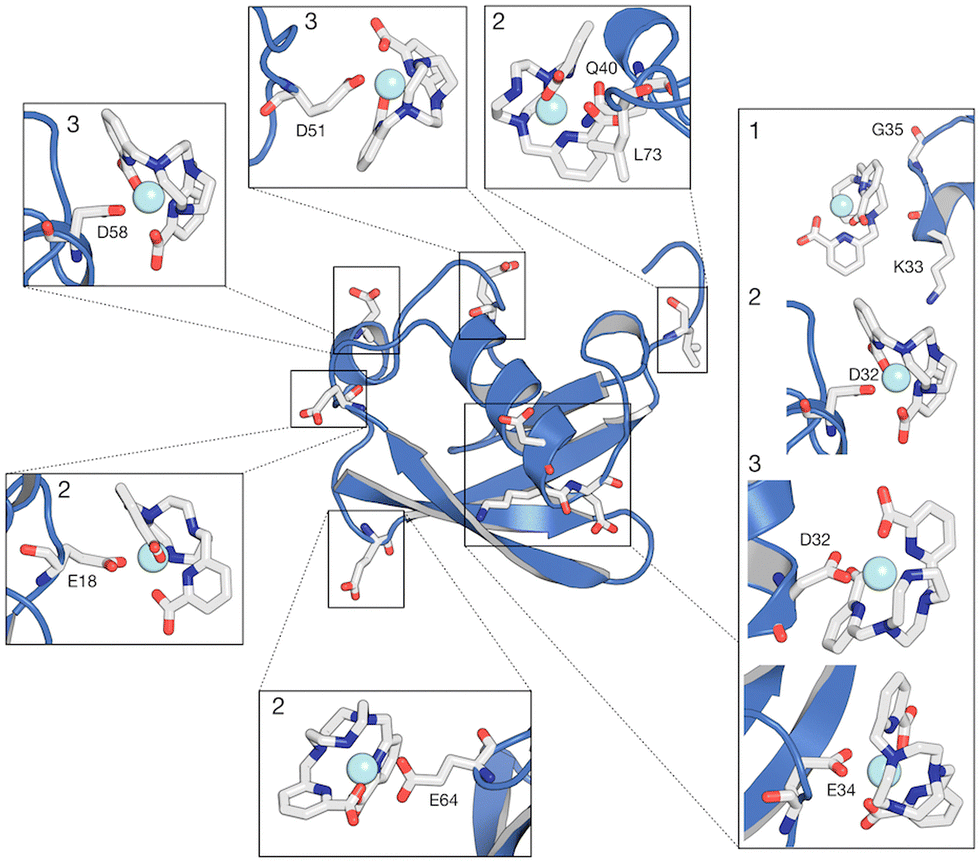 | ||
| Fig. 7 Most representative structures (1, 2 and 3) of the interaction between ubiquitin and [Ln–Xo4]+ obtained from cluster analysis. Within the same structure, different complexes are captured interacting with the protein. The relative population percentages are 56.5, 24 and 8% for structures 1, 2 and 3, respectively. | ||
Discussion
At this stage, it is important to establish the overall landscape of the interaction process as it can be addressed by the combination of NMR spectroscopy and MD simulations.Ubiquitin:[Ln(DPA)3]3− interaction
Table 1 summarizes for each identified interaction site in ubiquitin the groups of residues that are highlighted by PRE and PCS analyses on the one hand, and MD simulations on the other hand. Overall, we observe a good agreement between the various approaches, which allows us to highlight the role played by key residues in the different regions of ubiquitin.| PRE – [Gd(DPA)3]3− | PCS – [Yb(DPA)3]3− | Molecular dynamics | |
|---|---|---|---|
| Site #1 | A46/G47/K48/Q49/T66/H68 | I44/G47/K48/Q49/T66/L67/H68 | I44/A46/G47/K48/Q49/T66/L67/H68 |
| Site #2 | R42/R72/L73/R74 | R72/R74 | R42/R72/L73/R74 |
| Site #3 | K6/T12 | — | K6: K11/T12 |
For the first interaction site, H68 should be emphasized because the protonation state of its side chain, which is pH dependent, is known for regulating the ubiquitin-binding.35 This residue belongs to the main interaction site identified by Wei et al. for the same supramolecular system.27 Furthermore, the second interaction site located at the C-flexible tail contains two positively charged arginine residues R72 and R74. It should be noted here that these residues are known for interacting with aromatic groups via cation–π interactions.36,37 More generally, most of the proteins that have been shown to bind with lanthanide complexes present an arginine residue in their binding site.28 In this study, we observe that this C-flexible tail indeed plays a role in the binding of [Ln(DPA)3]3− to ubiquitin. Our molecular dynamic simulations also enlighten us on the role of R42 in this interaction site. The pair of residues R72 and R74 forms a molecular tweezer that is flexible enough to host two tris-dipicolinates, which are further stabilized by the interaction with R42.38 We note that R72 and R74 have also been investigated by Wei et al. They have determined that the affinity of these residues for DPA ligands decreases significantly at higher pH values (from 6.4 to 7.6), which prevented them from considering these amino-acids as a primary binding site.27 It is also worth noting that several lanthanide complexes were used in their study except [Yb(DPA)3]3−, i.e., with protons at positions ortho and para on the aromatic ring, as in the present work. For the third interaction site, our MD simulations also show that lysine residues such as K6 and K11 are able to make transient contacts with the complex, as evidenced by the interaction footprint obtained along our MD simulations (see Fig. S8, ESI†). Such contacts characterize a transient site residue, typically solvent-exposed, which is in general too weak to be reflected in chemical shift or relaxation perturbation (except for a small relaxation enhancement detected for K6). K33, R54 and K63 play this role as well, as shown in Fig. S10 (ESI†).
To perform a more quantitative analysis of the observed perturbations of TOCSY correlations, we have fitted the evolution of selected measured 1H shifts upon the addition of [Yb(DPA)3]3− with titration curves modeling the effect of the interaction process (Fig. S3, ESI†). Relying on the results of MD simulations and NMR measurements, for each interaction site described above we have selected a set of residues that have been identified as belonging to this site, and we have chosen to fit their 1H shift evolutions together to determine one unique dissociation constant (Kd) value, hence improving the accuracy of the parameter adjustment. We have also assumed that because the structure of ubiquitin is not altered by the interaction process, the three interaction sites are independent. From the analysis of these titration curves it was possible to determine the average values of the dissociation constant of the order of 19 ± 1 mmol L−1 for Site #1, 7.3 ± 0.7 mmol L−1 for Site #2, and 11 ± 8 mmol L−1 for Site #3, reflecting overall a rather weak association process between ubiquitin and [Yb(DPA)3]3−. Noteworthily, these values are higher than the one reported by Wei et al. (Kd = 0.3 mmol L−1).27 The average shifts measured for the different steps of the titration experiment and the resulting titration curves can be found in the ESI† (see Table S2 and Fig. S3).
Finally, our data provide an interesting insight into residues that do not belong to one of the interaction sites highlighted above but play a particular role in the stability of the protein, or even in the supramolecular interaction process itself. On the one hand, Q41 is a widely conserved, crucial residue in the ubiquitin family of proteins as it forms the beginning of the β-strand and its side chain is tightly packed against the α-helix.39 This residue is most rigidly locked in the core of the protein, with its amino protons forming stable hydrogen bonds with the backbone carbonyl oxygens of K27 and I36, respectively.40 Interestingly, the latter hydrogen bond I36–Q41 is known to be decisive in defining the native, folded state of ubiquitin.41 Moreover, the side chains of residues Q41, L43, and F45 point towards the hydrophobic core, while the side chains of R42 and I44 are solvent-exposed.38 Beyond being essential for protein stability as described above, our NMR data suggest that the region around Q41 also participates in the protein–[Ln(DPA)3]3− supramolecular interaction. Indeed, although Q41 does not directly interact with the tris-dipicolinate ligand, this region presents residues, such as Q40 and even R42, that can interact with the negatively charged [Ln(DPA)3]3−. Indeed, these positively charged residues have already been reported as bridge residues between binding sites, due to their ability to coordinate ligands at the protein surface.42–44 We surmise that the use of a non-polarizable force field may also lead to overshooting the formation of salt-bridges with respect to other non-covalent interactions. Our MD simulations in the μs-time range are not prone to capture all microsecond-to-millisecond motions. On the other hand, particular attention should also be paid to residues K11, K27, K29, E34, and D52 that participate in the salt bridges within the α-helix anchor to the β-sheet.45,46 The K11–E24 salt bridge is a second critical interaction defining the ubiquitin folded, native state. Interestingly, most of these residues – with the exception of K11 – are not affected by the presence of the paramagnetic center, which suggests that the protein structure is not significantly changed upon binding. Similarly, no signal perturbation was detected in the residues V17 to A28 that form the α-helix.
Ubiquitin:[Ln–Xo4]+ interaction
In contrast to the analysis of [Ln(DPA)3]3− interaction, paramagnetic NMR analysis does not provide a clear interaction site for [Ln–Xo4]+, and its interaction dynamics is more labile and thus challenging to analyse. Comparatively, while [Ln(DPA)3]3− interacts specifically around positively charged residues, [Ln–Xo4]+ appears to explore more dynamically the whole surface of the protein, as reflected by the rather homogeneous relaxation enhancement effect observed among the different amide proton signals from all the residues. Molecular dynamics analysis, in addition to confirming a broader ligand density at the protein surface (see Fig. 5 and Fig. S7, ESI†), reveals that [Ln–Xo4]+ interacts directly with negatively charged residues, including Q18, D32, E51, D58, E64 and G7, as well with hydrophobic contacts, such as K33, Q40 and L73. Noteworthily, these residues exhibit a lower probability of contact (Fig. S8, ESI†) and are physically distributed throughout the protein in such a way that no specific interaction region is created (Fig. 3B). The cluster analysis further reveals that there is a more likely conformational state for the ubiquitin:[Ln–Xo4]+ interaction, which represents more than 50% of the relative percentage (structure 1 in Fig. 7). This state corresponds to the interaction of [Ln–Xo4]+ with the negatively charged residues D32 and E34 (Fig. S9, ESI†). Interestingly, the interaction of the crystallophore complex in this region requires a certain flexibility at the end of the α-helix loop, with the positively charged residues K33 and Q35 possibly being electrostatically repelled by the positive charge in the lanthanide complex.Our combined NMR/MD investigation offers a direct comparison between the contrasted modes of action of [Ln(DPA)3]3−vs. [Ln–Xo4]+, which is fully consistent with the overall behavior of the crystallophore toward proteins described elsewhere, with a more versatile and tunable mode of action. Indeed, solid state crystallography data indicate that [Tb–Xo4]+ is able to induce a large variety of interactions with (i) the negatively charged residue via direct coordination to the lanthanide ion (i.e. carboxylate), (ii) positively charged residues (arginine and lysine) via hydrogen bonding and (iii) aromatic residues (tryptophan and phenylalanine) via hydrophobic interaction.29
Conclusions
In recent years, molecular dynamics has taken an increasingly important place in the process of analyzing experimental data, as the precision of the simulations and the duration of the calculated trajectories have progressed towards the description of events increasingly close to the reality of the molecular systems involved. In this context, the synergy that can occur between MD and NMR has been demonstrated for numerous systems and to characterize processes of supramolecular interactions involving objects whose flexibility until now constituted an obstacle to the interpretation of spectroscopic data. Here, we have presented a study combining paramagnetic NMR and molecular dynamics, which allows us to revisit the supramolecular interaction process between ubiquitin and lanthanide complexes. On the one hand, for the tris-dipicolinate [Ln(DPA)3]3− complex, we show that the contribution of molecular dynamics makes it possible to propose a point of view that completes the interpretation of this system made there a few years ago by Wei et al.27 and in particular to describe the properties of the secondary interaction sites that were difficult to probe using only NMR when experimental conditions such as pH modify the protonation state of amino acids and therefore their capacity to develop electrostatic type interactions that are fundamental for this type of complex. We have thus demonstrated a dynamic and non-trivial interaction mode involving three interaction sites.On the other hand, we have shown for the [Ln–Xo4]+ complex that molecular dynamics allows for describing the main characteristics of the sites of the protein, which are visited, even furtively, by the complex when it explores its surface. We have notably been able to understand that [Ln–Xo4]+ targets negatively charged functions such as carboxylate groups, which is coherent with our NMR observations even if their interpretation is more challenging for such highly dynamic association processes. This ability to offer a description, even qualitative, of very weak interaction processes is important, because it has been demonstrated, for example, that the remarkable crystallization properties of [Ln–Xo4]+ are linked to its ability to interact not too strongly with proteins. However, probing the direct environment around these chemical functions has been shown to be out of the reach of the NMR experiments that were chosen to monitor this interaction process, i.e. 2D 1H–1H TOCSY and 1H–15N HSQC. We note that 1H–13C HSQC spectra might give further information about possible interaction sites involving carboxylate groups, although this analytical strategy would require a 13C-labelled protein sample to address the sensitivity issue raised by the actual protein concentration. The use of lanthanide center with stronger paramagnetism is also explored in our group to address this issue.
The development of such an approach combining MD and NMR is therefore strategic, since it will pave the way for the modelling of not only the structural, but also the dynamic basis of supramolecular mechanisms such as the crystallization of proteins in the presence of this type of crystallization agent.
Experimental
NMR sample preparation
Uniformly 15N-labelled ubiquitin was obtained from Giotto Biotech and diluted to 0.5 mM in a 56 mM phosphate buffer (pH = 7) with 10% D2O and 0.02% NaN3. Then, 500 μL of ubiquitin solution was transferred to a 5 mm NMR tube. For the PRE assay, a solution of 25 mM lanthanide complex [Gd(DPA)3]3− (DPA = 2,6-pyridine dicarboxylate) was added to the ubiquitin sample to prepare solutions with the molecular ratios of 0.2, 0.4 and 0.8. For the PCS experiments, a solution with 25 mM [Yb(DPA)3]3− was added to the ubiquitin sample to obtain samples with the following ubiquitin:[Yb(DPA)3]3− ratios ranging from 0 to up to 10. A similar procedure was used for crystallophore complexes, with molecular ratios ranging from 0 to 1.5 equivalents for the ubiquitin–[Gd–Xo4]+ complex, and 0 to 4 molar equivalents for [Yb–Xo4]+. The detailed procedures for preparing tris-dipicolinate lanthanide complexes ([Na]3[Ln(DPA)3]˙·xH2O) have been published elsewhere.25NMR spectra acquisition
All NMR spectra and experiments were recorded at 300 K on a 600 MHz Bruker AVANCE IVDr spectrometer, equipped with a 5-mm 2H–1H–X BBI probe with a z field gradient coil. After each lanthanide complex addition, 1D 1H, 2D 1H–1H TOCSY, 1H–1H NOESY, and 1H–15N HSQC spectra were recorded and processed using TopSpin 4.1.4 software.The 2D 1H–1H TOCSY spectra were recorded using a MLEV-17 spin-lock sequence with a mixing time of 70 ms and a solvent suppression scheme. Typically, spectra were acquired with 256 t1 increments, 1024 data points, and a relaxation delay of 0.8 s and 8 with 32 scans.
The 2D 1H–15N HSQC spectra were obtained using a standard phase-sensitive edited HSQC sequence with gradients and adiabatic pulses. Typically, 256 t1 increments were acquired, with 4096 data points, a relaxation delay of 2 s, and 16 scans. All spectra were zero-filled in the F1 spectral dimension to 1024 data points followed by a forward linear prediction of 400 points. The baseline of the FID was corrected using a Gaussian function to further suppress the residual water signal. Finally, a square sine bell window function (SSB = 2) was applied in both dimensions prior to the Fourier transformation.
Computational methods
Classical all-atom molecular dynamics (MD) simulations were performed using the Amber18 package.47 The lanthanide complex ligand parametrization is described in our previous work15 for the [Ln(DPA)3]3− (Δ and Λ enantiomers) ligand and was used hereby.Binding site search onto the protein surfaces follows a computational protocol recently exploited.42–44,50 Ubiquitin monomer structure with 76 residues was taken from an unpublished, high-resolution PDB provided by Dr E. Girard IBS, Grenoble. Protonation states of ubiquitin residues were assigned using the H++ server48 at pH 6.5. Ubi–[Ln(DPA)3]3− and Ubi–LnXo4 with 10 ligands (5 Δ and 5 Λ enantiomers) were immersed in parallelepiped boxes containing 56940 and 65397 TIP3P49 water molecules, respectively. Na+ and Cl− counterions were added to correspond to a concentration of 0.15 M and to mimic the experimental ionic strength of about 0.17 M, leading to simulation boxes with dimensions of 130 × 123 × 123 Å3 and 134 × 130 × 128 Å3 (shown in Fig. S5, ESI†).
Rendering
Fig. 5 was rendered using visual molecular dynamics (VMD).51Fig. 6 and 7 were rendered using Pymol.Author contributions
N. G., E. D. and O. M. jointly conceptualised the work and acquired funding. N. G. administrated the project. K. D. S. and A. B. wrote the manuscript. A. R. and F. R. synthesized the lanthanide complexes. A. B., N. G., R. F. and E. D. performed MD simulations. K. D. S., E. L., Z. X., P. C. and X. C. carried out NMR experiments. M. B., G. B., C. C. and S. D. Q. supervised the NMR experiments. All authors contributed to data analysis and assisted with the drafting of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
KDS acknowledges support from École Doctorale Médicament, Toxicologie, Chimie, Imageries (MTCI ED563) for her PhD studentship funding. NG and ED acknowledge the support of the French Agence Nationale de la Recherche (ANR) under the grant ANR-22-CE29-0026-01 (project DREAMY). AB acknowledges région AuRA for his post-doctoral fellowship (Crystfrag project). NMR studies were performed at the MétaboParis Santé core facility of BioMedTech Facilities (INSERM US36 – CNRS UAR2009 – Université Paris Cité).Notes and references
- S. C. J. Meskers and H. P. J. M. Dekkers, J. Phys. Chem. A, 2001, 105, 4589–4599 CrossRef CAS.
- H. Tsukube and S. Shinoda, Chem. Rev., 2002, 102, 2389–2404 CrossRef CAS PubMed.
- R. Jastrząb, M. Nowak, M. Skrobańska, A. Tolińska, M. Zabiszak, M. Gabryel, L. Marciniak and M. T. Kaczmarek, Coord. Chem. Rev., 2019, 382, 145–159 CrossRef.
- M. Rajendran, E. Yapici and L. W. Miller, Inorg. Chem., 2014, 53, 1839–1853 CrossRef CAS PubMed.
- K. Y. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770–1839 CrossRef CAS PubMed.
- S. Engilberge, F. Riobé, S. Di Pietro, L. Lassalle, N. Coquelle, C.-A. Arnaud, D. Pitrat, J.-C. Mulatier, D. Madern, C. Breyton, O. Maury and E. Girard, Chem. Sci., 2017, 8, 5909–5917 RSC.
- É. Girard, M. Stelter, J. Vicat and R. Kahn, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2003, 59, 1914–1922 CrossRef PubMed.
- N. R. Silvaggi, L. J. Martin, H. Schwalbe, B. Imperiali and K. N. Allen, J. Am. Chem. Soc., 2007, 129, 7114–7120 CrossRef CAS PubMed.
- G. Pompidor, A. D’Aléo, J. Vicat, L. Toupet, N. Giraud, R. Kahn and O. Maury, Angew. Chem., Int. Ed., 2008, 47, 3388–3391 CrossRef CAS PubMed.
- R. E. McGovern, S. C. Feifel, F. Lisdat and P. B. Crowley, Angew. Chem., Int. Ed., 2015, 54, 6356–6359 CrossRef CAS PubMed.
- M. L. Rennie, G. C. Fox, J. Pérez and P. B. Crowley, Angew. Chem., Int. Ed., 2018, 57, 13764–13769 CrossRef CAS PubMed.
- A. Roux, R. Talon, Z. Alsalman, S. Engilberge, A. D’Aléo, S. Di Pietro, A. Robin, A. Bartocci, G. Pilet, E. Dumont, T. Wagner, S. Shima, F. Riobé, E. Girard and O. Maury, Inorg. Chem., 2021, 60, 15208–15214 CrossRef CAS PubMed.
- M. Allegrozzi, I. Bertini, M. B. L. Janik, Y.-M. Lee, G. Liu and C. Luchinat, J. Am. Chem. Soc., 2000, 122, 4154–4161 CrossRef CAS.
- I. Bertini, C. Luchinat, G. Parigi and R. Pierattelli, ChemBioChem, 2005, 6, 1536–1549 CrossRef CAS PubMed.
- X. Jia, H. Yagi, X.-C. Su, M. Stanton-Cook, T. Huber and G. Otting, J. Biomol. NMR, 2011, 50, 411–420 CrossRef CAS PubMed.
- G. Pintacuda, M. John, X.-C. Su and G. Otting, Acc. Chem. Res., 2007, 40, 206–212 CrossRef CAS PubMed.
- S. Denis-Quanquin, A. Bartocci, F. Szczepaniak, F. Riobé, O. Maury, E. Dumont and N. Giraud, Phys. Chem. Chem. Phys., 2021, 23, 11224–11232 RSC.
- A. Hershko and A. Ciechanover, Annu. Rev. Biochem., 1992, 61, 761–807 CrossRef CAS PubMed.
- S. Bergink and S. Jentsch, Nature, 2009, 458, 461–467 CrossRef CAS PubMed.
- A. Al-Hakim, C. Escribano-Diaz, M.-C. Landry, L. O’Donnell, S. Panier, R. K. Szilard and D. Durocher, DNA Repair, 2010, 9, 1229–1240 CrossRef CAS PubMed.
- Z. J. Chen and L. J. Sun, Mol. Cell, 2009, 33, 275–286 CrossRef CAS PubMed.
- R. Yau and M. Rape, Nat. Cell Biol., 2016, 18, 579–586 CrossRef CAS PubMed.
- N. Ouali, B. Bocquet, S. Rigault, P.-Y. Morgantini, J. Weber and C. Piguet, Inorg. Chem., 2002, 41, 1436–1445 CrossRef CAS PubMed.
- T. Saio, M. Yokochi, H. Kumeta and F. Inagaki, J. Biomol. NMR, 2010, 46, 271–280 CrossRef CAS PubMed.
- E. Dumont, G. Pompidor, A. D’Aléo, J. Vicat, L. Toupet, R. Kahn, E. Girard, O. Maury and N. Giraud, Phys. Chem. Chem. Phys., 2013, 15, 18235 RSC.
- S. Denis-Quanquin, F. Riobé, M.-A. Delsuc, O. Maury and N. Giraud, Chem. – Eur. J., 2016, 22, 18123–18131 CrossRef CAS PubMed.
- Z. Wei, Y. Yang, Q.-F. Li, F. Huang, H.-H. Zuo and X.-C. Su, Chem. – Eur. J., 2013, 19, 5758–5764 CrossRef CAS PubMed.
- X.-C. Su, H. Liang, K. V. Loscha and G. Otting, J. Am. Chem. Soc., 2009, 131, 10352–10353 CrossRef CAS PubMed.
- S. Engilberge, F. Riobé, T. Wagner, S. Di Pietro, C. Breyton, B. Franzetti, S. Shima, E. Girard, E. Dumont and O. Maury, Chem. – Eur. J., 2018, 24, 9739–9746 CrossRef CAS PubMed.
- G. Tetreau, M. R. Sawaya, E. De Zitter, E. A. Andreeva, A.-S. Banneville, N. A. Schibrowsky, N. Coquelle, A. S. Brewster, M. L. Grünbein, G. N. Kovacs, M. S. Hunter, M. Kloos, R. G. Sierra, G. Schiro, P. Qiao, M. Stricker, D. Bideshi, I. D. Young, N. Zala, S. Engilberge, A. Gorel, L. Signor, J.-M. Teulon, M. Hilpert, L. Foucar, J. Bielecki, R. Bean, R. De Wijn, T. Sato, H. Kirkwood, R. Letrun, A. Batyuk, I. Snigireva, D. Fenel, R. Schubert, E. J. Canfield, M. M. Alba, F. Laporte, L. Després, M. Bacia, A. Roux, C. Chapelle, F. Riobé, O. Maury, W. L. Ling, S. Boutet, A. Mancuso, I. Gutsche, E. Girard, T. R. M. Barends, J.-L. Pellequer, H.-W. Park, A. D. Laganowsky, J. Rodriguez, M. Burghammer, R. L. Shoeman, R. B. Doak, M. Weik, N. K. Sauter, B. Federici, D. Cascio, I. Schlichting and J.-P. Colletier, Nat. Commun., 2022, 13, 4376 CrossRef CAS PubMed.
- B. Vögeli, S. Engilberge, E. Girard, F. Riobé, O. Maury, T. J. Erb, S. Shima and T. Wagner, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3380–3385 CrossRef PubMed.
- S. Engilberge, T. Wagner, G. Santoni, C. Breyton, S. Shima, B. Franzetti, F. Riobé, O. Maury and E. Girard, J. Appl. Crystallogr., 2019, 52, 722–731 CrossRef CAS PubMed.
- H. Yagi, K. V. Loscha, X.-C. Su, M. Stanton-Cook, T. Huber and G. Otting, J. Biomol. NMR, 2010, 47, 143–153 CrossRef CAS PubMed.
- C. Nitsche and G. Otting, Prog. Nucl. Magn. Reson. Spectrosc., 2017, 98–99, 20–49 CrossRef CAS PubMed.
- K. Fujiwara, T. Tenno, K. Sugasawa, J.-G. Jee, I. Ohki, C. Kojima, H. Tochio, H. Hiroaki, F. Hanaoka and M. Shirakawa, J. Biol. Chem., 2004, 279, 4760–4767 CrossRef CAS PubMed.
- J. P. Gallivan and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459–9464 CrossRef CAS PubMed.
- K. Kumar, S. M. Woo, T. Siu, W. A. Cortopassi, F. Duarte and R. S. Paton, Chem. Sci., 2018, 9, 2655–2665 RSC.
- N. A. Lakomek, C. Farès, S. Becker, T. Carlomagno, J. Meiler and C. Griesinger, Angew. Chem., Int. Ed., 2005, 44, 7776–7778 CrossRef CAS PubMed.
- T. I. Igumenova, A. J. Wand and A. E. McDermott, J. Am. Chem. Soc., 2004, 126, 5323–5331 CrossRef CAS PubMed.
- C. Scheurer, N. R. Skrynnikov, S. F. Lienin, S. K. Straus, R. Brüschweiler and R. R. Ernst, J. Am. Chem. Soc., 1999, 121, 4242–4251 CrossRef CAS.
- S. Kitazawa, M. Yagi-Utsumi, K. Kato and R. Kitahara, Molecules, 2017, 22, 1414 CrossRef PubMed.
- A. Bartocci, N. Gillet, T. Jiang, F. Szczepaniak and E. Dumont, J. Phys. Chem. B, 2020, 124, 11371–11378 CrossRef CAS PubMed.
- A. Bartocci, G. Pereira, M. Cecchini and E. Dumont, J. Chem. Inf. Model., 2022, 62, 6739–6748 CrossRef CAS PubMed.
- A. Bartocci and E. Dumont, Probing the dynamical interaction of the para-sulfonato-calix[4]arene with an antifungal protein, Phys. Chem. Chem. Phys., 2023, 25, 18067–18074 RSC.
- J. J. Prompers, C. Scheurer and R. Brüschweiler, J. Mol. Biol., 2001, 305, 1085–1097 CrossRef CAS PubMed.
- E. Heid and D. Braun, Phys. Chem. Chem. Phys., 2019, 21, 4435–4443 RSC.
- D. Case; I. Ben-Shalom; S. Brozell; D. Cerutti; T. Cheatham III; V. Cruzeiro; T. Darden; R. Duke; D. Ghoreishi and M. Gilson, AMBER 2018, University of California, San Francisco, 2018 Search PubMed.
- R. Anandakrishnan, B. Aguilar and A. V. Onufriev, Nucleic Acids Res., 2012, 40, W537–W541 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- A. Bartocci and E. Dumont, Situating the phosphonated calixarene-cytochrome C association by molecular dynamics simulations, J. Chem. Phys., 2024, 160, 105101 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp00463a |
| ‡ K. Dos Santos and A. Bartocci contributed equally to this work as the first authors. |
| This journal is © the Owner Societies 2024 |