
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConformationally supramolecular chirality prevails over configurational point chirality in side-chain liquid crystalline polymers†
Xiaoxiao
Cheng
a,
Yijing
Gan
a,
Gong
Zhang
a,
Qingping
Song
*b,
Zhengbiao
Zhang
 *a and
Wei
Zhang
*ab
*a and
Wei
Zhang
*ab
aState and Local Joint Engineering Laboratory for Novel Functional Polymeric Materials, Jiangsu Engineering Laboratory of Novel Functional Polymeric Materials, Suzhou Key Laboratory of Macromolecular Design and Precision Synthesis, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, Suzhou, 215123, P. R. China. E-mail: zhangzhengbiao@suda.edu.cn; weizhang@suda.edu.cn
bSchool of Chemical and Environmental Engineering, Anhui Polytechnic University, Wuhu, 241000, P. R. China. E-mail: songqp@ahpu.edu.cn
First published on 18th April 2023
Abstract
In nature, the communication of primary amino acids in the polypeptides influences molecular-level packing, supramolecular chirality, and the resulting protein structures. In chiral side-chain liquid crystalline polymers (SCLCPs), however, the hierarchical chiral communication between supramolecular mesogens is still determined by the parent chiral source due to the intermolecular interactions. Herein, we present a novel strategy to enable the tunable chiral-to-chiral communication in azobenzene (Azo) SCLCPs, in which the chiroptical properties are not dominated by the configurational point chirality but by the conformationally supramolecular chirality that emerged. The communication of dyads biases supramolecular chirality with multiple packing preference, thereby overruling the configurational chirality of the stereocenter. The chiral communication mechanism between the side-chain mesogens is revealed through the systematic study of the chiral arrangement at the molecular level, including mesomorphic properties, stacking modes, chiroptical dynamics and further morphological dimensions.
Introduction
In nature, the most advanced functions of biological and non-biological systems emerge from homochiral primary structures,1–3 such as DNA replication, enzyme asymmetric catalysis, and biological evolution.4–6 The study of the chiral communication between primary structures can provide a theoretical basis for elucidating the origin of biological homochirality. Among this, artificial regulation of chiral communication in chiral polymeric supramolecules has been receiving ever-increasing attention due to its importance for both biological functions and application of chiral materials.7,8 In particular, in side-chain liquid crystalline polymers (SCLCPs),9–11 intensive research has been fueled by their actively tunable physicochemical properties and structural complexity, comparable to those of sophisticated natural materials.12,13 However, the conventional chirality regulation of polymers mainly relies on the modification of the chiral stereocenter or the external chiral sources.14–17 It is still a great challenge to regulate the chiral superstructures in different ways that can potentially program the helicity-switching in the opposite handedness.In 1989, Green et al. first induced nonracemic main-chain helical polyisocyanates by a small chiral bias and described the unusual chirality amplification of the helical backbone. Two general strategies, Sergeants-and-Soldiers Principle (chiral-to-achiral communication) and Majority Rule (chiral communication between enantiomers), were established and further developed in helical systems with low helix inversion barriers.18,19 Both effects are based on the accumulation of small local energy preferences in the helical main-chain, which results in an excess of helical conformation in completely dissolved polymer systems. In the case of an “abnormal” Sergeant-and-Soldiers effect,20 Suginome et al. successfully controlled the helicity of poly(quinoxaline-2,3-diyl)s by modulating the contents of chiral/achiral dyads in the polymer chain. The backbone helicity could be inverted by changing the distribution between the chiral–chiral and the chiral–achiral segments according to the Sato model.21 Furthermore, the combination of the chiral-sergeant and chiral-soldier dyads in the polymer chains could enable the selective interconversion of backbone helicity due to the differential expression of the two chiral components. This leads to pioneering exploration of chiral communication mechanisms, including chiral-to-chiral sergeants-and-soldiers effect,22 chiral coalition,23 chiral conflict,24 chiral accord,25 as well as synergistic chiral enhancement effect.26
Alternatively, modulating the stacking patterns of building blocks (i.e., intra-chain and inter-chain packing, J- and H-aggregation according to Kasha's exciton theory27) is another efficient strategy for controlling the chiroptical properties and predicting the handedness of suprastructures.28 Our previous studies described the multiple chiroptical inversion effects in SCLCP assemblies, which was driven by the transition of the different triple stacking modes (intra-chain stacking and inter-chain chiral transcription with the combination of H- and J-aggregation).29,30 However, once two chiral building block dyads are packed into one polymer chain with different sequences (random or block with different distributions),31–33 the synergistic effect of the supramolecular packing and the sequence switching will bring an unexpected chiral communication. Such a synergistic effect can guide the functional design of peptides, where the chiral communication of primary amino acid sequences in the polypeptides influences molecular-level packing, supramolecular chirality, and the resulting morphology.34–36
The elucidation of chiral communication in synthetic polymers is beneficial for the design of chiral materials, since the sequence of monomers in polymer chains can be easily regulated by living polymerization. Herein, we report the well-controlled chiral-to-chiral communication and morphological transition in SCLCP assemblies by copolymerizing two Azo dyads bearing the same stereocenter (Scheme 1). The relative ratio of the two chiral comonomer dyads, molecular weight of solvophobic blocks and the sequence of polymer structures have a significant effect on the chiral communication effect, resulting in tunable chiroptical properties of SCLCPs. This is collectively confirmed to be driven by multiple transitions of conformational supramolecular chirality in the chiral LC phase, ignoring the configuration of point chirality. The current sequence-controlled chiral communication strategy is different from typical chiral amplification effects in dissolved polymer systems, which do not involve stacking modes between chiral units.
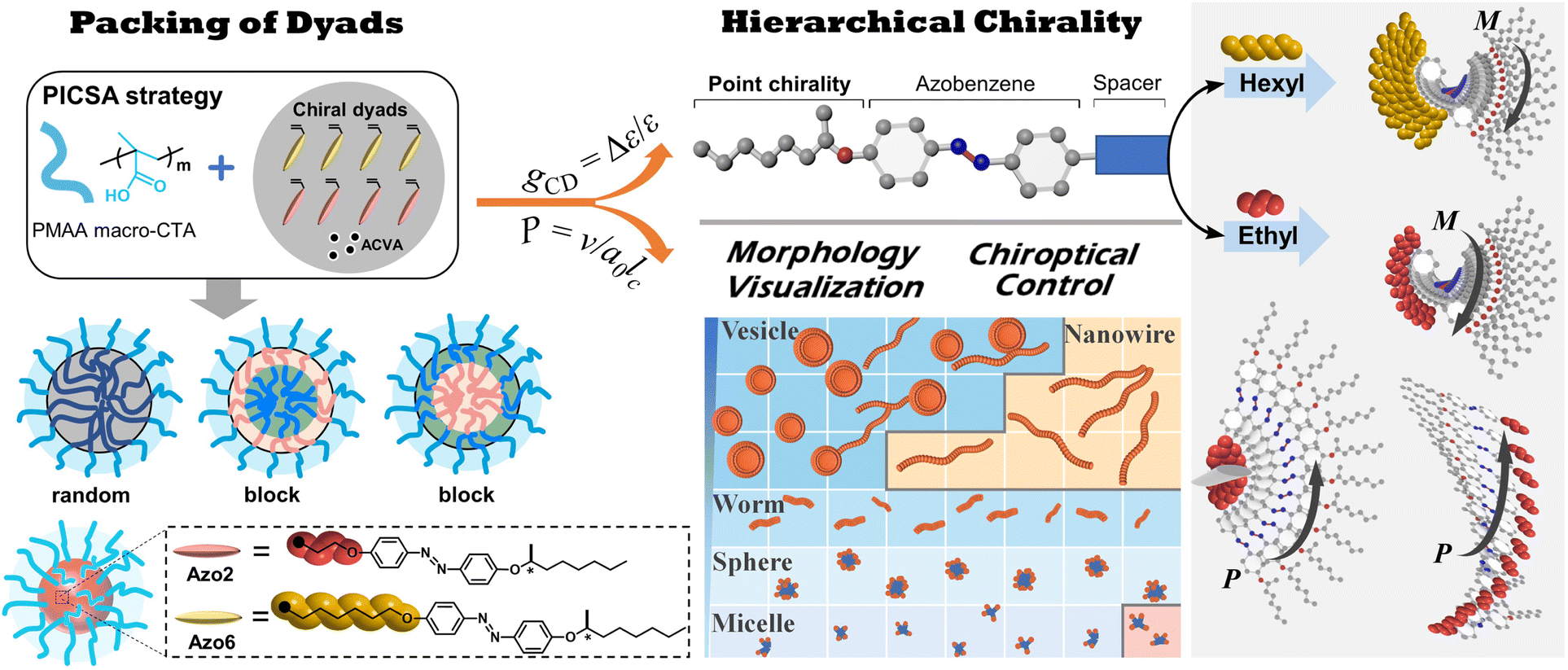 | ||
| Scheme 1 The chiroptical control and morphological visualization in sequence-dependent SCLCPs. In these systems, the conformationally supramolecular chirality determines the chiroptical properties of the assemblies, regardless of the configurational point chirality of the stereocenter. | ||
Results and discussion
Polymerization-induced chiral self-assembly (PICSA) of amphiphilic supramolecular assemblies
To explore this unprecedented chiral communication, judicious choice of monomer dyads is thereby critical. Two chiral Azo comonomers, Azo6 and Azo2 (Scheme 1), with different spacers but the same chiral tails were chosen, as the former only underwent chiral head-to-head H-aggregation,29 while the latter has been shown to undergo competitive DP-dependent stacking modes including intra-chain stacking, inter-chain H-aggregation and head-to-tail J-aggregation.30We firstly synthesized solvophilic poly(methacrylic acid) as the macromolecular chain transfer agent (PMAA macro-CTA) by reversible addition–fragmentation chain transfer (RAFT) polymerization. The DP of PMAA macro-CTA was 51, the Mn and Đ were 9000 g mol−1 and 1.08, according to 1H NMR spectra and GPC analysis (Fig. S1†), respectively. The purified PMAA51 macro-CTA was subsequently in situ chain-extended with Azo2 and Azo6 in selective ethanol. A series of amphiphilic PMAA51-b-P(Azo61−n-co-Azo2n), PMAA51-b-P(Azo61−n-b-Azo2n) and PMAA51-b-P(Azo2n-b-Azo61−n) copolymer assemblies composed of two chiral units in different ratios were prepared by the stepwise-feeding seeded RAFT polymerization (Fig. 1a and S2†). The conversion of the monomer was higher than 98%, and the linear Mn-conversion relationship (Fig. S3†) indicated the living nature of the polymerization-induced chiral self-assembly (PICSA) process.37–40 Nearly quantitative polymerization was achieved and the polymer dispersity of all copolymer sequences was relatively low (Đ < 1.25, Fig. S4–S6†). Their random and block distributions within the polymer chains were characterized by the Kelen–Tüdos method and 1H NMR spectra.41
In the PICSA process, the living polymerization, supramolecular stacking and self-assembly occurred simultaneously, allowing the preparation of the corresponding supramolecular SCLCP assemblies in situ. Fig. 1 shows the aggregation-induced circular dichroism (CD) that originated from exciton coupling between the interacting Azo units in side-chains. It should be noted that the stereocenter is located at the terminal tails of the side-chain, which has a low influence on the radical polymerization of the flexible backbone. In this aspect, the introduced alkyl spacers also maintain the flexibility of the main-chains. The involved polymer contains four important units: (1) the main-chain of methacrylate; (2) alkyl spacer; (3) Azo mesogens and (4) alkyl chiral tails. A polymethacrylate main-chain was chosen because of its flexibility in polymers. The polymethacrylate main-chains were prepared by radical polymerization, whose tacticity is inherently not stereospecific. To preserve the flexibility of the main-chains, alkyl linkers were also inserted between the main-chain and the Azo mesogens. The induced supramolecular chirality arises from superstructural LC chirality, i.e., helical structure via aggregation of side-chain Azo units, and is independent of the conformation of the polymer backbone.
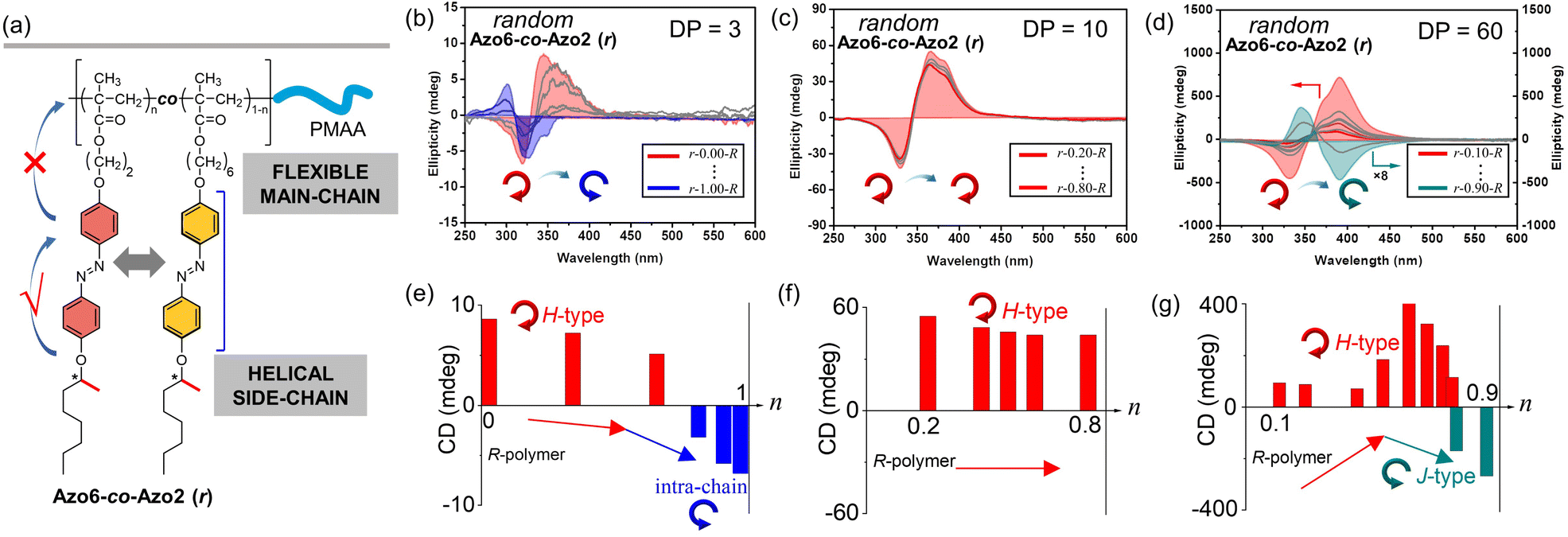 | ||
| Fig. 1 (a) The chemical structures of random Azo6-co-Azo2 SCLCP assemblies. CD spectra and maximum changes of random Azo6-co-Azo2 assemblies with (b) and (e) DP = 3, (c) and (f) DP = 10, (d) and (g) DP = 60 with the increase of Azo2 ratio (n). | ||
The stacking modes of 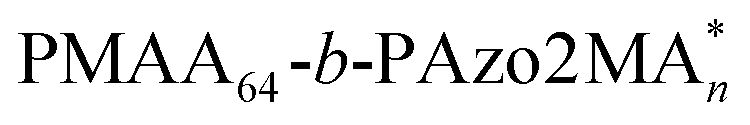 diblock copolymer have been clearly revealed by many studies,29,30,42 which are also presented in Fig. S7† for clarity. Broad bands containing four typical absorption peaks could be detected in the UV-vis spectra due to the aggregated interaction between adjacent azo units (Fig. S7a–e†). The local absorption maxima at ca. 320 nm, 340 nm, 357 nm and 377 nm can be assigned to the intra-chain stacking, inter-chain H-aggregation, non-associated Azo units and inter-chain J-aggregation, respectively. The type of supramolecular chirality can be determined by the dominated stacking modes by distinguishing the exciton coupling centers.
diblock copolymer have been clearly revealed by many studies,29,30,42 which are also presented in Fig. S7† for clarity. Broad bands containing four typical absorption peaks could be detected in the UV-vis spectra due to the aggregated interaction between adjacent azo units (Fig. S7a–e†). The local absorption maxima at ca. 320 nm, 340 nm, 357 nm and 377 nm can be assigned to the intra-chain stacking, inter-chain H-aggregation, non-associated Azo units and inter-chain J-aggregation, respectively. The type of supramolecular chirality can be determined by the dominated stacking modes by distinguishing the exciton coupling centers.
Chiroptical control by the synergistic effect of dyad ratios and sequence switching
CD, infrared spectroscopy (IR) and vibratory circular dichroism (VCD) were first employed to detect the conformation of the polymer backbone. As shown in Fig. S8,† CD signals of three copolymer sequences are obviously silent in the well-dissolved THF solvent. Meanwhile, three copolymer sequences exhibited same VCD silences of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band between 1750 and 1700 cm−1. The results demonstrate that the polymer backbone does not have a distinct helical conformation. Thus, aggregation-induced LC chirality is responsible for CD signals of assemblies in ethanol dispersion. Taking Azo assemblies with an R-configuration as examples, random Azo6-co-Azo2 SCLCP assemblies with DP = 3 showed a positive Cotton effect with a maximum at 347 nm when Azo2 ratio n = 0 (Fig. 1b and e, inter-chain H-type). However, as n gradually increased to 1.0, the CD spectra gradually decreased and were opposite in sign with similar shapes but blue-shifted Cotton band (intra-chain stacking). The opposite sign indicates that the preferred helicity of the Azo stack inverts between n = 0 and n = 1.0. When the DP of the random copolymers was 10, little changes in the CD sign and shape can be observed as n gradually increased (Fig. 1c and f, inter-chain H-type). Unexpectedly, for copolymers with a DP of 60, the CD properties of the assemblies appear significantly different (Fig. 1d and g). For example, a positive Cotton effect was first observed when n = 0.1 (inter-chain H-type). As n gradually increased to 0.9, the CD maxima first increased and gradually decreased to nearly zero, and then inverted to negative values with a red-shifted Cotton band (inter-chain J-type). These transitions depend on the ratio of Azo2 units, demonstrating that the Azo2/Azo6 dyads compete for the chiral stacking in the self-assembly process.
O stretching band between 1750 and 1700 cm−1. The results demonstrate that the polymer backbone does not have a distinct helical conformation. Thus, aggregation-induced LC chirality is responsible for CD signals of assemblies in ethanol dispersion. Taking Azo assemblies with an R-configuration as examples, random Azo6-co-Azo2 SCLCP assemblies with DP = 3 showed a positive Cotton effect with a maximum at 347 nm when Azo2 ratio n = 0 (Fig. 1b and e, inter-chain H-type). However, as n gradually increased to 1.0, the CD spectra gradually decreased and were opposite in sign with similar shapes but blue-shifted Cotton band (intra-chain stacking). The opposite sign indicates that the preferred helicity of the Azo stack inverts between n = 0 and n = 1.0. When the DP of the random copolymers was 10, little changes in the CD sign and shape can be observed as n gradually increased (Fig. 1c and f, inter-chain H-type). Unexpectedly, for copolymers with a DP of 60, the CD properties of the assemblies appear significantly different (Fig. 1d and g). For example, a positive Cotton effect was first observed when n = 0.1 (inter-chain H-type). As n gradually increased to 0.9, the CD maxima first increased and gradually decreased to nearly zero, and then inverted to negative values with a red-shifted Cotton band (inter-chain J-type). These transitions depend on the ratio of Azo2 units, demonstrating that the Azo2/Azo6 dyads compete for the chiral stacking in the self-assembly process.
The effect of the copolymer sequences on the chiral communication phenomena was then considered (Fig. 2a). As shown in Fig. 2b and c, the CD maxima of random Azo6-co-Azo2, block Azo6-b-Azo2 and block Azo2-b-Azo6 SCLCP assemblies were inverted twice and essentially dependent on the polymer sequences. Both chiral competition and sequence switching of Azo mesogens can change the hierarchical stacking modes and further control chiral-to-chiral communication in Azo side-chains (Fig. 2d). Therefore, we systematically investigated the dependence of chiroptical transitions on the Azo sequence, the ratio of Azo2 units (n) and DP of Azo cores (Fig. 3 and S9–S12†).
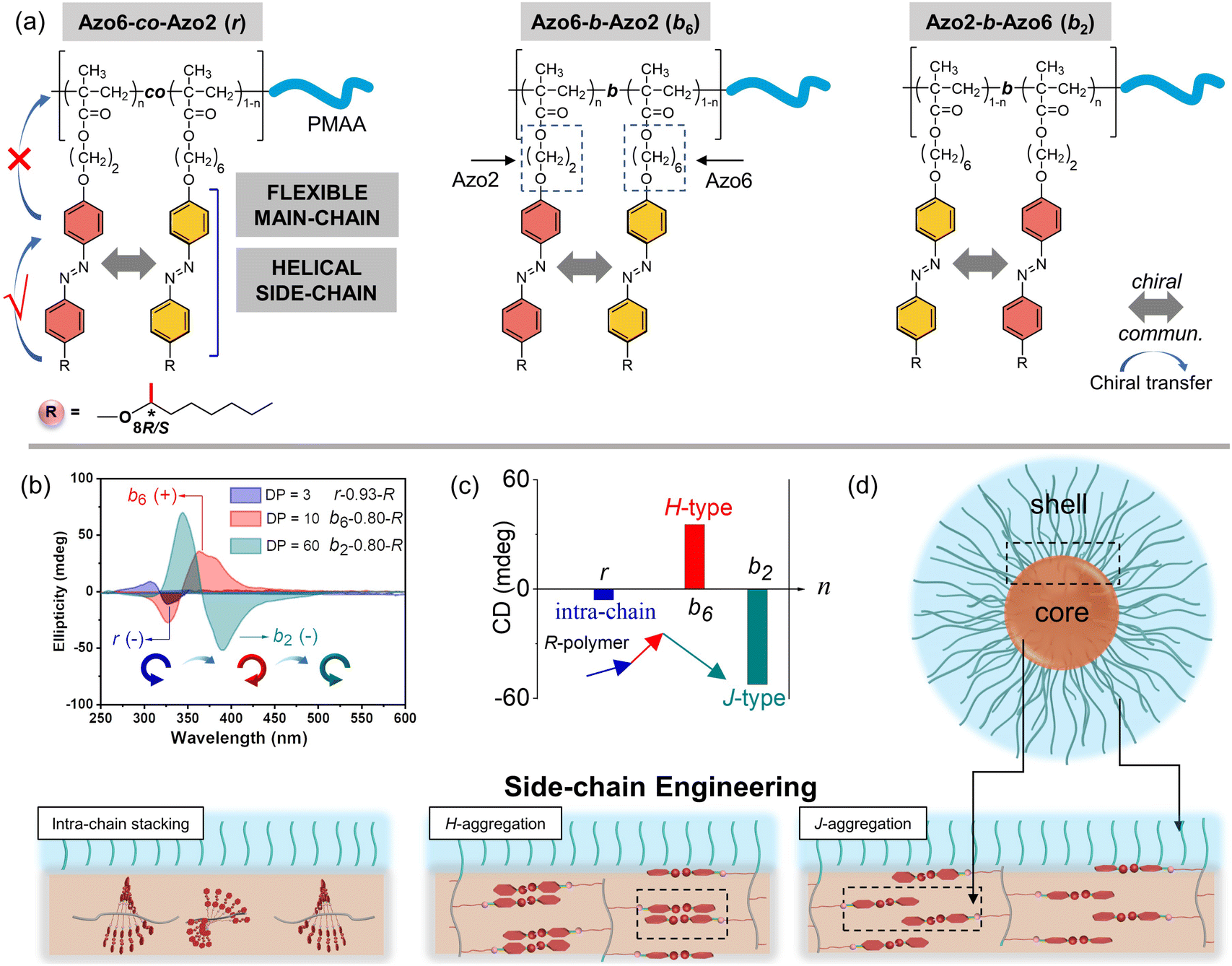 | ||
| Fig. 2 Synergistic effects of chiral competition and sequence switching between Azo mesogens toward controlling the hierarchical chiral-to-chiral communication. (a) The chemical structures of random Azo6-co-Azo2, block Azo6-b-Azo2 and block Azo2-b-Azo6 copolymers. (b) and (c) CD spectra of random Azo6-co-Azo2 (r, DP = 3, n = 0.93), block Azo6-b-Azo2 (b6, DP = 10, n = 0.80) and block Azo2-b-Azo6 (b2, DP = 60, n = 0.80). (d) Stacking modes inside the solvophobic core. | ||
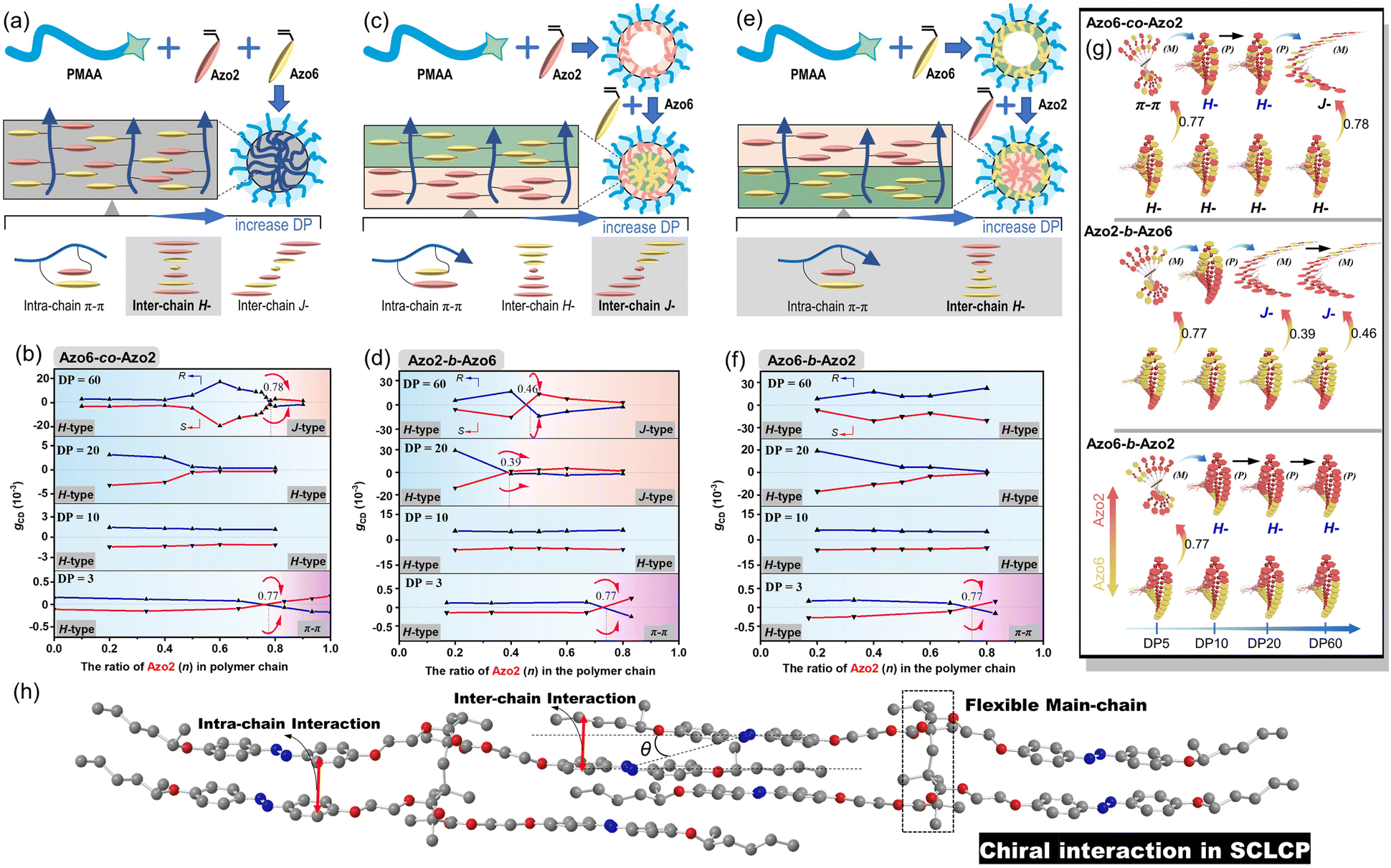 | ||
Fig. 3 Schematic representation of the PICSA procedure of (a) random Azo6-co-Azo2, (c) block Azo2-b-Azo6 and (e) block Azo6-b-Azo2 SCLCP assemblies. The gCD values and the transition of stacking modes of (b) random Azo6-co-Azo2, (d) block Azo2-b-Azo6, (f) block Azo6-b-Azo2 SCLCP assemblies with the increase of DP and Azo2 ratios. (g) The internal stacking presented by the yellow Azo6 and red Azo2 units, respectively. (h) The homopolymer poly(Azo2) exhibits supramolecular polymorphism, and the unique asymmetric stacking modes can be depicted by the slip angle θ according to Kasha's exciton theory. (dissymmetry factors, gCD = Δε/ε = [ellipticity/32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 980]/absorbance at the CD extremum). 980]/absorbance at the CD extremum). | ||
We separately analyzed the results extracted from different sequence systems; all discussions were based on Azo assemblies with an R-configuration (blue line) unless otherwise specified. For the random Azo6-co-Azo2 SCLCPs with Azo dyads randomly packed inside the core (Fig. 3a), the assemblies exhibited preferred positive gCD values when DP = 3 and n < 0.77 (Fig. 3b, bottom). However, the positive signals were completely inverted to negative values when n approached 1.0. From a mechanistic point of view, it should be noted that Azo6 units only have chiral H-aggregation preference, while the Azo2 unit can adopt changeable stacking modes with mutually inverted signals, such as π–π stacking along one polymer chain (intra-chain) or H- and J-aggregations of different polymer chains (inter-chain).30 The chiroptical inversion probably originated from the transition from inter-chain H-aggregation to intra-chain stacking, according to the chiral competition of Azo2/Azo6 dyads. For DPs of 10 and 20, the assemblies showed chiral H-aggregation without the chiroptical inversion, independent of the content of Azo2 units. Interestingly, the chiroptical signals were inversed from positive to negative signals when the DP of the core-forming blocks was equal to 60 (Fig. 3b, top), with the reverse point n at 0.78, indicating the transition from H- to J-aggregates.
The ratio-dependent transition was also observed in block Azo2-b-Azo6 SCLCP assemblies (Fig. 3c). When the DP was as low as 3 (Fig. 3d, bottom), the CD signals changed from positive to negative, indicating the transition from inter-chain H-aggregation to intra-chain stacking. The H-aggregates without reversal of gCD values consistently existed in assemblies with a DP of 10. However, the H-to-J transformation-induced chiroptical inversion was found when the DP was 20. A continuous increase of DP to 60 still resulted in a H-to-J transition that varied with the Azo2 ratio (Fig. 3d, top). The reversal points for DP20 and DP60 were n = 0.39 and 0.46, respectively. In this case, the Azo6 unit that prefers H-packing is induced by the Azo2 unit to arrange into a J-type stack, thereby inducing a reversal of gCD values in the spectra.
For the block Azo6-b-Azo2 SCLCP assemblies where the Azo6 segment is adjacent to the PMAA fragment (Fig. 3e), the chiroptical inversion also existed when the DP of Azo core-forming blocks was 3 (Fig. 3f, bottom), the reversal point n was 0.77, similar to that of Azo6-co-Azo2 and Azo2-b-Azo6. In contrast, the gCD values showed the same H-aggregation associated with a positive Cotton effect when DP exceeded 10. This result indicated that the H-stacking of Azo6 units commands the Azo2 units to adopt the same stacking patterns, essentially independent of Azo2 ratios and DP variation.
Meanwhile, the corresponding CD data also follow the changing tendency of gCD values (Fig. S12†). Mirror-image chiroptical changes and therefore similar stacking transitions were presented in assemblies with an S-configuration, indicating the control over the differentiations of the chiral communication. According to the above results, the multiple inversions of the SCLCP assemblies are independent of the stereocenter, suggesting that chiroptical properties are dominated not by the configurational point chirality but by the conformationally supramolecular chirality. The rich regulation of chiroptical properties in these SCLCP assemblies is impossible to realize in any previous main-chain helical polymers.
These multiple chiroptical inversions may follow a novel chiral-to-chiral communication mechanism, which is different from the traditional chiral communication effects (one is the chiral communication between the chiral sergeants and two enantiomeric soldiers, and the other is the classical “Sergeants-and-Soldiers Principle” composed of chiral sergeants and achiral soldiers). An explanation of those results can be envisioned by considering the chiral competition of the Azo2 units in the helical H-packing of the parent poly(Azo6) segments and their response to the DP of Azo blocks (schematic presentation in Fig. 3g and h). The Azo6 units tend to form a single preferred chiral H-aggregation. As a result, Azo6 units can induce Azo2 units to adopt preferential chiral H-aggregation in three sequences once small amounts of Azo2 units are copolymerized, which is independent of the DP of Azo blocks. However, as the ratio of Azo2 increased, the competition between Azo2 and Azo6 units that bias supramolecular helicity becomes more prominent; the more abundant Azo2 units can command Azo6 units to selectively adopt one of the three stacking modes. As a result, multiple chiroptical inversions can be observed in Azo6-co-Azo2 and Azo2-b-Azo6 SCLCP assemblies, although the transition of H- to J-aggregation occurs early in the latter. The block Azo6-b-Azo2 SCLCP assemblies are exceptional since they possess no J-type chiroptical property. This can be ascribed to the preferential nucleation of first Azo6 blocks, leading to the formation of thermodynamically stable H-aggregation. Thus, it is difficult for the second Azo2 blocks to transform the already formed H-cores into unstable J-aggregates even at high DP and n. Therefore, we could conclude that the chiral communication mechanism observed in Azo SCLCPs is derived from two features: (1) the comonomers must display different packing preferences, e.g., Azo2 with different stacking modes in self-assembly and Azo6 with only stable H-aggregation; (2) the SCLCP assemblies tend to express the chiroptical property of core-forming blocks that are firstly nucleated.
Helicity modulation and morphological visualization of polymer assemblies
The morphology of Azo assemblies may change due to the minimization of energetically unfavorable hydrophobe–solvent interactions.43,44 The packing parameter P (P = v/a0lc, a0 is the area of the PMAA hydrophilic segment, v and lc are the volume and length of the Azo hydrophobic segment) of amphiphilic Azo assemblies can be fine-tuned from P ≤ 1/3 for spherical micelles to 1/3 ≤ P ≤ 1/2 for cylindrical micelles and then to 1/2 ≤ P ≤ 1 for vesicles,45–47 as the ratio of solvophobic Azo6 units and the DP of the core-forming blocks increased. The morphological transition of these Azo SCLCP assemblies was further analyzed (Fig. 4 and S13–S16†). Taking assemblies with DP = 60 as an example (R-configuration), random Azo6-co-Azo2 assemblies gradually changed from vesicles to long nanowires as the ratio of Azo2 units increased (Fig. 4a). The average diameter![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) obtained from TEM first increased from 266 to 516 nm, and then decreased to 32 nm. This morphological transition was also observed in the case of block Azo2-b-Azo6 assemblies (Fig. 4b), while the continuously decreased from 187 to 34 nm. Interestingly, the inner stacking modes of these two sequences also changed from H-aggregation to J-aggregation when the vesicles were completely transformed into nanowires. Meanwhile, strong attenuation and reverse of CD signals from H-to-J transition were observed when vesicles were completely transformed into long nanowires (CD values in Fig. 4a and b). The CD changes can be ascribed to that the uniform helical twisting in smectic layers is forbidden by symmetry rules. The H-aggregates would first grow together with the orthogonal smectic layers (vide infra), inducing strong H-stacking helical bias, which would be then suppressed since the smectic layers become more ordered. Interestingly, the vesicular morphology of block Azo6-b-Azo2 assemblies was retained in all cases without morphological transition (Fig. 4c), which is consistent with the fact that only H-aggregated signals were observed in the CD spectra.
obtained from TEM first increased from 266 to 516 nm, and then decreased to 32 nm. This morphological transition was also observed in the case of block Azo2-b-Azo6 assemblies (Fig. 4b), while the continuously decreased from 187 to 34 nm. Interestingly, the inner stacking modes of these two sequences also changed from H-aggregation to J-aggregation when the vesicles were completely transformed into nanowires. Meanwhile, strong attenuation and reverse of CD signals from H-to-J transition were observed when vesicles were completely transformed into long nanowires (CD values in Fig. 4a and b). The CD changes can be ascribed to that the uniform helical twisting in smectic layers is forbidden by symmetry rules. The H-aggregates would first grow together with the orthogonal smectic layers (vide infra), inducing strong H-stacking helical bias, which would be then suppressed since the smectic layers become more ordered. Interestingly, the vesicular morphology of block Azo6-b-Azo2 assemblies was retained in all cases without morphological transition (Fig. 4c), which is consistent with the fact that only H-aggregated signals were observed in the CD spectra.
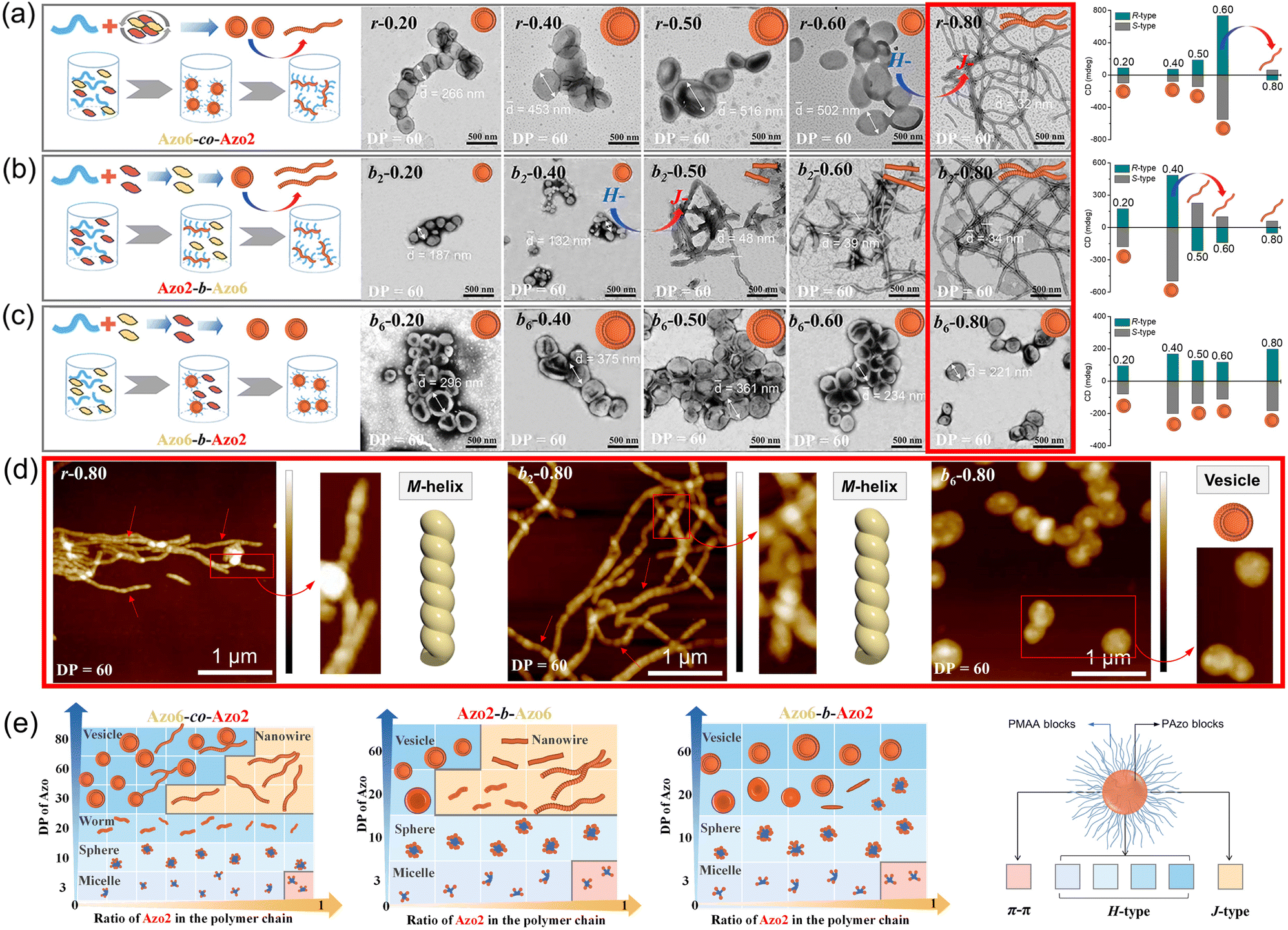 | ||
| Fig. 4 Solvophobic phase separation drives sharp morphological changes of assemblies with different Azo2 ratios. (a) Random Azo6-co-Azo2 (r), (b) block Azo2-b-Azo6 (b2) and (c) block Azo6-b-Azo2 (b6) of Azo assemblies with an R-configuration. (d) AFM images corresponding to samples shown in the red box of (a), (b) and (c), respectively. (e) Phase diagram of the self-assembled morphology with different DP and Azo2 unit ratios in these copolymer sequences. The packing modes are shown with gradated background colors representing intra-chain stacking (pink), inter-chain H-aggregation (blue) and J-aggregation (orange). | ||
Clearly, tuning the ratio of Azo2 units in polymer chains and thus the sequences of the resultant random or block copolymers effectively varies the morphologies of the assemblies. For SCLCP assemblies with identical DPs (DP = 60) and Azo2 ratios (n = 0.8) (red boxes in panels Fig. 4a–c), the sequence switching-driven morphological transition was further confirmed by AFM measurements (Fig. 4d and S16†). Exclusively left-handed (M-type) helical nanowires were observed both in random Azo6-co-Azo2 and block Azo2-b-Azo6 assemblies with an R-configuration, corresponding to the negative Cotton effect of J-aggregates. Meanwhile, isotropic vesicles of block Azo6-b-Azo2 were observed in the AFM images, and the corresponding CD values displayed positive signals related to H-aggregation. Considering the sequences of polymers was the only alteration in these SCLCPs; it was reasonable to infer that the morphological differences as well as above chiroptical changes were attributed to the sequence switching.
The growing DP of Azo core-forming blocks leads to an increase in the packing parameter P, thus a micelle-to-sphere-to-vesicle transition was observed for these SCLCP assemblies (Fig. 4e). The phase diagrams clearly showed preferential morphological transitions at a lower Azo2 ratio, regardless of the polymer sequences. However, the inner stacking modes are closely related to the morphological transition of the assemblies. For example, the packing mode of intra-chain stacking only existed in micelles (pink area). The change from vesicles to nanowires appeared in both Azo6-co-Azo2 and Azo2-b-Azo6 due to the transition from H-aggregation (blue area) to J-aggregation (orange area), whereas the vesicle is the main phase in Azo6-b-Azo2; in this case, only H-aggregates are present. Therefore, both the chiral communication and morphology visualization of SCLCP assemblies can be achieved through the transition of the conformationally supramolecular chirality of the Azo building blocks.
Dynamic chiral response controlled by UV/vis light and temperature
The conformational supramolecular chirality can be further regulated by external stimuli due to the nature of weak noncovalent interactions and the flexible trans–cis properties of Azo units.48–52 Herein, light and temperature were employed to switch the handedness of the helical structures. When the Azo assemblies are exposed to 365 nm UV light, significant decreases of the π–π* transition (ca. 350 nm) as well as increases of the n–π* transition (ca. 450 nm) were detected, regardless of the copolymer sequences and stacking modes (Fig. S17 and S18†). The CD values of both H- and J-aggregates decreased during the trans-to-cis processes (Fig. 5a–c), which completely disappeared in the photostationary state due to the loss of ordered helical suprastructures. The bent cis-form Azo units with various (Ph-NN-Ph) dihedral angles disrupted the efficient Azo helical stacks and led to disordered structures (Fig. 5d).
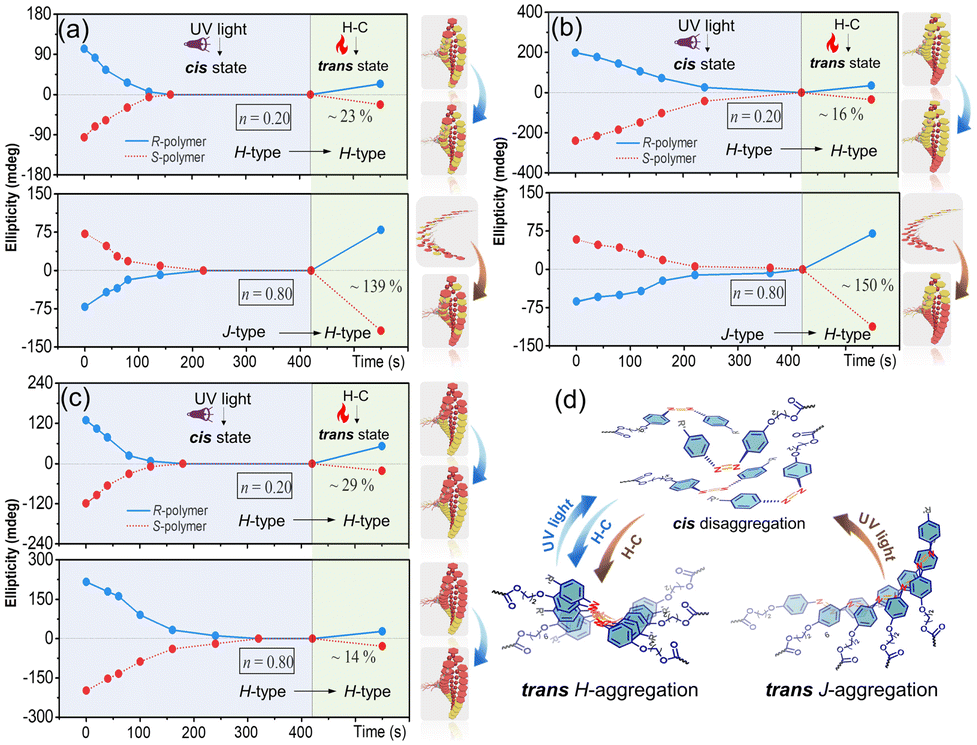 | ||
| Fig. 5 Changes of the CD maximum (first Cotton band) of Azo SCLCP assemblies upon 365 nm UV light irradiation and H–C treatment. (a) Random Azo6-co-Azo2, (b) block Azo2-b-Azo6 and (c) block Azo6-b-Azo2 with n = 0.2 and n = 0.8, respectively. (d) The helical order–disorder transition of H- and J-aggregations in the process of trans–cis–trans isomerization. The DP of the corresponding Azo core-forming blocks is 60. | ||
In the heating–cooling (H–C) assisted reassembly process, the destroyed Azo stacks in these copolymer assemblies underwent different pathways to recover their helical superstructures, as demonstrated by the reappearance of the CD signals. After heating and then cooling Azo6-co-Azo2 assemblies, recovery effectiveness of ∼23% for the H → H pathway was observed when n = 0.20 (Fig. 5a, top), while a chiroptical inversion of the J → H pathway with ∼139% chiroptical enhancement was detected when n = 0.80 (Fig. 5a, bottom). Analogous H–C treatment of Azo2-b-Azo6 showed similar trends to that of Azo6-co-Azo2, with recovery effectiveness of ∼16% for the H → H pathway and ∼150% enhancement for the J → H pathway (Fig. 5b). Nevertheless, only the H → H pathway with ∼29% (n = 0.20) and ∼14% (n = 0.80) recovery effectiveness was observed in Azo6-b-Azo2 (Fig. 5c). These results indicate that H-aggregates are more thermodynamically stable than J-aggregates (Fig. 5d). When cis-disaggregated Azo units recover to trans helical stacks, the H–C procedure could invert and enhance the helical asymmetry in the J → H transition while inhibiting the recovery of helical asymmetry in the H → H process. The effectiveness of the chiral recovery depends on the sequence differences and the chiral competition between Azo2/Azo6 dyads for generating the preferred noncovalent stacks.
Driving force and communication mechanism of chirality evolution
To investigate the underlying chiral communication mechanism, the changes in the mesomorphic properties and inner packing parameters of Azo copolymers were studied in detail. Our previous results demonstrated that the chirality of intra-chain stacking is dominant in the amorphous phase, while H-chirality and J-chirality are dominant in TGBA* and SmA* phases, respectively.30 For the assemblies containing intra-chain stacking, as shown in Fig. S19–S21,† only broad peaks with a normal distribution (2θ = 16.0°) were observed by wide-angle X-ray diffraction (WAXD) in the angle region where LC structures were expected. And no scattering peaks were detected in small-angle X-ray scattering (SAXS) patterns (Fig. S22–S24†), suggesting the amorphous nature of Azo assemblies with intra-chain stacking mode. The typical distance between intra-chain adjacent Azo units in chiral π–π stacking mode is 3.6 Å.Meanwhile, Azo assemblies with chiroptical properties of H- and J-aggregation modes showed a clear LC phase with periodic layered structures, regardless of the copolymer sequences. These can be distinguished by the clear diffraction peaks in WAXD patterns (Fig. S19–S21†) and two proportional scattering peaks in SAXS spectra (q1/q2 = 1/2, Fig. S22–S24†). The periodicity of layer spacing (d) and the distance of Azo units (r) were calculated according to SAXS and WAXD patterns, as presented in Fig. 6a and b. With the increase of Azo2 units, the layer spacing d decreased from ca. 5.5 nm to ca. 4.8 nm (Fig. 6a), but was less than twice of the fully extended Azo6 and Azo2 chain length (2 L = 5.88 nm for Azo6 and 2 L = 4.94 nm for Azo2 calculated by Gaussian 09, Fig. 6c). These results clearly suggested a double-layered smectic structure with an interdigitation of the side-chains.
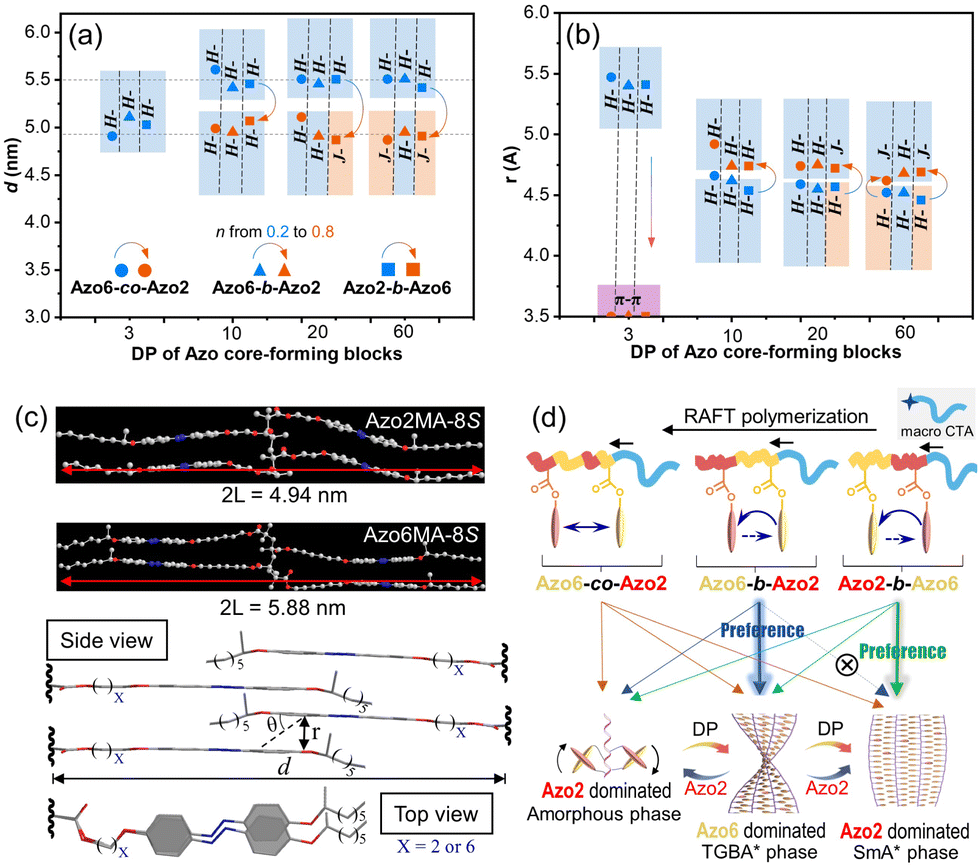 | ||
| Fig. 6 The mesomorphic properties and inner packing parameters in the Azo copolymer series. Plots of (a) the calculated layer spacing (d) and (b) the distances of Azo units (r) versus DP of Azo core-forming blocks (the increase of Azo2 ratio is presented as a color change from blue dots: n = 0.2 to red dots: n = 0.8). (c) The fully extended length of the Azo2MA-8S and Azo6MA-8S side-chains. Bottom: the proposed packing structures of the double-layered LC phase in the Azo SCLCP series. (d) Relationship between copolymer sequences and competitive phase transitions. | ||
In the LC phase, however, the distances of Azo units (r) increased with the increase of Azo2 units (Fig. 6b). Because Azo2 units with a shorter spacer have a weaker solvophobicity than Azo6 units, as the ratio of Azo2 units increased, the loosed microdomains and increased distances lead to a smaller slip angle θ of Azo units (Fig. 6c), resulting in the transition from H-aggregation (θ > magic angle 54.7°) to J-aggregation (θ < 54.7°). POM images also indicated the LC phase transition in three copolymer sequences, where the bright LC aggregates can be observed in a broad region (Fig. S25–S27†). The amorphous-to-TGBA*-to-SmA* phase transition allows the transition of three stacking modes, resulting in the mutually opposite chiroptical properties of exciton coupling between closely situated transition dipole moments on neighboring Azo units.
As a result, the LC phase structures and chiroptical transitions were dependent on the copolymer sequences (Fig. 6d). In Azo6-co-Azo2 and Azo2-b-Azo6, the preferential nucleation of Azo2 units allows phase transitions to occur, so multiple chiroptical inversions of the three stacking modes exist. For example, in Azo2-b-Azo6, the stepwise-feeding procedure favors the first Azo2 segment to form the SmA* phase before adding the second Azo6 segment. As a result, the chiroptical inversion from H- to J-aggregation occurs favorably, which can be reflected by the CD signal reversal at DP = 20 and n = 0.39.
In Azo6-b-Azo2, however, the Azo6 segments tend to adopt a preferred TGBA* phase responsible for inducing a solely chiral H-aggregation in the nucleus. Thus, the pre-formed H-aggregates of Azo6 units dominate the chiral preference of the core and command the neighbouring Azo2 units to adopt the same H-aggregation. As a result, the transition from H- to J-aggregation did not occur in these copolymer assemblies even at higher DP and n, as demonstrated by CD in Fig. 3f.
Conclusions
In summary, we have reported the regulation of conformationally supramolecular chirality in SCLCP assemblies composed of random Azo6-co-Azo2, block Azo2-b-Azo6 and block Azo2-b-Azo6, in which the chiral dyads possess the same configurational point chirality. Sequence switching can generate differences in the stacking preference and aggregation order of Azo2/Azo6 units, enabling control of the chiral-to-chiral communication and assembly morphologies. The ratio- and DP-dependent chiroptical properties and LC characterization indicate that the transition of mesomorphic properties, for example, the amorphous-to-TGBA*-to-SmA* phase transition, is the driving force for the chiral communication effect. Moreover, a multidimensional phase diagram from micelles to spheres, worms, helical nanowires and vesicles was observed, visually demonstrating the transition of the stacking modes. The helicity modulation and morphology visualization of SCLCP are achieved through the transition of the conformational supramolecular arrangements, which breaks the limitation of absolute configuration and will find practical application in the design of functions in complex LC materials.Data availability
The experimental data can be found in the ESI† and manuscript and is referred to throughout the manuscript.Author contributions
Prof. W. Zhang and X. X. Cheng conceived the research idea and co-wrote the original draft. Y. J. Gan, Q. P. Song and G. Zhang performed the synthesis and characterization. Prof. W. Zhang and Prof. Z. B. Zhang revised the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for the financial support from the National Nature Science Foundation of China (92056111, 21971180 and 21925107), China Postdoctoral Science Foundation (2022M722312), Key Laboratory of Polymeric Material Design and Synthesis for Biomedical Function, the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions and the Program of Innovative Research Team of Soochow University.Notes and references
- Y. Ma, X. Cheng, H. Ma, Z. He, Z. Zhang and W. Zhang, Chem. Sci., 2022, 13, 13623–13630 RSC.
- T. Ikai, M. Okubo and Y. Wada, J. Am. Chem. Soc., 2020, 142, 3254–3261 CrossRef CAS PubMed.
- J. Yuan, X. Lu, Q. Li, Z. Lu and Q. Lu, Angew. Chem., Int. Ed., 2021, 60, 12308–12312 CrossRef CAS PubMed.
- T. Miao, X. Cheng, H. Ma, Z. He, Z. Zhang, N. Zhou, W. Zhang and X. Zhu, Angew. Chem., Int. Ed., 2021, 60, 18566–18571 CrossRef CAS.
- T. Nakano and Y. Okamoto, Chem. Rev., 2001, 101, 4013–4038 CrossRef CAS PubMed.
- S. Cai, J. Chen, S. Wang, J. Zhang and X. Wan, Angew. Chem., Int. Ed., 2021, 60, 9686–9692 CrossRef CAS PubMed.
- W. Shang, X. Zhu, Y. Jiang, J. Cui, K. Liu, T. Li and M. Liu, Angew. Chem., Int. Ed., 2022, 61, e202210604 CAS.
- Y. Yan, K. Deng, Z. Yu and Z. Wei, Angew. Chem., Int. Ed., 2009, 48, 2003–2006 CrossRef CAS.
- T. Miao, X. Cheng, G. Zhang, Y. Wang, Z. He, Z. Wang and W. Zhang, Chem. Sci., 2023, 14, 1673–1678 RSC.
- L. Wang, L. Yin, W. Zhang, X. Zhu and M. Fujiki, J. Am. Chem. Soc., 2017, 139, 13218–13226 CrossRef CAS PubMed.
- Z. Zheng, Y. Li, H. K. Bisoyi, L. Wang, T. J. Bunning and Q. Li, Nature, 2016, 531, 352–356 CrossRef CAS PubMed.
- C. D. H. Alarcon, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- J. Babin, M. Pelletier, M. Lepage, J.-F. Allard, D. Morris and Y. Zhao, Angew. Chem., Int. Ed., 2009, 48, 3329–3332 CrossRef CAS PubMed.
- H. Goto and E. Yashima, J. Am. Chem. Soc., 2002, 124, 7943–7949 CrossRef CAS PubMed.
- T. Ikai, R. Ishidate, K. Inoue, K. Kaygisiz, K. Maeda and E. Yashima, Macromolecules, 2020, 53, 973–981 CrossRef CAS.
- G. Liu, X. Li, J. Sheng, P. Z. Li, W. K. Ong, S. Z. F. Phua, H. Agren, L. Zhu and Y. Zhao, ACS Nano, 2017, 11, 11880–11889 CrossRef CAS PubMed.
- K. Maeda, S. Wakasone, K. Shimomura, T. Ikai and S. Kanoh, Macromolecules, 2014, 47, 6540–6546 CrossRef CAS.
- M. M. Green, M. P. Reidy, R. J. Johnson, G. Darling, D. J. Oleary and G. Willson, J. Am. Chem. Soc., 1989, 111, 6452–6454 CrossRef.
- M. M. Green, N. C. Peterson, T. Sato, A. Teramoto, R. Cook and S. Lifson, Science, 1995, 268, 1860–1866 CrossRef CAS PubMed.
- Y. Nagata, T. Nishikawa and M. Suginome, J. Am. Chem. Soc., 2015, 137, 4070–4073 CrossRef CAS PubMed.
- S. Wang, J. Chen, X. Feng, G. Shi, J. Zhang and X. Wan, Macromolecules, 2017, 50, 4610–4615 CrossRef CAS.
- K. Cobos, E. Quinoa, R. Riguera and F. Freire, J. Am. Chem. Soc., 2018, 140, 12239–12246 CrossRef CAS PubMed.
- S. Arias, R. Rodriguez, E. Quinoa, R. Riguera and F. Freire, J. Am. Chem. Soc., 2018, 140, 667–674 CrossRef CAS PubMed.
- M. Alzubi, S. Arias, R. Rodriguez, E. Quinoa, R. Riguera and F. Freire, Angew. Chem., Int. Ed., 2019, 58, 13365–13369 CrossRef CAS PubMed.
- S. Sforza, T. Tedeschi, R. Corradini and R. Marchelli, Eur. J. Org. Chem., 2007, 5879–5885 CrossRef CAS.
- K. Cobos, R. Rodriguez, E. Quinoa, R. Riguera and F. Freire, Angew. Chem., Int. Ed., 2020, 59, 23724–23730 CrossRef CAS PubMed.
- G. Liu, J. Sheng, H. Wu, C. Yang, G. Yang, Y. Li, R. Ganguly, L. Zhu and Y. Zhao, J. Am. Chem. Soc., 2018, 140, 6467–6473 CrossRef CAS PubMed.
- F. Yang, X. Liu and Z. Yang, Angew. Chem., Int. Ed., 2021, 60, 14671–14678 CrossRef CAS PubMed.
- X. Cheng, T. Miao, L. Yin, Y. Ji, Y. Li, Z. Zhang, W. Zhang and X. Zhu, Angew. Chem., Int. Ed., 2020, 59, 9669–9677 CrossRef CAS PubMed.
- X. Cheng, T. Miao, Y. Ma, X. Zhu, W. Zhang and X. Zhu, Angew. Chem., Int. Ed., 2021, 60, 24430–24436 CrossRef CAS PubMed.
- L. Wang, Y. Ding, Q. Liu, Q. Zhao, X. Dai, X. Lu and Y. Cai, ACS Macro Lett., 2019, 8, 623–628 CrossRef CAS PubMed.
- R. K. Roy and J. F. Lutz, J. Am. Chem. Soc., 2014, 136, 12888–12891 CrossRef CAS PubMed.
- T. Wen and R. Ho, ACS Macro Lett., 2017, 6, 370–374 CrossRef CAS PubMed.
- E. G. Bellomo, M. D. Wyrsta, L. Pakstis, D. J. Pochan and T. J. Deming, Nat. Mater., 2004, 3, 244–248 CrossRef CAS PubMed.
- D. Ke, C. Zhan, A. D. Li and J. Yao, Angew. Chem., Int. Ed., 2011, 50, 3715–3719 CrossRef CAS PubMed.
- N. R. Lee, C. J. Bowerman and B. L. Nilsson, Biomacromolecules, 2013, 14, 3267–3277 CrossRef CAS PubMed.
- C. E. Boott, J. Gwyther, R. L. Harniman, D. W. Hayward and I. Manners, Nat. Chem., 2017, 9, 785–792 CrossRef CAS PubMed.
- J. Chen, S. Cai, R. Wang, S. Wang, J. Zhang and X. Wan, Macromolecules, 2020, 53, 1638–1644 CrossRef CAS.
- S. Jimaja, S. Varlas, Y. Xie, J. C. Foster, D. Taton, A. P. Dove and R. K. O'Reilly, ACS Macro Lett., 2020, 9, 226–232 CrossRef CAS PubMed.
- F. Lv, Z. An and P. Wu, Nat. Commun., 2019, 10, 1397 CrossRef PubMed.
- K. Maeda, M. Muto, T. Sato and E. Yashima, Macromolecules, 2011, 44, 8343–8349 CrossRef CAS.
- G. Iftime, F. L. Labarthet, A. Natansohn and P. Rochon, J. Am. Chem. Soc., 2000, 122, 12646–12650 CrossRef CAS.
- J. Yeow and C. Boyer, Adv. Sci., 2017, 4, 1700137 CrossRef PubMed.
- B. Zhang, X. Lv, A. Zhu, J. Zheng, Y. Yang and Z. An, Macromolecules, 2018, 51, 2776–2784 CrossRef CAS.
- R. Deng, M. J. Derry, C. J. Mable, Y. Ning and S. P. Armes, J. Am. Chem. Soc., 2017, 139, 7616–7623 CrossRef CAS PubMed.
- M. Douverne, Y. Ning, A. Tatani, F. C. Meldrum and S. P. Armes, Angew. Chem., Int. Ed., 2019, 58, 8692–8697 CrossRef CAS PubMed.
- S. Xu, J. Yeow and C. Boyer, ACS Macro Lett., 2018, 7, 1376–1382 CrossRef CAS PubMed.
- J. del Barrio, R. M. Tejedor, L. S. Chinelatto, C. Sanchez, M. Pinol and L. Oriol, J. Mater. Chem., 2009, 19, 4922–4930 RSC.
- S. Huang, Y. Chen, S. Ma and H. Yu, Angew. Chem., Int. Ed., 2018, 57, 12524–12528 CrossRef CAS PubMed.
- Y. J. Gan, H. B. Dai, Y. F. Ma, X. X. Cheng, Z. Wang and W. Zhang, Macromolecules, 2022, 55, 8556 CrossRef CAS.
- Z. Liu, Y. Yao, X. Tao, J. Wei and S. Lin, ACS Macro Lett., 2021, 10, 1174–1179 CrossRef CAS PubMed.
- X. Tong, L. Cui and Y. Zhao, Macromolecules, 2004, 37, 3101–3112 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc00975k |
| This journal is © The Royal Society of Chemistry 2023 |