
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiologically active drimane derivatives isolated from submerged cultures of the wood-inhabiting basidiomycete Dentipellis fragilis†
Nico Mitschke‡§
 *a,
Winnie Chemutai Sum§ab,
Khadija Hassanab,
Marco Kirchenwitzb,
Hedda Schreya,
Luca Gerhardsd,
Harald Kellnere,
Theresia E. B. Stradalc,
Josphat C. Matasyohf and
Marc Stadler*ab
*a,
Winnie Chemutai Sum§ab,
Khadija Hassanab,
Marco Kirchenwitzb,
Hedda Schreya,
Luca Gerhardsd,
Harald Kellnere,
Theresia E. B. Stradalc,
Josphat C. Matasyohf and
Marc Stadler*ab
aDepartment of Microbial Drugs, Helmholtz Centre for Infection Research GmbH, Inhoffenstrasse 7, 38124 Braunschweig, Germany. E-mail: nico.mitschke@uni-oldenburg.de
bInstitute of Microbiology, Technische Universität Braunschweig, Spielmannstraße 7, 38106 Braunschweig, Germany. E-mail: marc.stadler@helmholtz-hzi.de
cDepartment of Cell Biology, Helmholtz Centre for Infection Research, Inhoffenstrasse 7, 38124 Braunschweig, Germany
dDepartment of Physics, Carl von Ossietzky Universität Oldenburg, Carl-von-Ossietzky-Str. 9-11, 26129 Oldenburg, Germany
eDepartment of Bio- and Environmental Sciences, Technische Universität Dresden - International Institute Zittau, Markt 23, 02763 Zittau, Germany
fDepartment of Chemistry, Egerton University, P.O. Box 536, 20115, Njoro, Kenya
First published on 31st August 2023
Abstract
Four previously undescribed drimane sesquiterpenoids were isolated from submerged cultures of the wood-inhabiting basidiomycete Dentipellis fragilis along with two compounds that were previously reported as synthetic or biotransformation compounds but not as natural products. The constitution and relative configuration of these compounds was determined based on high-resolution electrospray ionization mass spectrometry as well as by 1D and 2D nuclear magnetic resonance spectroscopy. The absolute configurations were established based on exemplary calculation of circular dichroism spectra and comparison with measured data as well as on biogenetic considerations. The biological activities of the isolated compounds were assessed in antimicrobial, cytotoxicity and neurotrophic assays. 10-Methoxycarbonyl-10-norisodrimenin (3) exhibited weak activity against the Gram-positive bacterium Staphylococcus aureus and the zygomycete Mucor hiemalis with minimal inhibitory concentrations of 66.7 μg mL−1. In addition, compound 3 showed weak inhibition of the mammalian cell line KB3.1 (human endocervical adenocarcinoma) with a half maximal inhibitory concentration of 21.2 μM. The neurotrophic activities of 15-hydroxyisodrimenin (1) and 10-carboxy-10-norisodrimenin (5) were assed in neurite outgrowth and real-time quantitative reverse transcription polymerase chain reaction (RT-qPCR) assays. When supplemented with 5 ng mL−1 nerve growth factor (NGF), the drimanes 1 and 5 induced neurite outgrowth in PC-12 (rat pheochromocytoma) cells compared to cells solely treated with NGF. As evaluated by RT-qPCR, compounds 1 and 5 also increased NGF and brain-derived neurotrophic factor expression levels in 1321N1 astrocytoma cells. Interestingly, the current study only represents the second report on neurotrophic activities of this widespread class of terpenoids. The only other available study deals with Cyathus africanus, another basidiomycete that can produce drimanes and cyathanes, but is only distantly related to Dentipellis and the Hericiaceae.
Introdcution
Dentipellis fragilis, a hydnoid basidiomycete with resupinate fruiting bodies associated with white-rot,1,2 belongs to the family Hericiaceae (order Russulales).3 Various secondary metabolites isolated from the Hericiaceae family have been reported to possess among others neuritogenic, antimicrobial and cytotoxic activities.4–8 As part of our on-going research in novel drug discovery from the division Basidiomycota, we recently started to explore the secondary metabolism of D. fragilis and found eight previously undescribed cyathane-xylosides.8 During the evaluation of these compounds, we noted the presence of another group of secondary metabolites that were present in the same extracts and decided to isolate them as well. Here, we report on a follow-up study that led to the isolation and structure elucidation of six drimane-type sesquiterpenoids whose biological activities were assessed in antimicrobial, cytotoxicity and neurotrophic assays.Results and discussion
Identity of the producer strain
The producer strain was obtained from a specimen that had been collected in the Bavarian Forest (Germany) and identified from morphological characteristics of the basidiomes and sequencing of the nrDNA (5.8S gene region, the internal transcript spacer region 1 and 2, and parts of the large and small subunit regions of the rRNA) as described previously.8Structure elucidation of compounds 1–6
The HPLC-DAD/MS (high performance liquid chromatography coupled to diode array or mass spectrometric detection) screening of ethyl acetate crude extracts obtained from submerged cultures of D. fragilis recently revealed its potential for the production of unexpected secondary metabolites.8 Thus, a large-scale fermentation was performed that led to the discovery of the drimane sesquiterpenoids 1–6 (Fig. 1). The combined crude extract from mycelia and supernatant was purified in two steps by flash chromatography on silica and in a second step by reversed-phase HPLC. The constitution and relative configuration of these compounds was elucidated by nuclear magnetic resonance (NMR) spectroscopy and high-resolution electrospray ionization mass spectrometry (HR-ESIMS). The absolute configurations were established by exemplary calculation of electronic circular dichroism (ECD) spectra and comparison with experimental spectra as well as based on biogenetic considerations.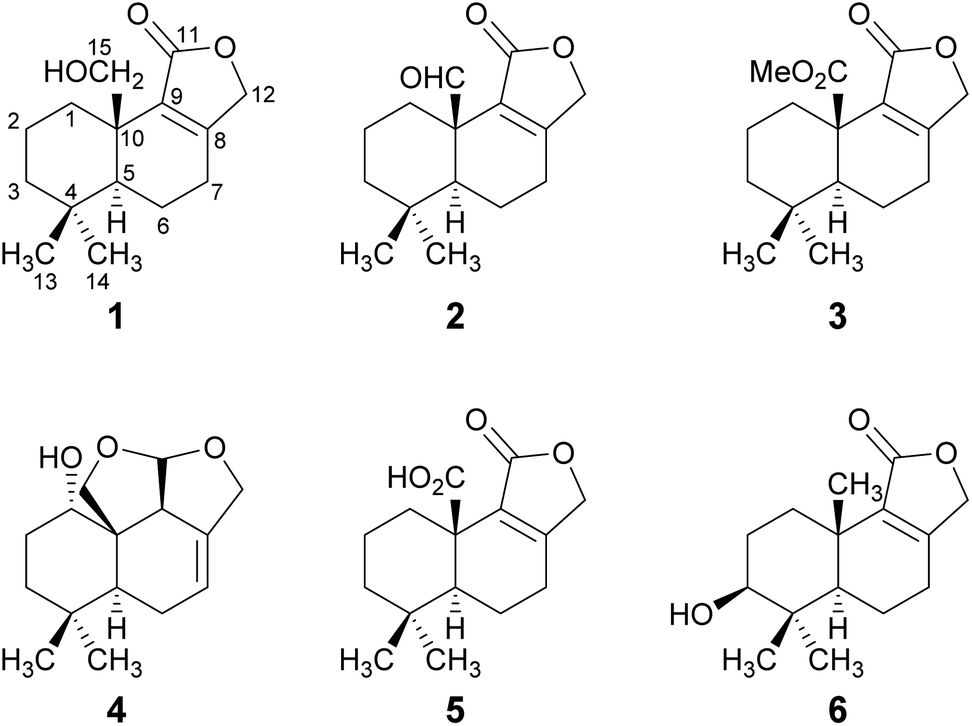 | ||
| Fig. 1 Structures of novel drimane sesquiterpenoids 1–4 and of the known drimanes 5 and 6 isolated from submerged cultures of D. fragilis. | ||
Compound 1 was isolated as a brown solid in a yield of 7.95 mg. The molecular formula was determined to be C15H22O3 based on the [M+H]+ ion at m/z 251.1640, consistent with 5 degrees of unsaturation. In accordance with the molecular formula, the 13C NMR spectrum (Fig. S4†) exhibited 15 carbon atoms that could be assigned along with the DEPT-HSQC spectrum (Fig. S5†) as five quaternary carbon atoms and two CH3, seven CH2 and one CH groups. Thus, compound 1 must possess one exchangeable proton. In total, three sp2 hybridized carbon atoms were identified, two that account for a double bond (δC 161.6 ppm [C-8]; δC 132.7 ppm [C-9]) and one that forms a carbonyl group (δC 174.8 ppm [C-11]). Considering the remaining three degrees of unsaturation, compound 1 must consist of three rings that are most likely to be annulated due to the H/C ratio. HMBC (for spectrum see Fig. S6†) and COSY (for spectrum see Fig. S7†) correlations (Fig. 2, left) finally established the constitution of compound 1, a tricyclic drimane lactone with a hydroxy group attached at C-15. HMBC correlations from H-15 (δH 3.66/4.14 ppm) to C-1 (δC 30.5 ppm), C-5 (δC 52.7 ppm), C-9 (δC 132.7 ppm) and C-10 (δC 40.7 ppm) confirmed the position of the exomethylene group OHCH2-15. It should be noted that drimane 1 exhibited a relatively unusual long-range 5J HMBC correlation between H-12α/β (δH 4.65/4.71 ppm) and C-15. Similar correlations were also observed for compounds 2, 3, 5 and 6 with compounds 5 and 6 being literature-known.9–11 However, the orientation of the lactone ring system was also secured by ROESY (for spectrum see Fig. S8†) correlations between H-12 and H-7 (δH 2.35/2.48 ppm, Fig. 2, right). This becomes even more evident when comparing the chemical shifts of the carbon atoms forming the double bond (C-8 and C-9) with those of further known drimanes, including isodrimenin12 and 7α-acetoxyisodrimenin,9 whose structures were secured by X-ray crystallography. In general, drimane lactones with the carbonyl group located at C-12 exhibited more down-field (δC ∼170 ppm) and high-field (δC ∼124 ppm) shifted chemical shift values for the carbon atoms C-9 and C-8 of the double bond, respectively, compared to drimanes in which the carbonyl is located at C-11 (C-8: δC ∼ 159 ppm, C-9: δC ∼ 135 ppm).9 Therefore, the chemical shift values of C-8 (δC 161.6 ppm) and C-9 (δC 132.7 ppm) of compound 1 are comparable to those of other compounds in which the carbonyl is located at C-11. The absolute configuration of the basic drimane carbon skeleton has been proven several times by X-ray crystallography.13,14 In accordance with that, C-15 is always β-orientated and H-5 always α-orientated in all known drimane sesquiterpenoids,13,14 e.g. also in drimanes derived from other species of the order Russulales.15–17
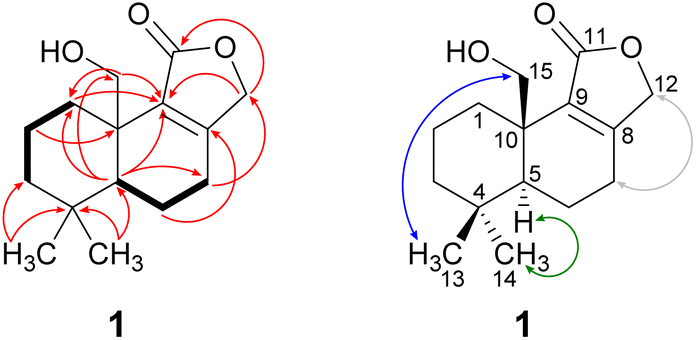 | ||
| Fig. 2 Key NMR correlations of 15-hydroxyisodrimenin (1). Bold bonds: COSY correlations; red arrows: 1H–13C HMBC correlations; blue arrows: ROESY correlations, indicating β-orientation; green arrows: ROESY correlations, indicating α-orientation and grey: arrows: ROESY correlations between H-7α/β and H-12α/β that secure the position of CH2-12. | ||
ROESY correlations between H-15 and H-13 (δH 0.87 ppm, Fig. 2, blue arrows) as well as between H-5 (δH 1.31 ppm) and H-14 (δH 0.96 ppm, Fig. 2, green arrows) established the relative configuration and as a consequence of the aforementioned considerations also the absolute configuration of drimane 1. To finally ensure the stereochemical assignment of compound 1, ECD spectra were calculated that agreed well with the experimental spectrum (Fig. 3A). Thus, the absolute configuration of 15-hydroxyisodrimenin (1) was determined to be 5S,10R.
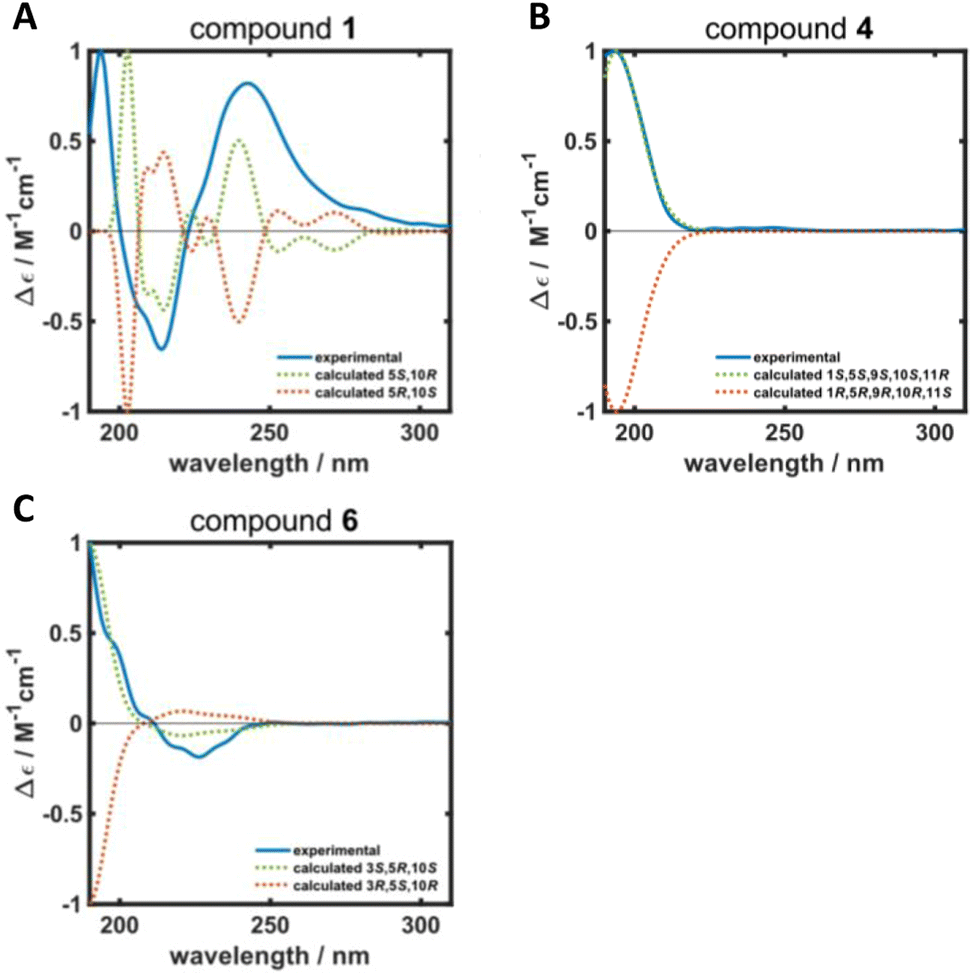 | ||
| Fig. 3 Calculated (dotted green and orange lines) and experimental (solid blue line) ECD spectra of (A) 15-hydroxyisodrimenin (1), (B) 1α-hydroxymarasmene (4) and (C) 3β-hydroxyisodrimenin (6). | ||
Compound 2 was isolated as a yellow oil in a yield of 0.45 mg. Based on the [M+H]+ ion at m/z 249.1483, the molecular formula was deduced to be C15H20O3, resulting in 6 degrees of unsaturation. The analysis of the NMR data (Fig. S13–S18†) revealed that drimane 2 was structurally closely related to compound 1 with the only difference that compound 2 showed an aldehyde signal for C-15 in the NMR spectra (δC 200.9 ppm, δH 9.93 ppm) and the former methylene signal (δC 65.5 ppm, δH 3.66/4.14 ppm) of compound 1 was missing. The position of the aldehyde was secured by HMBC correlations from H-15 to C-1 (δC 29.2 ppm) and C-10 (δC 49.1 ppm). Based on ROESY correlations between H-15/H-13 (δH 0.80 ppm) and H-5 (δH 1.51 ppm)/H-14 (δH 0.99 ppm), the relative configuration was established which was in agreement with the configuration of the basic drimane skeleton. The absolute configuration of compound 2 was assigned based on biogenetic considerations.
Compound 3 was isolated as a colorless oil in a yield of 0.71 mg. The molecular formula was deduced from the [M+H]+ ion at m/z 279.1591 to be C16H22O4 with 6 degrees of unsaturation. As for compound 2, the detailed analysis of the NMR data (Fig. S23–S28†) revealed that drimane 3 was structurally closely related to compound 1. In accordance with the molecular formula, an additional methyl group CH3-16 (δC 52.1 ppm, δH 3.68 ppm) was identified in the HSQC spectrum and the methylene group signal (δC 65.5 ppm, δH 3.66/4.14 ppm) of drimane 1 was missing. Instead, a quaternary carbon atom C-15 (δC 173.9 ppm) was present in compound 3. HMBC correlations from CH3-16 to C-15 connected both fragments to a methyl ester that was attached to C-10 (δC 44.3 ppm) as secured by HMBC correlations from H-1α/β (δH 1.10/3.25 ppm) and H-5 (δH 1.41 ppm) to C-15. The absolute configuration was established based on the same considerations and ROESY correlations as mentioned for the structurally closely related compounds 1 and 2.
Compound 4 was isolated as a yellow oil in a yield of 2.50 mg. Its molecular formula C15H22O3 with 5 degrees of unsaturation was determined from its [M+H]+ ion at m/z 251.1637. In contrast to the other isolated compounds, drimane 4 possessed only one double bond (δC 120.5 ppm [C-7]; δC 133.6 ppm [C-8], for NMR spectra see Fig. S33–S44†) and no further sp2-hydbridized carbon atom. Thus, its structure must contain four rings that were likely to be annulated due to the H/C ratio. The basic carbon skeleton was deduced from characteristic COSY and HMBC correlations (Fig. 4, left) as well as by the comparison of the NMR data of drimane 4 with those of the literature-known marasmene18 (structure as compound 4 but without hydroxy group) and 1α,15-dihydroxymarasmene18,19 (structure as compound 4 but with an additional hydroxy group attached to C-15) that had been isolated from other basidiomycetes. Considering the absolute configuration of the basic drimane skeleton, ROESY correlations between H-13 (δH 0.73 ppm) and H-15 (δH 3.58/3.82 ppm) suggested β-orientation, whereas correlations between H-14 (δH 0.95 ppm) and H-5 (δH 2.18 ppm) indicated α-orientation. In addition, the hydroxy group attached to C-1 must be α-orientated due to ROESY correlations from H-1 (δH 3.67 ppm in CD3OD) to one proton of CH2-15 (δH 3.58 ppm in CD3OD; Fig. 4, right, blue arrows). In addition, the orientation of H-1 was secured by J-coupling analysis. The signal of H-1 appears as a triplet (J = 2.9 Hz) in the 1H NMR spectrum measured in CD3OD where no signal overlapping occurred compared to the 1H NMR spectrum measured in CDCl3. The chair conformation as depicted in Fig. 4 (right) was found to be the most stable conformer for both orientations of the hydroxy group. Thus, only the β-orientation of H-1 would lead to a triplet due to similar dihedral angles between H-1 and H-2α and between H-1 and H-2β (both approx. 60°). An α-orientation of H-1 would cause a doublet of doublets because of different dihedral angles between H-1 and H-2α (approx. 60°) and between H-1 and H-2β (approx. 180°). The absolute configuration of compound 4 was deduced to be 1S,5S,9S,10S,11R based on biogenetic considerations and secured by calculation of ECD spectra (Fig. 3B).
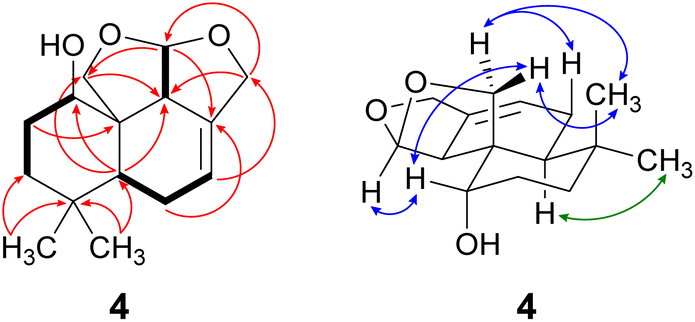 | ||
| Fig. 4 Key NMR correlations of 1α-hydroxymarasmene (4). Bold bonds: COSY correlations; red arrows: 1H–13C HMBC correlations; blue arrows: ROESY correlations, indicating β-orientation and green arrows: ROESY correlations, indicating α-orientation. | ||
Compound 5 was isolated as a brown solid in a yield of 18.96 mg and its molecular formula C15H20O4 with 6 degrees of unsaturation was deduced from its [M+H]+ ion at m/z 265.1433. A detailed comparison of its NMR spectroscopic properties (for NMR spectra see Fig. S49–S54†) with those of the literature-known 10-carboxy-10-norisodrimenin revealed structural identity.10 Notably, drimane 5 has never been described before as a natural product and was solely reported as a synthetic intermediate during the total synthesis of (−)-antrocin.10
Compound 6 was isolated as a white solid in a yield of 1.33 mg. The molecular formula was determined to be C15H22O3 based on the [M+H]+ ion at m/z 251.1642, consistent with 5 degrees of unsaturation. A detailed comparison of its NMR spectroscopic properties (for NMR spectra see Fig. S59–S64†) with those of the previously reported 3β-hydroxyisodrimenin, which had been obtained either by chemical derivatization from (−)-3β-acetoxyisodrimenin11 or derived by microbial hydroxylation of isodrimenin,9 revealed structural identity. The absolute configuration was ascertained by comparing the measured with calculated ECD spectra (Fig. 3C) and based on biogenetic considerations.
Physico-chemical properties of compounds 1–6
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) = 195 (1.89), 218 (2.07) nm; ECD (MeOH, 1.0 mg mL−1): λ (Δε) 194 (3.08), 214 (−2.02), 243 (2.52); NMR data (1H: 500 MHz, 13C: 125 MHz, CDCl3) see Table 1; HR-(+)ESIMS: m/z 233.1533 [M+H−H2O]+ (calcd 233.1536 for C15H21O2+), 251.1640 [M+H]+ (calcd 251.1642 for C15H23O3+), 273.1461 [M+Na]+ (calcd 273.1462 for C15H22NaO3+), 523.3028 [2M+Na]+ (calcd 523.3032 for C30H44NaO6+); tR = 8.6 min (HR-LC-ESIMS); tR = 45 min (preparative HPLC). C15H22O3 (250.34 g mol−1).
ε) = 195 (1.89), 218 (2.07) nm; ECD (MeOH, 1.0 mg mL−1): λ (Δε) 194 (3.08), 214 (−2.02), 243 (2.52); NMR data (1H: 500 MHz, 13C: 125 MHz, CDCl3) see Table 1; HR-(+)ESIMS: m/z 233.1533 [M+H−H2O]+ (calcd 233.1536 for C15H21O2+), 251.1640 [M+H]+ (calcd 251.1642 for C15H23O3+), 273.1461 [M+Na]+ (calcd 273.1462 for C15H22NaO3+), 523.3028 [2M+Na]+ (calcd 523.3032 for C30H44NaO6+); tR = 8.6 min (HR-LC-ESIMS); tR = 45 min (preparative HPLC). C15H22O3 (250.34 g mol−1).
| 1a | 2a | 3b | ||||
|---|---|---|---|---|---|---|
| δC | δH (mult., J in Hz) | δC | δH (mult., J in Hz) | δC | δH (mult., J in Hz) | |
| a Recorded at 500 MHz (1H) or 125 MHz (13C), CDCl3, 298 K, δH and δC in ppm.b Recorded at 700 MHz (1H) or 175 MHz (13C), CDCl3, 298 K, δH and δC in ppm.c Orientation could not be assigned with certainty. | ||||||
| 1 | 30.5, CH2 | 1.13 (tdd, 13.5, 4.1, 1.4, 1H, α) | 29.2, CH2 | 1.24 (td, 13.5, 4.2, 1H, α) | 31.9, CH2 | 1.10 (td, 13.4, 4.2, 1H, α) |
| 2.79 (dtd, 13.8, 3.5, 1.5, 1H, β) | 3.23 (dddd, 13.9, 3.5, 3.3, 1.4, 1H, β) | 3.25 (dtd, 13.6, 3.3, 1.5, 1H, β) | ||||
| 2 | 18.2, CH2 | 1.58 (ddddd, 14.4, 4.4, 4.1, 3.3, 3.1, 1H, α) | 18.4, CH2 | 1.68 (dtt, 14.7, 4.1, 3.2, 1H, α) | 19.4, CH2 | 1.58 (ddddd, 14.4, 4.6, 4.2, 3.0, 2.8, 1H, α) |
| 1.68 (qt, 13.7, 3.5, 1H, β) | 1.78 (qt, 14.0, 3.6, 1H, β) | 1.99 (qt, 13.7, 3.9, 1H, β) | ||||
| 3 | 41.5, CH2 | 1.25 (td, 13.4, 4.4, 1H, α) | 41.2, CH2 | 1.32 (td, 13.5, 4.2, 1H, α) | 41.9, CH2 | 1.28 (td, 13.6, 4.6, 1H, α) |
| 1.53 (dtd, 13.4, 3.4, 1.5, 1H, β) | 1.54 (m, 1H, β) | 1.49 (dddd, 13.4, 4.1, 2.8, 1.5, 1H, β) | ||||
| 4 | 33.0, Cq | 33.4, Cq | 33.4, Cq | |||
| 5 | 52.7, CH | 1.31 (dd, 12.7, 2.0, 1H, α) | 53.6 | 1.51 (dd, 11.2, 4.0, 1H, α) | 53.5, CH | 1.41 (dd, 12.5, 2.8, 1H, α) |
| 6 | 17.8, CH2 | 1.75 (dddd, 13.6, 12.7, 10.7, 6.2, 1H, β) | 17.5, CH2 | 2.01 (m, 2H) | 17.7, CH2 | 1.95 (m, 1H, α) |
| 1.95 (ddt, 13.6, 7.2, 1.6, 1H, α) | 2.40 (m, 1H, β) | |||||
| 7 | 25.2, CH2 | 2.35 (dddt, 19.2, 10.7, 7.2, 1.1, 1H, α) | 24.9, CH2 | 2.43 (m, 1H, α) | 24.6, CH2 | 2.39 (m, 1H, α) |
| 2.48 (ddt, 19.2, 6.2, 1.2, 1H, β) | 2.58 (ddd, 19.3, 5.1, 2.2, 1H, β) | 2.53 (m, 1H, β) | ||||
| 8 | 161.6, Cq | 164.6, Cq | 163.6, Cq | |||
| 9 | 132.7, Cq | 126.8, Cq | 129.2, Cq | |||
| 10 | 40.7, Cq | 49.1, Cq | 44.3, Cq | |||
| 11 | 174.8, Cq | 171.2, Cq | 171.4, Cq | |||
| 12 | 71.4, CH2 | 4.65c (dd, 17.1, 1.1, 1H) | 71.1 | 4.68c (d, 18.3, 1H) | 71.0, CH2 | 4.64c (d, 17.3, 1H) |
| 4.71c (dt, 17,1, 1.0, 1H) | 4.72c (d, 18.3, 1H) | 4.66c (d, 17.3, 1H) | ||||
| 13 | 21.7, CH3 | 0.87 (s, 3H, β) | 21.6 | 0.80 (s, 3H, β) | 19.7, CH3 | 0.76 (s, 3H, β) |
| 14 | 33.3, CH3 | 0.96 (s, 3H, α) | 31.7 | 0.99 (s, 3H, α) | 32.3, CH3 | 0.96 (s, 3H, α) |
| 15 | 65.5, CH2 | 3.66 (dd, 10.8, 1.4, 1H, β) | 200.9 | 9.93 (s, 1H, β) | 173.9, Cq | |
| 4.14 (d, 10.8, 1H, β) | ||||||
| 16 | 52.1, CH3 | 3.68 (s, 3H, β) | ||||
ε) = 220 (2.39) nm; ECD (MeOH, 1.0 mg mL−1): λ (Δε) 196 (11.31), 225 (−11.07), 307 (10.87); NMR data (1H: 500 MHz, 13C: 125 MHz, CDCl3) see Table 1; HR-(+)ESIMS: m/z 231.1374 [M+H−H2O]+ (calcd 231.1380 for C15H19O2+), 249.1483 [M+H]+ (calcd 249.1485 for C15H21O3+), 271.1304 [M+Na]+ (calcd 271.1305 for C15H20NaO3+), 497.2896 [2M+H]+ (calcd 497.2898 for C30H41O6+), 519.2719 [2M+Na]+ (calcd 519.2717 for C30H40NaO6+); tR = 9.2 min (HR-LC-ESIMS); tR = 50 min (preparative HPLC); tR = 52 min (semipreparative HPLC). C15H20O3 (248.32 g mol−1).ε) = 195 (1.97), 217 (2.15) nm; ECD (MeOH, 1.0 mg mL−1): λ (Δε) 242 (32.46); NMR data (1H: 700 MHz, 13C: 175 MHz, CDCl3) see Table 1; HR-(+)ESIMS: m/z 219.1376 [M+H–HCO2Me]+ (calcd 219.1380 for C14H19O2+), 279.1591 [M+H]+ (calcd 279.1591 for C16H23O4+), 301.1411 [M+Na]+ (calcd 301.1410 for C16H22NaO4+), 557.3108 [2M+H]+ (calcd 557.3109 for C32H45O8+), 579.2928 [2M+Na]+ (calcd 579.2928 for C32H44NaO8+); tR = 10.0 min (HR-LC-ESIMS); tR = 52 min (preparative HPLC). C16H22O4 (278.35 g mol−1).ε) = 201 (2.02) nm; ECD (MeOH, 1.0 mg mL−1): λ (Δε) 193 (9.00); NMR data (1H: 700 MHz, 13C: 175 MHz, see Table 2 (CDCl3) or Table S6† (CD3OD)); HR-(+)ESIMS: m/z 233.1531 [M+H−H2O]+ (calcd 233.1536 for C15H21O2+), 251.1637 [M+H]+ (calcd 251.1642 for C15H23O3+), 273.1458 [M+Na]+ (calcd 273.1461 for C15H22NaO3+), 501.3209 [2M+H]+ (calcd 501.3211 for C30H45O6+), 523.3028 [2M+Na]+ (calcd 523.3030 for C30H44NaO6+); tR = 7.3 min (HR-LC-ESIMS); tR = 42 min (preparative HPLC). C15H22O3 (250.34 g mol−1).
| 4b,c | 5a | 6b | ||||
|---|---|---|---|---|---|---|
| δC | δH (mult., J in Hz) | δC | δH (mult., J in Hz) | δC | δH (mult., J in Hz) | |
| a Recorded at 500 MHz (1H) or 125 MHz (13C), CDCl3, 298 K, δH and δC in ppm.b Recorded at 700 MHz (1H) or 175 MHz (13C), CDCl3, 298 K, δH and δC in ppm.c Also recorded at 700 MHz (1H) or 175 MHz (13C) in CD3OD (298 K) to prevent signal overlapping of H-15 and H-1 in the 1H NMR spectrum that was crucial for the elucidation of the stereochemistry (for NMR data see ESI Table S6).d Orientation could not be assigned with certainty. | ||||||
| 1 | 68.5, CH | 3.82 (m, 1H, β) | 31.9, CH2 | 1.11 (td, 13.4, 4.2, 1H, α) | 32.6, CH2 | 1.32 (tdd, 13.6, 3.8, 0.7, 1H, α) |
| 3.25 (dtd, 13.6, 3.4, 1.4, 1H, β) | 2.62 (dt, 13.6, 3.6, 1H, β) | |||||
| 2 | 26.6, CH2 | 1.65 (dq, 14.9, 3.3, 1H, α) | 19.3, CH2 | 1.59 (ddddd, 14.5, 4.4, 4.2, 3.1, 2.8, 1H, α) | 27.3, CH2 | 1.68 (tdd, 13.4, 11.7, 3.8, 1H, β) |
| 1.94 (dddd, 14.9, 14.3, 4.0, 2.5, 1H, β) | 2.00 (qt, 14.0, 3.9, 1H, β) | 1.79 (ddt, 13.4, 4.6, 3.6, 1H, α) | ||||
| 3 | 34.8, CH2 | 1.25 (ddd, 13.7, 4.0, 3.0, 1H, β) | 41.9, CH2 | 1.28 (td, 13.5, 4.4, 1H, α) | 78.6, CH | 3.29 (dd, 11.7, 4.6, 1H, α) |
| 1.74 (td, 14.0, 3.9, 1H, α) | 1.51 (dddd, 13.4, 4.0, 2.8, 1.5, 1H, β) | |||||
| 4 | 33.1, Cq | 33.6, Cq | 38.8, Cq | |||
| 5 | 38.0, CH | 2.18 (dd, 11.1, 6.9, 1H, α) | 53.3, CH | 1.43 (dd, 12.4, 2.6, 1H, α) | 51.5, CH | 1.17 (dd, 12.4, 1.8, 1H, α) |
| 6 | 23.6, CH2 | 2.03 (m, 1H, β) | 17.7, CH2 | 1.96 (m, 1H, α) | 18.0, CH2 | 1.62 (dddd, 13.6, 12.4, 11.3, 6.0, 1H, β) |
| 2.33 (m, 1H, α) | 2.42 (m, 1H, β) | 1.94 (ddt, 13.6, 7.0, 1.6, 1H, α) | ||||
| 7 | 120.5, CH | 5.75 (m, 1H) | 24.5, CH2 | 2.42d (m, 1H) | 25.4, CH2 | 2.30 (dddt, 19.0, 11.3, 7.0, 1.3, 1H, α) |
| 2.53d (m, 1H) | 2.43 (ddt, 19.0, 6.0, 1.2, 1H, β) | |||||
| 8 | 133.6, Cq | 164.3, Cq | 159.1, Cq | |||
| 9 | 49.0, CH | 3.36 (m, 1H, α) | 128.7, Cq | 135.2, Cq | ||
| 10 | 48.6, Cq | 44.2, Cq | 34.6, Cq | |||
| 11 | 105.5, CH | 5.68 (d, 4.4, 1H) | 171.3, Cq | 172.2, Cq | ||
| 12 | 72.3, CH2 | 4.39 (m, 2H) | 71.0, CH2 | 4.65d (d, 17.3, 1H) | 70.6, CH2 | 4.57d (dt, 16.9, 1.0, 1H) |
| 4.69d (d, 17.3, 1H) | 4.60d (dd, 16.9, 1.2, 1H) | |||||
| 13 | 20.2, CH3 | 0.73 (s, 3H, β) | 19.9, CH3 | 0.86 (s, 3H, β) | 15.4, CH3 | 0.87 (s, 3H, β) |
| 14 | 30.9, CH3 | 0.95 (s, 3H, α) | 32.3, CH3 | 0.97 (s, 3H, α) | 28.2, CH3 | 1.07 (s, 3H, α) |
| 15 | 69.6, CH2 | 3.58 (d, 8.6, 1H, α) | 178.2, Cq | 20.0, CH3 | 1.16 (d, 0.7, 3H, β) | |
| 3.82 (d, 8.6, 1H, β) | ||||||
ε) = 195 (2.08), 214 (2.32) nm; ECD (MeOH, 1.0 mg mL−1): λ (Δε) 193 (3.88), 211 (−9.27), 232 (13.95); NMR data (1H: 500 MHz, 13C: 125 MHz, CDCl3) see Table 2; HR-(+)ESIMS: m/z 219.1377 [M+H–HCO2H]+ (calcd 219.1380 for C14H19O2+), 265.1433 [M+H]+ (calcd 265.1434 for C15H21O4+), 287.1255 [M+Na]+ (calcd 287.1254 for C15H20NaO4+), 529.2797 [2M+H]+ (calcd 529.2796 for C30H41O8+), 551.2621 [2M+Na]+ (calcd 551.2615 for C30H40NaO8+); tR = 8.5 min (HR-LC-ESIMS); tR = 53 min (preparative HPLC). C15H20O4 (264.32 g mol−1). All spectroscopic and spectrometric data were in accordance with the literature.10ε) = 218 (1.24) nm; ECD (MeOH, 1.0 mg mL−1): λ (Δε) 226 (−1.29); NMR data (1H: 700 MHz, 13C: 175 MHz, CDCl3) see Table 2; HR-(+)ESIMS: m/z 251.1642 [M+H]+ (calcd 251.1642 for C15H23O3+), 273.1457 [M+Na]+ (calcd 273.1461 for C15H22NaO3+), 501.3212 [2M+H]+ (calcd 501.3211 for C30H45O6+), 523.3031 [2M+Na]+ (calcd 523.3030 for C30H44NaO6+); tR = 6.6 min (HR-LC-ESIMS); tR = 43 min (preparative HPLC). C15H22O3 (250.34 g mol−1). All spectroscopic and spectrometric data were in accordance with the literature.9,11Biological activities of compounds 1–6
Drimanes 1–6 were subjected to both, cytotoxicity and antimicrobial assays. In the antimicrobial assays against a panel of clinically relevant microorganisms, compound 3 had very weak inhibitory effects against the Gram-positive Staphylococcus aureus and the filamentous fungus Mucor hiemalis with minimal inhibitory concentrations (MICs) of 66.7 μg mL−1. Drimanes 1 and 4–6 were inactive whereas compound 2 was not tested due to its insufficient isolated amount (Tables S1 and S2†). The drimanes were also checked for activities against our standard mammalian cell lines, but no significant cytotoxic activities were observed (Table S3†). Only compound 3 showed very weak cytotoxic activities (IC50 21.2 μM) against KB3.1 (human endocervical adenocarcinoma) cells.Previous studies have already reported neurotrophic activities of drimane sesquiterpenoids.20 Thus, in addition to the aforementioned biological activity assays, drimanes 1 and 5 that were isolated in significant amounts were also assessed regarding their neurotrophic activities. For this purpose, the potential of these compounds to induce nerve growth factor (NGF) mediated neurite outgrowth in rat pheochromocytoma cells (PC-12) as well as to upregulate gene expression of NGF and brain-derived neurotrophic factor (BDNF) in human astrocytoma cells (132N1) were examined.
Neurite outgrowth assays are well-established to assess the neurotrophic potential of fungal metabolites.7,21–23 Upon NGF stimulation, PC-12 cells differentiate into neuronal cells.24 Thus, PC-12 cells were differentiated with or without supplementation of NGF in the presence or absence of compounds 1 and 5 and the mean neurite length after 24 h was measured. Neurite outgrowth was not observed when PC-12 cells were treated directly with DMSO or with the compounds (cf. Fig. S1†). However, when PC-12 cells were treated with compounds 1 or 5, supplemented with NGF at a concentration of 5 ng mL−1, neurite outgrowth was observed (Fig. 5A–C). NGF treatment alone only promoted slightly increase of neurite outgrowth, whereas the presence of compounds 1 and 5 significantly increased the neurite lengths of PC-12 cells (Fig. 5A–D).
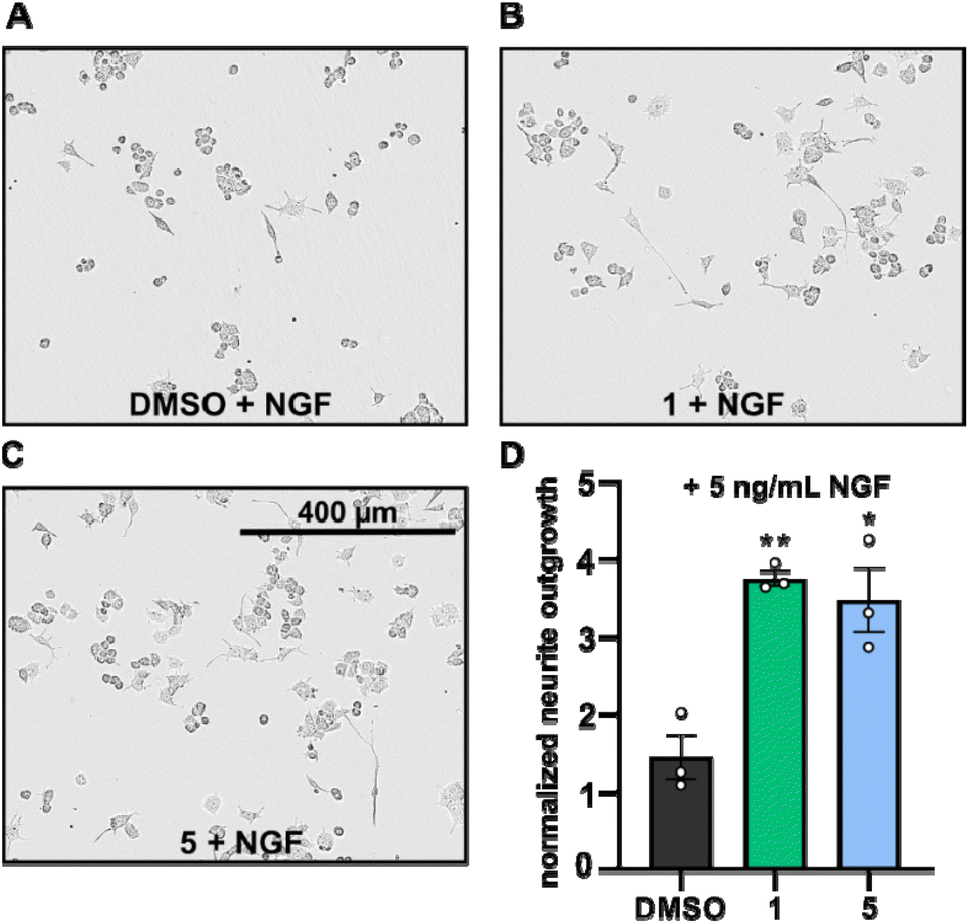 | ||
| Fig. 5 (A–C): Phase contrast images of PC-12 cells supplemented with NGF (5 ng mL−1) and treated with either DMSO (A, 5 μg mL−1, control), drimane 1 (B, 5 μg mL−1) or drimane 5 (C, 5 μg mL−1) after 24 h of incubation. (D): bar chart displaying the normalized neurite outgrowth of PC-12 cells supplemented with NGF (5 ng mL−1) treated with either DMSO, compound 1 (5 μg mL−1) or compound 5 (5 μg mL−1) after 24 h of incubation. Neurite outgrowths were normalized with respect to the neurite outgrowths detected after 0 h (t0) and are given in fold changes. Data are means ± S.E.M from three independent experiments. **p < 0.05, *p < 0.10 in unpaired t-test. | ||
In a second approach, the neurotrophic potential of drimanes 1 and 5 to upregulate gene expression of NGF and BDNF in human astrocytoma 132N1 cells was investigated. In physiological conditions, astrocytes produce and secrete NGF and BDNF for neuronal development and protection.25 Strikingly, real-time quantitative reverse transcription polymerase chain reaction (RT-qPCR) revealed that drimanes 1 and 5 upregulate the expression of NGF by approx. 4- and 2-fold and of BDNF by 3 and 2.5-fold, respectively (Fig. 6). Notably, compound 1 increased NGF and BDNF expression levels more profoundly than compound 5. Further investigations on whether drimanes 1 and 5 promote NGF-mediated neurite outgrowth by stimulating the NGF-synthesis or as substitutes for NGF (NGF-mimicking activity) are ongoing. Nonetheless, the results clearly demonstrated the neurotrophic potential of the drimanes 1 and 5.
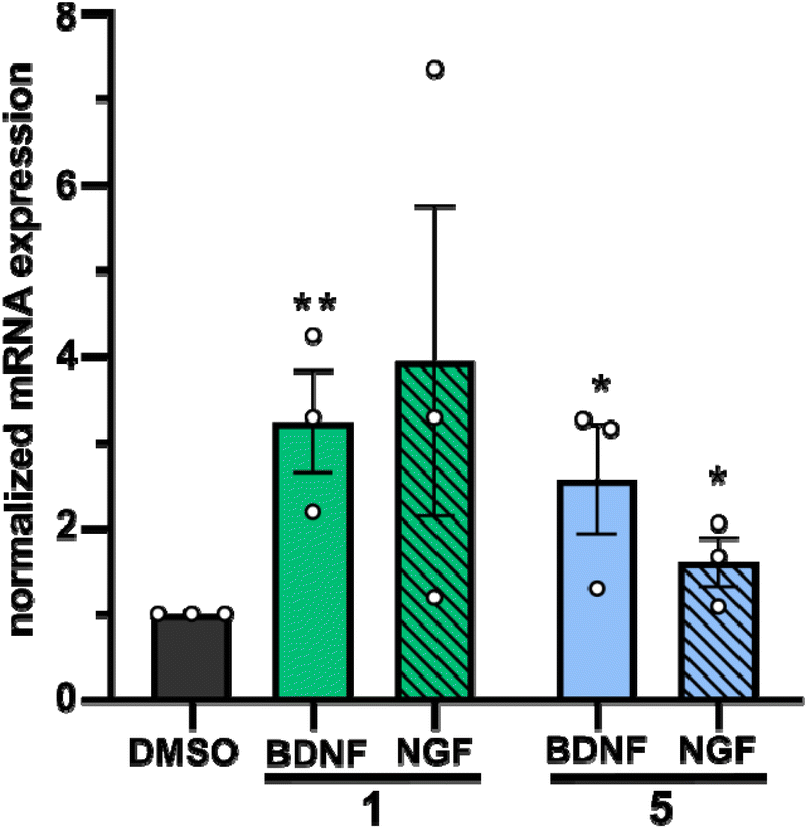 | ||
| Fig. 6 Quantification of NGF and BDNF mRNA levels by RT-qPCR in 1312N1 astrocytoma cells treated with either DMSO (negative control, 5 μg mL−1), compound 1 (5 μg mL−1), or compound 5 (5 μg mL−1) for 48 h. Expression levels are given in fold changes with respect to the DMSO control. Data are means ± S.E.M from three independent experiments. **p < 0.05, *p < 0.10 in unpaired t-test. | ||
Material and methods
General experimental procedures
HPLC-DAD/MS analysis was performed on an amaZon speed ETD ion trap mass spectrometer (Bruker Daltonics, Bremen, Germany) in positive and negative ionization modes. The HPLC system (Dionex UltiMate 3000 UHPLC, Thermo Fisher Scientific Inc., Waltham, MA, USA) was equipped with a C18 Acquity UPLC BEH column (Waters, Milford, MA, USA) as stationary phase. Deionized H2O + 0.1% formic acid (FA) (v/v) was used as solvent A and acetonitrile (ACN) + 0.1% FA (v/v) was used as solvent B. The gradient was operated at 5% B for 0.5 min, from 5% B to 100% B within 20 min and at 100% B for 10 min. The flow rate was set to 0.6 mL min−1 and the UV/vis detection to 190–600 nm.HR-ESIMS data were measured on a MaXis ESI-TOF (time-of-flight) mass spectrometer (Bruker Daltonics) operated in positive ionization mode and coupled to an Agilent 1260 series HPLC-UV system (Agilent Technologies, Santa Clara, CA, USA). The HPLC system was equipped with a C18 Acquity UPLC BEH column (waters) as stationary phase. Deionized H2O + 0.1% FA (v/v) was used as solvent A and ACN + 0.1% FA (v/v) was used as solvent B. The gradient was operated at 5% B for 0.5 min, from 5% B to 100% B within 19.5 min and at 100% B for 5 min. The flow rate was set to 0.6 mL min−1 and UV/vis detection to 200–600 nm. Molecular formulas of the isolated compounds were calculated with the Smart Formula algorithm of the Compass DataAnalysis software (Bruker Daltonics, version 4.4 SR1).
NMR spectra were measured on Avance III 500 (Bruker Biospin, Ettlingen, Germany, 1H: 500 MHz, 13C: 125 MHz) or Avance III 700 (Bruker Biopsin, 1H: 700 MHz, 13C: 175 MHz) instruments that were locked to the respective deuterium signal of the solvent. CDCl3 and CD3OD were used as deuterated solvents for the NMR measurement. Chemical shifts were reported in parts per million (ppm) and coupling constants in Hertz (Hz). The residual proton signal was used for the calibration of 1H NMR spectra with reference values of 7.27 ppm for CDCl3 and 3.31 ppm for CD3OD, respectively. 13C NMR spectra were calibrated using the 13C signal of the deuterated solvents with reference values of 77.00 ppm for CDCl3 and 49.15 ppm for CD3OD, respectively. HSQC spectra were recorded multiplicity-edited and carbon multiplicities were derived from these spectra. If 1H NMR chemical shifts could not be determined directly from the 1D 1H NMR spectrum due to overlapping signals, they were derived from the corresponding HSQC spectrum.
Optical rotations were measured with an Anton Paar MCP-150 Polarimeter (Graz, Austria) with sodium D line at 589 nm and 100 mm path length at a concentration of 1.0 mg mL−1 in MeOH.
UV/vis spectra were measured on a Shimadzu UV/vis 2450 spectrophotometer (Kyoto, Japan) at a concentration of 0.02 mg mL−1 in MeOH.
All ECD were measured on a J-815 spectropolarimeter (Jasco, Pfungstadt, Germany) at a concentration of 1.0 mg mL−1 in MeOH.
All solvents (analytical and HPLC grade) and chemicals were obtained from AppliChem GmbH (Darmstadt, Germany), Merck KGaA (Darmstadt, Germany), Avantor Performance Materials (Deventer, Netherlands) and Carl Roth GmbH & Co. KG (Karlsruhe, Germany). Deionized water was prepared in-house with a Purelab® flex water purification system (Veolia Water Technologies, Celle, Germany).
Fungal material
The fungus was collected, identified and cultured by Harald Kellner as described previously.8 A mycelial culture was deposited at the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) in Braunschweig under the designation number DSM 105465. The ITS nrDNA sequence of the strain was deposited at GenBank (accession number MK463979).Fermentation and extraction of metabolites
Fermentation and extraction had been carried out in YMG medium as described recently by Sum et al.8 The latter publication treated the isolation of cyathane diterpenoids from the same culture. We now report on another class of terpenoids that were obtained concurrently from the very same extract.Isolation of compounds 1–6
The pre-fractioning had been carried out previously on a Reveleris X2 flash chromatography system (W.R. Grace and Co., Columbia, MD, USA) using 40 g silica cartridges (Reveleris®) as stationary phase (for experimental parameters see Sum et al.8).Further purification was performed with a reverse-phase LC system (PLC 2020; Gilson, Middleton, WI, USA), equipped with a Synergi™ 10 μm Polar-RP 80 Å (250 × 50 mm) AXIA™ packed column (Phenomenex Inc., Aschaffenburg, Germany) as stationary phase. Deionized H2O + 0.1% formic acid (FA) (v/v) was used as solvent A and ACN + 0.1% FA (v/v) was used as solvent B. The gradient was operated from 5% B to 40% B within 5 min, from 40% B to 60% B within 20 min, from 60% B to 100% B within 5 min and at 100% B for 15 min. The flow rate was set to 20 mL min−1, and UV detection was carried out at 190, 210 and 280 nm, respectively. Fractions that contained the same compounds based on HPLC-DAD/MS screening were combined to furnish pure compounds 1 and 3–6. Compound 2 was only isolated highly enriched and thus further purified with a semipreparative Vanquish HPLC system (Thermo Fisher Scientific), equipped with a Synergi™ 4 μm Polar-RP 80 Å (250 × 4.6 mm) column (Phenomenex Inc., Torrance, CA, USA). Deionized H2O + 0.1% FA (v/v) was used as solvent A and ACN + 0.1% FA (v/v) was used as solvent B. The gradient was operated from 10% B to 100% B within 30 min and at 100% B for 10 min.
Calculation of ECD spectra
ECD spectra were calculated for compounds 1, 4 and 6. Conformers were determined with the program package CREST26,27 with a set temperature of 400 K and an energy window of 7 kcal mol−1. For compound 1, 16 conformers were found for each of the two possible enantiomers, whereas 8 and 7 conformers were found per enantiomer for compounds 4 and 6, respectively. All conformer structures were geometry-optimized using the hybrid density function CAM-B3LYP28 and the def2-TZVPD basis set29,30 as implemented in the software package ORCA (version 5.0.0).31 Here, the resolution of identity approximation RIJCOSX32,33 with an auxiliary Def2/J basis set was employed.34 The D4 correction was used to account for dispersion interactions.35 Furthermore, the implicit solvent model CPCM with methanol was included.36Time-dependent density functional theory (TDDFT) calculations were performed for each geometry-optimized conformer using the aforementioned parameters. 50 electronic excitations were calculated for each conformer. SpecDis (version 1.71) was used to construct additive spectra for each enantiomer by summing up the spectra of individual conformers according to their Boltzmann weightings (T = 298 K). In addition, discrete excitation spectra were converted into continuous spectra (σ = 0.23, 0.4 and 0.27 eV for compounds 1, 4 and 6, respectively) using SpecDis.37 To compare calculated and experimental ECD spectra, all spectra were scaled between molar circular dichroism values of −1 and 1, and calculated spectra were shifted by 62 nm, 5 nm and 11 nm for compounds 1, 4 and 6, respectively.
Determination of antimicrobial and cytotoxic activity
Minimum inhibitory concentrations (MICs) of pure compounds against different test microorganisms and their cytotoxic effects on mammalian cell lines were determined by serial dilution assays according to our standard protocol as described by Becker et al.,38 using the same organisms and test conditions.Assays for assessment of neurotrophic effects
Neurite outgrowth and RT-qPCR assays have been employed to test the neurotrophic activities of compounds 1 and 5 in parallel to the reported triterpenoids from other basidiomycetes.24 A detailed description is given by Hassan et al.,24 using exactly the same equipment and protocols. We therefore refrain from a lengthy description but only briefly summarize the experimental procedure.Rat pheochromocytoma cells (PC-12, adherent variant) were grown in RPMI-1640 (Gibco, Thermo Fisher Scientific) medium containing 10% horse serum (Capricorn Scientific GmbH, Ebsdorfergrund, Germany) and 5% heat-inactivated fetal bovine serum-FBS (Capricorn).23 Astrocytoma cells (1321N1) were cultured in DMEM (Thermo Fisher Scientific) containing 10% heat-inactivated FBS (Capricorn). The medium for 1321N1 cells was supplemented with penicillin (0.15 mM), streptomycin (86 μM) and glutamine (2 mM). Cells were incubated at 37 °C in a humidified environment of 7.5% CO2 and were routinely passaged every 3–4 days. Collagen type IV (Sigma-Aldrich, St. Louis, MO, USA) was coated on 96-well plates and left for at least 6 h, washed with phosphate-buffered saline and dried under the laminar floor to dryness prior to seeding PC-12 cells.
The neurite outgrowth assay was conducted on PC-12 cells and the RT-qPCR assay on 1321N1 cells as described previously.24 PC-12 cells or 1321N1 cells were treated with compounds 1 and 5 (each 5 μg mL−1) in the presence or absence of NGF (5 ng mL−1). A treatment with DMSO (5 μg mL−1) served as negative control. The neurite outgrowth of PC-12 cells was examined after 24 h using an IncuCyte S3 live-cell analysis system (Sartorius, Göttingen, Germany). Six random fields were examined in each well. Neurite length was measured using IncuCyte NeuroTrack Software Module (2019B Rev2 GUI). For RT-qPCR, total mRNA was extracted and RT-qPCR was carried out using the SensiFast™ SYBR® No-ROX one-step kit (Meridian Bioscience Inc., Cincinnati, OH, USA) according to the manufacturer's protocol. The following PCR primers were used for amplifying specific cDNA fragments: GAPDH (sense: 5′-ACCACAGTCCATGCCATCAC-3′; antisense: 5′-TCCACCACCCTGTTGCTGTA-3′; 451 bp), NGF (sense: 5′-CCAAGGGAGCAGTTTCTATCCTGG-3′; antisense: 5′-GGCAGTTGTCAAGGGAATGCTGAAGTT-3′; 189 bp), and BDNF (sense: 5′-TAACGGCGGCAGACAAAAAGA-3′; antisense: 5′-GAAGTATTGCTTCAGTTGGCCT-3′; 101 bp). The PCR reactions were analyzed and quantified using the LightCycler 96 (Roche Diagnostics International Ltd., Mannheim, Germany, version 1.1.0.1320) real-time PCR instrument. The reference gene GAPDH was used for normalization. Relative changes in gene expression were calculated by the 2−ΔΔCT method.
Statistical analyses
All statistical analyses were performed using the software Prism (version 9.4.0, Graphpad Software Inc., San Diego, CA, USA).Conclusion
The four new drimane sesquiterpenoids 1–4 were isolated from submerged cultures of the basidiomycete D. fragilis along with the two drimanes 5 and 6 that have already been reported previously. However, compounds 5 and 6 have only been described as biotransformation or synthetic compounds and have never been described to occur as natural products. In addition, this is the first report on drimanes that were isolated from the family Hericiaceae in general and from the genus Dentipellis in particular.The isolated compounds did not show any remarkable antimicrobial or cytotoxic effects. Following an observation by Kou et al.20 on neurotrophic activities of other drimanes isolated from Cyathus africanus, compounds 1 and 5, of which sufficient quantities were available, were assessed in similar assays that are established in our laboratory.
Compared to PC-12 (rat pheochromocytoma) cells supplied solely with NGF, those that were treated with NGF and compound 1 or 5, respectively, showed enhanced neurite outgrowth. In addition, metabolites 1 and 5 induced the expression of the neurotrophins NGF and BDNF in astrocytoma cells (1321N1). Notably, compound 1 was slightly more active in both assays. To date, the neurotrophic potential of drimane-type sesquiterpenoids has hardly been investigated, except for the study by Kou et al.,20 who only checked different drimanes in concentrations of 10 μM for their effect on neurite outgrowth in PC-12 cells by counting the percentages of differentiated cells in comparison to the DMSO control. These authors did not study the influence of the metabolites on BDNF and did not employ qPCR tests. Therefore, it is not possible to compare their results with ours. However, their work represents another example for the co-occurrence of cyathane and drimane type terpenoids in the same organism. Considering that the genus Cyathus (Bird's nest fungi) belongs to the Agaricales and is thus rather distantly related to the Hericiaceae (which belong to the order Russulales), it is puzzling how the analogous production of these two terpenoid classes has come about. Drimanes are widespread in plants and fungi but have also here been discovered from a species of Hericiaceae for the first time, even though some species of the latter family like Hericium erinaceus and H. coralloides can be considered to be very well studied. Nevertheless, it could be worthwhile to re-examine the species of Hericium, the related Laxitextum and other genera of Hericiaceae for the production of drimanes as co-metabolites of the ubiquitous cyathanes in order to look for possible chemotaxonomic correlations.
Author contributions
N. M.: conceptualization, screening, identification of the producer strain, isolation and structure elucidation of compounds, preparing the final draft; W. C. S.: conceptualization, screening, large scale fermentation, isolation and structure elucidation of compounds, bioassays, preparing the original draft; K. H. and M. K.: neurotrophic assays, statistical analysis; H. S.: supervision, editing and correcting the draft; L. G.: calculation of ECD spectra; H. K.: isolation of the producer strain; J. C. M.: supervision, reviewing and correcting the draft; T. E. B. S. and M. S.: supervision, funding acquisition, editing and correcting the draft. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors have no relevant financial or non-financial interests to disclose.Acknowledgements
This research benefitted from funding by the European Union’s Horizon 2020 Research Innovation and Staff Exchange program (RISE) under the Marie Skłodowska-Curie grant agreement No. 101008129, project acronym “Mycobiomics” (lead beneficiaries J. C. M. and M. S.). W. C. S. was supported by a doctoral scholarship funding from the German Academic Exchange Service (DAAD) program number 57507871. N. M. was financially supported by the VolkswagenStiftung within the framework of the project: “Global Carbon Cycling and Complex Molecular Patterns in Aquatic Systems: Integrated Analyses Powered by Semantic Data Management”. Calculations of CD spectra were performed on the HPC Cluster CARL, located at the University of Oldenburg (Germany) and funded by the DFG through its Major Research Instrumentation Programme (INST 184/157-1 FUGG) and the Ministry of Science and Culture (MWK) of the State of Lower Saxony. We deeply thank Silke Reinecke for technical assistance in the chromatography laboratory, Wera Collisi for assistance with the bioassays, Christel Kakoschke for conducting the NMR spectroscopic measurements and Aileen Gollasch as well as Esther Surges for performing LC-HR-ESIMS measurements.References
- M. A. Donk, Persoonia, 1962, 2, 217–238 Search PubMed.
- T. Niemelä and R. Saarenoksa, Karstenia, 1985, 25, 70–74 CrossRef.
- N. N. Wijayawardene, K. Hyde, D. Dai, B. T. Goto, R. Saxena, M. Erdoğdu, F. Selçuk, K. C. Rajeshkumar, A. Aptroot, J. Błaszkowski, N. Boonyuen, G. A. da Silva, F. A. de Souza, W. Dong, D. Ertz, D. Haelewaters, E. Jones, S. Karunarathna, P. Kirk, M. Kukwa, J. Kumla, D. Leontyev, H. Lumbsch, S. Maharachchikumbura, F. Marguno, P. Martínez-Rodríguez, A. Mešić, J. Monteiro, F. Oehl, J. Pawłowska, D. Pem, W. Pfliegler, A. Phillips, A. Pošta, M. He, J. Li, M. Raza, O. Sruthi, S. Suetrong, N. Suwannarach, L. Tedersoo, V. Thiyagaraja, S. Tibpromma, Z. Tkalčec, Y. Tokarev, D. Wanasinghe, D. Wijesundara, S. Wimalaseana, H. Madrid, G. Zhang, Y. Gao, I. Sánchez-Castro, L. Tang, M. Stadler, A. Yurkov and M. Thines, Mycosphere, 2022, 13, 53–453 CrossRef.
- H. Kawagishi, A. Shimada, R. Shirai, K. Okamoto, F. Ojima, H. Sakamoto, Y. Ishiguro and S. Furukawa, Tetrahedron Lett., 1994, 35, 1569–1572 CrossRef CAS.
- J. W. Shen, Y. Ruan and B. J. Ma, J. Basic Microbiol., 2009, 49, 242–255 CrossRef CAS PubMed.
- B. Thongbai, S. Rapior, K. D. Hyde, K. Wittstein and M. Stadler, Mycol. Prog., 2015, 14, 91 CrossRef.
- K. Wittstein, M. Rascher, Z. Rupcic, E. Löwen, B. Winter, R. W. Köster and M. Stadler, J. Nat. Prod., 2016, 79, 2264–2269 CrossRef CAS PubMed.
- W. C. Sum, N. Mitschke, H. Schrey, K. Wittstein, H. Kellner, M. Stadler and J. C. Matasyoh, Antibiotics, 2022, 11, 1072 CrossRef CAS PubMed.
- M. Maurs, R. Azerad, M. Cortés, G. Aranda, M. B. Delahaye and L. Ricard, Phytochemistry, 1999, 52, 291–296 CrossRef CAS PubMed.
- F. Z. Li, S. Li, P. P. Zhang, Z. H. Huang, W. Bin Zhang, J. Gong and Z. Yang, Chem. Commun., 2016, 52, 12426–12429 RSC.
- J. R. Sierra, J. T. López and M. J. Cortés, Phytochemistry, 1986, 25, 253–254 CrossRef CAS.
- C. Escobar and O. Wittke, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 154–156 CrossRef.
- D. Wenyu, Y. Qian, X. Hui-Min, D. Liao-Bin, D. Wenyu, Y. Qian, X. Hui-Min and D. Liao-Bin, Chin. J. Nat. Med. Press, 2022, 20, 1–12 Search PubMed.
- B. J. M. Jansen and A. De Groot, Nat. Prod. Rep., 2004, 21, 449–477 RSC.
- D. Zhi-Hui, D. Ze-Jun and L. Ji-Kai, Helv. Chim. Acta, 2001, 84, 259–262 CrossRef.
- W. A. Ayer and L. S. Trifonov, J. Nat. Prod., 1992, 55, 1454–1461 CrossRef CAS.
- Y. H. Kim, B. S. Yun, I. J. Ryoo, J. P. Kim, H. Koshino and I. D. Yoo, J. Antibiot., 2006, 59, 432–434 CrossRef CAS PubMed.
- W. A. Ayer and P. Craw, Can. J. Chem., 1989, 67, 1371–1380 CrossRef CAS.
- R. Velten, D. Klostermeyer, B. Steffan, W. Steglich, A. Kuschel and T. Anke, J. Antibiot., 1994, 47, 1017–1024 CrossRef CAS PubMed.
- R. W. Kou, S. T. Du, Y. X. Li, X. T. Yan, Q. Zhang, C. Y. Cao, X. Yin and J. M. Gao, J. Antibiot., 2019, 72, 15–21 CrossRef CAS PubMed.
- C. W. Phan, G. S. Lee, S. L. Hong, Y. T. Wong, R. Brkljača, S. Urban, S. N. Abd Malek and V. Sabaratnam, Food Funct., 2014, 5, 3160–3169 RSC.
- Z. Rupcic, M. Rascher, S. Kanaki, R. W. Köster, M. Stadler and K. Wittstein, Int. J. Mol. Sci., 2018, 19, 740 CrossRef PubMed.
- M. Rascher, K. Wittstein, B. Winter, Z. Rupcic, A. Wolf-Asseburg, M. Stadler and R. W. Köster, Biomolecules, 2020, 10, 1440 CrossRef CAS PubMed.
- K. Hassan, B. Matio Kemkuignou, M. Kirchenwitz, K. Wittstein, M. Rascher-Albaghdadi, C. Chepkirui, J. C. Matasyoh, C. Decock, R. W. Köster, T. E. B. Stradal and M. Stadler, Int. J. Mol. Sci., 2022, 23, 13593 CrossRef CAS PubMed.
- S. D. Skaper, Methods Mol. Biol., 2018, 1727, 1–17 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- S. Grimme, J. Chem. Theory Comput., 2019, 15, 2847–2862 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- B. Helmich-Paris, B. de Souza, F. Neese and R. Izsák, J. Chem. Phys., 2021, 155, 104109 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- E. Caldeweyher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2017, 147, 034112 CrossRef PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- T. Bruhn, A. Schaumlöffel, Y. Hemberger and G. Bringmann, Chirality, 2013, 25, 243–249 CrossRef CAS PubMed.
- K. Becker, A. C. Wessel, J. J. Luangsa-ard and M. Stadler, Biomolecules, 2020, 10, 805 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra04204a |
| ‡ Current address: Research Group for Marine Geochemistry (ICBM-MPI Bridging Group), Institute for Chemistry and Biology of the Marine Environment (ICBM), Carl von Ossietzky Universität Oldenburg, Carl-von-Ossietzky-Str. 9-11, 26129 Oldenburg, Germany. |
| § N. M. and W. C. S. contributed equally to this paper. |
| This journal is © The Royal Society of Chemistry 2023 |