
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective synthesis of CF3-containing spirocyclic-oxindoles using N-2,2,2-trifluoroethylisatin ketimines: an update
Biplob Borah
 a,
Naveena S. Veeranagaiah
b,
Samrita Sharma
a,
Mihir Patat
a,
Madavi S. Prasad
*c,
Raghavaiah Pallepogu
b and
L. Raju Chowhan
*a
a,
Naveena S. Veeranagaiah
b,
Samrita Sharma
a,
Mihir Patat
a,
Madavi S. Prasad
*c,
Raghavaiah Pallepogu
b and
L. Raju Chowhan
*a
aSchool of Applied Material Sciences, Centre for Applied Chemistry, Central University of Gujarat, Gandhinagar-382030, India. E-mail: rchowhan@cug.ac.in
bDepartment of Chemistry, Central University of Karnataka, Kalaburagi, Karnataka-585367, India
cDepartment of Chemistry, Asymmetric Synthesis and Catalysis Laboratory, Central University of Tamil Nadu (CUTN), Tiruvarur, 610 005, India
First published on 1st March 2023
Abstract
The introduction of fluorine-containing groups into organic molecules either changes or improves the characteristics of the target compounds. On the other hand, spirocyclic-oxindoles featuring C-3 functionalized sp3-hybridized carbon atoms of three-dimensional orthogonally shaped molecules were well recognized in the core structure of varied natural products and synthetic pharmaceutical targets. Consequently, the construction of spirooxindoles by an elegant synthetic approach with efficient stereocontrol has received tremendous interest over the past decades. In this context of the synergic combination of the features associated with fluorine-containing compounds and the synthetic and medicinal efficiency associated with spirooxindoles, the stereodivergent installation of CF3 groups embedded with spirooxindoles is of increasing academic and scientific interest. This mini-review article is dedicated to demonstrating a critical overview of the recent stereoselective synthesis of spirocyclic-oxindoles featuring trifluoromethyl groups by employing the reactivity of N-2,2,2-trifluoroethylisatin ketimines as an efficient and easily prepared synthon, and covers the literature reports from 2020 to date. Besides exploring the advancements accomplished in this area, we also investigate the limitations associated with reaction discovery, mechanistic rationalization, and future applications.
 Biplob Borah | Biplob Borah obtained his BSc degree in Chemistry from Nowgong College (Gauhati University), Assam in 2017, and received his Master's degree in Industrial Chemistry from the Central University of Gujarat, India in 2019. Then, he joined the School of Applied Material Sciences at the Central University of Gujarat as a PhD scholar under the guidance of Dr L. Raju Chowhan. His research interests include organocatalysis, asymmetric synthesis, radical chemistry mechanochemistry, MCRs, photochemistry, green chemistry, synthesis of medicinally privileged heterocycles, and hybrid molecules in the aqueous medium. |
 Naveena S. Veeranagaiah | Naveena S. Veeranagaiah obtained his BSc degree in Chemistry from Government First Grade College (Tumkur University) in 2015, and received his Master's degree in Chemistry from Government Science College at Mysore University in 2018. Then, he joined the Department of Chemistry at the Central University of Karnataka University as a PhD scholar under the guidance of Dr Raghavaiah Pallepogu. His research interests includes supramolecular chemistry, pharmaceutical co-crystallization, synthesis of small organic heterocyclic molecules with their crystal studies and DFT calculation. |
 Samrita Sharma | Samrita Sharma obtained her BSc degree in Chemistry from Rishi Bankim Chandra College (West Bengal State University) in 2021. Then, she joined the School of Applied Material Sciences, Centre for Applied Chemistry, Gandhinagar, Gujarat, India for her Master's degree in Industrial Chemistry. Currently, she is working on her MSc dissertation under the supervision of Dr L. Raju Chowhan. Her research interests include enantioselective synthesis, MCRs, photochemistry, the development of novel methodologies for the synthesis of heterocyclic compounds, metal-free catalysis in the aqueous medium, and green synthesis. |
 Mihir Patat | Mihir Patat obtained his BSc degree from Smt Devkiba College Silvassa at the University of Mumbai, in 2020. Then, he joined the School of Applied Material Sciences, Centre for Applied Chemistry, Gandhinagar, Gujarat, India for his Master's degree in Industrial Chemistry, and completed it in 2022. His research interests include domino and consecutive multicomponent reactions, developing novel methodologies for synthesizing heterocyclic compounds, metal-free catalysis for diverse organic synthesis in the aqueous medium, and green synthesis. |
 Madavi S. Prasad | Dr Madavi S. Prasad received his MSc degree in Physical Organic Chemistry from Osmania University, Hyderabad, India. He completed his doctoral studies in chemistry under Prof. Dhevalapally B. Ramachary, School of Chemistry, University of Hyderabad, Hyderabad, India. He joined the Chemistry Department at Central University of Tamil Nadu, Tiruvarur, India, in 2013, where he is currently an Assistant Professor (Senior scale). His research interests center around the asymmetric synthesis of small natural products, natural product mimics, and biologically potent scaffolds by exploiting catalysis. |
 Raghavaiah Pallepogu | Dr Raghavaiah Pallepogu obtained his Master's degree in Chemistry from the School of Chemistry, University of Hyderabad, Hyderabad, in 1995. He received his PhD from Goa University under the supervision of Prof. B. R. Srinivasan and Prof. K. S. Rane. After that, he worked as a post-doctoral fellow at the Centre for Supramolecular Chemistry Research, University of Cape Town (UCT) South Africa with Prof. Mino R. Caira. Currently, he is working as an Associate Professor at the Central University of Karnataka, India. His research interests include crystal engineering, pharmaceutical co-crystallization, solid-state chemistry, and investigation of symmetry breaking in small molecules to understand supramolecular chemistry. |
 L. Raju Chowhan | Dr L. Raju Chowhan obtained his BSc degree from Osmania University, Hyderabad, in 2005, and a Master's degree in Chemical Science from Hyderabad Central University, Hyderabad, in 2007. He received his PhD degree from CSIR-Indian Institute of Chemical Technology, in association with Hyderabad Central University. He joined as an Assistant Professor at the Centre for Applied Chemistry, Central University of Gujarat, Gandhinagar, on 5th September 2012. His research interests include the stereoselective synthesis of natural products, the development of novel methodologies for asymmetric synthesis, multicomponent reactions, and 1,3-dipolar cycloaddition reactions. |
1. Introduction
Recognizing the small atomic size, strong electron-withdrawing ability and greater electronegativity, fluorine-containing organic compounds play an essential role in medicinal and drug discovery as a consequence of their so-called fluorine scan in the development of drug candidates.1,2 Incorporating fluorine into pharmaceutically relevant molecules either as a single molecule or as a substituted molecule often changes or adds distinctive properties, such as increased binding interactions, metabolic stability and physical properties,3–5 that improve the characteristics of the particular compound and the generated molecules typically offered substantial or even unprecedented properties. Among diverse fluorine-containing compounds, the trifluoromethyl core especially mounted at the α-position of the nitrogen atom reduces the alkalinity of the amide group, which in turn impacts the binding potential of the drug receptor.6Approximately 20% of the top-selling small drug molecules and 30% of agrochemicals available on the market comprises one or more fluorine atoms in their structure, with the trifluoromethyl group being the most prevalent.7 The presence of fluorine efficiently influences the properties of a drug molecule via strong polar interactions.6 After the hydrogen (H) atom, fluorine (F) is the second smallest and most highly electronegative atom in the periodic table.8 The C–F bond is only 1.28 times longer than the C–H bond, so the implementation of a fluorine atom in a drug candidate in place of hydrogen does not significantly alter the parent structure, although the bond energy is substantially greater than that for the C–H bond (C–F bond: 105.4 kcal mol−1, C–H bond: 98.8 kcal mol−1).9 According to the Pauling scale (F: 4.0, O: 3.5, H: 2.1), fluorine is more akin to oxygen than hydrogen in terms of its electronic characteristics.9 Because of the C–F bond, which is the strongest bond that carbon can form, fluorine-containing pharmaceuticals are often more metabolically stable.10 Fluorine causes bond polarization, which may alter the proportions of lipophilicity and hydrophilicity of a compound.10 The introduction of a trifluoromethyl group instead of a trimethyl group improves various properties, such as the metabolic stability, lipophilicity, and selectivity in a drug molecule.11 In terms of size, CF3 groups are much more identical to the isopropyl group rather than a trimethyl group.12 As predicted, the basicity and pKa values of nearby functional groups (for instance, amines, carboxylic acid, and alcohols) are significantly impacted by the trifluoromethyl group functioning as an electron-withdrawing substituent, thereby affecting the bioavailability of drug molecules.13 On the other hand, it is a prevalent misperception that fluorination always increases lipophilicity.14 However, due to the fluorine's powerful ability to withdraw electrons, mono-fluorination or trifluoromethylation of saturated alkyl groups often results in a reduction in lipophilicity.14 Trifluoromethyl groups are thought to have a van der Waals volume that is comparable to that of an ethyl group while having a drastically different shape, which might cause a more severe steric change when added to a molecule.8 The strong electronegativity of the fluorine atom and these steric fluctuations can cause alterations in the preferable structural conformation.15 For example, the ground state conformations of methoxybenzene and trifluoromethoxy benzene will adopt different conformations in a complex molecule. The C–F bond also can participate in weak hydrogen bonding.14 Contrary to hydrogen bonding, fluorine is commonly acknowledged to participate in electrostatic interactions, thereby increasing the binding affinity to the drug receptors as compared to non-fluorinated analogs.13,14
On the other hand, spirocyclic-oxindoles featuring the C-3 functionalized sp3-hybridized carbon atom of a three-dimensional orthogonally shaped molecule have been recognized as a privileged structural motif in drug discovery and medicinal chemistry.16–19 These pharmaceutically potential and structurally functionalized compounds were well-organized in the architectures of a plethora of natural products and synthetic drug candidates, in addition to their immense significance in synthetic organic chemistry.20–22 Alternatively, with the presence of a tetrahedral quaternary sp3-hybridized carbon center at the C-3 position, they have even been known to provide outstanding binding potential with many drug receptors, exceptional lipophilicity, and stereochemical rigidity compared to the planar monocyclic systems.23 Compounds possessing the spirooxindole core were found to exhibit a broad spectrum of therapeutic applications, such as antibacterial, anticancer, anti-malarial, anti-platelet, anti-viral, MDM2 inhibitor, anti-HIV, analgesic, and anti-rhythmic. Some examples of therapeutically significant natural and synthetic drug candidates featuring spirooxindoles are shown in Fig. 1.17,24–29
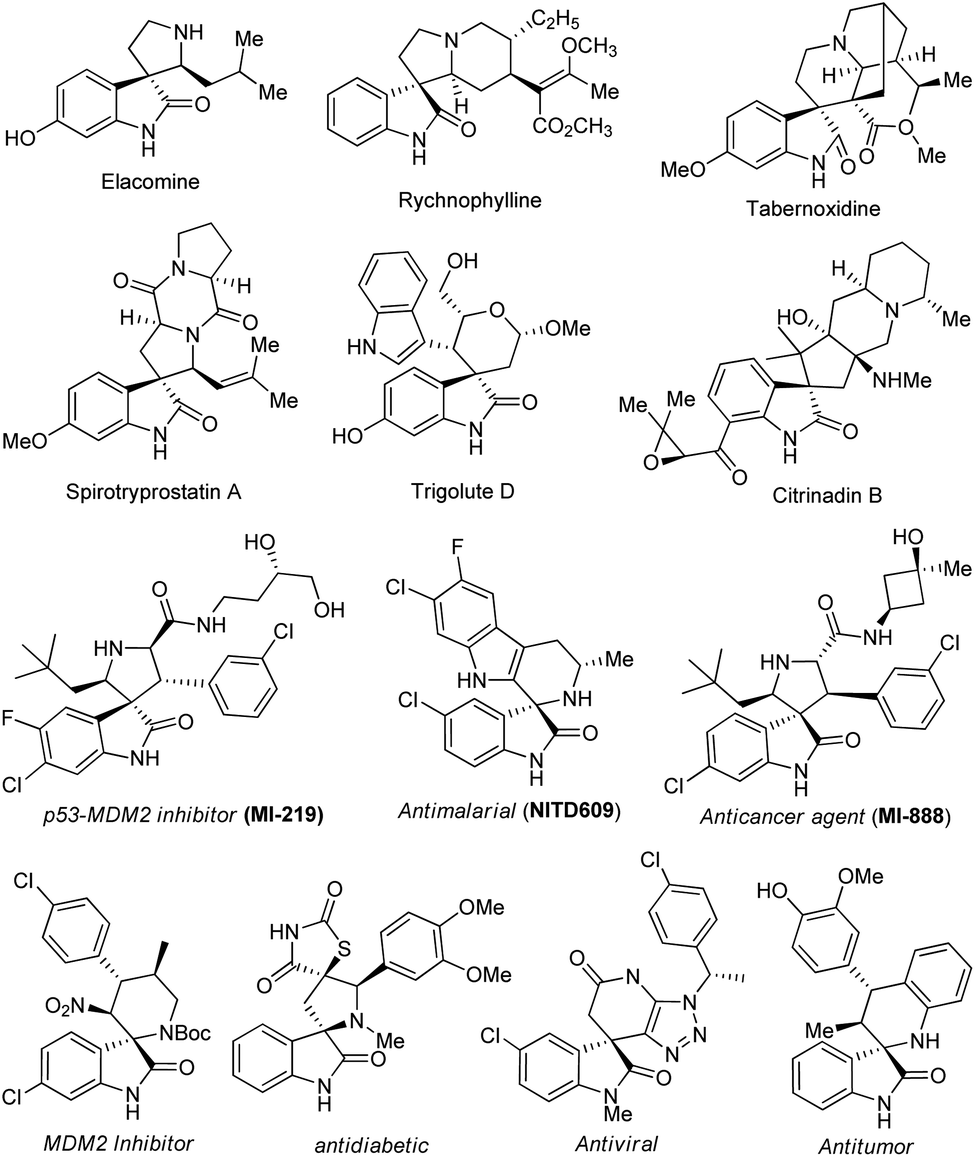 | ||
| Fig. 1 Representative demonstration of potentially therapeutic natural and synthetic molecules comprising the spirooxindole scaffold. | ||
Considering the widespread chemical landscape and broad-spectrum therapeutic significance and their utilization as a crucial partner for natural product synthesis, the construction of spirooxindoles has received special attention in organic chemistry, and has been well-established in the literature over the past decades.30–32 Although a lot of outstanding work on the creation of these spirocyclic compounds has been devised, the stereocontrol construction of such fascinating molecular structures with manifold contiguous stereogenic centers including two spiro quaternary centers has remained a great challenge in synthetic organic chemistry. This is presumably due to the strong steric hindrance facilitated by bulkier groups in one ring atom. Again, owing to the synergic combination of the features associated with fluorine-containing compounds and the synthetic and medicinal efficiency associated with spirooxindoles, the stereodivergent installation of CF3 groups embedded with spirooxindoles is of increasing academic and scientific interest. Consequently, the development of an efficient approach accessible to CF3-containing spirooxindoles with multiple vicinal stereocenters under mild reaction conditions is highly desired.
Intriguingly, a diverse set of organic transformations enabling efficient access to a broad-spectrum molecular structure in a stereocontrolled manner from easily available synthetic precursors via the creation of carbon–carbon and carbon-heteroatom bonds were devised. Among them, the 1,3-dipolar cycloaddition (1,3-DC) reactions comprise one of the successful and substantial approaches for realizing carbon–carbon, carbon–heteroatom bonds, and spirocyclic scaffolds in synthetic organic chemistry.33,34 Accordingly, various types of 1,3-dipoles (for instance, azomethine ylides,35–37 carbonyl ylides,38,39 and nitrones40,41) have been introduced and effectively utilized in cycloaddition reactions. After the pioneering work by Wang et al.,42 N-2,2,2-trifluoroethylisatin ketimines synthesized from the condensation of isatin and trifluoroethylamine hydrochloride with the help of 5 mol% p-toluenesulfonic acids (p-TSA) in toluene were recently recognized as stable, highly efficient, and reactive azomethine ylide precursors, and have been explored for the assembly of a broad variety of CF3-containing spirocyclic oxindoles (Scheme 1). While many groups devoted their efforts towards the successful application of this 1,3-dipole in the C–C, C–N bond formation reactions43–45 and synthesis of spirocyclic oxindoles, ample attention still needs to be paid for investigating the diverse mode of reactivity of this useful precursor for the asymmetric construction of value-added structural scaffolds of potential therapeutic interest.
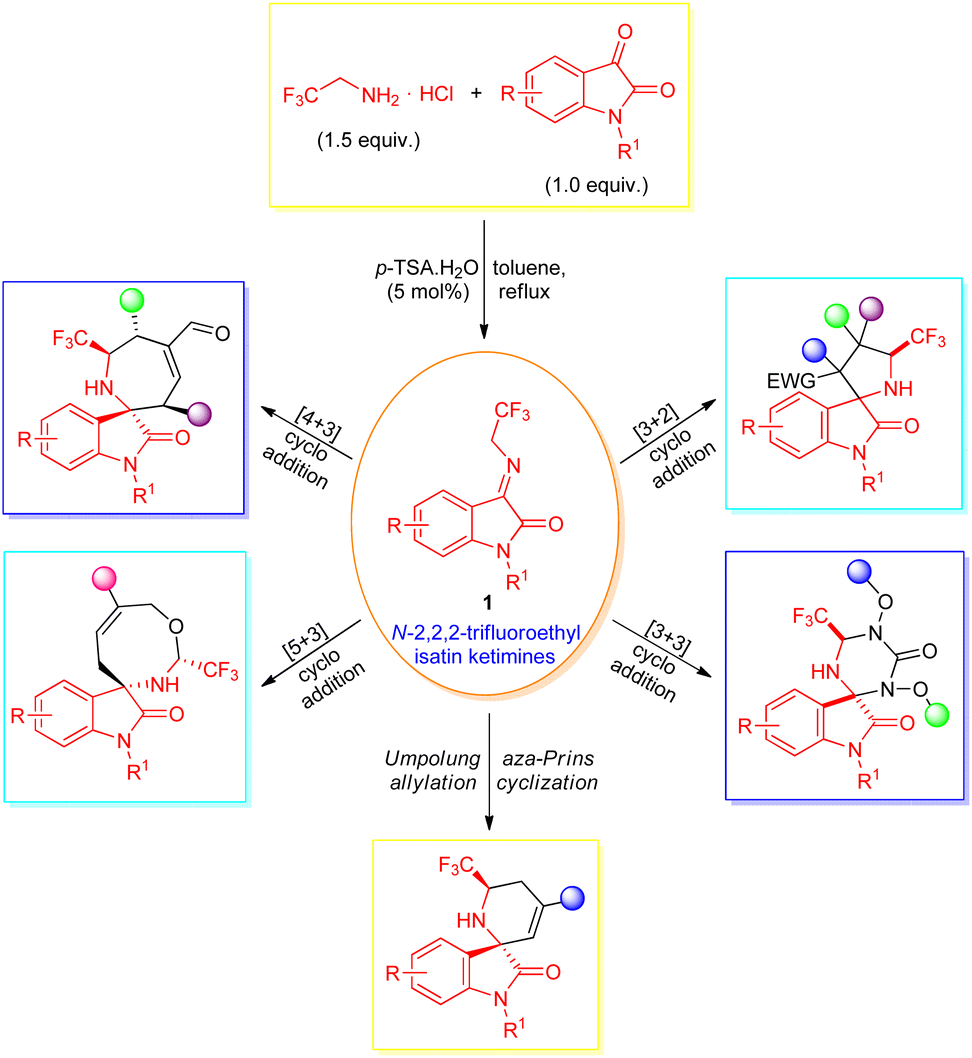 | ||
| Scheme 1 Reactivity of N-2,2,2-trifluoroethylisatin ketimines as 1,3-dipole in the assembly of CF3-containing spirooxindoles. | ||
In 2020, Wei, Shi, and co-workers reviewed some examples of the synthesis of the trifluoromethyl group containing spirooxindoles in a controlled manner.46 The 1,3-dipolar cycloaddition reactions for synthesizing diverse structurally functionalized molecular frameworks have been well-reviewed.47–49 Shankaraiah et al. reported the cycloaddition reactions of 3-methyleneindolinones for the synthesis of spirooxindoles.50 We also demonstrated the reactivity of isatin N,N′-cyclic azomethine imines towards spirooxindoles.51 This review article is dedicated to demonstrating the recent progress achieved in the stereodivergent assembly of CF3-containing spirocyclic oxindoles by employing the reactivity of N-2,2,2-trifluoroethylisatin ketimines as an efficient and easily prepared synthon, and covers reports from 2020 to the present date, including the contribution in this ongoing research field from our group. Along with emphasizing the advancements made in this field, we have made an effort to draw attention to the drawbacks and difficulties related to the reaction discovery, which will hopefully provoke further investigation.
2. Synthesis of spirocyclic oxindoles comprising trifluoromethyl group via [3+2] cycloaddition reactions
Jia, Hua, Wang, and co-workers devised a highly efficient asymmetric [3+2] cycloaddition catalyzed by a zinc metal complex towards the rapid construction of CF3-containing chiral spiro[benzofuran-pyrrolidine] derivatives (Scheme 2).52 With the help of 10 mol% of ligand L1 and 20 mol% of ZnEt2, the reaction of N-2,2,2-trifluoroethylisatin ketimines 1 with aurone 2a (X = O) or thioaurone 2b (X = S) was established to lead to the respective spirooxindole products 3 in moderate to excellent yield with excellent diastereoselectivity and enantioselectivity. The existence of electron-withdrawing and electron-releasing substances on the aryl ring of aurone had no greater influences, and were well tolerated for this reaction under identical conditions. Upon changing the aurone to thioaurone, the respective [3+2] cycloadduct was accomplished in good yield, albeit with good enantioselectivity as compared to aurone. Similarly, imines 1 possessing various substituents like chloro, fluoro, bromo, and methyl on the C-5, and C-7 position also provided the products with good yields and enantioselectivity. The synthetic potentiality of the protocol was further demonstrated by transferring the synthesized product 3 (R = R1 = H; X = O) to N-benzylated spirooxindole in 95% yield with 94% ee.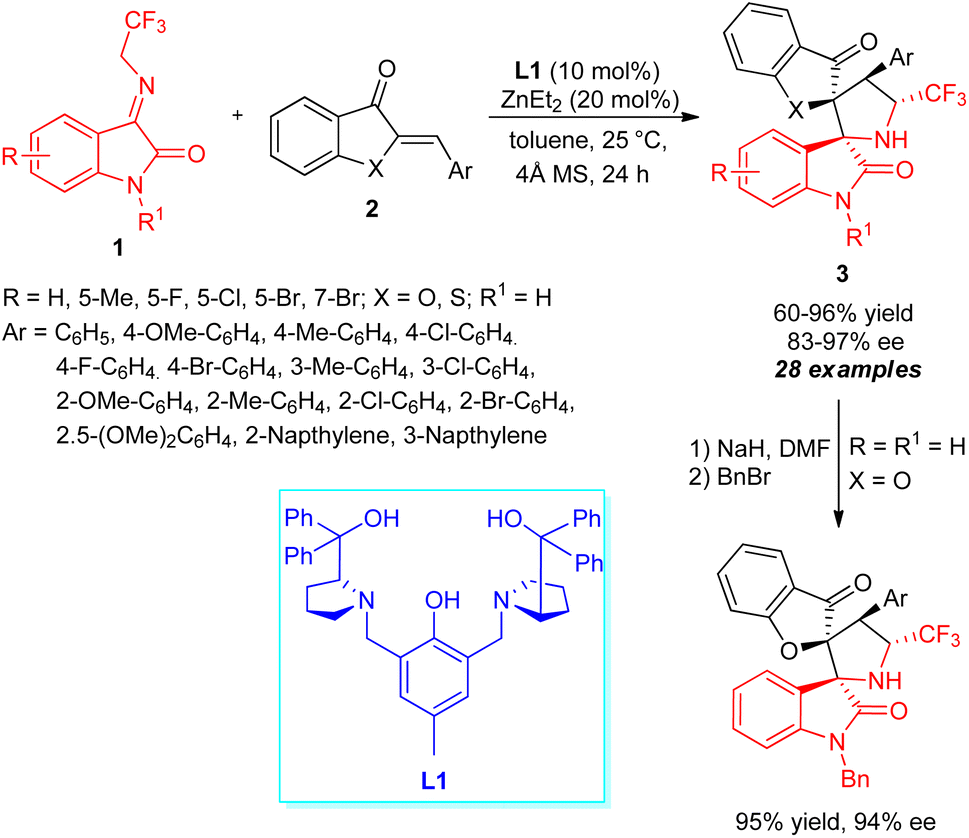 | ||
| Scheme 2 Dinuclear zinc-catalyzed enantioselective construction of CF3-containing spirooxindoles. | ||
Based on a set of experimental results, the mechanistic pathway for this transformation was postulated by the authors, which starts with the in situ generations of the dinuclear zinc species L1-Zn2Et (Int-1) from the reaction of 1 equivalent of L1 and 2 equivalents of ZnEt2 (Scheme 3). Under the influences of Int-1, N-2,2,2-trifluoroethylisatin ketimines 1 underwent deprotonation to provide activated azomethine ylide Int-2 with the subsequent removal of 1 equivalent of ethane. The most favored coordination of 2 from the less hindered side to both zinc atoms delivered the intermediate Int-3, which participated in the Michael addition reaction to afford Int-4. The subsequent intramolecular Mannich reaction of Int-4 provided Int-5, which can then afford the final product 3 by proton exchange with another imine 1, thereby restarting the catalytic cycle with the subsequent formation of azomethine ylide Int-2.
 | ||
| Scheme 3 Plausible mechanism for the dinuclear zinc-catalyzed [3+2] cycloaddition towards the formation of 3. | ||
An unexpected diastereoselectivity switchable formal [3+2] cycloaddition using mono gold to gold/silver bimetallic catalytic system for the construction of spirooxindoles was established by Xiao, Su, and co-workers (Scheme 4).53 By employing the reactivity of N-2,2,2-trifluoroethylisatin ketimines 1 in the initial reaction with enynones 4 in the presence of 5 mol% of [PPh3Au]NTf2 and 20 mol% of Et3N, the final CF3-containing spiro-pyrrolidine-oxindoles 5 were achieved in 38–98% yield with good to excellent diastereoselectivity (up to >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Both N-protected and N-unprotected ketimines having different substitution on the aryl ring were found to be suitable for this reaction. It is important to note that the presence of fluoro and chloro groups at the C-4, C-5, or C-7 positions decreases the yield of the products, and is likely due to the special electronic effect. Again, ketimines with a chloro group at the C-4 position lead to a decrease of diastereoselectivity of the product. Similarly, enynones with C-2, C-3, and C-4 substitutions were well tolerated under standard reaction conditions. On the other hand, upon changing the mono-catalytic system to a bimetallic system using 5 mol% of C-1 and AgSbF6, the respective diastereomers 7 were recognized to be formed in moderate to excellent yields. To establish the synthetic application of the protocol, the synthesized product 5 was further transformed into furan-fused spirooxindoles 8 by 1,2′-alkyl migration and also reduced to product 9 by Pd/C in good yield. The synthesis of both diastereomers of the spirooxindole products in quantitative yields by switching the catalytic system marks the highlights of this protocol.
:1). Both N-protected and N-unprotected ketimines having different substitution on the aryl ring were found to be suitable for this reaction. It is important to note that the presence of fluoro and chloro groups at the C-4, C-5, or C-7 positions decreases the yield of the products, and is likely due to the special electronic effect. Again, ketimines with a chloro group at the C-4 position lead to a decrease of diastereoselectivity of the product. Similarly, enynones with C-2, C-3, and C-4 substitutions were well tolerated under standard reaction conditions. On the other hand, upon changing the mono-catalytic system to a bimetallic system using 5 mol% of C-1 and AgSbF6, the respective diastereomers 7 were recognized to be formed in moderate to excellent yields. To establish the synthetic application of the protocol, the synthesized product 5 was further transformed into furan-fused spirooxindoles 8 by 1,2′-alkyl migration and also reduced to product 9 by Pd/C in good yield. The synthesis of both diastereomers of the spirooxindole products in quantitative yields by switching the catalytic system marks the highlights of this protocol.
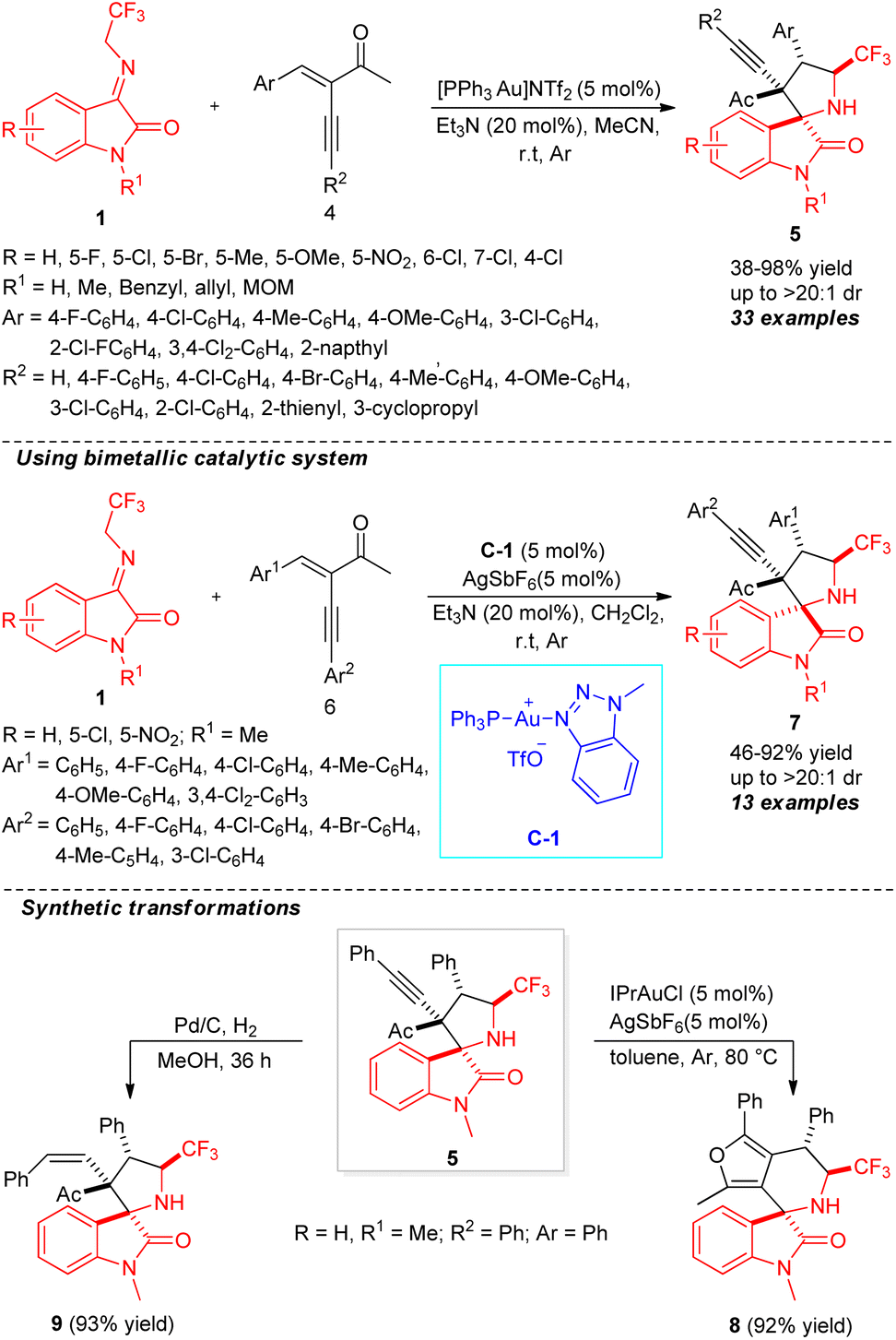 | ||
| Scheme 4 Diastereoselectivity switchable gold-catalyzed construction of CF3-containing spirooxindoles. | ||
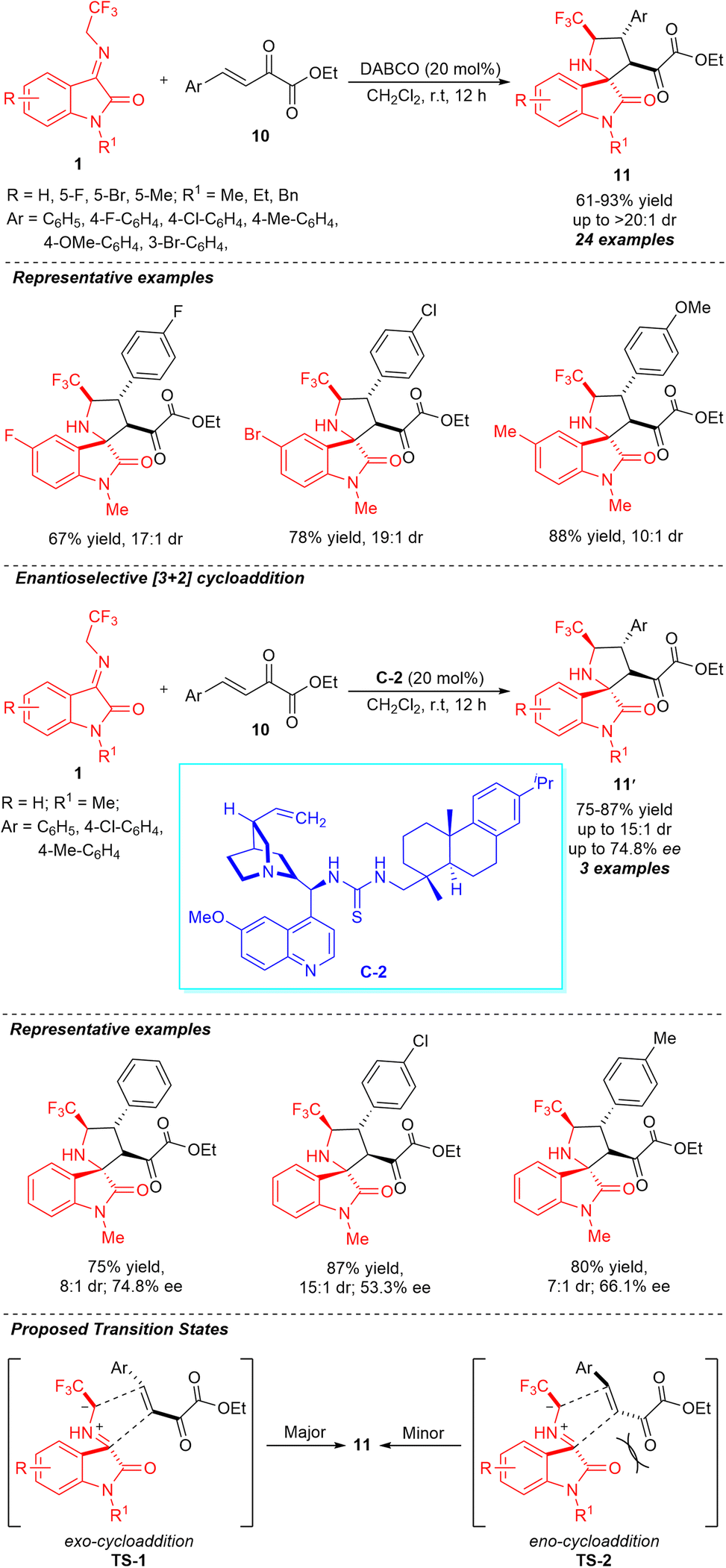
Enlightened by the chemistry and biology of fluorine-containing compounds and spiro-pyrrolidine-oxindoles, Wang, Liu, and co-workers constructed a series of various CF3-containing spirooxindoles possessing four contiguous stereocenters through a highly diastereoselective DABCO-catalyzed [3+2] cycloaddition (Scheme 5).54 With the assistance of 20 mol% of the catalyst, the treatments of N-2,2,2-trifluoroethylisatin ketimines 1 and β,γ-unsaturated α-ketoesters 10 delivered the respective products 11 in 61–93% yield with high diastereoselectivity (up to >20:1). To broaden the scopes of the reactions, a variety of differently substituted ketimines having halogenated groups and electron-donating groups were examined and recognized to be very efficient for this reaction. Similarly, the electronic properties of numerous substituents on different positions of the aromatic ring of β,γ-unsaturated α-ketoesters had no greater influence on the reactivity and diastereoselectivity of the reaction. Moreover, the authors demonstrated the enantioselective construction of spirooxindoles 11′ by employing 20 mol% of quinine-derived thiourea catalyst C-2 at room temperature. Although the products were attained in slightly good yields, the low enantioselectivity and diastereoselectivity of the present study call for further developments regarding the catalytic enantioselective sequence. The relative configuration of the synthesized products was confirmed to follow an exo-transition state (TS-1), rather than the endo-pathway (TS-2) by single crystal analysis.
 | ||
| Scheme 5 DABCO-catalyzed highly diastereoselective [3+2] cycloaddition of ketimines to β,γ-unsaturated α-ketoesters. | ||
A highly practical phase transfer catalytic approach for the diastereoselective assembly of CF3-containing spiro-fused[succinimide-pyrrolidine-oxindoles] was reported by Chen and co-workers (Scheme 6).55 With the help of 5 mol% of quaternary ammonium salt tetrahexylammonium bromide (THAB) as the phase transfer catalyst and 2 mol% of Cs2CO3 as the base, the respective products 13 derived from the [3+2] cycloaddition of N-2,2,2-trifluoroethylisatin ketimines 1 and maleimides 12 were accomplished in 69–99% yields with excellent diastereoselectivity (up to >99:1 dr) within a short duration of time. Investigations on the reactivity of ketimines with maleimides revealed that both N-H free and N-protected ketimines encompassing electron-deficient and electron-rich substances on diverse positions participated in the reaction very smoothly. The electronic characteristics of the substituents on the aromatic ring of maleimide had a pronounced effect on the yield of the product. While the electron-rich group at the C-2, C-3, C-4, or C-5 positions of the phenyl ring provided a better yield of the product, the corresponding products with electron-deficient groups gave diminished yield. The overall process was proposed to proceed through a cis-addition between the azomethine ylide (formed from 1) and maleimide 12 via transition state TS-3. Furthermore, by introducing THAB as the phase-transfer catalyst in the Pd(II)-catalyzed Suzuki–Miyaura cross-coupling reaction of the synthesized product 13 (R = 5-Br; R1 = Bn; R2 = Ph) with aryl/heteroaryl boronic acids, the corresponding more complex coupling products 14 was constructed in moderate yields. This achievement contributes to the key significance of the present study.
 | ||
| Scheme 6 Phase transfer-catalyzed stereoselective formation of spiro-fused[succinimide-pyrrolidine-oxindoles]. | ||
Taking advantage of the high reactivity of the phase transfer catalytic system and the aforementioned successful attempts, Chen and co-workers again demonstrated the potentiality of the quaternary ammonium salt phase transfer catalysis in the diastereoselective [3+2] cycloaddition reaction of N-2,2,2-trifluoroethylisatin ketimines 1 and (Z)-4-(chromone-3-yl)methylene oxazolones 15 (Scheme 7).56 With the aid of 5 mol% of tetrabutylammonium bromide (TBAB) along with K2CO3 as an inorganic base, the corresponding CF3-containing bispiro-[oxazolone-pyrrolidine-oxindoles] 16 bearing a chromone ring has been achieved in 51–94% yield with high diastereoselectivity. Ketimines 1 having an N-benzylated group provides a greater yield as compared to N-methyl, N-allyl, and N-H free substitutions. While various substitutions at the C-5, C-6, and C-7 positions of the aromatic ring of ketimines furnished the products with excellent yields, the substitution at the C-7 position by the CF3 group reduces the diastereoselectivity of the product. Conversely, the presence of electron-deficient groups in the oxazolone moiety gave lower yields.
 | ||
| Scheme 7 TBAB-catalyzed highly diastereoselective [3+2] cycloaddition of ketimines to access spirooxindoles. | ||
Based on the control experiments, the author realized a mechanistic rationalization for this reaction (Scheme 8). Initially, it was proposed that TBAB plays a role in transporting the carbonate across the phases, and the in situ generated tetrabutylammonium carbonate (TBAC) acts as an actual catalyst in this transformation. The basic carbonate of TBAC abstracts a proton from ketimines 1, thereby furnishing the carbanion intermediate Int-6, which is followed by retaking of the proton from ammonium bicarbonate to form the highly reactive azomethine ylide. The final major diastereomer was assumed to be formed via the favored anti-addition (TS-4) of oxazolone 15 to azomethine ylide, rather than the syn-addition (TS-5). This is probably owing to the dipole repulsion of the carbonyl groups of azomethine ylide and chromene oxazolone moiety.
 | ||
| Scheme 8 The suggested mechanism developed by Chen et al. for the TBAB-catalyzed formal [3+2] cycloaddition. | ||
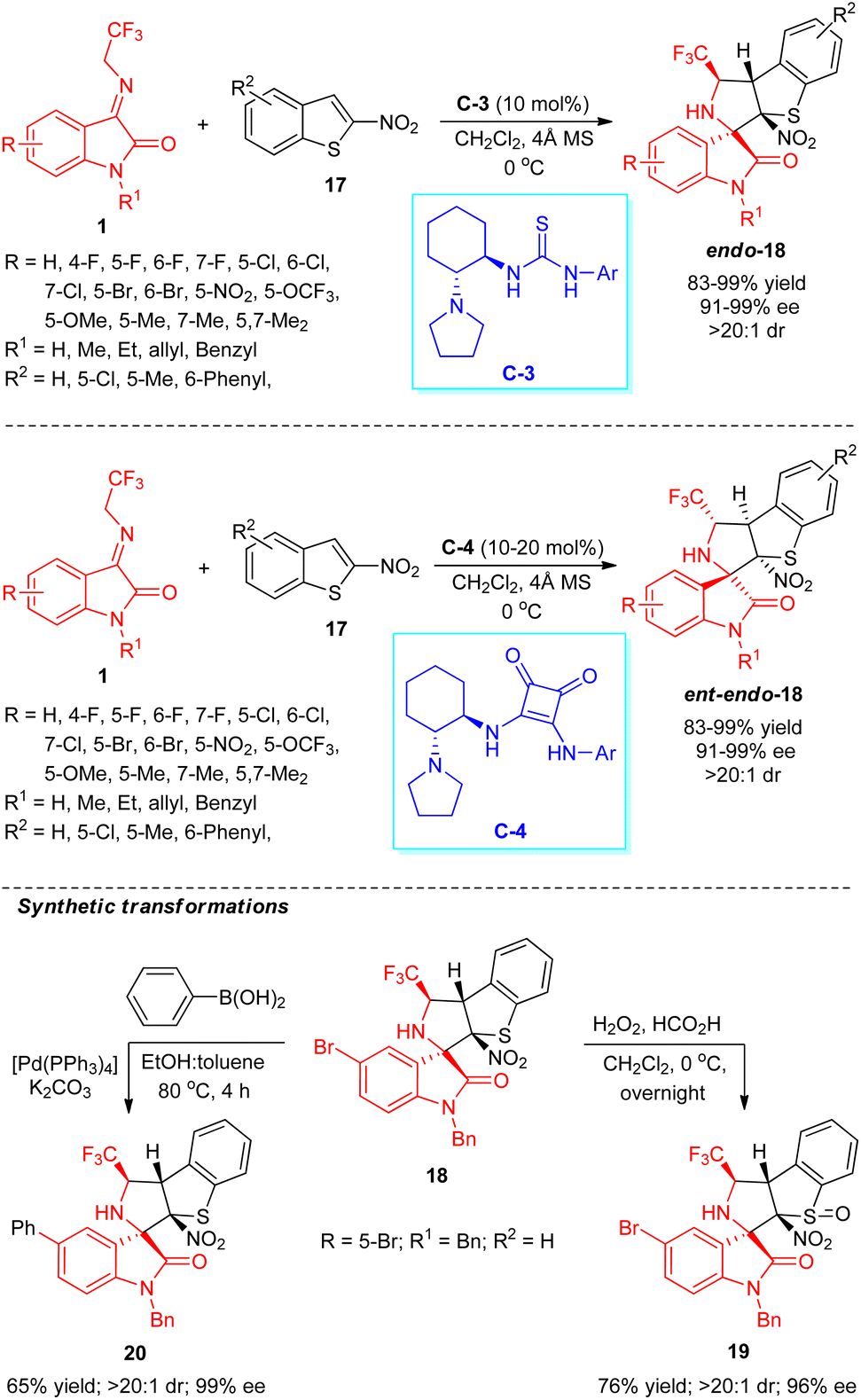
Zhao, Yuan, and co-workers explored the catalytic activity of the bifunctional hydrogen-bonding thiourea catalyst in the dearomative enantioselective [3+2] cycloaddition reaction for the assembly of CF3-embedded spirocyclic oxindoles (Scheme 9).57 With the assistance of 10 mol% of chiral organocatalyst C-3, the treatments of N-2,2,2-trifluoroethylisatin ketimines 1 and 2-nitrobenzothiophenes 17 lead to the formation of respective products 18 in 83–99% yield with excellent diastereoselectivity (>20:1 dr) and enantioselectivity (up to 99% ee). Ketimines 1 with broad spectrum substitutions on the aryl ring of the oxindole core by halogenated groups, strong-electron-withdrawing groups, and electron-donating groups have been well documented in this reaction, and had no greater effect on the selectivity of the products. Again, the reaction condition was recognized to be very compatible with both N-H free (R1 = H) and N-substituted ketimines (R1 = Me, Et, allyl, Bn). On the other hand, with the introduction of hydrogen-bonding squaramide catalyst C-4 having an identical chiral core, the enantioselectivity of the target compounds was established to be reversed. The significance and potentiality of their work were recognized by the synthetic transformation of the target compound 18 (R = 5-Br; R1 = Bn; R2 = H) to sulfur monoxide-containing product 19 in good yield without altering the selectivity. Furthermore, the Suzuki–Miyaura cross-coupling reaction of 18 with benzene boronic acid furnished the respective cross-coupling product 20 in good yield with no change in enantioselectivity.
 | ||
| Scheme 9 Enantioselective thiourea-catalyzed dearomative [3+2] cycloaddition towards the rapid access to spirooxindoles. | ||
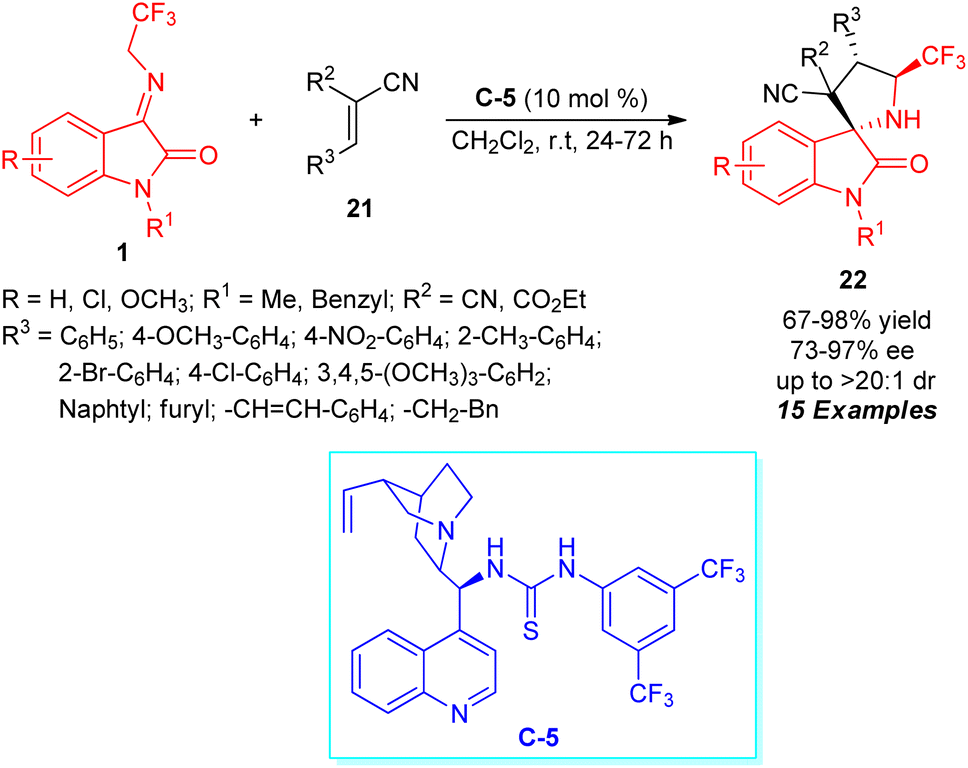
A highly efficient enantioselective approach for expedient access to a broad variety of CF3-embedded spirocyclic oxindoles having four contiguous stereocenters via a formal [3+2] cycloaddition sequence was realized by Knipe and co-workers (Scheme 10).58 By employing 10 mol% of cinchona-derived thiourea C-5 as the chiral organocatalyst, the respective products 22 derived from N-2,2,2-trifluoroethylisatin ketimines 1 and activated alkene dipolarophiles 21 have been achieved in 67–98% yield with excellent diastereoselectivity and moderate to outstanding enantioselectivity. Overall, 15 compounds comprising varied substitutions were constructed with identical conditions.
 | ||
| Scheme 10 Organocatalytic asymmetric [3+2] cycloaddition for the assembly of CF3-embedded spirooxindoles 22. | ||
The exploitation of isoxazoles 23 in the asymmetric [3+2] cycloaddition proceeding through a cascade Michael addition and Mannich reaction with N-2,2,2-trifluoroethylisatin ketimines 1 was demonstrated by Du and Li (Scheme 11).59 With the assistance of 5 mol% of squaramide catalyst C-6, the respective CF3-embedded spirooxindole products 24 possessing four contiguous stereocenters have been obtained in 42–99% yield with outstanding diastereoselectivity and moderate to excellent enantioselectivity. The existence of both electron-rich and electron-deficient moieties on the aromatic ring of the ketimines was well executed in this reaction. Notwithstanding the N-substitution (R1 = Me, Bn, allyl, 4-Br-benzyl) on the ketimines have been found to work efficiently, the N-unsubstituted ketimines (R1 = H) provided the respective product in good yield, albeit with low enantioselectivity. Alternatively, the installation of N-protected groups (R3 = Me, Et, Bn) on the isoxazole ring leads to the product in sufficient yield with good to excellent enantioselectivity, except for N-4-NO2-benzyl substitution, which reduces the yield of the product while retaining the enantioselectivity. Isoxazoles 23 with N-H free substitution (R3 = H) also diminished the enantioselectivity of the product, which again calls for the development of the catalytic system for further improvements of the enantioselectivity in the future. Furthermore, the reduction of the synthesized product 24 (R = R2 = H; R1 = R3 = Bn; R4 = Me) by tin chloride to product 25 points towards the synthetic application of the protocol.
 | ||
| Scheme 11 Squaramide-catalyzed domino Michael/Mannich 1,3-dipolar cycloaddition for synthesizing CF3-embedded spirooxindoles. | ||
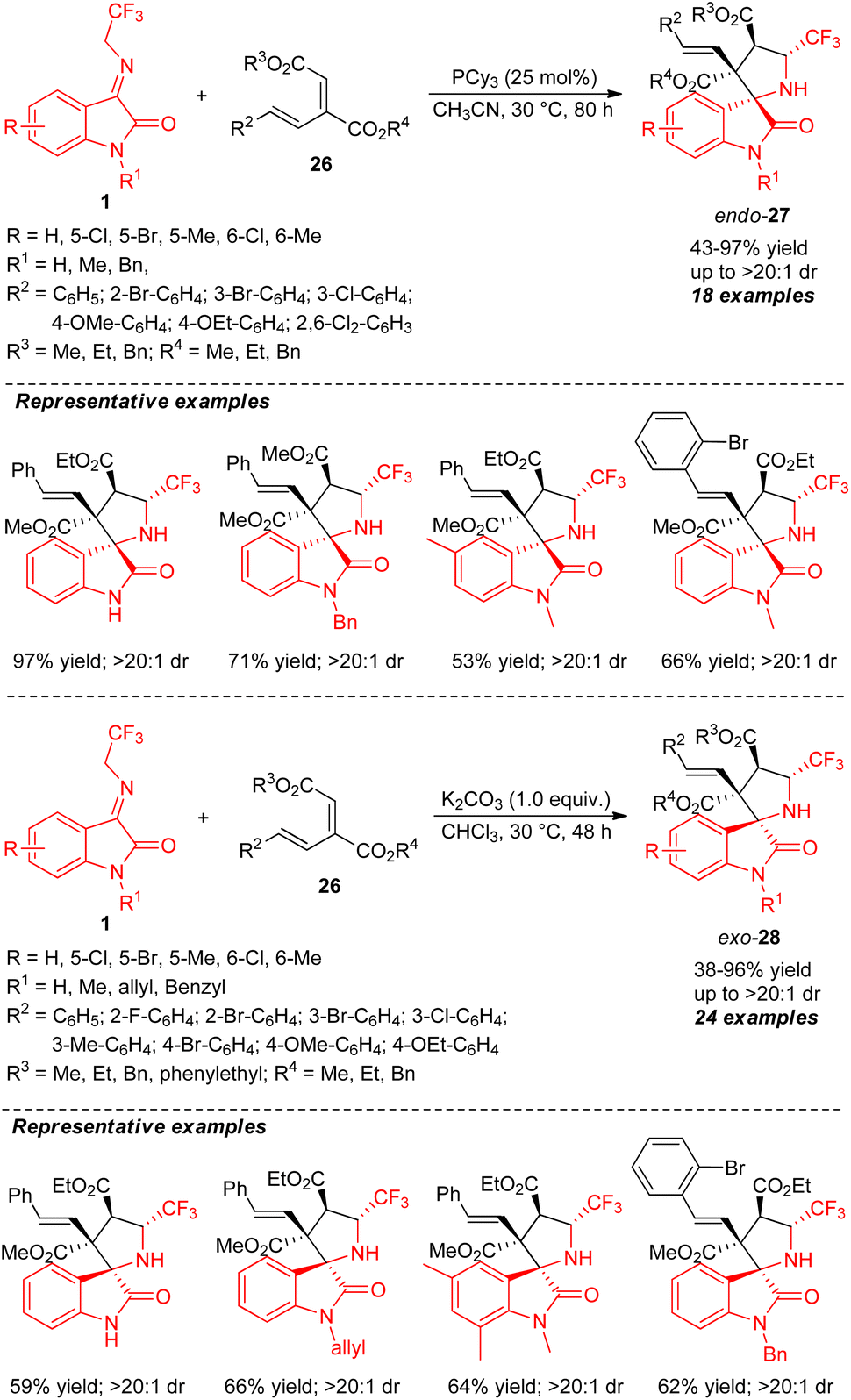
The stereocontrolled construction of a broad variety of CF3-embedded spirocyclic oxindoles by controlling the configuration of the stereocenters with two different catalytic systems was developed by Li, Duan, and co-authors (Scheme 12).60 With 25 mol% of tri-cyclohexyl phosphine (PCy3) as the Lewis base catalyst, the stereodivergent [3+2] cycloaddition of N-2,2,2-trifluoroethylisatin ketimines 1 and conjugated dienes 26 was found to lead to the formation of quaternary centered C-3 functionalized endo-products 27 in 43–97% yield with excellent diastereoselectivity (up to >20:1). The explorations of N-H free and N-substituted ketimines having different electronic properties on different positions of the aryl ring reacted smoothly under this condition with a variety of diene possessing various ester groups. Despite this, low diastereoselectivity was observed for diene with ethyl and benzyl groups at the R3 and R4 positions, respectively. Alternatively, switching the catalytic system from Lewis bases to Brønsted base K2CO3, the reaction of 1 and 26 afforded a different diastereomer of CF3-embedded spirocyclic oxindoles exo-28 in 38–96% yield with up to >20:1 diastereoselectivity. Intriguingly, the diastereoselectivity of the offered products derived from various N-protected ketimines seems to be retained, regardless of the electronic properties. N-Unprotected ketimines (R1 = H) also provided excellent diastereoselectivity, albeit with a low yield.
 | ||
| Scheme 12 Catalyst-controlled diastereodivergent construction of CF3-embedded spirocyclic oxindoles. | ||
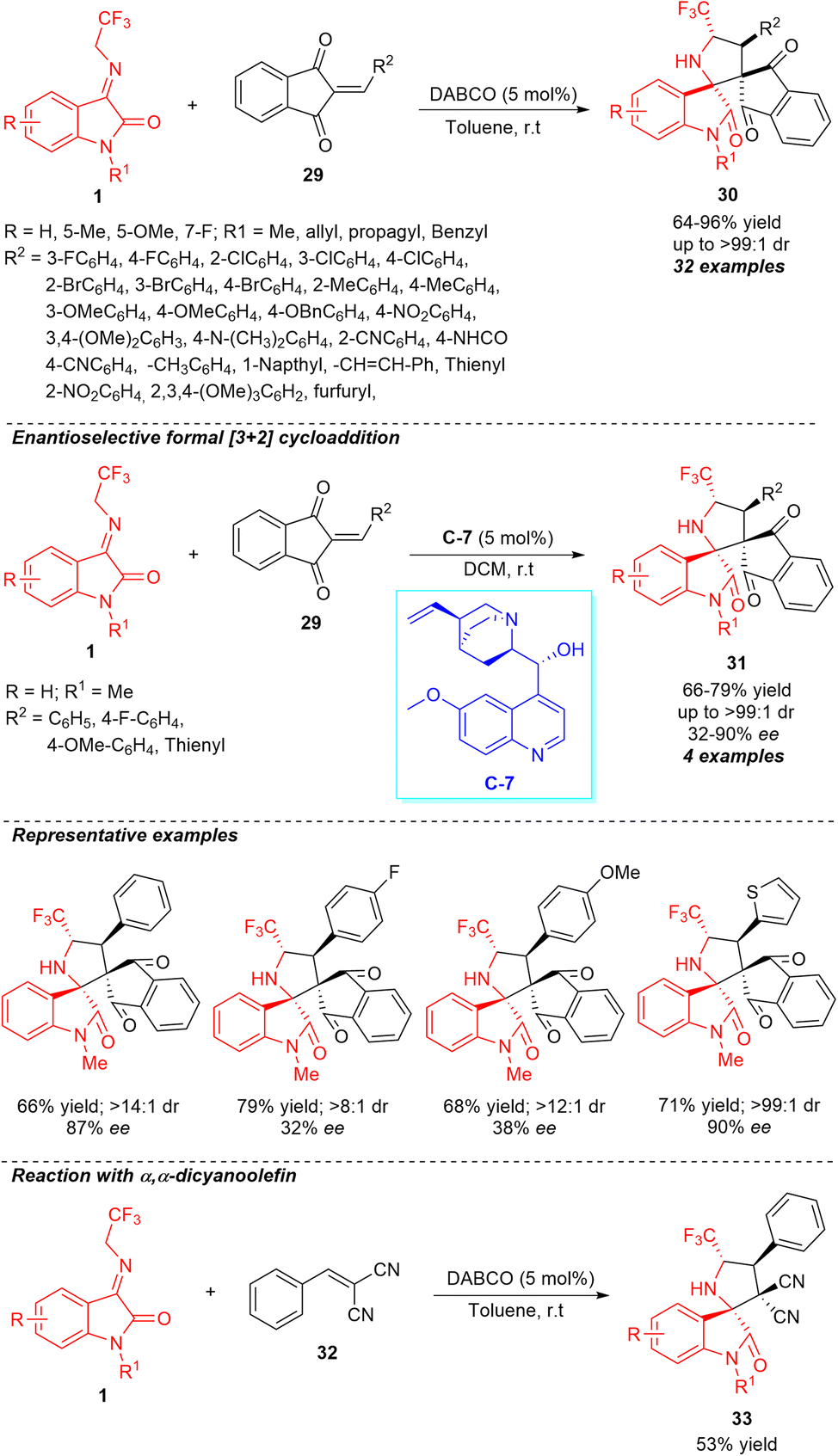
Recently, our group also demonstrated the highly diastereoselective construction of a broad variety of novel spirocyclic oxindoles encompassing the trifluoromethyl group from base-catalyzed [3+2] cycloaddition of N-2,2,2-trifluoroethylisatin ketimines 1 and 2-aryl/heteroarylidene-1H-indene-1,3(2H)-diones 29 as an activated olefin source (Scheme 13).61 Using 5 mol% of DABCO as the basic organocatalyst, we were delighted to accomplish the respective products 30 encompassing four contiguous stereocenters in moderate to excellent yield and diastereoselectivity. Notwithstanding the reaction showed good tolerances for a variety of electronic substituents present at the varied positions of the aromatic ring of ketimines, the R2 substitutions of 1H-indene-1,3(2H)-diones by aromatic compounds with electron-deficient and electron-rich groups were established to work efficiently, delivering the products in 64–96% yield with up to >99:1 diastereoselectivity. The reaction condition was found to be very compatible not only with aryl-substituted 1H-indene-1,3(2H)-diones, but also well executed with heteroaryl-substituted 1H-indene-1,3(2H)-diones. The enantioselective synthesis of spirooxindoles 31 with two adjacent spiro-quaternary chiral centers in 66–79% yield with up to >99:1 diastereoselectivity and 32–90% enantioselectivity was demonstrated using the ultralow loading (5 mol%) of quinine C-7 as the chiral organocatalyst at room temperature, showcasing the synthetic potentiality of the strategy. However, ample attention still needs to be paid to extend the scopes of the substrates with enhanced enantioselectivity by developing other catalytic conditions. In the same way, we executed a reaction between ketimines 1 and arylidene malononitrile 32 by employing the typical condition, which offered the product 33 in 53% yield. This result further authenticates the implications of the current approach.
 | ||
| Scheme 13 Utilization of 1H-indene-1,3(2H)-diones in [3+2] cycloaddition to access novel CF3-embedded spirooxindoles. | ||
3. Synthesis of spirocyclic oxindoles comprising trifluoromethyl group via [3+3] cycloaddition reaction
Although the reactivity of N-2,2,2-trifluoroethylisatin ketimines in the [3+2] cycloaddition with a broad-spectrum activated olefin source for recognizing 5-membered ring-bearing spirooxindoles that consist of trifluoromethyl groups was well established, the corresponding [3+3] cycloaddition of N-2,2,2-trifluoroethylisatin ketimines was limited. Consequently, a deep investigation for the demonstration of this azomethine ylide precursor is highly required.After the successful attempts of N-2,2,2-trifluoroethylisatin ketimines in the [3+3] cycloaddition by the Bi group,62 a new [3+3] cycloaddition strategy employing the umpolung reactivity of ketimines 1 and aza-Prins cyclization to produce diverse six-membered rings containing spirooxindoles encompassing the trifluoromethyl group was developed by Ko and co-workers (Scheme 14).63 With the aid of DBU as the basic catalytic system, the one-pot treatments of 1 and allyl bromide 34 in the presence of H2O and TMSBr enabled easy access to the respective products 35 in moderate to outstanding yield by varying the temperature of the reaction from −10 to 0 °C in the allylation step, and from room temperature to 40 °C for the cyclization step. The electronic properties of the substrates have a small effect on the yield of the products. On the other hand, substitutions on the allyl bromide ring by different alkyl and aryl-substituted groups resulted in the formation of products 36 and 36′ subsequently. Moreover, the asymmetric version of this umpolung allylation/aza-Prins cyclization was examined by the authors. In the presence of 10 mol% of Pd(PPh3)4 as the metal catalyst and 20 mol% of (R)-BINAP as the chiral ligand, the reaction of 1 (R = H; R1 = Me) with 37 afforded the chiral product 35 (R = H; R1 = Me) in 48% yield with only 19% enantioselectivity. This result indicates the possibility of enhancing the enantioselectivity of the reaction by introducing other catalytic asymmetric approaches.
 | ||
| Scheme 14 Umpolung allylation/aza-Prins cyclization towards the rapid access to six-membered containing spirooxindoles. | ||
4. Synthesis of spirocyclic oxindoles comprising the trifluoromethyl group via [4+3] cycloaddition reactions
The stereodivergent construction of polycyclic molecules greater than five- and six-membered rings via [4+3] cycloaddition remains a continuous challenge in synthetic organic chemistry due to the increased ring tension. On the other hand, the catalytic asymmetric [3+2] cycloaddition of α,β-unsaturated carbonyl compounds and 1,3-dipoles has been well recognized in the literature. The subsequent [4+3] annulation of substrates comprising an extended double bond was still unassigned and difficult, and underwent [3+2] cycloaddition rather than [4+3] cycloaddition.64Considering the aforementioned challenges, Chun and group realized the reactivity of α-vinyl α,β-unsaturated aldehydes as 4-C synthons in β,γ′-regioselective [4+3] annulations with N-2,2,2-trifluoroethylisatinimines 1 for constructing seven-membered spirooxindoles having the azepane motif by iminium ion-dienamine catalysis (Scheme 15).65 With the aid of 20 mol% of proline-derived organocatalyst C-8 in the presence of 20 mol% of benzoic acid, a broad spectrum of structurally functionalized spirocyclic oxindoles encompassing a trifluoromethyl group with a seven-membered azepane ring have been isolated in moderate to excellent yield with good to excellent enantioselectivity. A variety of β-aromatic or heteroaromatic substituents installed in the α-vinylenals ring were well reacted in this reaction and provided high to excellent enantioselectivity, while β-alkyl substituents showed reduced reactivity for this reaction. Similarly, the corresponding product 39 was not observed with substrates bearing a γ′-alkyl group. The synthesized product 39 (R = R3 = H; R1 = Me; R2 = Ph) comprising the α,β-unsaturated aldehyde moiety could be further transformed into a high molecular complex structure 41 in 87% yield with >19:1 diastereoselectivity and 90% enantioselectivity by a [3+3] cycloaddition with cyclohexane-1,3-dione 40. The β,γ′-regioselective [4+2] annulation pathway is assumed to proceed, rather than the traditional [3+2] pathway, due to the generation of a more hindered quaternary center by α-substitution.
 | ||
| Scheme 15 Enantioselective [4+3] cycloaddition of ketimines for synthesizing seven-membered CF3-embedded spirooxindoles. | ||
5. Conclusions
Considering the advantageous outcome produced from the introduction of fluorine or fluorine-containing molecules into bioactive compounds, the installation of fluorine (especially the trifluoromethyl core) at the α-position of a nitrogen atom has received immense attention in synthetic organic chemistry, as it affects the binding potential of the drug receptor by reducing the alkalinity of the amide group. On the other hand, spirocyclic oxindoles were well distributed in the core scaffolds of broad-spectrum natural products, and pharmaceutically potential synthetic drug candidates. In this regard, the stereocontrolled construction of spirocyclic oxindoles encompassing the trifluoromethyl group is of increasing academic and scientific interest. Consequently, after the pioneering work by Wang et al. in 2015, the reactivity of N-2,2,2-trifluoroethylisatin ketimines as an efficient and easily prepared azomethine ylide precursor in many 1,3-dipolar cycloaddition reactions for the construction of diverse CF3-containing spirocyclic oxindoles has been devised. This mini-review article is organized to provide a recent overview of the stereoselective assembly of spirocyclic oxindoles comprising the trifluoromethyl group by employing the reactivity of N-2,2,2-trifluoroethylisatin ketimines.From the aforementioned information presented in this review article and based on the literature review, it was found that most of the synthetic methodology implemented the reactivity of these azomethine ylide precursors through [3+2] cycloaddition reaction. The corresponding [3+3], [3+5], [4+2], and [4+3] cycloadditions were still not fully developed, and only a limited report existed. We hope that the information provided in this review article will help and stimulate the scientific community for further improvements in the selectivity of the products by introducing more catalytic asymmetric methodologies. We believe that the limited [3+3], [3+5], [4+2], and [4+3] cycloadditions of N-2,2,2-trifluoroethylisatin ketimines will also be investigated, and more unprecedented methodologies will be developed in the near future.
Author contributions
BB: conceptualization, investigation, software, writing original draft, writing – review & editing, resources. NSV: writing original draft. SS: writing original draft. MP: software. MSP: investigation. RP: investigation. RC: supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
B. B. and N. S. V. thank UGC-India for the fellowship. Author thanks the Central University of Gujarat and Central University of Karnataka for the infrastructure to carry out the work.Notes and references
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- W. K. Hagmann, J. Med. Chem., 2008, 51, 4359–4369 CrossRef CAS PubMed.
- T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed.
- K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed.
- A. S. Nair, A. K. Singh, A. Kumar, S. Kumar, S. Sukumaran, V. P. Koyiparambath, L. K. Pappachen, T. M. Rangarajan, H. Kim and B. Mathew, Processes, 2022, 10, 2054 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- H.-J. Böhm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Müller, U. Obst-Sander and M. Stahl, ChemBioChem, 2004, 5, 637–643 CrossRef PubMed.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294–8308 CrossRef CAS PubMed.
- J. A. MacPhee, A. Panaye and J.-E. Dubois, Tetrahedron, 1978, 34, 3553–3562 CrossRef CAS.
- T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- G. Yan, K. Qiu and M. Guo, Org. Chem. Front., 2021, 8, 3915–3942 RSC.
- M. Ganesh and S. Suraj, Org. Biomol. Chem., 2022, 20, 5651–5693 RSC.
- L.-M. Zhou, R.-Y. Qu and G.-F. Yang, Expert Opin. Drug Discovery, 2020, 15, 603–625 CrossRef CAS PubMed.
- A. J. Boddy and J. A. Bull, Org. Chem. Front., 2021, 8, 1026–1084 RSC.
- B. Borah, J. Bora, P. Ramesh and L. R. Chowhan, RSC Adv., 2022, 12, 12843–12857 RSC.
- B. Borah, K. D. Dwivedi and L. R. Chowhan, Polycyclic Aromat. Compd., 2022, 42, 5893–5937 CrossRef CAS.
- B. Borah, K. D. Dwivedi and L. R. Chowhan, Arkivoc, 2021, 2021, 273–328 Search PubMed.
- T. L. Pavlovska, R. Gr. Redkin, V. V. Lipson and D. V. Atamanuk, Mol. Diversity, 2016, 20, 299–344 CrossRef CAS PubMed.
- M. Aldeghi, S. Malhotra, D. L. Selwood and A. W. E. Chan, Chem. Biol. Drug Des., 2014, 83, 450–461 CrossRef CAS PubMed.
- N. Basarić, Ž. Marinić and M. Šindler-Kulyk, J. Org. Chem., 2006, 71, 9382–9392 CrossRef PubMed.
- M. Asad, M. N. Arshad, A. M. Asiri, S. A. Khan, M. Rehan and M. Oves, Polycyclic Aromat. Compd., 2022, 42, 5385–5397 CrossRef CAS.
- R. C. Elderfield and R. E. Gilman, Phytochemistry, 1972, 11, 339–343 CrossRef CAS.
- R. G. Geetha and S. Ramachandran, Pharmaceutics, 2021, 13, 1170 CrossRef CAS PubMed.
- B. Borah, M. Patat, V. Singh, M. Sivaprakash, M. S. Prasad and L. R. Chowhan, Org. Biomol. Chem., 2023, 21, 1518–1530 RSC.
- B. Borah, S. Sharma and L. R. Chowhan, Asian J. Org. Chem., 2023 DOI:10.1002/ajoc.202300020.
- X. Luo, W. Li, H. Lu, G. Deng, Y. Yang, C. Yang and Y. Liang, Chin. Chem. Lett., 2021, 32, 713–716 CrossRef CAS.
- M. Zhang, Y.-H. Liu, Z.-R. Shang, H.-C. Hu and Z.-H. Zhang, Catal. Commun., 2017, 88, 39–44 CrossRef CAS.
- M.-N. Chen, J.-Q. Di, J.-M. Li, L.-P. Mo and Z.-H. Zhang, Tetrahedron, 2020, 76, 131059 CrossRef CAS.
- T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366–5412 CrossRef CAS PubMed.
- M. Kissane and A. R. Maguire, Chem. Soc. Rev., 2010, 39, 845–883 RSC.
- G. Pandey, P. Banerjee and S. R. Gadre, Chem. Rev., 2006, 106, 4484–4517 CrossRef CAS PubMed.
- M. S. Reddy, L. R. Chowhan, N. Satish Kumar, P. Ramesh and S. B. Mukkamala, Tetrahedron Lett., 2018, 59, 1366–1371 CrossRef CAS.
- M. S. Reddy, N. S. Kumar and L. R. Chowhan, RSC Adv., 2018, 8, 35587–35593 RSC.
- H. Nambu, M. Hikime, J. Krishnamurthi, M. Kamiya, N. Shimada and S. Hashimoto, Tetrahedron Lett., 2009, 50, 3675–3678 CrossRef CAS.
- H. Suga, D. Ishimoto, S. Higuchi, M. Ohtsuka, T. Arikawa, T. Tsuchida, A. Kakehi and T. Baba, Org. Lett., 2007, 9, 4359–4362 CrossRef CAS PubMed.
- G.-H. Li, W. Zhou, X.-X. Li, Q.-W. Bi, Z. Wang, Z.-G. Zhao, W.-X. Hu and Z. Chen, Chem. Commun., 2013, 49, 4770 RSC.
- S.-I. Murahashi and Y. Imada, Chem. Rev., 2019, 119, 4684–4716 CrossRef CAS PubMed.
- M. Ma, Y. Zhu, Q. Sun, X. Li, J. Su, L. Zhao, Y. Zhao, S. Qiu, W. Yan, K. Wang and R. Wang, Chem. Commun., 2015, 51, 8789–8792 RSC.
- Y.-X. Song and D.-M. Du, J. Org. Chem., 2018, 83, 9278–9290 CrossRef CAS PubMed.
- Y. Lin, Y.-X. Song and D.-M. Du, Adv. Synth. Catal., 2019, 361, 1064–1070 CrossRef CAS.
- B. Li, F. Gao, X. Feng, M. Sun, Y. Guo, D. Wen, Y. Deng, J. Huang, K. Wang and W. Yan, Org. Chem. Front., 2019, 6, 1567–1571 RSC.
- H. Gui, Y. Wei and M. Shi, Chem.–Asian J., 2020, 15, 1225–1233 CrossRef CAS PubMed.
- J. Adrio and J. C. Carretero, Chem. Commun., 2014, 50, 12434–12446 RSC.
- L. M. Stanley and M. P. Sibi, Chem. Rev., 2008, 108, 2887–2902 CrossRef CAS PubMed.
- X. Fang and C.-J. Wang, Org. Biomol. Chem., 2018, 16, 2591–2601 RSC.
- R. Saeed, A. P. Sakla and N. Shankaraiah, Org. Biomol. Chem., 2021, 19, 7768–7791 RSC.
- B. Borah and L. R. Chowhan, Tetrahedron Lett., 2022, 104, 154014 CrossRef CAS.
- R.-L. Wang, S.-K. Jia, Y.-J. Guo, Y. Yi, Y.-Z. Hua, H.-J. Lu and M.-C. Wang, Org. Biomol. Chem., 2021, 19, 8492–8496 RSC.
- J. Xiao, X. Cheng, H. Peng, J. Liang, X. Luo and W. Su, Chem.–Asian J., 2021, 16, 2435–2438 CrossRef CAS PubMed.
- Y. Xiong, X.-X. Han, Y. Lu, H.-J. Wang, M. Zhang and X.-W. Liu, Tetrahedron, 2021, 87, 132112 CrossRef CAS.
- Z. Chen, W. Liang, Z. Chen and L. Chen, Eur. J. Org. Chem., 2021, 2021, 788–793 CrossRef CAS.
- L. Chen, H.-Y. Geng, Z.-J. Chen, W. Liang and W.-Y. Jiao, Tetrahedron Lett., 2021, 78, 153276 CrossRef CAS.
- J.-Q. Zhao, S. Zhou, L. Yang, H.-Y. Du, Y. You, Z.-H. Wang, M.-Q. Zhou and W.-C. Yuan, Org. Lett., 2021, 23, 8600–8605 CrossRef CAS PubMed.
- C. Duffy, W. E. Roe, A. M. Harkin, R. McNamee and P. C. Knipe, New J. Chem., 2021, 45, 22034–22038 RSC.
- T.-H. Li and D.-M. Du, Org. Biomol. Chem., 2022, 20, 817–823 RSC.
- K. Li, Z. Zhang, J. Zhu, Y. Wang, J. Zhao, E.-Q. Li and Z. Duan, Org. Chem. Front., 2022, 9, 19–24 RSC.
- M. S. Prasad, S. Bharani, S. M. Sharief, M. Ravi, M. Sivaprakash, B. Borah and L. R. Chowhan, RSC Adv., 2022, 12, 34941–34945 RSC.
- H.-W. Zhao, J.-M. Guo, L.-R. Wang, W.-Q. Ding, Z. Tang, X.-Q. Song, H.-H. Wu, X.-Z. Fan and X.-F. Bi, Org. Chem. Front., 2019, 6, 3891–3895 RSC.
- W. C. Jang, M. Jung and H. M. Ko, Org. Lett., 2021, 23, 1510–1515 CrossRef CAS PubMed.
- P. H. Poulsen, S. Vergura, A. Monleón, D. K. B. Jørgensen and K. A. Jørgensen, J. Am. Chem. Soc., 2016, 138, 6412–6415 CrossRef CAS PubMed.
- Y. Gao, X. Song, R.-J. Yan, W. Du and Y.-C. Chen, Org. Biomol. Chem., 2021, 19, 151–155 RSC.
| This journal is © The Royal Society of Chemistry 2023 |