
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Defect-free graphene enhances enzyme delivery to fibroblasts derived from patients with lysosomal storage disorders†
Yingxian
Chen
ab,
Tooba
Taufiq
ab,
Niting
Zeng
 c,
Neus
Lozano
d,
Angeliki
Karakasidi
ab,
Heather
Church
e,
Ana
Jovanovic
f,
Simon A.
Jones
e,
Adyasha
Panigrahi
c,
Igor
Larrosa
c,
Kostas
Kostarelos
abd,
Cinzia
Casiraghi
c and
Sandra
Vranic
*ab
c,
Neus
Lozano
d,
Angeliki
Karakasidi
ab,
Heather
Church
e,
Ana
Jovanovic
f,
Simon A.
Jones
e,
Adyasha
Panigrahi
c,
Igor
Larrosa
c,
Kostas
Kostarelos
abd,
Cinzia
Casiraghi
c and
Sandra
Vranic
*ab
aNanomedicine Lab, Faculty of Biology, Medicine and Health, The University of Manchester, AV Hill Building, Manchester M13 9PT, UK. E-mail: sandra.vranic@manchester.ac.uk
bNational Graphene Institute, The University of Manchester, Booth Street East, Manchester M13 9PL, UK
cDepartment of Chemistry, University of Manchester, Oxford Road, Manchester, UK
dCatalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC and BIST, Campus UAB, Bellaterra, 08193 Barcelona, Spain
eManchester Centre for Genomic Medicine, Manchester University NHS Foundation Trust, Oxford Road, Manchester, UK
fAdult Inherited Metabolic Department, Salford Royal NHS Foundation Trust, Manchester M6 8HD, UK
First published on 29th March 2023
Abstract
Enzyme replacement therapy shows remarkable clinical improvement in treating lysosomal storage disorders. However, this therapeutic approach is hampered by limitations in the delivery of the enzyme to cells and tissues. Therefore, there is an urgent, unmet clinical need to develop new strategies to enhance the enzyme delivery to diseased cells. Graphene-based materials, due to their dimensionality and favourable pattern of interaction with cells, represent a promising platform for the loading and delivery of therapeutic cargo. Herein, the potential use of graphene-based materials, including defect-free graphene with positive or negative surface charge and graphene oxide with different lateral dimensions, was investigated for the delivery of lysosomal enzymes in fibroblasts derived from patients with Mucopolysaccharidosis VI and Pompe disease. We report excellent biocompatibility of all graphene-based materials up to a concentration of 100 μg mL−1 in the cell lines studied. In addition, a noticeable difference in the uptake profile of the materials was observed. Neither type of graphene oxide was taken up by the cells to a significant extent. In contrast, the two types of graphene were efficiently taken up, localizing in the lysosomes. Furthermore, we demonstrate that cationic graphene flakes can be used as carriers for arylsulfatase B enzyme, for the delivery of the lacking enzyme to the lysosomes of Mucopolysaccharidosis VI fibroblasts. Arylsulfatase B complexed with cationic graphene flakes not only retained the enzymatic activity, but also exerted biological effects almost twice as high as arylsulfatase B alone in the clearance of the substrate in Mucopolysaccharidosis VI fibroblasts. This study lays the groundwork for the potential use of graphene-based materials as carriers for enzyme replacement therapy in lysosomal storage disorders.
 Dr Sandra Vranic | Dr Sandra Vranic is a Lecturer and leader of Nano-Cell-Biology team in the University of Manchester. Her team investigates the mechanisms of nanomaterial–cell interactions, using monocellular and human-organoid models, aiming to understand the safety profile of nanomaterials and discover new nanotherapeutic modalities. Sandra obtained her BSc and MRes Degrees in Molecular Biology from the University of Belgrade, Serbia. She completed her MRes and PhD in Toxicology at University Paris 7 – Diderot in 2013. She obtained a JSPS Postdoctoral Fellowship at Nagoya University and joined University of Manchester in 2015 as a Marie Curie Research Fellow. |
Introduction
Lysosomal storage disorders (LSDs) are a heterogeneous group of metabolic diseases characterised by the abnormal accumulation of non-degraded substrates in the lysosomes, caused mainly by the absence or deficiency of the specific lysosomal enzyme within the cell.1 Although each LSD is individually rare, the cumulative incidence of LSDs has an estimated frequency of 1 in 5000 births, with an even higher frequency in certain ethnic groups.2 Clinical features are associated with progressive and irreversible damage to multiple organs and body systems (e.g., central nervous system, skeletal system).2,3 Despite the great unmet clinical need, there is currently no cure for LSDs.3 Of the limited available treatments, enzyme replacement therapy (ERT) is showing the highest clinical benefit.3 The concept of ERT is to replace the defective/missing enzyme with the functional one.1 While ERT is now considered the standard therapy for several types of LSD, it still has significant drawbacks.4 Besides immunogenicity, insufficient delivery of intravenously administered enzymes to the required pathological sites is a major obstacle to effective ERT.4–6 For instance, the low stability of the recombinant enzyme in the blood, the lack of targetability, and complete reliance on endogenous receptors for enzyme internalisation are all limiting the effective delivery of the enzyme to its targets.4–6 Therefore, strategies to improve the efficacy of conventional ERT are urgently needed.Nanomaterials (NMs) as carriers for lysosomal enzymes hold great potential for ERT: further to offering the protection of the enzyme from premature degradation, NMs could prolong the circulation time, improve the enzyme release profile, and reduce immunogenicity.5,7 In addition, NMs could promote enzyme internalisation, typically hampered by the absence of receptors on the target cell. Instead of using receptor-mediated uptake pathways, NMs could be internalised via multiple uptake pathways depending on their physico-chemical properties.8
Among the variety of NMs, graphene-based materials (GBMs), such as graphene (Gr) and graphene oxide (GO) flakes, have drawn significant research interest for various biomedical applications due to their unique properties.9–11 However, GBMs have not yet been widely considered as potential candidates for LSD treatment despite their advantageous features, such as a large surface area, enabling high enzyme loading. In addition, graphene can be easily functionalised: the presence of different functional groups and the hydrophobic carbon basal plane provide different interaction routes for both covalent and non-covalent attachment of the enzyme. Furthermore, we have shown that GBMs have excellent biocompatibility and dispersibility in biological media and are also internalised by immortalised epithelial and fibroblast cells.12–14 In addition, the subcellular localization of GO in the lysosomes, after being taken up via multiple endocytic pathways, has also been confirmed in BEAS-2B cells.12 Finally, using GO as a siRNA carrier, we showed that the delivery of siRNA to the cytosol of primary mouse embryonic fibroblasts has not been successful due to the vesicle entrapment, which is, however, favourable for the delivery of lysosomal enzymes.15
At present, polymeric nanoparticles, owing to their biocompatibility and stability, are one of the most explored NMs in ERT.16–20 Enzyme encapsulation or surface coating have been proposed so far.20 However, this approach might involve complex chemistry due to surface modifications needed, in order to trigger cellular uptake and trafficking to the lysosomes.17–19 Thus, GBMs could provide a simpler option for both the loading and delivery of enzymes: high enzyme loading can be achieved through non-covalent interactions between the enzyme and the material, while subcellular localization within the lysosomes occurs through a natural intracellular trafficking route of GBMs.
This study aims to investigate the potential use of GBMs as carriers to enhance the delivery of lysosomal enzymes in patient-derived primary fibroblasts. Using human primary fibroblasts (HPFs) and LSD patient-derived fibroblasts (MPS VI and Pompe cells, characterised by the deficiency of arylsulfatase B and acid α-glucosidase, respectively21,22), we first looked at the cellular interaction profile of a panel of GBMs (including defect-free Gr with positive and negative surface charge and GO with two distinct lateral dimensions). Following the establishment of the interaction profile, we prepared and characterized GBM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :enzyme complexes and characterized the degradation of the primary storage substrate within the lysosomes of the diseased cells.
:enzyme complexes and characterized the degradation of the primary storage substrate within the lysosomes of the diseased cells.
Experimental
Graphene based materials – synthesis and characterization
Gr materials (Gr-TMA3 and Gr-PS1) were synthesized using liquid phase exfoliation in water, as already reported.13,25 GO materials (s-GO and us-GO) were synthesized using the modified Hummers’ method, as already reported.12,27 The lateral dimensions of the materials were measured by electron microscopy and DLS, while the surface charge was characterised by ELS. Electron micrographs were obtained using a Magellan 400L field emission scanning electron microscope (Oxford Instruments), at ICN2 Electron Microscopy Unit, with Everhart–Thornley secondary electron detector, using an acceleration voltage of 20 kV and a beam current of 0.1 nA. GBMs were drop-casted onto an ultrathin carbon film on a lacey carbon support film, 400 mesh. The Gr (Gr-TMA3 and Gr-PS1) thickness was characterized using a MultiMode 8-HR AFM (Bruker, UK) in ScanAsyst® mode. Silicon coated cantilevers with a resonance frequency of 70 kHz and a force constant of 0.4 N m−1 were used. For the thickness assessment of s-GO and us-GO, AFM height images were obtained using an Agilent 5500 AFM/SPM microscope at the ICN2 Scanning Probe Microscopy Facility, in tapping mode equipped with silicon cantilevers (Ted Pella) with a nominal force of 40 N m−1 and a resonance frequency of 300 kHz. For the GBM sample preparation, the following procedure was followed. In a 12 mm iron disk covered with carbon tape and cleaved mica, 20 μL of 0.01% of poly-L-lysine (Sigma-Aldrich, UK) were drop cast. After 1 min, the samples were washed with 1 mL of water, followed by drop casting 20 μL of GBMs at 50–100 μg mL−1. The same procedure was followed for the preparation of Gr-TMA3 with the poly-L-lysine step to be excluded. Any unbound material was removed by washing with 1 mL of water, twice. The samples were allowed to dry overnight and the acquired images were processed using Gwyddion 7.1 analysis software.Cell culture
HPFs were maintained in high glucose DMEM cell culture medium (D6429, Sigma-Aldrich, Merck Sigma, UK), supplemented with 10% FBS (Gibco, Thermo Fisher Scientific, UK), 1000 units of penicillin, and 1 mg mL−1 streptomycin (P4333, Sigma-Aldrich, Merck Sigma, UK) at 37 °C, in a humidified 5% CO2 incubator. The cell lines used in this study were obtained from a MPS VI severe phenotype patient and a severe infantile onset Pompe disease patient. Patient derived cells were kindly donated by the Manchester University NHS Foundation Trust and were maintained in L-glutamine contained MEM culture medium (M4665, Sigma-Aldrich, Merck Sigma, UK), supplemented with 10% FBS, 1% kanamycin sulfate (15160–054, Gibco, Thermo Fisher Scientific, UK), and 1% MEM non-essential amino acid (M7145, Sigma-Aldrich, Merck Sigma, UK) at 37 °C in a humidified 5% CO2 incubator. The cells were passaged using 0.05% Trypsin–EDTA solution (T3924, Sigma-Aldrich, Merck Sigma, UK) at monolayer confluence, and the activity of trypsin was stopped using 10% FBS. HPF, MPS VI and Pompe cells with passage numbers of 5–12, 11–16, and 4–11 were used, respectively.Cell treatment
HPF, MPS VI, and Pompe cells were seeded in 12-well plates (Corning, Costar, Sigma-Aldrich, Merck Sigma, UK) for cytotoxicity and uptake quantification assessments, using optical microscopy and flow cytometry. For uptake assessment by confocal microscopy, cells were seeded in CellView™ dishes (627870, Greiner Bio-One Ltd, UK) in cell-specific medium (as described in Cell culture) for at least 24 h and treated when reaching 60–80% confluence in RPMI-1640 medium (11835-030, Gibco, Thermo Fisher Scientific, UK) supplemented with 10% FBS, 1000 units of penicillin, and 1 mg mL−1 streptomycin (hereafter referred to as the treatment medium) at 37 °C in a humidified 5% CO2 incubator.Optical microscopy and flow cytometry
HPF, MPS VI, and Pompe cells were treated with Gr-TMA3, Gr-PS1, s-GO, and us-GO (25, 50, 75 and 100 μg mL−1, 1 mL per well) for 24 h. After treatment, optical images were taken with an EVOS FL microscope (10× objective), and then the cells were washed with PBS (+/+) (1 mL per well, ×2, D8662, Sigma-Aldrich, Merck Sigma, UK), detached with Trypsin–EDTA (300 μL per well, 15 min), neutralised with FBS (30 μL per well), collected in a 1.5 mL Eppendorf tube, and counted using the Trypan Blue exclusion dye (10 μL per 10 μL of cell sample, T8154, Sigma-Aldrich, Sigma, UK). After counting, the cells were centrifuged (1500 RPM, 5 min), re-suspended in annexin-binding buffer (1×, 200 μL per sample, V13246, Invitrogen, Thermo Fisher Scientific, UK), stained with annexin V Alexa Fluor™ 488 (1 μL per tube, 20 min, room temperature in the dark, A13201, Molecular Probes, Thermo Fisher Scientific, USA), stored on ice, and stained with propidium iodide (1 μL per tube, 1002294094, Sigma-Aldrich, Sigma, UK) just before analysis using a BD FACSVerse™ flow cytometer, performed using FITC-A (bandpass: 488-530/30) and PE-A (bandpass: 488-574/26) channels. A total population of 10000 cells was acquired, and the mean side-scattering value was used for uptake semi-quantification. The data were statistically analyzed using GraphPad Prism (version 9.3.1). For cell counting and PI/AV staining, the analysis of variance (two-way ANOVA [main effect only]) with Tukey's multiple comparisons test was used (n = 1 with duplicate). For semi-quantification of the material uptake, analysis of variance (two-way ANOVA [main effects only]) with Tukey's multiple comparisons test was used (n = 1 with duplicate). Differences with p < 0.05 were considered as statistically significant: ns = not significant, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Excitation/emission wavelength: AV = 495/519, PI = 493/636.
Confocal microscopy
HPF, MPS VI, and Pompe cells were treated with Gr-TMA3, Gr-PS1, s-GO and us-GO (25, 50, and 75 μg mL−1, 0.5 mL per well) for 24 h. After treatment, the supernatants were removed and replaced by the CellMask™ green plasma membrane stain (C37608, Invitrogen, Thermo Scientific, UK) with a dilution of 1:2500 (0.5 mL per well). For Gr-TMA3 and Gr-PS1 treated cells, the cells were washed using the treatment medium (0.5 mL per well, ×2), stained with the CellTracker™ Green CMFDA dye diluted in treatment medium (3 μM, 0.5 mL per well, C7025, Thermo Fisher Scientific, UK), and after 15 min incubation, replaced by the treatment medium (0.5 mL per well). The cells were analysed using a Zeiss 780 confocal laser scanning microscope using a 40× objective, and the acquired images were processed using the Zeiss microscope software ZEN. As established in our previous studies, the fluorescence quenching properties of Gr and the auto-fluorescence properties of GO were used to follow their cellular interactions in real-time by confocal imaging.14 Excitation/emission wavelengths: CellMask™ green plasma membrane dye = 488/520 nm, GO = 594/620–690 nm, CellTracker™ green CMFDA dye = 492/517 nm.
Establishment of the Gr-TMA3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :ARSB complex ratio and complex stability assessment
:ARSB complex ratio and complex stability assessment
The complexes were prepared in 10 μL of ultra-pure distilled water (10977035, Invitrogen, Thermo Scientific, UK) by pipetting Gr-TMA3 (75 μg mL−1) with different amounts of ARSB (0, 30, 15, 7.5, 3.75, 2.5, 1.875, 1.5, 0.75 and 0.5 μg mL−1, 4415-SU, R&D systems, Bio-Techne, UK) to obtain Gr-TMA3 and Gr-TMA3:ARSB complexes in concentration ratios of 150:60, 150:30, 150:15, 150:7.5, 150:5, 150:3.75, 150:3, 150:1.5 and 150:1. Then the Gr-TMA3 and complexes were further mixed (300 RPM, 2 h, 19 °C) using a Thermomixer-Mixer HC. Visual stability and dispersibility of the complexes were assessed by monitoring the signs of material sedimentation after mixing using a Thermomixer-Mixer HC and the appearance of the residual material on pipette tips.
Gr-TMA3:ARSB stability assessment
The complexes were prepared in 1 mL of ultra-pure distilled water (10977035, Invitrogen, Thermo Scientific, UK) by pipetting Gr-TMA3 (75 μg mL−1) with different amounts of ARSB (0, 1.5, 0.75 and 0.5 μg mL−1, 4415-SU, R&D systems, Bio-Techne, UK) to give the Gr-TMA3 control and Gr-TMA3:ARSB complexes in concentration ratios of 150:3, 150:1.5 and 150:1. Then Gr-TMA3 and complexes were further mixed (300 RPM, 2 h, 19 °C) using a Thermomixer-Mixer HC. The stability of the Gr-TMA3 control and complexes were characterised by DLS and zeta potential measurements, with ≥3 replicate measurements.
Enzymatic activity assay
A commercial sulfatase activity assay kit (MAK276, Sigma-Aldrich, Sigma, UK) was used to assess the enzymatic activity of Gr-TMA3:ARSB complexes, following the manufacturer's instructions. Briefly, an active enzyme hydrolyzes sulfate ester bonds of the provided substrate into 4-nitrocatechol, which was detected at OD 515 nm using a microplate reader. Prior to the assessment, the lowest detectable concentration of ARSB was confirmed by examining the enzymatic activity for different concentrations of ARSB (0.5, 1, 10, 30, and 67 μg mL−1) prepared in 10 μL of ultra-pure distilled water. The enzymatic activity of Gr-TMA3:ARSB complexes in the concentration ratios of 150:60, 150:3, 150:1.5 and 150:1 were respectively prepared in 10, 200, 400, and 600 μL of ultra-pure distilled water, mixed (300 RPM, 2 h, 19 °C) using a Thermomixer-Mixer HC, centrifuged (17 kg RCF, 30 min, 19 °C) and resuspended in 10 μL of ultra-pure distilled water for sulfatase activity assessment. These complexes were intentionally prepared in different volumes of water (150:60 (10 μL), 150:3 (200 μL), 150:1.5 (400 μL), and 150:1 (600 μL)) to ensure that in each sample, Gr-TMA3 was mixed with 30 μg mL−1 of ARSB, which was within the detection limit of the sulfatase assay kit. The potential interference of Gr-TMA3 on the outcome of the sulfatase activity measurement was assessed by examining the enzymatic activity of different concentrations of Gr-TMA3. Briefly, 75 μg mL−1 of Gr-TMA3 was prepared in 10, 200, 400, and 600 μL of ultra-pure distilled water, mixed (300 RPM, 2 h, 19 °C), centrifuged (17 kg RCF, 30 min, 19 °C) and resuspended in 10 μL of ultra-pure distilled water to give different final concentrations of Gr-TMA3 (75 μg mL−1, 1.5, 3.0, and 4.5 mg mL−1) for sulfatase activity assessment. The effect of centrifugation conditions on the sulfatase activity measurement of the Gr-TMA3:ARSB complex was also assessed. Firstly, 5 different sets of Gr-TMA3 (75 μg mL−1) and the Gr-TMA3:ARSB complex in a concentration ratio of 150:3 were prepared in 200 μL of ultra-pure distilled water, mixed (300 RPM, 2 h, 19 °C), centrifuged under different centrifugation conditions (17, 8.5, 5.68, 4.25, or 3.4 kg, 30 min, 19 °C), and monitored for residual materials on pipette tips after re-mixing of the centrifugated material/complex. Then the enzymatic activity of Gr-TMA3:ARSB complexes (150:3) collected under different centrifugation conditions (17, 8.5 or 4.25 kg, 30 min, 19 °C) was compared. All the generated OD spectra were baseline corrected using the minimal constant (for 4-nitrocatechol standard curve) or user-defined baseline correction function of the OriginPro 2021b software.
Uptake of Gr-TMA3:ARSB by MPS VI cells and degradation of the primary substrate
Gr-TMA3:ARSB complexes were prepared at 10× of treatment concentration, in the following concentration ratios: 150:1 (750 μg mL−1 Gr-TMA3: 5 μg mL−1 ARSB), 150:1.5 (750 μg mL−1 Gr-TMA3: 7.5 μg mL−1 ARSB), and 150:3 (750 μg mL−1 Gr-TMA3: 15 μg mL−1 ARSB), in 50 μL of ultra-pure distilled water and mixed (300 RPM, 2 h, 19 °C). Then the Gr-TMA3:ARSB complexes were added to 0.45 mL of RPMI medium with 10% FBS. MPS VI cells treated with Gr-TMA3 (75 μg mL−1, 0.5 mL), ARSB (0.5, 0.75, and 1.5 μg mL−1, 0.5 mL), and Gr-TMA3:ARSB complexes in the concentration ratios of 150:1 (75 μg mL−1 Gr-TMA3:0.5 μg mL−1 ARSB, 0.5 mL), 150:1.5 (75 μg mL−1 Gr-TMA3:0.75 μg mL−1 ARSB, 0.5 mL), and 150:3 (75 μg mL−1 Gr-TMA3:1.5 μg mL−1 ARSB, 0.5 mL), in RPMI treatment medium with 10% FBS for 24 or 48 h. After treatment, cells were washed with PBS (+/+) (0.5 mL per well, ×1, D8662, Sigma-Aldrich, Merck Sigma, UK), fixed with formaldehyde (4% in PBS (−/−), 15 min, 0.5 mL per well, 28908 Thermo Fisher Scientific, UK), washed with PBS (+/+) (0.5 mL per well, ×2), incubated with chondroitinase ABC (stock diluted in 0.01% BSA solution, treatment concentration of 0.1 U mL−1 in digestion buffer [Tris HCl 50 mM, sodium acetate 60 mM, 0.02% BSA, pH 8], 2 h, 37 °C, 0.5 mL per well, C3667-5UN, Sigma-Aldrich, Merck Sigma, UK), permeabilized with goat serum containing (5%, G9023, Sigma-Aldrich, Merck Sigma, UK) Triton™ X-100 (0.1% in PBS (−/−), 15 min, 0.5 mL per well, 9002-93-1, Sigma-Aldrich, Merck Sigma, UK), washed with PBS (+/+) (0.5 mL per well, ×1), treated with goat serum (5% in PBS (−/−), 0.5 mL per well, 1 h, room temperature), goat serum removed, stained with anti-chondrotin sulfate primary antibody (3.5 μg mL−1 prepared in PBS (−/−) diluted 5% goat serum, 100 μL per well on the inner glass surface of the CellView™ dishes, ab11570, Abcam, UK) and stored at 4 °C overnight, in the dark. Then the cells were washed with PBS (+/+) (0.5 mL per well, ×3, last wash for 10 min, room temperature), stained with goat anti-mouse Alexa Fluor™ 647 (in a dilution of 1:500 in PBS (−/−) diluted 5% goat serum, 100 μL per well on the inner glass surface, 2 h in the dark, room temperature, A21236, Thermo Fisher Scientific, UK), solution removed, stained with Hoechst 33342 (2 μg mL−1 in PBS (−/−), 100 μL per well on the inner glass surface, 10 min in the dark, room temperature, 62249, Thermo Fisher Scientific, UK), washed with PBS (+/+) (0.5 mL per well, ×3, last wash for 10 min, room temperature), mounted with a ProLong™ Gold Antifade Mountant (P36934, Thermo Fisher Scientific, UK) and stored at 4 °C until imaged. A Zeiss 780 confocal laser scanning microscope using a 40× objective, and the acquired images were processed using a Zeiss microscope software ZEN. Quantitative characterization of the intracellular chondroitin sulfate signal was carried out using the ImageJ software. For each condition, cells (n ≥ 30) from 2 or more fields of view (at 0.6× zoom) were randomly selected, and the intracellular chondroitin sulfate intensity was measured against the background intensity. Statistical analysis of the result was performed using GraphPad Prism (version 9.3.1) with analysis of variance (ordinary one-way ANOVA) and Tukey's multiple comparisons tests. Differences with p < 0.05 were considered as statistically significant: ns = not significant, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Results
Synthesis and characterisation of Gr and GO
GBMs have different physicochemical properties depending on how they are produced, which in turn determines how they interact with the biological environment.23,24 Therefore, in this study, a panel of GBMs were used, characterised by different size and thickness distributions as well as surface charge and chemistry. Defect-free Gr flakes were produced in water by liquid-phase exfoliation assisted by stabilisers.13,25,26 This non-covalent approach enables the structure of graphene to be retained while tuning its surface charge and chemistry by selecting the type of stabiliser. In our study, two stabilisers were used: pyrene bearing a linked trimethylammonium functionality (TMA3) and 1-pyrenesulfonic acid sodium salt (PS1), giving rise to cationic and anionic graphene dispersions (Gr-TMA3 and Gr-PS1),13,25,26 respectively. Defective graphene, in the form of GO, was produced utilising the modified Hummer's method.12,27 The size distribution of the flakes was selected by changing the sonication conditions, as reported in previous work,12,27 giving rise to two samples: small GO (s-GO) and ultra-small GO (us-GO).Table 1 and Fig. 1 show a summary of the main physicochemical properties of the four GBMs used. Gr-TMA3 and Gr-PS1 were characterised by similar size and thickness distributions but contained different surface charges and chemistry. As shown in Table 1, measurement by electrophoretic light scattering (ELS) showed that the zeta potentials of Gr-TMA3 and Gr-PS1 were +39 mV and −36 mV, respectively. In contrast, s- and us-GO were characterised by comparable surface charge and chemistry but different lateral dimensions: measurement by electron microscopy showed that s-GO and us-GO were characterised by average lateral dimensions of 477 ± 270 and 125 ± 70 nm, respectively (see Table 1 and Fig. 1). The difference in lateral dimensions between s- and us-GO was confirmed using dynamic light scattering (DLS), with the former and latter exhibiting hydrodynamic sizes of 509.1 ± 8.3 and 85.8 ± 0.8 nm in water, respectively (see Table 1). In addition, measurement by atomic force microscopy (AFM) showed that while Gr-TMA3 (average thickness = 5.7 ± 2.3 nm) and Gr-PS1 (average thickness = 6.8 ± 2.4 nm) consisted of mainly few-layer flakes, s-GO (94.4% ≤ 2 nm) and us-GO (99.2% ≤ 2 nm) consisted of mainly single and bilayer flakes (see Table 1 and Fig. 1).
 | ||
| Fig. 1 Physicochemical characterisation of (a and b) Gr-TMA3, (c and d) Gr-PS1, (e and f) s-GO and (g and h) us-GO: (a, c, e and g) electron and (b, d, f and h) AFM micrographs were obtained for lateral dimension and thickness measurements, respectively. | ||
| Properties | Technique | Gr-TMA3 | Gr-PS1 | s-GO | us-GO |
|---|---|---|---|---|---|
| a Preparation and measurement in water. b The percentage of carbon was calculated based on the reported percentage of oxygen, nitrogen and sulfur.25 Gr-TMA3 was referred to as TMA3 and TME3 in the studies of Shin et al.13 and Read et al.,26 respectively. Gr-PS1 was referred to as Py-1SO3 based graphene and PS1 in the studies of Yang et al.25 and Read et al.,26 respectively. | |||||
| Lateral dimensions (average) | Electron microscopy | 161 ± 96 nm | 169 ± 91 nm | 477 ± 270 nm | 125 ± 70 nm |
| Dynamic light scattering | Preparation and measurement in water | ||||
| Hydrodynamic size: 203.1 ± 0.8 nm | Hydrodynamic size: 170.9 ± 1.8 nm | Hydrodynamic size: 509.1 ± 8.3 nm | Hydrodynamic size: 85.8 ± 0.8 nm | ||
| Polydispersity index: 0.249 ± 0.014 | Polydispersity index: 0.263 ± 0.036 | Polydispersity index: 0.617 ± 0.108 | Polydispersity index: 0.219 ± 0.005 | ||
| Preparation in RPMI with 10% FBS and resuspended in water for measurement | |||||
| Hydrodynamic size: 280.5 ± 5.3 nm | Hydrodynamic size: 286.7 ± 3.6 nm | Hydrodynamic size: 1324.0 ± 104.0 nm | Hydrodynamic size: 461.3 ± 11.1 nm | ||
| Polydispersity index: 0.261 ± 0.006 | Polydispersity index: 0.216 ± 0.010 | Polydispersity index: 0.470 ± 0.114 | Polydispersity index: 0.543 ± 0.062 | ||
| Thickness | Atomic force microscopy | 2–20 nm (average: 5.7 ± 2.3 nm) | 2–20 nm (average: 6.8 ± 2.4 nm) | 94.4% ≤ 2 nm | 99.2% ≤ 2 nm |
| Mainly single and bilayer | Mainly single and bilayer | ||||
| Zeta potentiala | Electrophoretic light scattering | 39 mV | −36 mV | −62 mV | −58 mV |
| Surface chemistry | X-ray photoelectron spectroscopy | C: 95.6% | C: 93.6%b | C: 70.3% | C: 67.9% |
| O: 3.8% | O: 4.9% | O: 29.3% | O: 30.9% | ||
| N: 0.3% | N: 0.7% | (99.6% purity, C:O = 2.4) |
(98.9% purity, C:O = 2.2) |
||
| S: 0.2% (C:O = 25.2) |
S: 0.8% (C:O = 19.1) |
||||
Cytotoxicity and uptake profile of Gr and GO
The cytotoxicity and the uptake profiles of the four types of GBMs in HPF, MPS VI and Pompe cells were examined by exposing the cells to increasing concentrations of the material (up to 100 and 75 μg mL−1 for cytotoxicity and uptake profiles, respectively) in RPMI culture medium with 10% fetal bovine serum (FBS) for 24 h. It is necessary to acknowledge that upon contact with the serum-containing culture medium, the material surface is instantly covered with various serum proteins, hence affecting the way it interacts with cells.28,29 For instance, the presence of serum proteins could mitigate the cytotoxicity effects of GO by lowering plasma membrane ruffling caused by direct contact with the materials,14 but serum-coated NMs reported to display reduced cellular uptake compared to their non-serum-coated counterparts.30 Considering that conventional ERT is delivered intravenously and that the bloodstream is rich in various protein molecules, it is more clinically relevant to perform the cytotoxicity and uptake studies in the presence of serum. Hence, all treatments in this study were performed under serum-contained conditions.The cytotoxicity of the four types of GBMs was assessed by first monitoring the cell morphology using optical microscopy, counting alive cells using the trypan blue dye exclusion assay and finally, quantifying the double unstained alive cell percentages by propidium iodide (PI)/annexin V (AV) staining with flow cytometry. Fig. S1–S3,† respectively, show the optical images of HPF, MPS VI, and Pompe cells treated with the four types of GBMs. It is apparent that except for the cells treated with the positive control (20% DMSO), no evidence of cellular stress/toxicity was observed. For instance, DMSO treated cells were characterised by obvious changes in the cell morphology compared to untreated cells (e.g., the elongated cells became rounder), along with a noticeable reduction in cell density. However, the morphology of the cells treated with materials remained comparable to untreated cells. Further quantification using the trypan blue exclusion assay and dead cell staining with necrotic (PI) and apoptotic (AV) indicators confirmed no significant differences in cell viability between the untreated and material treated cells (Fig. 2 and S4†).
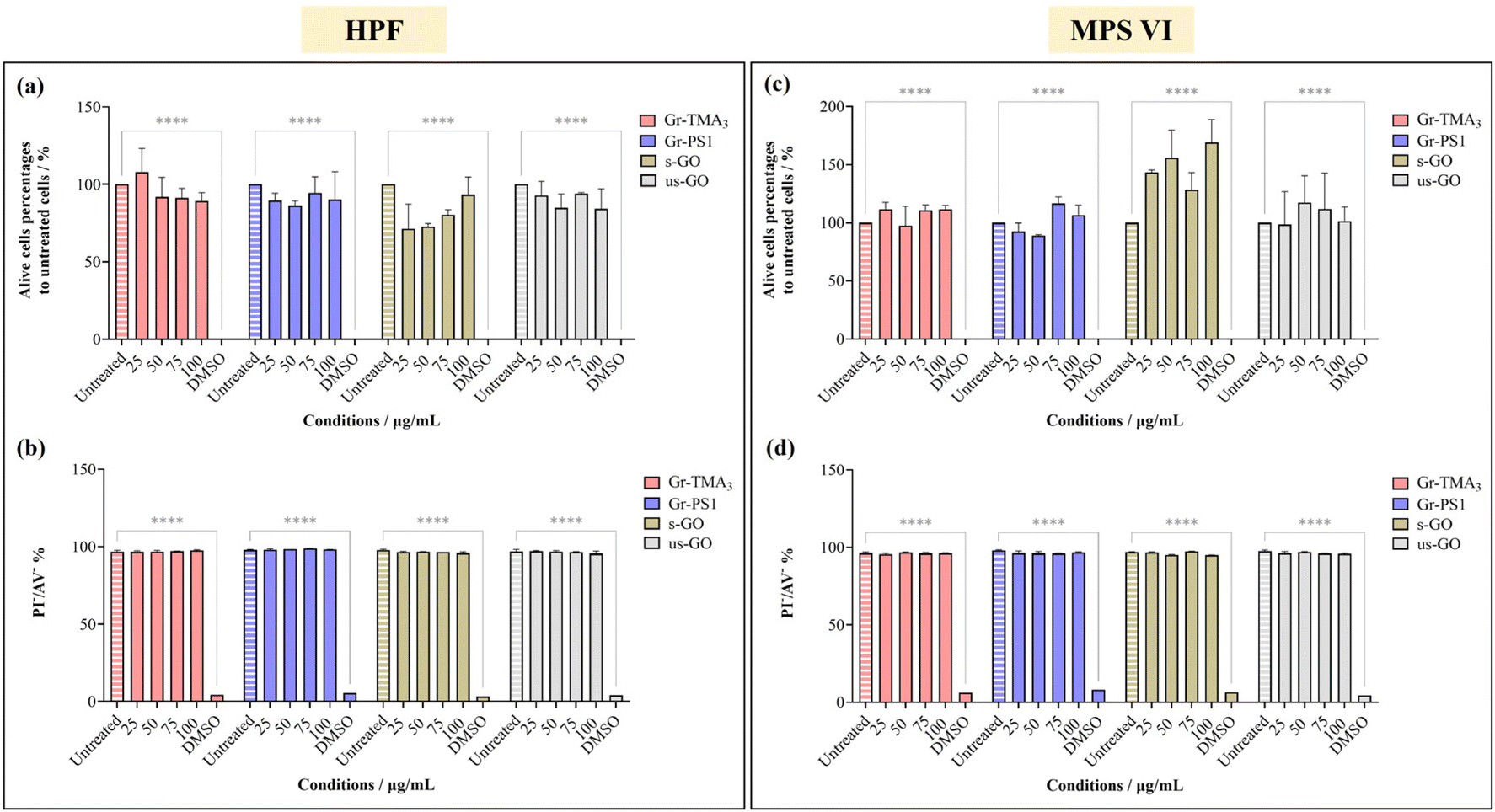 | ||
| Fig. 2 Cytotoxicity assessment of Gr-TMA3, Gr-PS1, s-GO and us-GO (25, 50, 75 and 100 μg mL−1, 24 h) in (a and b) HPF and (c and d) MPS VI cells by live-cell counting using (a and c) trypan blue exclusion dye and (b and d) double negative PI/AV staining. 20% DMSO was used as positive control for both assays. See Fig. S1 and S2† for the corresponding optical images after treatment with the four materials in HPF and MPS VI cells, respectively. The data were statistically analysed using analysis of variance (two-way ANOVA [main effects only]) with Tukey's multiple comparisons tests. n = 1 with duplicate. *Statistically different: ****p < 0.0001; material-treated cells showed no significant differences from the corresponding untreated cells. | ||
Using confocal microscopy and flow cytometry, we investigated the uptake profile of the four types of GBMs by HPF, MPS VI and Pompe cells. Fig. 3 and 4 and S5–S8† show the uptake profiles of the four types of material in HPF, MPS VI and Pompe cells, studied by confocal imaging. For Gr, the material appeared black, and the co-localization of the material in the brightfield with the quenched signal of the CMFDA labelled cells in the merged channel indicates material internalisation (see Fig. 3 and 4 and S7†). For GO, red-spotted signals found within the green plasma membrane labelled cells confirm the presence of the internalised material (see Fig. S5–S6 and S8†). The same trend in the uptake of Gr and GO was observed across the three cell lines studied. While we observed a minimal uptake of GO regardless of the cell model, Gr was taken up in a dose-dependent manner. However, the three cell lines were taking up Gr-TMA3 and Gr-PS1 to a different extent. HPF and MPS VI cells were taking up more of Gr-TMA3 than Gr-PS1 (see Fig. 3 and 4). In contrast, Pompe cells were taking up more of Gr-PS1 than Gr-TMA3 (see Fig. S7†).
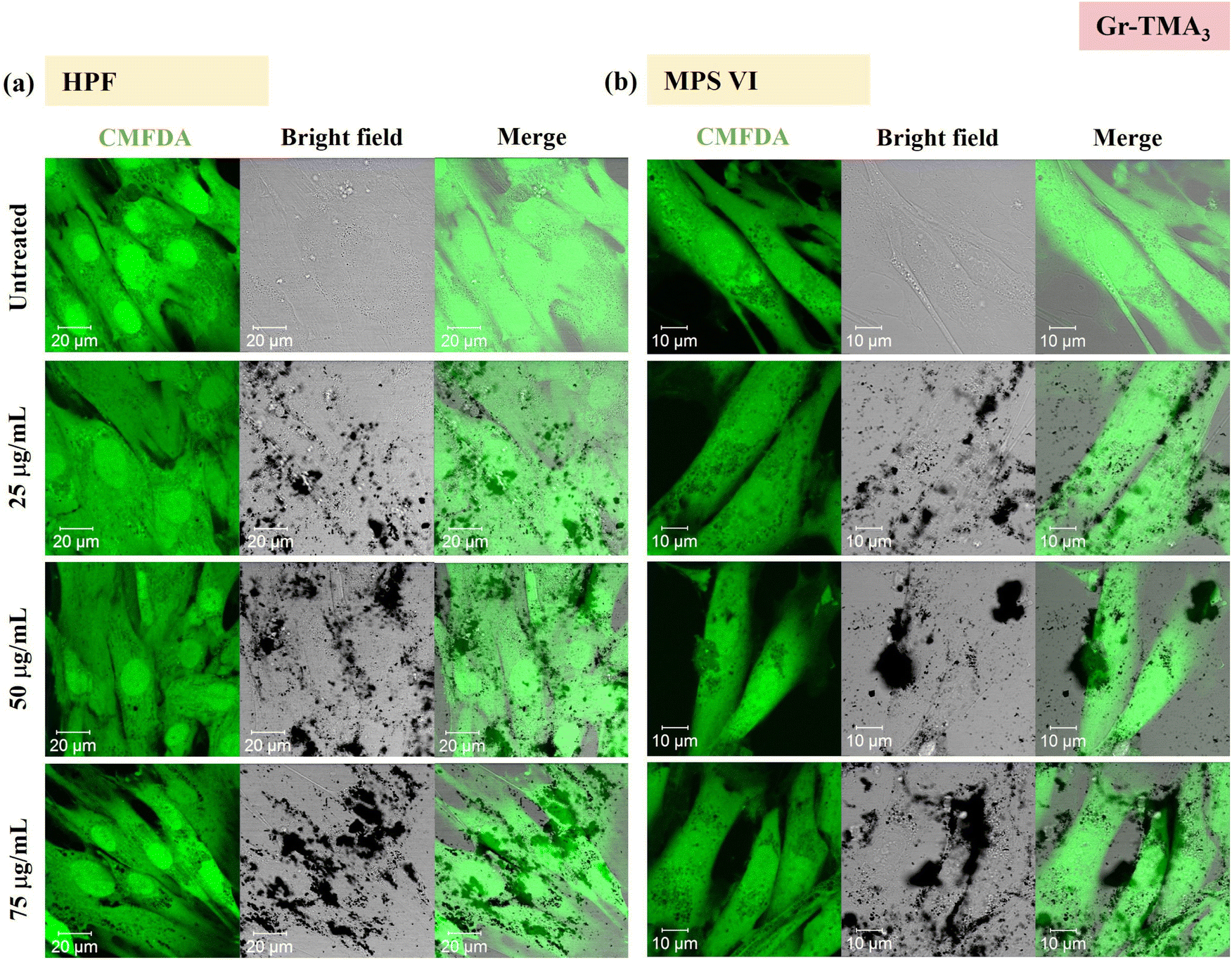 | ||
| Fig. 3 Uptake profile of Gr-TMA3 in (a) HPF and (b) MPS VI cells, analysed by confocal imaging (25, 50 and 75 μg mL−1, with FBS, 24 h). Middle sections of the confocal images are shown. Green = CMFDA dye labelled cells, black = Gr-TMA3. | ||
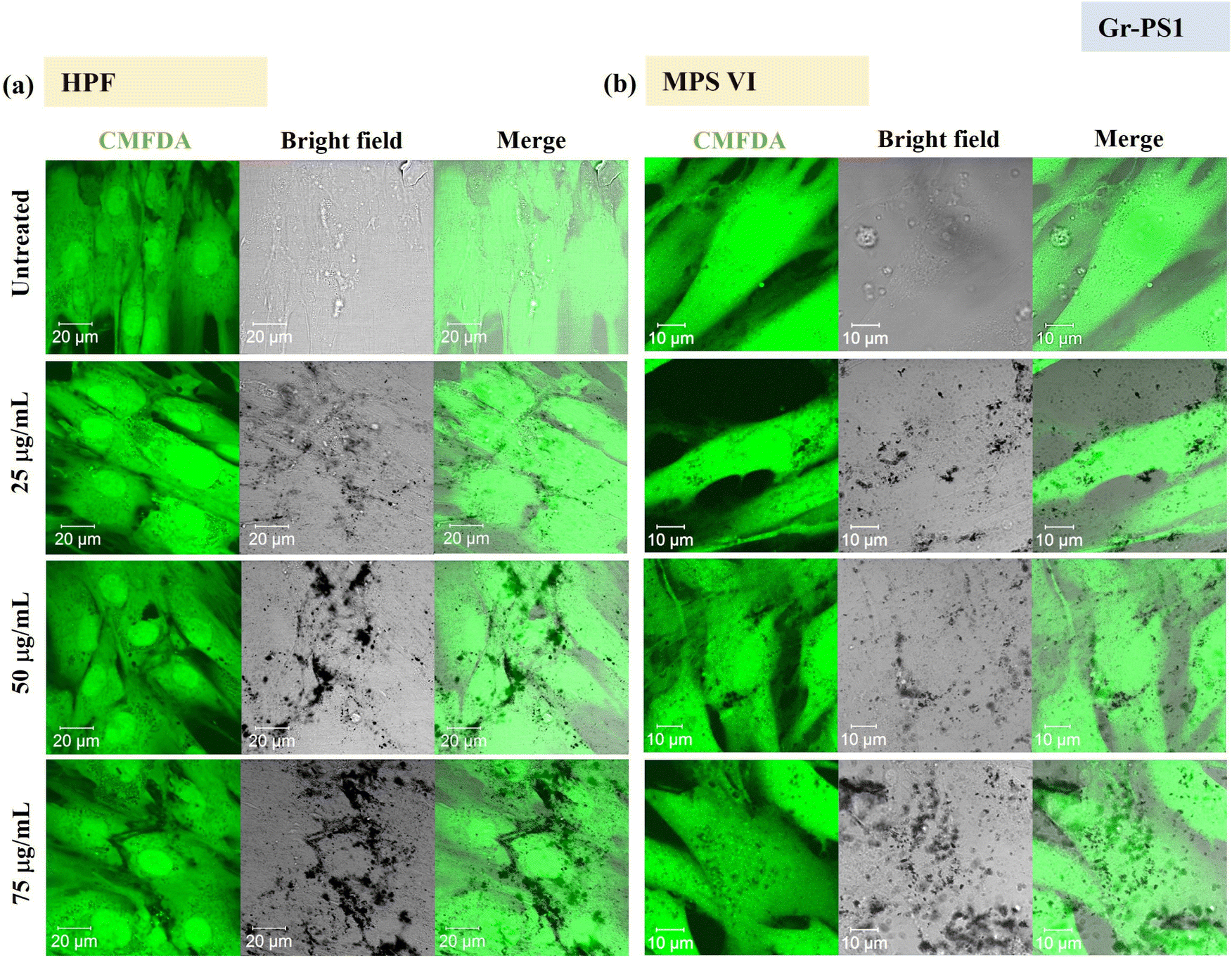 | ||
| Fig. 4 Uptake profile of Gr-PS1 in (a) HPF and (b) MPS VI cells, analysed by confocal imaging (25, 50 and 75 μg mL−1, with FBS, 24 h). Middle sections of the confocal images are shown. Green = CMFDA dye labelled cells, black = Gr-PS1. | ||
Flow cytometry was subsequently used to semi-quantify the uptake of Gr and GO in healthy and LSD fibroblasts. Cellular granularity was measured as the side scattering intensity (SSC) and reported as the SSC fold change normalized to untreated cells (see Fig. 5 and S9†). Although measurement by flow cytometry describes the total interaction of the materials with cells, including internalised and the material attached to the cell surface, the cells were washed before and after detachment from the plate. Therefore, complementary to observation by confocal imaging, measurement by flow cytometry represents a reasonable estimation of the uptake of the materials.
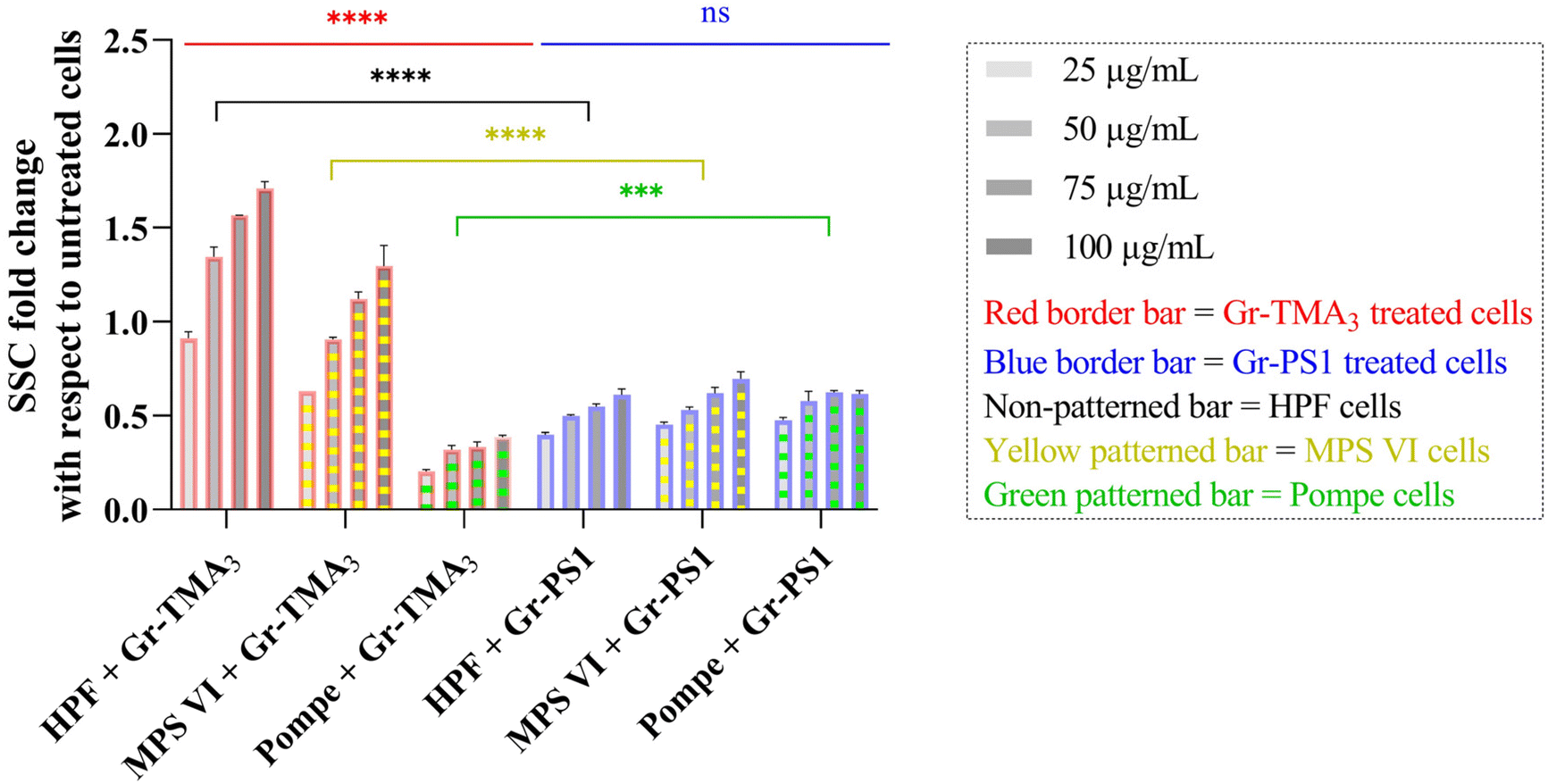 | ||
| Fig. 5 SSC fold changes of HPF, MPS VI, and Pompe cells treated with Gr-TMA3 or Gr-PS1 (25, 50, 75, and 100 μg mL−1) normalized to untreated cells. The flow cytometry data were statistically analysed using analysis of variance (two-way ANOVA [main effects only]) with Tukey's multiple comparisons tests. n = 1 with duplicate. *Statistically different ([red or blue significance levels] comparison between different cell models with the same material treatment at the same concentration; [black, yellow, or green significance levels] comparison between the same cell model with different material treatments at the same concentration): ns = not significant, ***p < 0.001 and ****p < 0.0001. | ||
As shown in Fig. 5, the cellular interaction of Gr was confirmed by flow cytometry, and the results are in agreement with confocal imaging. For example, the SSC fold change of Gr (Gr-TMA3 and Gr-PS1) treated cells (HPF, MPS VI, and Pompe cells) increased with treatment concentrations, hence confirming that the materials were taken up by the cells in a dose-dependent manner. More importantly, the results confirmed that the HPF and MPS VI cells interacted better with the positively charged Gr-TMA3 than the negatively charged Gr-PS1, and the opposite for Pompe cells. For instance, the mean SSC fold change for Gr-TMA3 treated HPF and MPS VI cells (at 25 to 100 μg mL−1, HPF: 0.913 to 1.71, MPS VI: 0.631 to 1.297) was significantly higher than that for HPF and MPS VI cells treated with the corresponding concentration of Gr-PS1 (at 25 to 100 μg mL−1, HPF: 0.397 to 0.611, MPS VI: 0.454 to 0.697). In contrast, the mean SSC fold change for Gr-TMA3 treated Pompe cells (at 25 to 100 μg mL−1: 0.203 to 0.387) was significantly lower than the corresponding conditions of Gr-PS1 treated Pompe cells (at 25 to 100 μg mL−1: 0.475 to 0.616). However, despite the three primary cells showing a differential preference toward the two Gr, HPF and MPS VI cells displayed a stronger preference for Gr-TMA3 than Pompe cells displayed for Gr-PS1. For example, the maximum mean SSC fold change differences between Gr-TMA3 and Gr-PS1 treated cells for HPF, MPS VI, and Pompe cells was 1.098 (observed at 100 μg mL−1), 0.600 (observed at 100 μg mL−1), and 0.289 (observed at 75 μg mL−1), respectively. In addition, while significant differences were found between the three primary cells with Gr-TMA3 treatment (SSC fold change: HPF cells > MPS VI cells > Pompe cells), no significant differences were found between the three primary cells with Gr-PS1 treatment. This revealed that the three primary cell types displayed a comparable level of interaction with the Gr-PS1.
Cellular interaction of s- and us-GO was validated in HPF and MPS VI cells by flow cytometry (see Fig. S9†). The results confirmed that cellular interactions with GO were minimal compared to Gr. For example, the greatest mean SSC fold change for GO treated HPF (MPS VI) cells was 0.136 (0.198), observed with us-GO treatment at 100 μg mL−1. Still, this was lower than the lowest mean SSC fold change of 0.399 (0.454) for Gr treated HPF (MPS VI) cells, observed with Gr-PS1 treatment at 25 μg mL−1 (see Fig. 5 and S9†). In addition, as shown in Fig. S9,† no significant differences were found between GO treated HPF and MPS VI cells, regardless of the size of GO. However, it should be noted that both HPF and MPS VI cells interacted better with us-GO compared to s-GO; SSC fold change for us-GO treated cells increased with treatment concentrations and was significantly higher than the SSC fold change for s-GO treated cells (see Fig. S9†).
Gr-TMA3 and ARSB complexation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :ARSB concentration ratios.
Given that ARSB has negatively charged regions,31 we investigated the capacity of Gr-TMA3 to complex with ARSB by non-covalent interactions. To determine the optimal mass ratio between Gr-TMA3 and ARSB, we examined the stability of complexes formed using different mass ratios. Gr-TMA3 with a fixed concentration of 75 μg mL−1 was directly mixed with variable concentrations of ARSB (30 to 0.5 μg mL−1) for 2 h. The stability of the complexes was examined visually by looking at the sedimentation of the material in the tube and the appearance of the residual material on the pipette tips, after re-mixing. This occurred due to the agglomeration/aggregation of the material upon interaction with the enzyme and these complexes were considered unstable (see Fig. 6(a) and S10†). As shown in Fig. S10,† Gr-TMA3:ARSB complexes in the ratios of 150:3, 150:1.5, and 150:1 were visually stable.
:ARSB concentration ratios.
Given that ARSB has negatively charged regions,31 we investigated the capacity of Gr-TMA3 to complex with ARSB by non-covalent interactions. To determine the optimal mass ratio between Gr-TMA3 and ARSB, we examined the stability of complexes formed using different mass ratios. Gr-TMA3 with a fixed concentration of 75 μg mL−1 was directly mixed with variable concentrations of ARSB (30 to 0.5 μg mL−1) for 2 h. The stability of the complexes was examined visually by looking at the sedimentation of the material in the tube and the appearance of the residual material on the pipette tips, after re-mixing. This occurred due to the agglomeration/aggregation of the material upon interaction with the enzyme and these complexes were considered unstable (see Fig. 6(a) and S10†). As shown in Fig. S10,† Gr-TMA3:ARSB complexes in the ratios of 150:3, 150:1.5, and 150:1 were visually stable.
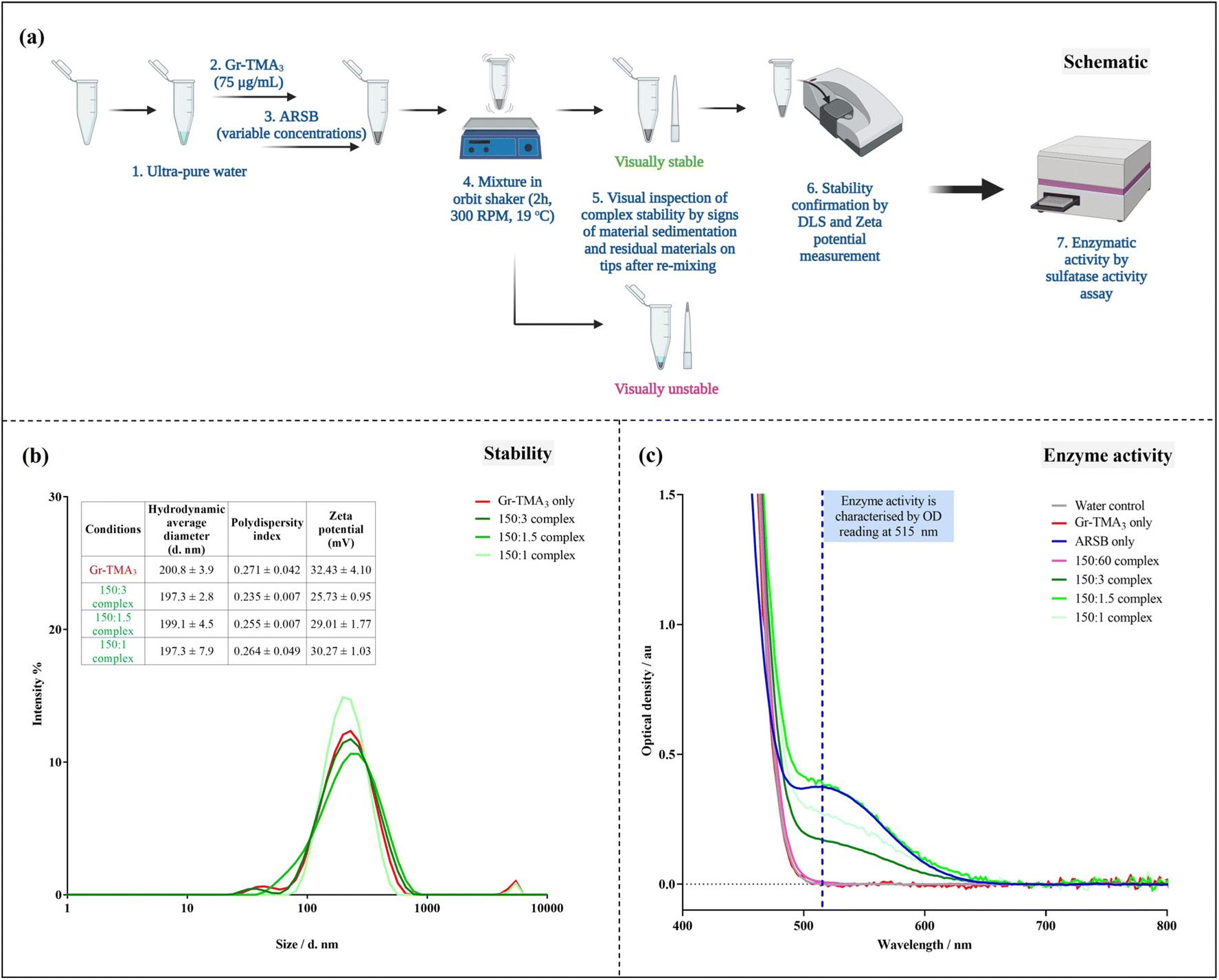 | ||
| Fig. 6 Gr-TMA3:ARSB complexation and enzymatic activity assessment using commercially available sulfatase activity assay. (a) Schematic of the protocol of complexation and complex characterisation. Please refer to Fig. S10† for images of Gr-TMA3:ARSB complexes. (b) Characterisation of the Gr-TMA3 (75 μg mL−1) and the visually stable Gr-TMA3:ARSB complexes (at concentration ratios of 150:3, 150:1.5, and 150:1) in water by DLS (hydrodynamic size distribution by intensity %) and zeta potential measurements. (c) Baseline-corrected optical density spectrum of visually stable (150:3, 150:1.5 and 150:1) and unstable (150:60) Gr-TMA3:ARSB complexes compared against water, Gr-TMA3 (75 μg mL−1), and ARSB (30 μg mL−1) controls using the sulfatase activity assay. Please refer to Fig. S12† for sulfatase activity calculation. | ||
Subsequently, we validated the stability of these complexes by hydrodynamic size and zeta potential measurements (see Fig. 6(b)). As illustrated in Fig. 6(b), the measured zeta potential of the Gr-TMA3 control (32.43 ± 4.10 mV) was higher than the 150:3 (25.73 ± 0.95 mV), 150:1.5 (29.01 ± 1.77 mV), and 150:1 (30.27 ± 1.03 mV) for Gr-TMA3:ARSB complexes. Although the magnitude of the zeta potential is indicative of a system's colloidal stability,32 which suggested the reduction in stability from high for the Gr-TMA3 control to moderate for the complexes, it may not fully reflect the system's colloidal stability. This is because the overall stability of a colloid system depends on both the attractive van der Waals and the repulsive electrostatic forces, and the zeta potential consists of information only for the latter force.32 Hence, we measured the size distribution of the samples for stability confirmation. As shown in Fig. 6(b), comparable hydrodynamic size distributions were measured between the Gr-TMA3 and Gr-TMA3:ARSB (150:3, 150:1.5 and 150:1) complexes. Similar mean hydrodynamic sizes and polydispersity indexes (PI*) were measured for the Gr-TMA3 (size = 200.8 ± 3.9 nm, PI* = 0.271 ± 0.042), 150:3 (size = 197.3 ± 2.8 nm, PI* = 0.235 ± 0.007), 150:1.5 (size = 199.1 ± 4.5 nm, PI* = 0.255 ± 0.007), and 150:1 (size = 197.3 ± 7.9 nm, PI* = 0.264 ± 0.049) Gr-TMA3:ARSB complexes, thus confirming that the Gr-TMA3:ARSB (150:3, 150:1.5 and 150:1) complexes were stable. The reduction in the zeta potential magnitude for the complexes can be explained by the complexation of ARSB with Gr-TMA3. As the zeta potential is also indicative of the nanoparticle's surface charge, the result suggested that Gr-TMA3 remained positively charged with ARSB complexation but became less positively charged with an increased ratios of ARSB, as expected.
:ARSB ratio that gave stable complexes (150:3, 150:1.5, and 150:1), we measured the enzymatic activity of the enzyme in the complexes, to confirm that the activity was preserved. This was done using a commercial sulfatase activity assay kit, which detects active sulfatase at an optical density (OD) of 515 nm. However, before assessing the enzymatic activity of the complexes, we assessed the enzymatic activity of Gr-TMA3 and ARSB (see Fig. S11†). This was performed to confirm whether Gr-TMA3 and ARSB had intrinsic sulfatase activity. As shown in Fig. S11(a and d),† both the Gr-TMA3 and ARSB controls showed positive readings at OD 515 nm in a dose-dependent manner. However, this result must be interpreted with caution as we did not anticipate Gr-TMA3 to have sulfatase activity. Hence, the OD spectra (400–800 nm) for the Gr-TMA3 and ARSB controls at different concentrations were plotted and compared (see Fig. S11(b and e)†). The OD spectra of Gr-TMA3 (see Fig. S11(b)†) revealed that regardless of its concentrations (75 μg mL−1 to 4.5 mg mL−1), the OD spectra appeared tilted and featureless in the region of 500–800 nm; this was comparable to the OD spectra of the water control, except that the baseline reading for the water control was lower. In contrast, the OD spectra of ARSB (see Fig. S11(e)†) at a concentration of ≥30 μg mL−1 showed a peak at 515 nm; this was comparable to the OD spectra of the positive control (provided in the commercial sulfatase activity assay kit). Therefore, the result confirmed that the Gr-TMA3 control does not possess sulfatase activity, and the lowest detectable concentration of ARSB obtained using the sulfatase activity kit was 30 μg mL−1. Therefore, the false positive sulfatase activity of Gr-TMA3 was removed with the baseline correction of the spectra (see Fig. S11(c)†). Since this approach allowed a more accurate comparison of the sulfatase activity of a sample (see Fig. S11(c and g)†), the subsequent OD spectra were all presented with the baseline correction.
:ARSB complexes.
As shown in Fig. 6(c), we compared the enzymatic activity of the Gr-TMA3:ARSB (150:60, 150:3, 150:1.5 and 150:1) complexes with the water, Gr-TMA3 (75 μg mL−1), and ARSB (30 μg mL−1). Interestingly, while the unstable 150:60 complex showed no sulfatase activity, the stable complexes (150:3, 150:1.5 and 150:1) showed various degrees of sulfatase activity (see Fig. 6(c) and S12† for calculation of the sulfatase activity, the calculations were based on the measurement at OD 515 nm). For example, the 150:1.5 complex showed the highest sulfatase activity (1261.958 nmol mg−1 min−1), followed by the 150:1 (869.091 nmol mg−1 min−1) and 150:3 (542.303 nmol mg−1 min−1) complexes. It is worth mentioning here that the calculated sulfatase activity of the three complexes was much higher than the sulfatase activity reported in previous literature for normal patients (4.427 to 15.538 nmol mg−1 min−1, n = 10).33 Considering the common issue of insufficient enzyme delivery, complexes with higher sulfatase activity offer a higher chance of delivering the effective dose of the enzyme. However, the measured sulfatase activity of the complexes from the assay may not fully reflect the amount of the enzyme loaded onto Gr-TMA3. This is because the Gr-TMA3:ARSB complexes (as well as Gr-TMA3) were collected by centrifugation for enzymatic activity assessment, and we noticed changes in the dispersibility of the complexes after centrifugation. As shown in Fig. S12,† residual materials were found in pipette tips after re-suspension of the complex pellets, which indicates impairment of the complex's dispersibility after centrifugation. The amount of the residual material matched the observed trend of calculated sulfatase activity.
By comparing the enzyme activity of the 150:3 complexes collected with different centrifugation conditions (see Fig. S13 and S14†), we confirmed that the enzyme activity measurement was affected by the centrifugation conditions. Thus, although the sulfatase activity assay was useful for indication of the enzymatic activity retained by the complex, it was not suitable for quantification of the amount of the enzyme complexed to the material. Nevertheless, the centrifugation step was not required to assess the biological effect in MPS VI cells. Thus, in the final part of the study, we investigated the effect of the Gr-TMA3:ARSB complexes on the degradation of the primary storage substrate of the MPS VI cells.
The biological effect of Gr-TMA3:ARSB in MPS VI cells
Chondroitin sulfate (CS) and dermatan sulfate glycosaminoglycan, the natural substrates of ARSB, are the primary substrates which excessively build up in MPS VI cells.21 In this study, the in vitro enzymatic activity of the Gr-TMA3:ARSB complexes was assessed via imaging of the intracellular CS degradation by immunocytochemistry. As shown in Fig. 7, the accumulation of intracellular CS after 24 h of treatment with Gr-TMA3:ARSB complexes (150:1, 150:1.5, and 150:3) was individually compared with the untreated cells and cells treated with the equivalent amount of ARSB (0.5, 0.75, and 1.5 μg mL−1) and Gr-TMA3 (75 μg mL−1). Further to imaging the CS-stained cells, the fluorescence intensity of intracellular Alexa 647 stained CS (mean ± SD) in each condition was quantified and statistically analysed.
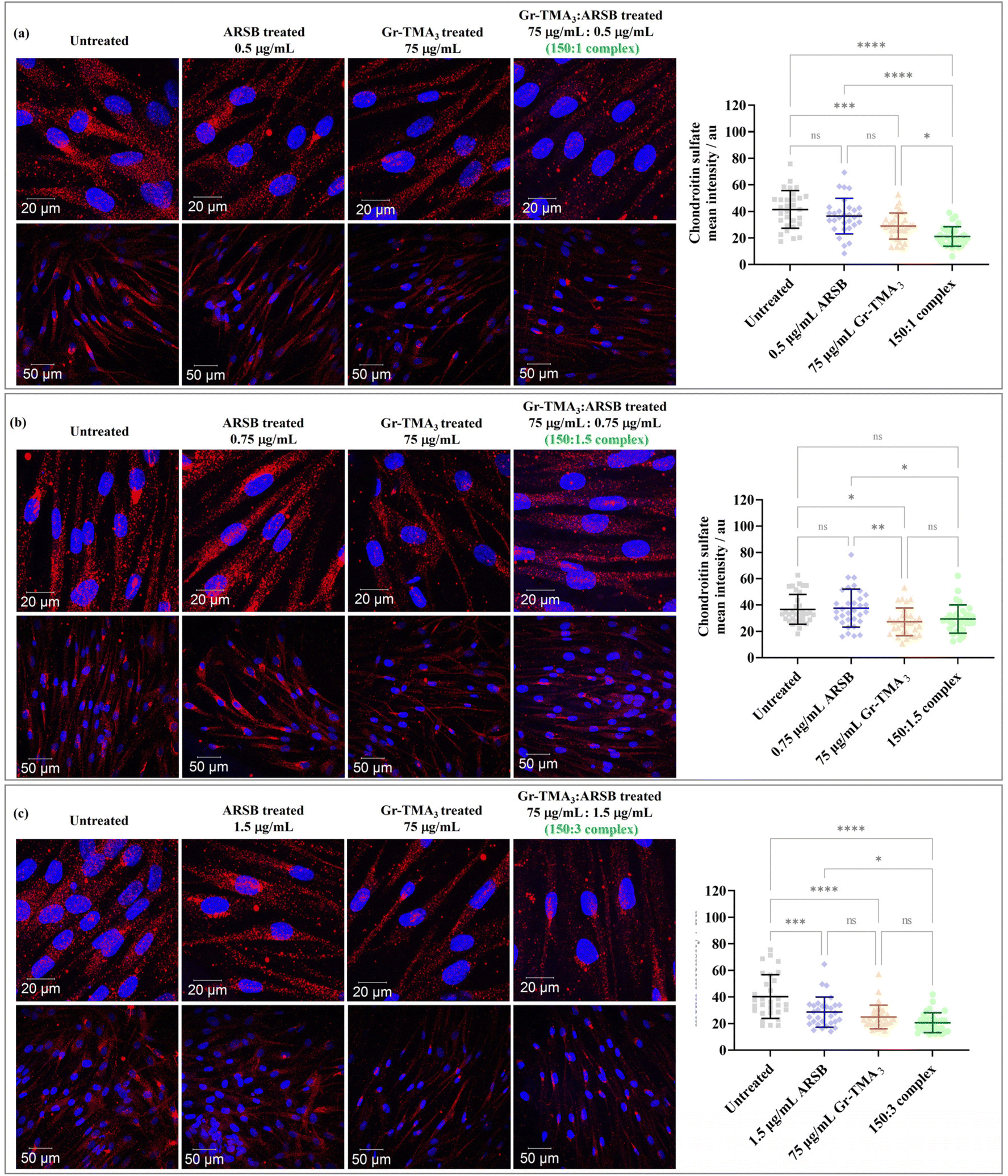 | ||
| Fig. 7 Confocal images (maximum intensity projection) of MPS VI cells treated with ARSB ((a) 0.5 μg mL−1, (b) 0.75 μg mL−1, and (c) 1.5 μg mL−1), Gr-TMA3 (75 μg mL−1), and Gr-TMA3:ARSB complexes in the ratio of (a) 150:1, (b) 150:1.5, and (c) 150:3 for 24 h. The chondroitin sulfate signals were quantified using ImageJ (n ≥ 30 cells from 2 or more fields of view at the lower magnification) and statistically analyzed using analysis of variance (one-way ANOVA) with Tukey's multiple comparisons tests. *Statistically different: ns = not significant, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. Blue = nucleus. Red = chondroitin sulfate. | ||
:ARSB.
Looking at the effect of the Gr-TMA3:ARSB complexes on MPS VI cells, the CS signal of all cells treated with complexes was found to be lower than in untreated cells. However, the significance level varied across the different ratios of the complexes. For example, the CS signal for cells treated with Gr-TMA3:ARSB complexes in the ratio of 150:1 (untreated: 41.49 ± 14.11, complex: 21.10 ± 7.39, P-value < 0.0001) and 150:3 (untreated: 40.26 ± 16.46, complex: 20.60 ± 7.53, P-value < 0.0001) was statistically lower compared to the corresponding control untreated cells. However, the CS signal for Gr-TMA3:ARSB in the ratio of 150:1.5 was not found to be statistically different to untreated cells (untreated: 36.69 ± 11.37, complex: 29.39 ± 10.70, P-value = 0.0738).
:ARSB treated cells compared to ARSB treated cells.
The CS signal of cells treated with the complex was found to be either lower or comparable to the corresponding ARSB treated cells, depending on the ARSB concentration. The 150:1 Gr-TMA3:ARSB complex showed the greatest difference compared to the ARSB treated cells (ARSB: 36.45 ± 13.40, complex: 21.10 ± 7.39, P-value < 0.0001). With a further increase in the ARSB concentration ratio, the 150:1.5 (ARSB: 37.62 ± 14.46, complex: 29.39 ± 10.70, P-value < 0.01) and 150:3 (ARSB: 28.61 ± 11.35, complex: 20.60 ± 7.53, P-value < 0.01) Gr-TMA3:ARSB complexes showed a reduced level of significance compared to the control ARSB.
:ARSB treated cells compared to Gr-TMA3 treated cells.
Finally, we compared the effect of the Gr-TMA3:ARSB complex with Gr-TMA3. For cells treated with the 150:1 Gr-TMA3:ARSB complex, the CS signal was statistically lower than cells treated with the control Gr-TMA3 (Gr-TMA3: 28.95 ± 9.85, complex: 21.10 ± 7.39, P-value < 0.01). However, for the 150:1.5 and 150:3 Gr-TMA3:ARSB complexes, no statistical differences were found when compared against the corresponding Gr-TMA3 control (Gr-TMA3/150:1.5 complex: 27.29 ± 10.55/29.39 ± 10.70, P-value = 0.9042; Gr-TMA3/150:3 complex: 24.89 ± 8.90/20.60 ± 7.53, P-value = 0.4796).
Discussion
Cytotoxicity profiles of Gr and GO
Understanding the cytotoxicity profile is a fundamental prerequisite for the use of any type of NM for biomedical applications. Apart from being non-cytotoxic, ideal carriers for lysosomal enzymes should be taken up by the cells and carry the functional enzyme inside the lysosomes. The four types of GBMs that we investigated showed excellent biocompatibility in HPF, MPS VI and Pompe cells in concentrations up to 100 μg mL−1 (see Fig. 2 and S1–S4†). In fact, the GBMs used in our study repeatedly show exceptional biocompatibility in many cell types.12–14 In contrast, many studies reported the concentration-dependent cytotoxicity of GBMs in primary fibroblasts, with the materials being toxic at concentrations up to 50 μg mL−1.34–37 Hence, further modification strategies like silanisation and PEGylation were employed to improve the biocompatibility of GBMs in primary and immortalised fibroblasts.38,39 The discrepancy in GBM biocompatibility could be attributed to intrinsic differences in the GBM properties, exfoliation agent used and treatment conditions. For instance, certain studies employed GBMs with lateral sizes of 1–5 μm35,37 and lacked clarification on the usage of serum proteins for treatment.36 However, the materials used in this study were much smaller (see Table 1), and all experiments were performed in the presence of serum. In our previous study, we found that both size and serum proteins were critical factors that influence the cytotoxicity of GBMs, with large GBMs (5–15 μm) exhibiting significantly higher cytotoxicity than small GBMs (50–200 nm) at 100 μg mL−1, and the absence of serum proteins could further elevate the toxicity effect.14 Thus, material biocompatibility depends strongly on its properties and treatment conditions.In addition, previous studies reported that the oxidation status of GBMs heavily influences biocompatibility.34–36 However, most of these studies employed defective graphene for cytotoxicity comparison with GO, and contradictory findings on the effect of oxidation on GBM biocompatibility emerged. For example, Gurunathan et al. reported that GO exhibited much higher cytotoxicity than defective graphene (produced via the reduction of GO by spinach leaf extracts) in primary mouse embryonic fibroblasts (MEFs).35 In contrast, also using MEFs, Zhang et al. reported that GO with a lower degree of oxidation (produced by modified Hummers’ method with a lower amount of KMnO4), which resembles defective graphene, induced significantly higher cytotoxicity than GO with a higher degree of oxidation.36 Likewise, Liao et al. reported that graphene sheets (produced via dehydration using GO) induced greater cytotoxicity than GO in human skin fibroblasts.34
These studies suggested different explanations for their results, such as the use of spinach leaf extracts as a reducing agent offered lower cytotoxicity,35 GO with an increase in the size of the aromatic domain facilitated better production of OH radicals,36 and greater aggregation and sedimentation of graphene had likely inhibited the availability of the nutrients required by cells.34 However, owing to inherent differences in the production of these materials, the employed graphene consists of distinct physicochemical properties with various levels of oxidation, which make it difficult to draw general conclusions about the effect of oxidation on GBM cytotoxicity. Here, using GO and defect-free Gr, we observed no obvious differences between the two types of materials, with both exhibiting excellent biocompatibility.
Uptake profiles of Gr and GO
Based on confocal imaging and flow cytometry, three primary cell models showed a bare minimum uptake of GO compared to Gr (see Fig. 3–5 and S5–S9†). Cellular uptake of nanoparticles is highly complicated and influenced by a combination of many factors, such as the physicochemical properties of the material, the characteristics of the cell model, and the treatment conditions employed.28,40 In a more complex but not uncommon scenario, the properties of the material and cell model can be altered during or upon interaction with each other, making it even harder to predict the outcome of the cell's response to the material.From the point of view of the employed cell model, a possible explanation for the lack of GO uptake can be attributed to the intrinsic characteristic of fibroblast cells. We previously found that s- and us-GO were internalised via multiple pathways in the established epithelial cell line, with macropinocytosis and clathrin-mediated endocytosis (CME) being the dominant pathways, respectively.12 Human fibroblasts, however, have been predicted to take up material predominantly by caveolae-mediated endocytosis (CavME) due to the enrichment of caveolin-1, which represents the major constituent of caveolae.41 Therefore, the minimum uptake of GO could possibly be due to the limited availability of the uptake pathway specific for GO in human fibroblasts. Furthermore, much less is known about the interaction of primary fibroblasts with GBMs, and it is still not known whether primary cells behave like immortalised cells when interacting with nanoparticles. Following the same treatment conditions, we previously found efficient internalisation of both s- and us-GO in immortalised mouse fibroblast cells (NIH/3T3).12
Currently, studies concerning the cellular interaction of GBMs have been largely performed using immortalised cell lines. Of the few studies that investigated the uptake of GO in primary fibroblasts, literature reported cellular uptake of GO in primary fibroblasts using TEM, regardless of the discrepancies in the material, cell model and treatment conditions.36,42 However, it is important to note that uptake studies solely based on TEM often only focus on the success or failure of uptake but are rarely concerned with the level of internalisation for a large population of cells. Also, visual comparisons of the TEM images between GO uptake in primary fibroblasts36,42 and NIH/3T3 cells12 showed that at a comparable concentration and exposure time, the primary fibroblasts take up GO in a much lower amount compared to NIH/3T3 cells, thus supporting the idea of minimum GO uptake by primary fibroblasts compared to the immortalised fibroblasts. A recent study by Xiao et al. showed that while human primary cells utilised one dominant uptake pathway for the internalisation of gold nanorods with different surface properties, the immortalised cells employed all three major uptake pathways (macropinocytosis, CME and CavME) for material internalisation.43 This further illustrates how uptake differences of NMs can arise between primary and immortalised cells.
There is a wide acceptance that LSD fibroblasts displayed altered endocytosis activities.44,45 For instance, abnormal accumulation of insoluble lipids, such as cholesterol, either by means of primary non-degradable substrates or triggered secondary to primary storage of other biomaterials, have been reported to directly impair vesicle trafficking and fusion, which in turn causes “traffic jam” to the endocytic pathway.1,44,45 In a study of the endocytic pathways for four different LSD fibroblasts with primary lipid storage (type A Niemann-Pick, type C Niemann-Pick, Fabry, and Gaucher diseases), Rappaport et al. confirmed that all LSD models showed diminished endocytosis activities compared to healthy primary skin fibroblasts, with CME affected the most and the other uptake pathways (CavME and macropinocytosis) affected to a different extent between LSD models.45 The present work agrees with the literature; the control HPFs displayed the highest uptake capacity toward Gr-TMA3, followed by the two LSD fibroblasts.
Also, the results showed a substantial difference between MPS VI and Pompe cells. For example, the total interaction of Gr-TMA3 with MPS VI was (on average) 29.02 ± 4.23% lower compared to HPFs, while for Pompe cells, it was 77.54 ± 1.19% lower compared to HPFs (see Fig. 5, calculated based on the average SSC fold change at different treatment concentrations). The finding suggests that the endocytosis activity of Pompe cells was affected to a greater extent than MPS VI cells. This result may be explained by the fact that glycogen is the primary storage material of Pompe cells, which is a form of carbohydrate, and it is known that lipid accumulation may arise secondary to the build-up of carbohydrates. In contrast, although an elevated level of gangliosides (GM2 and GM3), a type of sialic acid-containing glycosphingolipids, has been reported for MPS VI, much less is known on lipid accumulation due to MPS VI.1 In addition, uptake differences between MPS VI and Pompe cells may arise depending on the clinical severity of the MPS VI and Pompe patients who donated the cells.
We move now towards considering the influence of the material's physicochemical properties on the cellular uptake of the four types of materials. To date, very little is known about the effect of GBM's physicochemical properties on primary and LSD fibroblast internalisation. The present findings on the uptake profile of the four types of GBMs offer some important insights into the influence of key material parameters on the internalisation of GBMs in healthy primary and LSD fibroblasts, such as size and surface charge.
First, the limited uptake of GO with different size distributions confirmed that the material size was not the dominant parameter in driving the observed uptake profiles of the GBM. This result agrees with those obtained by Heo et al., who showed by TEM that GO with different size distributions of 150 nm–1.1 μm were taken up to a comparable extent by human dermal fibroblasts.42 On the other hand, the surface charge of the materials played a vital role in the uptake of GBMs. For instance, not only GO with minimum uptake was negatively charged, but HPF and MPS VI cells also showed a strong uptake preference for cationic Gr-TMA3 than anionic Gr-PS1. The result agrees with our previous findings, which showed that despite differences in surface chemistry, different cationic Gr dispersions were better internalised than the anionic Gr dispersion by immortalised cell lines.13 However, considering both GO and Gr-PS1 exhibited a negative surface charge, the uptake differences between GO and Gr were possibly attributed to the different surface chemistry, which determines the interaction with the cell.
Nevertheless, Gr-PS1 was better taken up than Gr-TMA3 in Pompe cells. Although cationic materials have been widely recognised to be taken up better than their anionic counterparts due to the favourable electrostatic attraction with the negatively charged plasma membrane, this is not always the case.46 The fundamental reason can be attributed to the fact that material internalisation does not rely on a single parameter, such as surface charge, but on a combination of various factors. For instance, Gr-PS1 and Gr-TMA3 also differed in surface chemistry. Thus, a combined effect of its surface charge and chemistry may contribute to their uptake profiles. Furthermore, considering that the uptake capacity of Pompe cells toward Gr-PS1 was at a comparable level to HPF and MPS VI cells, Pompe cells did not differentially take up Gr-PS1 better than HPF and MPS VI cells. In addition, the literature also showed that the uptake mechanism of small-size graphene (∼200 nm) is surface charge-dependent: the positively charged graphene utilised CME for internalisation, whereas negatively charged graphene utilised sulphate-receptor-mediated endocytosis.47 Hence, knowing that CME has not only been reported as the uptake pathway for positively charged graphene but also the mechanism most affected in LSD cells, another possible explanation of the result is that the uptake mechanism of Gr-TMA3 was impaired in Pompe cells.45
The biological effect of the Gr-TMA3:ARSB complex in MPS VI cells
:ARSB treated cells compared to untreated, ARSB or Gr-TMA3 treated cells.
From the comparison against untreated cells, the result suggested that the Gr-TMA3:ARSB complexes at ratios of 150:1 and 150:3 were most effective at reducing the intracellular CS signal (see Fig. 7). Yet, we also compared the effect of the complexes against the corresponding ARSB and Gr-TMA3 conditions. This is because the ideal complex should result in a more effective reduction of the accumulating substrate than the enzyme-alone control. In addition, since the Gr-TMA3 alone control also mediated the reduction of the CS signal, it is worth understanding the effect of the complex compared to the effect of the Gr-TMA3 control.
Compared with the ARSB treated cells, the enhanced effect of the complex was reduced with an increased ratio of ARSB. Hence, the Gr-TMA3:ARSB complex at a ratio of 150:1 was most effective at CS signal reduction compared to ARSB treated cells (see Fig. 7). A possible explanation of the result could be attributed to the alteration of the material's intrinsic properties with a higher concentration of ARSB. ARSB complexed with Gr-TMA3 could potentially increase the delivery of ARSB via triggering multiple uptake pathways, and the Gr-TMA3:ARSB complex bound to the cell surface could offer a concentrating effect of the enzyme in the proximity of the cells. However, considering that Gr-TMA3 exhibits excellent stability in serum-contained cell culture medium,13 complexation of a high concentration of ARSB to Gr-TMA3 in culture medium could result in the agglomeration of the material, which in turn affects the uptake profile of the complex. The stability of the complexes was examined in water (see Fig. 6(b) and S10†), and future works should characterise the physicochemical properties of the complex in the culture medium. Finally, the Gr-TMA3:ARSB complex at a ratio of 150:1 remained the most effective when compared to Gr-TMA3 treated cells. Also, the complex-mediated reduction of the CS signal was more likely due to the successful internalisation of the complex than due to the quenching of the signal by the complex localized extracellularly (Fig. S15†).
Conclusion
In this study, we investigated the potential use of GBMs as carriers to enhance the delivery of lysosomal enzymes in patient-derived primary fibroblasts. We used a panel of four types of GBMs, including defect-free Gr with positive or negative surface charges (Gr-TMA3 and Gr-PS1) and defective GO with distinct lateral dimensions (s-GO and us-GO), and two types of LSD-patient derived fibroblasts (MPS VI and Pompe cells) as cell models. As expected, all four types of GBMs exhibited excellent biocompatibility, up to 100 μg mL−1 in the studied cell lines. Higher uptake of positively charged Gr-TMA3 in HPFs was observed, followed by MPS VI and Pompe cells, compared to negatively charged Gr-PS1. This indicates that LSD fibroblasts may not only display impaired endocytic activities, but also the degree of endocytic impairment may vary according to the type of disease. The three types of primary fibroblasts studied showed no uptake of GO. Subsequently, we demonstrate that positively charged Gr could act as an enzyme carrier, upon non-covalent complexation of the ARSB enzyme. Whilst the quantification of the amount of the complexed enzyme remains to be further investigated, we confirmed the enzymatic activity of the complexes prior to the in vitro assessment of biological effects. Gr complexed with the enzyme successfully reduced the amount of accumulated substrate in MPS VI fibroblasts, exceeding the activity of the enzyme alone (when used at a concentration ratio of 150:1). Furthermore, Gr-TMA3 also demonstrated its capability of reducing the substrate signal. This study lays the groundwork for the potential use of two-dimensional nanomaterials as carriers for ERT in LSDs.
Author contributions
Y. C. designed and performed the experiments, analysed the data, and wrote the manuscript. T. T. contributed to the assessment of GBM toxicity and uptake and related data analysis. N. Z., N. L., A. P., and A. K. produced and characterized GBMs. K. K., I. L., and C. C. directed and supervised the production of GBMs. H. C., A. J., and S. A. J. provided patient-derived cell lines and provided intellectual input. S. V. initiated, designed, directed, and supervised the work, provided intellectual input, and wrote the manuscript. All authors contributed to the writing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr David Spiller for his assistance with confocal imaging. We thank Dr Livia Crica for the synthesis of GO, Dr Gloria Garcia Ortega and Dr Juliana C. Neves for the electron micrographs of GBMs and the atomic force imaging micrographs of GO, respectively. Y. C. and A. K. acknowledge the studentship from the UK Research and Innovation (UKRI) Engineering and Physical Sciences Research Council (EPSRC) Centre for Doctoral Training programme (Graphene NOWNANO CDT; EP/L01548X/1). C. C., N. Z., I. L., and A. P. acknowledge the support by the EPSRC in the framework of the project 2D Health (EP/P00119X/1). The authors also acknowledge the support from the Faculty of Biology, Medicine and Health at the University of Manchester, and in particular Dr Nigel Hodson, from the BioAFM Core Facility, for their expert advice and assistance. The authors acknowledge the Manchester Collaborative Centre for Inflammation Research (MCCIR) as the funding source for the FACSVerse. The authors acknowledge funding from EU-Horizon 2020-Graphene Flagship Core 2 and 3 projects (grant agreement ID: 785219). The ICN2 team is funded by the CERCA programme/Generalitat de Catalunya and is supported by the Severo Ochoa Centres of Excellence programme, funded by the Spanish Research Agency (AEI, grant no. SEV-2017-0706).References
- G. Parenti, D. L. Medina and A. Ballabio, The rapidly evolving view of lysosomal storage diseases, EMBO Mol. Med., 2021, 13(e12836), 1–21 Search PubMed.
- S. Muro, New biotechnological and nanomedicine strategies for treatment of lysosomal storage disorders, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2010, 2, 189–204 CrossRef CAS.
- A. Sun, Lysosomal storage disease overview, Ann. Transl. Med., 2018, 6, 476 CrossRef CAS PubMed.
- R. H. Lachmann, Enzyme replacement therapy for lysosomal storage diseases, Curr. Opin. Pediatr., 2011, 23, 588–593 CrossRef CAS PubMed.
- M. Solomon and S. Muro, Lysosomal enzyme replacement therapies: Historical development, clinical outcomes, and future perspectives, Adv. Drug Delivery Rev., 2017, 118, 109–134 CrossRef CAS PubMed.
- D. H. Kim, H. S. Lee, T.-W. Kwon, Y.-M. Han, N.-W. Kang, M. Y. Lee, D.-D. Kim, M. G. Kim and J.-Y. Lee, Single enzyme nanoparticle, an effective tool for enzyme replacement therapy, Arch. Pharmacal Res., 2020, 43, 1–21 CrossRef PubMed.
- B. Rathore, K. Sunwoo, P. Jangili, J. Kim, J. H. Kim, M. Huang, J. Xiong, A. Sharma, Z. Yang, J. Qu and J. S. Kim, Nanomaterial designing strategies related to cell lysosome and their biomedical applications: A review, Biomaterials, 2019, 211, 25–47 CrossRef CAS PubMed.
- M. Sousa De Almeida, E. Susnik, B. Drasler, P. Taladriz-Blanco, A. Petri-Fink and B. Rothen-Rutishauser, Understanding nanoparticle endocytosis to improve targeting strategies in nanomedicine, Chem. Soc. Rev., 2021, 50, 5397–5434 RSC.
- H. Zhao, R. Ding, X. Zhao, Y. Li, L. Qu, H. Pei, L. Yildirimer, Z. Wu and W. Zhang, Graphene-based nanomaterials for drug and/or gene delivery, bioimaging, and tissue engineering, Drug Discovery Today, 2017, 22, 1302–1317 CrossRef CAS PubMed.
- S. Goenka, V. Sant and S. Sant, Graphene-based nanomaterials for drug delivery and tissue engineering, J. Controlled Release, 2014, 173, 75–88 CrossRef CAS PubMed.
- M. Vincent, I. De Lázaro and K. Kostarelos, Graphene materials as 2D non-viral gene transfer vector platforms, Gene Ther., 2017, 24, 123–132 CrossRef CAS PubMed.
- Y. Chen, J. Rivers-Auty, L. E. Cricǎ, K. Barr, V. Rosano, A. E. Arranz, T. Loret, D. Spiller, C. Bussy, K. Kostarelos and S. Vranic, Dynamic interactions and intracellular fate of label-free, thin graphene oxide sheets within mammalian cells: Role of lateral sheet size, Nanoscale Adv., 2021, 3, 4166–4185 RSC.
- Y. Shin, S. Vranic, X. Just-Baringo, S. M. Gali, T. Kisby, Y. Chen, A. Gkoutzidou, E. Prestat, D. Belijonne, I. Larrosa, K. Kostarelos and C. Casiraghi, Stable, concentrated, biocompatible, and defect-free graphene dispersions with positive charge, Nanoscale, 2020, 12, 12383–12394 RSC.
- S. Vranic, A. F. Rodrigues, M. Buggio, L. Newman, M. R. H. White, D. G. Spiller, C. Bussy and K. Kostarelos, Live Imaging of Label-Free Graphene Oxide Reveals Critical Factors Causing Oxidative Stress-Mediated Cellular Responses, ACS Nano, 2018, 12, 1373–1389 CrossRef CAS PubMed.
- I. de Lázaro, S. Vranic, D. Marson, A. F. Rodrigues, M. Buggio, A. Esteban-Arranz, M. Mazza, P. Posocco and K. Kostarelos, Graphene oxide as a 2D platform for complexation and intracellular delivery of siRNA, Nanoscale, 2019, 11, 13863–13877 RSC.
- A. Mühlstein, S. Gelperina and J. Kreuter, Development of nanoparticle-bound arylsulfatase B for enzyme replacement therapy of mucopolysaccharidosis VI, Pharmazie, 2013, 68, 549–554 Search PubMed.
- S. Muro, E. H. Schuchman and V. R. Muzykantov, Lysosomal Enzyme Delivery by ICAM-1-Targeted Nanocarriers Bypassing Glycosylation- and Clathrin-Dependent Endocytosis, Mol. Ther., 2006, 13, 135–141 CrossRef CAS PubMed.
- J. Hsu, D. Serrano, T. Bhowmick, K. Kumar, Y. Shen, Y. C. Kuo, C. Garnacho and S. Muro, Enhanced endothelial delivery and biochemical effects of α-galactosidase by ICAM-1-targeted nanocarriers for Fabry disease, J. Controlled Release, 2011, 149, 323–331 CrossRef CAS PubMed.
- J. Hsu, L. Northrup, T. Bhowmick and S. Muro, Enhanced delivery of α-glucosidase for Pompe disease by ICAM-1-targeted nanocarriers: comparative performance of a strategy for three distinct lysosomal storage disorders, Nanomedicine, 2012, 8, 731–739 CrossRef CAS PubMed.
- E. Muntimadugu, M. Silva-abreu, G. Vives, M. Loeck, V. Pham, M. D. Moral, M. Solomon and S. Muro, Comparison between Nanoparticle Encapsulation and Surface Loading for Lysosomal Enzyme Replacement Therapy, Int. J. Mol. Sci., 2022, 23, 1–22 Search PubMed.
- F. Vairo, A. Federhen, G. Baldo, M. Riegel, M. Burin, S. Leistner-Segal and R. Giugliani, Diagnostic and treatment strategies in mucopolysaccharidosis VI, Appl. Clin. Genet., 2015, 8, 245–255 CAS.
- J.-A. Lim, L. Li and N. Raben, Pompe disease: from pathophysiology to therapy and back again, Front. Aging Neurosci., 2014, 6, 1–14 Search PubMed.
- L. Fusco, A. Gazzi, G. Peng, Y. Shin, S. Vranic, D. Bedognetti, F. Vitale, A. Yilmazer, X. Feng, B. Fadeel, C. Casiraghi and L. G. Delogu, Graphene and other 2D materials: A multidisciplinary analysis to uncover the hidden potential as cancer theranostics, Theranostics, 2020, 10, 5435–5488 CrossRef CAS PubMed.
- B. Fadeel, C. Bussy, S. Merino, E. Vázquez, E. Flahaut, F. Mouchet, L. Evariste, L. Gauthier, A. J. Koivisto, U. Vogel, C. Martín, L. G. Delogu, T. Buerki-Thurnherr, P. Wick, D. Beloin-Saint-Pierre, R. Hischier, M. Pelin, F. C. Carniel, M. Tretiach, F. Cesca, F. Benfenati, D. Scaini, L. Ballerini, K. Kostarelos, M. Prato and A. Bianco, Safety Assessment of Graphene-Based Materials: Focus on Human Health and the Environment, ACS Nano, 2018, 12, 10582–10620 CrossRef CAS PubMed.
- H. Yang, Y. Hernandez, A. Schlierf, A. Felten, A. Eckmann, S. Johal, P. Louette, J.-J. Pireaux, X. Feng, K. Müellen, V. Palermo and C. Casiraghi, A simple method for graphene production based on exfoliation of graphite in water using 1-pyrenesulfonic acid sodium salt, Carbon, 2013, 53, 357–365 CrossRef CAS.
- O. Read, Y. Shin, C.-X. Hu, M. Zarattini, M. Boyes, X. Just-Baringo, A. Panigrahi, I. Larrosa and C. Casiraghi, Insights into the exfoliation mechanism of pyrene-assisted liquid phase exfoliation of graphene from lateral size-thickness characterisation, Carbon, 2022, 186, 550–559 CrossRef CAS.
- A. F. Rodrigues, L. Newman, N. Lozano, S. P. Mukherjee, B. Fadeel, C. Bussy and K. Kostarelos, A blueprint for the synthesis and characterisation of thin graphene oxide with controlled lateral dimensions for biomedicine, 2D Mater., 2018, 5, 035020 CrossRef.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Understanding biophysicochemical interactions at the nano-bio interface., Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- C. Bussy and K. Kostarelos, Culture Media Critically Influence Graphene Oxide Effects on Plasma Membranes, Chem, 2017, 2, 322–323 CAS.
- G. Duan, S. Kang, X. Tian, J. A. Garate, L. Zhao, C. Ge and R. Zhou, Protein corona mitigates the cytotoxicity of graphene oxide by reducing its physical interaction with cell membrane, Nanoscale, 2015, 7, 15214–15224 RSC.
- C. S. Bond, P. R. Clements, S. J. Ashby, C. A. Collyer, S. J. Harrop, J. J. Hopwood and J. M. Guss, Structure of a human lysosomal sulfatase, Structure, 1997, 5, 277–289 CrossRef CAS PubMed.
- S. Bhattacharjee, DLS and zeta potential - What they are and what they are not?, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- D. A. Brooks, G. J. Gibson, L. Karageorgos, L. K. Hein, E. F. Robertson and J. J. Hopwood, An index case for the attenuated end of the mucopolysaccharidosis type VI clinical spectrum, Mol. Genet. Metab., 2005, 85, 236–238 CrossRef CAS PubMed.
- K.-H. Liao, Y.-S. Lin, C. W. MacOsko and C. L. Haynes, Cytotoxicity of Graphene Oxide and Graphene in Human Erythrocytes and Skin Fibroblasts, ACS Appl. Mater. Interfaces, 2011, 3, 2607–2615 CrossRef CAS PubMed.
- S. Gurunathan, J. W. Han, V. Eppakayala, A. A. Dayem, D.-N. Kwon and J.-H. Kim, Biocompatibility effects of biologically synthesized graphene in primary mouse embryonic fibroblast cells, Nanoscale Res. Lett., 2013, 8, 1–13 CrossRef PubMed.
- W. Zhang, L. Yan, M. Li, R. Zhao, X. Yang, T. Ji, Z. Gu, J.-J. Yin, X. Gao and G. Nie, Deciphering the underlying mechanisms of oxidation-state dependent cytotoxicity of graphene oxide on mammalian cells, Toxicol. Lett., 2015, 237, 61–71 CrossRef CAS PubMed.
- K. Wang, J. Ruan, H. Song, J. Zhang, Y. Wo, S. Guo and D. Cui, Biocompatibility of Graphene Oxide, Nanoscale Res. Lett., 2011, 6, 1–8 Search PubMed.
- M. Wojtoniszak, X. Chen, R. J. Kalenczuk, A. Wajda, J. Lapczuk, M. Kurzewski, M. Drozdzik, P. K. Chu and E. Borowiak-Palen, Synthesis, dispersion, and cytocompatibility of graphene oxide and reduced graphene oxide, Colloids Surf., B, 2012, 89, 79–85 CrossRef CAS PubMed.
- S. S. R. Vuppaladadium, T. Agarwal, S. Kulanthaivel, B. Mohanty, C. S. Barik, T. K. Maiti, S. Pal, K. Pal and I. Banerjee, Silanization improves biocompatibility of graphene oxide, Mater. Sci. Eng., C, 2020, 110, 110647 CrossRef CAS PubMed.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Cellular uptake of nanoparticles: journey inside the cell, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- T. Fujimoto, H. Kogo, R. Nomura and T. Une, Isoforms of caveolin-1 and caveolar structure, J. Cell Sci., 2000, 113, 3509–3517 CrossRef CAS PubMed.
- J. Heo, J. Tanum, S. Park, D. Choi, H. Jeong, U. Han and J. Hong, Controlling physicochemical properties of graphene oxide for efficient cellular delivery, J. Ind. Eng. Chem., 2020, 88, 312–318 CrossRef CAS.
- Y. Xiao, W. Xu, Y. Komohara, Y. Fujiwara, H. Hirose, S. Futaki and T. Niidome, Effect of Surface Modifications on Cellular Uptake of Gold Nanorods in Human Primary Cells and Established Cell Lines, ACS Omega, 2020, 5, 32744–32752 CrossRef CAS PubMed.
- M. A. Samie and H. Xu, Lysosomal exocytosis and lipid storage disorders, J. Lipid Res., 2014, 55, 995–1009 CrossRef CAS PubMed.
- J. Rappaport, R. L. Manthe, M. Solomon, C. Garnacho and S. Muro, A Comparative Study on the Alterations of Endocytic Pathways in Multiple Lysosomal Storage Disorders, Mol. Pharmaceutics, 2016, 13, 357–368 CrossRef CAS PubMed.
- E. Fröhlich, The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef PubMed.
- Z. Tu, K. Achazi, A. Schulz, R. Mülhaupt, S. Thierbach, E. Rühl, M. Adeli and R. Haag, Combination of Surface Charge and Size Controls the Cellular Uptake of Functionalized Graphene Sheets, Adv. Funct. Mater., 2017, 27, 1701837 CrossRef.
- P. R. Harmatz and R. Shediac, Mucopolysaccharidosis VI: Pathophysiology, diagnosis and treatment, Front. Biosci., 2017, 22, 385–406 CrossRef CAS PubMed.
- J. A. Taylor, G. J. Gibson, D. A. Brooks and J. J. Hopwood, Human N-acetylgalactosamine-4-sulphatase biosynthesis and maturation in normal, Maroteaux-Lamy and multiple-sulphatase-deficient fibroblasts, Biochem. J., 1990, 268, 379–386 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr04971f |
| This journal is © The Royal Society of Chemistry 2023 |