
Accurate determination of rare earth elements in small volumes of porewater from marine sediments by laser ablation solution sampling ICP-MS†
Xiuhong
Liao
a,
Wen
Zhang
 *a,
Tao
Yang
b,
Zhaochu
Hu
a,
Tao
Luo
a,
Xianli
Zeng
a and
Lian
Zhou
a
*a,
Tao
Yang
b,
Zhaochu
Hu
a,
Tao
Luo
a,
Xianli
Zeng
a and
Lian
Zhou
a
aState Key Laboratory of Geological Processes and Mineral Resources, China University of Geosciences, Wuhan, 430074, PR China. E-mail: tuyaken@hotmail.com; Fax: +86 27 67885096; Tel: +86 27 61055600
bState Key Laboratory for Mineral Deposits Research, School of Earth Sciences and Engineering, Nanjing University, Nanjing, 210023, PR China
First published on 17th November 2022
Abstract
Rare earth elements (REEs) in porewater can be useful proxies in the studies of marine geochemistry. The main challenges for the determination of REEs in porewater samples from marine sediments lie in three aspects—low concentration, high salinity, and limited sample sizes (usually only a few millilitres). In this study, a method based on laser ablation (LA) solution sampling ICP-MS for REEs analysis of small volumes (2–4 mL) of porewater is established. With the help of matrix removal and REEs preconcentration from natural liquids by a miniaturized Fe(OH)3 co-precipitation method, a high enrichment factor (10–20 fold) was achieved with only 2 mL of the original liquid, which was preconcentrated to be as small as 100–200 μL. Ultratrace (sub-pg mL−1) analysis was realized through the use of high laser ablation pulse frequency (up to 250 Hz) and based on the high transport efficiency of aerosol particles by the LA system. Due to the almost “dry” ICP condition generated by LA and nitrogen addition (5 mL min−1) to the plasma, the CeO+/Ce+ ratio was reduced to approximately 0.05%, and the total yields of BaO+ and BaOH+ ions were reduced to 0.003%, which means that the potential biases induced by oxide and hydroxide interferences could be negligible. Method validation was achieved through the analysis of the seawater reference material NASS-7 and a spike recovery test with quantitative recoveries of 94–101% for REEs. The application results of porewater samples characterized by no or slight Ce anomalies together with significantly positive Eu anomalies may be indicative of highly reducing and neutral to basic pH conditions. Reagents consumed by the LA-ICP-MS method were significantly reduced, making it a more environment-friendly method. Moreover, the simplicity in sample preparation and lack of need for special reagents or devices give the proposed method great potential for wide applications in the determination of REEs in natural water samples.
1. Introduction
Porewater, often called interstitial water, is bottom water trapped in sediment grains when they deposit.1,2 In this work, it refers exclusively to porewater of marine sediments. Porewater can provide key information in marine geochemistry, and the rare earth elements (REEs, La–Lu) in it are especially sensitive indicators.2 The series of REEs have unique similarities in physicochemical properties and exist dominantly in the trivalent state, thus being chemically coherent, except for Ce(IV) and Eu(II).3 In the process of early diagenesis with accumulating sediments and compaction during burial, porewater is squeezed upward, traveling through the fractures and pores of the sediments or even upward to the sediment–water interface.1 REEs in porewater are thus altered by diagenetic release from sediments, adsorption onto particles, and incorporation into authigenic phases (e.g., nodules and phosphates);4–7 meanwhile, fractionation would occur during the process due to the systematically changed properties based on the decreasing ionic radius from La to Lu. For example, REEs in porewater exists mainly in the form of carbonate complexes, while there is an increase in carbonate complexation strength with increasing atomic number, which leads to the preferential scavenging of light REEs (La–Gd) compared to heavy REEs (Tb–Lu) by particles.8 In particular, with the employment of the depth profile of REEs or normalized REEs pattern, the enrichment or depletion of REEs in porewater can be easily recognized,3,7 which could provide important information on the diagenetic processes, sedimentary environment, sources of trace elements to the ocean, and the enrichment mechanism of REEs in marine sediments. However, only a few studies on porewater REEs3–7,9–13 have been reported because of analytical challenges. Similar to REEs in seawater, these elements exist in porewater at a concentration of pg mL−1 level and with a complex matrix of high salinity (about 35 g kg−1). Even using inductively coupled plasma-mass spectrometry (ICP-MS), the analytical method is characterized with extremely low limits of detection (LOD, as low as sub-picogram level)14 and the direct determination remains challenging because when solution samples containing high salt contents are analyzed, the dissolved solids can deposit on the nebulizer and cone orifices, leading to signal suppression and instability and even clogging.15 Moreover, matrix effects are also a limitation in ICP-MS functioning with such samples having complex matrices. Usually, a reduction in the REE sensitivity induced by ionization suppression would occur due to the large amounts of easily ionized matrix elements (e.g., Na, Mg, K, and Ca) loaded into the plasma.15 A further challenge for REEs analysis is that the relatively high concentrations of Ba and light REEs may produce serious polyatomic interferences on Eu and heavy REE, mainly in the forms of oxide and hydroxide species.16,17 What adds to the difficulty in porewater analysis is the limited sample volume, with only several microlitres usually available for each sample.18,19 This is especially the case for the pore fluid extracted from pelagic sediments with low porosity and moisture content.1,2,13To date, REEs concentrations in porewater have been commonly determined by ICP-MS,3,13,20 which is attributed to its relatively simple spectra, wide linear range, multi-element analysis ability, and, most importantly, high sensitivity.21,22 Due to the analytical difficulties mentioned above, preconcentration and matrix separation are often necessary. Common preconcentration/separation methods include solid phase extraction (SPE)3,7,11,23 and co-precipitation.9,13 The employed methods were all developed originally for seawater REEs analysis. SPE is preferred in porewater analysis partially because it usually consumes less sample solution. For example, among the SPE method adopted for the extraction of REEs from porewater/seawater, the commercially available seaFAST system, using columns filled with the chelating resins with both ethylenediaminetriacetic and iminodiacetic acid functional groups, consumes only 7 mL of seawater sample per analysis.17,24,25 For the preconcentration of REEs in porewater using the co-precipitation method, there are only limited reports.9,13 A major impediment in this method is that its sample amount generally needs to be more than 25 mL,13,26 while such a volume of the porewater sample is not always available. Moreover, the limited ability of the co-precipitation method for matrix removal could result in high concentrations of the salt contents retained in the final dissolved solution, posing risks of spectral interferences (e.g., BaO+ and BaOH+ interfering with Eu+ determination) and also matrix suppression.27–29 On the other hand, co-precipitation exhibits the advantages of simple and easy operation (e.g., no need of extremely careful pH adjustment, which is indispensable in the SPE method), being cost-effective due to the employment of common reagents and without the need for expensive pretreatment equipment.21,29,30 Unlike huge volume of reagents required for rinsing and elution in the SPE method, a small amount of carrier material (such as Fe or Ga) and ammonia solution are sufficient to prepare the sample completely using co-precipitation, which is more in line with the principles of Green Chemistry.21,31
Laser ablation inductively coupled plasma-mass spectrometry (LA-ICP-MS) is a powerful microanalytical technique. Although it is mainly applied for solid samples,32 LA-ICP-MS has also been demonstrated to be suitable for liquid sample analysis.33–36 For previously reported co-precipitation methods, a final volume of 2–5 mL was obtained to fulfil the requirement of conventional solution nebulization ICP-MS.13,27 Amazingly, for solution sample introduction by LA, only 1 μL of the liquid is consumed per analysis (∼50 s of laser ablation, 160 μm spot size, and 10 Hz pulse frequency, respectively).34 In view of the micro sample consumption feature by LA-ICP-MS analysis, it would be completely sufficient even if the volume of the final dissolved sample solution is only a few tens of microliters, which is an unprecedented reduction in the requirement for sample sizes.
In addition to sample consumption, oxides and hydroxides polyatomic interferences are also of great concern for REEs measurements in porewater samples.17 N2 addition into argon plasma for ICP-MS has long been recognized as one of the strategies for polyatomic interference attenuation.37,38 For the LA-ICP-MS analysis of solid samples, it was reported that oxide ratios (ThO+/Th+) could be reduced by one order of magnitude.38 For conventional solution nebulization ICP-MS analysis, Lam and Horlick39 reported that 20% nitrogen in the carrier gas (Ar) removed almost all of the LaO+ signal, whereas others reported that no significant effects on oxides (e.g., 140Ce16O+ and 238U16O+) were observed with N2 addition.40
In this study, we aimed to establish a method for REEs determination in marine porewater using laser ablation solution sampling ICP-MS for small sample volumes obtained from a modified iron co-precipitation pretreatment process. The effects of addition of N2 to the makeup gas of LA-ICP-MS for solution sampling were investigated in detail (particularly concerning oxides and hydroxides interferences). Analytical performances, including sensitivity, matrix effects, limits of detection, analytical precision, and recoveries, were investigated for the proposed method.
2. Experimental
2.1. Reagents and standards' preparation
Deionized water with a resistivity of 18.2 MΩ cm was produced by a Milli-Q water purification system (Merck Millipore; Darmstadt, Germany). Commercially available nitric acid (68%, GR grade, Sinopharm Chemical Reagent Co., Ltd, China) was further purified with a sub-boiling distillation system. NH3·H2O solution (≥25%, chromatographic grade, Shanghai Aladdin Biochemical Technology Co., Ltd, China) was used without further purification. NIST 610 glass was purchased from National Institute of Standards and Technology, USA. The mixed REEs stock solution (10 μg mL−1, Multi-element Calibration Standard 1) was purchased from Agilent Technologies, USA, while In, Ba, and Ce single-element stock solutions (1000 μg mL−1) were purchased from National Center for Analysis and Testing of Steel Materials, China. Using these stock solutions, a set of REEs calibration standards at concentrations of 0.1, 1, 2, and 5 ng mL−1 were prepared by stepwise dilution in 2% HNO3, and with In added to each solution at a concentration of 5 ng mL−1. Indium single-element standard solution (10 ng mL−1, 2% HNO3) was also prepared, which could be used in the following steps of sample preparation. Ba and Ce single-element standard solutions were diluted to a concentration of 1 μg mL−1 in 2% HNO3. An iron (Fe3+) solution (1000 μg mL−1) was prepared using the analytical balance by dissolving 50 mg of iron foil (IRMM-524A) in 10 mL 10% HNO3 and diluting to a final mass of 50 g with Milli-Q water. The seawater reference material NASS-7 was purchased from the National Research Council Canada (NRC).2.2. Instrument and parameters
An Agilent 7900 ICP-MS (Agilent Technologies, USA) coupled with a femtosecond laser ablation system (NWR-Femto, ESL, USA) was employed for REEs measurements in this study. All the experiments were conducted at the State Key Laboratory of Geological Processes and Mineral Resources (GPMR), China University of Geosciences, Wuhan. The analytical balance used for sample weighing and gravimetric dilution was XP205 (d = 0.01 mg, Mettler-Toledo, Switzerland). In this study, the spot size for all sample analyses was fixed to 60 μm. Nitrogen was added with a simple “T” junction downstream from the sample cell at a flow rate of 5 mL min−1 into the makeup gas flow for the reduction of oxides and hydroxides interferences (see schematic diagram in Fig. S1†). The instrument was tuned for robustness plasma conditions41 following a daily process by the ablation of NIST 610 glass (32 μm, 5 Hz) to obtain a maximum signal intensity for 238U+ while keeping the 232Th16O+/232Th+ ratio < 0.2% and the 238U+/232Th+ ratio close to 1. After instrument tuning, CeO+/Ce+ was checked by the ablation of the Ce single-element standard solution (1 μg mL−1, 2% HNO3), ensuring the formation rate to be about 0.05%. REEs quantification in this study was accomplished using isotopes of 139La, 140Ce, 141Pr, 146Nd, 147Sm, 151Eu, 158Gd, 159Tb, 163Dy, 165Ho, 166Er, 169Tm, 172Yb, and 175Lu, in consideration of analyte isotopic abundances and potential interferences. 115In was used as the internal standard, and 137Ba was monitored for the assessment of its oxide formation rates. Typical operation conditions of LA-ICP-MS are presented in Table 1.| ICP-MS (Agilent 7900) | |
|---|---|
| RF power (W) | 1450 |
| Plasma gas flow rate (L min−1) | 15.0 |
| Auxiliary gas flow rate (L min−1) | 1.0 |
| Sampling depth (mm) | 7.0 |
| Isotopes | 137Ba, 115In, 139La, 140Ce, 141Pr, 146Nd, 147Sm, 151Eu, 158Gd, 159Tb, 163Dy, 165Ho, 166Er, 169Tm, 172Yb, 175Lu |
| Sampler | 1 mm Ni |
| Skimmer | 0.45 mm Ni |
| Ion optical settings | Typical |
| Dwell time per isotope (ms) | 10 |
| Detector mode | Dual |
| Oxide formation (CeO+/Ce+, BaO+/Ba+, ThO+/Th+) | 0.05%, 0.003%, 0.2% |
| Acquisition mode | Time-resolved analysis |
| Laser ablation system | |
|---|---|
| Laser type | Yb:YAG femtosecond laser |
| Wavelength | 257 nm |
| Pulse length | 300 fs |
| Energy density | 2.5–3.5 J cm−2 |
| Carrier gas | 0.60 L per min He |
| Makeup gas | 0.75 L per min Ar |
| Spot size | 60 μm |
| Pulse frequency | 20–250 Hz |
2.3. Sample preparation
The preconcentration method used in this study was adapted from the method proposed by Shaw et al.,42 with the modification of using Fe(OH)3 instead of Mg(OH)2 as the co-precipitation carrier phase, mainly in the quest for higher analytical sensitivity (with details discussed in Section 3.1). For each seawater/porewater sample (2–4 mL) in a 5 mL centrifuge tube, Fe3+ was added at a concentration of 20 μL mL−1 (according to Section 3.1). To be more specific, 40 μL of Fe3+ solution (1000 μL mL−1) should be added when 2 mL of the original sample is involved, while 80 μL of the Fe3+ solution should be used for 4 mL of the original sample. After thorough mixing, 10% NH3·H2O solution was added dropwise until the pH reached ∼8 (with the colourless solution turning light yellow and becoming cloudy a few minutes later).43 The sample solutions were allowed to stand for 12 h (to ensure that REEs were fully scavenged onto the Fe-hydroxide phase) and then centrifuged for 15 min at 4000 rpm. Since REEs were retained on the precipitate, the supernatant was decanted from the centrifuge tube. The precipitate was then washed twice with Milli-Q water to remove the matrix elements trapped in the precipitate plug. Finally, the precipitate was added with 100 μL of the internal standard solution (indium: 10 ng mL−1, 2% HNO3), and then 10% HNO3 (approximately 30 μL) was added dropwise to dissolve the precipitate completely. After the pretreatment process, each of the final dissolved sample solutions had a volume of 100–200 μL prior to analysis, achieving an enrichment factor of 10 to 40 times for the use of 2–4 mL of the original sample. The procedural blank was obtained by subjecting one aliquot of Milli-Q water to the entire sample preparation process. All the chemical processes were conducted in a “class-100” laboratory condition and all the containers were pre-cleaned with 5% HNO3 and rinsed by Milli-Q water.2.4. Analytical strategy and quantification method
Each pretreated sample solution was pipetted into tiny holes (60 μL in volume) on a Teflon sheet and sealed with a parafilm “M” laboratory film (Bemis Company, Inc., USA). It should be noted that the final volume of the dissolved sample solutions ranged from 100 to 200 μL, which were 2 to 3 times of the amount loaded into the liquid target. The remaining volume of the pretreated solution can be used for repeat tests if there is any doubt regarding the obtained results. The liquid target was then put into the ablation cell and analyzed by the LA-ICP-MS instrument. Before the samples were ablated for data acquisition, “pre-ablation” (with the corresponding spot size for sample analysis) for a few seconds was carried out to remove the parafilm and expose the solution surface; then, the aerosol generated by LA was transported with the carrier gas (helium) into the ICP-MS for analysis. Each data acquisition process lasted 90 s: ∼20 s for “gas background” signal acquisition (used for background correction),44 and subsequent 50 s for sample solution, followed by ∼20 s washout times (Fig. S2†). The calibration curves were built by LA-ICP-MS analysis of a set of reference standard solutions at concentrations of 0.1, 1, 2, and 5 ng mL−1, with 5 ng mL−1 of In as the internal standard (Fig. S3†). All these standards were prepared in 2% HNO3 without NaCl matrix matching because we found that there was no need to match the sample matrix (see details in Section 3.3). Elemental concentrations in the studied samples were quantified by calibration against a set of standard solutions combined with In used for internal standardization; the equations used for concentration calculation, mainly adapted from Longerich et al.,44 are provided in the ESI.†3. Results and discussion
3.1. Optimization of the pretreatment procedure
The general carrier phases used for REEs co-precipitation include mainly Mg(OH)2, Fe(OH)3, and Ga(OH)3.21 In this study, Ga(OH)3 was not taken into account because of its scarcity and comparatively higher cost. Both porewater and seawater contain a large amount of Mg2+ (about 1200 μg mL−1 in seawater45); therefore, the simple addition of aqueous ammonia could induce the formation of Mg(OH)2 precipitates that co-precipitate REEs.26,42 The drawback is that the relatively high concentration of the co-precipitate carrier would cause signal suppression, especially in the case with 2–4 mL of original porewater/seawater samples enriched into a final volume of 100–200 μL, which means that the concentration of Mg in the final dissolved solution could be 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–48000 μg mL−1 (assuming 1200 μg mL−1 of Mg in the original sample). When Fe(OH)3 was employed as the carrier phase for REEs co-precipitation, only a small amount of iron solution was added and thus a relatively low concentration of Fe3+ was retained in the final dissolved solution. A comparison on the LA-ICP-MS signal intensities of co-precipitation using Mg(OH)2 and Fe(OH)3 was performed, wherein two aliquots (4 mL for each aliquot) of seawater were spiked separately with the same amounts of REEs (each at 24 pg mL−1), the pH was adjusted to about 11 for Mg2+ precipitation and 8 (the optimal pH) for Fe3+ precipitation using 10% NH3·H2O. Both precipitates were dissolved in 200 μL 10% HNO3. As illustrated in Fig. 1a, it is evident that 2–3 times higher signal intensities were obtained by Fe(OH)3 co-precipitation than by Mg(OH)2 co-precipitation using LA-ICP-MS. As a result, Fe(OH)3 was employed as the carrier phase for REEs co-precipitation in this method.
000–48000 μg mL−1 (assuming 1200 μg mL−1 of Mg in the original sample). When Fe(OH)3 was employed as the carrier phase for REEs co-precipitation, only a small amount of iron solution was added and thus a relatively low concentration of Fe3+ was retained in the final dissolved solution. A comparison on the LA-ICP-MS signal intensities of co-precipitation using Mg(OH)2 and Fe(OH)3 was performed, wherein two aliquots (4 mL for each aliquot) of seawater were spiked separately with the same amounts of REEs (each at 24 pg mL−1), the pH was adjusted to about 11 for Mg2+ precipitation and 8 (the optimal pH) for Fe3+ precipitation using 10% NH3·H2O. Both precipitates were dissolved in 200 μL 10% HNO3. As illustrated in Fig. 1a, it is evident that 2–3 times higher signal intensities were obtained by Fe(OH)3 co-precipitation than by Mg(OH)2 co-precipitation using LA-ICP-MS. As a result, Fe(OH)3 was employed as the carrier phase for REEs co-precipitation in this method.
 | ||
| Fig. 1 (a) Comparison of REEs signals in seawater samples co-precipitated using Mg(OH)2 and Fe(OH)3, respectively. (b) Recoveries of spiked REEs (24 pg mL−1 of each REE spiked) in seawater obtained by co-precipitation with different amounts of Fe3+ solution. | ||
The amount of Fe3+ needed for complete REEs co-precipitation by Fe-hydroxide was investigated. In this experiment, three aliquots (2 mL of seawater each) of mixed solutions containing 24 pg mL−1 REEs were added with 20 μL, 40 μL, and 60 μL of the Fe3+ solution (1000 μg mL−1). The rest of the sample preparation was the same as that described earlier in the section “Sample preparation”. As illustrated in Fig. 1b, the recoveries of the spiked REEs were 90% for 20 μL of Fe3+ solution, and 100% recoveries were achieved for both 40 μL and 60 μL of Fe3+ solution, which means a mere concentration of 20 μL mL−1 Fe3+ would be sufficient for complete REEs co-precipitation. Thus, 40 μL of the Fe3+ solution (1000 μg mL−1) was selected for all subsequent experiments with 2 mL of the original sample processed. It was also found that adding the internal standard either before the co-precipitation step or after at dissolving the precipitate step had no significant effect on the recoveries of REEs. If the internal standard solution was added before the co-precipitation step, more aqueous ammonia solution for pH adjustment and more HNO3 for dissolving the precipitate would be required. Hence, the latter approach was selected because smaller amounts of reagents were needed. The presented method, in general, offers a miniaturized Fe(OH)3 co-precipitation procedure with REEs enriched by 10–40 fold while processing 2–4 mL of the original sample. In the previously reported works about REEs analysis of porewater, the required sample volume for co-precipitation generally ranged from 25 mL to 40 mL.9,13 Thus, the amount of sample required for the proposed method is reduced approximately 10 times compared to that of the conventional method. This is of great importance for porewater samples with limited sample volume. In addition, the miniaturized process indicates less consumption of reagents, easier transportation, and storage of samples, which caters more to the increasing drive for green chemistry.21,30
3.2. Oxides and hydroxides interferences and the effect of N2 addition
Despite the fact that a large amount of Ba in the matrix can be eliminated during the Fe(OH)3 co-precipitation pretreatment stage, it is necessary to keep these polyatomic interferences at low yields because residual barium in the final dissolved solution could induce interferences (i.e., 135Ba16O+ and 134Ba16O1H+ on 151Eu+, 136Ba16O1H+ and 137Ba16O+ on 153Eu).29 In addition, oxides and hydroxides of light REEs are sometimes problematic for heavy REEs determination.46A single Ce (1 μg mL−1, 2% HNO3) solution was analyzed by LA-ICP-MS to investigate the oxide and hydroxide formation rates of CeO+/Ce+ and CeOH+/Ce+. It is widely acknowledged that the oxide and hydroxide interferences formation rates for Ce are among the highest for all the REEs from La to Lu.16 As shown in Fig. 2, CeO+/Ce+ was found to be at approximately 0.45% in the normal mode and was largely reduced to approximately 0.07% in the N2 addition mode (5 mL min−1) under optimal conditions that produced the highest signals, whereas the formation rate of CeOH+/Ce+ remained almost constant at 0.01%. Usually, the makeup gas flow rate was optimized to be slightly lower than that yielding the highest signal intensity, wherein the CeO+/Ce+ ratio was approximately 0.05%. Under this condition, the signal intensity was still approximately 1.5 times higher than the highest signal achieved in the normal mode without N2 addition. Because mass 151 was selected for Eu determination in this study, (135Ba16O+ + 134Ba16O1H+)/137Ba+ was monitored by measuring a single Ba (1 μg mL−1, 2% HNO3) solution and found to be approximately 0.003%. It should be noted that the concentration (1 μg mL−1) utilized for the investigation of interferences formation rates was much higher than those in the standards and the samples. This is because no significant signal would be observed on the interfering masses if solutions of low concentration (e.g., at the concentration levels of ng mL−1 or sub-ng mL−1) were analyzed, let alone the precise determination of the interferences formation rates.
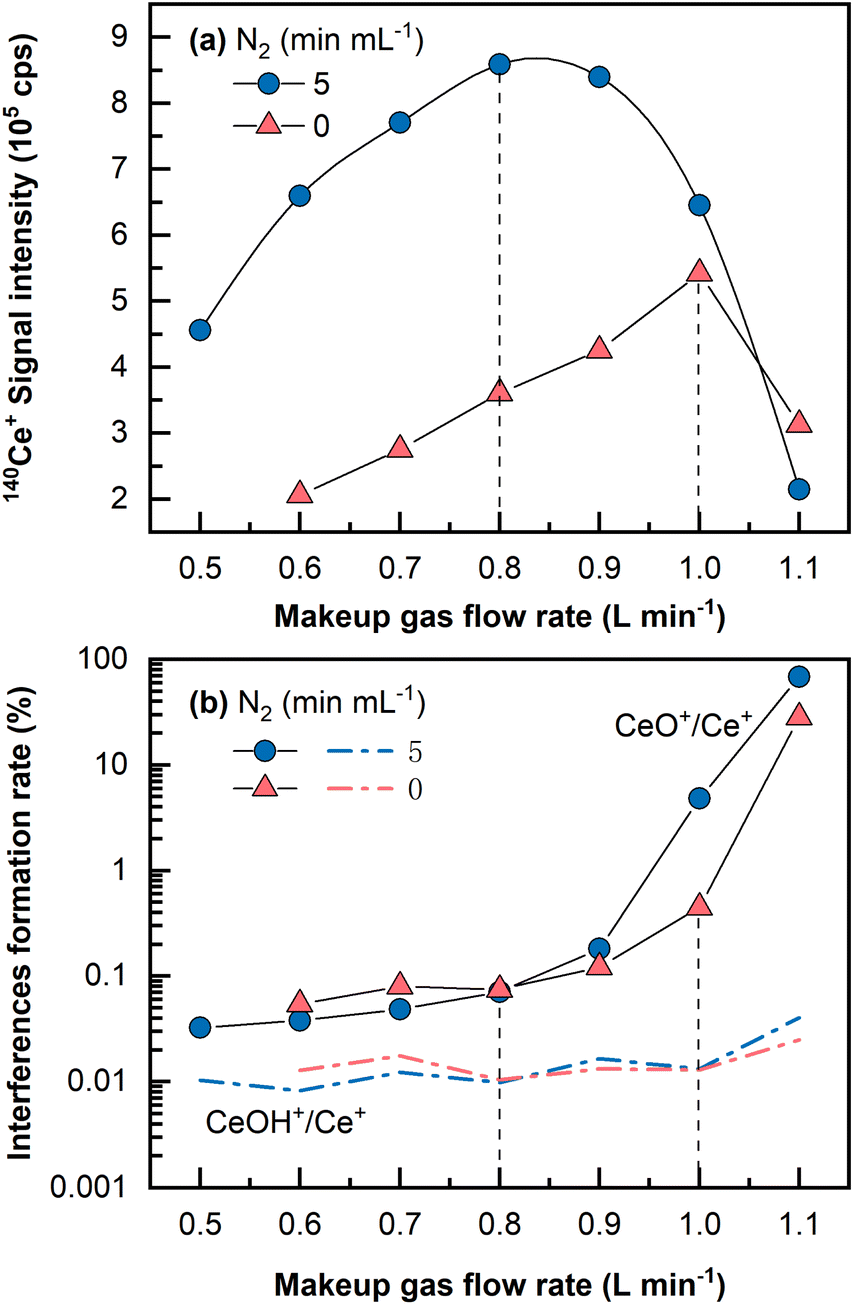 | ||
| Fig. 2 Effect of makeup gas flow rate on (a) Ce+ signal intensity and (b) CeO+/Ce+ and CeOH+/Ce+ under normal or N2 addition modes (test solution: 1 μg mL−1 Ce in 2% HNO3). The vertical dashed lines show the makeup gas flow rates yielding the maximum signal intensity for REEs. | ||
The CeO+/Ce+ formation rate (0.45%) obtained even under the normal mode without N2 addition by LA-ICP-MS was still much lower than that obtained in conventional solution nebulization ICP-MS (usually 1–3%47). This might be due to a “dry” plasma condition achieved by the low amount of oxygen and hydrogen sampled by LA into ICP, significantly reducing the oxides and hydroxides formation rates.33,36 Furthermore, with the assistance of N2 addition and the optimized makeup gas flow rate, the oxide formation rate of CeO+/Ce+ was further reduced to 0.05%, approximately two magnitudes lower than that in conventional solution nebulization ICP-MS, and lower than that obtained using the aerosol dilution method or membrane-desolvation sample introduction system, usually in the range between 0.1 and 0.4%.27,28,47 In a previous study, sample solution droplets were dried with an IR lamp on a PTFE filter membrane prior to REEs analysis by LA-ICP-MS.30 Our proposed LA-ICP-MS method achieves a very low level of oxide and hydroxide species without such a solidification process, reduces the contamination risk, and uses a small sample volume (2–4 mL versus 40 mL sample used by Hsieh et al.30).
3.3. Matrix effects
Because the capability of matrix removal by co-precipitation is limited,24 the potential matrix effects on the LA-ICP-MS measurement were investigated.Two sample solutions were prepared, one consisting of 4 mL seawater matrix and the other consisting of 4 mL 2% HNO3 matrix; both of the two samples were spiked with 100 μL of 1 ng mL−1 REEs (i.e., at the concentration of 24 pg mL−1). Then, the samples were pretreated according to Section 2.3 and analyzed by LA-ICP-MS. Since all REEs performed similarly, the analytical results of Eu and Lu, including signal intensities and the internal standard normalized intensity ratios, are shown as the examples (represent light REEs and heavy REEs respectively). As illustrated in Fig. 3, seawater matrix (approximately 3.5% NaCl) only induced slight signal suppression (10%) on REE+ sensitivities during the tested pulse frequency range of LA (Fig. 3a); the intensity ratios for REE+/In+ obtained in the sample with the original matrix of seawater remained almost consistent with those obtained in the sample with the original matrix of 2% HNO3 (Fig. 3b). This was also confirmed by applying independent sample t-test to the two sets of data, with p > 0.05 obtained for both Eu+/In+ and Lu+/In+, which meant the differences in the average values for REE+/In+ obtained from the two samples of different original matrices were statistically insignificant. The results indicated that no significant matrix effects resulting from the residual salts in the final dissolved solution would be observed with the proposed laser ablation solution sampling ICP-MS analytical method. Note that quantification in this method was realized through internal standardization by In. Therefore, what matters for quantification is the normalized signal intensity ratio (REE+/In+). Now that REE+/In+ remained consistent in the samples of totally different original matrices, quantification can be carried out using standards in 2% HNO3 without the need for matrix matching (i.e., 3.5% NaCl). Furthermore, both signal intensities of the analyte and internal standard increased proportionally as the ablation pulse frequency (20–250 Hz) increased, and the normalized signal ratio (REE+/In+) remained unchanged, confirming that ablation frequency can be flexible during measurement. In this method, porewater/seawater sample analysis was done using higher laser ablation pulse frequency (100–250 Hz), while the calibration curves were obtained using 20 Hz. This is because REEs in these actual samples were significantly lower than those in the calibration standards (0.1 to 5 ng mL−1), despite being preconcentrated.
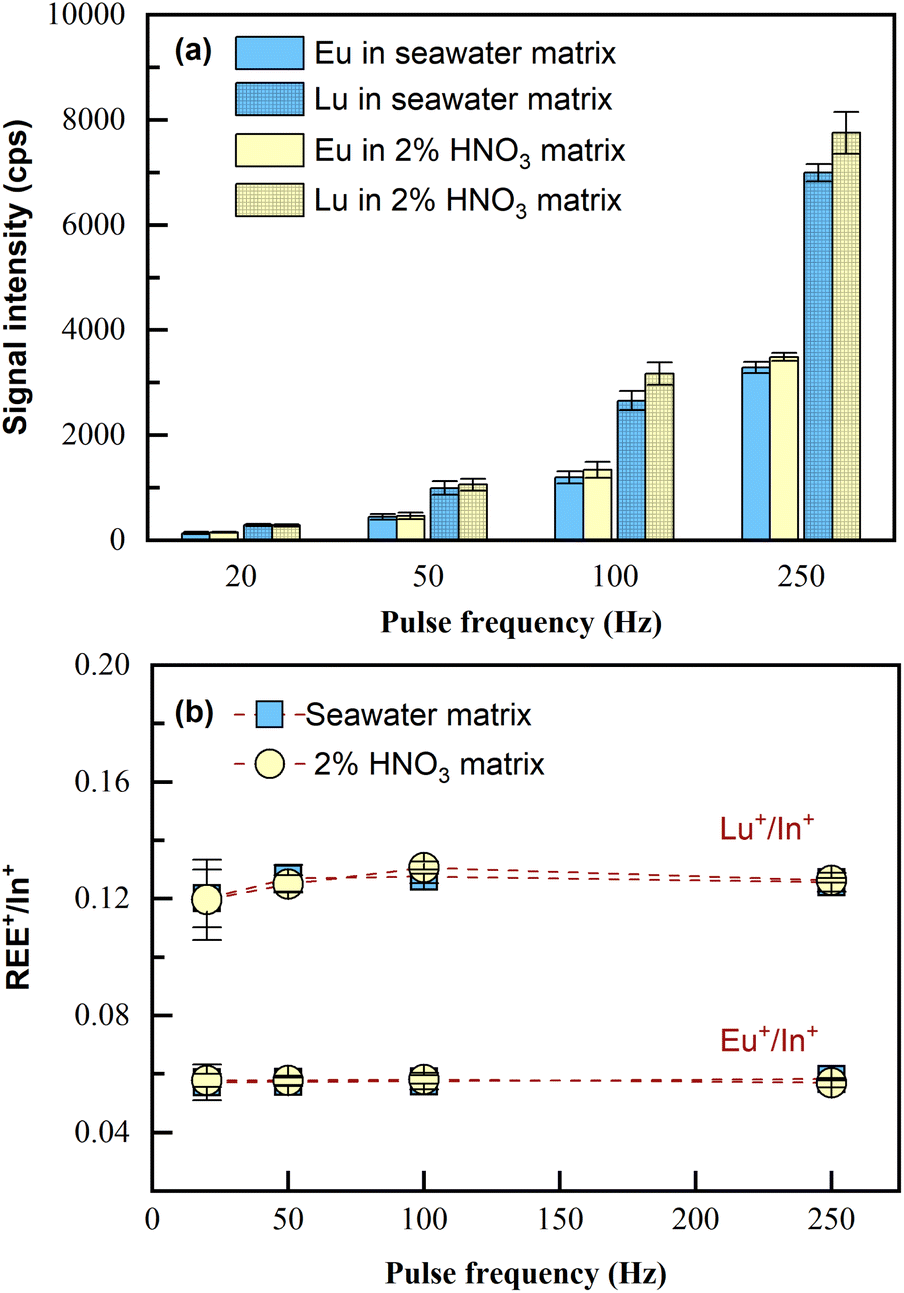 | ||
| Fig. 3 (a) Signal intensities and (b) the normalized intensity ratio REE+/In+ in seawater matrix and in 2% HNO3 matrix analyzed by LA-ICP-MS under various ablation pulse frequencies. Eu and Lu are shown as the representatives. The laser ablation spot size was fixed to 60 μm. The original samples were prepared by REEs spiked in different matrices and with In added as the internal standard. Error bars represent SD (n = 3). | ||
3.4. Limit of detection
Following the equations for limit of detection (LOD) calculation by Longerich et al.,44 LODs of REEs under different laser ablation pulse frequencies (20–250 Hz) were calculated, and the results are presented in Fig. 4 and ESI data Table S1.† Clearly, the LODs decreased significantly with increasing laser ablation repetition rate. Using higher repetition rates (100–250 Hz), LODs could be improved to sub-pg mL−1. For example, under ablation conditions of 60 μm and 250 Hz, LODs of 0.1 pg mL−1 to 0.7 pg mL−1 were obtained for REEs. It should be noted that these LODs were calculated without taking the enrichment factors into account.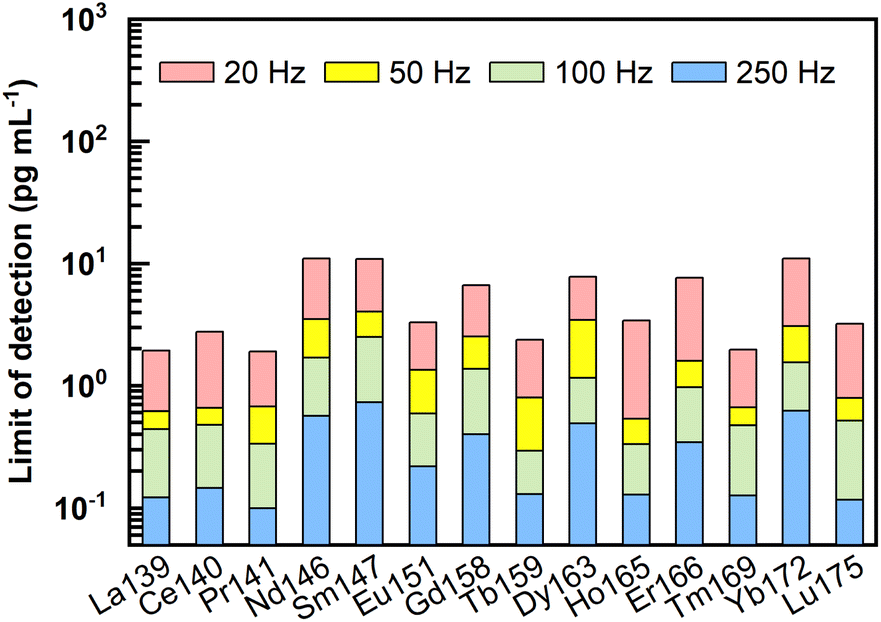 | ||
| Fig. 4 Limits of detection for REEs analyzed by LA-ICP-MS under different laser ablation frequencies (20, 50, 100, 250 Hz), with the laser ablation spot size fixed to 60 μm. Note that the LODs were calculated without taking the enrichment factors into account. | ||
LODs represent the lowest concentration of REEs that could be detected in the final dissolved solutions by LA-ICP-MS. While considering the lowest concentration of REEs in the original natural liquid that can be measured by the proposed method (referred to as method detection limits in the following text), the enrichment factor achieved through the pretreatment process should be considered. For the proposed methodology with 2–4 mL of the original samples enriched into a final volume of 100–200 μL, the enrichment factor obtained would be between 10 and 40 times. Method detection limits (pg mL−1) of REEs considering the potential enrichment factors analyzed by LA-ICP-MS under the ablation condition of 60 μm and 250 Hz are listed in Table S2.† Specifically, when faced with the case of a 4 mL initial sample pretreated and dissolved into a final volume of 100–200 μL, the lowest concentration of REEs that can be detected in the original sample should be 0.003–0.005 pg mL−1 for La, Ce, Pr, Tb, Ho, Tm, and Lu, 0.005–0.01 pg mL−1 for Eu, 0.008–0.015 pg mL−1 for Er, 0.01–0.02 pg mL−1 for Gd, 0.013–0.025 pg mL−1 for Dy, 0.015–0.03 pg mL−1 for Nd and Yb, and 0.018–0.035 pg mL−1 for Sm. Conclusively, such low method detection limits make the proposed method suitable for accurate REEs analysis in porewater.
3.5. Method validation by natural seawater reference material
Since porewater from marine sediments forms essentially by seawater trapped in the pore space when the sediments are deposited, porewater thus shares similar characteristics with seawater, containing low REEs concentrations and high salt matrix. One notable difference is that REEs in porewater can be up to 10 times more enriched than those in the overlying seawater,7,13 which means that if the method is effective for the determination of REEs in seawater, it will be inevitably suitable for the analysis of REEs in porewater. Because porewater certified reference material is lacking, seawater reference material was used to assess the precision and accuracy of the proposed method. Although the REEs concentrations of the certified seawater reference materials NASS-5 and NASS-6 have been reported in a few studies, they are no longer commercially available. A seawater CRM NASS-7, a substitution of NASS-6, distributed by the National Research Council of Canada,48 was used in this study. To the best of our knowledge, only one set of REEs values has been reported but without analytical uncertainties.48 Therefore, the validation of the proposed method was demonstrated from two aspects—by the comparison between the measured REEs concentrations and the reference values, and by the recoveries of the REEs in the spiked seawater samples.Two aliquots of 2 mL of NASS-7, with one of them spiked with 24 pg mL−1 REEs, were prepared, as described in the “Sample preparation” section. Quantification was carried out by calibration against the set of reference standard solutions, in conjunction with In used for internal standardization, and data acquisition for each sample was executed ten times. The calibration curves (Fig. S3†) as well as the equations used for concentration calculation are provided in the ESI.† Note that the above sample preparation and measurements were repeated three times within three months, and the average values are shown in Table 2. All the measured REEs values (0.18 to 8.27 pg mL−1) are similar to previously reported values (0.11 to 6 pg mL−1).48 The measurement precisions (relative standard deviation, RSD) of all REEs in the seawater reference material NASS-7 were less than 10%, except for Yb with an RSD of approximately 16%. Importantly, the spike recoveries obtained were 94–101%, with RSDs ranging from 0.7% to 2.7% (n = 3), confirming the accuracy of the proposed method.
| Element | Procedural blank | NASS-7 | REEs-spiked sample | ||
|---|---|---|---|---|---|
| Measured | Ref. 48 | Recovery (%) | RSD (%) | ||
| a Results below the limit of detection (LOD). | |||||
| 139La | 0.08 ± 0.02 | 8.27 ± 0.49 | 6 | 98 | 0.8 |
| 140Ce | 0.20 ± 0.08 | 3.42 ± 0.23 | 3 | 101 | 1.1 |
| 141Pr | 0.03 ± 0.03 | 1.19 ± 0.04 | 2 | 96 | 1.2 |
| 146Nd | 0.08 ± 0.04 | 5.17 ± 0.26 | 3 | 95 | 2.3 |
| 147Sm | 0.05 ± 0.04 | 0.97 ± 0.01 | 2 | 98 | 2.7 |
| 151Eu | 0.07 ± 0.04 | 0.25 ± 0.01 | 0.6 | 97 | 0.8 |
| 158Gd | 0.05 ± 0.05 | 1.20 ± 0.05 | 1.4 | 94 | 1.0 |
| 159Tb | —a | 0.18 ± 0.02 | 0.3 | 98 | 1.0 |
| 163Dy | 0.02 ± 0.03 | 1.31 ± 0.03 | 1.3 | 101 | 1.6 |
| 165Ho | —a | 0.34 ± 0.02 | 1 | 98 | 0.6 |
| 166Er | 0.24 ± 0.02 | 1.07 ± 0.02 | 1.2 | 101 | 1.3 |
| 169Tm | —a | 0.16 ± 0.01 | 0.11 | 97 | 1.2 |
| 172Yb | 0.03 ± 0.04 | 1.35 ± 0.21 | 1 | 96 | 1.7 |
| 175Lu | —a | 0.18 ± 0.01 | 0.2 | 101 | 0.7 |
3.6 Application to porewater sample analysis
REEs in 4 porewater samples were analyzed by LA solution sampling ICP-MS, and each sample consumed 2 mL of porewater. These porewater samples were collected from a gravity piston core sampled from the northeastern Dongsha area of the South China Sea. The results are shown as Post-Archean Australian Shale (PAAS)-normalized49 REE patterns in Fig. 5, and detailed contents are presented in Table 3. The determined REEs in these porewater samples ranged from 0.10 to 32.75 pg mL−1, with most RSDs less than 10%. As illustrated in Fig. 5, unlike REEs in seawater (e.g., NASS-7), which is characterized by a pronounced negative Ce anomaly, no or slight Ce anomalies were found in this set of porewater samples. Meanwhile, significantly positive Eu anomalies were found in these four samples. In this study, interference-related Eu anomalies can be excluded because the total of the Ba-related oxides and hydroxides (i.e., 135Ba16O + 134Ba16O1H) interfering on 151Eu was only 0.003% (relative to 137Ba), while 137Ba+ signals detected in the final samples were typically no more than 105 cps level. This phenomenon might be indicative of highly reducing and neutral to basic pH conditions.50,51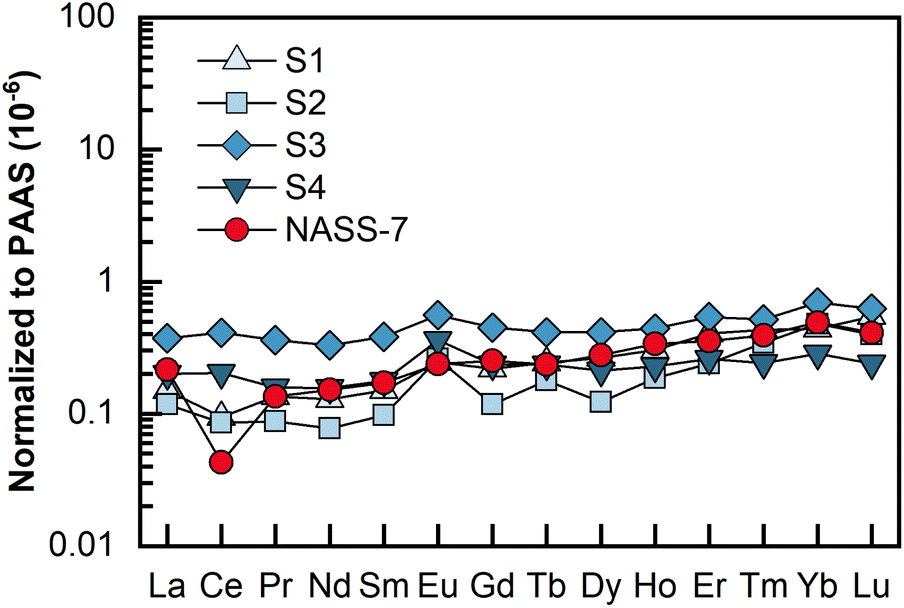 | ||
| Fig. 5 PAAS-normalized REE patterns for the four porewater samples and the seawater reference material NASS-7. The PAAS values (mg kg−1) used are La: 38.2; Ce: 79.6; Pr: 8.83; Nd: 33.9; Sm: 5.55; Eu: 1.08; Gd: 0.774; Dy: 4.68; Ho: 0.991; Er: 2.85; Tm: 0.405; Yb: 2.82; Lu: 0.433.49 | ||
| Element | S1 | S2 | S3 | S4 |
|---|---|---|---|---|
| 139La | 5.72 ± 0.16 | 4.48 ± 0.12 | 14.22 ± 0.42 | 7.63 ± 0.29 |
| 140Ce | 7.50 ± 0.39 | 6.80 ± 0.14 | 32.75 ± 0.38 | 16.20 ± 0.28 |
| 141Pr | 1.19 ± 0.05 | 0.77 ± 0.04 | 3.18 ± 0.12 | 1.39 ± 0.10 |
| 146Nd | 4.34 ± 0.33 | 2.63 ± 0.16 | 11.18 ± 0.46 | 5.29 ± 0.34 |
| 147Sm | 0.83 ± 0.06 | 0.54 ± 0.04 | 2.12 ± 0.13 | 0.98 ± 0.09 |
| 151Eu | 0.26 ± 0.02 | 0.28 ± 0.02 | 0.61 ± 0.04 | 0.39 ± 0.03 |
| 158Gd | 1.02 ± 0.08 | 0.55 ± 0.06 | 2.10 ± 0.10 | 1.09 ± 0.06 |
| 159Tb | 0.19 ± 0.02 | 0.14 ± 0.01 | 0.32 ± 0.03 | 0.18 ± 0.01 |
| 163Dy | 1.24 ± 0.09 | 0.57 ± 0.06 | 1.93 ± 0.11 | 0.99 ± 0.08 |
| 165Ho | 0.30 ± 0.03 | 0.18 ± 0.02 | 0.44 ± 0.03 | 0.22 ± 0.02 |
| 166Er | 1.15 ± 0.10 | 0.68 ± 0.06 | 1.54 ± 0.08 | 0.74 ± 0.05 |
| 169Tm | 0.17 ± 0.02 | 0.14 ± 0.01 | 0.21 ± 0.01 | 0.10 ± 0.01 |
| 172Yb | 1.24 ± 0.11 | 1.35 ± 0.12 | 1.97 ± 0.14 | 0.80 ± 0.05 |
| 175Lu | 0.24 ± 0.02 | 0.17 ± 0.01 | 0.27 ± 0.01 | 0.10 ± 0.01 |
4. Conclusion
This study established a method for REEs determination in small volumes of marine porewater by LA-ICP-MS. With samples pretreated with Fe(OH)3 co-precipitation and precipitates dissolved in 100–200 μL prior to analysis, the high salt contents were removed and REEs were enriched by 10–40 fold with only 2–4 mL of original porewater. The required sample volume of the proposed method is reduced approximately 10 times compared to that of the conventional method using co-precipitation and solution nebulization ICP-MS. N2 addition to the makeup gas flow demonstrated an improvement in the analytical performance of LA-ICP-MS for liquid samples. The addition of 5 mL min−1 N2 into the makeup gas flow significantly reduced the 140Ce16O/140Ce from 0.45% to 0.05%, while the sensitivities for REEs were improved by 1.5-fold. The 140Ce16O/140Ce ratio obtained by the proposed method is approximately two orders of magnitude lower than that of the conventional solution nebulization ICP-MS analytical method and lower than those achieved with the membrane-desolvation sample introduction system or aerosol dilution method. Good detection limits of 0.1 pg mL−1 to 0.7 pg mL−1 for REEs were obtained under ablation conditions of 60 μm and 250 Hz, which would be significantly lower than the typical REEs concentrations in natural seawater, taking into account the enrichment factors (10–40 fold). The proposed method was applied for the determination of REEs in the newly distributed seawater reference material NASS-7. The measured REEs contents ranging from 0.18 to 8.27 pg mL−1 with good repeatability (RSD < 10%) were in agreement with those reported in the literature. The application of the proposed method to the determination of REEs in porewater samples helped to find positive Eu anomalies in the northeastern Dongsha area of the South China Sea. In addition to the much lower sample volume required for accurate analysis, which provides a practical solution for porewater REEs determination with limited sample amount, the miniaturized sample pretreatment is greener chemistry because of its significant decrease in the consumption of various reagents. The proposed method is accurate, effective, and has great application potential.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the two anonymous reviewers for their constructive comments, which greatly helped to improve the manuscript. This research was supported by the National Science Fund for Distinguished Young Scholars (41725013), the National Natural Science Foundation of China (41873029), the Natural Science Foundation of Hubei Province (2020CFA045) and the MOST Special Fund from the State Key Laboratories of Geological Processes and Mineral Resources.References
-
D. E. Hammond, in Encyclopedia of Ocean Sciences, ed. J. K. Cochran, H. J. Bokuniewicz and P. L. Yager, Academic Press, Oxford, Third edn, 2019, pp. 82–89 Search PubMed
.
-
R. H. Byrne and E. R. Sholkovitz, in Handbook on the Physics and Chemistry of Rare Earths, Elsevier, 1996, vol. 23, pp. 497–593 Search PubMed
- A. N. Abbott, B. A. Haley, J. McManus and C. E. Reimers, Geochim. Cosmochim. Acta, 2015, 154, 186–200 CrossRef CAS
- Y. N. Deng, Q. J. Guo, C. Q. Liu, G. W. He, J. Cao, J. L. Liao, C. H. Liu, H. F. Wang, J. H. Zhou, Y. F. Liu, F. L. Wang, B. Zhao, R. F. Wei, J. Zhu and H. J. Qiu, Sci. Adv., 2022, 8, eabn5466 CrossRef CAS PubMed
- H. Elderfield and E. R. Sholkovitz, Earth Planet. Sci. Lett., 1987, 82, 280–288 CrossRef CAS
- E. R. Sholkovitz, D. J. Piepgras and S. B. Jacobsen, Geochim. Cosmochim. Acta, 1989, 53, 2847–2856 CrossRef CAS
- B. A. Haley, G. P. Klinkhammer and J. McManus, Geochim. Cosmochim. Acta, 2004, 68, 1265–1279 CrossRef CAS
- Y.-R. Luo and R. H. Byrne, Geochim. Cosmochim. Acta, 2004, 68, 691–699 CrossRef CAS
- G. Bayon, D. Birot, L. Ruffine, J. C. Caprais, E. Ponzevera, C. Bollinger, J. P. Donval, J. L. Charlou, M. Voisset and S. Grimaud, Earth Planet. Sci. Lett., 2011, 312, 443–452 CrossRef CAS
- U. Schacht, K. Wallmann and S. Kutterolf, J. Geochem. Explor., 2010, 106, 176–187 CrossRef CAS
- T.-O. Soyol-Erdene and Y. Huh, Chem. Geol., 2013, 358, 75–89 CrossRef CAS
- E. R. Sholkovitz, T. J. Shaw and D. L. Schneider, Geochim. Cosmochim. Acta, 1992, 56, 3389–3402 CrossRef CAS
- Y. N. Deng, J. B. Ren, Q. J. Guo, J. Cao, H. F. Wang and C. H. Liu, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed
- K. L. Linge and K. E. Jarvis, Geostand. Geoanal. Res., 2009, 33, 445–467 CrossRef CAS
- C. Agatemor and D. Beauchemin, Anal. Chim. Acta, 2011, 706, 66–83 CrossRef CAS PubMed
- P. Dulski, Fresenius' J. Anal. Chem., 1994, 350, 194–203 CrossRef CAS
- M. K. Behrens, J. Muratli, C. Pradoux, Y. Z. Wu, P. Boning, H. J. Brumsack, S. L. Goldstein, B. Haley, C. Jeandel, R. Paffrath, L. D. Pena, B. Schnetger and K. Pahnke, Mar. Chem., 2016, 186, 110–120 CrossRef CAS
- B. A. Haley and G. P. Klinkhammer, Mar. Chem., 2003, 82, 197–220 CrossRef CAS
- H. N. Liu, L. Li, X. J. Wang, Y. J. Ren and X. F. Shi, Arch. Environ. Contam. Toxicol., 2021, 81, 553–563 CrossRef CAS PubMed
- X.-P. Yan, R. Kerrich and M. Jim Hendry, J. Anal. At. Spectrom., 1999, 14, 215–221 RSC
- A. Fisher and D. Kara, Anal. Chim. Acta, 2016, 935, 1–29 CrossRef CAS PubMed
- B. Zawisza, K. Pytlakowska, B. Feist, M. Polowniak, A. Kita and R. Sitko, J. Anal. At. Spectrom., 2011, 26, 2373–2390 RSC
- T. Himmler, B. A. Haley, M. E. Torres, G. P. Klinkhammer, G. Bohrmann and J. Peckmann, Geo-Mar. Lett., 2013, 33, 369–379 CrossRef CAS
- E. C. Hathorne, B. Haley, T. Stichel, P. Grasse, M. Zieringer and M. Frank, Geochem., Geophys., Geosyst., 2012, 13, 1–12 CrossRef
- K. C. Crocket, E. Hill, R. E. Abell, C. Johnson, S. F. Gary, T. Brand and E. C. Hathorne, Front. Mar. Sci., 2018, 5, 1–22 CrossRef
- N. Freslon, G. Bayon, D. Birot, C. Bollinger and J. A. Barrat, Talanta, 2011, 85, 582–587 CrossRef CAS PubMed
- Y. T. Li, W. Guo, Z. W. Wu, L. L. Jin, Y. Q. Ke, Q. H. Guo and S. H. Hu, Microchem. J., 2016, 126, 194–199 CrossRef CAS
- C.-H. Chung, I. Brenner and C.-F. You, Spectrochim. Acta, Part B, 2009, 64, 849–856 CrossRef
- Y. B. Zhu, K. Inagaki, H. Haraguchi and K. Chiba, J. Anal. At. Spectrom., 2010, 25, 364–369 RSC
- H.-F. Hsieh, Y.-H. Chen and C.-F. Wang, Talanta, 2011, 85, 983–990 CrossRef CAS PubMed
- C. Bendicho, I. Lavilla, F. Pena-Pereira and V. Romero, J. Anal. At. Spectrom., 2012, 27, 1831–1857 RSC
- R. C. Machado, D. F. Andrade, D. V. Babos, J. P. Castro, V. C. Costa, M. A. Sperança, J. A. Garcia, R. R. Gamela and E. R. Pereira-Filho, J. Anal. At. Spectrom., 2020, 35, 54–77 RSC
- X. H. Liao, Z. C. Hu, T. Luo, W. Zhang, Y. S. Liu, K. Q. Zong, L. Zhou and J. F. Zhang, J. Anal. At. Spectrom., 2019, 34, 1126–1134 RSC
- X. H. Liao, T. Luo, S. H. Zhang, W. Zhang, K. Q. Zong, Y. S. Liu and Z. C. Hu, J. Anal. At. Spectrom., 2020, 35, 1071–1079 RSC
- M. A. Kokh, B. Luais, L. Truche, M. C. Boiron, C. Peiffert and A. Schumacher, Geostand. Geoanal. Res., 2021, 45, 341–358 CrossRef CAS
- X. H. Liao, Z. C. Hu, W. Zhang, Y. T. Feng, T. Luo, Z. C. Wang, M. Li, K. Q. Zong, Y. S. Liu and S. H. Hu, Anal. Chem., 2022, 94, 1286–1293 CrossRef CAS PubMed
- S. F. Durrant, Fresenius' J. Anal. Chem., 1993, 347, 389–392 CrossRef CAS
- Z. C. Hu, S. Gao, Y. S. Liu, S. H. Hu, H. H. Chen and H. L. Yuan, J. Anal. At. Spectrom., 2008, 23, 1093–1101 RSC
- J. W. H. Lam and G. Horlick, Spectrochim. Acta, Part B, 1990, 45, 1313–1325 CrossRef
- J. S. Wang, E. H. Evans and J. A. Caruso, J. Anal. At. Spectrom., 1992, 7, 929–936 RSC
- D. Günther and B. Hattendorf, TrAC, Trends Anal. Chem., 2005, 24, 255–265 CrossRef
- T. J. Shaw, T. Duncan and B. Schnetger, Anal. Chem., 2003, 75, 3396–3403 CrossRef CAS PubMed
- M. Raso, P. Censi and F. Saiano, Talanta, 2013, 116, 1085–1090 CrossRef CAS PubMed
- H. P. Longerich, S. E. Jackson and D. Günther, J. Anal. At. Spectrom., 1996, 11, 899–904 RSC
- H. T. Yue, C. Ling, T. Yang, X. B. Chen, Y. L. Chen, H. T. Deng, Q. Wu, J. C. Chen and G.-Q. Chen, Biotechnol. Biofuels, 2014, 7, 108 CrossRef
- T. C. C. Rousseau, J. E. Sonke, J. Chmeleff, F. Candaudap, F. Lacan, G. Boaventura, P. Seyler and C. Jeandel, J. Anal. At. Spectrom., 2013, 28, 573–584 RSC
- C. Duyck, R. R. Alves Peixoto, A. A. Rocha, H. Guilhermond de Souza Severino, P. V. Oliveira, R. Damasceno and R. Lorençatto, J. Anal. At. Spectrom., 2022, 37, 474–496 RSC
- L. Yang, K. Nadeau, J. Meija, P. Grinberg, E. Pagliano, F. Ardini, M. Grotti, C. Schlosser, P. Streu, E. P. Achterberg, Y. Sohrin, T. Minami, L. Zheng, J. Wu, G. Chen, M. J. Ellwood, C. Turetta, A. Aguilar-Islas, R. Rember, G. Sarthou, M. Tonnard, H. Planquette, T. Matoušek, S. Crum and Z. Mester, Anal. Bioanal. Chem., 2018, 410, 4469–4479 CrossRef CAS PubMed
- S. M. McLennan, Rev. Mineral. Geochem., 1989, 21, 169–200 CAS
- N. D. MacRae, H. W. Nesbitt and B. I. Kronberg, Earth Planet. Sci. Lett., 1992, 109, 585–591 CrossRef CAS
- L. Ge, S.-Y. Jiang, R. Swennen, T. Yang, J.-H. Yang, N.-Y. Wu, J. Liu and D.-H. Chen, Mar. Geol., 2010, 277, 21–30 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ja00236a |
| This journal is © The Royal Society of Chemistry 2023 |