
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Co and Ni single sites on the (111)n surface of γ-Al2O3 – a periodic boundary DFT study†
Jiande
Gu
*,
Jing
Wang
and
Jerzy
Leszczynski
 *
*
Interdisciplinary Nanotoxicity Center, Department of Chemistry, Physics and Atmospheric Sciences, Jackson State University, Jackson, MS 39217, USA. E-mail: jiande@icnanotox.org; jerzy@icnanotox.org
First published on 10th January 2023
Abstract
The influences of increasing the number of d-electrons in the single metal (Fe-like) substituted (111)n surface of γ-Al2O3 on its possible catalytic effects were explored. The energetic properties, local structures, and in-site electron configurations of the most active tri-coordinated Co and Ni single-site (111)n surface of γ-Al2O3 have been studied using the density functional theory (DFT) approach under periodic boundary conditions. The replacement of Al by a Co or Ni atom on the I position of the (111)n surface leads to significant elongations of metal–O distances. The energy released from the substitution process on the AlI site of the (111)n surface follows the sequence NiI (164.85 kcal mol−1) > CoI (113.17 kcal mol−1) > FeI (44.30 kcal mol−1). The triplet and quintet (ground state) of the CoI substituted complex are energy degenerate. Also, the doublet and quartet (ground state) of the NiI substituted complex have the same stable energy. This energy degeneracy comes from the α–β electron flipping on the p-orbital of the neighboring O that is next to the substituted CoI or NiI site on the (111)n surface of γ-Al2O3. Different from the FeI substituted single-site (111)n surface, in which the electron configuration of FeI varies according to its spin-multiplicity state, substituted NiI has a unique d8 electron configuration in all three spin states, and similarly, CoI has a unique d7 electron configuration in all three open shell spin states. An increase of the population of d-electrons in the single metal substituted (111)n surface of γ-Al2O3 is likely to provide a more stable electron configuration in the metal catalytic center.
Keywords: Co substituted surface of γ-Al2O3; Ni substituted surface of γ-Al2O3; (111)n surface; Periodic boundary DFT approach; Metal catalytic center.
1 Introduction
Single metal atoms have been widely applied and extensively advanced in catalytic processes.1–21 Surface-supported single-site metal atoms have wide-ranging applications in important chemical processes such as catalyzing the oxidation of CO,1,5–17 in reforming reactions,18,19 and in generating biofuels and biomass-derived chemicals by conversion of oxygen-rich biomass through hydro-deoxygenation processes.20 ZnO surface supported single Pt and Au atom-centers have exhibited remarkable catalytic activity for reformation of methanol to H2 and CO2.18 Catalysts with Fe deposited on SiO2 surfaces have been demonstrated to have notable activity and selectivity for direct nonoxidative conversion of methane.21 Heterogeneous catalysts such as single site Pd species framed in mesoporous organosilica have been effectively used for oxidative Heck reaction22 and a single site Ni-modified Zn-MOF has been exemplified to have high selectivity for dimerization of ethylene.23 A series of transition metal single-atom catalysts (i.e., Co, Ni, Fe, Mn, Cu) has been developed. Extensive research on Ni-based and Co-based single-atom catalysts resulted in various applications in electro- and photocatalysis, batteries, solar cells, natural gas conversion, oxidations, hydrogenations, and dehydrogenation reactions.24Alumina (Al2O3) has been recognized as a promising support for single-site atom catalysts.1–6,16,17 As an effective support, Al2O3 has the function of stabilizing single atoms and even improving the catalytic activities and the selectivity of these single site atoms in catalytic processes.16,17,25–30 Gamma-alumina (γ-Al2O3) is one of the important phases of this oxide. It has been well established and widely applied in experimental investigations.31,32 Routinely, γ-Al2O3 has been defined as a defective spinel structure in which Al vacancy sites are distributed within the lattice.32–34
Both experimental modeling and theoretical modeling on low-index surfaces indicate that penta-coordinated Al sites26,27,29,30,35,36 and tri-coordinated Al centers on the surfaces are especially important in the formation of active centers of catalysts.25,30,34,37–43 The highly reactive acid–base pairs created at the tri-coordinated Al on the γ-Al2O3 surfaces enable low-energy pathways for the heterolytic splitting of C–H bonds of methane.37,38 Recent studies have demonstrated that the most stable (110) surface of γ-Al2O3 provides three tetra-coordinated Al/Fe sites and one tri-coordinated Al/Fe site. On the other hand, a newly uncovered form of the (111) surface of γ-Al2O3 provides support for three structural stable tri-coordinated single Al/Fe sites on the γ-Al2O3 surface.30,44
The catalytic characteristics of single metal atoms on various supporting backgrounds have been reported to be very sensitive to the electronic and geometric surroundings of the support.45 The electronic structures of single-atom sites can be affected by the interacting atoms encircled, which might lead to alternations in the catalytic behaviors as compared to single-atom sites on nonmetallic supports.45–54 The variation of electronic and geometric surroundings of supports can provide new perceptions for activity and selectivity as well as for catalyzing effects.
As an elaboration of our recent studies on the formation of γ-Al2O3 supported tri-coordinated Fe single-site complexes,29,30 here we report the results of computational investigations on the local coordination and the in-site electronic structures of the Co and Ni single-atom sites on the most active tri-coordinated Al/Fe single-site (111) surface of γ-Al2O3. Compared to that of Fe, the electronic structures of the related Co and Ni compounds increase the population of d-electrons by 1 (Co) and 2 (Ni), respectively. Therefore, a theoretical investigation of these systems enables the elucidation of the effects of variations of d-electron population on their energetic properties and their microstructural characteristics. Such studies can provide a deep-rooted understanding and new outlooks for the activities, selectivity, and possible catalytic effects of these γ-Al2O3 supported single-atom metal systems.
2 Results and discussion
2.1 Co single site on the (111)n surface of γ-Al2O3
Among three different Al sites exposed on the (111)n surface (MI, AlII, and AlIII as shown in Fig. 1), MI has been recognized to be the most stable site to host Fe in a previous study.30 The energy released from this most stable substitution Fe3+ (sextet) + [(111)n surface of γ-Al2O3] → Al3+ + [FeI (doublet) on the (111)n surface of γ-Al2O3] has been calculated to be −44.30 kcal mol−1. This provides an additional (by about 22.82 and 26.56 kcal mol−1, respectively) energy gain compared to predictions for the two other possible Fe replaced sites for the ground state.30 Consequently, this most stable substitution site is our focal point for the present research.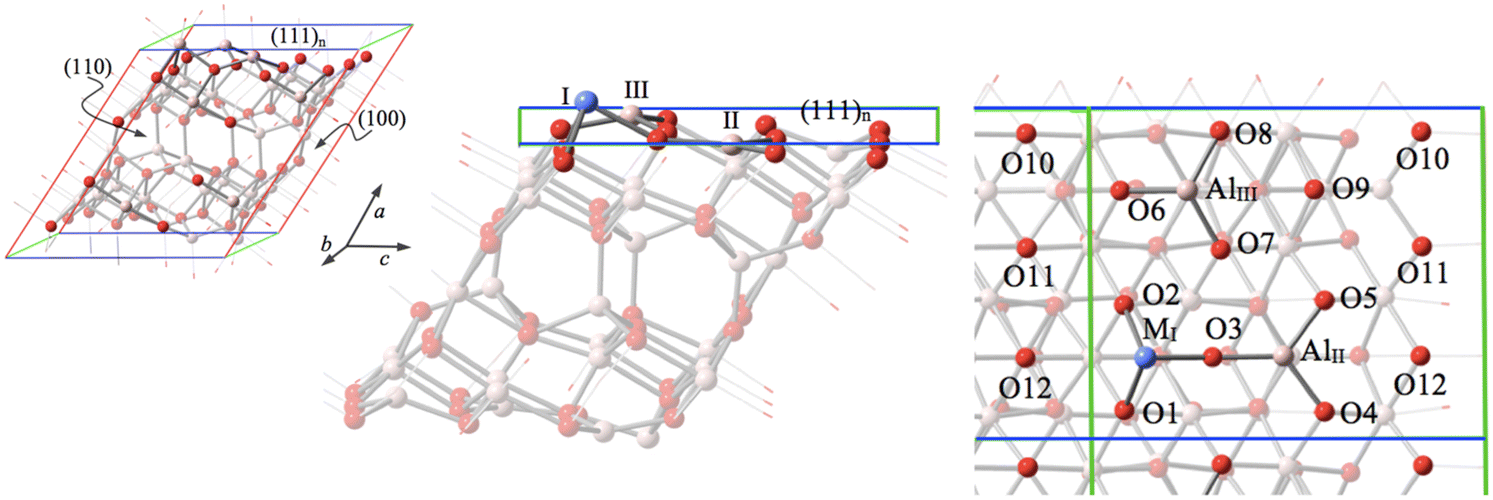 | ||
| Fig. 1 The fully optimized supercell (left) and the (111)n surface structure (side view and top view). The supercell model consists of two units along the (111)n surface direction (8 atomic slabs) adopted in this study. The upper (111)n surface layer consists of three metal atoms (MI, AlII, and AlIII, where MI represents Al, Co, and Ni) and 12 oxygen atoms. The bottom (111)n surface layer consists of three Al atoms (AlI, AlII, AlIII) and 12 oxygen atoms. Color legend: red for O, light pink for Al, and blue for Co or Ni. | ||
In this study, the spin multiplicity of Co was considered to vary from singlet (with 2S + 1 = 1) to triplet, quintet, and septet, respectively. The quintet (2S + 1 = 5) has been found to be the ground state for the Co replacements on the AlI site of the (111)n surface of γ-Al2O3. The energy released from this Co substitution process Co3+ (quintet) + [(111)n surface of γ-Al2O3] → Al3+ + [CoI (spin-state) on the (111)n surface of γ-Al2O3] has been computed to be −113.17 kcal mol−1 for the quintet, about −68.87 kcal mol−1 higher than the energy gain in the corresponding Fe substitution. The relative stable energies of the CoI substituted surface (111)n of gamma alumina with different spin states are summarized in Table 1. It is important to note that the triplet is virtually the same as the quintet in terms of energy. The ground state is therefore degenerate. Since the energy difference between ion Co3+ (quintet, ground state) and ion Co3+ (triplet) amounts to 44.33 kcal mol−1 in the present calculation, this degeneracy of the γ-Al2O3 supported CoI complex implies that the coordination of the γ-Al2O3 surface may affect the peripheral electronic structure around the hosted Co3+.
Table 2 displays the main geometric parameters of the optimized structure of the CoI substituted (111)n surface of the supercell. Replacement of AlI by Co on the (111)n surface results in elongations of metal–O bond lengths, especially for the high spin states, as compared to the pure γ-Al2O3. The CoI–O distances amount to 1.93–1.95 Å for the ground state (quintet) of the CoI substituted (111)n complex, about 0.18 Å longer than the corresponding AlI–O distance for the un-substituted (111)n surface. Coherent to the energetic degeneracy, nearly the same CoI–O distances (1.93–1.95 Å) are identified for the corresponding triplet state. A similar local structure is also unveiled for the system with the highest spin state. The related CoI–O distances are predicted to be 1.94–1.95 Å for the septet state. On the other hand, the CoI–O distances are around 1.77–1.78 Å for the low spin state (singlet) of the CoI substituted (111)n surface of γ-Al2O3, ∼0.01 Å longer than the corresponding AlI–O distance of the un-substituted (111)n surface only. The CoI–O bond distances are strongly correlated to the spin multiplicity of the coordinated Co on the (111)n surface of γ-Al2O3. This MI–O bond distance–spin multiplicity correlation can also be found in the FeI substituted (111)n surface of γ-Al2O3, in which the FeI–O bond distances are reported to be around 1.78 Å for the low-spin state (doublet) and 1.91–1.94 Å for the high-spin states (quartet and sextet), accordingly.
| Non-substituted | Co-substituted | ||||
|---|---|---|---|---|---|
| Singlet | Singlet | Triplet | Quintet | Septet | |
| a RI-i: Co–O atomic distance for substituted surfaces (Al–O atomic distance for non-substituted surface) in Å; DijkI: dihedral angle in (°); for labels, see Fig. 1. | |||||
| RI-1 | 1.766 | 1.772 | 1.946 | 1.947 | 1.942 |
| RI-2 | 1.767 | 1.774 | 1.948 | 1.948 | 1.945 |
| RI-3 | 1.763 | 1.778 | 1.929 | 1.932 | 1.939 |
| D1123 | 51.29 | 58.10 | 57.97 | 58.10 | 58.36 |
| Singlet | Triplet | Quintet | Septet | |||||
|---|---|---|---|---|---|---|---|---|
| Charge | Spin | Charge | Spin | Charge | Spin | Charge | Spin | |
| a Analysis based on the density calculated using the MN12-L functional with basis set SV. For labels, see Fig. 1. The numbers in the parentheses are for the AlI site in the non-substituted system. | ||||||||
| CoI | 1.4711 | 0.000 | 1.5467 | 2.724 | 1.5463 | 2.724 | 1.5478 | 2.724 |
| (1.7668) | (0.000) | |||||||
| O1 | −1.0557 | 0.000 | −1.2100 | 0.076 | −1.2096 | 0.081 | −1.2117 | 0.082 |
| (−1.2165) | (0.000) | |||||||
| O2 | −1.0517 | 0.000 | −1.2082 | 0.076 | −1.2080 | 0.081 | −1.2097 | 0.082 |
| (−1.2158) | (0.000) | |||||||
| O3 | −1.0992 | 0.000 | −1.1853 | 0.073 | −1.1855 | 0.073 | −1.1905 | 0.071 |
| (−1.1686) | (0.000) | |||||||
| O12 | −1.1294 | 0.000 | −0.8021 | −0.721 | −0.8024 | 0.724 | −0.7957 | 0.731 |
For the singlet state, the analysis of the projected density of states (PDOS, Fig. 2) reveals the typical closed-shell character, with symmetric α–β spin density of states.
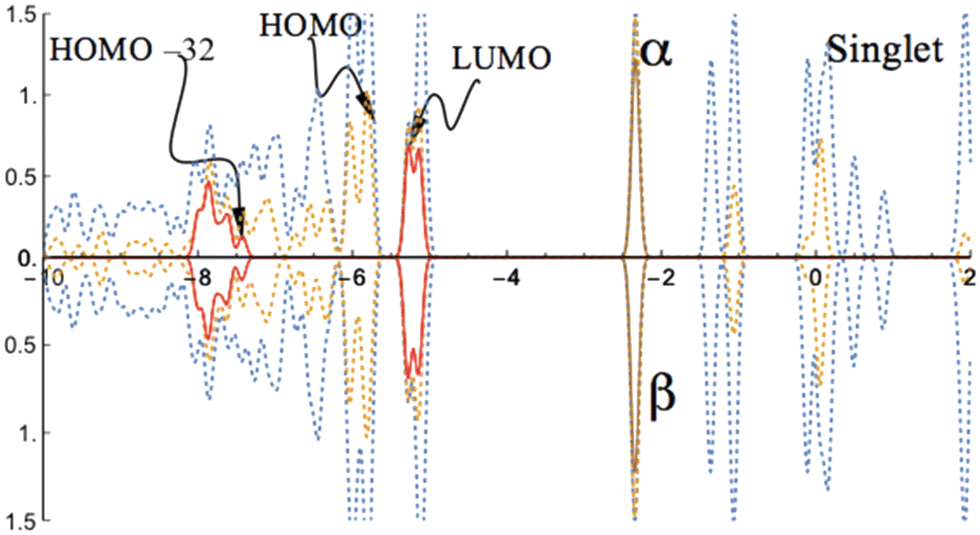 | ||
| Fig. 2 PDOS of the CoI substituted (111)n surface phase of γ-Al2O3 in the singlet state. The surface layer consists of one Co atom, two Al atoms, and twelve O atoms (CoI, AlII, AlIII, On, n = 1 to 12; see Fig. 1). The dotted blue lines are from both CoI substituted and un-substituted surface layers; the dotted orange lines are the contribution from the CoI substituted surface layer; the solid red lines are the contribution from d-type orbitals of CoI; the solid brown lines are the contribution from s-type orbitals of CoI. | ||
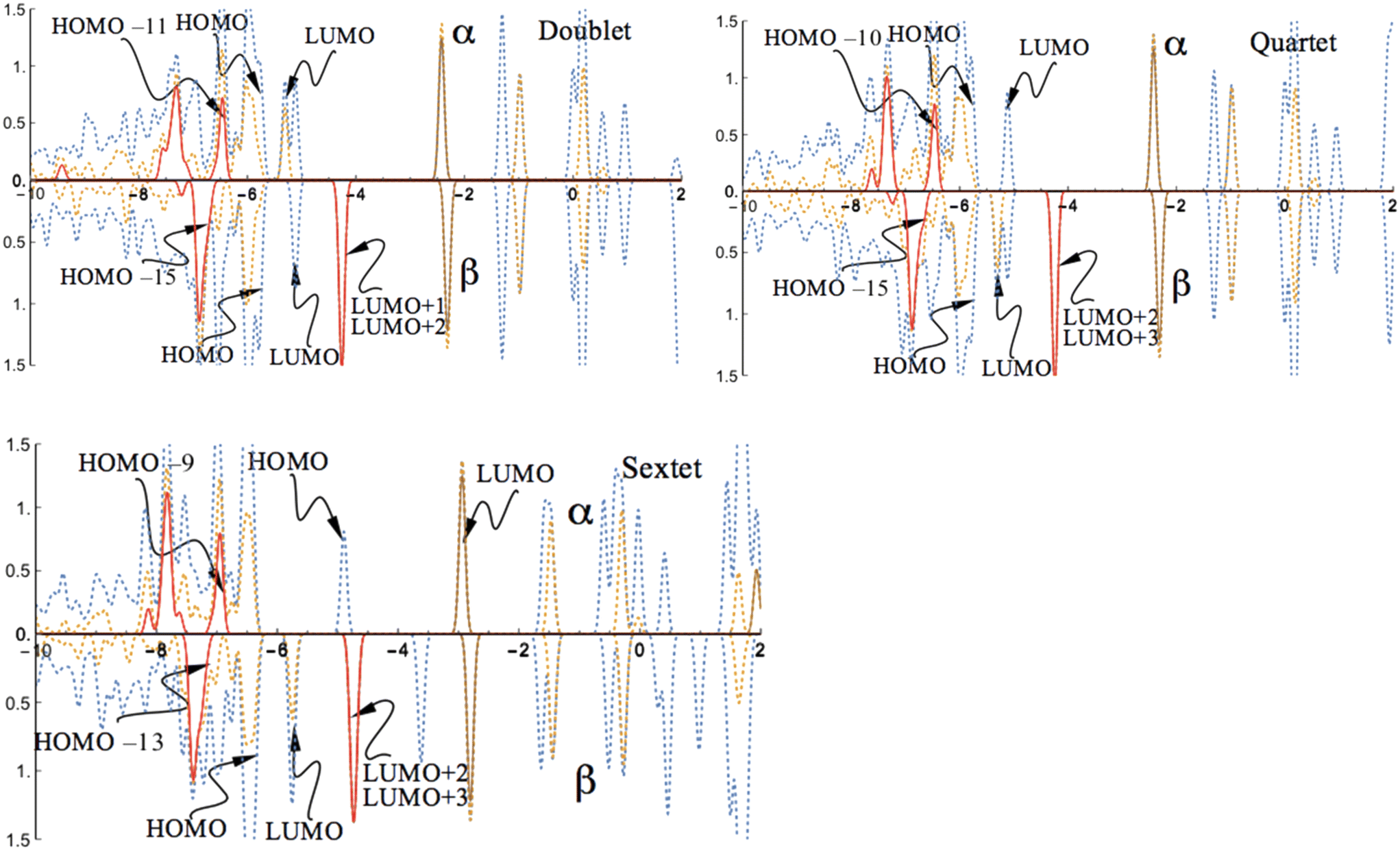
While the unoccupied states closest to the band gap are mainly determined by the d-orbitals of the coordinated Co atom, the occupied states near the band gap are dominated by the orbitals of the O atoms in the surface layer. The band gap (HOMO–LUMO gap) is predicted to be 0.49 eV for the singlet state of the CoI substituted (111)n surface system. This band gap is about 0.17 eV smaller than that of the un-substituted (111)n surface of γ-Al2O3 (0.66 eV).30,44 As a comparison, the minimum direct band gap (β-spin) for the doublet state of the FeI substituted (111)n surface was reported to be 0.47 eV.30
The corresponding MO analysis reveals that the p-type orbitals of O12 of both CoI substituted and un-substituted (the bottom layer of the supercell in Fig. 3 of the HOMO) surfaces dominate the HOMO. Thus, the HOMO is not localized entirely around the metal supporting site; it spreads to both substituted and the un-substituted surfaces.
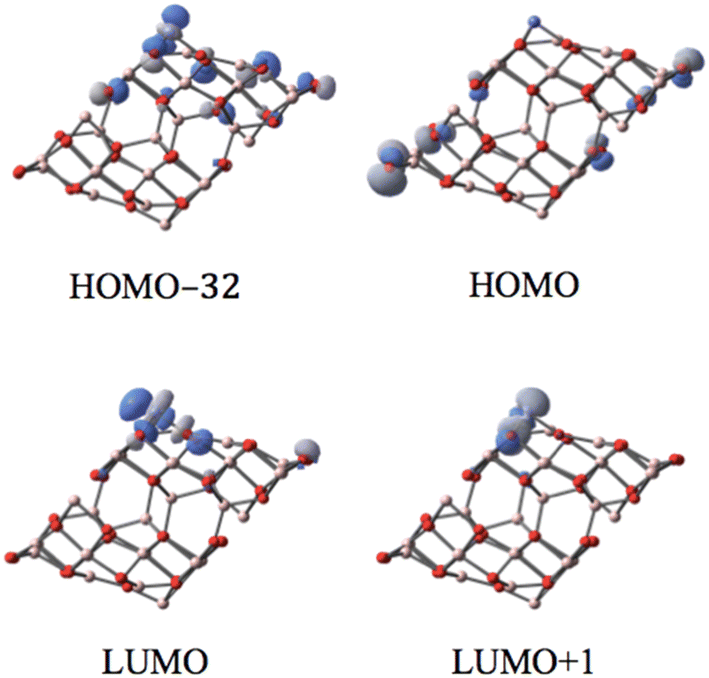 | ||
| Fig. 3 The MOs associated with the band gap for the singlet state of the CoI substituted (111)n surface of γ-alumina. | ||
Three fully occupied d-orbitals of CoI have been identified to contribute to the HOMO−32 and molecular orbitals below. These occupied d-type related MOs are highly non-localized. In contrast, the LUMO (also LUMO+1) is well localized on the d-type orbital of the surface coordinated CoI and the p-orbital of the related supporting O atoms. Hence, these two d-orbitals of CoI are totally unoccupied. On the (111)n surface, the supported CoI has the closed-shell d6 electron configuration in the singlet state.
Consistent with the charge and spin density distributions, the PDOSs of the high spin states exhibit nearly the same band character of the coordinated CoI for both triplet and quintet states. A similar feature is also shown in the band of the septet state. There are no contributions near the band gap from the d- or s-orbitals of the γ-Al2O3 surface supported CoI for these high spin states (Fig. 4).
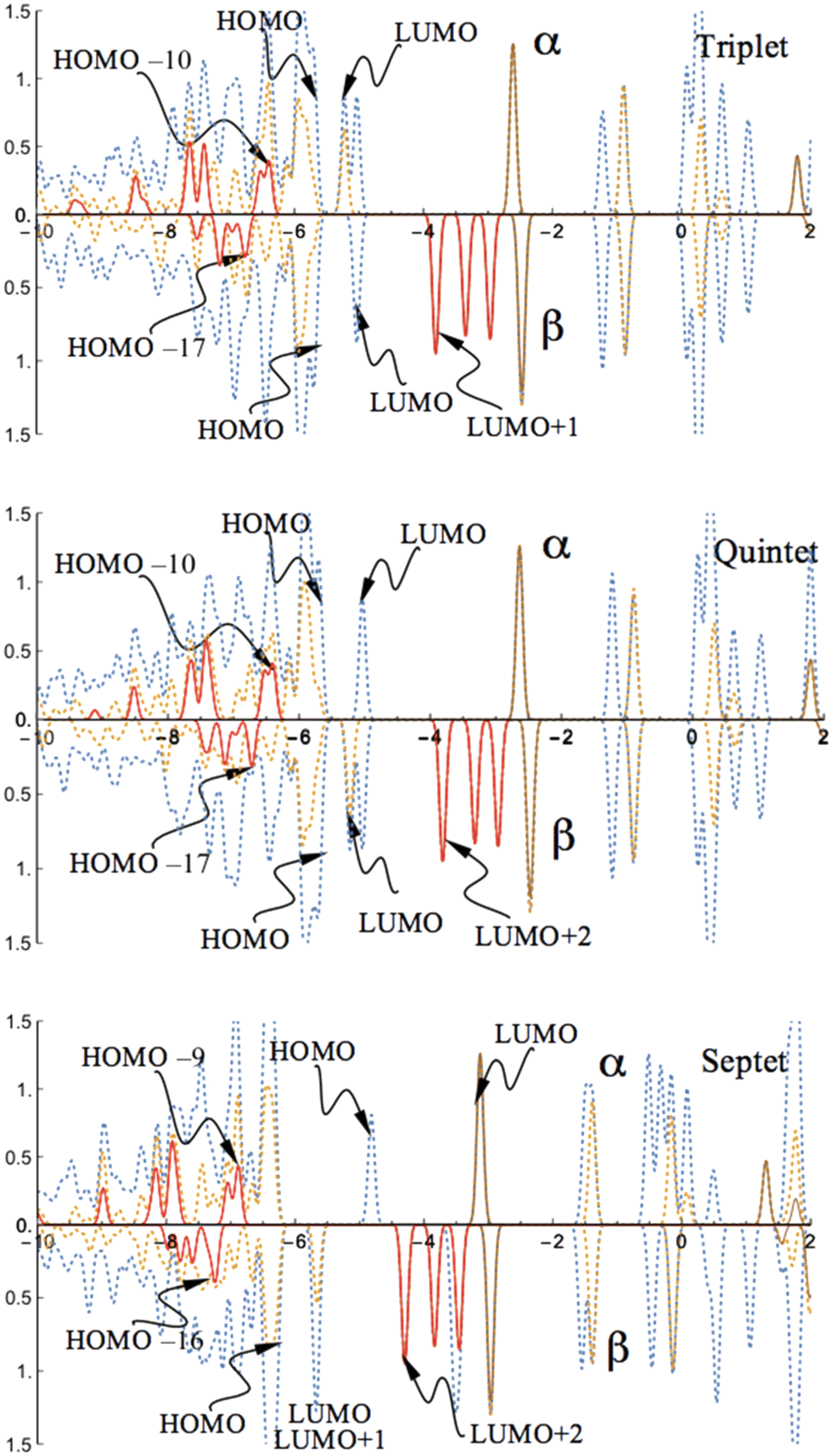 | ||
| Fig. 4 PDOS of the CoI substituted (111)n surface phase of γ-Al2O3 in the triplet, quintet, and septet states. The surface layer consists of one Co atom, two Al atoms, and twelve O atoms (CoI, AlII, AlIII, On, n = 1 to 12; see Fig. 1). The dotted blue lines are from both CoI substituted and un-substituted surface layers; the dotted orange lines are the contribution from the CoI substituted surface layer; the solid red lines are the contribution from d-type orbitals of CoI; the solid brown lines are the contribution from s-type orbitals of CoI. | ||
The state densities closest to the band gap are mainly determined by the sp3-orbitals of Al of the un-substituted (111)n surface and the p-orbitals of the O atoms in the surface layer. The band gaps (HOMO–LUMO gap) are evaluated to be 0.47 eV (α-spin) and 0.66 eV (β-spin) for the triplet state, and 0.66 eV (α-spin) and 0.48 eV (β-spin) for the quintet state, respectively. A large band gap is found for the α-spin contribution in the septet state. For this state, the band gap is 1.76 eV for α-spin and 0.65 eV for the β-spin part. Interestingly, the band gap for the highest spin state (sextet) of the FeI substituted (111)n surface was reported to be 0.66 eV for α-spin and 0.27 eV for β-spin.30 Taking into account that the s-type orbital (4s) of Co dominates the LUMO in the α-spin part for the septet state, this significant variation in the band gap is expected.
MOs of the triplet and quintet states depicted in Fig. 5 reveal the considerable similarity between the nearly degenerate triplet and quintet states. The sole difference is the components of the corresponding LUMO. In the triplet state, the p-type orbital with α-spin of O12 of the CoI substituted surface is the main contribution of the LUMO with α-spin, while in the quintet state, it mainly arises from the LUMO with β-spin. The former suggests that in the triplet state the corresponding β component of the p-type orbital is occupied (providing a spin density of −0.72, and as a result of balancing 2.72 of spin density on Co, the total spin density amounts to 2). The latter implies that in the quintet, the α-spin component of this p-type orbital is occupied (the resulting spin density amounts to 0.72, leaving the corresponding β-spin orbital un-occupied). The energy degeneracy of the triplet and quintet states is due to this α–β electron flipping on the p-orbital of O12.
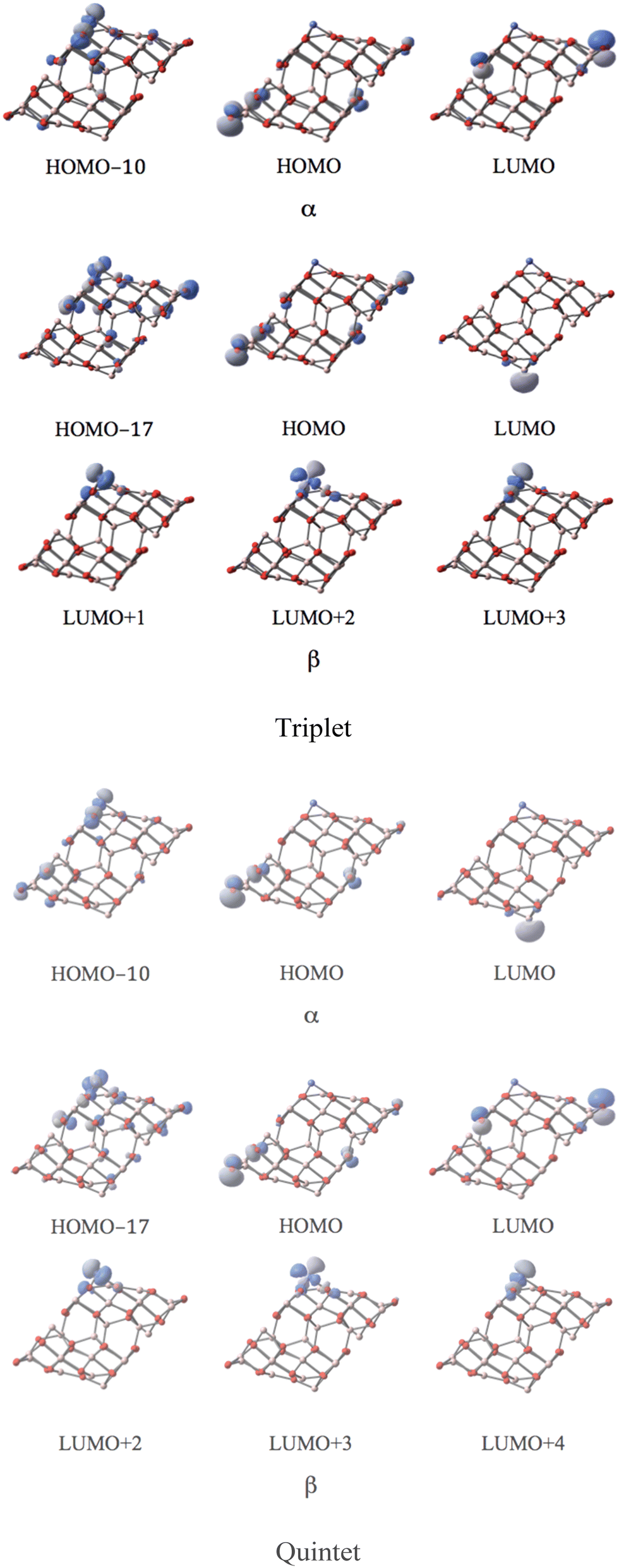 | ||
| Fig. 5 The MOs associated with the band gap for the degenerate triplet and quintet states of the CoI substituted (111)n surface of γ-alumina. | ||
MOs of the septet state in Fig. 6 reveal the participation of s-orbital components of AlI (HOMO) and CoI (LUMO) in the large band gap in α-spin contributions.
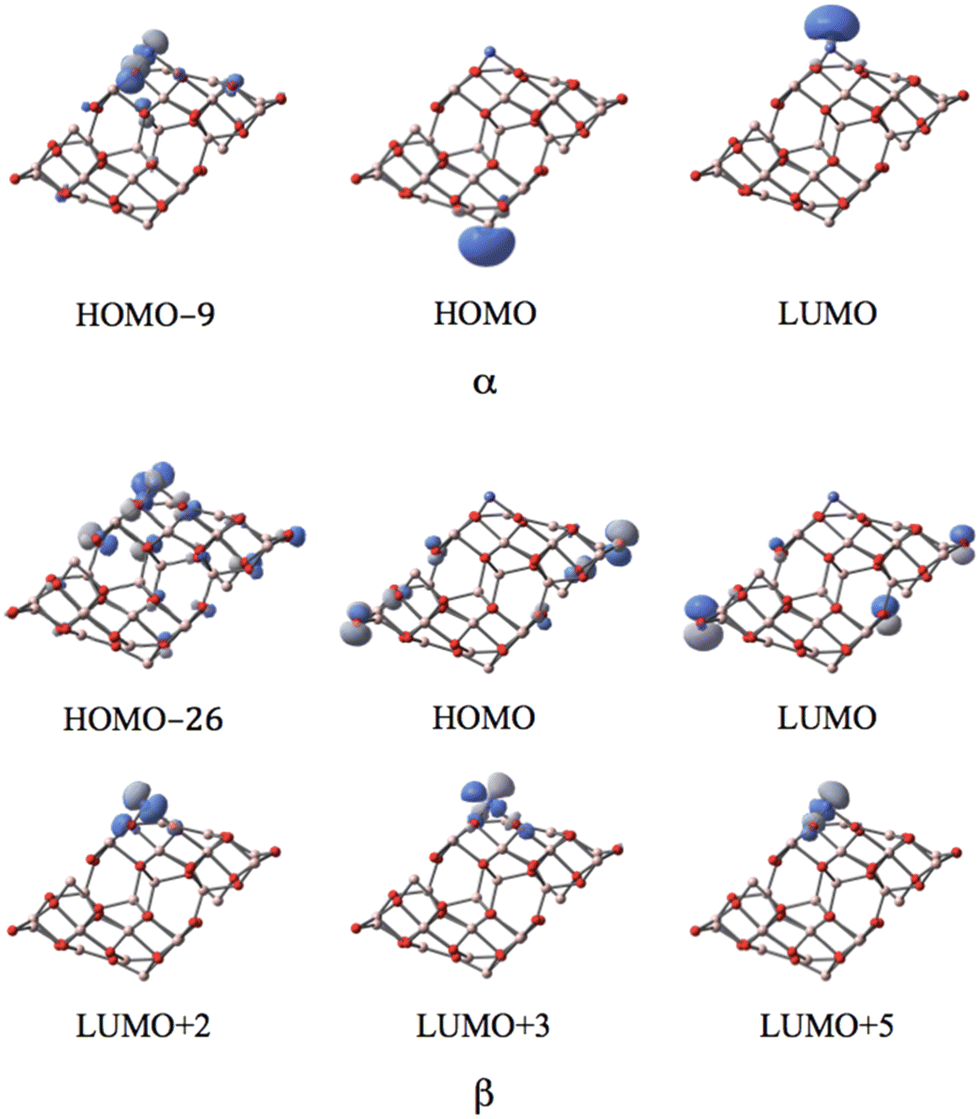 | ||
| Fig. 6 The MOs associated with the band gap for the septet state of the CoI substituted (111)n surface of γ-alumina. | ||
Three well-localized d-orbitals of CoI can be identified in the β-spin component as unoccupied orbitals for all high spin states. They are the LUMO+1, LUMO+2, and LUMO+3 for the triplet, LUMO+2, LUMO+3, and LUMO+4 for the quintet, and LUMO+2, LUMO+3, and LUMO+5 for the septet state. Thus, three d-orbitals of CoI are only half-occupied. Consequently, the coordinated CoI on the (111)n surface of γ-Al2O3 has the open-shell d7 electron configuration in the high spin states.
2.2 Ni single site on the (111)n surface of γ-Al2O3
Three spin states were revealed for Ni on the substituted (111)n surface of γ-alumina, ranging from doublet (with 2S + 1 = 2) to quartet (2S + 1 = 4) and sextet (2S + 1 = 6), respectively. The quartet is the ground state for the replacements of Ni on the AlI site of the (111)n surface of γ-Al2O3. The energy released from the Ni substitution process Ni3+ (quartet) + [(111)n surface of γ-Al2O3] → Al3+ + [NiI (quartet) on the (111)n surface of γ-Al2O3] amounts to −164.85 kcal mol−1, much larger than that in the corresponding Co substitution process (−113.17 kcal mol−1) and in the corresponding Fe substitution (−44.30 kcal mol−1). The details of relative stable energies of the NiI substituted surface (111)n of γ-Al2O3 with different spin states are provided in Table 4. It is clear that the doublet is virtually the same as the quartet state, in terms of energy. Thus, similar to the case of the CoI complex, the ground state for the single NiI substituted (111)n surface of γ-Al2O3 is nearly degenerate. The predicted energy difference between ion Ni3+ (quartet, ground state) and ion Ni3+(doublet) amounts to 30.49 kcal mol−1. The degeneracy of the ground state of the γ-Al2O3 supported NiI complex implies that the coordination of the γ-Al2O3 surface may disturb its electron structure around the hosted Ni3+.Table 5 summarizes the main geometric parameters of the optimized structure of the NiI substituted (111)n surface of γ-Al2O3. Replacement of AlI by Ni on the (111)n surface leads to elongations of metal–O bond lengths as compared to the pure γ-Al2O3. The NiI–O distances amount to 1.90–1.91 Å for all three spin states of the NiI substituted (111)n complex, approximately 0.14 Å longer than the corresponding AlI–O distance of the un-substituted (111)n surface. Consistent with the energetic degeneracy, nearly the same NiI–O bond lengths (∼1.91 Å) are identified for both doublet and quartet states. A similar local structure is also unveiled for the system with the highest spin state. The related NiI–O distances are computed to be ∼1.91 Å for the sextet state. The local geometric feature of the NiI substituted surface (111)n of gamma alumina resembles those of the CoI substituted surface (111)n in high spin states (triplet, quintet, and septet). On the other hand, the structure corresponding to short CoI–O and FeI–O distances (1.77–1.78 Å) for the low spin state of the CoI and FeI substituted (111)n surface of γ-Al2O3 has not been revealed for the NiI substituted (111)n surface of γ-Al2O3.
| Non-substituted | Ni-substituted | |||
|---|---|---|---|---|
| Singlet | Doublet | Quartet | Sextet | |
| a RI-i: Ni–O atomic distance for substituted surfaces (Al–O atomic distance for non-substituted surface) in Å; DijkI: dihedral angle in (°); for labels, see Fig. 1. The numbers in parentheses are those of the FeI substituted surface (111)n of gamma alumina, computed at the same level of theory.30 | ||||
| RI-1 | 1.766 | 1.907 | 1.908 | 1.905 |
| (1.778) | (1.906) | (1.942) | ||
| RI-2 | 1.767 | 1.901 | 1.910 | 1.906 |
| (1.779) | (1.908) | (1.941) | ||
| RI-3 | 1.763 | 1.905 | 1.904 | 1.905 |
| (1.812) | (1.908) | (1.947) | ||
| D1123 | 51.29 | 58.16 | 58.08 | 58.31 |
| (53.74) | (58.52) | (58.51) | ||
| Doublet | Quartet | Sextet | ||||
|---|---|---|---|---|---|---|
| Charge | Spin | Charge | Spin | Charge | Spin | |
| a Analysis based on the density calculated using the MN12-L functional with basis set SV. For labels, see Fig. 1. The numbers in parentheses are those of the FeI substituted surface (111)n, computed at the same level of theory.30 | ||||||
| NiI | 1.4581 | 1.619 | 1.4574 | 1.623 | 1.4592 | 1.623 |
| (1.5595) | (1.067) | (1.5769) | (3.489) | (1.5455) | (3.692) | |
| O1 | −1.1930 | 0.110 | −1.1936 | 0.115 | −1.1958 | 0.116 |
| (−1.0696) | (−0.015) | (−1.1466) | (0.057) | (−1.1857) | (0.073) | |
| O2 | −1.1908 | 0.110 | −1.1913 | 0.115 | −1.1940 | 0.115 |
| (−1.0683) | (−0.015) | (−1.1800) | (0.122) | (−1.2098) | (0.091) | |
| O3 | −1.1778 | 0.100 | −1.1788 | 0.100 | −1.1829 | 0.099 |
| (−1.1045) | (−0.023) | (−1.1660) | (0.066) | (−1.2107) | (0.091) | |
| O12 | −0.7999 | −0.723 | −0.8026 | 0.725 | −0.7955 | 0.7322 |
| (−1.1341) | (−0.017) | (−1.0533) | (−0.244) | (−0.8082) | (0.712) | |
For comparison, the corresponding Mulliken charge and spin density of the FeI substituted surface (111)n are also listed in Table 6. In the FeI substituted complex, one spin electron is assigned to FeI in its doublet state, while more than three spin electrons are located on FeI in the related quartet and sextet states.30 Moreover, only in its sextet state, the neighboring O12 site of the surface layer hosts roughly one spin electron (spin density of 0.72, α-spin).
The PDOSs of the doublet and quartet states demonstrate nearly the same band character of the coordinated NiI, as expected from their similar charge and spin density distributions and from their energy degeneracy. No contributions from the d- or s-orbitals of the γ-Al2O3 surface near the band gap are revealed for these two degenerate states (Fig. 7).
 | ||
| Fig. 7 PDOS of the NiI substituted (111)n surface phase of γ-Al2O3 in the doublet, quintet, and sextet states. The surface layer consists of one Ni atom, two Al atoms, and twelve O atoms (NiI, AlII, AlIII, On, n = 1 to 12; see Fig. 1). The dotted blue lines are from both NiI substituted and un-substituted surface layers; the dotted orange lines are the contribution from the NiI substituted surface layer; the solid red lines are the contribution from d-type orbitals of NiI; the solid brown lines are the contribution from s-type orbitals of NiI. | ||
The state densities flanking the band gap are mainly determined by the p-orbitals of the O atoms in the surface layer and the sp3-orbitals of Al of the un-substituted (111)n surface. The band gaps are evaluated to be 0.47 eV (α-spin) and 0.66 eV (β-spin) for the doublet state, and 0.65 eV (α-spin) and 0.46 eV (β-spin) for the quartet state, respectively. This band gap pattern of the NiI substituted surface (111)n is nearly the same as that of the CoI substituted surface (111)n in the energy degenerate triplet and quintet spin states and hence, both NiI substituted and CoI substituted systems have similar band gaps.
For the sextet state, the s-type orbital (4s) of Ni serves as the LUMO in the α-spin component, and therefore the band gap is expected to change. A large band gap, 2.06 eV, is found for the α-spin contribution in the sextet state. Meanwhile, for the β-spin component, the band gap is dominated by the p-orbitals of the O atoms in the surface layer and the band gap value amounts to 0.65 eV for the β-spin component.
MOs of the doublet and quartet states depicted in Fig. 8 reveal the high similarity between the nearly degenerate doublet and quartet states of the NiI substituted surface. The unique difference is revealed in the case of the corresponding LUMO. While the p-type orbital of O12 of the coordinated NiI represents the LUMO with α-spin in the doublet, in the quartet, it swaps the spin to form a β-spin LUMO. The former suggests that in the doublet state, the corresponding p-type orbital is occupied in its β-spin component (spin density of −0.72 in Mulliken population analysis; balancing 1.62 of spin density on Ni results in a total spin density of 1), In the quartet state, the α-spin component of the p-type orbital is occupied (spin density of 0.72), leaving the corresponding β-spin orbital un-occupied, This α–β electron flipping on the p-orbital of O12 leads to the energy degeneracy between the doublet and quartet states.
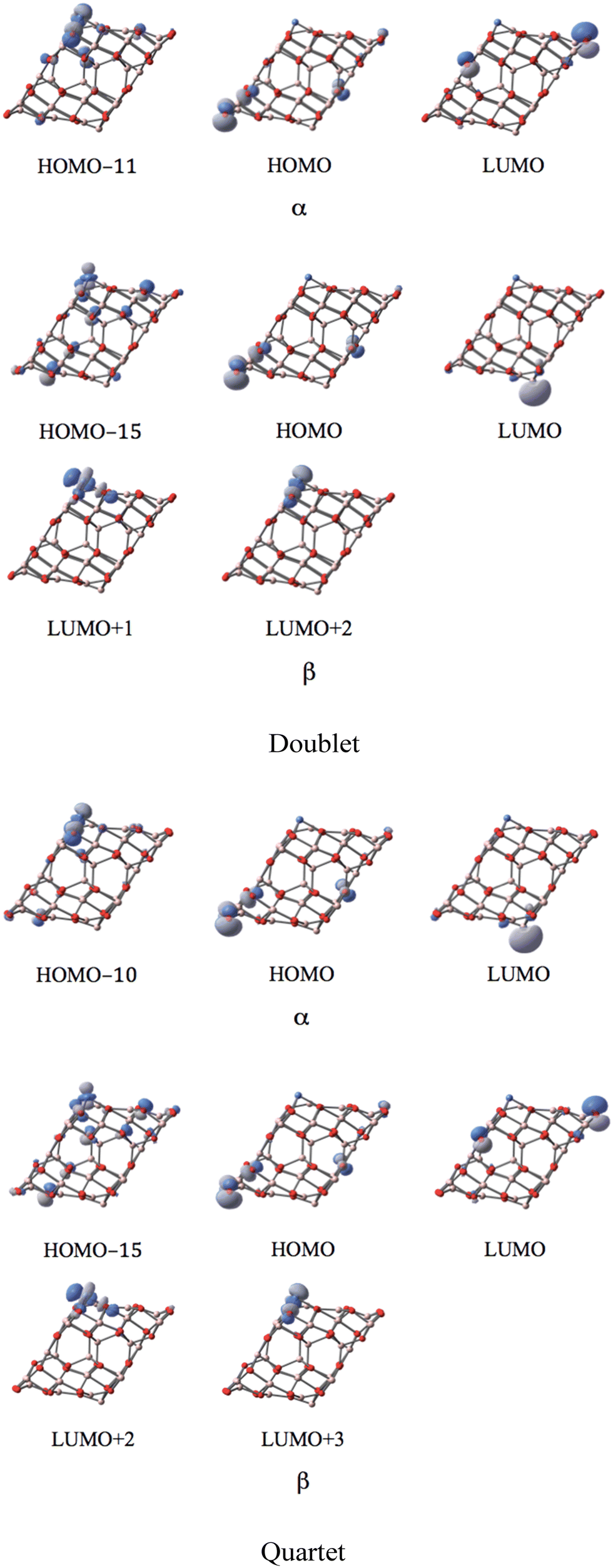 | ||
| Fig. 8 The MOs associated with the band gap for the degenerate doublet and quartet states of the NiI substituted (111)n surface of γ-alumina. | ||
Five d-type orbitals of NiI are located below the HOMO−11 (ranging from HOMO−11 to HOMO−37) in the α-spin component for the doublet state, and below the HOMO−10 (ranging from HOMO−10 to HOMO−39) for the quartet state. In the β-spin state, only three d-orbitals of NiI can be identified below the HOMO−15 (ranging from HOMO−11 to HOMO−23) for the doublet state, and below the HOMO−15 (ranging from HOMO−10 to HOMO−24) for the quartet state. Two localized d-orbitals of NiI appear in the β-spin component as unoccupied orbitals for both doublet and quartet. These are the LUMO+1 and LUMO+2 in the doublet state, and the LUMO+2 and LUMO+3 in the quintet state. Consequently, the coordinated NiI on the (111)n surface of γ-Al2O3 has the open-shell d8 electron configuration in the doublet and quartet spin states.
MOs of the sextet state in Fig. 9 reveal the participation of s-orbital components of AlI (HOMO) and NiI (LUMO) in the large band gap in α-spin, which resembles the case of coordinated CoI on the (111)n surface.
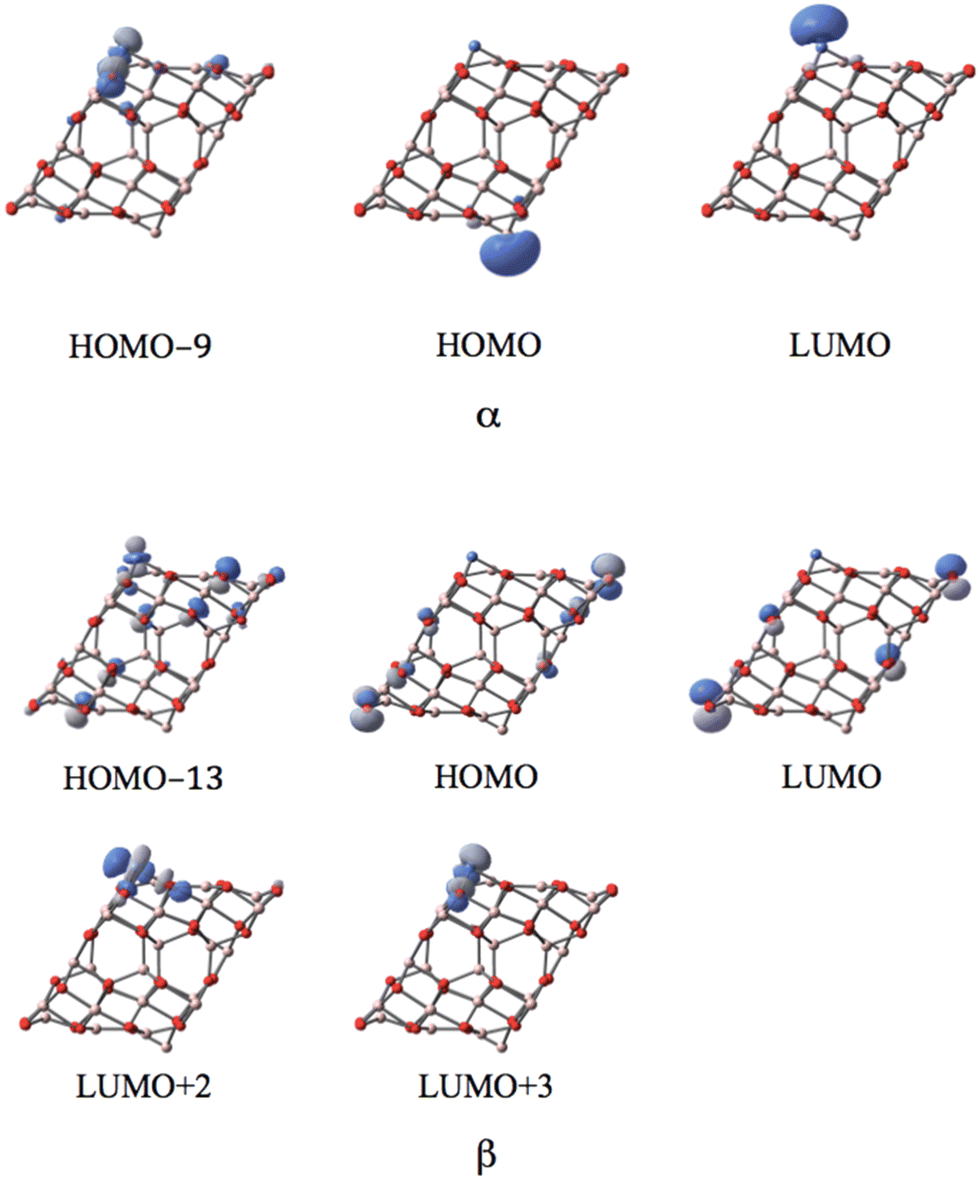 | ||
| Fig. 9 The MOs associated with the band gap for the sextet state of the NiI substituted (111)n surface of γ-alumina. | ||
In the sextet state, five occupied d-type orbitals of NiI can be found below the HOMO−9 (ranging from HOMO−9 to HOMO−38) in the α-spin component. Meanwhile, three occupied d-orbitals of NiI can be identified below the HOMO−13 (ranging from HOMO−13 to HOMO−21) for the sextet state. Two unoccupied d-orbitals of NiI appear in the corresponding β-spin component (LUMO+2 and LUMO+3). Consequently, the coordinated NiI on the (111)n surface of γ-Al2O3 also has the open-shell d8 electron configuration in the sextet spin state.
3 Conclusions
A computational study of coordination of Co and Ni single-atom sites on the most active tri-coordinated Al/metal single-site (111)n surface of γ-Al2O3 reveals that the energy released from the single atom substitution process on the AlI site of (111)n surface follows the sequence NiI (164.85 kcal mol−1) > CoI (113.17 kcal mol−1) > FeI (44.30 kcal mol−1).The ground states of both CoI and NiI substituted single-site (111)n surfaces of γ-Al2O3 are energy degenerate. The triplet and quintet states of the CoI substituted complex have the same stable energy. The same energy degeneracy is predicted for the doublet and quartet states of the NiI substituted complex. This arises from the α–β electron flipping on the p-orbital of the neighboring O12 that is next to the substituted transition metal site on the (111)n surface of γ-Al2O3. As shown in our previous study on the CO/CO2 conversion mechanism on the calcium ferrite (CFO) surface, these energy degenerate CoI or NiI sites on the (111)n surface of γ-Al2O3 could serve as effective catalytic centers for the CO/CO2 conversion.
Different from the FeI substituted single-site (111)n surface, in which the electron configuration of FeI varies according to its spin-multiplicity state, NiI has a unique d8 electron configuration in all three spin states of the NiI substituted single-site (111)n surface. Analogously, CoI has a unique d7 electron configuration in all three open shell spin states of the CoI substituted single-site (111)n surface. Only in the singlet state, CoI adopts a d6 electron configuration on the supportive (111)n surface of γ-Al2O3. Consequently, in the state with the highest spin multiplicity, the spin density extends to the un-substituted (111)n surface for the CoI and NiI substituted single-site (111)n systems. In contrast, in the case of Fe substitution, the change of spin multiplicity only leads to the alternation of the electron configuration of FeI, and the spin density distribution is limited to the FeI substituted site and its close neighboring O on the substituted surface.
The replacement of Al by a Co or Ni atom on the I position of the (111)n surface leads to significant elongations of metal–O distances. Different from the FeI substitution case, these atomic distance elongations are less affected by the variation of the corresponding spin-multiplicity states of CoI and NiI. This is due to the unique d-electron configuration of the substituted CoI (d7) and NiI (d8) atoms at the surface site. Thus, an increase of d-electrons (compared to the case of iron atom) on the metal substituted (111)n surface of γ-Al2O3 results in the formation of stable local structures and steady d-electron configurations for the CoI and NiI substituted complexes. A previous study indicated that the most stable FeI adopts a d5 electron configuration,30 so one might expect a d6 and d7 electron configuration for the CoI and NiI complexes, respectively. However, the present study illustrates that by increasing the number of d-electrons by one and two, the most stable electron configuration becomes d7 for CoI and d8 for NiI. It is interesting to understand the effects of reducing the number of d-electrons on the FeI substituted (111)n surface of γ-Al2O3, as is the case in the CrI and MnI substituted surface systems. These systems are currently studied in our laboratory.
4 Methods of computation
The DFT method along with the functional by Peverati and Truhlar (MN12-L)55 has been used in the present study. Different from traditional surface studies using the plane wave basis such as in the Vienna ab initio simulation package (VASP),56,57 here the localized basis set, namely the split valence basis set SV by Ahlrichs and coworkers,58 was applied. Reasonable estimations of the cell volume were achieved using the MN12-L functional (with double and triple zeta basis sets, i.e. SV and TZV) as reported in our previous studies.29,30,44 Compared to the experimental value of the cell volume of 46.39 (Å3/Al2O3), a cell volume of 46.94 (Å3/Al2O3) calculated at the MN12-L/SV level with the PBC approach is better than the VASP result of 47.40 (Å3/Al2O3).29,30,44 It is important to note that the application of 2-dimensional periodic boundary conditions implies an infinite vacuum above the studied surface. Therefore, the artificial long-range interactions between the surfaces are excluded by this method. The application of the MN12-L functional is an efficient approach for the computational investigation of gamma alumina systems. As a reliable compromise between accurateness and computational feasibility, the MN12-L/SV approach is applied in the reported calculations. Periodic boundary conditions were applied for the description of the bulk phase. The total k-point numbers amount to 4 × 6 × 6 (for the supercell) for the bulk phase. For the computations of the surface phases, 2-dimensional periodic boundary conditions were applied. The total k-point number is 10 × 10 in the surface phase computations. For the study of the surface, the positions of all of the atoms were fully optimized while the corresponding lattice vectors were kept fixed. It is important to note that application of 2-dimensional periodic boundary conditions implies an infinite vacuum above the studied surface. Therefore, the artificial long-range interactions between the surfaces are excluded by this method. The Gaussian-09 package of programs59 was applied for all computations.The hexagonal cell unit model described in our previous study (ref. 30 and 44, consisting of (111)n, (100) and (110) planes) was adapted in the present investigation of the (111)n surface. To ensure that there is a sufficient bulk environment underneath the investigated surface, a supercell model consisting of two units along the (111)n surface direction (8 atomic slabs) has been adapted (see Fig. 1).
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This study was supported by the NSF PREM grant #1826886. The computer time was provided by the Extreme Science and Engineering Discovery Environment (XSEDE) by the National Science Foundation Grant Number OCI-1053575 and XSEDE award allocation number DMR110088 and by the Mississippi Center for Supercomputer Research.References
- L. L. Liu and A. Corma, Metal catalysts for heterogeneous catalysis: from single atoms to nanoclusters and nanoparticles, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- C. S. Maldonado, J. R. De la Rosa, C. J. Lucio-Ortiz, A. Hernández-Ramírez, F. F. C. Barraza and J. S. Valente, Low concentration Fe-doped alumina catalysts using sol-gel and impregnation methods: the synthesis, characterization and catalytic performance during the combustion of trichloroethylene, Materials, 2014, 7, 2062–2086 CrossRef CAS PubMed.
- S. Mosallanejad, B. Z. Dlugogorski, E. M. Kennedy and M. Stockenhuber, On the chemistry of iron oxide supported on γ-alumina and silica catalysts, ACS Omega, 2018, 3, 5362–5374 CrossRef CAS PubMed.
- T. Yang, R. Fukuda, S. Hosokawa, T. Tanaka, S. Sakaki and M. Ehara, A theoretical investigation on CO oxidation by single-atom catalysts M1/γ-Al2O3 (M=Pd, Fe, Co, and Ni), ChemCatChem, 2017, 9, 1222–1229 CrossRef CAS PubMed.
- J. Wang, J. Gu, A. Rony, M. Fan and J. Leszczynski, Theoretical DFT study on the mechanisms of CO/CO2 conversion in chemical looping catalyzed by calcium ferrite, J. Phys. Chem. A, 2021, 125, 8159–8167 CrossRef CAS PubMed.
- Q. Fu, W.-X. Li, Y. Yao, H. Liu, H.-Y. Su, D. Ma, X.-K. Gu, L. Chen, Z. Wang, H. Zhang, B. Wang and X. Bao, Interface-confined ferrous centers for catalytic oxidation, Science, 2010, 328, 1141–1144 CrossRef CAS PubMed.
- M. Haruta, Size- and support-dependency in the catalysis of gold, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- B. Qiao, J.-X. Liang, A. Wang, C.-Q. Xu, J. Li, T. Zhang and J. J. Liu, Ultrastable single-atom gold catalysts with strong covalent metal- support interaction (CMSI), Nano Res., 2015, 8, 2913–2924 CrossRef CAS.
- B. Qiao, J. Liu, Y.-G. Wang, Q. Lin, X. Liu, A. Wang, J. Li, T. Zhang and J. Liu, Highly efficient catalysis of preferential oxidation of CO in H-rich stream by gold single-atom catalysts, ACS Catal., 2015, 25, 6249–6254 CrossRef.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Single-atom catalysis of CO oxidation using Pt1/ FeOx, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- M. Moses-DeBusk, M. Yoon, L. F. Allard, D. R. Mullins, Z. Wu, X. Yang, G. Veith, G. M. Stocks and C. K. Narula, CO oxidation on supported single Pt atoms: experimental and ab initio density functional studies of CO interaction with Pt atom on θ-Al2O3(010) surface, J. Am. Chem. Soc., 2013, 135, 12634–12645 CrossRef CAS PubMed.
- L. DeRita, S. Dai, K. Lopez-Zepeda, N. Pham, G. W. Graham, X. Pan and P. Christopher, Catalyst architecture for stable single atom dispersion enables site-specific spectroscopic and reactivity measure- ments of CO adsorbed to Pt atoms, oxidized Pt clusters, and metallic Pt clusters on TiO2, J. Am. Chem. Soc., 2017, 139, 14150–14165 CrossRef CAS PubMed.
- J. D. Kistler, N. Chotigkrai, P. Xu, B. Enderle, P. Praserthdam, C. Y. Chen, N. D. Browning and B. C. Gates, A single-site platinum CO oxidation catalyst in zeolite KLTL: microscopic and spectroscopic determination of the locations of the platinum atoms, Angew. Chem., Int. Ed., 2014, 53, 8904–8907 CrossRef CAS PubMed.
- J. Saavedra, T. Whittaker, Z. Chen, C. J. Pursell, R. M. Rioux and B. D. Chandler, Controlling activity and selectivity using water in the Au-catalysed preferential oxidation of CO in H2, Nat. Chem., 2016, 8, 584–589 CrossRef CAS PubMed.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. Pereira Hernandez, Y. Wang and A. K. Datye, Thermally stable single-atom platinum-on-ceria catalysts via atom trapping, Science, 2016, 353, 150–154 CrossRef CAS PubMed.
- Z. Zhang, Y. Zhu, H. Asakura, B. Zhang, J. Zhang, M. Zhou, Y. Han, T. Tanaka, A. Wang and T. Zhang, Thermally stable single atom Pt/m-Al2O3 for selective hydrogenation and CO oxidation, Nat. Commun., 2017, 8, 16100 CrossRef CAS PubMed.
- E. J. Peterson, A. T. DeLaRiva, S. Lin, R. S. Johnson, H. Guo, J. T. Miller, J. Hun Kwak, C. H. Peden, B. Kiefer, L. F. Allard, F. H. Ribeiro and A. K. Datye, Low-temperature carbon monoxide oxidation catalysed by regenerable atomically dispersed palladium on alumina, Nat. Commun., 2014, 5, 4885 CrossRef CAS PubMed.
- X.-K. Gu, B. Qiao, C.-Q. Huang, W.-C. Ding, K. Sun, E. Zhan, T. Zhang, J. Liu and W.-X. Li, Supported single Pt1/Au1 atoms for methanol steam reforming, ACS Catal., 2014, 4, 3886–3890 CrossRef CAS.
- Y. Zhu, A. An and J. He, Single-atom and small-cluster Pt induced by Sn (IV) sites confined in an LDH lattice for catalytic reforming, J. Catal., 2016, 341, 44–54 CrossRef CAS.
- S. De, B. Saha and R. Luque, Hydrodeoxygenation processes: advances on catalytic transformations of biomass-derived platform chemicals into hydrocarbon fuels, Bioresour. Technol., 2015, 178, 108–118 CrossRef CAS PubMed.
- X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng and M. Wei, Direct, nonoxidative conversion of methane to ethylene, aromatics, and hydrogen, Science, 2014, 344, 616–619 CrossRef CAS PubMed.
- H. Duan, M. Li, G. Zhang, J. R. Gallagher, Z. Huang, Y. Sun, Z. Luo, H. Chen, J. T. Miller and R. Zou, Single-site palladium(II) catalyst for oxidative heck reaction: catalytic performance and kinetic investigations, ACS Catal., 2015, 5, 3752–3759 CrossRef CAS.
- E. D. Metzger, C. K. Brozek, R. J. Comito and M. Dinca, Selective dimerization of ethylene to 1-butene with a porous catalyst, ACS Cent. Sci., 2016, 2, 148–153 CrossRef CAS PubMed.
- S. K. Kaiser, Z. Chen, D. F. Akl, S. Mitchell and J. Peŕez-Ramírez, Single-atom catalysts across the periodic table, Chem. Rev., 2020, 120, 11703–11809 CrossRef CAS PubMed.
- R. Wisechert, P. Florian, C. Copéret, D. Massiot and P. Sautet, Visibility of Al surface sites of γ-alumina: a combined computational and experimental point of view, J. Phys. Chem. C, 2014, 118, 15292–15299 CrossRef.
- J. H. Kwak, D. Mei, C. H. F. Peden, R. Rousseau and J. Szanyi, (100) Facets of γ- Al2O3: the active surfaces for alcohol dehydration reactions, Catal. Lett., 2011, 141, 649–655 CrossRef CAS.
- J. H. Kwak, J. Hu, D. Mei, C.-W. Yi, D. H. Kim, C. H. F. Peden, L. F. Allard and J. Szanyi, Coordinatively unsaturated Al3+ centers as binding sites for active catalyst phases of platinum on γ- Al2O3, Science, 2009, 325, 1670–1673 CrossRef CAS PubMed.
- J. R. Copeland, X.-R. Shi, D. S. Sholl and C. Sievers, Surface interactions of C2 and C3 polyols with γ-Al2O3 and the role of Co-adsorbed water, Langmuir, 2012, 29, 581–593 CrossRef PubMed.
- J. Gu, J. Wang and J. Leszczynski, Single site Fe on the (110) surface of γ-Al2O3: insights from a DFT study including the periodic boundary approach, Phys. Chem. Chem. Phys., 2021, 23, 7164–7177 RSC.
- J. Gu, J. Wang and J. Leszczynski, Single Fe site on the surface of γ-Al2O3: insights from density functional theory periodic boundary approach, J. Phys. Chem. C, 2020, 124, 20931–20941 CrossRef CAS.
- S. J. Wilson, The dehydration of boehmite, γ-AlOOH, to γ-Al2O3, J. Solid State Chem., 1979, 30, 247–255 CrossRef CAS.
- R. Goswami, C. S. Pande, N. Bernstein, M. D. Johannes, C. Baker and G. Villalobos, A high degree of enhancement of strength of sputter deposited Al/Al2O3 multilayers upon post annealing, Acta Mater., 2015, 95, 378–385 CrossRef CAS.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, Use of DFT to achieve a rational understanding of acid–basic properties of γ-alumina surfaces, J. Catal., 2004, 226, 54–68 CrossRef CAS.
- M. Digne, P. Sautet, P. Raybaud, P. Euzen and H. Toulhoat, Hydroxyl groups on γ-alumina surfaces: a DFT study, J. Catal., 2002, 211, 1–5 CrossRef CAS.
- H. P. Pinto, R. M. Nieminen and S. D. Elliott, Ab initio study of γ−Al2O3 surfaces, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 125402 CrossRef.
- L. Kovarik, A. Genc, C. Wang, A. Qiu, C. H. F. Peden, J. Szanyi and J. H. Kwak, Tomography and high-resolution electron microscopy study of surfaces and porosity in a plate-like γ-Al2O3, J. Phys. Chem. C, 2013, 117, 179–186 CrossRef CAS.
- R. Wisechert, C. Copéret, F. Delbecq and P. Sautet, Optimal water coverage on alumina: a key to generate lewis acid–base pairs that are reactive towards the C—H bond activation of methane, Angew. Chem., Int. Ed., 2011, 50, 3202–3205 CrossRef PubMed.
- J. G. Larson and W. K. Hall, Studies of the hydrogen held by solids. VII. the exchange of the hydroxyl groups of alumina and silica-alumina catalysts with deuterated methane, J. Phys. Chem., 1965, 69, 3080–3089 CrossRef CAS.
- P. J. Robertson, M. S. Scurrell and C. Kemball, Exchange of alkanes with deuterium over γ-alumina. aBrønsted linear free energy relationship, J. Chem. Soc., Faraday Trans., 1975, 71, 903–912 RSC.
- J. S. J. Hargreaves, G. J. Hutchings, R. W. Joyner and S. H. Taylor, A study of the methane–deuterium exchange reaction over a range of metal oxides, Appl. Catal., A, 2002, 227, 191–200 CrossRef CAS.
- L. Quanzhi and Y. Amenomiya, Exchange reaction of methane on some oxide catalysts, Appl. Catal., 1986, 23, 173–182 CrossRef.
- H. Knozinger and P. Ratnasamy, Catalytic aluminas: surface models and characterization of surface sites, Catal. Rev.: Sci. Eng., 1978, 17, 31–70 CrossRef.
- J. Joubert, A. Salameh, V. Krakoviack, F. Delbecq, P. Sautet, C. Coperet and J.-M. Basset, Heterolytic splitting of H2 and CH4 on γ-alumina as a structural probe for defect sites, J. Phys. Chem. B, 2006, 110, 23944–23950 CrossRef CAS PubMed.
- J. Gu, J. Wang and J. Leszczynski, Structure and energetics of (111) surface of γ-Al2O3: insights from DFT including periodic boundary approach, ACS Omega, 2018, 3, 1881–1888 CrossRef CAS PubMed.
- S. Zhang, L. Nguyen, J.-X. Liang, J. Shan, J. Liu, A. I. Frenkel, A. Patlolla, W. Huang, J. Li and F. Tao, Catalysis on singly dispersed bimetallic sites, Nat. Commun., 2015, 6, 7938 CrossRef CAS PubMed.
- L. Nguyen, S. Zhang, L. Wang, Y. Li, H. Yoshida, A. Patlolla, S. Takeda, A. I. Frenkel and F. Tao, Reduction of nitric oxide with hydrogen on catalysts of singly dispersed bimetallic sites Pt1Com and Pd1Con, ACS Catal., 2016, 6, 840–850 CrossRef CAS.
- Y. Han, Y.-G. Wang, W. Chen, R. Xu, L. R. Zheng, J. Zhang, J. Luo, R.-A. Shen, Y. Zhu and W.-C. Cheong, Hollow N-doped carbon spheres with isolated cobalt single atomic sites: superior electrocatalysts for oxygen reduction, J. Am. Chem. Soc., 2017, 139, 17269–17272 CrossRef CAS PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong and Z. Deng, Single cobalt atoms with precise N- coordination as superior oxygen reduction reaction catalysts, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- Q. Cheng, L. Yang, L. Zou, Z. Zou, C. Chen, Z. Hu and H. Yang, Single cobalt atom and N codoped carbon nanofibers as highly durable electrocatalyst for oxygen reduction reaction, ACS Catal., 2017, 7, 6864–6871 CrossRef CAS.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S. Z. Qiao, Molecule-level g-C3N4 coordinated transition metals as a new class of electrocatalysts for oxygen electrode reactions, J. Am. Chem. Soc., 2017, 139, 3336–3339 CrossRef CAS PubMed.
- Y. Chen, S. Ji, Y. Wang, J. Dong, W. Chen, Z. Li, R. Shen, L. Zheng, Z. Zhuang and D. Wang, Isolated single iron atoms anchored on N-doped porous carbon as an efficient electrocatalyst for the oxygen reduction reaction, Angew. Chem., Int. Ed., 2017, 56, 6937–6941 CrossRef CAS PubMed.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Direct atomic-level insight into the active sites of a high-performance PGM-free ORR catalyst, Science, 2017, 357, 479–484 CrossRef CAS PubMed.
- C. Zhu, S. Fu, J. Song, Q. Shi, D. Su, M. H. Engelhard, X. Li, D. Xiao, D. Li and L. Estevez, Self-assembled Fe-N-doped carbon nanotube aerogels with single-atom catalyst feature as high- efficiency oxygen reduction electrocatalysts, Small, 2017, 13, 1603407 CrossRef PubMed.
- Z. Zhang, X. Gao, M. Dou, J. Ji and F. Wang, Biomass derived N-doped porous carbon supported single Fe atoms as superior electrocatalysts for oxygen reduction, Small, 2017, 13, 1604290 CrossRef PubMed.
- R. Peverati and D. G. Truhlar, An improved and broadly accurate local approximation to the exchange–correlation density functional: the MN12-L functional for electronic structure calculations in chemistry and physics, Phys. Chem. Chem. Phys., 2012, 10, 13171–13174 RSC.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- A. Schaefer, H. Horn and R. Ahlrichs, Fully optimized contracted Gaussian-basis sets for atoms Li to Kr, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, M. A. Scuseria, J. R. Robb, G. Cheeseman, V. Scalmani, B. Barone, G. A. Mennucci and G. E. Petersson, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2im00039c |
| This journal is © Institute of Process Engineering of CAS 2023 |