 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA mechanochemical approach to the synthesis of sydnones and derivatives†
Nicolas
Pétry
 a,
Florian
Luttringer
a,
Xavier
Bantreil
*ab and
Frédéric
Lamaty
*a
a,
Florian
Luttringer
a,
Xavier
Bantreil
*ab and
Frédéric
Lamaty
*a
aIBMM, Université de Montpellier, CNRS, ENSCM, Montpellier, France. E-mail: xavier.bantreil@umontpellier.fr; frederic.lamaty@umontpellier.fr
bInstitut Universitaire de France (IUF), France
First published on 4th July 2022
Abstract
Sydnones are heterocyclic compounds which display important biological activities, including their abilities to react in 1,3-dipolar additions for applications in the development of new prodrugs. Capitalizing on our preliminary work on the mechanosynthesis of sydnones, an extension of this work to two related families of molecules, diarylsydnones and iminosydnones is reported. A ball-milling approach towards the synthesis of diaryl sydnones was developed, a necessary step for the synthesis of potential sydnone-based ligands of metal complexes. A mechanochemistry-based synthesis of iminosydnones was optimized, including the preparation of active pharmaceutical ingredients (API) related to feprosidnine, linsidomine, mesocarb and molsidomine. This work demonstrated that the ball-milling procedures were efficient and time saving through avoiding purification steps, and reduced the use of organic solvents.
Introduction
Sydnones are mesoionic compounds which have been intensively studied in the last decades1 because of their biological activities and their propensity to smoothly undergo 1,3-dipolar cycloaddition.2 More recently, iminosydnones, aza-derivatives of sydnones known for their biological activities, have found applications in the preparation of new pro-drugs (Fig. 1).3 Driven by our will to promote sustainable synthetic methods and relying on our expertise in mechanochemistry and ball-milling techniques to prepare organic compounds and metal complexes, we have recently developed a solvent-free mechanochemical approach to access aryl sydnones and the corresponding sydnone–metal complexes.4,5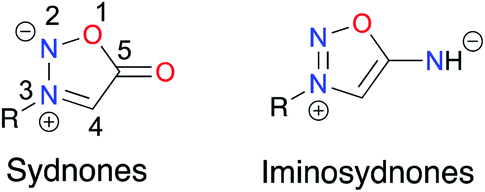 | ||
| Fig. 1 General structure of sydnones and iminosydnones. | ||
We decided to expand this work in two directions, namely developing a solventless method for the preparation of (i) N3,C4-diarylsubstituted sydnones, potential ligands in metallic complexes, and (ii) iminosydnones (ImSyds) as active pharmaceutical ingredients (API).
Sydnones as ligand precursors for metallic complexes
Sydnones were discovered in 1935 at the University of Sydney6 and were named according to their city of invention. Since then, many groups developed procedures to synthesize them from N-substituted amino-acids.7 To date, their implication in coordination chemistry, while showing interesting features, remains scarce.5 Sydnones, as ligands in metal complexes, can coordinate the metal directly, either through their C4 after deprotonation or modification with a halide atom (Hg, Pd, Cu, Zn, Fe, Ni, Pt), or through their exocyclic oxygen after introduction of a second coordinating site (Pd, Cu, Zn, Co). Another possibility is to use the sydnone as a core and introduce different moieties (phosphines, pyridine) to coordinate the metal (Hg, Pd).Iminosydnones and derivatives as active pharmaceutical ingredients
ImSyds were first synthesized by two groups in parallel in 1957 8,9 and quickly received much attention because of their rich biological properties,10 in particular in the cardiovascular field.11 As exogeneous NO donating agents they can modulate several functions of the cellular metabolism12–14 and besides their main therapeutic indications as antithrombotic, antihypertensive and antianginal drugs,15 ImSyds have more recently been investigated as selective dopamine reuptake inhibitors,16,17 antimicrobial18 and antifungal19 compounds and plant growth stimulants.20 ImSyd prodrugs have also been designed for the treatment of glaucomatous optic neuropathy and showed promising properties for a potential topical ocular formulation.21Molsidomine and mesocarb are two of the most famous drugs based on the iminosydnone scaffold (Fig. 2). The former was discovered by the Takeda Company22 and has been prescribed for its antianginal properties since 1977,17 whereas the latter has been developed as a psychotropic stimulating agent,16 both being nonenzymatic exogeneous NO donors.
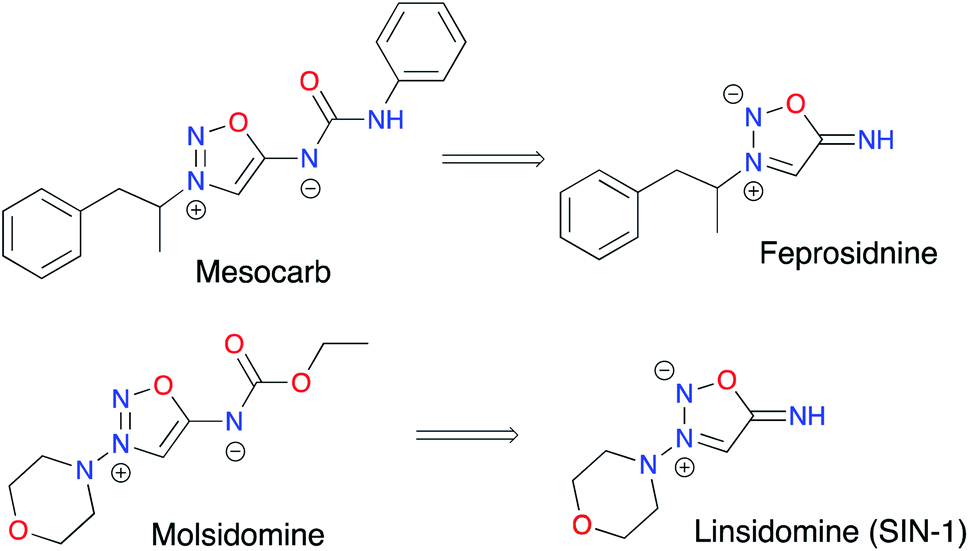 | ||
| Fig. 2 Drugs based on the iminosydnone scaffold. | ||
Linsidomine (SIN-1) has been identified as the active metabolite of molsidomine.23 Indeed, deacetylation by liver esterase followed by nonenzymatic hydrolysis at physiological pH (pH > 5) leads to ring opening of the heterocycle. The resulting N-nitroso derivative then undergoes oxidation to generate a radical cation which finally releases nitric oxide.24,25 Thus, SIN-1 demonstrated effective vasorelaxing and antiplatelet properties.26 On the other hand, mesocarb and feprosidinine were shown to exhibit two different pharmacological effects: the first is reported as an antidepressive drug (a strong inhibitor of monoamine oxidase) whereas the second is a potent psychostimulant (sympathomimetic effect).
Herein, we will present the latest results of our work, i.e. the efficient mechanosynthesis of several sydnone derivatives (N-aryl and N-alkyl sydnones) which can serve as ligands for making metal-complexes or as organic synthons for further derivatization (including the preparation of active pharmaceutical ingredients). In addition to drastically reducing the amount of solvent used, the mechanochemical approach proved to be very efficient, with improved selectivity and purity, avoiding degradation in purification steps and allowing higher yields of targeted molecules. This point will be discussed in detail.
Mechanosynthesis of organic molecules
The development of solventless methods for the preparation of organic molecules has increased dramatically in recent years and has been extensively reviewed.27–39 Mostly, the use of devices such as ball-mills in organic synthesis has expanded to many families of molecules. The major impact of this approach is the decrease in using toxic and polluting solvents, but also to provide the possibility to develop efficient and high-yielding methods and avoid tedious and costly purifications.Experimental
General information
All reagents were purchased from Sigma Aldrich, Fluka, Acros or Alfa Aesar. The milling treatments were carried out in a vibrating Retsch Mixer Mill 200 or 400 (vbm) operated at 25 Hz or 30 Hz. Milling load (ML) is defined as the ratio between the mass of the reactant and the free volume of the jar.NMR analyses were performed at the ‘Laboratoire de Mesures Physiques’ (IBMM, Université de Montpellier). 1H NMR spectra were recorded on a Bruker AVANCE 400 MHz, a Bruker AVANCE III 500 MHz or a Bruker AVANCE III 600 MHz and are reported in ppm using deuterated solvents (CDCl3 at 7.26 ppm or DMSO-d6 at 2.50 ppm or acetone-d6 at 2.05 ppm) as internal standards. Data are reported as s = singlet, d = doublet, t = triplet, q = quadruplet, qt = quintuplet, sept = septuplet, m = multiplet; coupling constant in Hz; integration. 13C NMR spectra were recorded on a Bruker AVANCE 101 MHz, a Bruker AVANCE III 126 MHz or a Bruker AVANCE III 151 MHz and are reported in ppm using deuterated solvents (CDCl3 at 77.2 ppm or DMSO-d6 at 39.5 ppm or acetone-d6 at 29.8 ppm) as internal standards.
Mass spectra were obtained by LC-MS with ESI using a Waters Alliance 2695 for LC, coupled to a Waters ZQ spectrometer with electrospray source, a simple quadrupole analyzer and a UV Waters 2489 detector. HRMS analyses were performed on a UPLC Acquity H-Class from Waters connected to a Synapt G2-S mass spectrometer with a dual ESI source from Waters.
HPLC conversion was measured on an Agilent Technologies 1220 Infinity LC using a Chromolith® high resolution RP-18e 50–4.6 mm column and a linear gradient of 0 to 100% CH3CN/0.1% TFA in H2O/0.1% TFA over 3 min, detection at 214 nm. Flow rate: 1 mL min−1.
Flash chromatography was performed by using prepacked silica columns on a Biotage® Isolera™ Four system.
Representative experiment for mechanochemical reactions (mechanosynthesis of compound 4)
Sydnone 1 (207.1 mg, 1 eq., 1.277 mmol) was added to a screw-top ZrO2 milling jar along with NIS (316.2 mg, 1.1 eq., 1.405 mmol) and AcOH (73 μL, 1 eq., 1.277 mmol). A ZrO2 ball (10 mm diameter) was added to the reactor, which was then closed and sealed with parafilm. The reactor was milled for 1.5 h at 30 Hz. The reaction mixture was taken up with EtOAc and H2O, and extracted with EtOAc (3 times). The organic phase was washed with NaOH (1 M, 3 times) and brine, dried over MgSO4 and evaporated, yielding the title compound as a dark red solid (m = 273.3 mg, 74% yield).Results and discussion
Synthesis of N3,C4-diphenylsydnone
Driven by the desire to obtain organometallic complexes with sydnone derivatives acting as bidentate ligands, our group has already been working on the C4 functionalization of sydnones.4 First, we aimed at developing a general and efficient mechanochemical method for the preparation of model compound N3,C4-diphenyl substituted sydnone. Direct functionalization at C4 of N3-phenyl sydnone (or halogenated derivatives) was considered (Scheme 1). | ||
| Scheme 1 Strategies for the mechanochemical C4-arylation of sydnones. | ||
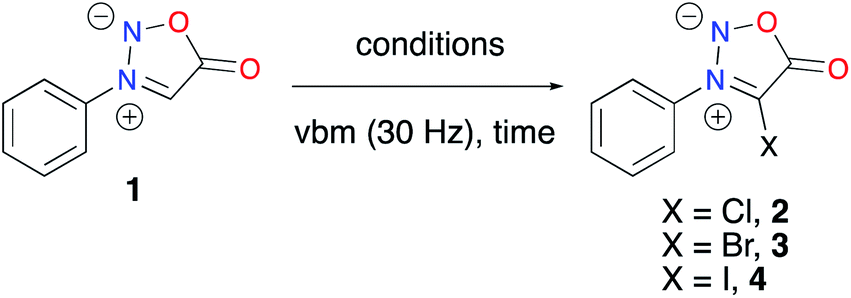
First, we explored the C4 halogenation of sydnones that would allow their use in cross-coupling reactions. In the literature, several methods were developed in solution to perform such halogenations. Chlorination of sydnones has been realized with Cl2,40 KClO3/HCl,41 PhICl2,42 NCS/DMF,43 DABCO-Cl2,44 and ICl/DCM.45 Bromination has been achieved with Br2/NaHCO3,46 Br2/Ac2O,47 KBrO3/HBr,41N-bromosuccinimide (NBS),48 or 1,3-dibromo-5,5-dimethylhydantoin (DBH).49 Finally, direct iodination of sydnones has been described with N-iodosuccinimide (NIS) in AcOH,50 or ICl in AcOH.51
Based on these data, using a vibratory ball-mill (vbm) operated at 30 Hz, we milled in a ZrO2 milling jar sydnone 1 with 1.1 eq. of N-chlorosuccinimide. However, no conversion was observed. When 1 eq. of acetic acid was added, a conversion of 40% into compound 2 was observed after 5 h milling (Table 1, entry 1). Milling for more than 5 hours resulted in the conversion of 2 into an unidentified compound. Bromination using N-bromosuccinimide (NBS) was revealed to be more successful (Table 1, entry 2). A mixture of 4 products was obtained, sydnone 3 being the major one (63% HPLC conversion). Work-up with basic washing with NaOH (1 M) and purification of 3 by recrystallisation using CH2Cl2/pentane furnished a mixture of two major products that recrystallized together, leading to a tedious purification. A filtration and a second recrystallisation were necessary to obtain pure 3 in 38% yield. Based on literature data, our first attempt to perform a mechanochemical iodination of 3-phenylsydnone 1 with NIS and AcOH led to complete conversion into the desired 4-iodosydnone within 1.5 h of milling (Table 1, entry 3). A simple extraction and a basic washing (NaOH, 1 M) gave the target compound 4 in 74% yield without the need for further purification. As an alternative to NIS, ICl was used under different conditions. In the literature, in solution, a mixture of 4 and 4-chlorosydnone 2 was obtained when 1 eq. of ICl (1 M in dry CH2Cl2, commercial solution) was used and 4 was the only observed product when 3 eq. of ICl (1 M in dry CH2Cl2) were used.8 Under mechanochemical conditions, when pure ICl (1 eq.) was reacted with 1 in a ZrO2 milling jar, only 4 was observed, albeit with incomplete conversion (Table 1, entry 4). Full conversion was obtained when using 3 eq. of pure ICl under LAG (liquid-assisted grinding)52 conditions (dry CH2Cl2), giving 4 in 90% yield after work-up (Table 1, entry 5). Thus, two mechanochemical strategies have been developed to access 4. When comparing them, the first one using NIS is simpler than the latter using ICl, which needs to be handled in an inert atmosphere, but on the other hand, ICl offers a substantial advantage in terms of atom economy.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | X | Conditions | Additive | Reaction time (h) | Conv. (%) | Yield (%) |
| a n.d. = not determined. b Liquid-assisted grinding, η = 0.5 μL mg−1. | ||||||
| 1 | Cl | NCS (1.1 eq.), AcOH (1 eq.) | — | 5 | 40 | n.d.a |
| 2 | Br | NBS (1.1 eq.) | 1.5 | 63 | 38 | |
| 3 | I | NIS (1.1 eq.), AcOH (1 eq.) | — | 1.5 | 100 | 74 |
| 4 | I | ICl, NaOAc (1 eq.) | — | 2.5 | 83 | n.d.a |
| 5 | I | ICl, NaOAc (3 eq.) | CH2Cl2b | 1.5 | 100 | 90 |
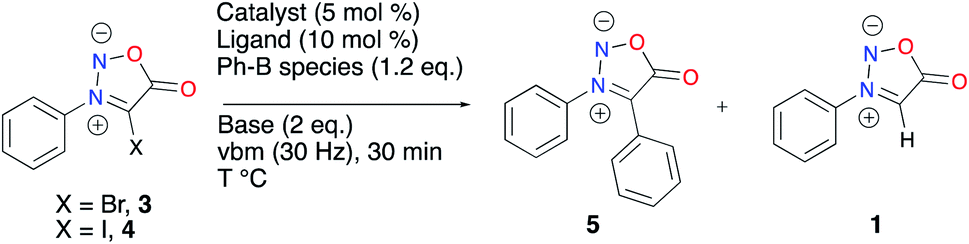
The ease of isolation of 4 led us to test it in a model Suzuki–Miyaura C–C coupling reaction with phenyl boronic acid (Table 2). We transposed to mechanochemistry reaction conditions optimized for the in-solution microwave-activated Suzuki–Miyaura coupling of 4-bromo-3-phenylsydnone 3.46 These mechanochemical conditions, with a similar catalytic system in a stainless steel jar heated with an external heatgun set to 100 °C, gave 47% of the desired sydnone 5 and 39% of the protodehalogenation compound 1 (Table 2, entry 1). Of note, a high quantity of biphenyl, resulting from the homocoupling of the boronic acid was observed, thus hampering the desired cross-coupling reaction. Increasing the temperature to 140 °C led to a decrease in the formation of 5 with a concomitant increase of protodehalogenation (Table 2, entry 2). A screening of the literature concerning mechanochemical Suzuki–Miyaura cross-coupling made us look at different catalytic systems. Pd(OAc)2 and RuPhos were employed along with CsF as base and H2O and 1,5-COD as additives.53 Protodehalogenation and homocoupling products were again observed, along with 24% of 5 (Table 2, entry 3). A rapid screening of ligands revealed DavePhos to be ineffective in the conditions used (Table 2, entry 4). Interestingly, the use of BrettPhos furnished a selective conversion into sydnone 1, with no desired coupling nor homocoupling (Table 2, entry 5). XPhos was thus used for the rest of our tests, screening different boron species, additives and bases. Using a phenylboronic acid pinacol ester or potassium phenyltrifluoroborate gave a low conversion into the expected product 5 and no conversion respectively (Table 2, entries 6 and 7). Adding boronic acid in excess gave the same results as entry 1 with 47% of the desired C–C coupling, also with an incomplete conversion (Table 2, entry 8). Using a reductant such as sodium formate did not improve the conversion into 5 (Table 2, entry 9). Metal additives were tested to improve the transmetalation step, but neither CuI nor Ag2O could prevent side reactions from happening (Table 2, entries 10 and 11). A rapid screening of bases didn’t result in any improvement in the product ratio observed (Table 2, entries 12 and 13). 3 (X = Br) was also tested in the cross-coupling conditions (Table 2, entry 14). In that case, protodehalogenation became predominant and a 23% conversion of 3 into 5 was obtained.
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Conditions | B species (eq.) | Base | Additive | Ratio (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:1)b :5:1)b |
|
a Heatgun set to 100 °C unless otherwise noted. X = I.
b Evaluated by HPLC.
c Heatgun temperature set to 140 °C.
d Heatgun temperature set to 170 °C.
e X = Br. 3:5:1 ratio is given.
|
|||||
| 1 | Pd(OAc)2/XPhos | Ph-B(OH)2 | Cs2CO3 | — | 14/47/39 |
| 2c | Pd(OAc)2/XPhos | Ph-B(OH)2 | Cs2CO3 | 1,5-COD (0.13 μL mg−1) | 0/44/56 |
| 3 | Pd(OAc)2/RuPhos | Ph-B(OH)2 | Cs2CO3 | H2O (3, 7 eq.) 1,5-COD (0.13 μL mg−1) | 0/24/76 |
| 4 | Pd(OAc)2/DavePhos | Ph-B(OH)2 | Cs2CO3 | — | Traces |
| 5 | Pd(OAc)2/BrettPhos | Ph-B(OH)2 | Cs2CO3 | — | 15/0/85 |
| 6 | Pd(OAc)2/XPhos | Ph-Bpin (1.2) | Cs2CO3 | — | 68/5/27 |
| 7 | Pd(OAc)2/XPhos | Ph-BF3K (1.2) | Cs2CO3 | — | 97/0/3 |
| 8 | Pd(OAc)2/XPhos | Ph-B(OH)2 (4) | Cs2CO3 | — | 13/47/40 |
| 9 | Pd(OAc)2/XPhos | Ph-B(OH)2 | Cs2CO3 | HCO2Na | 0/40/60 |
| 10 | Pd(OAc)2/XPhos | Ph-B(OH)2 | Cs2CO3 | CuI | 0/49/51 |
| 11 | Pd(OAc)2/XPhos | Ph-B(OH)2 | Cs2CO3 | Ag2CO3 | 46/40/14 |
| 12 | Pd(OAc)2/XPhos | Ph-B(OH)2 | KOtBu | — | 9/19/72 |
| 13d | Pd(OAc)2/XPhos | Ph-B(OH)2 | KOH | — | 0/26/74 |
| 14e | Pd(OAc)2/XPhos | Ph-B(OH)2 | Cs2CO3 | — | 27/23/50 |
With these unsatisfying results in hand, we sought another way to obtain our target compound 5. C–H activation appeared as a good alternative to afford C4 coupled sydnones while preventing the side-reactions observed in the Suzuki–Miyaura cross-coupling (i.e. protodehalogenation and homocoupling) from occurring (Scheme 2).54 When reacting 1 and iodobenzene in the presence of Cs2CO3 and Pd(OAc)2/XPhos as the catalytic system at 200 °C (preset temperature of the heatgun), 49% conversion into the coupling product was obtained after 30 min of milling. Gratifyingly, total conversion was obtained after 2.5 h of milling. Unfortunately, extraction and filtration on a short pad of silica afforded 5 in only 22% yield. Further developments for increasing the isolated yield of 5 are still ongoing in our laboratory.
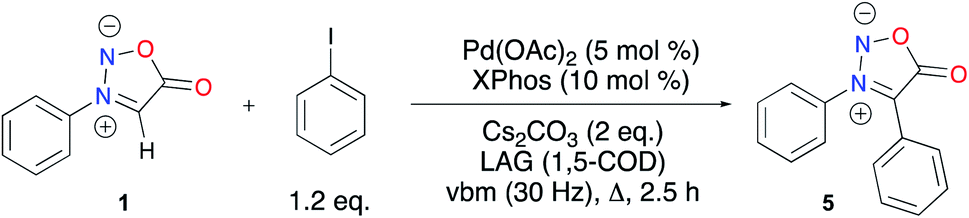 | ||
| Scheme 2 Mechanochemical palladium-catalyzed C–H activation. | ||
To conclude this part, a mechanochemistry-mediated approach was explored for the preparation of N3,C4 aryldisubstituted sydnones. While bromination and iodination of sydnones in a ball-mill were successful, the use of the corresponding halogenated product in Suzuki–Miyaura cross-coupling was low yielding. On the other hand, palladium catalyzed direct activation of N3 phenylsubstituted sydnone, combining ball-milling and heating provided the diphenyl sydnone with high conversion, albeit with a low yield, but further developments will open the way to the preparation of more analogues including potential ligands for metal complexes.
Synthesis of norfeprosidnine and linsidomine
Targeting more efficient and “greener” access to biologically active ImSyds like molsidomine and mesocarb, we envisioned transposing our solvent-free mechanosynthesis previously developed for sydnones.4 To validate our hypothesis, we first aimed at the preparation of a non-substituted ImSyd model compound, 3-phenyl-iminosydnone 8.Inspired by our previous results on the alkylation of anilines with ethylbromoacetate,4 we implemented a solvent-free procedure to obtain N-phenylaminoacetonitrile. Nucleophilic substitution of aniline on bromoacetonitrile was performed by grinding an equimolar mixture of both reagents in the presence of 1 equivalent of K2CO3 as a base for 90 min in a vibratory ball mill. Interestingly, no dialkylated product was detected and the expected aminonitrile 6 was obtained pure in 86% yield after work-up (Scheme 3). Then, we focused on the nitrosylation step. The desired nitroso-aminoacetonitrile 7 was easily formed during a solventless transformation using NaNO2 as nitroso agent and KHSO4 as a solid proton donor. Indeed, contrary to the case of N-aryl-amino acids which can be nitrosylated in a ball-mill by the sole action of NaNO2 thanks to the acid proton of the substrate, aminoacetonitrile 6 required a proton source to reach reaction completion. The desired nitroso compound 7 was isolated pure in 90% yield after only 1 h of milling and simple work-up.
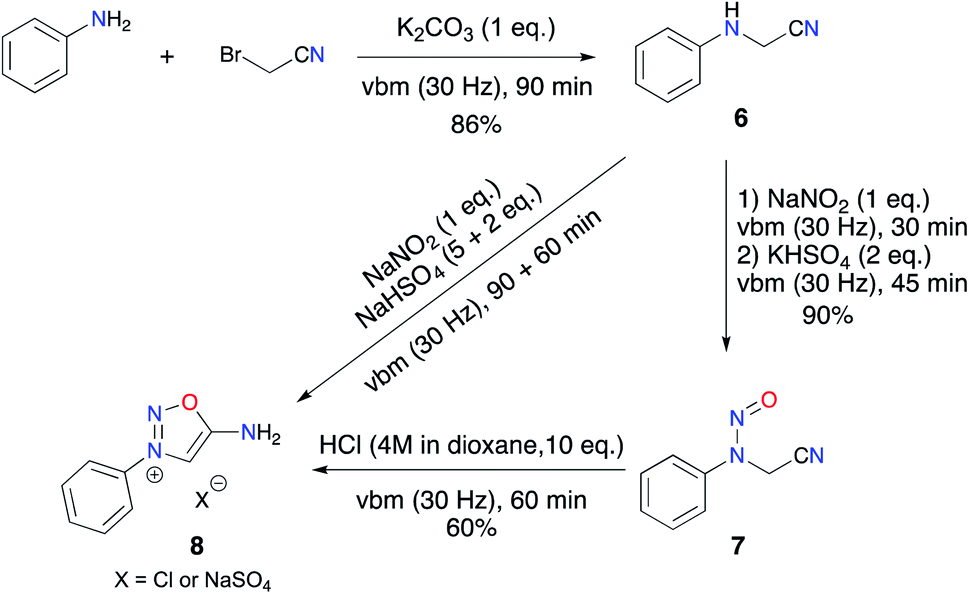 | ||
| Scheme 3 Mechanochemical iminosydnone synthesis. | ||
With this result in hand, we carried out exploratory trials to achieve the cyclization step with different sources of protons by mechanochemistry and to our delight, grinding 7 with an excess of acid (4 M HCl in dioxane) allowed reaching complete formation of the iminosydnone core. However, considering the potential toxicity of the nitroso intermediates, we moved directly to a one-pot two-step procedure and replaced KHSO4 with NaHSO4, which is less hygroscopic and easier to handle. In the first experiment, 5 equiv. of NaHSO4 were insufficient to reach complete conversion but an addition of 2 more equiv. allowed 8 to be obtained as the only product formed. However, neither organic nor aqueous work-up led to the isolation of 8 without residual salts.
To address this problem, we first optimized the amount of proton donor to 6 equiv. and then carried out anion metathesis in the ball mill using KPF6. This solvent-free method was previously applied in our group for the isolation of imidazolium hexafluorophosphates as NHC precursors.55,56 This additional step permitted recovery of the desired ImSyd 8 as a hexafluorophosphate salt after simple extraction with EtOAc in 93% yield (Scheme 4). To the best of our knowledge, this is the first example of isolation of an ImSyd with a non-coordinating counter-ion. In addition, this one-pot procedure with only solid reagents avoids resorting to hazardous strong mineral acids (HNO3, HCl…) and toxic organic solvents (THF, methanol, dioxane…).
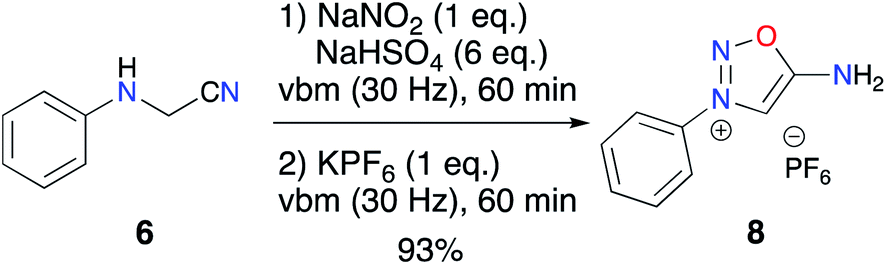 | ||
| Scheme 4 One-pot two-step iminosydnone mechanosynthesis. | ||
Having demonstrated the possibility of synthesizing ImSyd 8 by mechanochemistry, we turned our attention to the preparation of feprosidnine and linsidomine in a ball-mill. Concerning feprosidnine, because of the regulatory constraints regarding drug precursors and the difficulty of accessing amphetamine, we decided to target an analog of the original molecule, norfeprosidnine, and started from phenethylamine. The first experiments confirmed the possibility of obtaining the aminoacetonitrile 9 by the alkylation route. However, with phenethylamine being more prone to dialkylation than aniline, a quick optimization of our previous procedure was required. Finally, using an excess of K2CO3 allowed 9 to be obtained in 93% yield and with a satisfactory purity after work-up. Then, mechanochemical formation of the ImSyd core was accomplished as previously in a ball-mill, a slight excess of NaNO2 being necessary this time to obtain complete conversion, and the desired ImSyd 10 was obtained in high yield without the need for a purification step (Scheme 5).
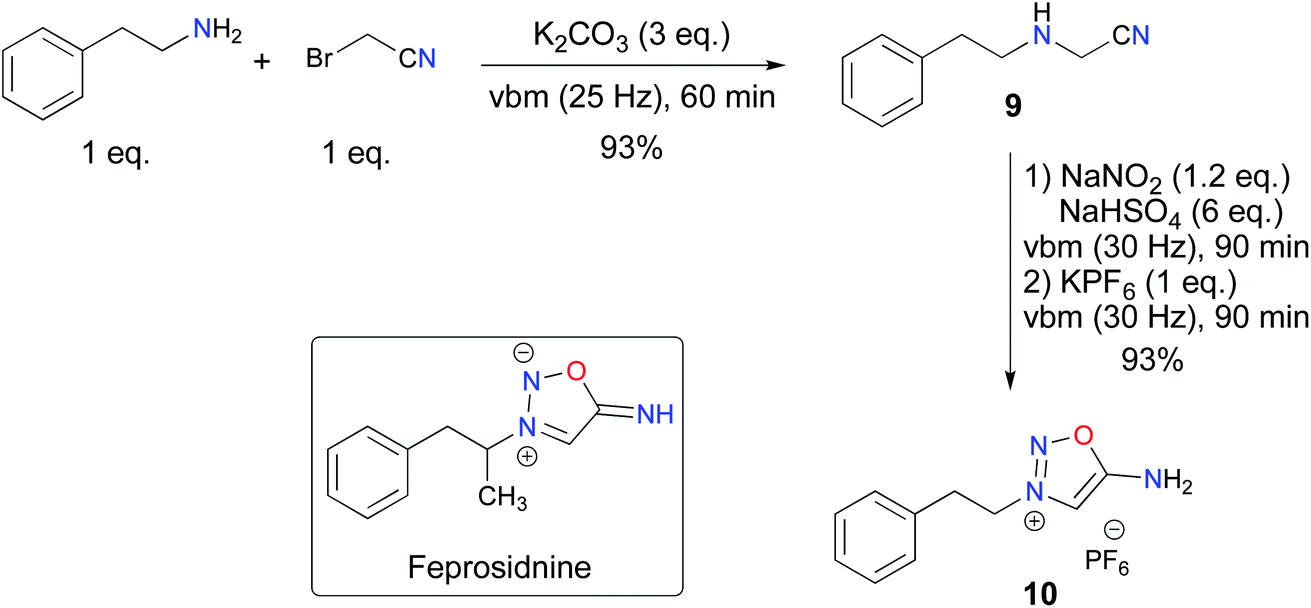 | ||
| Scheme 5 Mechanochemical access to feprosidnine analogue 10. | ||
Next, we considered the preparation of linsidomine. In the literature, only Strecker type reactions with 4-aminomorpholine and different forms of formaldehyde are described for the synthesis of the aminoacetonitrile 11.57 A test experiment confirmed to us that the alkylation of 4-aminomorpholine by bromoacetonitrile was unsuccessful. As Hernández and Bolm had already set up procedures to perform Strecker reactions by mechanochemistry,58,59 we confidently envisioned transposing the preparation of 11 in a ball mill (Scheme 5). However, the published reports described mechanochemical Strecker reactions with various aldehydes (aromatic, heteroaromatic, aliphatic) and amines (aliphatic, heterocyclic and anilines) but no examples were published using formaldehyde and hydrazines. We began our study by identifying the different forms of formaldehyde commercially available and we chose to select aqueous formaldehyde solution, 1,3,5-trioxane, sodium formaldehyde bisulfite and paraformaldehyde.
We started with the bisulfite derivative, used by Masuda in the original preparation of 3-dialkylaminosydnonimines.60,61 However, grinding in a vbm an equimolar mixture of 4-aminomorpholine and bisulfite adduct resulted in no imine formation according to 1H NMR monitoring, and adding KCN (caution: KCN is an extremely toxic chemical) from the start did not have any effect (Table 3, entry 1). As it was shown that addition of SiO2 as a solid additive improved dramatically mechanochemical Strecker reactions,58 6 equiv. of silica were employed in a new experiment, but no trace of the expected imine was detected. The same outcome was observed using 1,3,5-trioxane as a formaldehyde source. As the bisulfite reagent was usually used in aqueous solution, we investigated the effect of a small amount of water as a liquid additive but once again no conversion was observed (Table 3, entry 2). Then, free formaldehyde in solution was reacted and gratifyingly, an excellent result was obtained. Indeed, 1H NMR analysis of an aliquot showed complete formation of the imine after 60 min of milling of 4-aminomorpholine with 1 equiv. of formaldehyde in the presence of SiO2, and an additional hour of milling after addition of KCN furnished the expected aminonitrile 11 in quantitative yield, albeit impure (Table 3, entry 3). Traces of residual by-products in the NMR spectra and our will to go a step further led us to test in this transformation the use of less toxic paraformaldehyde. The first trials with this reagent proved unsuccessful. Although the imine was formed, cyanide addition did not occur and no trace of the expected aminonitrile was detected (Table 3, entries 4 and 5). However, using 6 equiv. of SiO2 as a solid additive gave satisfactory results. When the 2 steps of the transformation were carried out successively, the desired product 11 was obtained with excellent yield and good purity (Table 3, entry 6), whereas when KCN was introduced at the start of the experiment, the aminonitrile was obtained as the main compound but with a less satisfactory 1H NMR spectrum. Finally, we tried to use another solid additive (NaHSO4) but more by-products were again detected, and with a non-toxic cyanide source,62,63 K3(Fe(CN)6), the nitrile adduct did not form (Table 3, entry 7). It is also important to note that work-up is very straightforward. The reaction mixture was suspended in EtOAc and filtered, and the final product was obtained after evaporation under vacuum. Purification of the reaction mixture by flash chromatography did not bring any improvement to the method: the final yield was slightly lower and the 1H NMR spectrum was very similar to the one of the crude product (Table 3, entry 8).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Formaldehyde source | CN− source (eq.) | Additive (eq.) | Milling conditions | Conv. (%) | Yield of 11 (%) |
| a KCN was directly added to the reaction mixture. b n.d. = not determined. c Purity >90%, determined by 1H NMR. d Formation of intermediate imine but no cyanide addition. e Yield after purification on silica gel. | ||||||
| 1 | HOCH2SO3Na | KCN (1.1) | — | 25 Hz, 1 h | 0 | 0 |
| 2 | HOCH2SO3Na | KCN (1.1)a | H2O (LAG) | 25 Hz, 1 h | 0 | 0 |
| 3 | Formaldehyde (37% in H2O) | KCN (1.1) | SiO2 (6) | 25 Hz, 1 + 1 h | 100 | n.d.b,c |
| 4 | Paraformaldehyde | KCN (1.1) | — | 25 Hz, 1 + 1 h | 100 | 0d |
| 5 | Paraformaldehyde | KCN (1.1) | H2O (LAG) | 25 Hz, 1 + 1 h | 100 | 0d |
| 6 | Paraformaldehyde | KCN (1.1) | SiO2 (6) | 25 Hz, 0.5 + 1.5 h | 100 | 93 |
| 7 | Paraformaldehyde | K3(Fe(CN6)) (1.0) | SiO2 (6) | 30 Hz, 45 + 60 min | 100 | 0d |
| 8 | Paraformaldehyde | KCN (1.1) | SiO2 (6) | 30 Hz, 45 + 60 min | 100 | 75e |
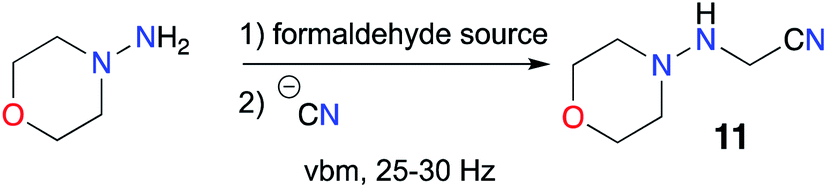
So, an efficient and solventless Strecker reaction was successfully developed in a ball mill for the preparation of the linsidomine precursor 11 (Table 3, entry 6). To the best of our knowledge, this is the first instance of a mechanochemical Strecker reaction using paraformaldehyde and our result shows that under ball milling conditions the polymeric form of the aldehyde does not hamper the progress of the expected transformation.
Finally, we implemented the previously devised one-pot two-step procedure which allowed formation of the ImSyd hexafluorophosphate 12 with a very good yield (Scheme 6).
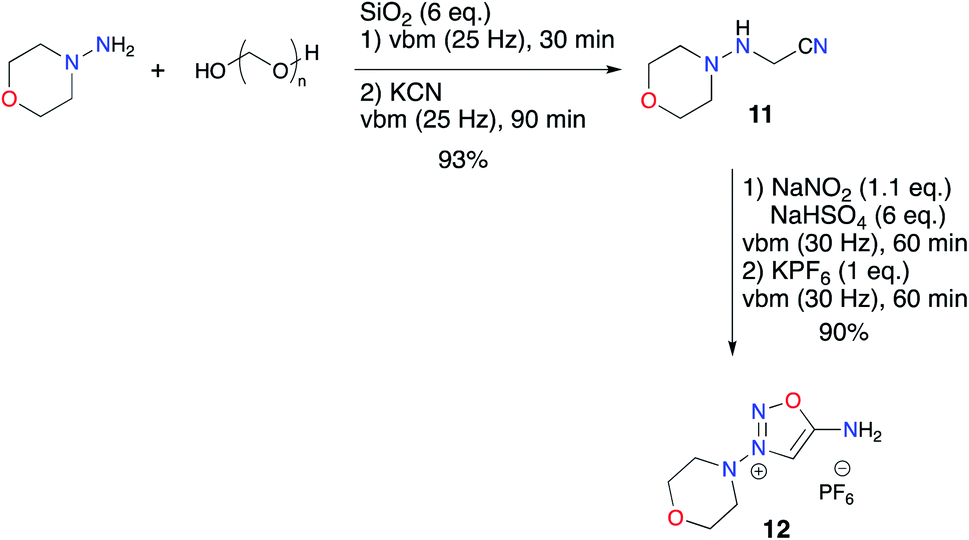 | ||
| Scheme 6 Mechanochemical synthesis of linsidomine 12. | ||
Hence, an unprecedented solventless and mechanochemical synthesis of several ImSyds, known for their biological activities, was developed. The aminoacetonitriles required for the preparation of these ImSyds were synthesized either by alkylation of an aryl/alkyl primary amine or by a Strecker reaction using only solid reagents, both approaches being carried out in a ball mill, with simplified purification steps. Attempts to replace the potassium cyanide source with less toxic sources were not successful. We showed that the ImSyd ring can be formed during a one-pot two-step solid state reaction with NaNO2 as nitrosylating agent and NaHSO4 as solid proton donor. An additional anion metathesis step gave access to ImSyds as hexafluorophosphate salts which were recovered by simple extraction.
Conclusions
The innovative mechanosynthesis of sydnones and iminosydnones, reported herein, provides particularly easy, safe and efficient methods for the preparation of these molecules. Each step was optimized to provide a maximized conversion towards the targeted molecule, hence improving the purification procedures. This opens the way to the facilitated synthesis of sydnone-based ligands and iminosydnone derived biologically active molecules. Most of all, the use of toxic and harmful solvents is drastically reduced not only because of the reaction conditions, but also because it avoids the use of column chromatography as a purification method. Compared to previously reported procedures, this work marks a significant improvement in terms of safety and environmental impact, two aspects of great value for the current chemical and pharmaceutical industries. Further work is underway to provide alternatives to the use of toxic cyanide.Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
We gratefully acknowledge financial support from the CNRS, the University of Montpellier and Institut Universitaire de France.Notes and references
- D. L. Browne and J. P. A. Harrity, Tetrahedron, 2010, 66, 553–568 CrossRef CAS.
- E. Decuypère, L. Plougastel, D. Audisio and F. Taran, Chem. Commun., 2017, 53, 11515–11527 RSC.
- K. Porte, B. Renoux, E. Péraudeau, J. Clarhaut, B. Eddhif, P. Poinot, E. Gravel, E. Doris, A. Wijkhuisen, D. Audisio, S. Papot and F. Taran, Angew. Chem., Int. Ed., 2019, 58, 6366–6370 CrossRef CAS PubMed.
- N. Pétry, T. Vanderbeeken, A. Malher, Y. Bringer, P. Retailleau, X. Bantreil and F. Lamaty, Chem. Commun., 2019, 55, 9495–9498 RSC.
- X. Bantreil, N. Pétry and F. Lamaty, Dalton Trans., 2019, 48, 15753–15761 RSC.
- J. C. Earl and A. W. Mackney, J. Chem. Soc., 1935, 899–900 RSC.
- J. Applegate and K. Turnbull, Synthesis, 1988, 1011–1012 CrossRef.
- P. Brookes and J. Walker, J. Chem. Soc., 1957, 4409–4416 RSC.
- H. Kato, M. Hashimoto and M. Ohta, Nippon Kagaku Zasshi, 1957, 78, 707 CrossRef.
- V. G. Yashunskii and L. E. Kholodov, Russ. Chem. Rev., 1980, 49, 28–45 CrossRef.
- K. Kikuchi, M. Hirata and Y. Aramaki, Jpn. J. Pharmacol., 1970, 20, 23–43 CrossRef CAS.
- K. Schönafinger, Il Farmaco, 1999, 54, 316–320 CrossRef PubMed.
- V. Levina, N. V. Grigor’ev and V. G. Granik, Chem. Heterocycl. Compd., 2004, 40, 507–509 CrossRef.
- E. Y. Khmel’nitskaya, V. I. Levina, L. A. Trukhacheva, N. B. Grigoriev, V. N. Kalinin, I. A. Cherepanov, S. N. Lebedev and V. G. Granik, Russ. Chem. Bull., 2004, 53, 2840–2844 CrossRef.
- K. Rehse, M. Kämpfe and K.-J. Schleifer, Arch. Pharm., 1993, 326, 483–487 CrossRef PubMed.
- H. Chen, M. Liu, M. Sathyamoorthy, Q. Su, L. Leary and W. S. Zhong, WO2011069051, 2011.
- H. Chen, M. Liu, Q. Su and M. Raisinghani, WO2008112968, 2008.
- J. L. Ellis, D. S. Garvey and C.-E. Lin, WO2007086884, 2007.
- H. D. Navadiya, A. R. Jivani, N. K. Undavia and B. S. Patwa, Indian J. Heterocycl. Chem., 2009, 19, 87–88 Search PubMed.
- I. A. Cherepanov, Y. Y. Spiridonov, S. K. Moiseev and E. F. Khirazov, Russia Pat., RU2656212C1, 2018.
- S. Acharya, P. Rogers, R. R. Krishnamoorthy, D. L. Stankowska, H. V. Dias and T. Yorio, Bioorg. Med. Chem. Lett., 2016, 26, 1490–1494 CrossRef PubMed.
- K. Masuda and Y. Imashiro, Germany Pat., DE1659897, 1967.
- Y. Asahi, K. Shinozaki and M. Nagaoka, Chem. Pharm. Bull., 1971, 19, 1079–1088 CrossRef.
- M. Feelisch, J. Ostrowski and E. Noack, J. Cardiovasc. Pharmacol., 1989, 14, S13–S22 CrossRef.
- L. Soulère, P. Hoffmann and F. Bringaud, J. Heterocycl. Chem., 2003, 40, 943–947 CrossRef.
- J. Reden, J. Vasc. Res., 1990, 27, 282–294 CrossRef PubMed.
- P. Ying, J. Yu and W. Su, Adv. Synth. Catal., 2021, 363, 1246–1271 CrossRef.
- M. Solares-Briones, G. Coyote-Dotor, J. C. Páez-Franco, M. R. Zermeño-Ortega, C. M. de la O Contreras, D. Canseco-González, A. Avila-Sorrosa, D. Morales-Morales and J. M. Germán-Acacio, Pharmaceutics, 2021, 13, 790 CrossRef CAS.
- K. Kubota and H. Ito, Trends Chem., 2020, 2, 1066–1081 CrossRef CAS.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef.
- D. Tan and T. Friščić, Eur. J. Org. Chem., 2018, 18–33 CrossRef CAS.
- J. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef PubMed.
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS.
- J. G. Hernández and T. Friščić, Tetrahedron Lett., 2015, 56, 4253–4265 CrossRef.
- G.-W. Wang, Chem. Soc. Rev., 2013, 42, 7668–7700 RSC.
- L. Takacs, Chem. Soc. Rev., 2013, 42, 7649–7659 RSC.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- O. Bento, F. Luttringer, T. Mohy El Dine, N. Pétry, X. Bantreil and F. Lamaty, Eur. J. Org. Chem., 2022, e202101516 CAS.
- R. A. Eade and J. C. Earl, J. Chem. Soc., 1948, 2307–2310 RSC.
- J. C. Earl, Recl. Trav. Chim. Pays-Bas, 1956, 75, 1080–1082 CrossRef.
- S. Ito and K. Turnbull, Synth. Commun., 1996, 26, 1441–1446 CrossRef.
- H.-J. Tien, M.-Y. Yeh and C.-Y. Huang, J. Chin. Chem. Soc., 1985, 32, 461–465 CrossRef.
- S. Salmanpoor, M. Tajbakhsh, S. Habibzadeh, D. Azarifar and H. Ghasemnejad-Bosra, J. Chem. Res., 2008, 2008, 662–663 CrossRef.
- I. F. Nashashibi, J. M. Tumey, B. L. Owens and K. Turnbull, Org. Prep. Proced. Int., 2017, 49, 59–63 CrossRef.
- D. L. Browne, J. B. Taylor, A. Plant and J. P. A. Harrity, J. Org. Chem., 2009, 74, 396–400 CrossRef PubMed.
- W. Baker, W. D. Ollis and V. D. Poole, J. Chem. Soc., 1950, 1542–1551 RSC.
- E. N. Beal and K. Turnbull, Synth. Commun., 1992, 22, 1515–1522 CrossRef.
- D. Azarifar and H. Ghasemnejad-Bosra, Synthesis., 2006, 1123–1126 CrossRef.
- D. C. Brown and K. Turnbull, Synth. Commun., 2013, 43, 3233–3237 CrossRef.
- F. Dumitrascu, C. Draghici, D. Dumitrescu, L. Tarko and D. Raileanu, Liebigs Ann., 1997, 2613–2616 CrossRef.
- G. A. Bowmaker, Chem. Commun., 2013, 49, 334–348 RSC.
- T. Seo, T. Ishiyama, K. Kubota and H. Ito, Chem. Sci., 2019, 10, 8202–8210 RSC.
- Z. Yao, X. Wu, X. Zhang, Q. Xiong, S. Jiang and Z. Yu, Org. Biomol. Chem., 2019, 17, 6777–6781 RSC.
- A. Beillard, F. Quintin, J. Gatignol, P. Retailleau, J.-L. Renaud, S. Gaillard, T.-X. Métro, F. Lamaty and X. Bantreil, Dalton Trans., 2020, 49, 12592–12598 RSC.
- A. Beillard, X. Bantreil, T.-X. Métro, J. Martinez and F. Lamaty, Dalton Trans., 2016, 45, 17859–17866 RSC.
- P. G. Wang, M. Xian, X. Tang, X. Wu, Z. Wen, T. Cai and A. J. Janczuk, Chem. Rev., 2002, 102, 1091–1134 CrossRef CAS PubMed.
- J. G. Hernández, M. Turberg, I. Schiffers and C. Bolm, Chem.–Eur. J., 2016, 22, 14513–14517 CrossRef.
- S. Dabral, M. Turberg, A. Wanninger, C. Bolm and J. G. Hernandez, Molecules, 2017, 22, 146 CrossRef.
- K. Masuda, T. Kamiya, Y. Imashiro and T. Kaneko, Chem. Pharm. Bull., 1971, 19, 72–79 CrossRef CAS.
- K. Masuda, Y. Imashiro and T. Kaneko, Chem. Pharm. Bull., 1970, 18, 128–132 CrossRef CAS.
- C. Bolm, R. Mocci, C. Schumacher, M. Turberg, F. Puccetti and J. G. Hernandez, Angew. Chem., Int. Ed., 2018, 57, 2423–2426 CrossRef CAS PubMed.
- C. Grundke and T. Opatz, Green Chem., 2019, 21, 2362–2366 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2fd00096b |
| This journal is © The Royal Society of Chemistry 2023 |