
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phenylimido complexes of rhenium: fluorine substituents provide protection, reactivity, and solubility†
Guilhem
Claude
a,
Erika
Kulitzki
a,
Adelheid
Hagenbach
a,
Maximilian
Roca Jungfer
 a,
Joshua S.
Figueroa
*b and
Ulrich
Abram
*a
a,
Joshua S.
Figueroa
*b and
Ulrich
Abram
*a
aFreie Universität Berlin, Institute of Chemistry and Biochemistry, Fabeckstr. 34/36, 14195 Berlin, Germany. E-mail: ulrich.abram@fu-berlin.de
bUniversity of California San Diego, La Jolla, Department of Chemistry and Biochemistry, California 92093, USA. E-mail: jsfig@ucsd.edu
First published on 8th March 2023
Abstract
Reactions of [Re(NPhF)Cl3(PPh3)2] ({NPhF}2− = p-fluorophenylimide) with a variety of alkyl and aryl isocyanides have been studied. Different reactivity patterns and products have been obtained depending on the steric and electronic properties of the individual ligands. This involves the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2 exchange products of Re(V) with the general formulae mer-[Re(NPhF)Cl3(PPh3)(isocyanide)] and cis- or trans-[Re(NPhF)Cl3(isocyanide)2]. The stability of the obtained products is correlated with the substitution pattern of the isocyanide ligands. The products have been studied by single-crystal X-ray diffraction and spectroscopic methods, including IR and multinuclear NMR spectroscopy as well as mass spectrometry. The use of partially fluorinated starting materials and ligands allows the modulation of the solubilities of the starting materials and the products as well as the monitoring of the reactions by means of 19F NMR. The attachment of the CF3 or F substituent on the isocyanides gives control over the steric bulk and the electronic properties of the ligands and, thus, their reactivity.
:1 and 1:2 exchange products of Re(V) with the general formulae mer-[Re(NPhF)Cl3(PPh3)(isocyanide)] and cis- or trans-[Re(NPhF)Cl3(isocyanide)2]. The stability of the obtained products is correlated with the substitution pattern of the isocyanide ligands. The products have been studied by single-crystal X-ray diffraction and spectroscopic methods, including IR and multinuclear NMR spectroscopy as well as mass spectrometry. The use of partially fluorinated starting materials and ligands allows the modulation of the solubilities of the starting materials and the products as well as the monitoring of the reactions by means of 19F NMR. The attachment of the CF3 or F substituent on the isocyanides gives control over the steric bulk and the electronic properties of the ligands and, thus, their reactivity.
Introduction
Fluorine possesses a series of peculiar properties. Its very high electronegativity (3.98 on the Pauling scale), as well as the strong hyperconjugation resulting from a low lying σ*(C–F) combined with its small atomic radius, has been shown to have profound structural and electronic effects on both organic and inorganic compounds.1,2 Fluorine can therefore be introduced in biologically active molecules to increase their bioavailability and/or efficacy, which led to a significant growth of the number of small, fluorine-containing molecules considered for pharmaceutical applications. It was estimated that already in 2012 fluorinated compounds made 40% of all phase III drug candidates.3In diagnostic nuclear medicine, the radioactive isotope 18F is a common tracer in positron-emission tomography (PET), e.g. by replacing a “cold” fluorine atom by 18F in an established pharmacologically active compound.4–6 A hitherto relatively little considered option is the use of fluorine-substituted ligand systems for the modulation of the pharmacokinetic properties of metal-containing radiotracers, e.g. of the matched-pair 99mTc/188Re.6–8 For the development of corresponding compounds, however, some fundamental work is required to learn more about the influence, which can be expected by partially fluorinated ligands in terms of the solubility and lipophilicity of the products, but also with regard to possible electronic effects. Such studies are commonly performed with natural rhenium and the long-lived technetium isotope 99Tc (weak β− emitter with Emax = 0.3 MeV, t1/2 = 2.1 × 105 years), which is available in macroscopic amounts and allows the use of conventional spectroscopic methods including 19F NMR spectroscopy, which is indicative of convenient monitoring of reactions.
Some recent work in our group with nuclear medically relevant metals such as indium, technetium or rhenium demonstrated that the structure and reactivity of metal complexes can markedly be influenced by (partial) fluorination of the ligands.2,9–12 The observed changes in properties were found to be dependent on both the number of fluorine atoms and their exact position. The addition of sometimes even a single fluorine atom on a ligand also led to new reactivity patterns for both rhenium and technetium complexes. Thus, the use of p-fluorophenyl isocyanide instead of unsubstituted phenyl isocyanide resulted in completely different substitution patterns of fac-tricarbonyl complexes of these metals.10,11
Isocyanides are interesting ligands for nuclear medical procedures with 99mTc and probably also for 188Re. 99mTc-Sestamibi (Cardiolite), an octahedral technetium(I) complex with six methoxy-substituted isocyanides, is one of the most used diagnostic radiopharmaceuticals worldwide.12–15 Isocyanide complexes with high-valent rhenium and technetium centers are rare and only a few examples of oxidorhenium(V) complexes are known,16–18 while the corresponding {TcO}3+ core is prone to reduction by isocyanides and no such compounds could be isolated until now.
More stable against reduction is the isoelectronic phenylimido core and some technetium isocyanide complexes with a central {Tc(NPh)}3+ unit could be isolated.19,20 A number of interesting reactivity features have been observed during this study, which makes it interesting to have a look at the related chemistry of rhenium.
The phenylimido complex [Re(NPh)Cl3(PPh3)2] is isoelectronic to the oxidorhenium(V) compound [ReOCl3(PPh3)2], which is frequently used as a common precursor for ligand exchange procedures.21 A similar use of the phenylimido compound as the starting material may give access to a wide variety of phenylimido complexes. Unfortunately, the rhenium compound is significantly less soluble than its technetium analog. Thus, predominately reactions with strong chelators or with robust ligands,22–45 which resist harsh reaction conditions, give pure products in good yields. The corresponding bromido complex [Re(NPh)Br3(PPh3)2] is slightly more soluble and is, thus, occasionally used as a better suitable starting material.37,40,44,45 Alternatively, substitutions on the arylimido ligand with the carboxylic, hydroxylic or amine group provide an enhanced solubility.46–50 Such residues, however, sometimes undergo undesired reactions with co-ligands and/or solvents (e.g. esterification, formation of Schiff bases, etc.), which frequently causes undesired side-reactions and/or lower yields. A modulation of the solubility of the phenylimido starting material without significant interference with the chemical behaviour of other ligands has recently been demonstrated with the use of p-fluoro-substituted phenylimido ligands in [M(NPhF)Cl3(PPh3)2] complexes, M = Tc or Re (1).51,52
In the present report, we present a series of (fluorinated) phenylimidorhenium(V) complexes with a variety of different alkyl and aryl isocyanides (Fig. 1).
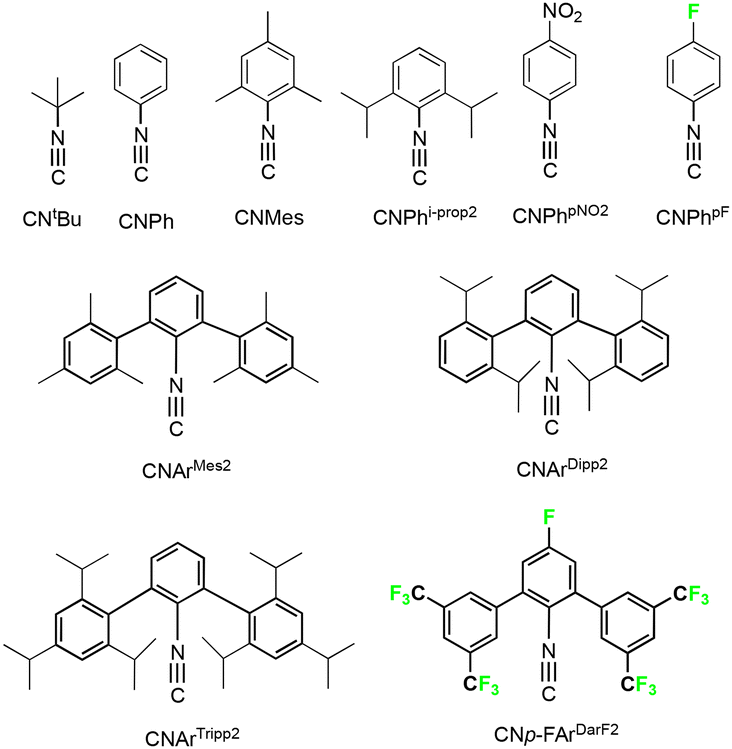 | ||
| Fig. 1 Isocyanides used throughout this paper. | ||
Results and discussion
The attachment of a fluorine atom to the phenylimido ligand of [Re(NPh)Cl3(PPh3)2] increases the room temperature-solubility by a factor of 5 in both dichloromethane and acetonitrile. This allows for milder reaction conditions and lower temperatures, which is particularly beneficial for sensitive isocyanides such as CNPh, CNMes or CNPhpNO2 of the present study. These ligands as well as other common isocyanides such as CNtBu or CNPhi-prop2 react with [Re(NPhF)Cl3(PPh3)2] (1) with the formation of mono-substituted complexes of the composition [Re(NPhF)Cl3(PPh3)(CNR)] (Scheme 1). The reactions have first been performed with an excess of ligands and for the more stable ligands (CNtBu and CNPhi-prop2) also prolonged reactions at higher temperatures have been tested. In both cases, exclusively 1:1 ligand exchange products could be isolated. The gradual (not metal-driven) decomposition of the isocyanides, however, caused significant problems during the isolation of pure products in these cases. For this reason, relatively short reaction times and only a slight excess of the ligands have been used in optimized procedures. Even so, only a small amount of pure products could be obtained from some of the sensitive isocyanides.
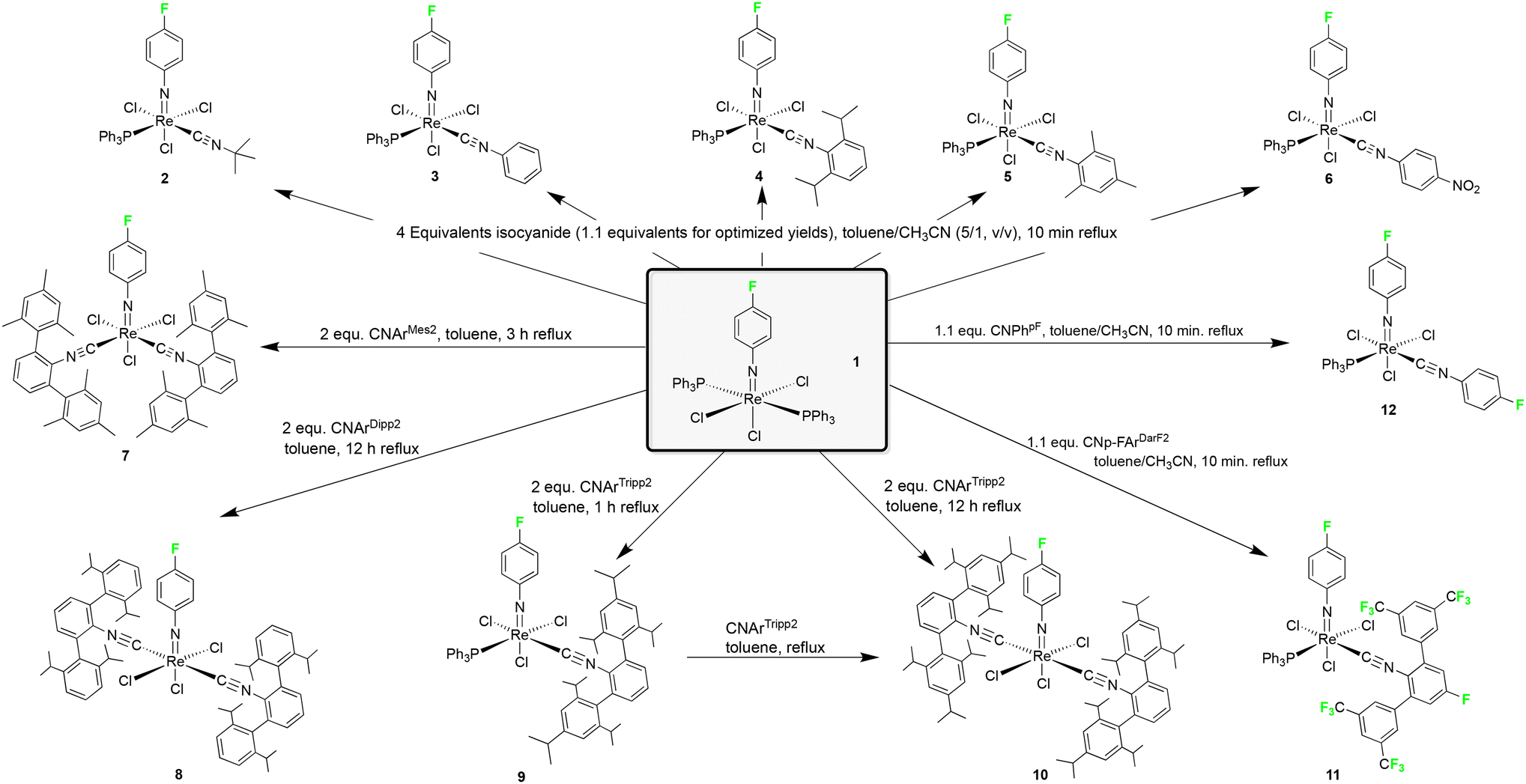 | ||
| Scheme 1 Reaction of [Re(NPhF)Cl3(PPh3)2] with various isocyanides. | ||
The formation of 1:1 complexes with common isocyanides has also been observed for reactions of [Tc(NPh)Cl3(PPh3)2],20 and for reactions of robust isocyanides with the corresponding phenyl- or tolyl- or xylylimido complexes.46,53–55 These findings are not necessarily trivial since the ligand exchange behaviour of isocyanides has been found to be strongly dependent on steric and electronic factors. In particular, electronic effects due to electron withdrawing or electron donating substituents on (phenyl) isocyanides have been underestimated in the coordination chemistry of these compounds. Systematic studies about this point are rare, which is attributed to the preferred use of commercially available and stable ligands such as e.g. tert-butyl or cyclohexyl isocyanide in many papers about the related chemistry. Approximately 40 percent of all studies with isocyanides are done with these two representatives. A nice overview of this point is given in ref. 56. This, finally, resulted in a (not fully justified) generalization of their chemical behaviour for all isocyanides. After the exploration of the coordination chemistry of the bulky (but also electronically diverse) m-terphenyl isocyanides,57–69 it became evident that isocyanides are more than bulky surrogates of carbonyl ligands. This also includes studies on group 7 elements, which have been stabilized in oxidation states ranging from ‘−1’ to ‘+5’.10,11,20–22,70,71 A DFT-based sum parameter describing the electronic potential on the accessible VdW surface of the isocyanide carbon atom has recently been derived for a number of isocyanides.10 The so-called SADAP (surface-averaged donor acceptor potential) parameter is a combined descriptor of the steric and electrostatic properties of the potential ligand. Table 2 contains selected SADAP parameters for the isocyanides of the present study. Details of the calculations and the derived parameter comprising a large number of isocyanides are outlined in ref. 10. Such parameters nicely describe the reactivity of a number of carbonyltechnetium(I) complexes, where ligands with progressively electron-deficient properties readily replace carbonyl ligands, while those with a large negative potential at the carbon atoms are σ-donors with predominantly negligible back-donation properties.10 Similar considerations have also been done for phenylimidotechnetium(V) compounds, d2 systems, where π-acceptor behaviour should be negligible.20
The formation of 1:1 complexes during reactions of [Re(NPhF)Cl3(PPh3)2] with the relatively electron-deficient isocyanides as shown in Fig. 1 is in line with such considerations. The νC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N IR stretches of the complexes appear between 2158 and 2192 cm−1. These values are significantly higher (between 39 and 72 cm−1, for individual values see the Experimental section) than those in the spectra of uncoordinated isocyanides, which means that there is no sign for π-backdonation into anti-bonding orbitals of the ligands in the d2 complexes under study.
N IR stretches of the complexes appear between 2158 and 2192 cm−1. These values are significantly higher (between 39 and 72 cm−1, for individual values see the Experimental section) than those in the spectra of uncoordinated isocyanides, which means that there is no sign for π-backdonation into anti-bonding orbitals of the ligands in the d2 complexes under study.
Ellipsoid plots of the molecular structures of the [Re(NPhF)Cl3(PPh3)(CNR)] complexes (R = tBu, Ph, Phi-prop2, Mes, PhpNO2) are depicted in Fig. 1. Selected bond lengths and angles are summarized in Table 1. The Re–N bonds are clearly in the range of double bonds and the slightly bent phenylimido units (Re–N–C angles between 164 and 173°) are a common feature of such compounds.22–45 The replacement of one of the PPh3 ligands of [Re(NPhF)Cl3(PPh3)2] goes along with a rearrangement of the coordination sphere of rhenium. The incoming isocyanides are found in cis position to phosphine in all 1:1 complexes.
| 2 | 3 | 4 | 5 | 6 | 11 | 12 | |
|---|---|---|---|---|---|---|---|
| a Values of two crystallographically independent species. | |||||||
| Re–N10 | 1.715(3), 1.715(3) | 1.731(3) | 1.722(3) | 1.712(3) | 1.711(3) | 1.721(3) | 1.705(4) |
| N10–C11 | 1.391(4), 1.382(4) | 1.377(4) | 1.385(4) | 1.399(5) | 1.390(5) | 1.380(4) | 1.395(7) |
| Re–P1 | 2.4593(9), 2.4473(9) | 2.4539(9) | 2.4476(9) | 2.463(1) | 2.454(1) | 2.4494(8) | 2.470(2) |
| Re–C1 | 2.020(4), 2.045(4) | 2.028(4) | 2.023(3) | 2.030(4) | 2.020(5) | 2.022(3) | 2.024(6) |
| C1–N1 | 1.151(5), 1.146(5) | 1.165(5) | 1.147(4) | 1.151(6) | 1.162(6) | 1.151(4) | 1.156(7) |
| Re–N10–C11 | 164.4(3), 166.7(2) | 168.7(3) | 172.1(2) | 166.0(3) | 171.2(3) | 170.4(2) | 172.9(4) |
| Re–C1–N1 | 172.9(3), 173.8(8) | 177.7(3) | 170.6(3) | 176.9(3) | 176.6(4) | 174.6(3) | 176.9(4) |
| N10–Re–P1 | 93.1(1), 91.42(9) | 89.99(9) | 94.15(9) | 92.0(1) | 94.2(1) | 91.96(8) | 94.2(2) |
| N10–Re–C1 | 86.0(1), 88.1(1) | 87.7(1) | 88.1(1) | 85.7(1) | 89.4(2) | 88.5(1) | 91.8(2) |
Two isocyanide ligands could be bonded to a {ReNPhF}3+ unit, when more electron-rich isocyanides such as CNArMes2, CNArDipp2 or CNArTripp2 were used. The steric bulk of the individual ligands, however, has an influence on the isomers formed. The reaction of the less bulky CNArMes2 gives the 1:2 complex fac-[Re(NPhF)Cl3(CNArMes2)2] (7) with the two isocyanide ligands in cis position to each other, while the corresponding trans complexes are formed by the sterically more encumbered ligands CNArDipp2 and CNArTripp2 (Scheme 1). The formation of cis complexes with CNArMes2 is a common feature of this ligand and has also been observed for oxido and nitrido complexes of rhenium and technetium.18,19 There are even some rare cases where three CNArMes2 ligands are accommodated in octahedral complexes of cobalt,72 molybdenum,58,73 and manganese.62 Such flexibility regarding the number of ligands and their coordination positions, however, is also the source of the appearance of a fluxional behaviour in solution, which could be detected preferably by 19F NMR, with subsequent problems for the isolation of satisfactory amounts of the individual species in pure form. This is mainly due to the almost identical solubility of the formed species and also applies to the phenylimido compounds under study with the consequence that only a relatively small amount of complex 7 could be isolated in crystalline form. Although the isolated blue needles of 7 were identical as has been tested by X-ray diffraction on several species, they quickly isomerize in solution and again several 19F NMR signals are detected. This behaviour is unlike that of the complexes with the more bulky CNArDipp2 or CNArTripp2 ligands, which form inert mer-[Re(NPhF)Cl3(isocyanide)2] complexes that contain the isocyanides necessarily in trans position to each other.
Ellipsoid plots of the molecular structures of complex fac-[Re(NPh)Cl3(CNArMes2)2] (7) and mer-[Re(NPhF)Cl3(CNArDipp2)2] (8) are shown in Fig. 3. All common features of the central {Re(NPhF)}3+ unit discussed above for compounds 2–6 (Re–N double bond, slightly bent Re–N10–C11 axis) also apply for the bis complexes 7 and 8. The rhenium-carbon bonds in the cis isomer 7 (2.034–2.044 Å) are slightly shorter than in the trans isomer 8 (2.055–2.076 Å). This comes not unexpected with regard to the sterically favoured trans compound. Similar effects have been observed previously for the cis and trans isomers of [Re(CO)3Br(CNArDipp2)2], where the Re–C (isocyanide) bonds are elongated by approximately 0.02 Å in trans position to carbonyl ligands,70 for which, however, mainly electronic effects shall be responsible.
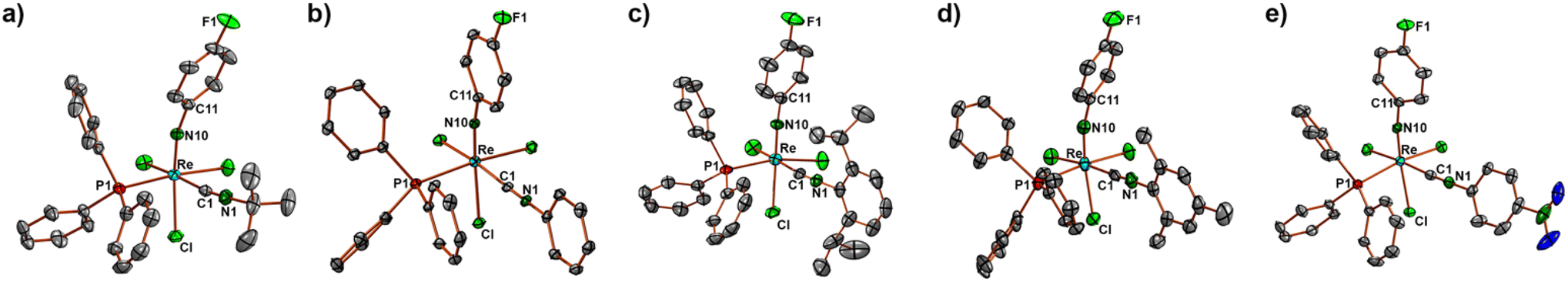 | ||
| Fig. 2 Molecular structures of (a) [Re(NPhF)Cl3(PPh3)(CNtBu)] (2), (b) [Re(NPhF)Cl3(PPh3)(CNPh)] (3), (c) [Re(NPhF)Cl3(PPh3)(CNPhi-prop2)] (4), (d) [Re(NPhF)Cl3(PPh3)(CNMes)] (5) and (e) [Re(NPhF)Cl3(PPh3)(CNPhpNO2)] (6). | ||
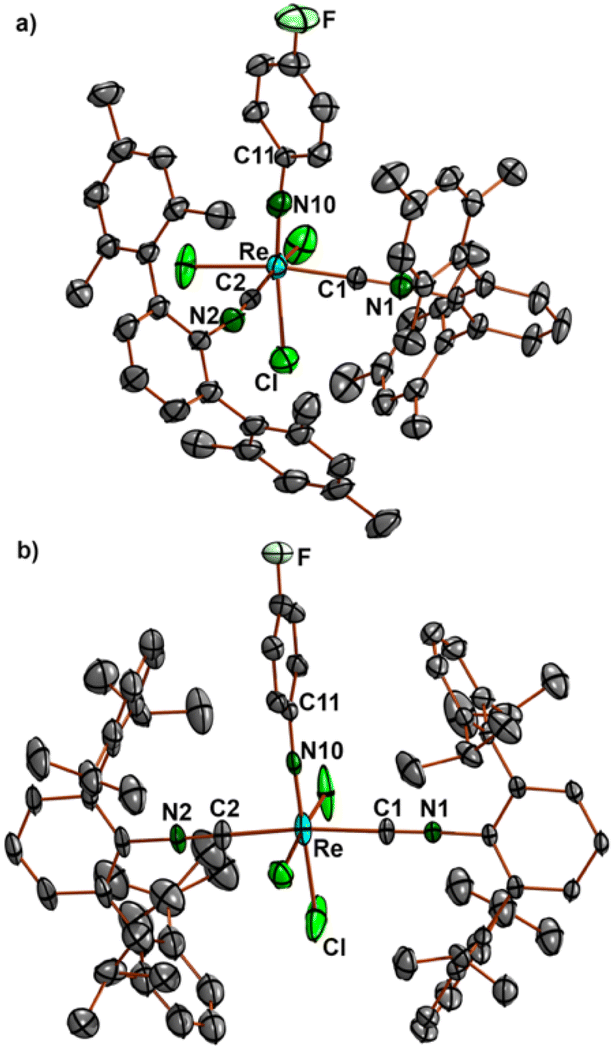 | ||
| Fig. 3 Molecular structures of (a) fac-[Re(NPh)Cl3(CNArMes2)2] (7) and (b) mer-[Re(NPhF)Cl3(CNArDipp2)2] (8) (selected bond lengths and angles of 7: Re–N10 1.724(3), Re–C1 2.043(4), Re–C2 2.034(4) Å, Re–N10–C11 173.0(3), Re–C1–N1 176.6(3), and Re–C2–N2 174.2(4)°; selected bond lengths and angles of 8: Re–N10 1.728(7) and 1.722(6), Re–C1 2.076(6) and 2.066(6), Re–C2 2.055(7) and 2.055(7) Å, Re–N10–C11 177.4(5) and 171.5(5), Re–C1–N1 179.1(7) and 178.2(6), and Re–C2–N2 176.6(7) and 178.3(6)°. The values of compound 8 refer to two independent molecular species. | ||
The exchange of the PPh3 ligands occurs stepwise and can nicely be monitored by 19F NMR. Fig. 4 shows 19F NMR spectra recorded for a corresponding reaction of [Re(NPhF)Cl3(PPh3)2] (1) with CNArDipp2 in toluene. The formation of two transient compounds can readily be detected by their 19F NMR signals in addition to the signals of the (not yet consumed) starting material 1 in the initial phase (first hour) of the reaction. Upon subsequent heating, the intensities of these signals and of compound 1 decrease in favour of the formation of the final product of the reaction mer-[Re(NPhF)Cl3(CNArDipp2)2] (8). The tentative assignment of the transient signals A and B is possible in two ways: (1) to a mixture of cis- and trans-[Re(NPhF)Cl3(PPh3)(CNArDipp2)] or (2) to a mixture of cis-[Re(NPhF)Cl3(PPh3)(CNArDipp2)] and [Re(NPhF)Cl3(PPh3)(CNArDipp2)2]. Evidence for the latter, seven-coordinate species is given with the detection of an intense peak at m/z = 1349.600 (simulation: 1349.309) in the ESI + mass spectrum of the reaction mixture after 10 min. Up to now, we could not isolate these two or at least one of these complexes in crystalline form for an unambiguous proof of one of these assumptions. This, however, was possible for the corresponding reaction with CNArTripp2.
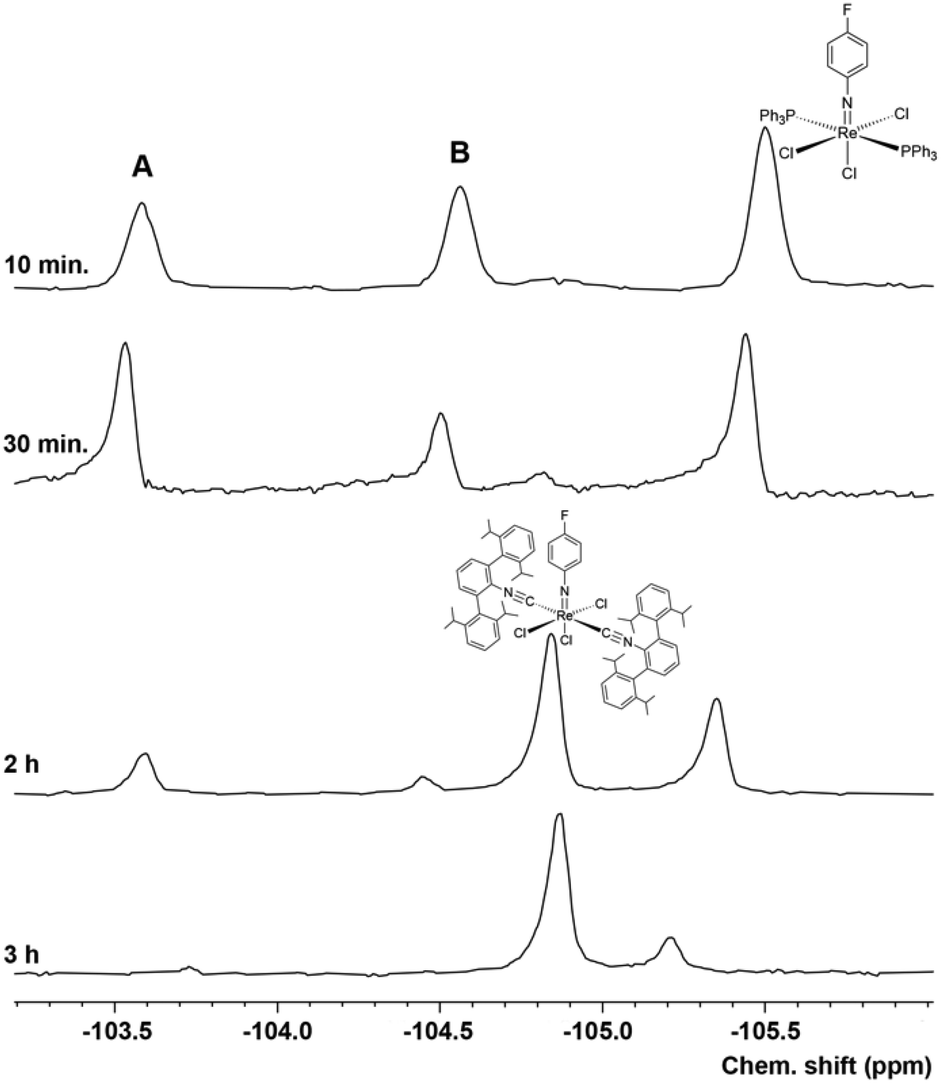 | ||
| Fig. 4 19F NMR monitoring of a reaction mixture of [Re(NPhF)Cl3(PPh3)2] and two equivalents of CNArDipp2 in boiling toluene. Note the narrow chemical shift range depicted and that the chemical shifts slightly differ from the values given in the Experimental part due to solvent effects. | ||
The 19F NMR monitoring of the reaction of 1 with CNArTripp2 (see the ESI, Fig. S57†) shows that essentially only one major transient compound is formed. The concentration of this compound reaches a maximum after approximately 90 min and when the reaction is stopped at this point, a small amount of this intermediate can be separated from the bulk of the starting material 1 and crystallized. An X-ray diffraction study on these single crystals confirms the formation of fac-[Re(NPhF)Cl3(PPh3)(CNArTripp2)] (9) as the (isolated) intermediate of this reaction. Prolonged heating of the reaction mixtures gives the bis complex [Re(NPhF)Cl3(CNArTripp2)2] (10) similar to the reaction with CNArDipp2. The molecular structures of compounds 9 and 10 are depicted in Fig. 5. Bond lengths and angles in the products with the extremely sterically encumbered CNArTripp2 ligand are similar to those observed in the other 1:1 complexes or in the analogous bis complex with CNArDipp2 discussed above. Also in compound 10, the Re–C bonds are slightly longer than in the complexes of the composition [Re(NPhF)Cl3(PPh3)(CNR)], where the isocyanides are in trans positions to chlorido ligands.
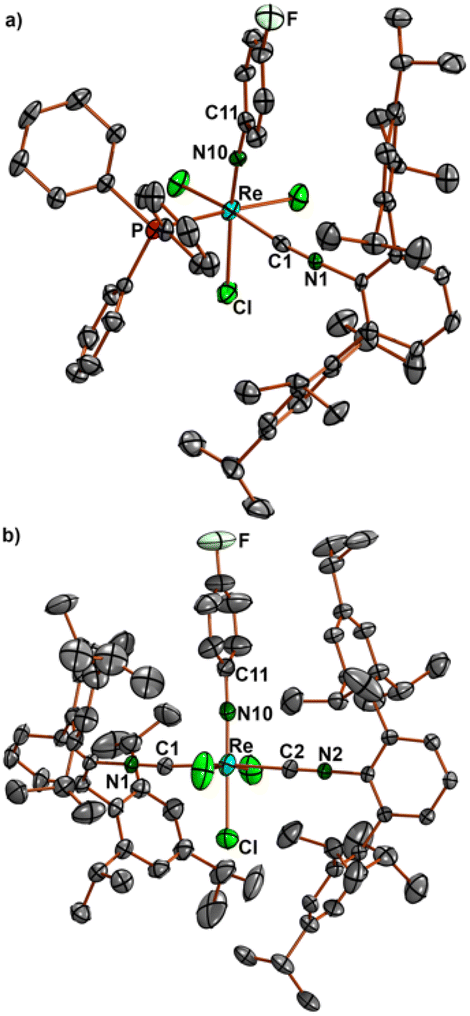 | ||
| Fig. 5 Molecular structures of (a) fac-[Re(NPh)Cl3(PPh3)(CNArTripp2)] (9) and (b) mer-[Re(NPhF)Cl3(CNArTripp2)2] (10). Selected bond lengths and angles of 9: Re–N10 1.711(4), Re–P 2.460(2), Re–C1 2.027(5), C1–N1 1.167(5) Å; Re–N10–C11 168.7(3), and Re–C1–N1 170.4(4)°. Selected bond lengths and angles of 10: Re–N10 1.698(6) and 1.711(5), Re–C1 2.093(6) and 2.090(6), Re–C2 2.084(6) and 2.076(6), C1–N1 1.135(7) and 1.139(7), C2–N2 1.135(7) and 1.148(7) Å; Re–N10–C11 177.1(5) and 178.7(5), Re–C1–N1 175.3(6) and 178.2(6), and Re–C2–N2 175.4(5) and 179.5(7)°. The values of compound 10 refer to two independent molecular species. | ||
CNArDipp2 and CNArTripp2 are structurally distinguished by the presence of additional isopropyl substituents at the peripheral phenyl rings. At first glance this seems to be a minor difference, especially since this substitution is in the 4-position and, thus, relatively far away from the coordinating carbon atom. This means, that a similar electronic potential should be expected for both ligands, while CNArTripp2 should be slightly more sterically hindered, which is finally also reflected by the SADAP parameters in Table 2 and the formation of 1:2 complexes of the present study. Nevertheless, there is a striking difference between these two isocyanides regarding their steric bulk. This can nicely be seen in Fig. 6, where the solid state structures of [Re(NPhF)Cl3(CNArDipp2)2] (8) and [Re(NPhF)Cl3(CNArTripp2)2] (10) are shown along the planes formed by their phenylimido ligands. The central phenyl rings of the two CNArDipp2 ligands are almost perfectly co-planar (twisting angle: 7.1(6)°) in a perpendicular arrangement to the phenylimido ring. The larger bulk of CNArTripp2 results in a torsion of 52.3(2)° of the isocyanide ligand around its coordination axis. This is a direct effect of the ‘outer-sphere’ substitution on this ligand and may underline the influence of such relatively small modifications in the ligand design on structural factors, but also on the reactivity of the respective metal ion. It shall be noted that the proton NMR resonances of CNArTripp2 appear as one set of signals suggesting that a rotation is possible in solution.
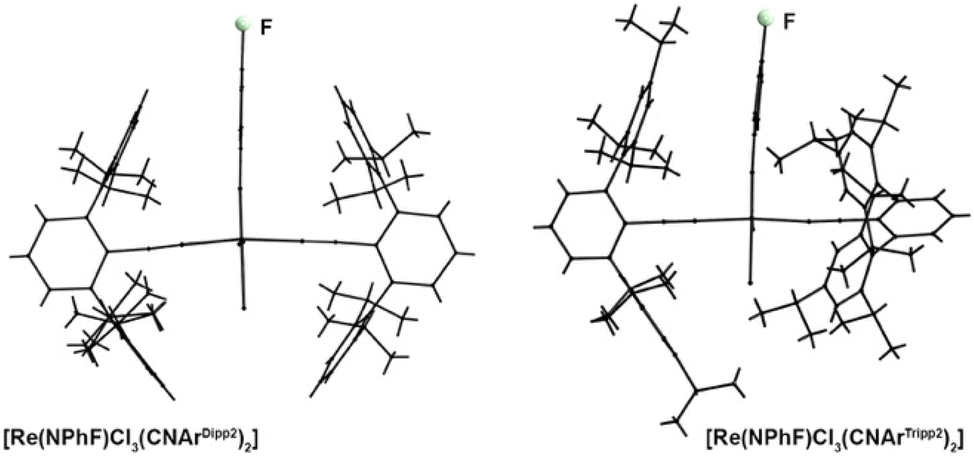 | ||
| Fig. 6 Wire-frame presentation of the structures of mer-[Re(NPh)Cl3(CNArDipp2)2] (8) and mer-[Re(NPhF)Cl3(CNArTripp2)2] (10) along the planes formed by their phenylimido ligands. | ||
| Isocyanide | SADAP |
|---|---|
| CNArTripp2 | −3.33 |
| CNArDipp2 | −3.28 |
| CNPhi-prop2, CNMes, CNtBu | −2.38, −2.26, −2.15 |
| CNPh, CNPhpNO2 | −1.64, −0.95 |
| CNPhpF | −1.25 |
| CNp-FArDarF2 | +2.73 |
The steric effects discussed above shall always be regarded together with potential electronic effects due to the introduced residues and the electronic configuration of the metal ion. An example of the complex interplay of such influences shall be discussed with the use of the fluorinated isocyanides CNp-FArDarF2 and CNPhpF.
The steric bulk of CNp-FArDarF2 due to the CF3 residues in meta positions of the terphenyl rings takes mainly effect in the outer sphere of the complex similar to CNArTripp2, but the fluorinated ligand is more flexible in terms of a spheric arrangement around the metal ions. Thus, it allows the coordination of up to four of the ligands around a metal ion. This has been demonstrated recently for carbonyl complexes of manganese, technetium and rhenium, where compounds of the compositions [M−I(CO)(CNp-FArDarF2)4]− (M = Tc, Re), [M0(CO)(CNp-FArDarF2)4] (M = Tc, Re) and [MI(CO)(CNp-FArDarF2)4] (M = Tc, Re) have been isolated in crystalline form.71,74 The replacement of carbonyl ligands from tricarbonylrhenium(I) or technetium(I) centers is not common and can be attributed to the special electronic situation on the donor carbon atom due to the strong electron withdrawing capacity of the fluorine atom at the central phenyl ring of CNp-FArDarF2.
For isocyanides, it is reasonable to assume that electron-deficient regions on the surface of the coordinating carbon atom would enable improved π-back-donation, while electron-rich regions on the surface of the same carbon atom are responsible for a better σ-donation. Steric restraints on the donor carbon atom can be partially included in such an approach by averaging the obtained potential energies over the accessible surface of the potential donor atoms.
Thus, sterically demanding isocyanides such as CNArDipp2 or CNArTripp2 show less overall accessible surface area, while the less encumbered isocyanides have a larger overall accessible carbon surface. A more comprehensive discussion of this approach is shown in ref. 10. In the sterically encumbered, but fluorine-substituted CNp-FArDarF2 ligand the steric effects are overruled by the electron withdrawing capacity of fluorine, which make this ligand a powerful π-acceptor when it reacts with electron rich metal ions such as rhenium(I) or technetium(I) and explains the reactivity with the corresponding carbonyl compounds.10,11,20 In the electron-deficient rhenium(V) complexes of the present study, however, π-back-donation plays practically no role, which is supported by the blue-shift of the IR νCN frequencies. Consequently, the reactivity of CNp-FArDarF2 in such cases is identical with those isocyanides with predominantly σ-donor properties and [Re(NPhF)Cl3(PPh3)(CNp-FArDarF2)] (11) is formed as the sole product in a reaction with 1. The corresponding reactivity is well reflected by the SADAP parameters in Table 2.
A similar behavior is observed for CNPhpF, where the fluorine substitution at the central ring is retained, but the steric bulk due to the peripheral residuals is removed. Indeed, it behaves with electron-rich metal ions as a powerful π-acceptor, as has been demonstrated by the complete replacement of carbonyl and halido ligands in [Tc2(CO)6Cl3]2− and the formation of a [Tc(CNPhpF)6]+ cation.20 Expectedly, this is not observed for the d2 complexes of the present study and the 1:1 complex [Re(NPhF)Cl3(PPh3)(CNPhpF)] (12) was isolated from a corresponding reaction with compound 1.
The structures of compounds 11 and 12 are shown in Fig. 7. All main structural features are close to those of the other 1:1 complexes presented in Fig. 2 and Table 1 and shall not be discussed here in detail.
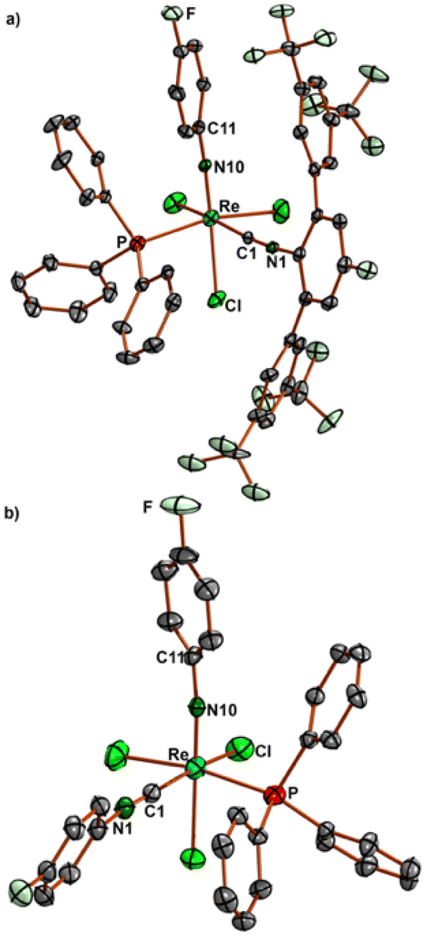 | ||
| Fig. 7 Molecular structures of a) fac-[Re(NPh)Cl3(PPh3)(CNArDarF2)2] (11) and b) mer-[Re(NPhF)Cl3(PPh3)(CNPhpF)2] (12). Selected bond lengths and angles for 11: Re–N10 1.721(43), Re–P 2.4494(9), Re–C1 2.022(3), C1–N1 1.151(4) Å; Re–N10–C11 170.4(2), and Re–C1–N1 174.6(43)°; selected bond lengths and angles of 12: Re–N10 1.705(4), Re–P 2.470(2), Re–C1 2.024(6), C1–N1 1.156(7) Å; Re–N10–C11 172.9(4), and Re–C1–N1 176.9(5)°. | ||
Experimental
General considerations
Unless otherwise stated, reagent-grade starting materials were purchased from commercial sources and either used as received or purified by standard procedures. Solvents were dried according to standard procedures. The reactions have been performed in air unless otherwise stated. [Re(NPhpF)Cl3(PPh3)] (1) as well as CNArDipp2, CNArTripp2, CNp-FArDarF2, CNPhpF, and CNPhi-prop2 were prepared according to literature procedures.10,51,56,58,59Physical measurements
NMR spectra were recorded with JEOL 400 MHz ECS or ECZ or JEOL ECZ 600 or Bruker AV700 multinuclear spectrometers. IR spectra were recorded with an ATR spectrometer (Nicolet iS10, Thermo Scientific). Intensities are classified as vs = very strong, s = strong, m = medium, w = weak, vw = very weak, and sh = shoulder. Electrospray ionization mass spectrometry (ESI MS) was carried out with the ESI MSD time-of-flight (TOF) unit of an Agilent 6210 TOF liquid chromatography/mass spectrometry system. Elemental analyses of carefully dried samples of the bulk were performed using a Vario MICRO cube CHNS elemental analyser.X-ray crystallography
The intensities for X-ray determinations were collected on STOE IPDS II or Bruker D8 Venture instruments with Mo Kα radiation. The space groups were determined by the detection of systematic absences. Absorption corrections were carried out by multi scan or integration methods.75,76 Structure solution and refinement were performed with the SHELX program package using OLEX 2.77–79 Hydrogen atoms were derived from the final Fourier maps and refined or placed at calculated positions and treated with the ‘riding model’ option of SHELXL. The representation of molecular structures was done using the program DIAMOND 4.2.2.80Additional information on the structure determinations is presented in the ESI† and has been deposited with the Cambridge Crystallographic Data Centre.
Synthesis
:1 toluene/acetonitrile mixture (6 mL) and the corresponding isocyanide (0.11 mmol) was added. The mixture was heated under reflux for 10 min in a pre-heated oil bath. The starting material dissolved and the solution turned blue or green. Volatiles were removed under reduced pressure and the residue was resuspended in diethyl ether (5 mL) and filtered. The obtained solid was washed with diethyl ether (3 × 5 mL) and dried under reduced pressure.
N), 1584 (vs). 1H NMR (CD2Cl2, ppm): δ = 7.75 (mc, 6H), 7.42 (mc, 9H), 7.02 (mc, 2H,), 6.82 (t, J = 8.0 Hz, 2H), 1.43 (s, 9H). 19F NMR (CD2Cl2, ppm): δ = −101.9 (mc). 13C{1H} NMR (CD2Cl2, ppm): δ = 162.83 (d, 1J(13C–19F) = 260 Hz), 151.32, 135.01 (d, J(13C–31P) = 8 Hz), 133.73 (d, 1J(13C–31P) = 53 Hz), 131.74, 128.90 (d, J(13C–31P) = 10 Hz), 127.09 (d, 3J(13C–19F) = 10 Hz), 117.55 (d, 2J(13C–19F) = 25 Hz), 60.30, 31.24. ESI + MS: m/z = 628.0141 [M − CNtBu − Cl]+ (calc.: 628.0158), 711.0855 [M − Cl]+ (calc.: 711.0894), 769.0463 [M + Na]+ (calc.: 769.0494), 1517.069 [2M + Na]+ (calc.: 1517.0999).
N). ESI + MS: m/z = 731.0669 [M − Cl]+ (calc.: 731.0581), 789.0183 [M + Na]+ (calc.: 789.01622), 804.9920 [M + K]+ (calc.: 804.9901), 1557.0502 [2M + Na]+ (calc.: 1557.0417).
C). 1H NMR (CD2Cl2, ppm): δ = 7.81 (mc, 6H), 7.39 (mc, 3H), 7.34 (mc, 6H), 7.26–7.19 (m, 3H), 7.03(mc, 2H), 6.86 (mc, 2H), 2.71 (h, 3J(1H, 1H) = 7 Hz, 2H), 1.00 (d, 3J(1H, 1H) = 7 Hz, 6H), 0.92 (d, 3J(1H, 1H) = 7 Hz, 6H). 19F NMR (CD2Cl2, ppm): δ = −101.9 (mc). 13C{1H} NMR (CD2Cl2, ppm): δ = 162.86 (d, 1J(13C,19F) = 260 Hz), 151.45 (t, 1J(13C,14N) = 3 Hz), 145.48, 135.02 (d, J(13C,31P) = 9 Hz), 133.45 (d, 1J(13C,31P) = 45 Hz), 131.87 (d, J(13C,31P) = 2 Hz), 131.30, 129.01 (d, J(13C,31P) = 11 Hz), 126.93 (d, 3J(13C,19F) = 10 Hz), 123.41, 117.58 (d, 2J(13C,19F) = 25 Hz), 29.74, 23.51, 22.86. ESI + MS: m/z = 873.109 [M + Na]+ (calc.: 873.112), 889.084 [M + K]+ (calc.: 889.086), 1725.233 [2M + Na]+ (calc.: 1723.234), 1739.209 [2M + K]+ (calc.: 1739.208).
C). 1H NMR (CD2Cl2, ppm): δ = 7.78 (mc, 6H), 7.34 (mc, 9H), 7.04 (mc, 2H), 6.92 (s, 2H), 6.83 (dd, 3J(1H, 1H) = 8 Hz, 2H), 2.42 (s, 3H), 2.01 (s, 6H). ESI + MS: m/z = 773.098 [M − Cl]+ (calc.: 773.1051), 1656.115 [2M + K]+ (calc.: 1656.1019), 1657.115 [2M + K]+ (calc.: 1657.1097).
C), 1735 (m, νN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). 1H NMR (acetone-d6, ppm): δ = 8.32 (mc, 2H), 7.79 (mc, 6H), 7.42–7.32 (m, 9H), 7.12–7.05 (m, 4H), 6.88 (dd, 3J(1H, 1H) = 8 Hz, 6H). 19F NMR (acetone-d6, ppm): δ = −100.4 (mc). ESI + MS: m/z = 834.003 [M + Na]+ (calc.: 834.003), 1647.013 [2M + Na]+ (calc.: 1645.017).
C). 1H NMR (acetone-d6, ppm): δ = 8.22 (s, 4H), 7.76 (s, 2H), 7.73 (d, 3J(1H, 19F) = 8 Hz, 2H), 7.49 (mc, 9H), 7.35 (mc, 3H,), 7.22 (mc, 6H), 6.75 (mc, 2H), 6.62 (mc, 2H). 19F NMR (acetone-d6, ppm): δ = −62.6 (s, 12F), −103.1 (mc, 1F), −109.9 (t, 3J(19F,1H) = 8 Hz, 1F). 13C{1H} NMR (acetone-d6, ppm): δ = 163.42 (d, 1J(13C,19F) = 260 Hz), 163.31 (d, 1J(13C,19F) = 260 Hz), 150.41, 148.13, 139.71 (d, J = 10 Hz), 138.30, 135.12 (d, J(13C,31P) = 10 Hz), 133.88 (d, 1J(13C,31P) = 52 Hz), 132.46 (q, 2J(13C,19F) = 33 Hz), 131.78, 131.14, 128.92 (d, J(13C,31P) = 10 Hz), 128.62 (d, 3J(13C,19F) = 10 Hz), 124.00 (q, 1J(13C,19F) = 260 Hz), 123.77, 119.98, 119.04 (d, 2J(13C,19F) = 26 Hz), 117.61 (d, 2J(13C,19F) = 26 Hz). ESI + MS: m/z = 1231.015 [M + Na]+ (calc.: 1231.020), 1246.989 [M + K]+ (calc.: 1246.995), 2441.037 [2M + Na]+ (calc.: 2441.043), 2457.010 [2M + K]+ (calc.: 2457.017).
C). 1H NMR (CD2Cl2, ppm): δ = 7.80 (mc, 6H), 7.39 (mc, 9H), 7.18 (mc, 2H), 7.08 (mc, 2H), 7.03 (mc, 2H), 6.85 (mc, 2H). 19F NMR (CD2Cl2, ppm): δ = −101.4 (mc, 1F), −106.9 (mc, 1F). 13C{1H} NMR (CD2Cl2, ppm): δ = 164.12 (d, (1J(13C–19F) = 260 Hz), 163.17 (d, (1J(13C–19F) = 260 Hz), 150.98, 140.81, 134.93 (d, J(13C–31P) = 8 Hz), 133.16 (d, 1J(13C–31P) = 54 Hz), 131.87, 129.49 (d, 3J(13C–19F) = 10 Hz), 129.05 (d, J(13C–31P) = 10 Hz), 127.65 (d, 3J(13C–19F) = 10 Hz), 122.79, 117.70 (d, 2J(13C–19F) = 25 Hz), 117.17 (d, 2J(13C–19F) = 25 Hz). ESI + MS: m/z = 806.990 [M + Na]+ (calc.: 807.008), 822.964 [M + K]+ (calc.: 822.983), 1590.993 [2M + Na]+ (calc.: 1591.028), 1607.002 [2M + K]+ (calc.: 1606.966).
N), 2163 (s, νCN). 1H NMR (CD2Cl2, ppm): δ = 7.52 (dd, 3J(1H,1H) = 8 Hz, 2H), 7.27 (d, 3J(1H,1H) = 8 Hz, 4H), 7.85 (dd, 3J(1H,1H) = 8 Hz, 2H), 6.83 (s, 4H), 6.74 (s, 4H), 6.43(mc, 2H), 2.22 (s, 12H), 2.01 (s, 12H), 1.98 (s, 12H). 19F NMR (CD2Cl2, ppm): δ = −101.1 (mc). 13C{1H} NMR (CD2Cl2, ppm): δ = 163.39 (d, 1J(13C,19F) = 260 Hz), 150.20 (br. S), 140.13, 138.08, 136.17 (d, 3J(13C,19F) = 14 Hz), 135.21, 133.13, 131.61, 130.27, 128.89, 128.72, 128.58 (d, 4J(13C,19F) = 9 Hz), 125.03, 117.14 (d, 2J(13C,19F) = 24 Hz), 21.38, 20.69, 20.29. ESI + MS: m/z = 1044.3266 [M − Cl]+ (calc.: 1044.3224), 1102.2871 [M + Na]+ (calc.: 1102.2850), 2183.5903 [2M + Na]+ (calc.: 2183.5718).
C). 1H NMR (CD2Cl2, ppm): δ = 7.47 (dd, 3J(1H,1H) = 7 Hz), 7.28 (d, 3J(1H,1H) = 8 Hz, 4H), 7.12 (dd, 3J(1H,1H) = 8 Hz), 7.06 (mc, 2H), 7.02–6.98 (m, 10H), 2.42 (hept, 3J(1H,1H) = 7 Hz), 1.05 (d, 3J(1H,1H) = 8 Hz, 24H), 1.03 (d, 3J(1H,1H) = 8 Hz, 24H). 19F NMR (CD2Cl2, ppm): δ = −103.4(mc). 13C{1H} NMR (CD2Cl2, ppm): δ = 160.43 (d, 1J(13C,19F) = 260 Hz), 153.48, 146.77, 140.32, 133.35, 130.83, 130.17, 129.57, 125.34, 124.25 (d, 3J(13C,19F) = 10 Hz), 123.17, 117.84 (d, 2J(13C,19F) = 25 Hz), 31.54, 24.57, 24.13. ESI + MS: m/z = 1270.4715 [M + Na]+ (calc.: 1270.46951).
C). 1H NMR (CD2Cl2, ppm): δ = 7.47 (dd, 3J(1H,1H) = 8 Hz, 2H), 7.31 (d, 3J(1H,1H) = 8 Hz, 4H), 7.07 (mc, 2H), 6.93 (s, 8H), 6.86 (mc, 2H), 2.82 (h, 3J(1H,1H) = 7 Hz, 4H), 2.38 (h, 3J(1H,1H) = 7 Hz, 8H), 1.28 (d, 3J(1H,1H) = 8 Hz, 24H), 1.02 (d, 3J(1H,1H) = 8 Hz, 24H), 0.95 (d, 3J(1H,1H) = 8 Hz, 24H). 19F NMR (CD2Cl2, ppm): δ = −104.1 (mc). 13C{1H} NMR (CD2Cl2, ppm): δ = 160.43 (d, 1J(13C,19F) = 260 Hz), 153.96, 149.74, 146.40, 141.49, 131.18, 130.76, 130.46, 126.09, 123.59 (d, 3J(13C,19F) = 10 Hz), 121.75, 117.52 (d, 2J(13C,19F) = 24 Hz), 34.99, 31.67, 24.91, 24.53, 24.09. ESI + MS: m/z = 1438.644 [M + Na]+ (calc.: 1436.657), 1454.618 [M + K]+ (calc.: 1454.631).
O). 1H NMR (acetone-d6, ppm): δ = 8.32 (mc, 2H), 7.79 (mc, 6H), 7.42–7.32 (m, 9H), 7.12–7.05 (m, 4H), 6.88 (dd, 3J(1H, 1H) = 8 Hz, 6H). 19F NMR (acetone-d6, ppm): δ = −100.4 (mc). ESI + MS: m/z = 834.003 [M + Na]+ (calc.: 834.003), 1647.013 [2M + Na]+ (calc.: 1645.017).
C). 1H NMR (acetone-d6, ppm): δ = 8.22 (s, 4H), 7.76 (s, 2H), 7.73 (d, 3J(1H, 19F) = 8 Hz, 2H), 7.49 (mc, 9H), 7.35 (mc, 3H,), 7.22 (mc, 6H), 6.75 (mc, 2H), 6.62 (mc, 2H). 19F NMR (acetone-d6, ppm): δ = −62.6 (s, 12F), −103.1 (mc, 1F), −109.9 (t, 3J(19F,1H) = 8 Hz, 1F). 13C{1H} NMR (acetone-d6, ppm): δ = 163.42 (d, 1J(13C,19F) = 260 Hz), 163.31 (d, 1J(13C,19F) = 260 Hz), 150.41, 148.13, 139.71 (d, J = 10 Hz), 138.30, 135.12 (d, J(13C,31P) = 10 Hz), 133.88 (d, 1J(13C,31P) = 52 Hz), 132.46 (q, 2J(13C,19F) = 33 Hz), 131.78, 131.14, 128.92 (d, J(13C,31P) = 10 Hz), 128.62 (d, 3J(13C,19F) = 10 Hz), 124.00 (q, 1J(13C,19F) = 260 Hz), 123.77, 119.98, 119.04 (d, 2J(13C,19F) = 26 Hz), 117.61 (d, 2J(13C,19F) = 26 Hz). ESI + MS: m/z = 1231.015 [M + Na]+ (calc.: 1231.020), 1246.989 [M + K]+ (calc.: 1246.995), 2441.037 [2M + Na]+ (calc.: 2441.043), 2457.010 [2M + K]+ (calc.: 2457.017).
C). 1H NMR (CD2Cl2, ppm): δ = 7.80 (mc, 6H), 7.39 (mc, 9H), 7.18 (mc, 2H), 7.08 (mc, 2H), 7.03 (mc, 2H), 6.85 (mc, 2H). 19F NMR (CD2Cl2, ppm): δ = −101.4 (mc, 1F), −106.9 (mc, 1F). 13C{1H} NMR (CD2Cl2, ppm): δ = 164.12 (d, (1J(13C–19F) = 260 Hz), 163.17 (d, (1J(13C–19F) = 260 Hz), 150.98, 140.81, 134.93 (d, J(13C–31P) = 8 Hz), 133.16 (d, 1J(13C–31P) = 54 Hz), 131.87, 129.49 (d, 3J(13C–19F) = 10 Hz), 129.05 (d, J(13C–31P) = 10 Hz), 127.65 (d, 3J(13C–19F) = 10 Hz), 122.79, 117.70 (d, 2J(13C–19F) = 25 Hz), 117.17 (d, 2J(13C–19F) = 25 Hz). ESI + MS: m/z = 806.990 [M + Na]+ (calc.: 807.008), 822.964 [M + K]+ (calc.: 822.983), 1590.993 [2M + Na]+ (calc.: 1591.028), 1607.002 [2M + K]+ (calc.: 1606.966).
N), 2163 (s, νCN). 1H NMR (CD2Cl2, ppm): δ = 7.52 (dd, 3J(1H,1H) = 8 Hz, 2H), 7.27 (d, 3J(1H,1H) = 8 Hz, 4H), 7.85 (dd, 3J(1H,1H) = 8 Hz, 2H), 6.83 (s, 4H), 6.74 (s, 4H), 6.43(mc, 2H), 2.22 (s, 12H), 2.01 (s, 12H), 1.98 (s, 12H). 19F NMR (CD2Cl2, ppm): δ = −101.1 (mc). 13C{1H} NMR (CD2Cl2, ppm): δ = 163.39 (d, 1J(13C,19F) = 260 Hz), 150.20 (br. S), 140.13, 138.08, 136.17 (d, 3J(13C,19F) = 14 Hz), 135.21, 133.13, 131.61, 130.27, 128.89, 128.72, 128.58 (d, 4J(13C,19F) = 9 Hz), 125.03, 117.14 (d, 2J(13C,19F) = 24 Hz), 21.38, 20.69, 20.29. ESI + MS: m/z = 1044.3266 [M − Cl]+ (calc.: 1044.3224), 1102.2871 [M + Na]+ (calc.: 1102.2850), 2183.5903 [2M + Na]+ (calc.: 2183.5718).
C). 1H NMR (CD2Cl2, ppm): δ = 7.47 (dd, 3J(1H,1H) = 7 Hz), 7.28 (d, 3J(1H,1H) = 8 Hz, 4H), 7.12 (dd, 3J(1H,1H) = 8 Hz), 7.06 (mc, 2H), 7.02–6.98 (m, 10H), 2.42 (hept, 3J(1H,1H) = 7 Hz), 1.05 (d, 3J(1H,1H) = 8 Hz, 24H), 1.03 (d, 3J(1H,1H) = 8 Hz, 24H). 19F NMR (CD2Cl2, ppm): δ = −103.4(mc). 13C{1H} NMR (CD2Cl2, ppm): δ = 160.43 (d, 1J(13C,19F) = 260 Hz), 153.48, 146.77, 140.32, 133.35, 130.83, 130.17, 129.57, 125.34, 124.25 (d, 3J(13C,19F) = 10 Hz), 123.17, 117.84 (d, 2J(13C,19F) = 25 Hz), 31.54, 24.57, 24.13. ESI + MS: m/z = 1270.4715 [M + Na]+ (calc.: 1270.46951).
C). 1H NMR (CD2Cl2, ppm): δ = 7.47 (dd, 3J(1H,1H) = 8 Hz, 2H), 7.31 (d, 3J(1H,1H) = 8 Hz, 4H), 7.07 (mc, 2H), 6.93 (s, 8H), 6.86 (mc, 2H), 2.82 (h, 3J(1H,1H) = 7 Hz, 4H), 2.38 (h, 3J(1H,1H) = 7 Hz, 8H), 1.28 (d, 3J(1H,1H) = 8 Hz, 24H), 1.02 (d, 3J(1H,1H) = 8 Hz, 24H), 0.95 (d, 3J(1H,1H) = 8 Hz, 24H). 19F NMR (CD2Cl2, ppm): δ = −104.1 (mc). 13C{1H} NMR (CD2Cl2, ppm): δ = 160.43 (d, 1J(13C,19F) = 260 Hz), 153.96, 149.74, 146.40, 141.49, 131.18, 130.76, 130.46, 126.09, 123.59 (d, 3J(13C,19F) = 10 Hz), 121.75, 117.52 (d, 2J(13C,19F) = 24 Hz), 34.99, 31.67, 24.91, 24.53, 24.09. ESI + MS: m/z = 1438.644 [M + Na]+ (calc.: 1436.657), 1454.618 [M + K]+ (calc.: 1454.631).
Conclusions
The results of the present study may demonstrate that the use of fluorine-substituted ligand systems can have a number of benefits apart from their use in pharmaceutical approaches, but also for fundamental chemical studies. (1) The introduction of the fluorine or CF3 substituent modulates the solubility e.g. of the starting materials, (2) reactions with diamagnetic compounds can readily be monitored (and thus optimized) by means of 19F NMR, (3) the use of a peripheral m,m′-(CF3)2Ar group is a suitable instrument to control the steric bulk of ligand systems and (4) fluorine substituents are well suitable to control the electronic properties of ligand systems.Author contributions
Conceptualization: U. A., J. S. F., and G. C.; methodology: M. R. J. and A. H; validation: G. C. and E. K.; formal analysis: G. C., E. K, A. H., and M. R. J.; investigation: G. C., E. K., and U. A.; resources: U. A.; writing – original draft preparation: U. A.; writing – review and editing: U. A., J. S. F., G. C., M. R. J., and E. K.; visualization: U. A. and G. C.; supervision: U. A., J. S. F., and G. C.; project administration: G. C. and U. A.; funding acquisition: U. A. and J. S. F. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the DFG (Deutsche Forschungsgemeinschaft: Graduate School BIOQIC), the U.S. National Science Foundation (International Supplement to CHE-1802646), and the Alexander von Humboldt Foundation (fellowship to J. S. F.). We acknowledge the assistance of the Core Facility BioSupraMol supported by the DFG and of the High-Performance-Computing (HPC) Centre of the Zentraleinrichtung für Datenverarbeitung (ZEDAT) of the Freie Universität Berlin for computational time and support.References
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med Chem., 2015, 58, 8315 CrossRef CAS PubMed.
- F. Salsi, G. Bulhões Portapilla, S. Simon, M. Roca Jungfer, A. Hagenbach, S. de Albuquerque and U. Abram, Inorg. Chem., 2019, 58, 10129 CrossRef CAS PubMed.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. Luis Aceña, V. A. Soloshonok, K. Izawa and H. Liu, Chem. Rev., 2016, 116, 422 CrossRef CAS PubMed.
- O. Jacobsen, D. O. Kiesewetter and X. Chen, Bioconjugate Chem., 2015, 26, 1–18 CrossRef PubMed.
- R. Halder and T. Ritter, J. Org. Chem., 2021, 86, 13873–13884 CrossRef CAS PubMed.
- U. Abram, Innovative PET and SPECT tracers, in Quantification of Biophysical Parameters in Medical Imaging, ed. I. Sack and T. Schaeffter, Springer International, 2017, pp. 255–279 Search PubMed.
- M. D. Bartholomä, A. S. Louie, J. F. Valliant and J. Zubieta, Chem. Rev., 2010, 110, 2903 CrossRef PubMed.
- C. S. Cutler, H. M. Hennkens, N. Sisay, S. Huclier-Markai and S. S. Jurisson, Chem. Rev., 2013, 113, 858 CrossRef CAS PubMed.
- F. Salsi, M. Roca Jungfer, A. Hagenbach and U. Abram, Eur. J. Inorg. Chem., 2020, 13, 1222 CrossRef.
- G. Claude, J. Genz, D. Weh, M. Roca Jungfer, A. Hagenbach, M. Gembicky, J. S. Figueroa and U. Abram, Inorg. Chem., 2022, 61, 16163 CrossRef CAS PubMed.
- G. Claude, D. Weh, A. Hagenbach, J. S. Figueroa and U. Abram, Z. Anorg. Allg. Chem., 2022, e202200320 Search PubMed.
- M. C. Gil Valenzuela, J. Környei, M. Mikolajzak, K. Ozker, M. R. A. Pillai, M. Venkatesh, E. B. Araujo, M. Dondi, S. C. Gomes, R. Koga, E. Lavie, Z. F. Luo, J. Mustansar, D. V. S. Narasimhan, W. Paragulla, A. Robles and S. Verdera, Technetium-99m Radiopharmaceuticals: Manufacture of Kits, IAEA Technical Reports Series No. 466, International Atomic Energy Agency, Vienna, Austria, 2008, pp. 126–129 Search PubMed.
- L. W. Herman, V. Sharma, J. F. Kronauge, E. Barbaric, L. Herman and D. Piwnica-Worms, J. Med. Chem., 1995, 38, 2955–2963 CrossRef CAS PubMed.
- D. Piwnica-Worms, J. F. Kronauge, B. L. Holman, A. Davison and A. G. Jones, Invest. Radiol., 1989, 24, 25–29 CrossRef CAS PubMed.
- J. F. Kronauge and D. J. Mindiola, Organometallics, 2016, 35, 3432–3435 CrossRef CAS.
- F. E. Hahn, L. Imhof and T. Lügger, Inorg. Chim. Acta, 1998, 269, 347–349 CrossRef CAS.
- J. Bryan, R. E. Stenkamp, T. H. Tulip and J. M. Mayer, Inorg. Chem., 1987, 26, 2283–2288 CrossRef CAS.
- J. S. Figueroa and U. Abram, Z. Anorg. Allg. Chem., 2020, 646, 909–914 CrossRef CAS.
- G. Claude, F. Salsi, A. Hagenbach, M. Gembicky, M. Neville, C. Chan, J. S. Figueroa and U. Abram, Organometallics, 2020, 39, 2287–2294 CrossRef CAS.
- G. Claude, L. Zeh, M. Roca Jungfer, A. Hagenbach, J. S. Figueroa and U. Abram, Molecules, 2022, 27, 8546 CrossRef CAS PubMed.
- U. Abram and Rhenium, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, 2003, vol. 5, pp. 271–403 Search PubMed.
- G. K. Lahiri, S. Goswami, L. R. Falvello and A. Chakravorty, Inorg. Chem., 1987, 26, 3365–3370 CrossRef CAS.
- C. M. Archer, J. R. Dilworth, P. Jobanputra, M. E. Harman, M. B. Hursthouse and A. Karaulov, Polyhedron, 1991, 10, 1539–1543 CrossRef CAS.
- M. Bakir, S. Paulson, P. Goodson and B. P. Sullivan, Inorg. Chem., 1992, 31, 1127–1129 CrossRef CAS.
- Yu.-P. Wang, C.-M. Che, K.-Y. Wong and S.-M. Peng, Inorg. Chem., 1993, 32, 5827–2832 CrossRef CAS.
- M. A. Masood and D. J. Hodgson, Inorg. Chem., 1994, 33, 2488–2490 CrossRef CAS.
- M. A. Masood, B. P. Sullivan and D. J. Hodgson, Inorg. Chem., 1994, 33, 5360–5362 CrossRef CAS.
- H. Luo, I. Setyawati, S. J. Rettig and C. Orvig, Inorg. Chem., 1995, 34, 2287–2299 CrossRef CAS.
- M. Bakir and B. P. Sullivan, J. Chem. Soc., Dalton Trans., 1995, 1733–1738 RSC.
- M. T. Ahmet, B. Coutinho, J. R. Dilworth, J. R. Miller, S. J. Parrott, Y. Zheng, M. Harman, M. B. Hursthouse and A. Malik, J. Chem. Soc., Dalton Trans., 1995, 3041–3048 RSC.
- M. Bakir, J. A. M. McKenzie and B. P. Sullivan, Inorg. Chim. Acta, 1997, 254, 9–17 CrossRef CAS.
- F. Refosco, C. Bolzati, F. Tisato and G. Bandoli, J. Chem. Soc., Dalton Trans., 1998, 923–930 RSC.
- M. A. Masood, B. P. Sullivan and D. J. Hodgson, Inorg. Chem., 1999, 38, 5425–5430 CrossRef CAS.
- A. L. Suing, C. R. Dewan, P. S. White and H. H. Thorp, Inorg. Chem., 2000, 39, 6080–6085 CrossRef CAS PubMed.
- X. Couillens, M. Gressier, R. Turpin, M. Dartiguenave, Y. Coulais and A. L. Beauchamp, J. Chem. Soc., Dalton Trans., 2002, 914–924 RSC.
- M. Porchia, F. Tisato, F. Refosco, C. Bolzati, M. Cavazza-Ceccato, G. Bandoli and A. Dolmella, Inorg. Chem., 2005, 44, 4766–4776 CrossRef CAS PubMed.
- H. Braband, D. Przyrembel and U. Abram, Z. Anorg. Allg. Chem., 2006, 632, 779–785 CrossRef.
- B. Kuhn and U. Abram, Z. Anorg. Allg. Chem., 2008, 634, 2982–2988 CrossRef CAS.
- B. Kuhn and U. Abram, Z. Anorg. Allg. Chem., 2011, 637, 242–245 CAS.
- H. H. Nguyen, C. T. Pham and U. Abram, Polyhedron, 2015, 99, 216–222 CrossRef CAS.
- S. Majumder, J. P. Naskar, A. Bhattacharya, R. Ganguly, P. Saha and S. Chowdhury, J. Coord. Chem., 2015, 68, 599–615 CrossRef CAS.
- P. Saha, J. P. Naskar, A. Bhattacharya, R. Ganguly, B. Saha and S. Chowdhury, J. Coord. Chem., 2016, 69, 303–317 CrossRef CAS.
- M. Roca Jungfer, A. Hagenbach, E. Schulz Lang and U. Abram, Eur. J. Inorg. Chem., 2019, 4974–4984 CrossRef CAS.
- A. P. Borges, B. Possato, A. Hagenbach, A. E. H. Machado, V. M. Deflon, U. Abram and P. I. S. Maia, Inorg. Chim. Acta, 2021, 516, 120110 CrossRef CAS.
- N. H. Huy and U. Abram, Z. Anorg. Allg. Chem., 2008, 634, 1560–1564 CrossRef CAS.
- X. Schoultz, T. I. A. Gerber, E. Hosten, R. Betz, L. Rhyman and P. Ramasami, Polyhedron, 2015, 96, 6–15 CrossRef CAS.
- A. Barandov and U. Abram, Polyhedron, 2009, 28, 1155–1159 CrossRef CAS.
- L. Wei, J. Zubieta and J. W. Babich, Inorg. Chem., 2004, 43, 6445–6454 CrossRef CAS PubMed.
- T. I. A. Gerber, D. Luzipo and P. Mayer, Inorg. Chim. Acta, 2004, 357, 429–435 CrossRef CAS.
- J. B. Arterburn, K. V. Rao, D. M. Goreham, M. V. Valenzuela, M. S. Holguin, K. A. Hall, K. C. Ott and J. C. Bryan, Organometallics, 2000, 19, 1789–1795 CrossRef CAS.
- C. Scholtysik, M. Roca Jungfer, A. Hagenbach and U. Abram, Z. Anorg. Allg. Chem., 2018, 644, 1451 CrossRef CAS.
- C. Scholtysik, C. Njiki Noufele, A. Hagenbach and U. Abram, Inorg. Chem., 2019, 58, 5241 CrossRef CAS PubMed.
- G. Bandoli, T. I. A. Gerber, J. Perilis and J. G. H. du Preetz, Inorg. Chim. Acta, 1998, 278, 96–100 CrossRef CAS.
- M. T. Ahmet, B. Coutinho, J. R. Dilworth, J. R. Miller, S. J. Parrott and Y. Zheng, Polyhedron, 1996, 15, 2041–2050 CrossRef CAS.
- T. D. Lohrey, E. A. Cortes, J. I. Fostvedt, A. L. Oanta, A. Jain, R. G. Bergman and J. Arnold, Inorg. Chem., 2020, 59, 11096–11107 CrossRef CAS PubMed.
- P. Patil, M. Ahmadian-Moghaddam and A. Dömling, Green Chem., 2020, 22, 6902–6911 RSC.
- L. A. Labios, M. D. Millard, A. L. Rheingold and J. S. Figueroa, J. Am. Chem. Soc., 2009, 131, 11318–11319 CrossRef CAS PubMed.
- T. B. Ditri, B. J. Fox, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Inorg. Chem., 2009, 48, 8362–8375 CrossRef CAS PubMed.
- B. J. Fox, M. D. Millard, A. G. DiPasquale, A. L. Rheingold and J. S. Figueroa, Angew. Chem., Int. Ed., 2009, 48, 3473–3477 CrossRef CAS PubMed.
- B. J. Fox, Q. Y. Sun, A. G. DiPasquale, A. R. Fox, A. L. Rheingold and J. S. Figueroa, Inorg. Chem., 2008, 47, 9010–9020 CrossRef CAS PubMed.
- G. W. Margulieux, N. Weidemann, D. C. Lacy, C. E. Moore, A. L. Rheingold and J. S. Figueroa, J. Am. Chem. Soc., 2010, 132, 5033–5035 CrossRef CAS PubMed.
- M. A. Stewart, C. E. Moore, T. B. Ditri, L. A. Labios, A. L. Rheingold and J. S. Figueroa, Chem. Commun., 2011, 47, 406–408 RSC.
- D. W. Agnew, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Organometallics, 2017, 36, 363–371 CrossRef CAS.
- C. C. Mokhtarzadeh, C. E. Moore, A. L. Rheingold and J. S. Figueroa, J. Am. Chem. Soc., 2018, 140, 8100–8104 CrossRef CAS PubMed.
- D. W. Agnew, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Angew. Chem., Int. Ed., 2015, 54, 12673–12677 CrossRef CAS PubMed.
- A. E. Carpenter, C. C. Mokhtarzadeh, D. S. Ripatti, I. Havrylyuk, R. Kamezawa, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Inorg. Chem., 2015, 54, 2936 CrossRef CAS PubMed.
- D. W. Agnew, M. D. Sampson, C. E. Moore, A. L. Rheingold, C. P. Kubiak and J. S. Figueroa, Inorg. Chem., 2016, 55, 12400–12408 CrossRef CAS PubMed.
- D. W. Agnew, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Dalton Trans., 2017, 46, 6700–6707 RSC.
- M. J. Drance, J. D. Sears, A. M. Mrse, C. E. Moore, A. L. Rheingold, M. L. Neidig and J. S. Figueroa, Science, 2019, 363, 1203–1205 CrossRef CAS PubMed.
- F. Salsi, M. Neville, M. Drance, A. Hagenbach, C. Chan, J. S. Figueroa and U. Abram, Chem. Commun., 2020, 56, 7009–7013 RSC.
- F. Salsi, M. Neville, M. Drance, A. Hagenbach, J. S. Figueroa and U. Abram, Organometallics, 2021, 40, 1336–1343 CrossRef CAS.
- N. Weidemann, G. W. Margulieux, C. E. Moore, A. L. Rheingold and J. S. Figueroa, Inorg. Chim. Acta, 2010, 364, 238–245 CrossRef CAS.
- T. B. Ditri, A. E. Carpenter, D. S. Ripatti, E. Moore, A. L. Rheingold and J. S. Figueroa, Inorg. Chem., 2013, 52, 132216–113229 CrossRef PubMed.
- F. Salsi, S. Wang, C. Teutloff, M. Busse, M. L. Neville, A. Hagenbach, R. Bittl, J. S. Figueroa and U. Abram, Angew. Chem., Int. Ed., 2023 DOI:10.1002/anie.202300254.
- G. M. Sheldrick, SADABS, University of Göttingen, Germany, 1996 Search PubMed.
- P. Coppens., The Evaluation of Absorption and Extinction in Single-Crystal Structure Analysis. Crystallographic Computing, Copenhagen, Muksgaard, 1979 Search PubMed.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., 2015, 71, 3 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- H. Putz and K. Brandenburg, DIAMOND, Crystal and Molecular Structure Visualization Crystal Impact, version 4.6.5, GbR, Bonn, Germany, 2021 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2236802–2236812. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00446e |
| This journal is © The Royal Society of Chemistry 2023 |