
Selective conversion of methane to ethane and hydrogen over In/molecular-sieve-3A catalyst†
Ayumi
Nakaya
a,
Ayako
Suzuki
a,
Shoji
Iguchi
 ab and
Ichiro
Yamanaka
*a
ab and
Ichiro
Yamanaka
*a
aDepartment of Chemical Science and Engineering, Tokyo Institute of Technology, Ookayama, Meguro, Tokyo 152-8552, Japan. E-mail: yamanaka.i.aa@m.titech.ac.jp
bDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Kyoto, 615-8510, Japan
First published on 17th October 2023
Abstract
Direct conversion of methane to valuable chemicals has gained much attention. Besides, silica-supported liquid–metal indium catalysts (In/SiO2) for the direct dehydrogenative conversion of methane (DCM) to higher hydrocarbons with 4% yield and 70% selectivity at 1173 K have been reported. In this work, In catalyst was found to be an excellent catalyst for the DCM reaction and selective formation of ethane and hydrogen without carbon deposition at the lower temperature of 873 K. The catalytic activity of In was strongly depended on supports at 873 K, and the molecular-sieve-3A (MS3A)-support calcined at 1123 K was more effective than the SiO2 support. Selective conversion of methane to ethane and hydrogen proceeded on the In/MS3A catalyst. The role of the MS3A support in the DCM reaction is discussed.
1. Introduction
The conversion of CH4 as a major component of natural gas into useful chemicals is one of the hot topics to curb the depletion of oil. Currently, CH4 is used as a source of thermal energy and electric power. Except for methanol synthesis and the Fischer–Tropsch process, the catalytic technology for the direct conversion of CH4 into useful chemicals does not exist. Therefore, studies on new catalysts and reactions are essential for developing the direct conversion process of CH4.Several chemists have been studying catalysts and reactions for the conversion of CH4; however, strong C–H bonds and a symmetric molecular form of CH4 are the major problems for the activation of CH4.1 Two scientific fields involving pioneering works are in existence, one is the oxidative coupling of CH4 (OCM) to C2H6 and C2H4 with O2 (ref. 2–4) and the other is a dehydrogenative conversion of CH4 (DCM) or none-oxidative coupling of CH4 (NOCM) to higher hydrocarbons.5–8 In the latter field, most of the studies have focused on catalysis involving transition metal elements (Mo, Fe, Ni, etc.) because of their strong catalysis of the activation of C–H bonds. In our study, we focused on the post-transition metal elements (Ga, In, Bi, Sn) because of their mild catalysis of the activation of C–H bonds and found that the SiO2-supported indium catalyst (In/SiO2) showed higher catalytic activity for the DCM reaction.7
The In/SiO2 catalyst showed a good result with 70% combined selectivity to hydrocarbons (ethylene, ethane, propylene, benzene, and toluene) with 4% conversion of CH4 at 1173 K. The particular nature of the In/SiO2 catalyst is that the metallic liquid of In shows catalysis for the activation of CH4. This unique property of the In metal liquid for the DCM reaction has been studied by kinetic studies, isotope effects studies, first-principle DFT MD calculations,9 and XAFS observations.10 The major conclusions from our previous studies are as follows; (i) In metal liquid is thermally activated, (ii) the activated In species, In–In cluster models, can cleave a C–H bond of CH4 to CH3–In and H–In species, then, couplings of the two CH3–In species produce C2H6 and of the two H–In species produce H2, and (iii) the onset temperature of the activation of CH4 by In liquid metal was observed approximately at 850 K using temperature programed reaction (TPR) studies and mass spectra analysis, but a quantitative analysis could not be performed.11
In this study, we found that the In catalyst showed significant catalytic activity in the DCM reaction at a lower temperature of 873 K and the support materials strongly affected the In catalysis; especially, molecular sieve 3A (MS3A) was effective. This work aimed to clarify the effects of the MS3A support on In catalysis approximately at 873 K and to propose functions of the MS3A support to enhance the catalytic activity of In in the DCM reaction.
2. Experimental
In/SiO2 catalyst was prepared by a conventional impregnation method.7,11 Several SiO2 supports (CARiACT Q-3, CARiACT Q-30, Admafine SO-E6, AEROSIL 300) were calcined in air at 1173 K before the catalyst preparations (ESI†). The SiO2 support was added to In(NO3)·nH2O solution, and this mixture was strongly stirred using a magnetic spin-bar. The suspension was dried at 353 K under stirring. The catalyst-precursor powder was calcined in air for 3 h at 773 K. After the calcined powder was reduced in a stream of H2 at 1173 K for 3 h, In/SiO2 catalysts were obtained. A loading of In0 was 10 wt%.The In/MS3A catalyst was prepared following a similar method to the In/SiO2 catalyst. Different procedures were used for the calcination of MS3A (Wako Pure Chemical Industries) at 1023 K and reduction of InOx/MS3A with H2 at 1023 K. A loading of In0 was the same as 10 wt%.
We prepared different types of In/MS3A catalysts as follows; the catalyst precursor of InOx/MS3A was held in saturated water vapor at 45 °C. This humidified InOx/MS3A material was reduced with pure H2 at 1173 K under the same reduction procedure of In/SiO2 and In/MS3A catalysts. The prepared catalyst was called In/MS3A(humid). In a similar way, In/MS4A(humid), In/LTA(humid), and In/SiO2(humid) catalysts were prepared.
A fixed-bed quartz reactor (I.D. 12 mm) was used for the DCM reaction with 100 mg of the catalyst (Fig. S1†). After Ar was flown through the reactor to replace air, H2 was flown at 20 mL min−1 and the temperature was raised to 873 K at 25 K min−1 and holding for 60 min. The reduction temperature was raised to 1023 K and lowered to the reaction temperature. After H2 was replaced with Ar for 60 min, CH4 was flown at 10 mL min−1and the DCM reaction was initiated.7,8,11
Products were analyzed using a gas chromatograph (GC). CH4, C2H6, and C2H4 were analyzed using a GC with a frame ionization detector (GC-8A-FID, Shimadzu) equipped with an Unibeads 1S column (3φ, 2 m). H2 was analyzed using a GC with a thermal conductivity detector (GC-8A-TCD, Shimadzu) equipped with an activated carbon column (3φ, 2 m). Conversion of CH4, selectivity to C2H6, and selectivity to C2H4 in the DCM reaction were calculated from eqn (1)–(3), respectively. These calculations were based on the hydrogen balance of the output gas mixture.7,8,11
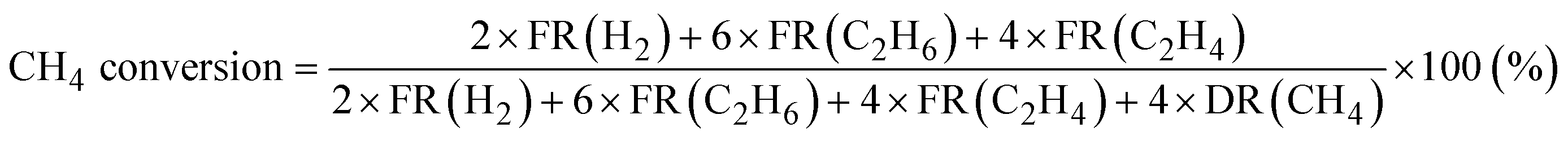 | (1) |
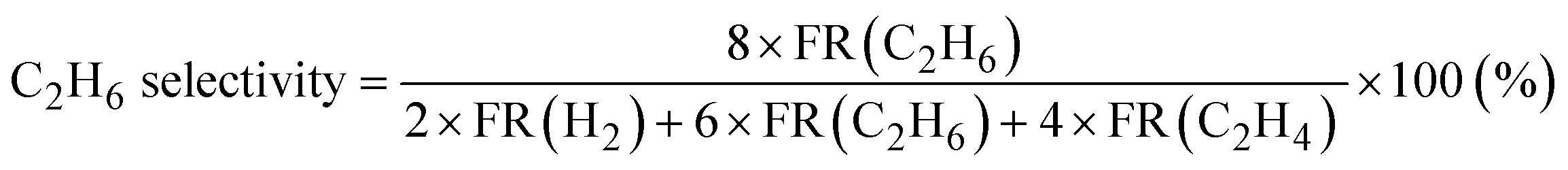 | (2) |
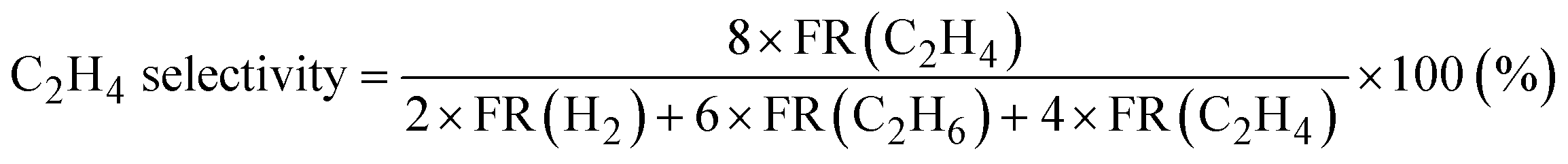 | (3) |
Brief characterization studies for the catalysts were conducted by XRD analysis (MiniFlex-600/TISS, Rigaku Co.) and gas-adsorption measurement (BELSORP-mini II, MicrotracBEL Co.).
3. Results and discussion
3.1 Catalysis of In(l) at a lower temperature
As mentioned in the introduction section, the previous results of the temperature-programed-reaction study for CH4 conversion on the In/SiO2 catalyst obtained using a mass spectrometer suggested that the activation of C–H bonds of CH4 would proceed at a lower temperature approximately at 850 K than at 1173 K, which was a typical condition for the DCM reaction producing higher hydrocarbons.7,11 Therefore, the DCM reaction was studied under steady reaction conditions at 873 K to clarify the formation of products at 873 K by the GC analysis.Fig. 1 shows the time course for the formation of C2H6 and H2 over the In/SiO2 catalyst at 873 K. At a blank test, without the In/SiO2 catalyst, no formation of C2H6 and H2 was not observed at 873 K. The formation rate of C2H6 gradually decreased with process time until 120 min and the steady formation was observed after 150 min. The formation rate of H2 gradually decreased similar to that of C2H6. The formation rate of C2H6 was lower than that of H2 for 120 min. In contrast, both formation rates were almost equal from 150 min to 240 min. The change in the formation rates impacted the selectivity to C2H6 as shown in Fig. 1. In the first half, until 120 min, an apparent selectivity to C2H6 was about 95% and the selectivity was almost 100% in the second half after 180 min. The stoichiometric conversion of CH4 to C2H6 and H2 continuously proceeded after 180 min (eqn (4)). The selectivity below 100% means that there were un-identified hydrocarbons, or carbon deposition forms (eqn (5)). The average results during 180 min are indicated in run 1 of Table 1.
| 2CH4 → C2H6 + H2 ΔH0 = +66 kJ mol−1 | (4) |
| CH4 → C(s) + 2H2 ΔH0 = +75 kJ mol−1 | (5) |
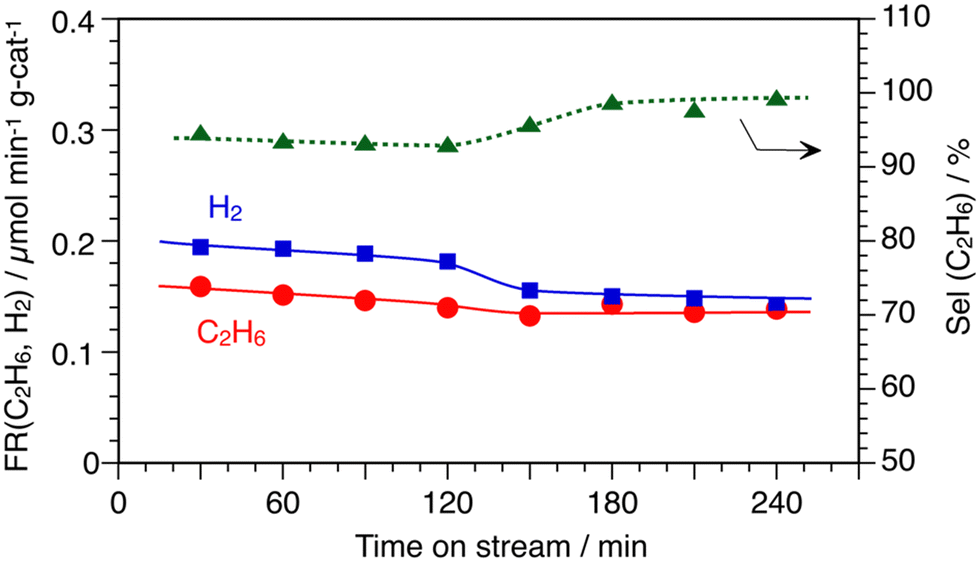 | ||
| Fig. 1 Time courses of DCM reaction by In/SiO2 catalyst at 873 K. CH4: 1 atm and 10 mL min−1, catalyst: 10 wt% In/SiO2. | ||
| Run | Catalyst | Form. rate (μmol min−1 gcat−1) | Select. (%) | |
|---|---|---|---|---|
| C2H6 | H2 | C2H6 | ||
| CH4: 1 atm and 10 mL min−1, catalysts: 10 wt% In loading and 100 mg in the reactor. | ||||
| 1 | In/SiO2 | 0.15 | 0.18 | 96 |
| 2 | In2O3/SiO2 | 0.05 | 1.82 | 10 |
| 3 | In/Al2O3 | 0.03 | 1.80 | 6 |
| 4 | In/TiO2 | 0.16 | 0.87 | 47 |
| 5 | In/Nb2O5 | 0.05 | 0.47 | 32 |
| 6 | In/ZrO2 | 0.08 | 3.08 | 10 |
| 7 | In/CeO2 | 0.23 | 2.41 | 30 |
| 8 | In/MgO | 0.19 | 25.4 | 3 |
When In2O3/SiO2 material, as a precursor of the In/SiO2 catalyst before the reduction, was used in the DCM reaction, a lower formation rate of C2H6 and a higher formation rate of H2 were observed at run 2, as shown in Table 1. This clearly indicated that In0(l) was the active phase for the selective C2H6 formation at a lower temperature of 873 K and SiO2 (CARiACT Q-3) is a good support for In0(l) catalyst.10 To increase the catalytic activity of In0(l) catalyst, other oxide supports were tested for the DCM reaction, runs 3–8 in Table 1. All the oxide supports were calcined in air at 1173 K before the preparation of In/oxide support catalysts. The formation rates of C2H6 and H2 were strongly affected by oxide supports. In the case of CeO2 and MgO supports, the formation rates of C2H6 were enhanced but that of H2 was strongly accelerated. On other supports, such as Al2O3, TiO2, Nb2O5, and ZrO2, the formation rates of C2H6 were suppressed and the decomposition rates were accelerated. Excess formation of H2 was due to the decomposition of CH4 to carbon deposits and H2 formation. Except for the SiO2 support, oxide supports were not suitable to achieve the selective conversion of CH4 to C2H6 and H2 by the In0(l) catalyst.
In0/SiO2 (CARiACT Q-3) catalyst showed excellent catalysis for the DCM reaction with over 96% selectivity to C2H6 and H2 at 873 K. In order to clarify the nature of SiO2 support for the DCM reaction, three SiO2 materials, CARiACT Q-30, Admafine SO-E6, and AEROSIL 300, were tested as the support. All SiO2 materials were calcined in air at 1173 K before In(NO3)3 impregnation, as mentioned in the experimental section. Fig. 2 shows the relationship between the formation rates of C2H6 and specific surface areas of SiO2 supports calcined at 1173 K. As you can see clearly in Fig. 2, a SiO2 support having a lower surface area was suitable for the selective formation of C2H6 by In0(l) catalyst. In contrast, decomposition of CH4 to H2 and carbon deposition proceeded on a SiO2 support having a higher surface area. Of course, we confirmed that no decomposition of CH4 occurred on the SiO2 materials without the In0(l) catalyst at 873 K.
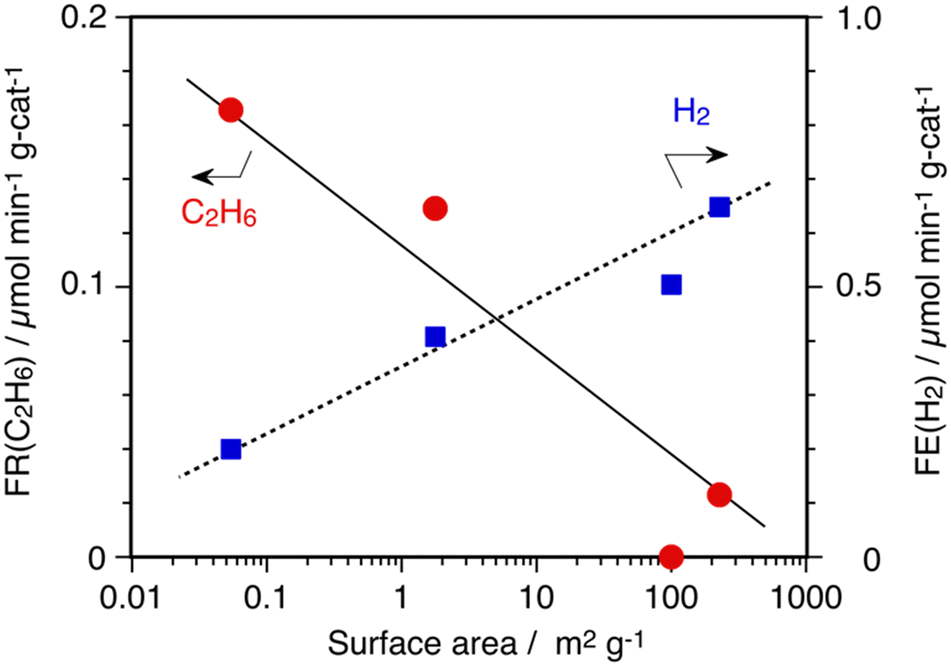 | ||
| Fig. 2 Influence of surface area of SiO2 support on DCM reaction by 10 wt% In/SiO2 catalysts at 873 K. Amount of catalyst: 100 mg, CH4: 1 atm and 10 mL min−1. | ||
We have already reported the conversion mechanism of CH4 to C2H6 and higher hydrocarbons catalyzed by the In/SiO2 catalysts at a higher temperature of 1173 K, as mentioned in the introduction.9,11 The brief reaction mechanism is as follows; (i) In liquid metal catalyzes the activation of the C–H bond of CH4, (ii) CH3–In(l) and H–In(l) species are produced, then, (iii) two CH3–In(l) species on the liquid surface were coupled and produced C2H6, (iv) H species on In diffused into a bulk phase of In(l), (v) two H species were dissolved in the bulk In(l) were coupled at the surface and desorbed as H2.
When a SiO2 support with a lower surface area was used, the same reaction mechanism could be applied on the In0(l) catalyst at 873 K, and C2H6 and H2 were selectively produced. Whereas, we propose a side reaction path over the SiO2 support with higher surface area in that CH3 species on In(l) migrate to the surface of the SiO2 support before the coupling reaction and decompose to carbon and hydrogen by surface catalysis of SiO2. If this mechanism was true then an inert surface of supports should be suitable to promote selective coupling of CH3 species on the surface of In(l).
3.2 Acceleration of In(l) catalysis by molecular sieve 3A
As mentioned above, the In0/SiO2 (CARiACT Q-3) catalyst showed high selectivity to the conversion of CH4 to C2H6 and H2; however, the conversion of CH4, as shown in Fig. 1 was only 0.004%, which was calculated from the product yields by GC (eqn (1)). The thermodynamic equilibrium conversion of this reaction was about 0.9% at 873 K. We have considered that the formation rate of C2H6 through the coupling of CH3 species was suppressed by the equilibrium between CH3 species and H species on the surface of In0(l).11 In other words, if we could accelerate the elimination of H species from the surface of In0(l), the recombination reaction between CH3 and H species decelerated and the coupling reaction of CH3 species accelerated.How to accelerate the elimination of H species on In0(l) is an essential topic and we came up with an idea for the acceleration. Our idea is the utilization of the function of the molecular sieve for the elimination of H species, that is to separate CH3 and H species using a molecular sieve. A molecular sieve-3A (MS3A) has micropores of about 0.3 nm in which H species can be introduced but not the CH4 and CH3 species. If this hypothesis is true, H species on and in In0(l) will spill over and diffuse into the MS3A and coupling reaction of the CH3 species to accelerate the formation of C2H6.
In/MS3A catalyst was prepared by the impregnation method similar to that of the In/SiO2 catalyst. The MS3A support was calcined in air at 1023 K before the preparation of the catalyst, as described in the Experimental section. The catalytic activity of In/MS3A at 873 K is indicated in run 9 shown in Table 2. Comparing runs 1 and 9, the formation rate of C2H6 on the In/MS3A catalyst was significantly accelerated, and it was 5 times higher than that on the In/SiO2 catalyst. In addition, C2H4 formed on the In/MS3A catalyst at 873 K though the selectivity was only 2%. The total selectivity of C2H6 and C2H4 was 98% and a stoichiometric conversion of CH4 to C2H6, C2H4, and H2 was performed.
| Run | Catalyst | Format. rate (μmol min−1 gcat−1) | Select. (%) | ||
|---|---|---|---|---|---|
| C2H6 | C2H4 | H2 | C2H6 + C2H4 | ||
| CH4: 1 atm and 10 mL min−1, catalysts: 100 mg in the reactor. | |||||
| 1 | 10 wt% In/SiO2 | 0.15 | 0.00 | 0.18 | 96 |
| 9 | 10 wt% In/MS3A | 0.89 | 0.05 | 0.52 | 98 |
| 10 | 10 wt% In/MS3A(humid) | 1.25 | 0.08 | 1.16 | 104 |
| 11 | 10 wt% In/SiO2(humid) | 0.28 | 0.01 | 0.42 | 90 |
| 12 | 10 wt% In/MS4A(humid) | 0.30 | 0.01 | 0.65 | 79 |
| 13 | 10 wt% In/H-LTA(humid) | 0.41 | 0.02 | 0.95 | 78 |
| 14 | 5 wt% In/MS3A(humid) | 0.15 | 0.00 | 1.07 | 41 |
| 15 | 20 wt% In/MS3A(humid) | 1.74 | 0.12 | 1.27 | 109 |
| 16 | 30 wt% In/MS3A(humid) | 0.96 | 0.05 | 0.95 | 102 |
To improve the catalytic activity of the In/MS3A catalyst, we studied influences of the preparation conditions of the catalyst, calcination temperature and time, reduction temperature and time, partial pressure of H2 and reduction temperature, etc. We have found that humidity around the catalyst precursor, InOx/MS3A, influenced the catalytic activity of the In/MS3A catalyst and this was named In/MS3A(humid). Preparation procedures are mentioned in the Experimental section. It was observed that for run 10, the formation rate of C2H6 was accelerated 1.4 times by the humidifying treatment and the total selectivity of C2H6 and C2H4 was 104%, as calculated from eqn (2). The selectivity of over 100% indicated less formation of H2 in comparison to the hydrocarbon formation. As a reference, the In/SiO2(humid) catalyst was applied in the DCM reaction. As indicated, at run 11, a little acceleration of C2H6 formation was observed by the humidification treatment of InOx/SiO2.
To reveal the unique function of the MS3A support, MS4A and LTA materials were also applied for the supports.12,13 In/MS4A(humid) and In/LTA(humid) catalysts were prepared using the above procedures and the results are shown as runs 12 and 13 in Table 2, respectively. Accelerations for the C2H6 formation rate by MS4A and LTA supports were clear from the data of the runs 10–13. In addition, the selectivity to C2H6 decreased using MS4A and LTA supports, and carbon deposition should proceed.
Effects of In loadings from 5 to 30 wt% over the MS3A support on the DCM reaction were studied, as shown in runs 10, 14–16. The 5 wt% In/MS3A(humid) catalyst showed a lower formation rate of C2H6 and a good rate for H2, corresponding to a lower selectivity to C2H6. 20 wt% In/MS3A(humid) catalyst showed the highest catalytic activity and the highest total selectivity for C2H6 and C2H4 over 100%.
Time courses for the total formation of C2H6 and C2H4 on the In/MS3A(humid), In/MS4A(humid), and In/SiO2(humid) catalysts are shown in Fig. 3. The formation rate of C2H6 and C2H4 over the In/MS3A(humid) catalyst was far higher than that of the In/SiO2(humid) catalyst at the initial stage of the reaction and decreased with the process time. However, significant acceleration of the C2H6 formation on the In/MS3A(humid) catalyst was observed at 180 min. The selectivity to C2H4 was constant during the reaction; 7% on In/MS3A(humid), and 3% on In/MS4A(humid) and In/SiO2(humid) catalysts.
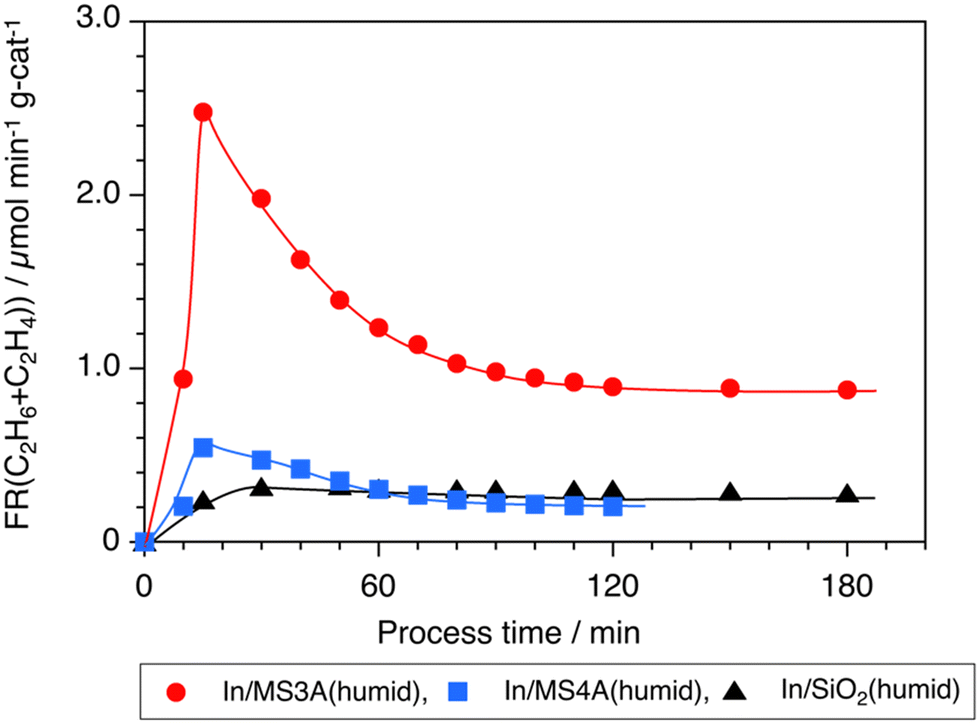 | ||
| Fig. 3 Time courses of the formation rate of the sum of C2H6 and C2H4 by 10 wt% In/MS3A(humid), 10 wt% In/MS4A(humid), and 10 wt% In/SiO2(humid) catalysts. Amount of catalyst 100 mg, CH4: 1 atm and 10 mL min−1. | ||
Fig. 4 shows the effects of the reaction temperatures on (a) the formation rate of the total C2H6 and C2H4 and (b) their selectivity on In/MS3A(humid), In/MS4A(humid), and In/SiO2(humid) catalysts. The In/MS3A(humid) catalyst showed significant catalytic activity and a higher selectivity of 90% to C2H6 at a lower temperature of 773 K, at which the In/MS4A(humid) and In/SiO2(humid) catalysts did not work. In addition, the In/MS3A(humid) catalyst showed catalytic activity even at 723 K through a lower selectivity. An advantage of In/MS3A(humid) catalyst against In/MS4A(humid) and In/SiO2(humid) catalysts for the activation and conversion of CH4 was clear. Apparent activation energies over In/MS3A(humid), In/MS4A(humid), and In/SiO2(humid) catalysts were calculated from the Arrhenius plots and were 16.2, 15.4, and 20.6 kJ mol−1, respectively (Fig. S2†). These apparent activation energies were too low to compare the cleavage energy of 463 kJ mol−1 in the C–H bond of CH4. When the rate-determining step was the C–H bond activation in the DCM reaction, higher activation energies of 170 kJ mol−1 for the In/SiO2 catalyst11 and 250 kJ mol−1 for the Ni2P/SiO2 catalyst8 were reported at higher reaction temperatures over 1000 K. In other words, the diffusion process should affect the apparent lower activation energies over the In/MS3A(humid), In/MS4A(humid), and In/SiO2(humid) catalysts below 873 K.
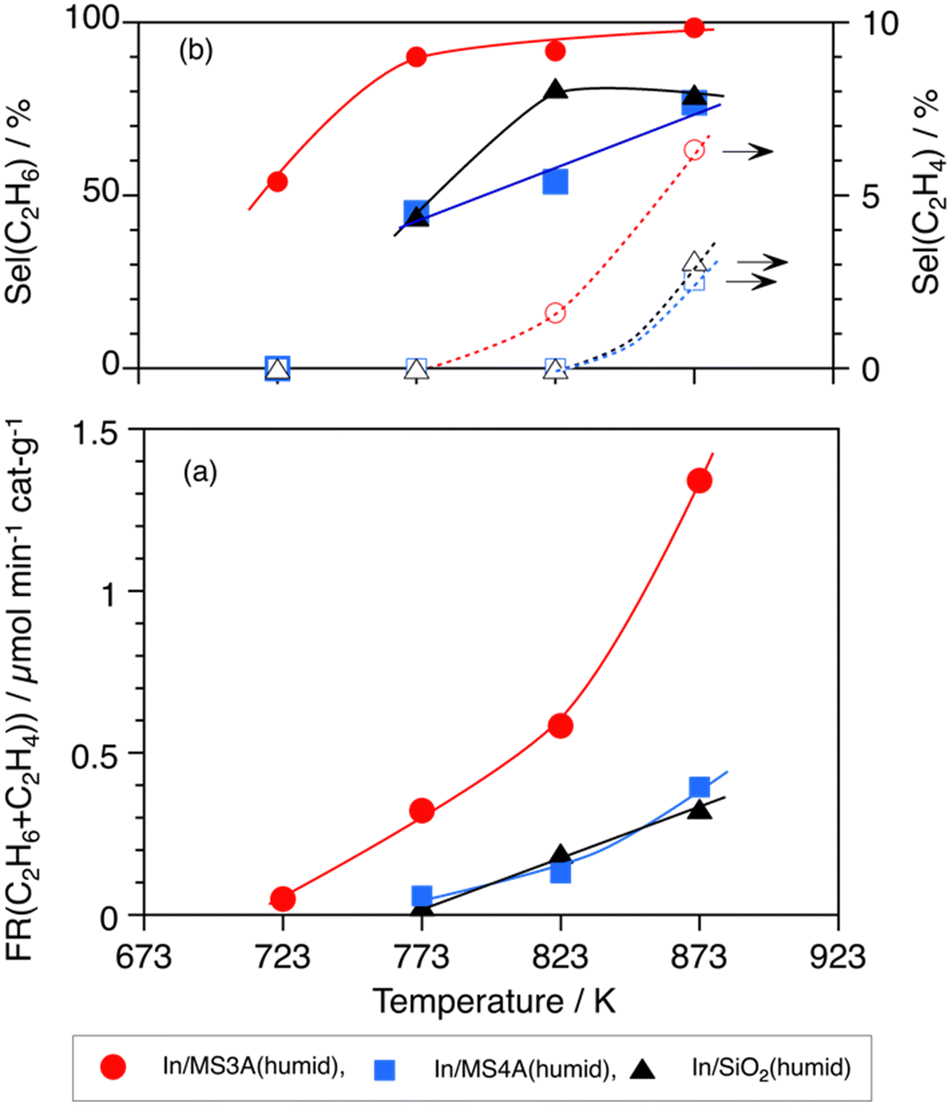 | ||
| Fig. 4 Effects of reaction temperatures on the DCM reaction by 10 wt% In/MS3A(humid), 10 wt% In/MS4A(humid), and 10 wt% In/SiO2(humid) catalysts. (a) Formation rate of the sum of C2H6 and C2H4; and (b) selectivity to C2H6 and C2H4. Amount of catalyst: 100 mg, CH4: 1 atm and 10 mL min−1. | ||
As mentioned above, the In/MS3A(humid) catalyst showed significant catalytic activity for the DCM reaction, a high formation rate, and a high selectivity of over 100%. The selectivity higher than 100% means a lower formation rate of H2 compared with the total formation of C2H6 and C2H4. The selectivity lower than 100% means carbon deposition or undetected product formation. In the former case, the lower formation of H2 is very strange because the stable form of hydrogen is only H2. If counter materials (molecules) were coupled with hydrogen species, H˙, H+, and H− species were stabilized. For example, H˙ (metal surface), H+ (anion species), and H− (cation species) forms are stable. We can accurately quantify H2 by the GC analysis, but H˙, H+, and H− species on the support cannot be quantified. In the latter case, the apparent selectivity to the C2H6 and C2H4 total was over 100% because hydrogen species were held on the catalyst.
4. Discussion
Basic characterization studies for In/MS3A(humid), In/MS4A(humid), and In/SiO2(humid) catalysts were conducted using XRD and N2 adsorption measurements. Fig. 5 shows the XRD patterns of the three catalysts. Diffraction patterns of In0 were clearly observed for the three catalysts. Original diffraction patterns indicated that MS-3A and MS-4A were not observed for the In/MS3A(humid) and In/MS4A(humid) catalysts. Diffraction patterns of MS3A and MS4A calcined at 1123 K were measured and particular diffraction patterns of MS3A and MS4A disappeared. Outer surface areas of MS3A and MS4A measured by BET method using N2 decreased from 18.5 to 0.4 m2 g−1 and from 9.8 to 1.2 m2 g−1, respectively, by calcination at 1023 K. These observations suggest that the original crystal structures of MS3A and MS4A were decomposed by calcination; however, a part of the original pores of MS3A and MS4A would remain after the calcination. Therefore, we can observe differences in the catalytic activities between the In/MS3A(humid), In/MS4A(humid), and In/SiO2(humid) catalysts for the DCM reaction.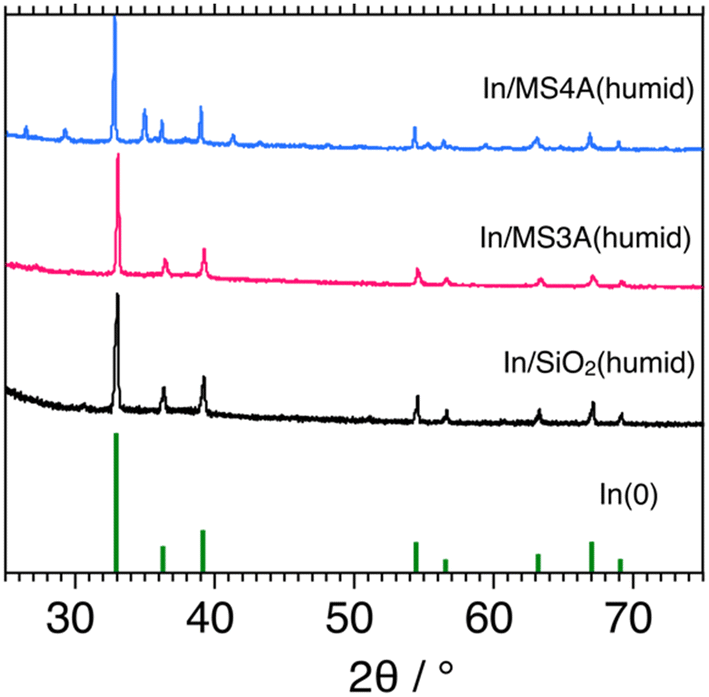 | ||
| Fig. 5 XRD patterns of 10 wt% In/MS3A(humid), 10 wt% In/MS4A(humid), and 10 wt% In/SiO2(humid) flesh catalysts. | ||
Effective pore diameters of the original MS3A, MS4A, and LTA materials were 0.3, 0.4, and 0.5 nm, respectively. As mentioned above, we raised the idea of separating CH3 and H species by the function of a molecular sieve. The micropores of MS3A were 0.3 nm in size, in which H species (<0.3 nm) can be introduced but CH4 (3.8 nm) and maybe CH3 species cannot be introduced. If this model is true, the recombination of CH3 species and H species is suppressed, and the coupling of two CH3 species is accelerated producing C2H6. On the other hand, both H and CH3 species can diffuse into a wide pore of MS4A and the recombination between H and CH3 species proceeds reproducing CH4. This situation is similar to that of the SiO2 support. Therefore, catalytic activities on the In/MS4(humid) and In/SiO2(humid) catalysts were very similar, as shown in Fig. 3 and Table 2. We consider that the separation of H and CH3 species on the In(l) surface by the micropores of MS3A (0.3 nm) is essential for C2H6 formation, as described above. We speculate that H species accumulate in the micropores and acceleration of H species removal in the micropores is essential to enhance the formation rate of C2H6. We have a plan to prove the accumulation of hydrogen species in micropores of the In/MS3A(humid) catalyst by applying TPD experiments in future work.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JST-CREST projects (Grant Number JPMJCR15P4).References
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- G. E. Keller and M. M. Bhasin, J. Catal., 1982, 73, 9–19 CrossRef CAS.
- T. Ito and J. H. Lunsford, Nature, 1985, 314, 721–722 CrossRef CAS.
- K. Otsuka, K. Jinno and A. Morikawa, Chem. Lett., 1985, 499–500 CrossRef CAS.
- L. Wang, L. Tao, M. Xie, G. Xu, J. Huang and Y. Xu, Catal. Lett., 1993, 21, 35–41 CrossRef CAS.
- X. Guo, G. Fang, G. Li, H. Ma, H. Fan, L. Yu, C. Ma, X. Wu, D. Deng and M. Wei, Science, 2014, 344, 616–619 CrossRef CAS PubMed.
- Y. Nishikawa, H. Ogihara and I. Yamanaka, ChemistrySelect, 2017, 2, 4572–4576 CrossRef CAS.
- A. L. Dipu, S. Ohbuchi, Y. Nishikawa, S. Iguchi, H. Ogihara and I. Yamanaka, ACS Catal., 2020, 10, 375–379 CrossRef CAS.
- Y. Ohtsuka, Y. Nishikawa, H. Ogihara, I. Yamanaka, A. Nakayama and J. Hasegawa, J. Phys. Chem. A, 2019, 123, 8907–8912 CrossRef CAS PubMed.
- U. Kashaboina, Y. Nishikawa, Y. Wakisaka, N. Sirisit, S. Nagamatsu, D. Bao, H. A. Miwa, S. Takakusagi, Y. Inami, F. Kuriyama, A. L. Dipu, H. Ogihara, S. Iguchi, I. Yamanaka, T. Wada and K. Asakura, Chem. Lett., 2019, 48, 1145–1147 CrossRef CAS.
- Y. Nishikawa, Y. Ohtsuka, H. Ogihara, R. Rattanawan, M. Gao, A. Nakayama, J. Hasegawa and I. Yamanaka, ACS Omega, 2020, 5, 28158–28167 CrossRef CAS PubMed.
- W. Fan, S. Shirato, F. Gao, M. Ogura and T. Okubo, Microporous Mesoporous Mater., 2006, 89, 227–234 CrossRef CAS.
- R. Abid, G. Delahay and H. Tounsi, J. Mater. Cycles Waste Manage., 2019, 21, 1188–1196 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cy01217d |
| This journal is © The Royal Society of Chemistry 2023 |