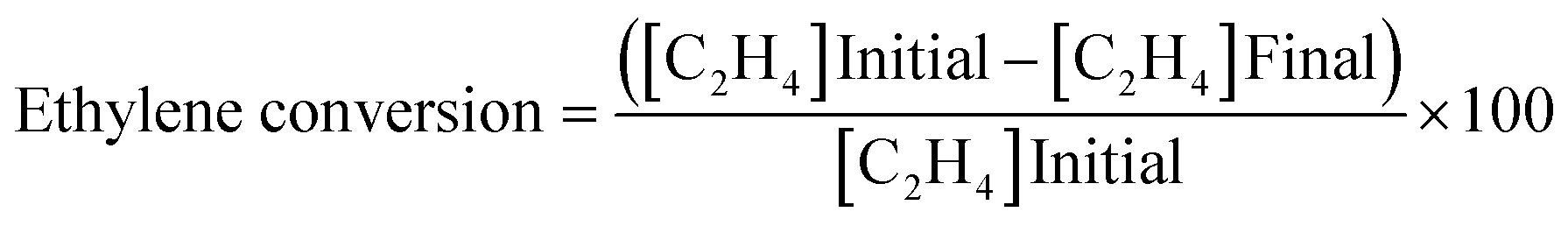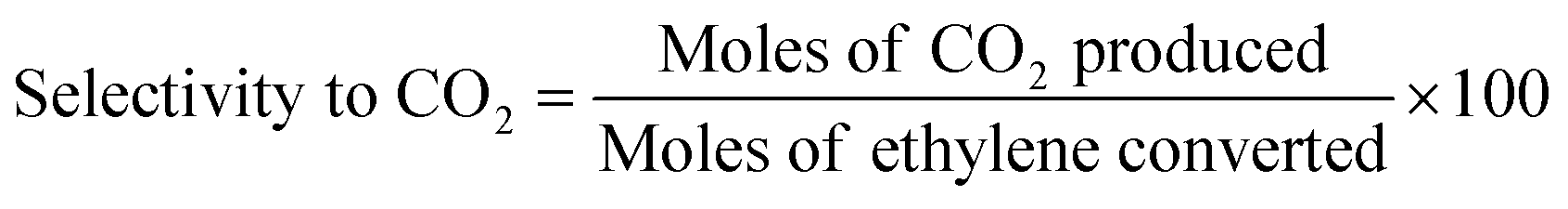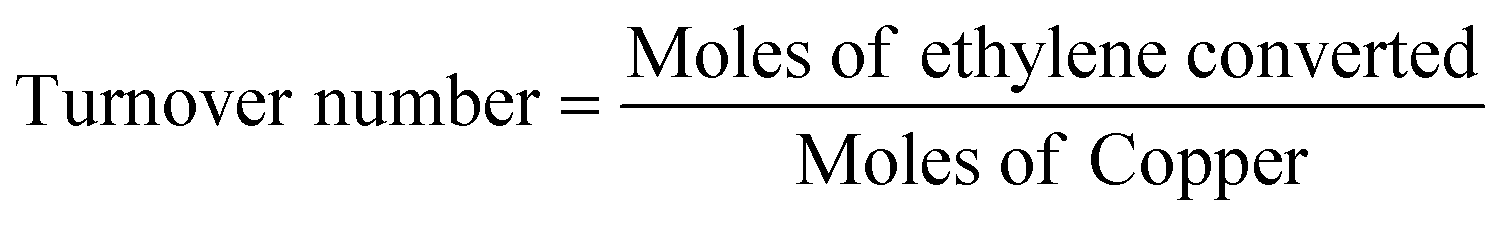Oxidation of ethylene by Cu/TiO2: reducibility of Cu2+ in TiO2 as a possible descriptor of catalytic efficiency†
Received
27th December 2022
, Accepted 9th March 2023
First published on 30th March 2023
Abstract
Catalytic oxidation using non-noble metal-based catalysts is a promising approach to mitigate pollution due to VOCs in the air. In this work, mesoporous Cu/TiO2 catalysts containing different concentrations of Cu2+ (0.2, 1, 3, and 4 wt% Cu w.r.t. Ti) were synthesized using the sol–gel technique. The catalysts were characterized using inductively coupled plasma-optical emission spectrometry, XRD, Raman spectroscopy, N2 physisorption, cyclic voltammetry, H2-TPR and electron microscopy to understand the structure and composition. The thermal catalytic gas phase oxidation of ethylene was studied by heating a mixture of ethylene (1.5 vol%) and air (5.9 vol%) in the presence of the Cu/TiO2 samples in the temperature range of 298 to 773 K. Cu/TiO2 showed a higher catalytic activity compared to TiO2 for the thermal oxidation of ethylene, indicating a strong promotion by doped copper ions. A volcanic behaviour in the catalytic activity was observed with different concentrations of Cu doping, with 1% Cu/TiO2 showing a 99.5% ethylene conversion at 673 K and 100% selectivity to CO2. The activity of 1% Cu/TiO2 remained consistent without deactivation for 24 h. At low dopant concentrations of Cu (0.2 and 1% Cu/TiO2), the reduction of Cu2+ to Cu+ was observed. An interplay of oxygen vacancies (OV), Cu+, Cu2+ and Ti4+ may be involved in controlling the activity. DRIFT studies indicated the formation of surface bidendate carbonate as a possible intermediate.
1. Introduction
In 2022, the Global Burden of Diseases reported air pollution as the single highest risk factor for tracheal, bronchial, and lung cancer among vulnerable populations.1 In recent years, various anthropogenic activities have led to a substantial increase in the concentration of atmospheric volatile organic compounds (VOCs).2–4 Particularly, the presence of ethylene in air is closely linked to the high likelihood for the formation of photochemical smog. Besides, ethylene in air is also responsible for the large-scale spoilage of fruits and vegetables during storage in warehouses. Mitigation of VOCs such as ethylene is therefore a problem with a wider social and economic impact.5 Complete mitigation of ethylene by chemical oxidation to CO2 (and not to CO or ethylene oxide) by catalytic technology is a promising approach.6–8 Catalysts based on noble metals such as Ru, Pd, Pt and Au have been reported to have the best rates of ethylene oxidation in the temperature range of 323–493 K.9–12 However, the high costs and vulnerability to poisoning of noble metals remain as bottlenecks to their scalability. As a result, catalysts based on transition metals and their oxides have been actively considered as viable alternatives.6,8,13,14 In this work, we have explored the catalytic activity of Cu doped TiO2 for ethylene oxidation.
Catalytic oxidation of a VOC by a metal oxide operates via the Mars–van Krevelen mechanism, which necessitates the involvement of oxygen vacancies (OV).15 In addition to OV, the Lewis acidity of cations may also be helpful to anchor the incoming unsaturated hydrocarbon.16 TiO2 is a thermally stable material that is commonly used as a support for active metal catalysts in reactions.17–20 With motivation to develop new transition metal oxide-based catalysts, we suspected that doping a low valent transition metal ion such as Cu2+ into the matrix of a reducible oxide such as TiO2 could lead to an interesting catalytic system. The imbalance in charge and size between Cu2+ and Ti4+ ions can create labile O atoms in the vicinity of the dopant, thereby decreasing the energy of formation of OV.21 Besides, the Lewis acidity of the cations (Cu2+ and Ti4+) could increase the adsorption of ethylene. Cu2+ was chosen as a dopant due to Cu–O bonds on the catalyst surface being able to activate C–H bonds.22,23 Although many Cu based catalysts have been used in the past for ethylene oxidation, a majority of them were used in combination with highly redox active metals such as Ce and Mn.8,13,14 In this work, we have synthesized Cu/TiO2 with various concentrations of Cu2+ ions (0.2, 1, 3, 4 wt% Cu w.r.t. Ti) and systematically studied the role of Cu in creating active sites for ethylene oxidation. It is demonstrated that Cu2+ can promote TiO2 resulting in an enhancement in the oxidation of ethylene to CO2 in the range of 500–600 K. To the best of our knowledge, there has not been any report on the catalytic thermal oxidation of ethylene using Cu doped TiO2 (Cu/TiO2).
2. Experimental section
2.1 Materials
Titanium butoxide Ti(OC4H9)4 97%, glacial acetic acid CH3COOH 99%, ethanol C2H5OH 99.5%, hydrochloric acid HCl 37%, Pluronic F127, and copper sulphate pentahydrate CuSO4·5H2O 99.995%. The above chemicals were obtained from Sigma-Aldrich and NICE.
2.2 Synthesis of materials
The sol–gel method was used for the synthesis of Cu/TiO2. In 30 mL ethanol, 1.6 g of Pluronic F127 was added along with 2.3 mL acetic acid and 0.74 mL HCl. Following this, 3.5 mL titanium butoxide and CuSO4·5H2O (Table S1†) were mixed. The mixture was stirred vigorously for 1 h and then transferred to a Petri dish, which was later kept in an oven for 12 h at 313 K and at 338 K for 24 h for aging. The sample was pulverized and calcined at 773 K for 4 h at a ramp rate of 1 °C min−1 to remove the surfactants to get mesoporous Cu/TiO2.
2.3 Ethylene oxidation activity measurement
Ethylene oxidation was carried out using 0.08 g (0.2, 1, 3, 4 wt%) Cu/TiO2 loaded in a 1/4′′ stainless steel tube in a vertical furnace. A total flow rate of 10 mL min−1 with 7.15 mL min−1 for ethylene (1.5 vol%) and 2 mL min−1 for air (5.9 vol%) was maintained using digital mass flow controllers. A hourly space velocity of 3157.9 h−1 was maintained throughout the experiment. The unreacted ethylene and products were analysed using an Agilent 7890B gas chromatograph (GC) equipped with a thermal conductivity detector (TCD) and a flame ionization detector (FID). The ethylene conversion, selectivity to CO2 and turnover number were calculated using eqn (1), (2) and (3), respectively.| |
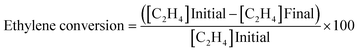 | (1) |
| |
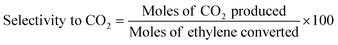 | (2) |
| |
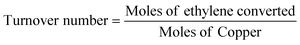 | (3) |
2.4 Characterization of materials
A Rigaku Smart Lab X-ray diffractometer was used for the analysis of the crystalline phase of the catalysts with Cu Kα radiation (λ = 0.154 nm) in the range of 2θ = 20–80 degrees, operated at a current of 200 mA and a voltage of 45 kV. Raman spectra of the catalysts were examined using a Horiba Labram spectrometer with a 532 nm ultra-low frequency laser source. A Quantachrome Chemstar TPX chemisorption instrument was used to check the reducibility of the catalysts. Temperature programmed reduction (H2-TPR) of samples was carried out in a 10% H2/Ar flow (used as the reducing atmosphere) and heated from room temperature to 1073 K at 10 K min−1. The H2 consumption of 0.6 mg of catalyst for the reduction process was monitored using a TCD at different temperature intervals. The surface elemental composition of the catalyst was analysed by X-ray photoelectron spectroscopy (XPS) using a K ALPHA+ (Thermo Fisher Scientific Instruments, UK). Monochromatic Al Kα was used as the X-ray source at a 6 mA beam current and 12 kV. Inductively coupled plasma-optical emission spectrometry (ICP-OES) was used to calculate the wt% of Cu in TiO2. N2 physisorption was performed at liquid N2 temperature using a Quantachrome Autosorp IQ-MP and Brunauer–Emmett–Teller (BET) analysis was carried out to determine the surface area, average pore size and total pore volume. Transmission electron microscopy (TEM) was used for the analysis of the microstructure of the material. The Cu/TiO2 samples were electrochemically investigated by performing cycling voltammetry studies on a CHI60038 electrochemical workstation. The setup has a 3-electrode system in which a Cu/TiO2 coated glassy carbon electrode (GCE) (diameter 3 mm, area 0.07 cm2) was used as the working electrode, a Pt wire as the counter electrode, and a saturated calomel electrode (SCE) as the reference electrode. A catalyst ink for the electrochemical studies was prepared by sonicating 10 mg of the Cu/TiO2 catalyst in a water–ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture containing 100 μL of Nafion (0.5 wt% in a solution of 1:1 water–ethanol) to a final concentration of 10 mg mL−1 of the catalyst. Thereafter, 6 μL of the catalyst ink (0.06 mg) was drop cast onto a GCE and allowed to dry under ambient conditions. Cyclic voltammetry studies were carried out at a scan rate of 50 mV s−1 with 0.50 M Na2SO4 as the electrolyte at room temperature. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was performed on a FT-IR spectrometer (Shimadzu IR Tracer 100), equipped with a diffuse reflectance accessory and a DLATGS (deuterated L-alanine doped triglycine sulphate) detector. The DRIFTS reaction chamber is made of stainless steel with a ZnSe window.
:1) mixture containing 100 μL of Nafion (0.5 wt% in a solution of 1:1 water–ethanol) to a final concentration of 10 mg mL−1 of the catalyst. Thereafter, 6 μL of the catalyst ink (0.06 mg) was drop cast onto a GCE and allowed to dry under ambient conditions. Cyclic voltammetry studies were carried out at a scan rate of 50 mV s−1 with 0.50 M Na2SO4 as the electrolyte at room temperature. In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was performed on a FT-IR spectrometer (Shimadzu IR Tracer 100), equipped with a diffuse reflectance accessory and a DLATGS (deuterated L-alanine doped triglycine sulphate) detector. The DRIFTS reaction chamber is made of stainless steel with a ZnSe window.
3. Results and discussion
3.1 Characterization of Cu doped TiO2 (Cu/TiO2)
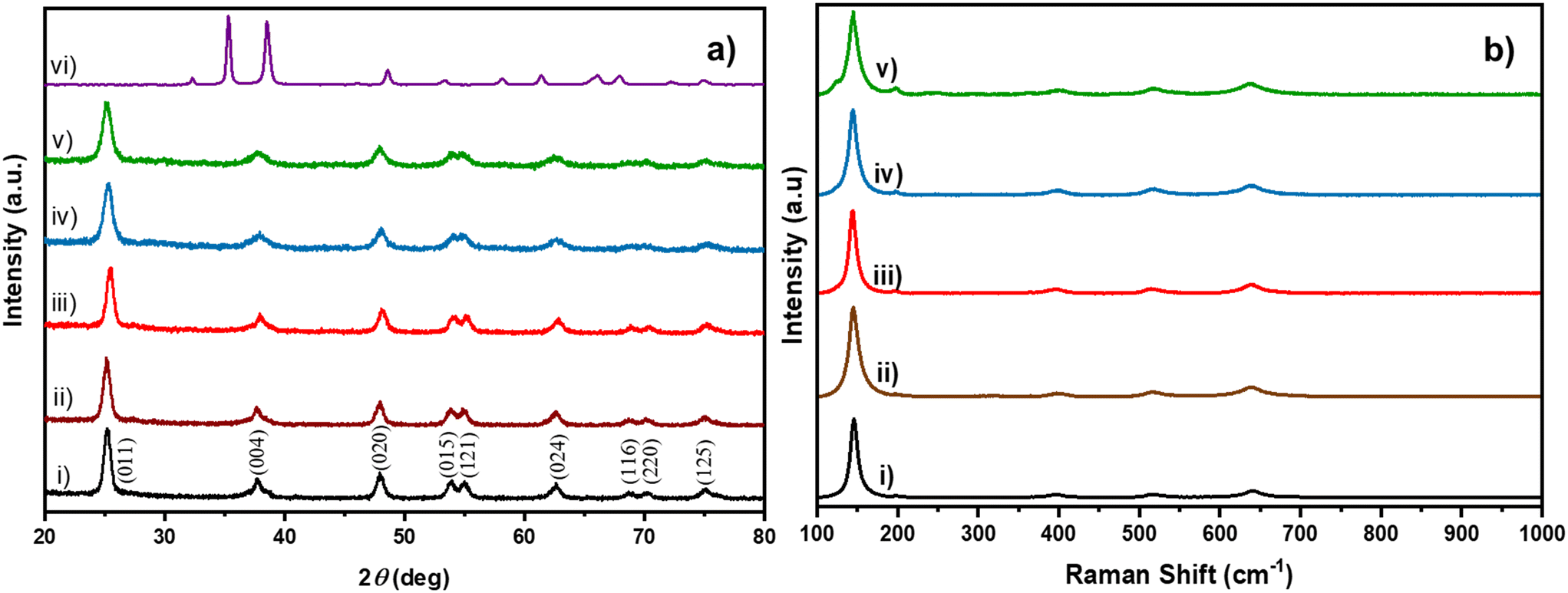
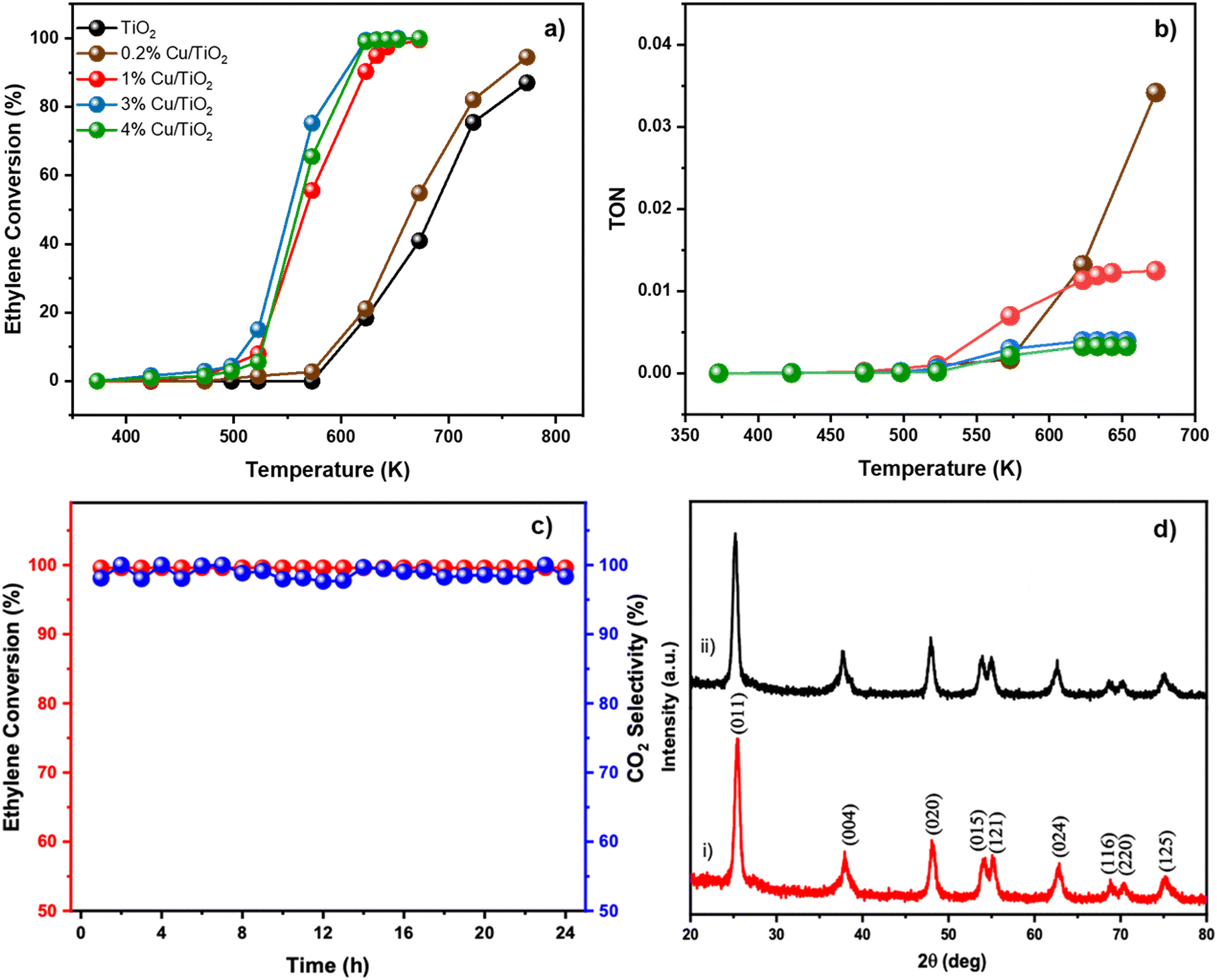
Cu/TiO2 catalysts containing different concentrations of the Cu dopant were prepared using the sol–gel method. In Fig. 1a trace (i), the powder XRD peaks revealed the anatase phase of TiO2 (ICSD code: 98-000-5224) with the (011), (004), (020), (015), (121), (024), (116), (220) and (125) planes. Interestingly, none of the peaks in the XRD patterns of Cu/TiO2 samples (traces ii to v) could be indexed to any of the bulk CuO phases. This observation has been reported by earlier researchers too.24 This indicated that either the CuO phase was too small to be detected by XRD or Cu existed in the form of highly dispersed ions in the TiO2 matrix. The suspicion behind the high dispersion of Cu in the TiO2 matrix is justified due to the excellent stabilization of Cu2+ during the condensation of the precursors in sol–gel synthesis.25,26 An interesting observation was the broadening of the (011) peak of the Cu/TiO2 samples. From the width of the XRD peak, it was estimated that the average crystallite size decreased from 10.3 nm for pure TiO2 to 7.4 nm for 4% Cu/TiO2 as shown in Table S2.† A slight shift of 0.2 degrees in two theta of the (011) plane was observed in 1% Cu/TiO2. Generally, a shift in the peak position may be attributed to a change in lattice parameters of the unit cell which could arise due to the doping of an ion, whose radius may vary by more than 10–15% of the radii of the native ions of the lattice, rTi4+ (0.68 Å) < rCu2+ (0.72 Å).27Fig. 1b elucidates the Raman spectra of undoped TiO2 and Cu/TiO2 samples. Trace i) corresponding to pure TiO2 displayed bands at 144.9, 197.4, 399.2, 516.6 and 639.2 cm−1, which are indexed to the anatase structure of titania.28 The Cu/TiO2 samples with different concentrations of Cu gave identical bands in the Raman spectra, indicating that the anatase phase of TiO2 was retained in all cases. Interestingly, the Eg mode at 144.9 cm−1, which is assigned to the symmetric stretching of Ti–O bonds, showed a redshift with increasing concentration of Cu in TiO2 (see Fig. S1†). The reduction in the particle size probably created more under-coordinated surface sites.24 This shrinkage of crystallite size with increasing concentration of Cu corroborated well with the XRD results. The effect of Cu doping resulted in the formation of new bonds such as Cu–O–Ti or Cu–O–Cu. An imbalance of charges across the new bonds can create oxygen vacancies.21 As a result, the Raman spectra clearly indicated the softening of the vibrational modes in the form of a red shift of the Eg mode. Similar results were observed earlier too.24
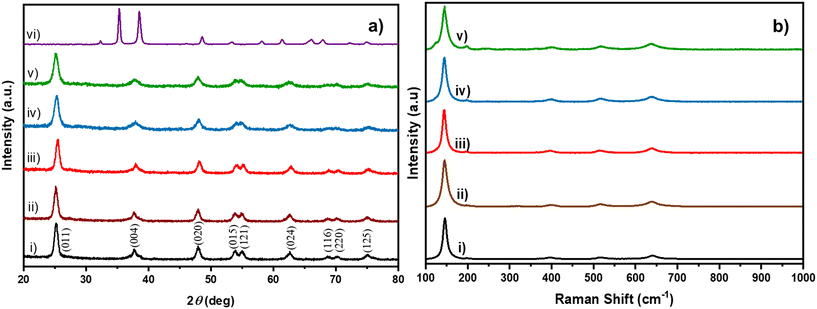 |
| | Fig. 1 a) XRD patterns of i) TiO2 (black), ii) 0.2% Cu/TiO2 (brown), iii) 1% Cu/TiO2 (red), iv) 3% Cu/TiO2 (blue), v) 4% Cu/TiO2 (green) and vi) bulk CuO (violet). b) Raman spectra of i) TiO2 (black), ii) 0.2% Cu/TiO2 (brown), iii) 1% Cu/TiO2 (red), iv) 3% Cu/TiO2 (blue) and v) 4% Cu/TiO2 (green). | |
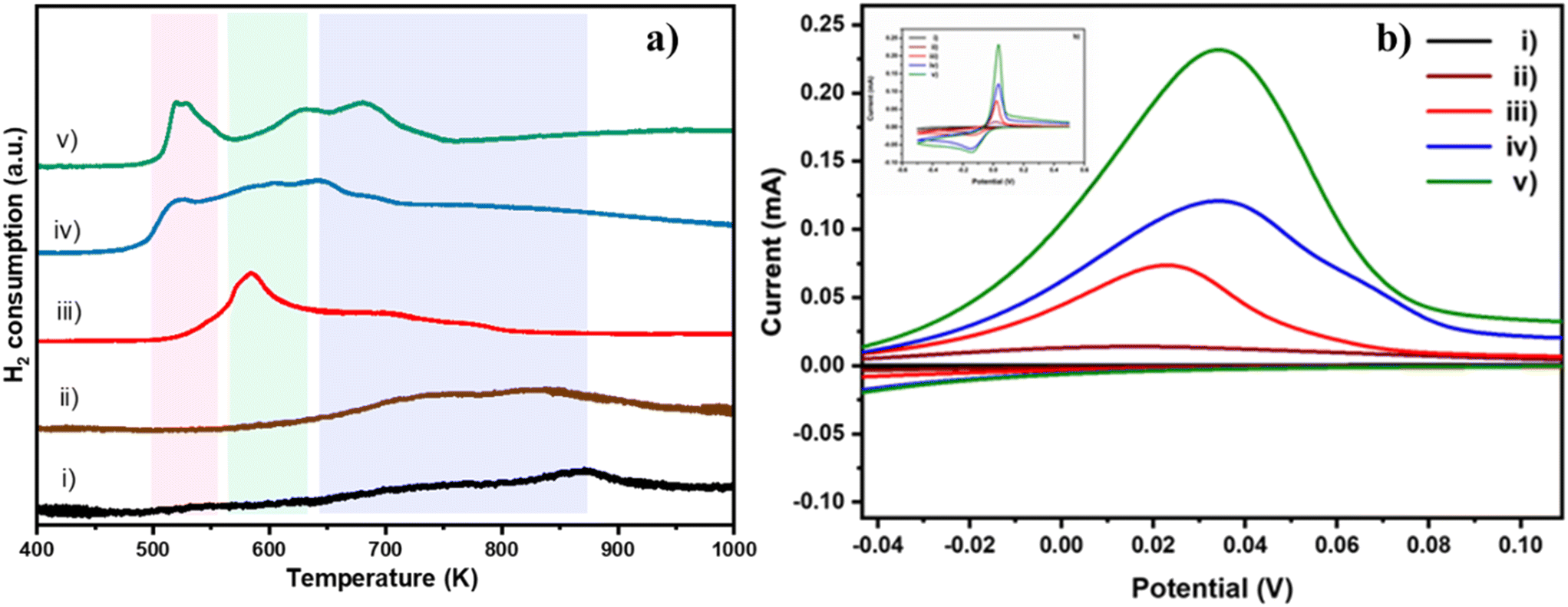
Having confirmed that doping of Cu neither affected the phase purity in TiO2 nor caused any phase separation, the temperature programmed chemisorption technique was employed to probe the nature of Cu and its interaction with the TiO2 matrix. H2-TPR was carried out to understand the ease of reduction of Cu2+ ions in the TiO2 matrix. H2-TPR profiles of bare TiO2 and Cu doped TiO2 samples are shown in Fig. 2a. Three thermally induced reduction events occur in the temperature range of 400 to 1000 K. The reduction of Cu2+ to Cu+ occurs in the temperature range of 400–550 K (pink region), while that of Cu+ to Cu0 occurs in the range of 550–650 K (green region) and Ti4+ to Ti3+ above 700 K (as a doublet, purple region).26,29 The curve corresponding to 0.2% Cu/TiO2 (trace ii) did not show significant difference in features compared to that of pure TiO2 (trace i). Although the concentration of Cu2+ may not be high enough to cause a noticeable reduction peak, the presence of traces of Cu0 after H2 reduction (during the H2-TPR experiment) may accelerate the reduction of Ti4+ to Ti3+.30 As a result, one can note that the positions of doublet peaks (Ti4+ to Ti3+) migrate to a lower temperature with increasing concentration of Cu. The narrower peak in the 550–650 K region in 1% Cu/TiO2 (trace iii) is indicative of the reduction of Cu+ to Cu0. Although not very distinct, there could also be the reduction of Cu2+ to Cu+ happening, as seen by the asymmetry of the peak.31 Importantly, as shown in Table S3 in the ESI,† the low temperature peak due to the reduction of copper ions is shifted to further lower temperatures as the concentration of Cu increased, suggesting that the reduction of Cu2+ is a more favourable process at higher concentrations. A concomitant shift in the position of the doublet also supports our observation that the availability of Cu0 becomes higher as the concentration of Cu is increased.30 Analysis of the ratio of Cu2+/Cu+ based on the volume of H2 consumed (Table S4 in the ESI†) revealed that at low dopant concentration, Cu2+ is reduced to Cu+ due to strong metal–support interaction. However, as the concentration increases, in addition to the existence of Cu+, the stabilization of Cu2+ is gradually improved leading to the co-existence of Cu+ and Cu2+. A slight shift in the position of reduction peaks of both Cu2+ and Ti4+ to higher temperature (for 4% Cu/TiO2) indicated that Cu2+ was probably stabilized making its reduction difficult.
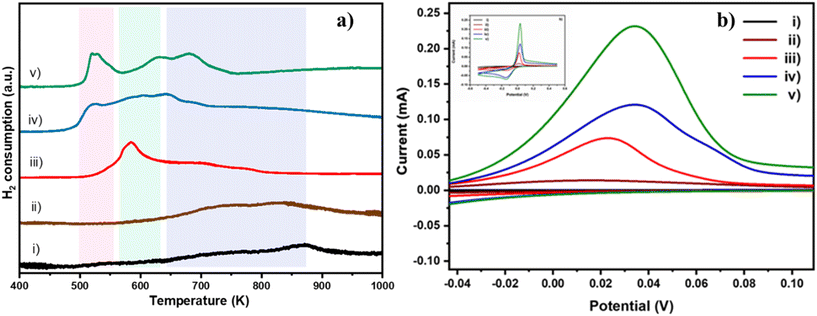 |
| | Fig. 2 a) H2-TPR curves of i) TiO2, ii) 0.2% Cu/TiO2, iii) 1% Cu/TiO2, iv) 3% Cu/TiO2 and v) 4% Cu/TiO2. Colour bands represent the following reduction events: pink band on the left side (Cu2+ to Cu+), green band in the middle (Cu+ to Cu0) and purple band on the right (Ti4+ to Ti3+). b) CV profiles at a scan rate of 50 mV s−1 for the different samples: i) TiO2 (black); ii) 0.2% Cu/TiO2 (brown); iii) 1% Cu/TiO2 (red); iv) 3% Cu/TiO2 (blue); v) 4% Cu/TiO2 (green). Inset shows the CV profiles of oxidation and reduction potential of the samples recorded at 50 mV s−1 over the potential range −0.5 to +0.5 V. | |
This concentration dependent redox behaviour of Cu in TiO2 was also followed using cyclic voltammogram (CV) studies (Fig. 2b). CVs were obtained in a cathodic sweep at a scan rate of 50 mV s−1, followed by an anodic sweep at the same scan rate. The anodic and cathodic peaks are attributed to the oxidation of Cu+ to Cu2+ and reduction of Cu2+ to Cu+ respectively.32 As expected, pure TiO2 did not exhibit any distinct redox peaks in the scanning range of −0.5 V to 0.5 V. The oxidation peak occurred at 0.016 V in the 0.2% Cu/TiO2 curve (ii). In the 1% Cu/TiO2 curve (iii), the oxidation peak shifted by 5 mV to 0.021 V. In the 3% Cu/TiO2 curve (iv), it increased further to 0.034 V. A very slight shift to a lower potential was observed for 4% Cu/TiO2, which was consistent with the H2-TPR data curve (v). The data suggested that the ease of oxidation of Cu+ to higher oxidation states increased with decreasing concentration of the dopant from 3 to 0.2% Cu/TiO2. Both H2-TPR and CV studies suggested the better redox characteristics of Cu/TiO2 compared to those of pure TiO2. The ease of reducibility of Cu2+ increased with the increase in the concentration of the dopant. The redox behaviour of Cu/TiO2 is a promising feature of the oxidation ability of the catalysts.
3.2 Catalytic activity of Cu/TiO2
Fig. 3a shows the catalytic performance of Cu/TiO2 for the oxidation of ethylene at various temperatures. The oxidation of ethylene over undoped TiO2 began at 573 K but did not reach 100% conversion even after reaching 750 K. In contrast, the ethylene conversion by Cu/TiO2 typically started at 423 K and improved to 70% conversion at 550 K. At 623 K, 3% Cu/TiO2 showed a 99% ethylene conversion, while 1% and 4% Cu/TiO2 gave a 90% and 99% conversion, respectively. Most importantly, in all the samples, CO2 was formed with 100% selectivity. Although the temperature dependent activity was more or less similar for the various Cu/TiO2 samples, the 3% and 4% samples achieved complete conversion at a slightly lower temperature. 3% Cu/TiO2 is the most active catalyst in all the temperature ranges. The oxidation ability of the Cu/TiO2 catalyst corroborated well with the H2-TPR and CV studies. Interestingly, even the subtle change in the trend of the reducibility of Cu2+ in the 3% and 4% samples could be correlated with an observable outcome in terms of catalytic activity. The efficiency of the catalyst was further understood by plotting the turnover number (TON) which represents the number of moles of ethylene converted by a mole of copper at a given temperature (Fig. 3b). At 573 K, where the activity began, 1% Cu/TiO2 showed a high TON of 6.9 × 10−3 compared to that of 0.2% (1.6 × 10−3), 3% (2.9 × 10−3) and 4% (2.1 × 10−3). Expectedly, as the temperature increased, the TON increased too and reached a saturation value at around 623 K. The TON value for 1%, 3% and 4% Cu/TiO2 was 1.13 × 10−2, 3.91 × 10−3 and 3.26 × 10−3 respectively. This temperature also correlated with a 100% conversion of ethylene. Interestingly, the activity of 0.2% Cu/TiO2 started at around 573 K but it reached a high TON of 1.3 × 10−2 at 623 K. At 673 K, when most of the other compositions had reached a saturation in conversion, the TON of 0.2% Cu/TiO2 reached 3.4 × 10−2, which was three times higher than that of 1% Cu/TiO2. Intriguingly, the TON value for 0.2% did not reach saturation even beyond 673 K. Although the temperature of conversion of 0.2% Cu/TiO2 was higher compared to those of other Cu/TiO2 compositions, the interesting observation presents scope for more in-depth structural correlation studies. Convincingly, the results indicated that the activity of Cu/TiO2 was significantly higher than that of pure TiO2 for the oxidation of ethylene suggesting the promotional effect of Cu on TiO2.
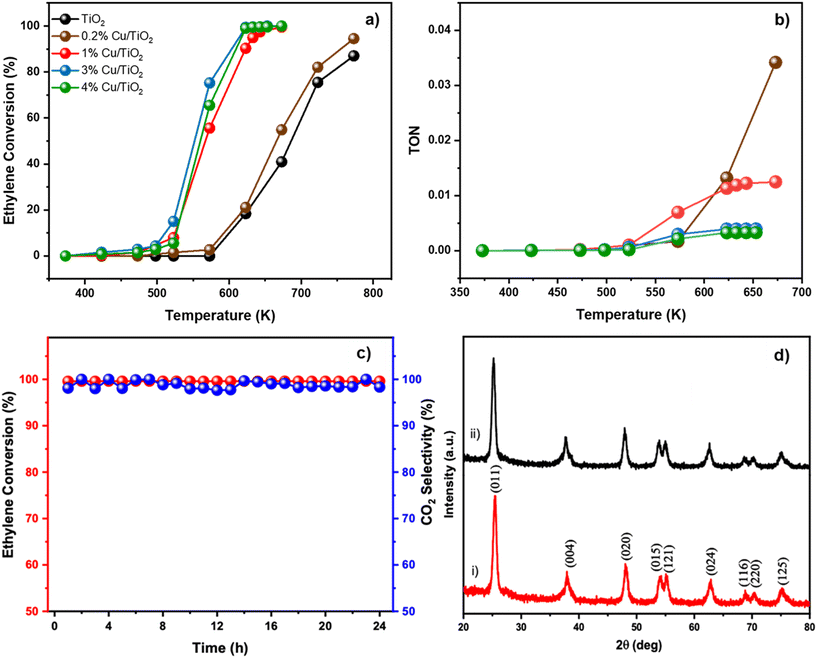 |
| | Fig. 3 a) Plots of ethylene conversion (%) against temperature (K) for TiO2 and Cu doped TiO2 samples (0.2%, 1%, 3% and 4%). Inset shows the colour coding for the various samples TiO2 (black), 0.2% Cu/TiO2 (brown), 1% Cu/TiO2 (red), 3% Cu/TiO2 (blue) and 4% Cu/TiO2 (green). b) Plots of turnover number (TON) of Cu doped TiO2 samples against temperature (K). The colour legend is same as that in a). c) Thermal stability of 1% Cu/TiO2 demonstrated by monitoring the conversion of ethylene at 673 K for 24 h. d) XRD patterns of 1% Cu/TiO2 i) before reaction and ii) after reaction for 24 h. | |
The thermal stability of 1% Cu/TiO2 was studied by carrying out the ethylene conversion at 673 K over 24 h continuously. In Fig. 3c, the conversion of ethylene and selectivity to CO2 remained at 100% throughout the time on stream. This indicated that no deactivation happened during the continuous operation at elevated temperatures. In Fig. 3d, the XRD pattern of 1% Cu/TiO2 after reaction for 24 h was compared with the XRD pattern before the reaction. There was no sign of phase separation indicating the excellent thermal stability of the catalyst. Considering the practicality of the process, 1% Cu/TiO2 displayed the best TON at a reasonable temperature. The structure of 1% Cu/TiO2 was examined using TEM. Spherical nanoparticles (Fig. S3†) were observed in the TEM micrographs. The average particle size of 1% Cu/TiO2 varied from 8–12 nm, which is close to the 9.76 nm crystallite size of 1% Cu/TiO2 as calculated from XRD using the Debye–Scherrer equation (Table S2†). The d-spacing was calculated as 0.35 nm from the lattice fringes, corresponding to the (011) plane of the anatase TiO2 phase in XRD.
3.3 Probing the active sites
One of the important impacts of doping Cu2+ in TiO2 is the creation of OV. Substitution of a larger Cu2+ ion (72 pm) for Ti4+ (65 pm) in TiO2 is likely to introduce strain as well as charge imbalance in the lattice, which may be alleviated by forming an OV.24,33,34 To confirm the presence of OV, XPS spectra were obtained for all the Cu/TiO2 compositions. Two peaks were observed in the Ti spectra (Fig. S4†) at 458.4 eV and 464.08 eV, corresponding to 2p3/2 and 2p1/2, respectively. This confirms the presence of Ti4+ in all the samples of Cu/TiO2.35
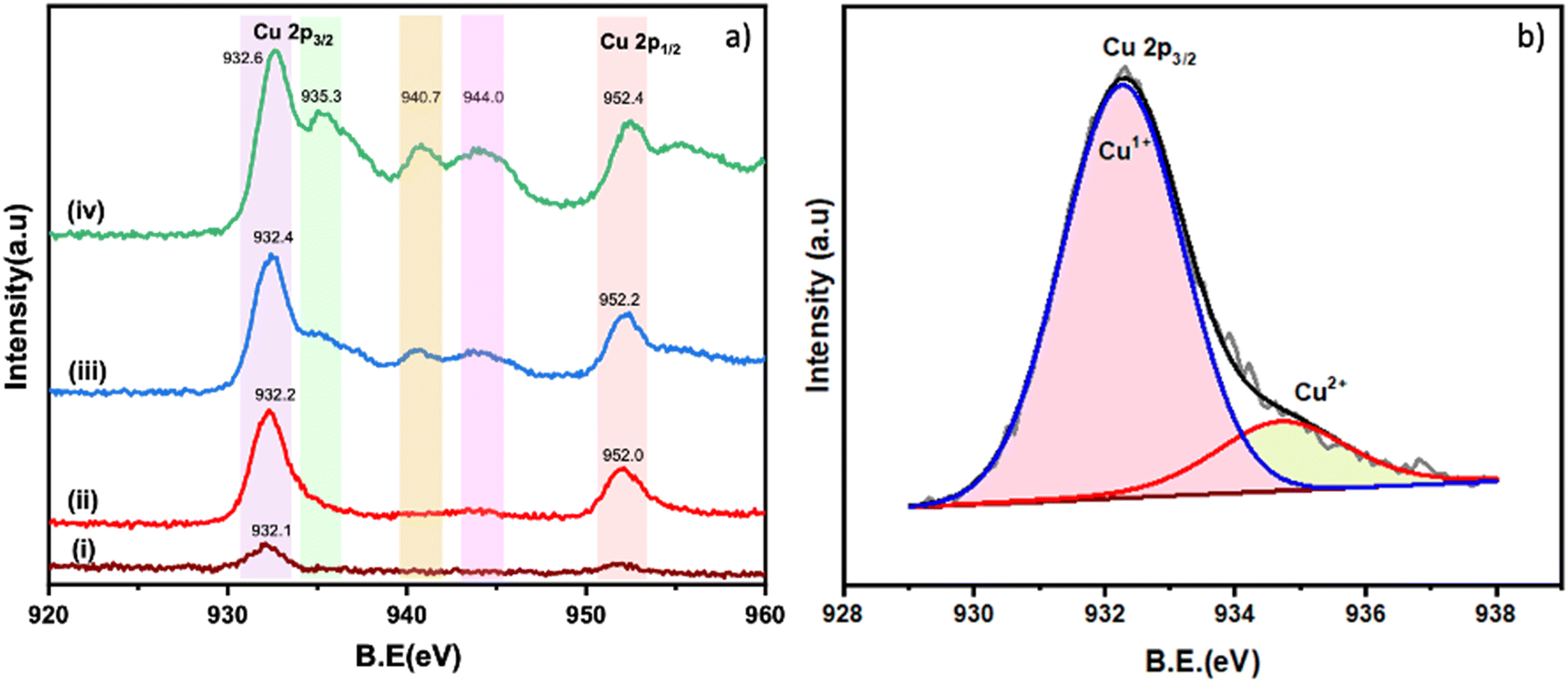
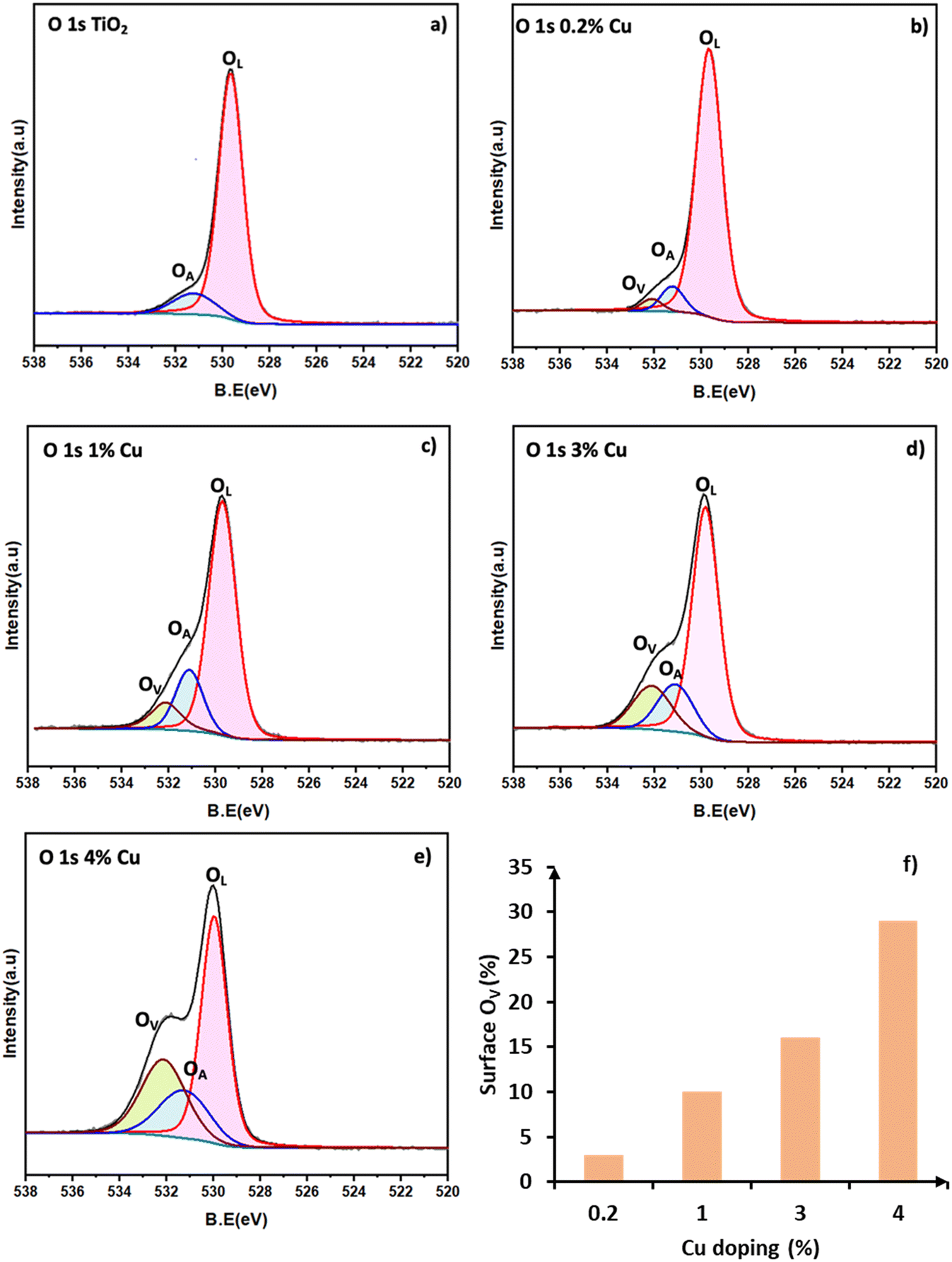
The Cu 2p XPS spectra of the different Cu/TiO2 samples are shown in Fig. 4a. In the literature, peaks at 932.5 eV for Cu 2p3/2 and 952.0 eV for Cu 2p1/2 are taken as confirmation of the presence of Cu+ species.35 Similarly, peaks at 935.0 eV (Cu 2p3/2) with satellite peaks at 940.7 and 944.0 eV are a confirmation of the presence of Cu2+. In the 0.2% Cu/TiO2 sample trace (i), the Cu species are clearly in +1 state as the satellite peaks which are characteristic of Cu2+ are absent. In the 1% Cu/TiO2 sample, Cu+ could be the dominant species. A hump of very low intensity in the satellite peak region (Cu2+) and a small tail peak at 935 eV (as shown in Fig. 4b) in 1% Cu/TiO2 raise suspicion about the presence of Cu2+ as well. With further increasing Cu concentration, the presence of Cu2+ is established beyond doubt in TiO2 along with Cu+. The valence ratio (Cu2+/Cu+) of 1%, 3%, and 4% Cu/TiO2 as calculated using XPS was 0.2, 0.5, and 0.9, respectively (Table S6†). An increase in the valence ratio (Cu2+/Cu+) was observed with the increase of Cu doping into TiO2. The data suggest a strong metal–support interaction between Cu2+ and the TiO2 matrix resulting in the reduction of Cu2+ to Cu+ at low concentrations, which is firmly in agreement with the H2-TPR and CV data. However, as the concentration increases, the interaction with the support is probably weakened leading to a higher presence of Cu2+ as well. The O 1s XPS spectra of the pure TiO2 and Cu/TiO2 samples are shown in Fig. 5a–e. The peak observed at 529.7 eV corresponds to the lattice oxygen (OL) in the TiO2 matrix. The second peak at 531.1 eV is due to the hydroxyl groups adsorbed on the surface (OA).24 The peak appearing at 532.1 eV is due to the chemisorbed oxygen created due to the presence of OV.24Fig. 5a suggests that the undoped TiO2 sample does not have OV sites. On doping Cu into the TiO2 lattice, the formation of OV was observed. The percentage area due to OV calculated with respect to the total area of the O 1s spectrum for the different samples (in brackets) suggested an increase in the surface OV with the doping of Cu: 3 (0.2%), 10 (1%), 16 (3%) and 29 (4%). There is a direct correlation between the concentration of the surface OV and the concentration of Cu in the TiO2 matrix (Fig. 5f).
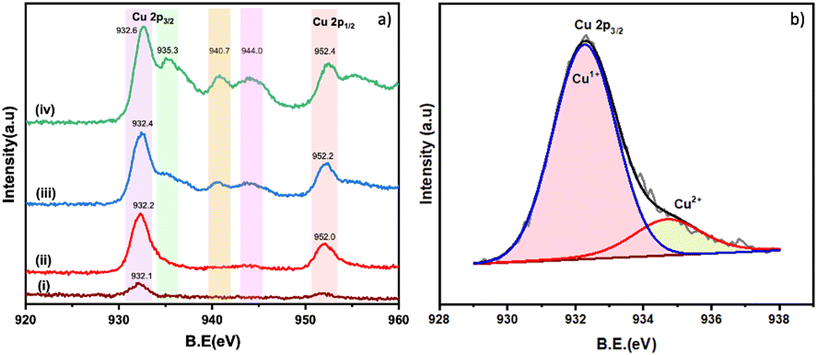 |
| | Fig. 4 a) XPS spectra of Cu 2p: i) 0.2% Cu/TiO2, ii) 1% Cu/TiO2, iii) 3% Cu/TiO2 and iv) 4% Cu/TiO2; b) deconvoluted spectrum of Cu 2p3/2 of 1% Cu/TiO2. | |
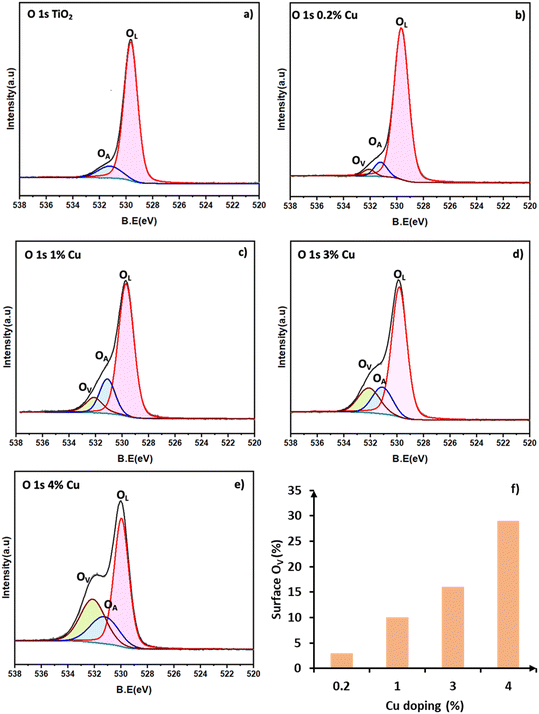 |
| | Fig. 5 X-ray photoelectron spectra of O 1s: a) TiO2, b) 0.2% Cu/TiO2, c) 1% Cu/TiO2, d) 3% Cu/TiO2 and e) 4% Cu/TiO2. f) Correlation between surface OV (normalized area in XPS) and doping concentration. | |
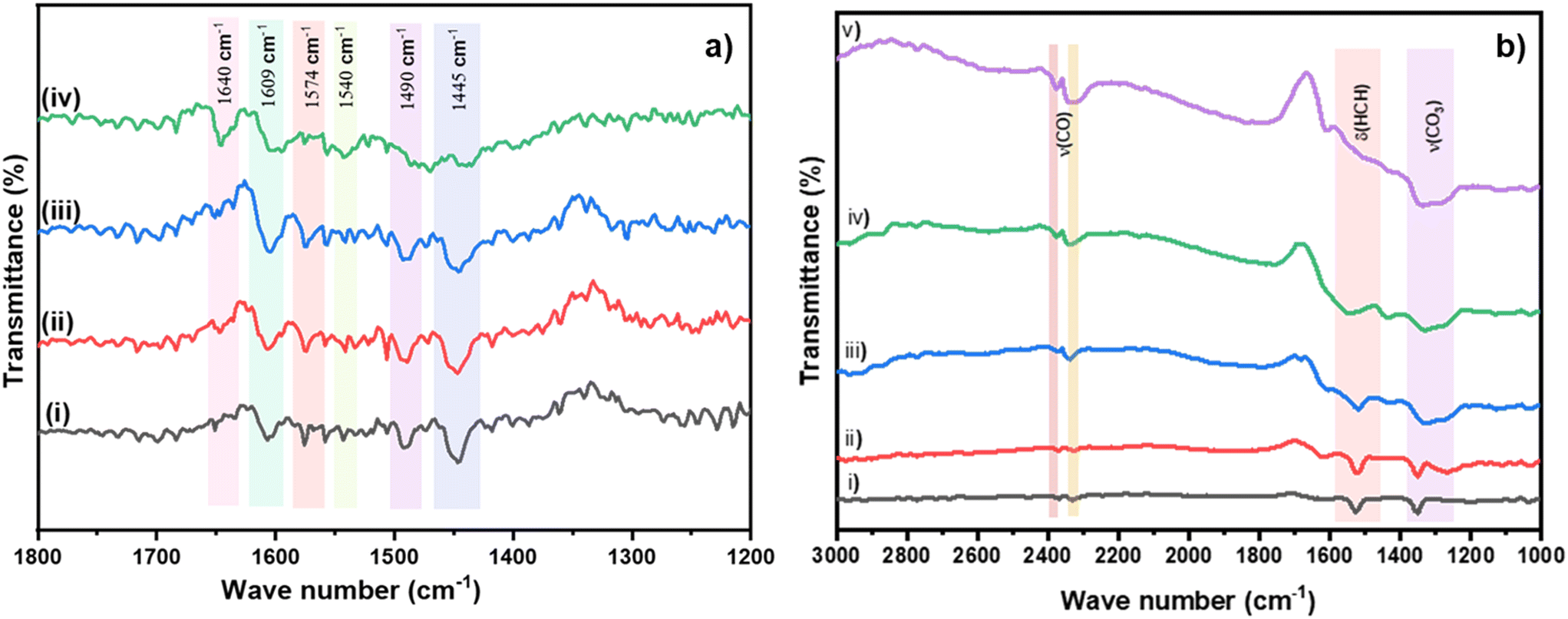
The acidity of the sample could also aid in a better adsorption of ethylene resulting in increased oxidation.16 Pyridine DRIFTS was performed to probe the acidic sites on the most active 1% Cu/TiO2 sample.36–39 Initially, the sample was heated at 473 K in an inert atmosphere to remove the impurities on the surface followed by pyridine injection at 323 K. The spectrum is shown in Fig. 6a(i). The major peaks observed at 1445 cm−1, 1574 cm−1, and 1609 cm−1 correspond to Lewis acidic sites.36,38 The peaks at 1540 and 1640 cm−1 represent the presence of Brønsted acidic sites. The band at 1490 cm−1 indicates both Lewis and Brønsted acidic sites.37 The bands were monitored by an increase in the temperature from 323 to 573 K under a N2 atmosphere. The desorption of pyridine was not observed even at high temperatures (Fig. 6a(ii) to (iv)), suggesting the presence of strong acidic sites in 1% Cu/TiO2.37 Importantly, pyridine DRIFTS carried out on bare TiO2 did not reveal the presence of strong acidic sites, clearly indicating the impact of doping of Cu into TiO2 (Fig. S7†). In the next step, ethylene/air-DRIFTS was performed to probe the surface species that form on the Cu/TiO2 during the reaction (Fig. 6b). Initially, the surface was purged with a N2 atmosphere for an hour at 373 K to avoid signals due to impurities. A mixture of ethylene (1.5 vol%) and air (5.9 vol%) having an identical feed ratio to those in the reactions was used. The IR spectrum was recorded from 298 to 673 K in a continuous flow of feed gas. The band observed at around 1524 cm−1 corresponds to the scissoring mode δ(HCH) of the ethylene molecules at 298 K. Trace (ii) clearly showed the band corresponding to the adsorption of ethylene on the surface of the catalyst. The bands at 2330 and 2370 cm−1 indicate the stretching mode of gaseous CO2. Moreover, the peak at 2350 cm−1 indicates the presence of adsorbed CO2 on metal atoms.40 As the temperature of the catalyst was increased, the band of δ(HCH) corresponding to the ethylene peaks disappeared and the band of ν(COC) and adsorbed CO2 increased confirming the complete oxidation of ethylene to CO2. The peak observed in the region of 1220 and 1350 cm−1 corresponds to the symmetric stretching of adsorbed bicarbonate and bidentate carbonate species on the metal oxide.41 The same band started to disappear when the temperature was increased from 298 to 673 K. However, it was not clear whether carbonates were formed due to the re-adsorption of CO2 or as an intermediate during the oxidation of ethylene. CO2 gas was passed over the catalyst surface and spectra were recorded at different temperatures (Fig. S5†). No peaks corresponding to the formation of carbonates were observed from RT to 673 K in CO2 DRIFTS.42 The results indicate that carbonates were formed as an intermediate during the oxidation, not by the re-adsorption of CO2. Moreover, the peak corresponding to –OH stretching in H2O is shown in Fig. S6.† This could be due to the adsorption of water formed during the oxidation of ethylene. Despite this observation, the activity did not decrease up to 24 h. This indicates that despite the interaction of H2O with the surface of the catalyst, the activity of the catalyst was unaffected. Although detailed surface studies are needed to understand the mechanism of ethylene oxidation, the DRIFT studies strongly indicate the oxidation occurring via the carbonate pathway.
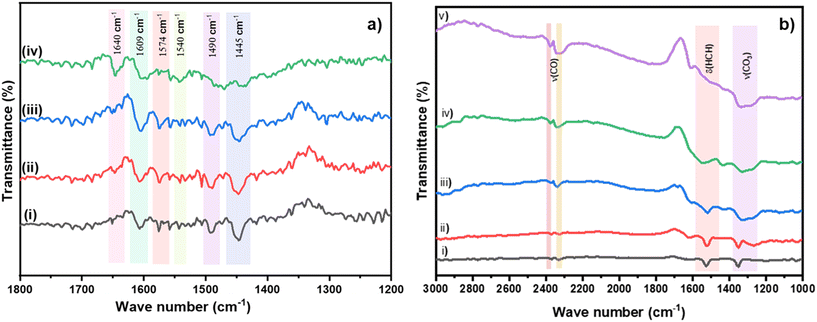 |
| | Fig. 6 a) Pyridine DRIFTS spectra of Cu/TiO2 at (i) 323 K, (ii) 373 K, (iii) 473 K and (iv) 573 K. b) DRIFTS measurements during oxidation of ethylene using 1% Cu/TiO2 with a mixture of ethylene (1.5 vol%) and oxygen (5.9 vol%) at (i) 298 K; (ii) 373 K; (iii) 473 K; (iv) 573 K; (v) 673 K. | |
The role of OV in an oxidation reaction as explained by the Mars–van Krevelen mechanism is to improve the mobility of lattice oxygen to the surface and facilitate the adsorption of gaseous O2 onto the lattice. According to the crude electron pair Lewis theory, OV can be Lewis basic because of the electrons that are left behind at the site when the oxide ion is removed to create the vacancy.21 This also facilitates the creation of a reactive lattice oxygen species.43,44 Although the concentration of OV on the surface increases with the concentration of doped Cu, the activity of oxidation catalysis seems to be dependent on the reducibility of Cu2+ (as shown by TPR, CV and XPS), making it an important descriptor of the activity. If the reducibility of Cu2+ is due to the acceptance of electrons left behind by the lattice oxygen at the OV sites, then the basicity of OV could be the signboard for reactivity. Thus, OV are necessary for the reactions, but electrons available at the OV sites (as indicated by the reducibility of Cu2+) could also be necessary. In addition to this, strong Lewis acidic sites like Cu2+ or Cu+ (Fig. 6 and S7†) on the surface can chemisorb ethylene, which can be eventually oxidized to CO2 by the reactive lattice O in a series of steps involving a carbonate intermediate. OV are eventually annihilated by the O2 in the feed during the process oxidizing cations.44
4. Conclusion
Thermal oxidation of ethylene using a Cu/TiO2 system has been studied in detail with varying concentrations of Cu. The doping of Cu neither caused any phase separation nor affected the phase purity of TiO2. Doping of Cu2+ created OV which played an important role in the oxidation catalyst. There is a direct correlation between the concentration of doped copper and concentration of surface OV. A strong metal–support interaction probably reduced doped Cu2+ to Cu+ at lower concentrations. As the concentration of doped Cu increased, a significant increase in the stability of Cu2+ was noticed. A correlation between the reducibility of Cu2+ and its reactivity was observed. The combination of surface cationic sites and OV created due to the doping of Cu played a crucial role in the enhanced catalytic oxidation by Cu/TiO2. 1% Cu/TiO2 showed the most promising activity with the highest turnover number compared to others (0.2%, 3%, and 4% Cu/TiO2). At 673 K, 1% Cu/TiO2 gave a 99.5% ethylene conversion and 100% selectivity to CO2. The reusability of 1% Cu/TiO2 was demonstrated by a stable conversion in a stream of ethylene and air at 673 K for 24 h without any deactivation. DRIFT studies indicated that the oxidation proceeded via carbonate intermediates. In conclusion, Cu/TiO2 is a promising non-noble metal oxide based catalyst for the mitigation of pollution by ethylene. Cu/TiO2 is a promising non-noble metal system as it is active and stable for the oxidation of ethylene.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The author K. R. would like to thank the Ministry of Education and IIT Palakkad for an HTRA fellowship. The authors would like to thank SAIF (Sophisticated Analytical Instruments Facility) in IIT Madras for the ICP-OES analysis. The authors acknowledge the Central Instrumentation Facility (CIF) in IIT Palakkad for the chemical and material characterization. The funding of the projects 2019-017-CY-DIJ-DFRL-SP and 2020-036-CY-DIJ-SERB-SP is acknowledged. The authors thank Dr. C. P. Priyakumari (IIT Palakkad) for useful discussions.
References
- K. B. Tran, J. J. Lang, K. Compton, R. Xu, A. R. Acheson, H. J. Henrikson, J. M. Kocarnik, L. Penberthy, A. Aali, Q. Abbas and B. Abbasi, Lancet, 2022, 400, 563–591 CrossRef PubMed.
- L. K. Sahu, Curr. Sci., 2012, 102, 1645–1649 CAS.
-
J. Williams and R. Koppmann, Volatile Organic Compounds in the Atmosphere, Blackwell Publishing Ltd, Oxford UK, 2007, vol. 1, pp. 1–32 Search PubMed.
- S. Sawada and T. Totsuka, Atmos. Environ., 1986, 20, 821–832 CrossRef CAS.
- A. Falls and J. Seinfeld, Environ. Sci. Technol., 1978, 12, 1398–1406 CrossRef CAS.
- W. Li, Z. Zhang, J. Wang, W. Qiao, D. Long and L. Ling, Chem. Eng. J., 2016, 293, 243–251 CrossRef CAS.
- A. Kolman, M. Chovanec and S. Osterman-Golkar, Mutat. Res., Rev. Mutat. Res., 2002, 512, 173–194 CrossRef CAS PubMed.
- H. Chen, X. Tong and Y. Li, Appl. Catal., A, 2009, 370, 59–65 CrossRef CAS.
- H. G. Ahn, B. M. Choi and D. J. Lee, J. Nanosci. Nanotechnol., 2006, 6, 3599–3603 CrossRef CAS PubMed.
- D. Kumar, Y. F. Han and D. W. Goodman, Top. Catal., 2007, 46, 169–174 CrossRef CAS.
- R. J. Isaifan and E. A. Baranova, Catal. Today, 2015, 241, 107–113 CrossRef CAS.
- M. Wang, L. Zhang, W. Huang, Y. Zhou, H. Zhao, J. Lv, J. Tian, X. Kan and J. Shi, RSC Adv., 2017, 7, 14809–14815 RSC.
- E. C. Njagi, H. C. Genuino, C. K. King'ondu, S. Dharmarathna and S. L. Suib, Appl. Catal., A, 2012, 421, 154–160 CrossRef.
- R. Dziembaj, M. Molenda, L. Chmielarz, M. Zaitz, Z. Piwowarska and A. Rafalska-Łasocha, Catal. Today, 2011, 169, 112–117 CrossRef CAS.
- M. Zhao, J. Deng, J. Liu, Y. Li, J. Liu, Z. Duan, J. Xiong, Z. Zhao, Y. Wei, W. Song and Y. Sun, ACS Catal., 2019, 9, 7548–7567 CrossRef CAS.
- H. Yang, C. Ma, Y. Li, J. Wang, X. Zhang, G. Wang, N. Qiao, Y. Sun, J. Cheng and Z. Hao, Chem. Eng. J., 2018, 347, 808–818 CrossRef CAS.
- S. Bagheri, N. Muhd Julkapli and S. Bee Abd Hamid, Sci. World J., 2014, 2014, 1–21 CrossRef PubMed.
- T. Barakat, J. C. Rooke, M. Franco, R. Cousin, J. F. Lamonier, J. M. Giraudon, B. L. Su and S. Siffert, Eur. J. Inorg. Chem., 2012, 2012, 2812–2818 CrossRef CAS.
- T. Barakat, J. C. Rooke, H. L. Tidahy, M. Hosseini, J. F. Cousin, J. M. Lamonier, G. Giraudon, B. L. De Weireld and S. Su, ChemSusChem, 2011, 4, 1420–1430 CrossRef CAS PubMed.
- P. Qiao, S. Xu, D. Zhang, R. Li, S. Zou, J. Liu, W. Yi, J. Li and J. Fan, Chem. Commun., 2014, 50, 11713–11716 RSC.
- E. W. McFarland and H. Metiu, Chem. Rev., 2013, 113, 4391–4427 CrossRef CAS PubMed.
- S. Grundner, M. A. Markovits, G. Li, M. Tromp, E. A. Pidko, E. J. Hensen, A. Jentys, M. Sanchez-Sanchez and J. A. Lercher, Nat. Commun., 2015, 6, 7546 CrossRef PubMed.
- J. Ren, K. H. Song, Z. Li, Q. Wang, J. Li, Y. Wang, D. Li and C. K. Kim, Appl. Surf. Sci., 2018, 456, 174–183 CrossRef CAS.
- K. Bhattacharyya, G. P. Mane, V. Rane, A. K. Tripathi and A. K. Tyagi, J. Phys. Chem. C, 2021, 125, 1793–1810 CrossRef CAS.
- V. Krishnakumar, S. Boobas, J. Jayaprakash, M. Rajaboopathi, B. Han and M. Louhi-Kultanen, J. Mater. Sci.: Mater. Electron., 2016, 27, 7438–7447 CrossRef CAS.
- G. Córdoba, M. Viniegra, J. L. Fierro, J. Padilla and R. Arroyo, J. Solid State Chem., 1998, 138, 1–6 CrossRef.
- M. Ikram, E. Umar, A. Raza, A. Haider, S. Naz, D. A. Ul-Hamid, J. Haider, I. Shahzadi, J. Hassan and S. Ali, RSC Adv., 2020, 10, 24215–24233 RSC.
- S. K. S. Patel, N. S. Gajbhiye and S. K. Date, J. Alloys Compd., 2011, 509, S427–S430 CrossRef CAS.
- H. Zhu, Z. Qin, W. Shan, W. Shen and J. Wang, J. Catal., 2004, 225, 267–277 CrossRef CAS.
- S. Yuan, P. Mériaudeau and V. Perrichon, Appl. Catal., B, 1994, 3, 319–333 CrossRef CAS.
- I. S. Petrik, G. V. Krylova, O. O. Kelyp, L. V. Lutsenko, N. P. Smirnova and L. P. Oleksenko, Himia, Fizika ta Tehnologia Poverhni, 2015, 6, 179–189 CrossRef.
- M. Liu, X. Qiu, K. Hashimoto and M. Miyauchi, J. Mater. Chem. A, 2014, 2, 13571–13579 RSC.
- J. Navas, A. Sánchez-Coronilla, T. Aguilar, N. C. Hernández, M. Desireé, J. Sánchez-Márquez, D. Zorrilla, C. Fernández-Lorenzo, R. Alcántara and J. Martín-Calleja, Phys. Chem. Chem. Phys., 2014, 16, 3835–3845 RSC.
- B. Choudhury, M. Dey and A. Choudhury, Int. Nano Lett., 2013, 3, 1–8 CrossRef CAS.
- L. Díaz, V. D. Rodríguez, M. González-Rodríguez, E. Rodríguez-Castellón, M. Algarra, P. Núñez and E. Moretti, Inorg. Chem. Front., 2021, 8, 3491–3500 RSC.
- Q. Ying, Y. Liu, Y. Zhang and Z. Wu, Catal. Sci. Technol., 2021, 11, 2280–2291 RSC.
- B. T. Loveless, A. Gyanani and D. S. Muggli, Appl. Catal., B, 2008, 84, 591–597 CrossRef CAS.
- A. P. Kulkarni and D. S. Muggli, Appl. Catal., A, 2006, 302, 274–282 CrossRef CAS.
- L. K. Noda, R. M. de Almeida, N. S. Gonçalves, L. F. D. Probst and O. Sala, Catal. Today, 2003, 85, 69–74 CrossRef CAS.
- M. R. D'Oliveira, J. Rabelo, A. G. Veiga, C. A. Chagas and M. Schmal, Catal. Lett., 2020, 150, 3036–3048 CrossRef.
- H. Takano, Y. Kirihata, K. Izumiya, N. Kumagai, H. Habazaki and K. Hashimoto, Appl. Surf. Sci., 2016, 388, 653–663 CrossRef CAS.
- L. Liliana, N. Yigit, R. Rameshan, E. Kolar, D. Teschner, M. Hävecker, A. Knop-Gericke, R. Schlögl, K. Föttinger and G. Rupprechter, ACS Catal., 2018, 9, 8630–8641 Search PubMed.
- J. C. Mora, Y. C. M. Nederstigt, J. M. Hill and S. Ponnurangam, J. Phys. Chem. C, 2021, 125, 14299–14310 CrossRef CAS.
- A. Bielański and J. Haber, Catal. Rev.: Sci. Eng., 1979, 19, 1–41 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Mandeep
Sharma
a,
Mandeep
Sharma