
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in externally controlled ring-opening polymerisations
Sandeep
Kaler
* and
Matthew D.
Jones
 *
*
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: mj205@bath.ac.uk; sk2372@bath.ac.uk
First published on 10th December 2021
Abstract
Switchable catalysis is a powerful tool in the polymer chemist's toolbox as it allows on demand access to a variety of polymer architectures. Switchable catalysts operate by the generation of a species which is chemically distinct in behaviour and structure to the precursor. This difference in catalytic activity has been exploited to allow spatiotemporal control over polymerisations in the synthesis of (co)polymers. Although switchable methodologies have been applied to other polymerisation mechanisms for quite some time, for ring opening polymerisation (ROP) reactions it is a relatively young area of research. Despite its infancy, the field is accelerating rapidly. Here, we review recent developments for selected external stimuli for ROP, including redox chemistry, light, allosteric and mechanical control. Furthermore, a brief review on switch catalysis involving exogeneous gases will also be provided, although this area differs from traditional switchable catalysis techniques. An outlook on the future of switchable catalysis is also provided.
1. Introduction
Switchable catalysis involves the use of an external stimulus to switch between multiple states of a single species allowing enhanced control of a system.1–3 Precisely controlled polymer synthesis to generate materials with highly regular structures remains a formidable challenge for polymer chemists. The preparation of well-defined polymers is dependent on a high degree of control since even slight changes to microstructural features i.e., tacticity, chain sequence and topology, can have a profound influence on the properties of these materials.4,5 Switchable catalysis is a viable solution to some of the challenges faced in this area and for this reason it has been expanding rapidly over the past two decades. There have been reports of several methods that allow spatial and temporal regulation to be achieved including light,6–9 electrical current,10,11 redox control,12–15 and mechanical control.16 Switchable catalysis offers the possibility of achieving control over activity and regio-, chemo-, or -stereoselectivities by the incorporation of stimulus responsive units into the catalyst structure. An ideal switchable system includes at least two reversible forms of the molecule which respond to an external stimulus, fast response times, thermal stability, and fatigue resistance. Traditionally, these catalysts have been designed to include a distinct active and inactive species which allows the reaction to be toggled between an “on” or “off” state. Externally controlled polymerisation methodologies could facilitate one-pot reactions featuring several substrates for the synthesis of copolymers. More recent developments in the area include tandem catalysis where multiple mechanistic pathways can occur i.e., photocontrolled ROP and radical polymerisations.172. Recent advances in selected external stimuli
2.1 Redox control
Redox controlled ROP systems have attracted considerable attention since the first report by Gibson and Long in 2006,18 whereby a redox switchable titanium salen bis(isopropoxide) complex (1ox/red) was used to alter the rate of ROP of rac-lactide (Scheme 1). Redox switchable catalysts can be designed so that the redox responsive unit is incorporated into the ligand backbone, which is distant to the catalytically active metal centre, or alternatively a change in oxidation state can occur on the active metal centre itself. Reports of redox switchable polymerisation catalysts have also increasingly been complemented with theoretical studies to unravel complex mechanistic and structural aspects, therefore some of the recent advances in computational studies will also be reviewed.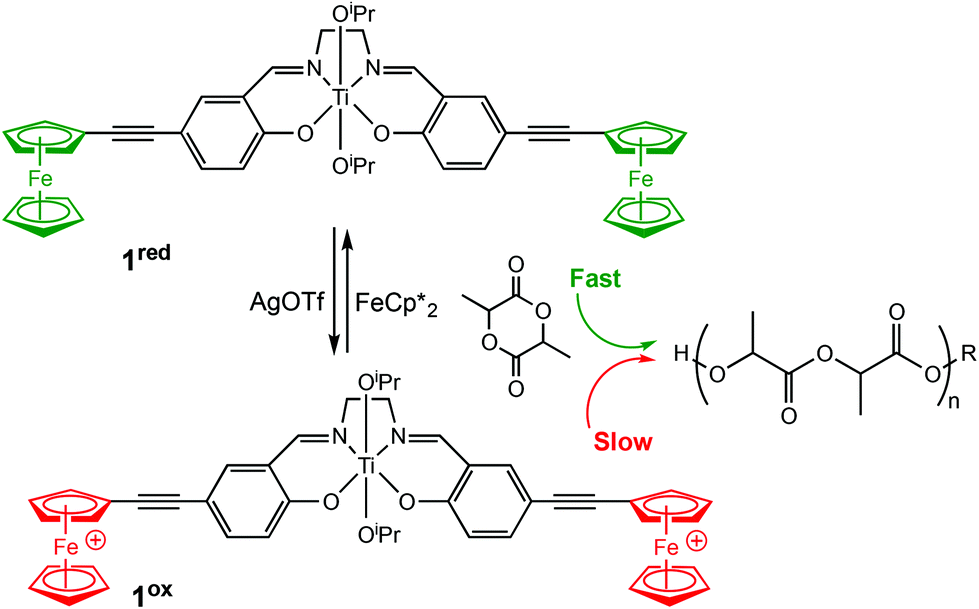 | ||
| Scheme 1 Switchable ROP of rac-lactide by a titanium salen bis(isopropoxide) complex with the reduced form 1red and the oxidised form 1ox.18 | ||
Redox “non-innocent” ligands have garnered popularity over the past decade since altering the electronic properties of the ligand can be used to control the reactivity of the metal centre i.e., change in electron density and Lewis acidity.19,20 In the context of ROP, ferrocene has undoubtedly been the most vastly utilised redox moiety, as is highlighted in a comprehensive review by Diaconescu and coworkers.21
Diaconescu et al. reported the first redox-switchable copolymerisation for L-lactide (L-LA) and ε-caprolactone (CL) with a titanium(IV) catalyst in 2014.22 A series of group IV catalysts were synthesised and characterised (Scheme 2). The reduced forms (2a-cred) exhibited higher activity towards L-LA, whereas the oxidised forms (2a-cox) were more active towards CL. For example, 2ared polymerised 90% L-LA in 2 h, whilst the oxidised form, 2aox afforded <5% conversion in the same period. A reverse activity trend was observed for CL with <5% conversion in 24 h, compared to 98% for the reduced form. The switching behaviour of the zirconium complexes could not be extended to the formation of block copolymers because the polymerisation of CL did not occur when L-LA was initially polymerised. This was presumably due to the strong coordination of the LA monomer to the metal centre. However, the less electrophilic titanium-based complex allowed formation of a diblock copolymer of L-LA and CL, although the activity was low.
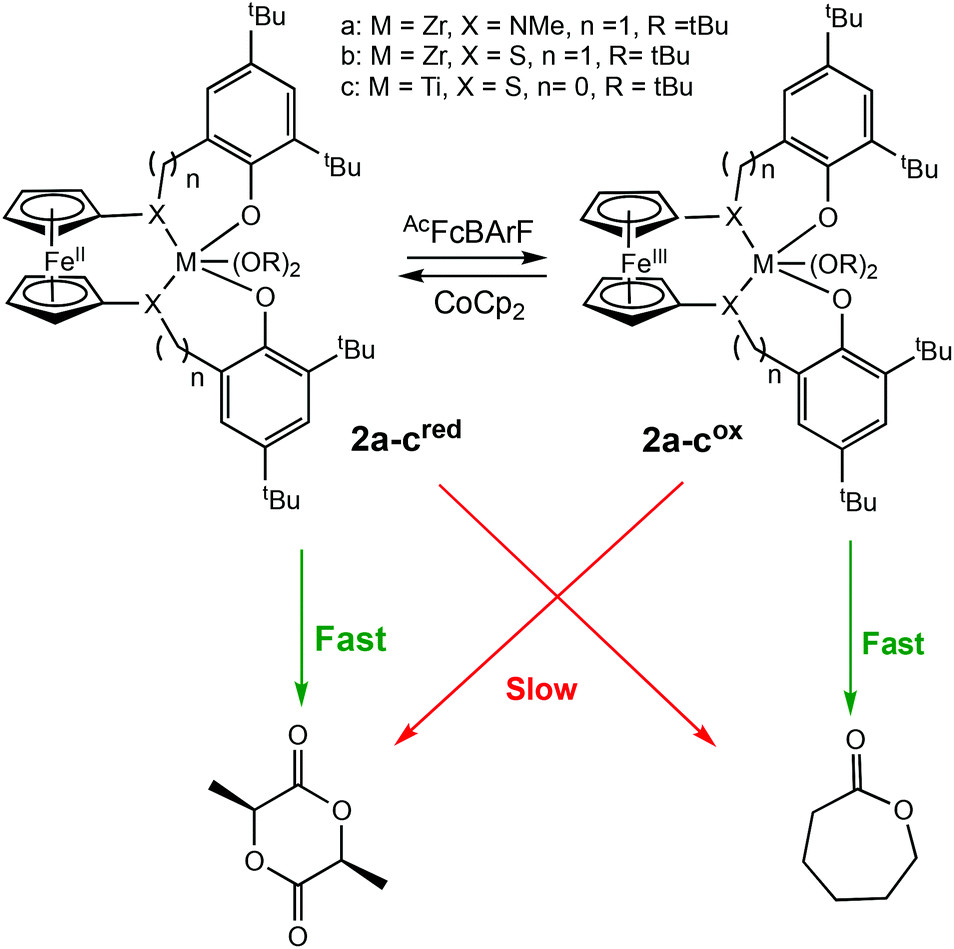 | ||
| Scheme 2 A redox switchable zirconium complex for ROP of L-LA and CL.22 The reduced forms 2a-cred displaying higher activity in LA ROP compared to CL ROP and a reversed trend observed for 2a-cox. | ||
A copolymerisation study followed in 2016 where the polymerisation of several cyclic esters and epoxides was investigated using (salfan)Zr(OtBu)2 (2ared) (salfan = 1,1′-di(2-tert-butyl-6-N-methylmethylenephenoxy)ferrocene) to form diblock and triblock copolymers.12 In 2017, a mechanistic study of the zirconium (2a-cred/ox) complex was reported using density functional theory (DFT) to probe the cyclohexene oxide (CHO) polymerisation mechanism and also the influence of one monomer over another in copolymerisations.23
In 2019, Diaconescu and coworkers, reported the redox activity of a zirconium complex bearing a fully conjugated ligand, (salfen)Zr(OiPr)2 (salfen = N,N′-bis(2,4di-tert-butylphenoxy)-1,1′-ferrocenediimine) (3red/ox) (Scheme 3).24 For LA polymerisation, the reduced form of the complex was able to convert 71% of the monomer, whereas the oxidised form afforded <3% conversion. For the polymerisation of epoxides, the reduced form was inactive in the homopolymerisations of cyclohexene oxide (CHO) and propylene oxide (PO), whereas the oxidised form converted 100% of CHO in 0.2 h. Although a direct comparison between (salfan)Zr(OtBu)2 (2ared/ox) and (salfen)Zr(OiPr)2 (3red/ox) is difficult due to different alkoxide groups,22 (salfan)Zr(OtBu)2 displayed superior activity in LA homopolymerisation, whilst [(salfen)Zr(OiPr)2][BArF] was faster for epoxide homopolymerisations. Furthermore, the redox switch was used to successfully synthesise PLA-PCHO block copolymer.
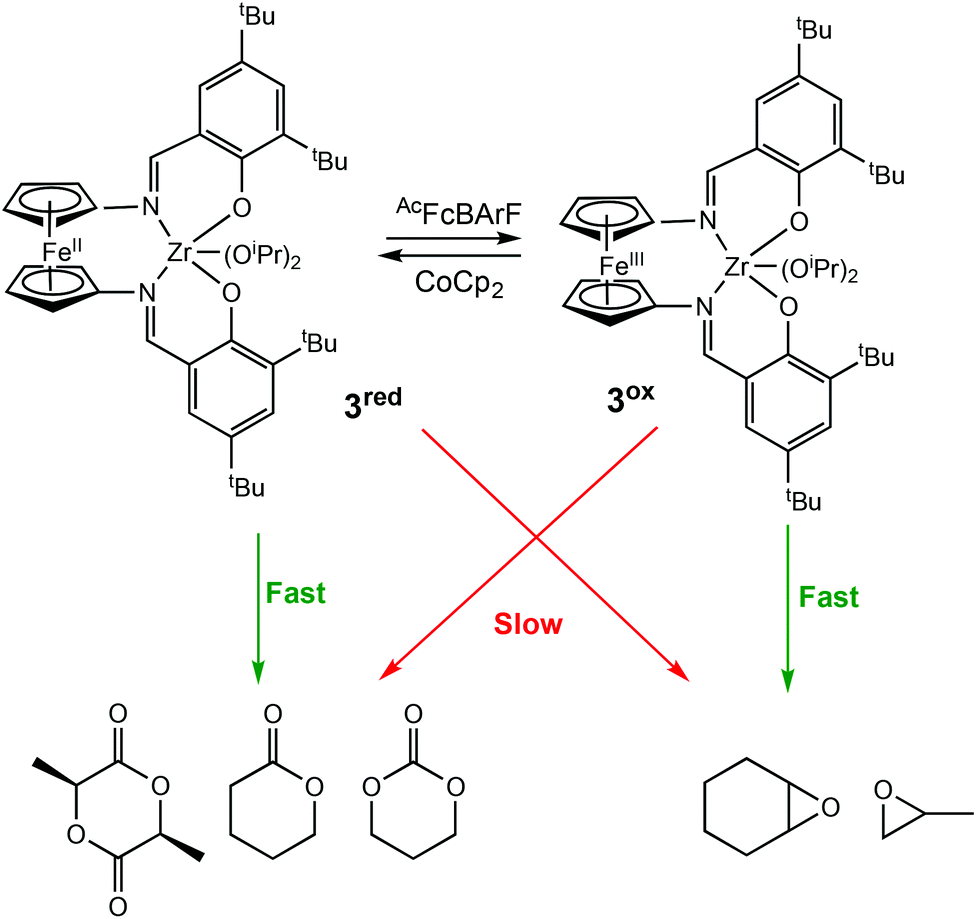 | ||
| Scheme 3 A zirconium complex bearing a fully conjugated ligand for the preferential ROP of several cyclic esters and epoxides.24 | ||
Aluminium complexes for ROP and their mechanistic studies are widely reported in the literature,25–29 however this metal remains underutilised in redox switchable catalysis.
In 2017, Diaconescu et al. reported a combined experimental and computational study of a redox switchable aluminium complex (Scheme 4) (4red/ox), (thiolfan*)Al(OtBu) (thiolfan = 1,1′-di(2,4-di-tert-butyl-6-thiophenoxy)ferrocene).30 The polymerisation of several cyclic monomers including L-LA, CL, δ-valerolactone (VL) and trimethylene carbonate (TMC) were investigated using both the reduced and oxidised species. For L-LA, the reduced form (4red) afforded 88% conversion after 24 h at 70 °C, whilst the oxidised form (4ox) displayed <5% conversion at 100 °C in 24 h. For CL, VL and TMC, both the reduced and oxidised forms were capable of polymerisation and the differences in activity were less pronounced compared to that of L-LA. DFT studies also revealed that chelation of the monomer plays a key role in polymerisation. Moreover, L-LA is more easily controlled than other monomers because the propagation is usually much slower than initiation.
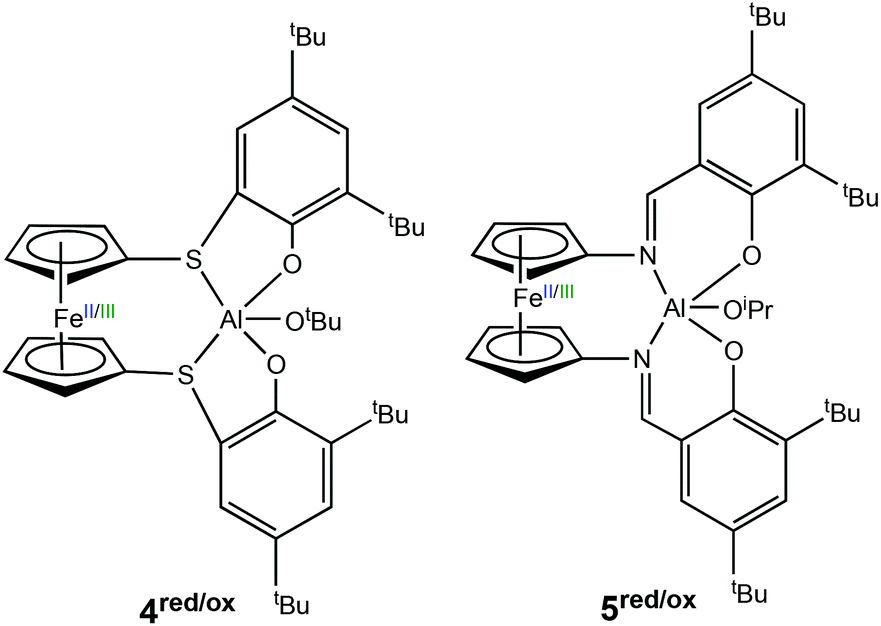 | ||
| Scheme 4 Redox switchable aluminium complexes reported for ROP reported by Diaconescu and coworkers.13,30 | ||
Following the findings of a computational study,31 Diaconescu and coworkers recognised that ferrocene supported aluminium complexes were virtually unexplored compared to other metal analogues. Subsequently, the group reported a redox switchable Al(III) iso-propoxide complex (5red/ox) for ROP.13 The reduced state was active towards ROP of LA, TMC and β-butyrolactone, whilst the in situ oxidised form was active towards CHO. The reduced species could convert 64% of LA in 24 h, whilst the oxidised form was inactive. On the other hand, no activity was observed in CHO polymerisation using the reduced species after 24 h at 70 °C, however the oxidised form converted 99% of 200 equivalents of CHO in 6 minutes at room temperature. For some cyclic esters, including CL and VL, there was no selectivity between reduced and oxidised states. Although the incorporation of TMC in the presence of LA by metal complexes has proven to be difficult,32,33 the group attempted AB and ABC-type copolymers of LA, CHO and TMC. It is important to add that the incorporation of CHO in this case was not redox controlled. A triblock copolymer of (PTMC/PLA)-PCHO-PLA-OiPr was synthesised, as well as a diblock copolymer of PTMC-PLA-OiPr which took 15 days to prepare.
In 2021, Fors and co-workers reported temporal control over the cationic ROP of cyclic esters by ferrocene-derived acid catalysts.34 The acidity of a ferrocenyl unit could be adjusted by changes to the oxidation state which was exploited in switchable ROP. The ferrocenyl acids were inactive for ROP of ε-CL, however the addition of an oxidant (AgBF4) efficiently initiated ROP, whilst the addition of a chemical reductant (CoCp2) could be used to halt the polymerisation and achieve switching.
The ability to switch between multiple catalytically active states i.e., more than one or two oxidation states of the same catalyst, is highly advantageous since it could facilitate the polymerisation of several monomers in a one pot system. Stepwise oxidation to form three redox active species has been previously reported for olefin polymerisations,35 however this approach has only been reported very recently for ROP reactions.36
Diaconescu and coworkers have described a redox-switchable ferrocene containing dimeric yttrium complex, which displayed three oxidation states, the first of its kind in ROP reactions (Scheme 5).36 Based on cyclic voltammetry experiments and NMR studies, the authors proposed two distinct oxidised species, a mono-oxidised [(salfen)Y(OPh)]2+ (6ox1), and a doubly oxidised [(salfen)Y(OPh)]22+ (6ox2), generated by stepwise oxidation. The reduced form, [(salfen)Y(OPh)]2 (6red) showed the highest activity in the ROP of L-LA with 73% conversion in 30 minutes. The three states of the dimeric yttrium complex were also investigated in copolymerisation of L-LA and TMC. It had previously been demonstrated that the use of a dimeric zinc complex lowered the high insertion barrier associated in the formation of multiblock copolymers of LLA-TMC.37,38 Copolymerisation was carried out by the sequential addition of each monomer. The addition of L-LA first resulted in 55% conversion, after which time 1 equiv. of the oxidant was introduced to generate the mono-oxidised species, followed by addition of TMC. After 24 h, complete conversion of L-LA was observed, and 53% conversion of TMC, resulting in a tapered block copolymer.
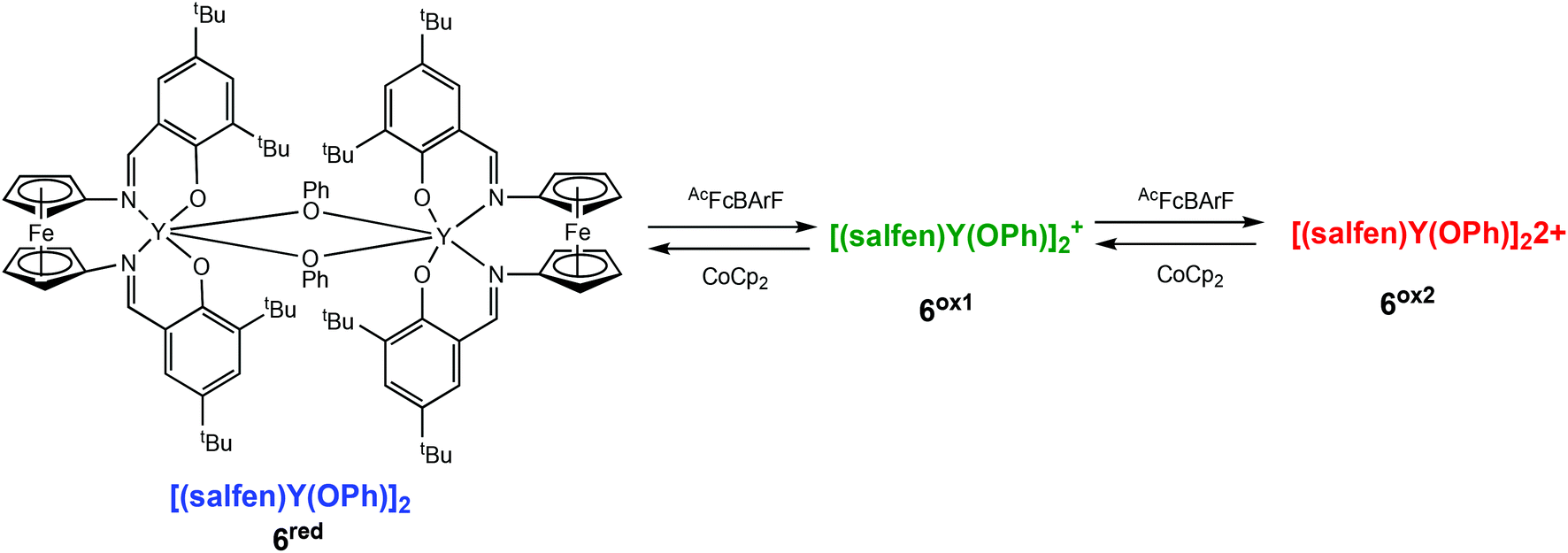 | ||
| Scheme 5 A redox switchable dimeric yttrium complex displaying three oxidation states upon stepwise oxidation leading to a mono-oxidised (6ox1) and a doubly oxidised species (6ox2).36 | ||
Most of the reports of redox switchable catalysts have involved redox control of the ligand, as is demonstrated with ferrocene derivatives. Reports of systems where redox control of the metal centre are not as widespread, even though these types of catalysts would permit a wider range of ligands to be used.
Diaconescu and co-workers described the first redox switchable ROP catalyst system where the change in oxidation state occurred on the metal rather than on the ligand.39 A cerium complex bearing tetradentate salen-type ligands could be switched between Ce(III) and Ce(IV) to control the rate of L-LA polymerisation. The neutral species polymerised 29% of L-LA within 15 min at ambient temperature after which the addition of an oxidising agent ceased activity. Okuda and co-workers have also reported a cerium-based system with [OSSO]-type ligands where switching between Ce(III) and Ce(IV) was used to control the ROP of meso-LA.40
Iron based redox systems form the basis of many biological pathways and chemists designing synthetic systems have sought inspiration from nature.41,42 Iron-based ROP catalysts are a highly attractive option due to their low cost, earth abundance, relatively low toxicity, and most importantly their wide range of redox potentials, which can be easily tuned with the appropriate choice of ligand.43
The Byers group has long been interested in redox switchable ROP iron catalysts.14,15,44,45 In 2013, they reported the first such catalyst for LA polymerisation.14 The oxidised species iron(III) bis(imino)pyridine bisalkoxide complex was completely inactive towards the ROP of LA and could therefore be used to effectively activate and deactivate the polymerisation, whilst retaining molecular weight control through redox switching events.
In 2016, the same group extended the use of iron bis(imino)pyridine bisalkoxide complexes to other monomers to form block copolymers.44 Generally, oxidised systems are more active towards CHO polymerisation compared to the reduced states.12,23,30,46 A cationic iron(III) complex (7ox) displayed activity towards epoxides, whereas the neutral iron(II) complexes (7red) were inactive (Scheme 6). An opposite reactivity trend was observed for LA polymerisation. The redox switchable behaviour in ROP of CHO showed that activity ceased upon addition of CoCp2 and resumed upon addition of an oxidising agent (Fig. 1). This change in chemoselectivity induced by the oxidation state of the catalyst could be used to form block copolymers of CHO and LA.
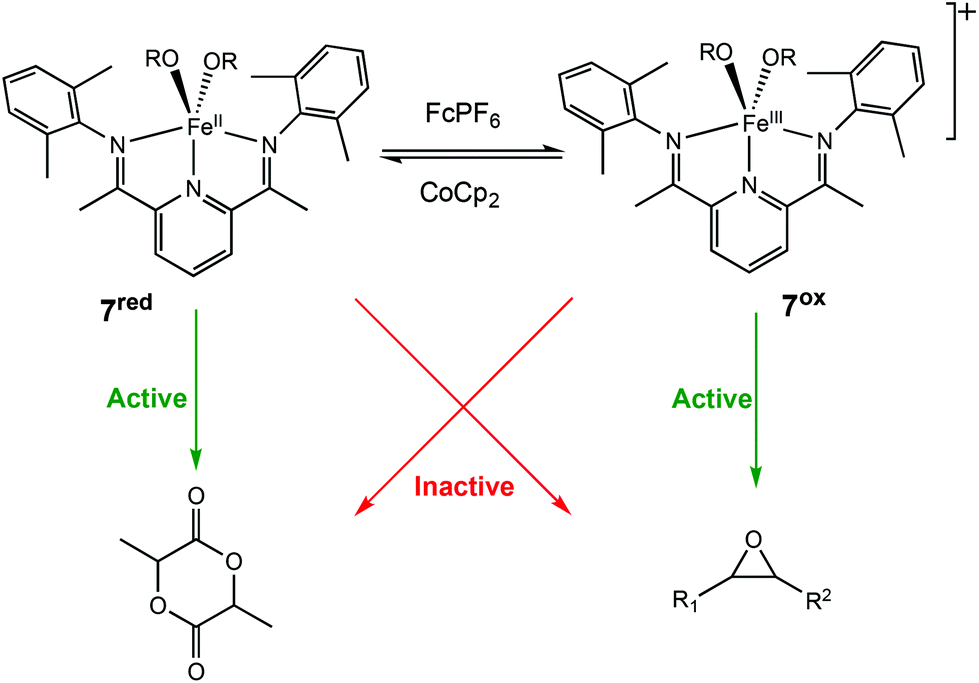 | ||
| Scheme 6 Redox switchable Iron(II)/(III) complexes for ROP of LA and CHO.44 | ||
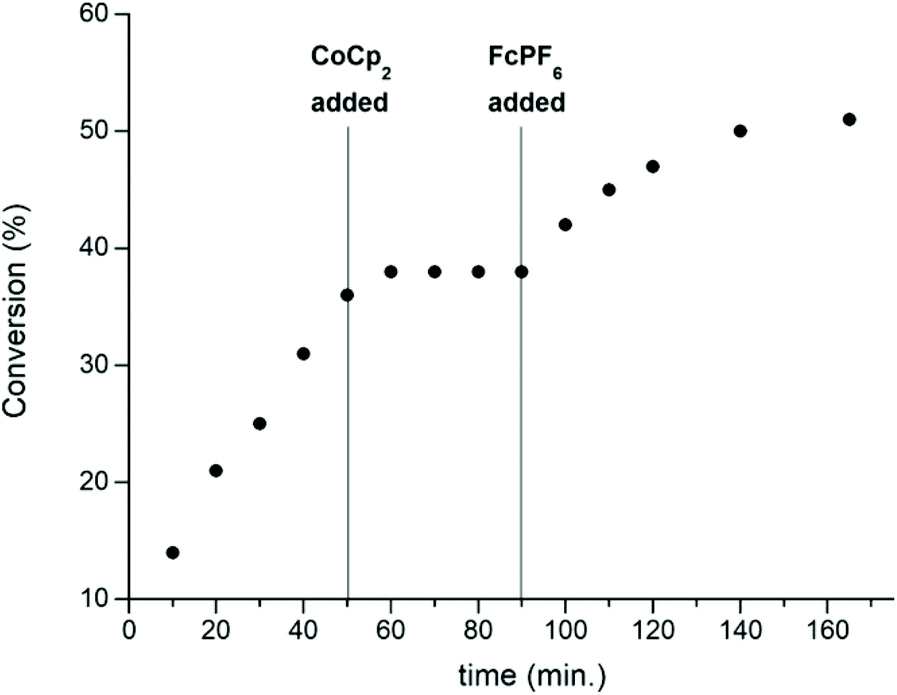 | ||
| Fig. 1 A plot of conversion vs. time for the polymerisation of CHO using 7red/ox (2 mol%). [Copyright (2016) Wiley. Used with permission from ref. 44]. | ||
The Byers’ group also described the synthesis of poly(lactic acid) (PLA) cross-linked polymers in 2016, using redox switchable iron bis(imino)pyridine alkoxide complexes (Fig. 2).45 Cross-linking PLA usually requires impractical methods involving high energy light or electron-beam irradiation.47 ROP of LA occurred with the iron(II) complex, with oxidation to the iron(III) complex triggering CHO polymerisation, resulting in chemical cross-linking of the polymer.
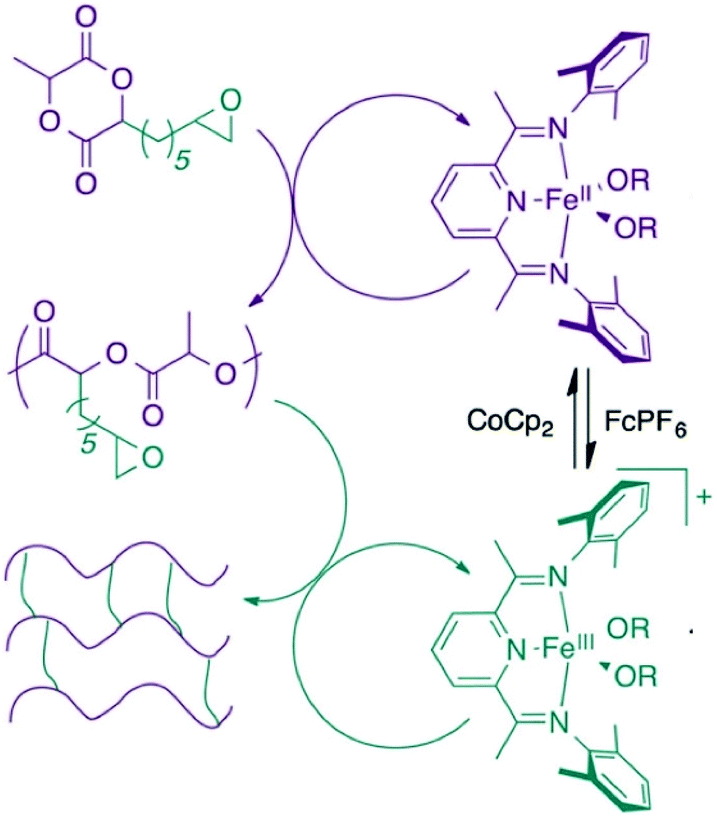 | ||
| Fig. 2 Cross-linked PLA polymer produced from a redox-switchable iron bis(imino)pyridine alkoxide complex.45 Reproduced from ref. 45 with permission from the Royal Society of Chemistry. | ||
There are several factors which govern the polymerisation activity of redox switchable catalysts including supporting ligands, metal centre, and oxidation state. For that reason, several groups have turned their efforts towards studies focusing on structure and mechanism, to rationalise and design experiments.
In 2018, Byers et al. reported a combined experimental and density functional theory (DFT) study of iron alkoxide catalysts bearing bis(imino)pyridine ligands for ROP of lactones.48 The influence of the alkoxide initiator, spin state and oxidation state on polymerisation activity was investigated. The initiation step of ROP was computed to investigate the influence of spin state which showed that high spin complexes were found to be more Lewis acidic than low spin complexes, and therefore had a greater propensity to initiate the polymerisation. Furthermore, the overall reactivity of a particular system is influenced largely by coordination of the monomer.
Structural changes to redox switchable catalysts based on ferrocene derivatives have been reported by Fey, Diaconescu and co-workers.31 A map of chemical space was generated featuring a total of 64 complexes containing a ferrocene ligand with varying metal centres, ferrocene oxidation states and ligand structure. Put succinctly, structural changes in the redox switchable catalysts studied were more greatly impacted by variation of the metal centre and the ligand structure than by oxidation state changes on ferrocene, in agreement with some of the literature already presented in this review. Most of the computed regions have already been reported, with complexes such as (salfen)Zr(OiPr)2 and (salfen)In(OtBu).24,49 Surprisingly, reports of redox switchable aluminium complexes are not as widespread, and hence inspired Diaconescu and coworkers to investigate a novel aluminium redox switchable complex.13
In 2020, Long and coworkers investigated the effects of metal centre and number of redox active units on catalytic performance and switchability.50 The catalytic activity of previously reported symmetrical titanium catalysts bearing two redox moieties (1red/ox),18 and newly synthesised non-symmetrical analogues bearing [ONN] ligands with one redox moiety were compared (Scheme 7). The ability of the catalyst to differentiate catalytic activity as a function of ligand-based oxidation state was not largely impacted by the number of ferrocene units, however overall polymerisation rates are more likely to be impacted by number of ferrocene units and ligand denticity. The switchability of zirconium complex (8bred/ox) was evaluated in the ROP of L-LA by the addition of oxidation and reducing agents at specific points (Fig. 3). 8bred yielded 30% monomer conversion after 2.5 h, after which the oxidised species (8box) was generated in situ and no increase in conversion was detected. It is also suggested that the metal centre has a drastic effect on catalytic activity, stability and switchability. The differentiation in catalytic activity of zirconium complex 8b was around double that of the Ti analogue 8a.
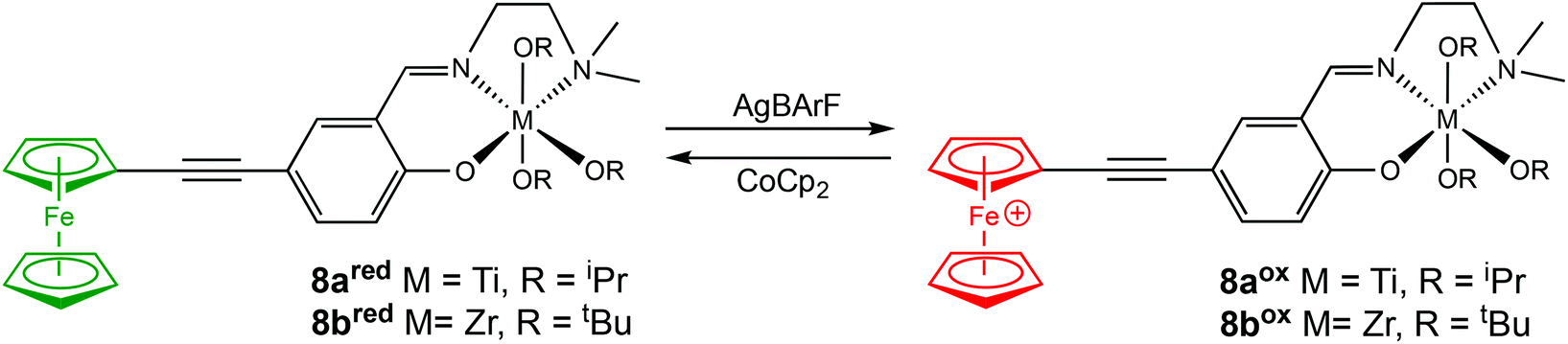 | ||
| Scheme 7 Non-symmetrical redox switchable [ONN]-type group IV metal complexes bearing no ferrocene units (7a/b) and one ferrocene unit (8a/bred/ox).50 | ||
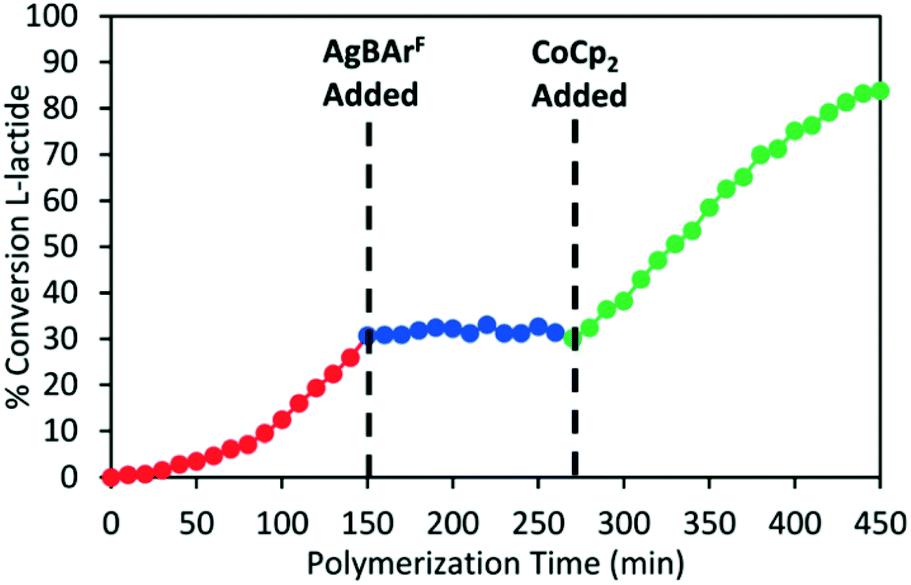 | ||
| Fig. 3 Time vs. conversion plot of redox switchable ROP of L-LA using Zr complex 8bred/ox.50 The oxidised species 8box was generated in situ by addition of AgBArF (blue circles) and the reduced species was regenerated by CoCp2 (green circles). Reproduced from ref. 50 with permission from the Royal Society of Chemistry. | ||
Luo, Luo, Diaconescu and coworkers described a theoretical study of redox switchable group IV complexes bearing [OSSO]-type ligands for CL polymerisation.51 A series of complexes with similar ligand frameworks but differing metal centres were investigated (Fig. 4). The oxidised and reduced forms of these complexes had previously been shown to display differences in catalytic activity in CL polymerisation.12,23,46,52 DFT and experimental results indicated that a higher Lewis acidity at the titanium metal centre accounts for the higher activity of the oxidised species (9ox). The higher energy barrier of the oxidised form of the Zr complex (10ox) is caused by higher stability of the monomer coordination intermediates compared to the reactants. The effect of metal centre Lewis acidity and ligand framework was computationally modelled using a new set of [OSSO]-type bis(phenolato) ferrocene-based group IV complexes (12/13red/ox). It was proposed that a hafnium analogue (12red/ox) may be a more superior redox switchable catalyst than the zirconium analogue (10red/ox). Also, switching the donors in the supporting ligand from S to Se (13red/ox) presented a larger energy barrier difference between the reduced and oxidised states which could translate to greater differentiation in catalytic activity between the two forms.
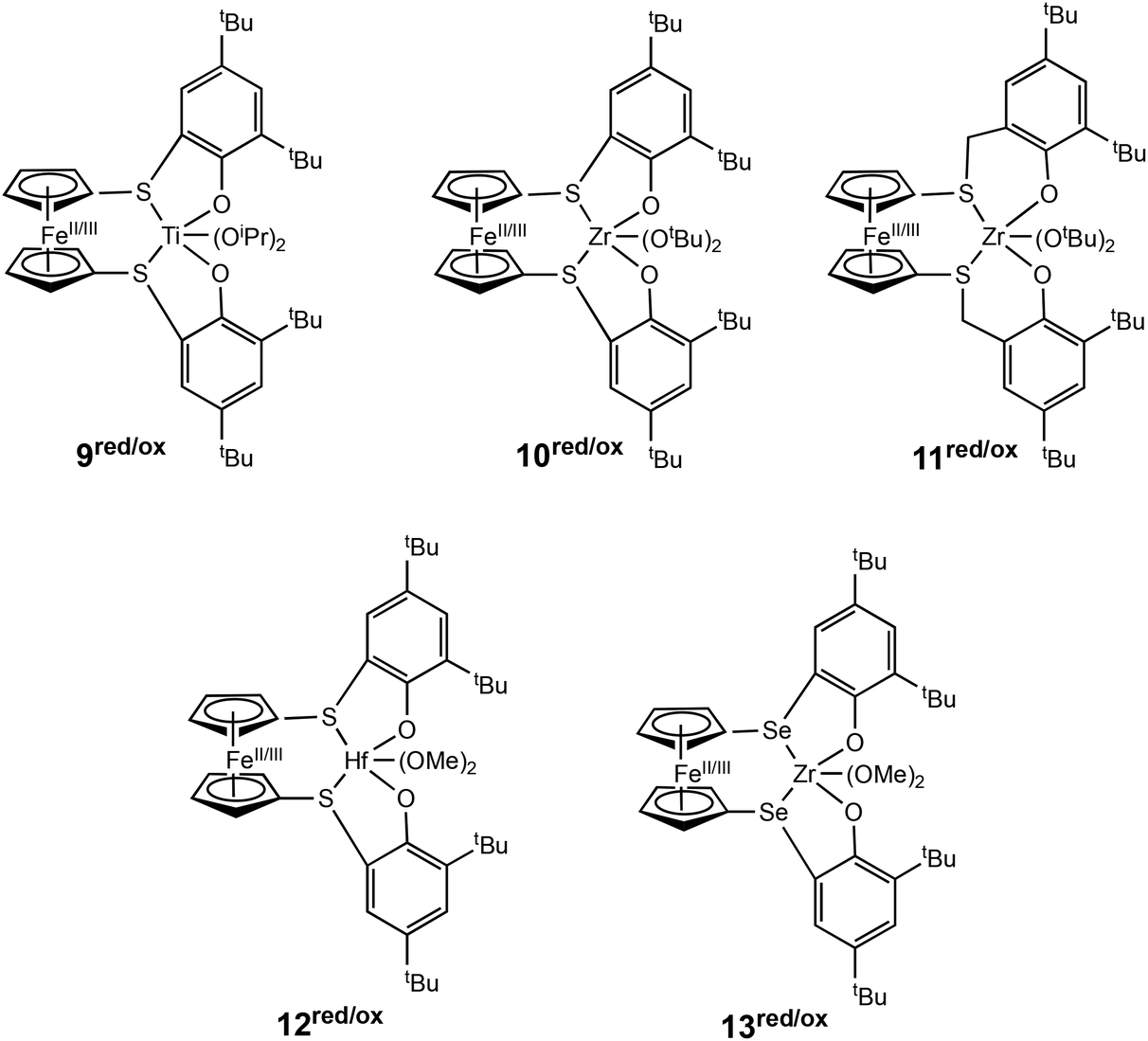 | ||
| Fig. 4 Group IV metal catalysts investigated experimentally and computationally for CL polymerisation (9–11red/ox).51 A series of new complexes computationally modelled as superior catalysts for ROP of CL (12–13red/ox). | ||
Luo et al. reported DFT investigations of the opposite reactivity of iron bis(imino)pyridine complexes towards CHO and LA.53 The energy profiles of the reduced and oxidised forms showed little difference in the free energies and transition states of the reduced/oxidised complexes until formation of a tetrahedral complex following alkoxide insertion, at which point the reduced form is more stable compared to the oxidised complex. Further analysis of the structures and free energies showed that the CHO monomer is more tightly bound to the metal centre on the transition state of the oxidised form compared to the reduced form. Furthermore, CHO is more sensitive towards the Lewis acidity of the metal centre compared to LA, due to its greater nucleophilicity.
These theoretical studies provide valuable insight into redox switching mechanisms, as well as structural factors affecting catalytic activity and redox switchability i.e., metal centre, auxiliary ligand, oxidation states and spin states, which should be taken into consideration when designing new redox switchable systems for ROP.
2.2 Electrochemical control
Redox switchable systems have traditionally involved the addition of oxidising and reducing agents which present several drawbacks including environmental problems and difficulties in upscaling. The application of electrochemistry helps to alleviate some of the challenges presented in using stoichiometric amounts of redox agents.54 Furthermore, electrochemistry has wider functional group tolerance and removes the need for extra purification steps associated with redox agents.In this context, Byers et al. reported the first redox switchable ROP system using electrochemistry to supply the redox equivalents required for switching in ROP of LA and CHO using a bis(imino)pyridine alkoxide complex (Scheme 8).11 Application of a reducing potential was used to trigger LA polymerisation with 14red, whereas the reverse redox process was capable of polymerising CHO with the generation of 14ox. This switching ability was further used to form 88% block copolymer, with the other 12% being homopolyether. The polymerisation of LA could be stopped as soon as the redox state was switched, with the ROP of epoxide commencing immediately.
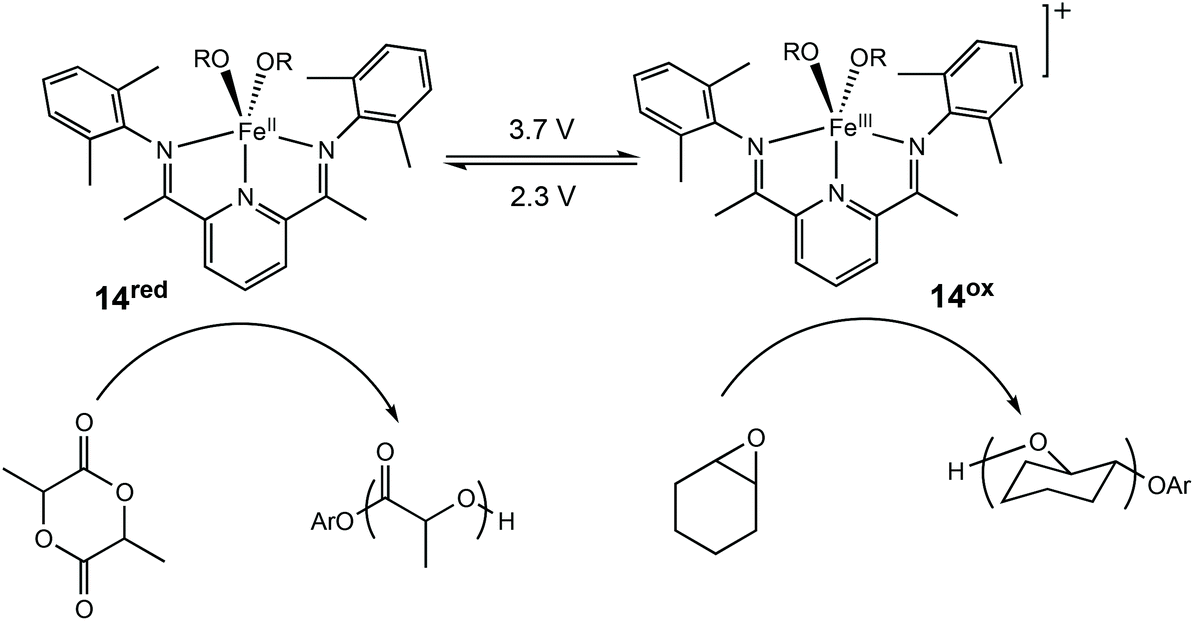 | ||
| Scheme 8 Electrochemically switchable ROP of CHO and LA with a bis(imino)pyridine iron alkoxide complex.11,14 | ||
Similarly, in 2020 Tong and co-workers reported a stereoselective Co/Zn catalytic system for electrochemical ROP of O-carboxyanhydrides to prepare high molecular weight polymers (>140 kDa).10 Remarkably, they also demonstrated an electrochemically stereoselective polymerisation of racemic O-carboxyanhydrides by modulation of the ligands on the metal centre making these catalysts either isoselective or syndioselective in the formation of block copolymers.
In 2021, Byers et al. reported the ROP of LA and epoxides to produce surface-initiated polymer brushes appended on to solid state surfaces using a redox controlled process (Fig. 5).55 Research interest into polymer brushes has flourished over the last two decades due to their mechanical and chemical robustness and ease of functionalisation,56–59 however reports of surface initiated ROP of cyclic esters are uncommon.60,61 Byers et al. used their previously reported bis(imino)pyridine iron bisalkoxide complexes14,44,62 supported on semi-conducting surfaces to produce a hybrid system, operating under both homogeneous and heterogeneous catalysis conditions. Deposition of the functionalised nanoparticles on fluorine doped tin oxide, which acted as the conductive surface, resulted in a system which could be electrochemically controlled. Iron(II) complexes were active towards LA, although at slower rates than the fully homogenous system previously reported,14 but still displaying characteristics of a living polymerisation. The addition of oxidising agent, FcPF6, produced iron(III) functionalised nanoparticles, which were completely inactive towards LA but reactive towards CHO, affording 33% conversion overnight.
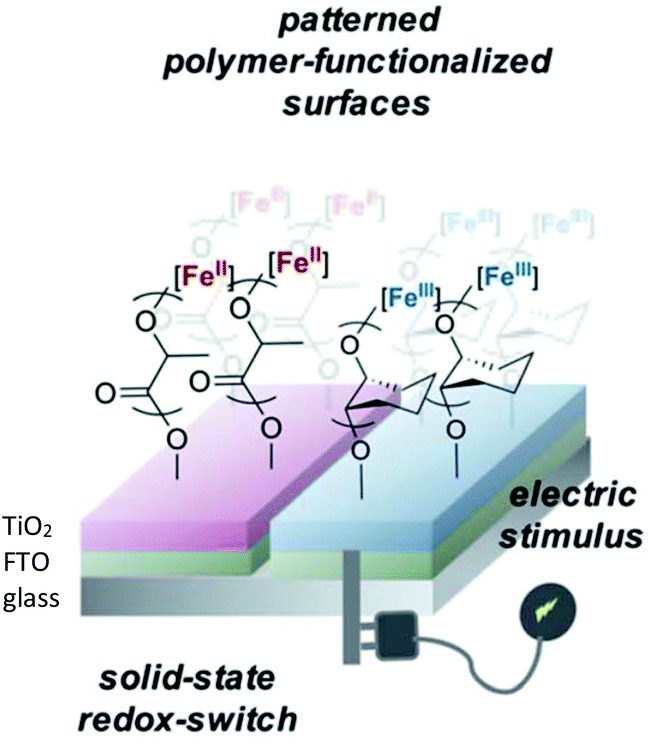 | ||
| Fig. 5 Surface anchored bis(imino)pyridine iron catalysts for ROP allowing patterning of surfaces with polymer brushes through application of an electrical stimulus.55 Reproduced from ref. 55 with permission from the Royal Society of Chemistry. | ||
2.3 Mechanochemical control
Although mechanochemical control offers many advantages in the way of green chemistry, for polymerisation processes it is a rather unconventional stimulus. The application of mechanical force in the production of polymers is generally viewed as a destructive approach as excessive force on polymeric materials can cause degradation and impedes access to high molecular weight materials. To our knowledge, only one report for mechanochemically controlled ROP of cyclic esters exists in the literature which was reported by Kim et al. in 2017.16 Polylactic acid (PLA) was synthesised by a solvent-free ball-milling procedure using the organocatalyst DBU (1,8-diazabicylo[5.4.0]undec-7-ene). Unfortunately, after some time Mn growth levelled out as the process became destructive to the polymer and resulted in degradation. Following this, liquid-assisted grinding was investigated, whereby a small amount of solvent was added to the reaction to enable sufficient energy dissipation and overcome the destructive nature of solvent-free ball-milling. After 2 hours of milling with toluene, high molecular weight poly(D-lactic acid) (Mn = 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) was prepared.
000 g mol−1) was prepared.
2.4 Allosteric control
Allosteric regulation is evident in many biological functions including protein activity modulation.63,64 The term allosteric translates to ‘other-space’ in Greek and is fittingly used for this mechanism because effectors operate by binding to a site other than the active site which can induce conformational changes capable of directly modulating activity.63Allosteric control has been applied to switchable ROP by Mirkin and coworkers in 2010 with a cleverly designed triple-layer catalyst consisting of Rh(I)Cl side arms with Cl− playing the role of the effector (Scheme 9).65 In the presence of Cl−, the active metal centre of the complex was accessible, however removal of Cl− anions from the Rh(I) centres enabled chelation of the phosphinoamine ligands. The aromatic groups of the phospinoamine moiety π-stacked above and below the Al(III) centre resulting in a “closed” form. The “semi-open” form (15o) and “closed” form (15c) of the catalyst exhibited different activity towards the ROP of ε-CL, with the closed form (15c) remaining inactive for the first 100 hours. On the other hand, the “semi-open” form (15o) was active and produced well-defined polymers with narrow dispersities (Đ = 1.10–1.20).
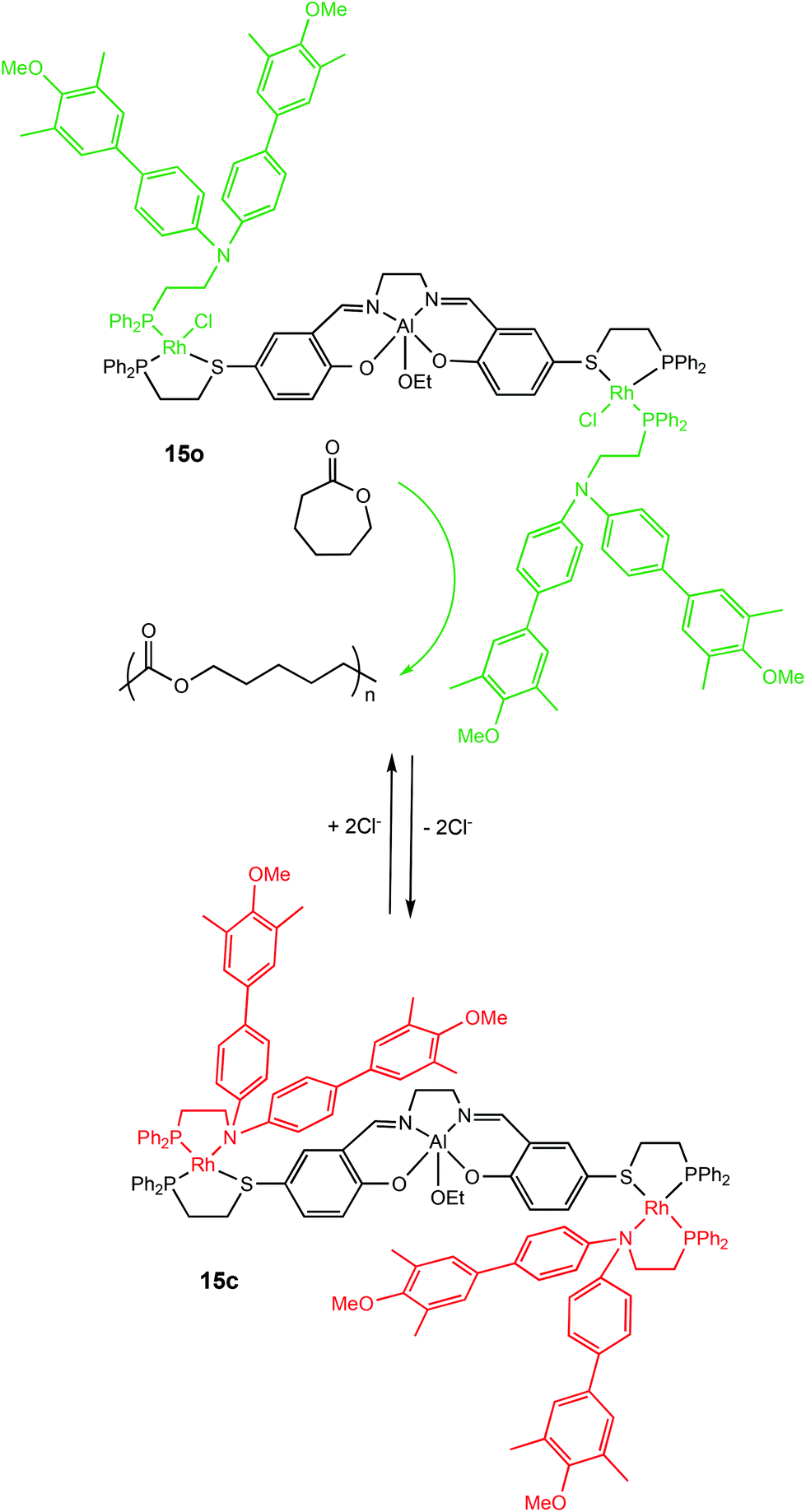 | ||
| Scheme 9 Allosteric control of a triple-layer Al(III)-salen catalyst for ROP of CL.65 | ||
In 2016, the same group briefly described the ROP of L-LA as proof of principle for a heteroligated Pt(II) complex containing both a hemilabile squaramide–piperidine-based catalytic ligand and a sodium sulfonate hydrogen-bond-accepting ligand, which could be toggled between a semi-open state and a fully closed state.66 The semi-open form was inactive towards ROP of L-LA, whereas the fully closed form afforded quantitative conversion.
2.5 Photochemical control
The use of light as an external trigger to control a reaction presents many advantages over other stimuli and provides many of the necessary characteristics of an ideal system, such as its non-invasive nature, easy handling, and the ability to exert high levels of control by simple manipulation of wavelength or intensity of the light source. The benefits of light have been manifested across both heterogeneous and homogeneous catalysis for a wide array of chemical transformations,67–70 however the field of light controlled ROP reactions has only recently started to gain momentum.In 2013, Bielawski and coworkers reported a photoswitchable organocatalyst for the ROP of cyclic esters (Scheme 10).71 Prior to this, reports of photocontrolled polymerisation processes were limited mostly to other processes, such as atom-transfer radical polymerisations.72–75 A photochromic dithienylethene (DTE)-annulated N-heterocyclic carbene (NHC) precatalyst could be switched reversibly between a ring-opened (16o) and ring-closed (16c) state by UV/Vis irradiation. Irradiation of the active ring-opened form with UV light (λ = 313 nm) generated the inactive ring-closed form. The ring opened form polymerised 95% of 100 eq. of CL at room temperature. In contrast, the increased conjugation present in the backbone of the ring-closed form resulted in an electron-deficient carbenoid centre which was inactive towards CL (<5% conversion). The photoswitchability of the catalyst was investigated in a series of light on–off experiments showing a highly responsive system (Fig. 6).
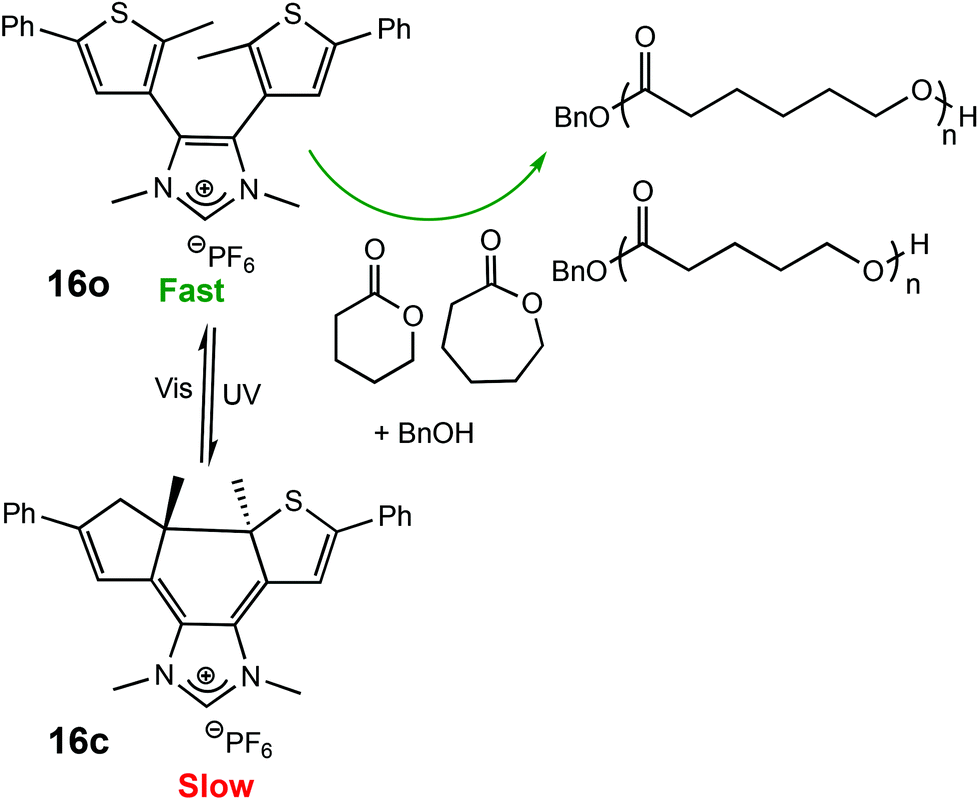 | ||
| Scheme 10 Photocyclisation of a DTE-annulated NHC ROP catalyst which generated a ring-closed inactive species upon irradiation with UV light.71 | ||
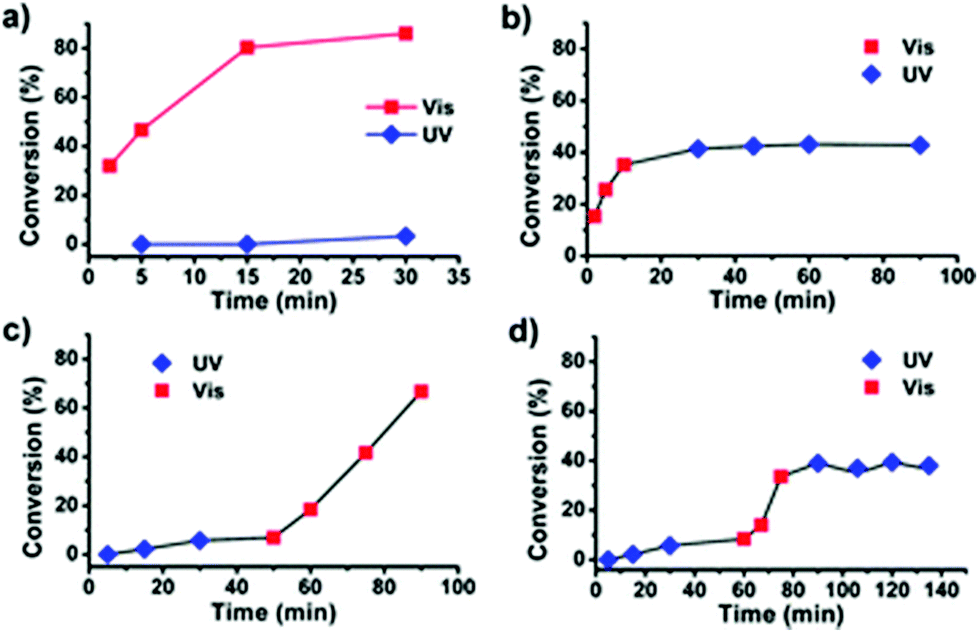 | ||
| Fig. 6 Time vs. conversion plots for light-on off experiments using 16o/c for ROP of VL.71 (a) Two concurrent reactions, one under visible light and another under UV light. (b) Reaction proceeded in visible light for 10 minutes before UV exposure. (c) Reaction proceeded under UV light for 60 minutes before exposure to visible light. (d) Reaction proceeded under UV light for 60 minutes before irradiation with visible light for 45 minutes, and UV light for the remainder of the run. Reproduced from ref. 71 with permission from the Royal Society of Chemistry. | ||
In 2018, Hecht et al. described a photoswitchable organocatalyst for the ROP of L-LA, as well as the photoregulation of trimethylene carbonate (TMC) and VL copolymerisation.6 A diarylethene functionalised with a phenol moiety could undergo light-controlled keto–enol tautomerism which was used to effectively turn the catalyst “on” and “off” (Scheme 11). The ring opened form containing a phenol moiety (17o) could participate in hydrogen bonding for carbonyl activation of the incoming monomer. Irradiation with UV light generated the metastable ring closed enol form which could thermally tautomerise to form a ketone (17c), incapable of carbonyl activation. ROP of L-LA with the open form resulted in 45% conversion in 24 h with good molecular weight control, whereas the closed form was around 4 times slower in the same period. The use of light as a stimulus is perhaps of most interest for the preparation of copolymers where the desired properties of materials can only be achieved by highly controlled systems. The ring closed form afforded a ratio of 10 and 7 for copolymerised TMC and VL. On the other hand, an inverted pattern of incorporation was observed for the ring open phenol form with 15 vs. 20 units of TMC and VL, respectively. This system described by Hecht et al. was the first report of monomer sequence control using a single catalyst which could be controlled in situ.
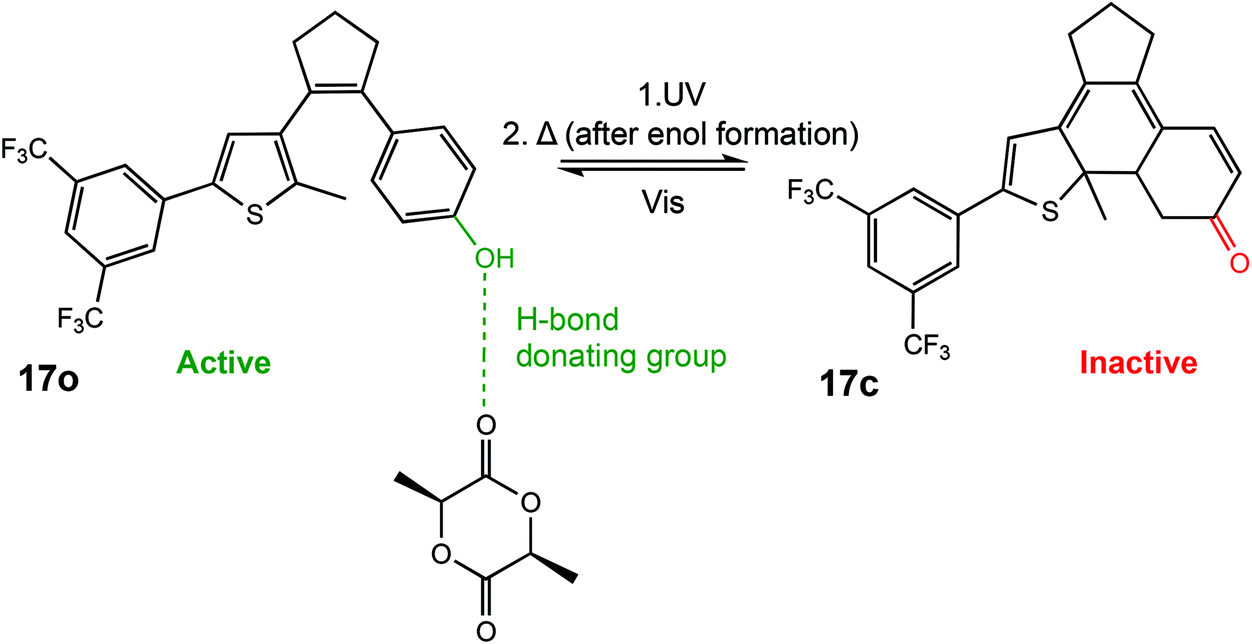 | ||
| Scheme 11 A photoswtichable diarylethene funcionalised with a phenol moiety undergoes cyclization to produce an inactive ketone form incapable of hydrogen bonding in the ROP of L-LA.6 | ||
Photocaged organocatalysis in ROP enhances the possibility of control by allowing the careful release of active species at precise points within a reaction using light as the stimulus.76 The first report of a photocaged organocatalyst was reported by Wang and coworkers in 2008, who generated the salt of 1,5,7-triaza-bicyclo[4.4.0]dec-5-ene (TBD) which was inactive in polymerisations,77 however irradiation with UV light (λ = 254 nm) generated a free base which was active in the ROP of ε-CL.
A few years later, Dove and co-workers reported a triarylsulfonium hexafluorophosphate salt which could undergo rearrangement under UV irradiation (λ = 365 nm) to generate an acid which was active in the cationic polymerisation of several cyclic esters including ε-CL, δ-VL and TMC, however the generated acid was incapable of polymerising LA.78
In 2018, the same group reported a photocaged tetramethyl guanidine capable of polymerising L-LA (Scheme 12).79 Irradiation of 2-(nitrophenyl)propoxycarbonyl-1,1,3,3-tetramethylguanidine (NPPOC-TMG) (18) with 320–400 nm light released TMG (19), an organocatalyst only previously reported for ROP of N-butyl N-carboxyanhydride.80 NPPOC-TMG was inactive towards L-LA, however switching to UV light for 15 minutes released the active TMG species, which converted 90% of the monomer in 3 h. The photoinduced ROP of δ-VL was also attempted with the photocaged catalyst, although the addition of a thiourea cocatalyst was necessary for monomer activation.81 After a period of 76 h, 34% monomer conversion was observed which was considerably slower than using the free TMG base devoid of the photolabile protecting NPPOC group (93% conversion in 23 h).
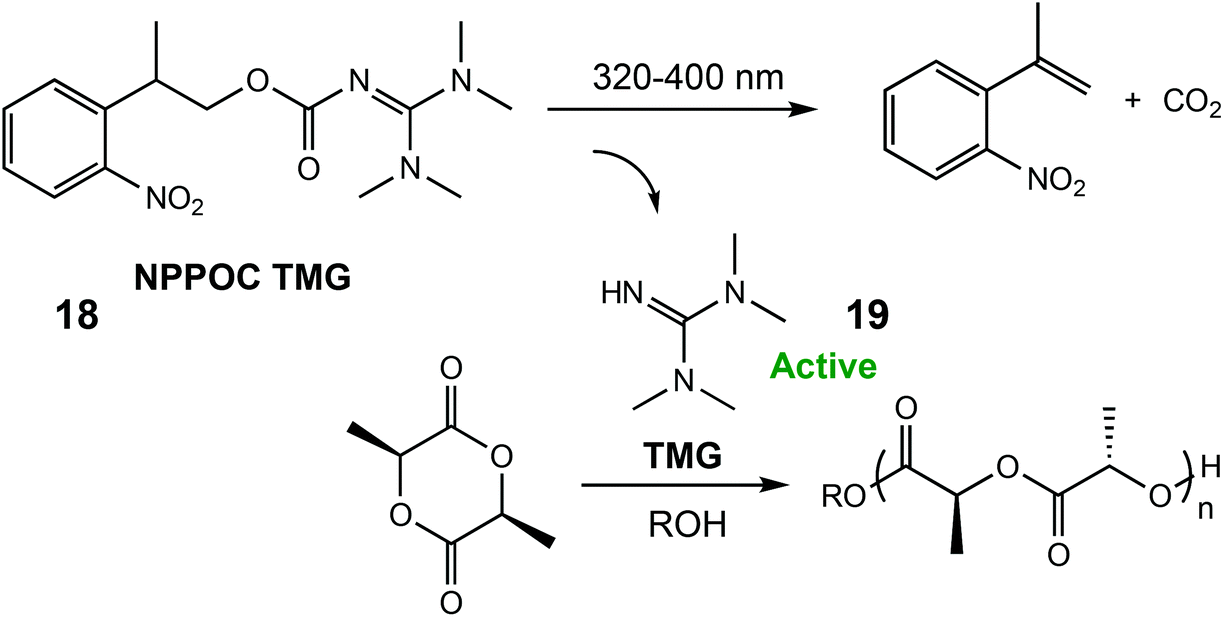 | ||
| Scheme 12 A photocaged tetramethyl guanidine which releases the active species TMG for ROP of L-LA.79 Reproduced from ref. 79 with permission from the Royal Society of Chemistry. | ||
Photoacids are molecules which generate a strong acid upon irradiation with light of an appropriate wavelength and have been used to achieve spatiotemporal control across several disciplines.82
Xu and Boyer have reported a visible-light mediated process for the proton-catalysed ROP of cyclic esters using a photoacid.17 The photoacid (20o) could be reversibly activated by visible light (λ = 460 nm) resulting in cyclisation (20c) and proton dissociation to form a strong acid, which could effectively catalyse the ROP of cyclic esters (Scheme 13). For the ROP of VL, polymerisation under visible light with 50% of the photoacid afforded 88% conversion in 22 h with good molecular weight control (D = 1.18). More interestingly, the light regulated ROP could be used in an orthogonal polymerisation method. Photoinduced electron/energy transfer-reversible addition–fragmentation chain transfer radical (PET-RAFT) catalysed by zinc tetraphenyl porphyrin,83–85 was combined with photo-ROP to selectively polymerise either VL or methacrylate to produce a well-defined PVL-b-PMA block copolymer (Mn 4610 g mol−1, Mw/Mn = 1.15), as well as a graft copolymer (PMA-g-PVL).
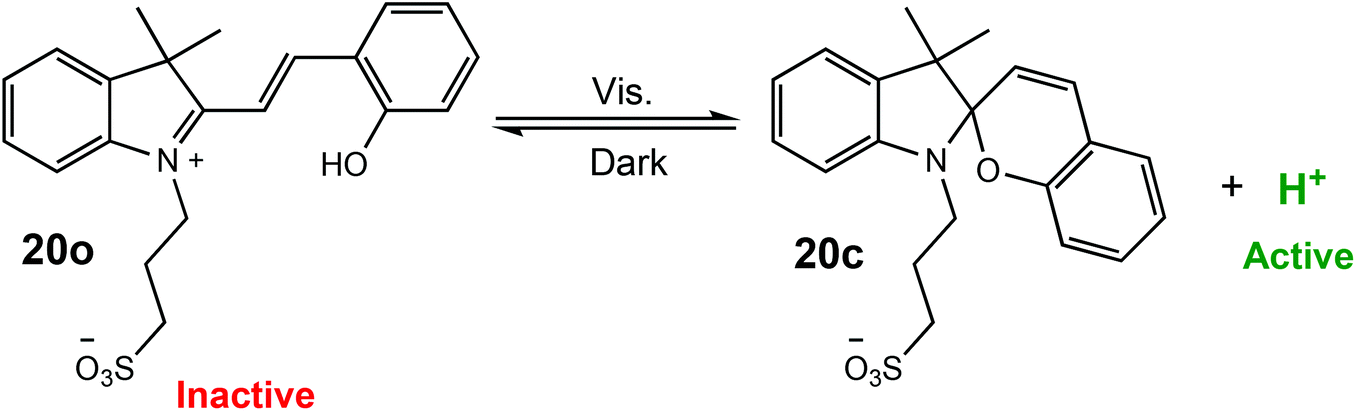 | ||
| Scheme 13 Reversible activation of a photoacid and proton dissociation resulting in the formation of a strong acid capable of effectively catalysing the ROP of cyclic esters.17 | ||
In 2020, You and coworkers developed a series of composite photoacid generators for the living/controlled cationic ROP (CROP) of lactones under visible light.86 The system consisted of a common photocatalyst and an onium salt. Irradiation with light generated radicals from the excited photocatalyst/onium salt which could abstract a proton (H+) from the monomer or solvent. H+ could catalyse the living cationic ring opening polymerisation of lactones. Moreover, the generation of radical species allowed a simultaneous CROP/reversible-addition–fragmentation chain transfer reaction. It is important to note that light was not used to switch between two states once the polymerisation started and light simply acted as a trigger for generating a more active species (H+) which allowed polymerisation to commence.
Similarly, in 2021 Liao and coworkers reported a visible light regulated ROP using an aromatic alcohol as the photocatalyst (Scheme 14).87 The irradiation of aromatic alcohols is known to generate a high-acidity excited state which is more catalytically active.82 The same group had previously explored this area and reported that naphthols were inefficient for this purpose due to weak absorption of visible light in the absence of photosensitisers.88 1-Hydroxypyrene (PyOH) (21) could catalyse the ROP of lactones under visible light without the need for expensive metal based photosensitisers. Irradiation of PyOH increased acidity and activation of the lactone whilst the weaker acid (21) was not active for ROP. The system was tested in a light on–off experiment in the ROP of VL and observed attenuation of catalytic activity once the light source was turned off (Fig. 7). For example, conducting the polymerisation under purple light afforded 23% conversion, which was reduced to 8% once the polymerisation proceeded in the dark.
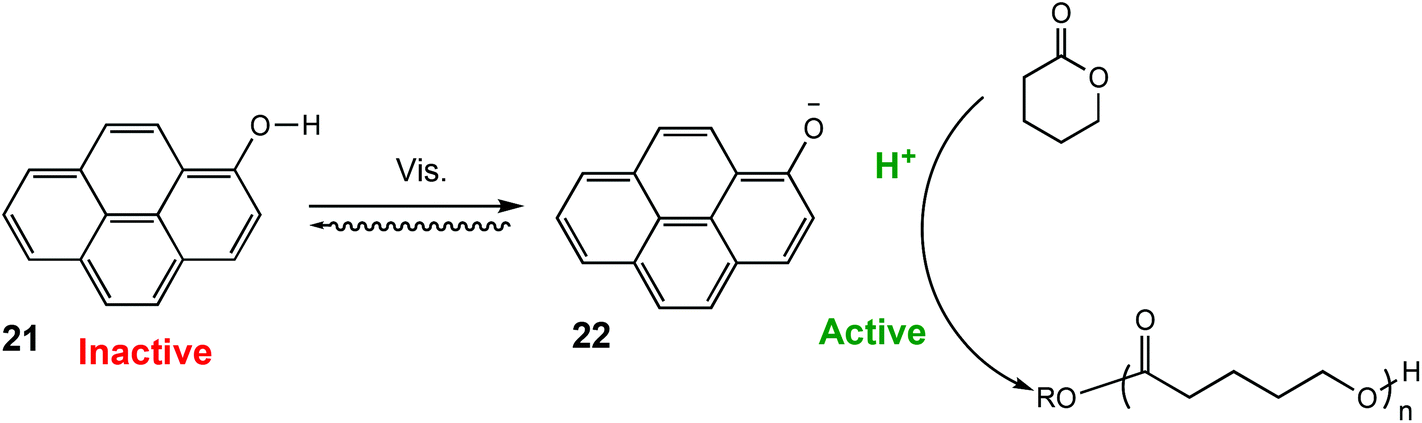 | ||
| Scheme 14 ROP catalysed by PyOH under light to generate a stronger acid.87 | ||
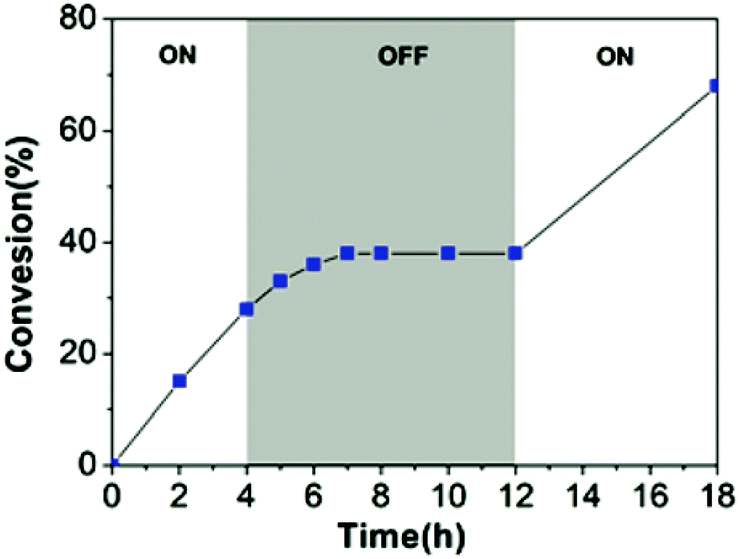 | ||
| Fig. 7 Time vs. conversion plot for ROP of VL in light on–off experiments.87 Reproduced from ref. 87 with permission from the Royal Society of Chemistry. | ||
The reports mentioned so far have focused on light responsive organocatalysts to turn a polymerisation “on” or “off” through the generation of a distinct species which displays either an increase or decrease in activity compared to the native form. Few reports to date have utilised metal complexes for light controlled polymerisations,7,8 even though some of the most active ROP catalysts reported have been metal based.89,90
Photoresponsive metal complexes can be designed so that the chromophore is incorporated into the ligand backbone, similar to a ferrocene derivate in redox switchable catalysts. Azobenzenes present many of the characteristics required for photoswitchable catalysts including high photochemical conversions, reversibility and high fatigue resistance.91 Azobenzenes undergo trans–cis isomerisation upon irradiation with UV light, producing a large geometrical change which can be used to manipulate activity around a metal centre (Scheme 15). The meta stable cis isomer can be converted back to the thermodynamically stable trans isomer, either by irradiation with UV light, or thermally. This reversibility allows easy tunability of a system through the generation of two distinct species and as a result their application has been widespread including in polymeric materials,92–94 molecular machines,95,96 and catalysis.97–99
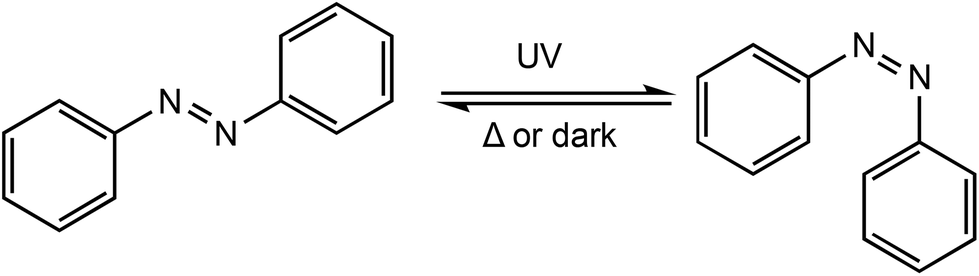 | ||
| Scheme 15 E/Z isomerisation of azobenzenes. | ||
In 2019, Chen and co-workers reported a light controlled ROP method using zinc-based catalysts functionalised with a light responsive azobenzene chromophore (Scheme 16).8 For the ROP of ε-CL, reaction rates up to six times faster were reported for the polymerisations catalysed by the cis isomers. For example, 23-E afforded 22% conversion (ktrans = 1.43 × 10−3 min−1), whilst the cis form 23-Z converted 60% of ε-CL (kcis = 8.22 × 10−3 min−1). A reversed reactivity difference was observed towards LA with higher rate constants for the trans isomers. ROP of TMC, β-oxetanone, VL, cyclopentadecanolide and 5-methyl-5-propyl-1,3-dioxan-2-one also displayed higher reactions rates for cis-Zn–H. The isomers 24E/Z displayed monomer discrimination, for example the amount of CL incorporated in a TMC/CL copolymerisation catalysed by 24-E incorporated 32% ε-CL, whereas 24-Z resulted in 26% incorporation. The initial rate constant for the conversion of TMC (2.73 × 10−3 min−1) and CL (1.89 × 10−3 min−1) under ambient light conditions was similar. Upon switching to UV irradiation, a greater disparity in rate constants was observed, with the rate constant for TMC (6.74 × 10−3 min−1) being much larger than CL (0.99 × 10−3 min−1).
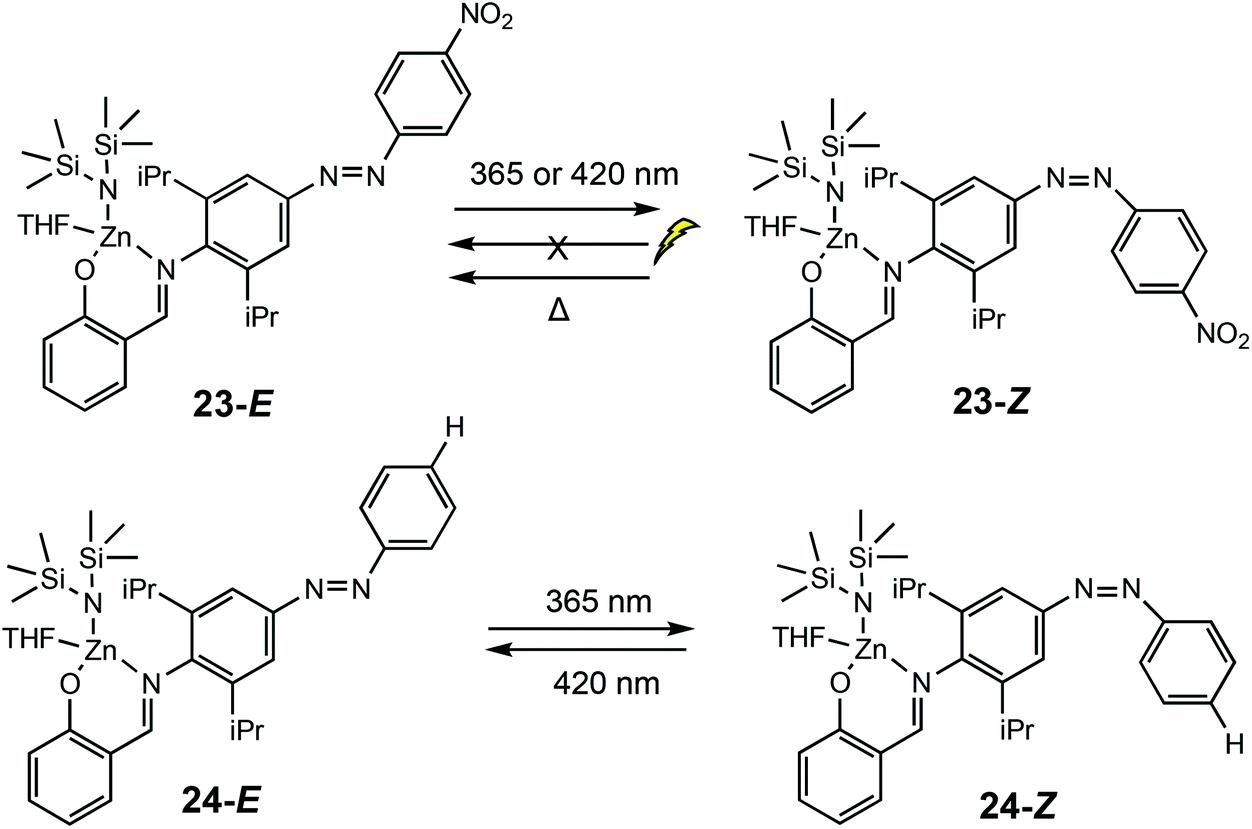 | ||
| Scheme 16 Photoswitchable salicylaldimine Zn(II) complexes which undergo reversible trans–cis isomerisation upon light/thermal treatment.8 | ||
Recently, our group has reported a series of photoresponsive Al(III) and Zn(II) complexes bearing azobenzene Schiff-base ligands for ROP of rac-LA and ε-CL (Scheme 17).7 Al(III) complexes were synthesised to include either one or two azobenzene units. For ROP of CL at room temperature, an increase in activity was observed when the polymerisations were carried out under UV light for all Al(III) complexes (λ = 365 nm), indicating 25–29-Z were the more active isomers. For example, ROP with 25-E under ambient light afforded 33% conversion after 12 h, whereas the 25-Z resulted in 56% conversion in the same period, although with marginally higher than expected molecular weights. ROP of rac-LA did not display any switching due to the requirement of heat which thermally isomerised the cis isomer to the more stable trans isomer. Complexes bearing one azobenzene containing ligand displayed higher activity presumably due to a less sterically congested metal centre.
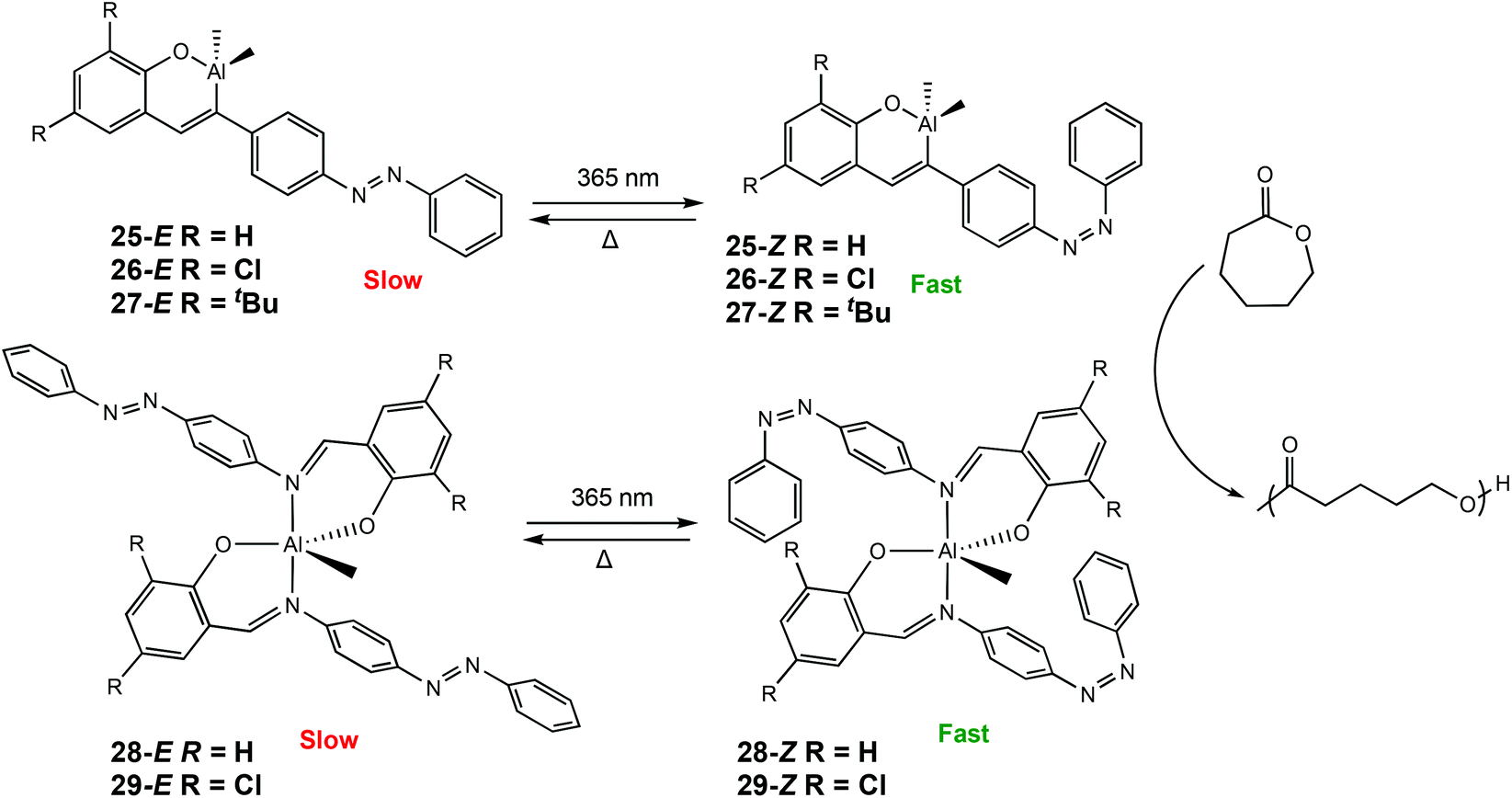 | ||
| Scheme 17 Photoswitchable Al(III) complexes bearing azobenzene Schiff-base ligands which undergo trans–cis isomerisation for ROP of CL.7 | ||
In 2021, Coulembier and coworkers reported a photoresponsive bis(15-crown-5 ether) containing an azobenzene linkage which could acts as a molecular tweezer in the light regulated ROP of L-LA (Fig. 8).100 The same group had previously reported the use of crown ethers to accelerate the ROP of L-LA through the in situ generation of free ions.101 The use of azobenzene as a bridge could allow a controllable system where more active species are generated “on-demand”. Light was used to transform the K+−OAc ion pair-like into free ion catalysts through E/Z isomerisation. The ROP of L-LA which proceeded under UV light resulted in increased polymerisation rates (kfast/kslow ∼ 10).
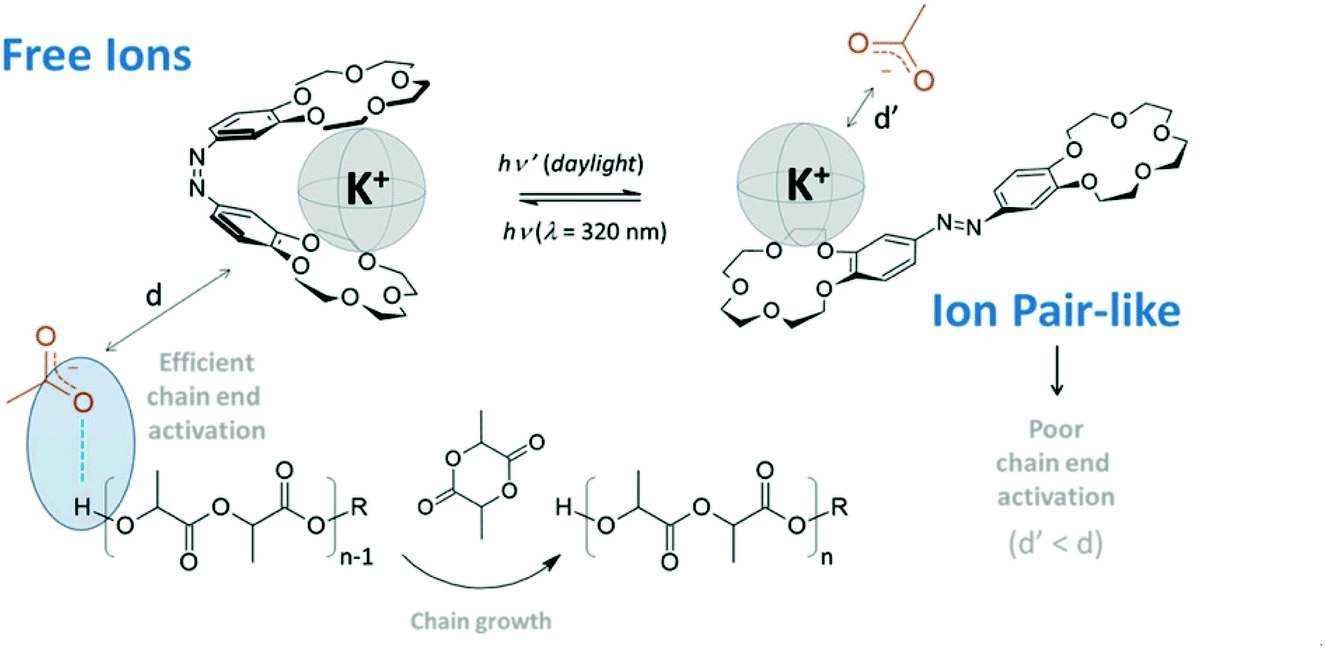 | ||
| Fig. 8 A photoresponsive azobenzene incorporated bis(15-crown-5-ether) which transforms the K+−OAc ion pair like into free ions through a butterfly movement by E/Z isomerisation of the azo linkage resulting in increased polymerisation rates.100 Reproduced from ref. 100 with permission from Royal Society of Chemistry. | ||
2.6 Gas control
Recently gas-controlled systems have gained attention for switchable polymerisation catalysis because they appeal to greener chemistry approaches and can be used to synthesise sequence-controlled block polymers from a mixture of monomers. A key advantage of these systems is the easy removal of exogeneous gases which enables switching between multiple polymerisation pathways. Removal of these exogeneous gases often requires a simple purge of the system, without any further complex purification required. The Williams group have long been interested in switch catalysis;102,103 however, it is important to note that the type of switch catalysis explored here differs from the examples mentioned elsewhere in this review. Here, the term ‘switch’ catalysis refers to switching between multiple catalytic cycles, as opposed to switching between an active and dormant state through the application of an external stimulus. In 2014, Williams and co-workers reported a switchable system based on a dizinc catalyst capable of forming block copolymers from ROCOP of CO2/CHO and ROP of ε-CL (Fig. 9).104 In the presence of CO2, poly(cyclohexene carbonate) was formed, and after removal of CO2 a metal alkoxide-species was generated which catalysed ROP to afford polycaprolactone. This reactivity was explained by the zinc chain end groups controlling selectivity. ROCOP or ROP was catalysed by the zinc alkoxide chain end group, whereas the zinc carboxylate/carbonate chain end group only catalysed ROCOP.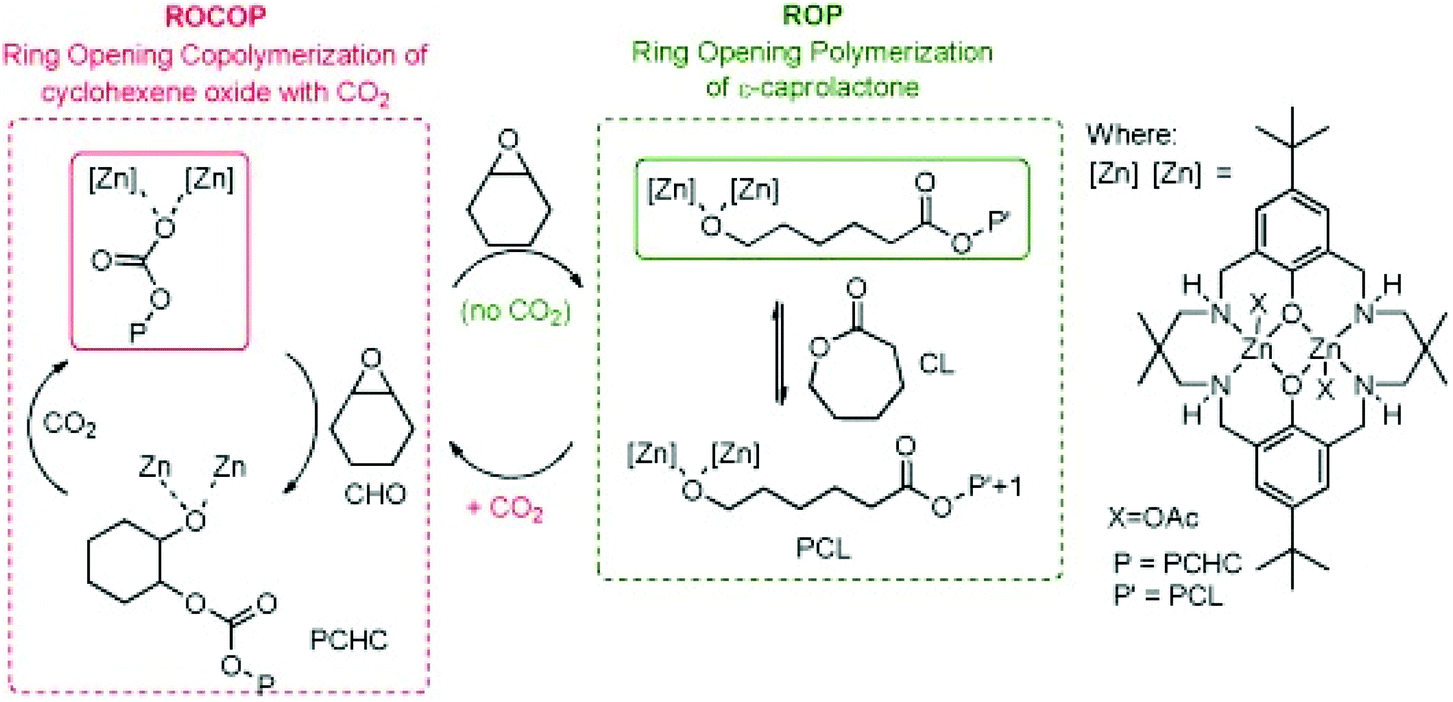 | ||
| Fig. 9 Williams and coworkers switchable dizinc catalyst for ROP and ROCOP. Copyright (2014) Wiley. Used with permission from ref. 104. | ||
Subsequently, ABA triblock co(polyester-b-carbonate-b-ester)s were prepared from CO2/epoxide and lactone using a similar dizinc catalyst.105 The ROCOP of epoxide/CO2 occurs first to produce a polycarbonate polyol, with the removal of CO2 triggering the selective ROP of the lactone. In 2017, Reiger and co-workers reported the pressure controlled terpolymerisation of rac-β-butyrolactone, CHO and CO2 using a Lewis acid BDICF3-Zn-N(SiMe3) catalyst. Adjusting the pressure of CO2 resulted in the activation of different reaction pathways, with the formation of terpolymer with a statistical composition under 3 bar CO2. Switching to 40 bar CO2 afforded copolymerisation of CHO/CO2. In 2021, Williams and coworkers reported a series of Mg(II) heterodinuclear catalysts for the preparation of triblock (ABA), pentablock (BABAB) and heptablock (ABABABA) polymers.106 Of the synthesised catalysts, the heterodinuclear Mg(II)–Co(II) catalyst displayed the highest activity and was used in gas switching experiments to prepare the required multiblock polymers. Three polymerisation pathways could be accessed from a mixture of CO2, anhydride (tricyclic anhydride) and CHO. Recently, the Williams group has exploited switchable polymerisation catalysis to improve the properties of poly(L-lactide) using a commercially relevant Sn(II) alkoxide catalyst.107 A three-step process was used to synthesise tapered block polyesters. The three stages were: (1) propylene oxide (PO)/maleic anhydride (MA) ROCOP, followed by (2) simultaneous L-LA ROP and PO/MA ROCOP, and finally, (3) ROP of L-LA until complete consumption of LA.
3. Prospects
Significant effort has been expended into the area of switchable catalysis since some of the earliest works and the literature presented here clearly indicates that there is great potential in this area. Whilst it is difficult to make direct comparisons between external stimuli, areas such as redox control and photocontrol have forged ahead of others and provide the greatest potential for highly controllable system, however there is still much work needed to fulfil the requirement of systems capable of controlling one-pot polymerisations with several monomers. The infancy of the field means that most reports are based on simply designed experiments merely involving “on/off” or “fast/slow” species, although there is evidence to suggest that research groups in this area are setting themselves more complex objectives e.g., orthogonal polymerisation methods combining multiple mechanisms. The stringent criteria required for switchable catalysts to be highly effective i.e., high conversions, fatigue resistance and a sizeable differentiation in catalytic activity between distinct states, means that the rational design of new catalysts should also consider mechanistic and structural aspects. Gas controlled systems have rapidly advanced over the last decade with highly selective catalysts capable of forming multiblock polymers from mixtures of monomers, and we expect that this area will experience huge growth in the coming years. Theoretical calculations and studies have already proven to be insightful in several areas of switchable catalysis i.e., redox control and have guided several research groups in designing new systems. Given that most of the examples outlined in this review are based on homogeneous catalysis, a more transformative approach may be to bring aspects of heterogeneous catalysis into the mix to create even more robust systems. Future studies should also consider the scalability of switchable catalysts to industrial settings as this is often completely overlooked. Despite the enormous growth displayed in this area, there are still some technical and chemical challenges which need to be addressed. We are convinced that reports covering these future works are likely to be published in the very near future.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We would like to thank the EPSRC for funding for a PhD studentship to SK (EP/L016443/1).References
- S. M. Guillaume, E. Kirillov, Y. Sarazin and J.-F. Carpentier, Chem. – Eur. J., 2015, 21, 7988–8003 CrossRef CAS PubMed.
- F. A. Leibfarth, K. M. Mattson, B. P. Fors, H. A. Collins and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 199–210 CrossRef CAS PubMed.
- A. J. Teator, D. N. Lastovickova and C. W. Bielawski, Chem. Rev., 2016, 116, 1969–1992 CrossRef CAS PubMed.
- N. Badi, D. Chan-Seng and J.-F. Lutz, Macromol. Chem. Phys., 2013, 214, 135–142 CrossRef CAS.
- Y. Yagci and M. A. Tasdelen, Prog. Polym. Sci., 2006, 31, 1133–1170 CrossRef CAS.
- F. Eisenreich, M. Kathan, A. Dallmann, S. P. Ihrig, T. Schwaar, B. M. Schmidt and S. Hecht, Nat. Catal., 2018, 1, 516–522 CrossRef CAS.
- S. Kaler, P. McKeown, B. D. Ward and M. D. Jones, Inorg. Chem. Front., 2021, 8, 711–719 RSC.
- M. Li, P. Zhang and C. Chen, Macromolecules, 2019, 52, 5646–5651 CrossRef CAS.
- B. M. Neilson and C. W. Bielawski, ACS Catal., 2013, 3, 1874–1885 CrossRef CAS.
- Y. Zhong, Q. Feng, X. Wang, J. Chen, W. Cai and R. Tong, ACS Macro Lett., 2020, 9, 1114–1118 CrossRef CAS.
- M. Qi, Q. Dong, D. Wang and J. A. Byers, J. Am. Chem. Soc., 2018, 140, 5686–5690 CrossRef CAS PubMed.
- S. M. Quan, X. Wang, R. Zhang and P. L. Diaconescu, Macromolecules, 2016, 49, 6768–6778 CrossRef CAS.
- A. Lai, Z. C. Hern and P. L. Diaconescu, ChemCatChem, 2019, 11, 4210–4218 CrossRef CAS.
- A. B. Biernesser, B. Li and J. A. Byers, J. Am. Chem. Soc., 2013, 135, 16553–16560 CrossRef CAS PubMed.
- K. R. Delle Chiaie, A. B. Biernesser, M. A. Ortuño, B. Dereli, D. A. Iovan, M. J. T. Wilding, B. Li, C. J. Cramer and J. A. Byers, Dalton Trans., 2017, 46, 12971–12980 RSC.
- N. Ohn, J. Shin, S. S. Kim and J. G. Kim, ChemSusChem, 2017, 10, 3529–3533 CrossRef CAS PubMed.
- C. Fu, J. Xu and C. Boyer, Chem. Commun., 2016, 52, 7126–7129 RSC.
- C. K. A. Gregson, V. C. Gibson, N. J. Long, E. L. Marshall, P. J. Oxford and A. J. P. White, J. Am. Chem. Soc., 2006, 128, 7410–7411 CrossRef CAS PubMed.
- V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- S. Blanchard, E. Derat, M. Desage-El Murr, L. Fensterbank, M. Malacria and V. Mouriès-Mansuy, Eur. J. Inorg. Chem., 2012, 3, 376–389 CrossRef.
- J. Wei and P. L. Diaconescu, Acc. Chem. Res., 2019, 52, 415–424 CrossRef CAS PubMed.
- X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 11264–11267 CrossRef CAS PubMed.
- S. M. Quan, J. Wei and P. L. Diaconescu, Organometallics, 2017, 36, 4451–4457 CrossRef CAS.
- R. Dai and P. L. Diaconescu, Dalton Trans., 2019, 48, 2996–3002 RSC.
- N. Spassky, M. Wisniewski, C. Pluta and A. Le Borgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 CrossRef CAS.
- S. M. Kirk, G. Kociok-Köhn and M. D. Jones, Organometallics, 2016, 35, 3837–3843 CrossRef CAS.
- P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688–2689 CrossRef CAS PubMed.
- F. E. Kohn, J. G. Van Ommen and J. Feijen, Eur. Polym. J., 1983, 19, 1081–1088 CrossRef CAS.
- F. M. García-Valle, V. Tabernero, T. Cuenca, M. E. G. Mosquera, J. Cano and S. Milione, Organometallics, 2018, 37, 837–840 CrossRef.
- J. Wei, M. N. Riffel and P. L. Diaconescu, Macromolecules, 2017, 50, 1847–1861 CrossRef CAS.
- A. Lai, J. Clifton, P. L. Diaconescu and N. Fey, Chem. Commun., 2019, 55, 7021–7024 RSC.
- W. Guerin, M. Helou, J.-F. Carpentier, M. Slawinski, J.-M. Brusson and S. M. Guillaume, Polym. Chem., 2013, 4, 1095–1106 RSC.
- M. Abubekerov, J. Wei, K. R. Swartz, Z. Xie, Q. Pei and P. L. Diaconescu, Chem. Sci., 2018, 9, 2168–2178 RSC.
- M. J. Supej, E. A. McLoughlin, J. H. Hsu and B. P. Fors, Chem. Sci., 2021, 12, 10544–10549 RSC.
- M. Zhao and C. Chen, ACS Catal., 2017, 7, 7490–7494 CrossRef CAS.
- S. Deng and P. L. Diaconescu, Inorg. Chem. Front., 2021, 8, 2088–2096 RSC.
- M. Abubekerov, V. Vlček, J. Wei, M. E. Miehlich, S. M. Quan, K. Meyer, D. Neuhauser and P. L. Diaconescu, iScience, 2018, 7, 120–131 CrossRef CAS PubMed.
- F. Santulli, I. D'Auria, L. Boggioni, S. Losio, M. Proverbio, C. Costabile and M. Mazzeo, Organometallics, 2020, 39, 1213–1220 CrossRef CAS.
- E. M. Broderick, N. Guo, T. Wu, C. S. Vogel, C. Xu, J. Sutter, J. T. Miller, K. Meyer, T. Cantat and P. L. Diaconescu, Chem. Commun., 2011, 47, 9897 RSC.
- A. Sauer, J.-C. Buffet, T. P. Spaniol, H. Nagae, K. Mashima and J. Okuda, ChemCatChem, 2013, 5, 1088–1091 CrossRef CAS.
- S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed.
- P. K. Sasmal, C. N. Streu and E. Meggers, Chem. Commun., 2013, 49, 1581–1587 RSC.
- A. Fürstner, ACS Cent. Sci., 2016, 2, 778–789 CrossRef PubMed.
- A. B. Biernesser, K. R. Delle Chiaie, J. B. Curley and J. A. Byers, Angew. Chem., Int. Ed., 2016, 55, 5251–5254 CrossRef CAS PubMed.
- K. R. Delle Chiaie, L. M. Yablon, A. B. Biernesser, G. R. Michalowski, A. W. Sudyn and J. A. Byers, Polym. Chem., 2016, 7, 4675–4681 RSC.
- M. Y. Lowe, S. Shu, S. M. Quan and P. L. Diaconescu, Inorg. Chem. Front., 2017, 4, 1798–1805 RSC.
- M. Bednarek, K. Borska and P. Kubisa, Molecules, 2020, 25, 4919 CrossRef CAS PubMed.
- M. A. Ortuño, B. Dereli, K. R. D. Chiaie, A. B. Biernesser, M. Qi, J. A. Byers and C. J. Cramer, Inorg. Chem., 2018, 57, 2064–2071 CrossRef PubMed.
- S. M. Quan and P. L. Diaconescu, Chem. Commun., 2015, 51, 9643–9646 RSC.
- A. M. Doerr, J. M. Burroughs, N. M. Legaux and B. K. Long, Catal. Sci. Technol., 2020, 10, 6501–6510 RSC.
- X. Xu, G. Luo, Z. Hou, P. L. Diaconescu and Y. Luo, Inorg. Chem. Front., 2020, 7, 961–971 RSC.
- L. A. Brown, J. L. Rhinehart and B. K. Long, ACS Catal., 2015, 5, 6057–6060 CrossRef CAS.
- X. Xu, H. Lu, G. Luo, X. Kang and Y. Luo, Inorg. Chem. Front., 2021, 8, 1005–1014 RSC.
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358–3375 RSC.
- M. Qi, H. Zhang, Q. Dong, J. Li, R. A. Musgrave, Y. Zhao, N. Dulock, D. Wang and J. A. Byers, Chem. Sci., 2021, 12, 9042–9052 RSC.
- S. Edmondson, V. L. Osborne and W. T. S. Huck, Chem. Soc. Rev., 2004, 33, 14 RSC.
- J. O. Zoppe, N. C. Ataman, P. Mocny, J. Wang, J. Moraes and H.-A. Klok, Chem. Rev., 2017, 117, 1105–1318 CrossRef CAS PubMed.
- D. K. Schneiderman and M. A. Hillmyer, Macromolecules, 2017, 50, 3733–3749 CrossRef CAS.
- C. Feng and X. Huang, Acc. Chem. Res., 2018, 51, 2314–2323 CrossRef CAS PubMed.
- J. Lahann and R. Langer, Macromol. Rapid Commun., 2001, 22, 968–971 CrossRef CAS.
- G. Carrot, D. Rutot-Houzé, A. Pottier, P. Degée, J. Hilborn and P. Dubois, Macromolecules, 2002, 35, 8400–8404 CrossRef CAS.
- M. Qi, Q. Dong, D. Wang and J. A. Byers, J. Am. Chem. Soc., 2018, 140, 5686–5690 CrossRef CAS PubMed.
- D. Kern and E. R. P. Zuiderweg, Curr. Opin. Struct. Biol., 2003, 13, 748–757 CrossRef CAS PubMed.
- E. Guarnera and I. N. Berezovsky, Structure, 2019, 27, 866–878 CrossRef CAS PubMed.
- H. J. Yoon, J. Kuwabara, J.-H. Kim and C. A. Mirkin, Science, 2010, 330, 66–69 CrossRef CAS PubMed.
- C. M. McGuirk, J. Mendez-Arroyo, A. I. D'Aquino, C. L. Stern, Y. Liu and C. A. Mirkin, Chem. Sci., 2016, 7, 6674–6683 RSC.
- F. Parrino and G. Palmisano, Molecules, 2021, 26, 23 CrossRef CAS PubMed.
- S. Gisbertz and B. Pieber, ChemPhotoChem, 2020, 4, 456–475 CrossRef CAS.
- B. König, Eur. J. Org. Chem., 2017, 2017, 1979–1981 CrossRef.
- D. Friedmann, A. Hakki, H. Kim, W. Choi and D. Bahnemann, Green Chem., 2016, 18, 5391–5411 RSC.
- B. M. Neilson and C. W. Bielawski, Chem. Commun., 2013, 49, 5453 RSC.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC.
- X. Liu, Y. Ni, J. Wu, H. Jiang, Z. Zhang, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2018, 9, 584–592 RSC.
- S. Shanmugam, J. Xu and C. Boyer, Macromol. Rapid Commun., 2017, 38, 1700143 CrossRef PubMed.
- R. S. Stoll and S. Hecht, Angew. Chem., Int. Ed., 2010, 49, 5054–5075 CrossRef CAS PubMed.
- X. Sun, J. P. Gao and Z. Y. Wang, J. Am. Chem. Soc., 2008, 130, 8130–8131 CrossRef CAS PubMed.
- I. A. Barker and A. P. Dove, Chem. Commun., 2013, 49, 1205 RSC.
- P. K. Kuroishi and A. P. Dove, Chem. Commun., 2018, 54, 6264–6267 RSC.
- B. A. Chan, S. Xuan, M. Horton and D. Zhang, Macromolecules, 2016, 49, 2002–2012 CrossRef CAS.
- S. S. Spink, O. I. Kazakov, E. T. Kiesewetter and M. K. Kiesewetter, Macromolecules, 2015, 48, 6127–6131 CrossRef CAS PubMed.
- Y. Liao, Acc. Chem. Res., 2017, 50, 1956–1964 CrossRef CAS PubMed.
- S. Shanmugam, J. Xu and C. Boyer, Angew. Chem., Int. Ed., 2016, 55, 1036–1040 CrossRef CAS PubMed.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- J. Xu, K. Jung, N. A. Corrigan and C. Boyer, Chem. Sci., 2014, 5, 3568 RSC.
- L. Xia, Z. Zhang and Y.-Z. You, Polym. J., 2020, 52, 1323–1331 CrossRef CAS.
- X. Zhang, Q. Ma, Y. Jiang, S. Hu, J. Li and S. Liao, Polym. Chem., 2021, 12, 885–892 RSC.
- X. Zhang, S. Hu, Q. Ma and S. Liao, Polym. Chem., 2020, 11, 3709–3715 RSC.
- O. Santoro, X. Zhang and C. Redshaw, Catalysts, 2020, 10, 800 CrossRef CAS.
- M. Labet and W. Thielemans, Chem. Soc. Rev., 2009, 38, 3484 RSC.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- S. Sun, S. Liang, W.-C. Xu, G. Xu and S. Wu, Polym. Chem., 2019, 10, 4389–4401 RSC.
- P. Weis and S. Wu, Macromol. Rapid Commun., 2018, 39, 1700220 CrossRef PubMed.
- J. M. Andjaba, C. J. Rybak, Z. Wang, J. Ling, J. Mei and C. Uyeda, J. Am. Chem. Soc., 2021, 143, 3975–3982 CrossRef CAS PubMed.
- I. Aprahamian, ACS Cent. Sci., 2020, 6, 347–358 CrossRef CAS PubMed.
- S. Silvi, M. Venturi and A. Credi, Chem. Commun., 2011, 47, 2483–2489 RSC.
- S. P. Ihrig, F. Eisenreich and S. Hecht, Chem. Commun., 2019, 55, 4290–4298 RSC.
- E. Léonard, F. Mangin, C. Villette, M. Billamboz and C. Len, Catal. Sci. Technol., 2016, 6, 379–398 RSC.
- T. H. L. Nguyen, N. Gigant and D. Joseph, ACS Catal., 2018, 8, 1546–1579 CrossRef CAS.
- Q. De Roover, T. Vucko, S. P. Vincent, J. De Winter and O. Coulembier, Catal. Sci. Technol., 2021, 11, 6048–6052 RSC.
- S. Moins, C. Henoumont, Q. De Roover, S. Laurent, J. De Winter and O. Coulembier, Catal. Sci. Technol., 2021, 11, 4387–4391 RSC.
- A. C. Deacy, G. L. Gregory, G. S. Sulley, T. T. D. Chen and C. K. Williams, J. Am. Chem. Soc., 2021, 143, 10021–10040 CrossRef CAS PubMed.
- T. Stößer, T. T. D. Chen, Y. Zhu and C. K. Williams, Philos. Trans. R. Soc., A, 2018, 376, 20170066 CrossRef PubMed.
- C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS PubMed.
- S. Paul, C. Romain, J. Shaw and C. K. Williams, Macromolecules, 2015, 48, 6047–6056 CrossRef CAS.
- G. Rosetto, A. C. Deacy and C. K. Williams, Chem. Sci., 2021, 12, 12315–12325 RSC.
- N. Yuntawattana, G. L. Gregory, L. P. Carrodeguas and C. K. Williams, ACS Macro Lett., 2021, 10, 774–779 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |