
The dynamicity of light-up aptamers in one-pot in vitro diagnostic assays
Marimuthu
Citartan

Advanced Medical & Dental Institute (AMDI), Universiti Sains Malaysia, Bertam, 13200, Kepala Batas, Penang, Malaysia. E-mail: citartan@usm.my
First published on 3rd December 2021
Abstract
Light-up aptamers are aptamers that ignite the fluorescence emission of certain dyes upon binding. Widely harnessed in in vivo imaging, the binding capacity of the light-up aptamers can also be deployed in in vitro diagnostic assays, engendering a mix-and-read format. Intrigued by this, I intend to provide an overview of the various formats of diagnostic assays developed using light-up aptamers from the direct modulation of the light-up aptamers, split aptamer-based configuration, strand displacement, in vitro transcription-based one-pot diagnostic assay, CRISPR-Cas system to the measurement of the ion reliance. The incorporation of the light-up aptamers into each configuration is expounded and further supported by describing the exemplary assays developed thus far. It is anticipated that the present study can be enlightening to any researchers who aspire to embark on the development of one-pot in vitro diagnostic assays based on light-up aptamers.
1. Introduction
Light-up aptamers are a class of aptamers that are able to impart significant changes in the fluorescence of certain fluorogenic dyes upon binding. Generated by systematic evolution of ligands by the exponential enrichment, the first light-up aptamer was developed against Malachite green in 2003. This aptamer was able to bind to Malachite Green and enhances the fluorescence by over 2000-fold.1 Spinach is another light-up aptamer that is able to bind and incite the fluorescence emission of 3,5-difluoro-4-hydroxybenzylidene imidazolinone (DFHBI), a small molecule derivative of green fluorescent protein (GFP). In the unbound state, the molecule is non-fluorescent but upon binding to the aptamer, the fluorescence improves by 20-fold.2 The aptamer was used for the imaging of RNA in mammalian cells but however still suffers from drawbacks such as low thermal instability and poor folding efficiency in vivo. To address these limitations, an improvised variant was generated, termed Spinach2, which has a better thermal stability, isolated via systematic mutation of Spinach.3 Despite its thermal stability, its reliance on high Mg2+ concentrations and susceptibility to enzymatic degradation limit its applications and in fact pave the way for further efforts in improvising the aptamer. As such, different variants of Spinach were generated and one such variant is Baby Spinach, which exhibits higher resistance to degradation as compared to Spinach. One particular variant known as Broccoli, is a robust variant of Spinach, which was isolated by FACS-SELEX process. The resulting aptamer is much shorter than Spinach/Spinach2 and produces bright green fluorescence upon binding to DFHBI or the improvised version of this fluorophore, (Z)-4-(3,5-difluoro-4- hydroxybenzylidene)-2-methyl-1-(2,2,2-trifluoroethyl)-1Himidazol-5(4H)-one (DFHBI-1 T). The folding efficiency of this aptamer is similar to that of Spinach2 but has a much lower dependency on the Mg2+ for folding and has a much elevated thermal stability.4 In addition to the Spinach family, a number of other light-up aptamers are Mango RNA aptamer, which activates the fluorescence of thiazole-orange (TO)-derived fluorophores such as thiazole orange derivative TO-1-biotin,5 Corn, RNA aptamer that is specific against 3,5-difluoro-4-hydroxybenzylidene imidazolinone-2-oxime (DFHO)6 and RNA aptamer the recognizes dimethylindole red (DIR).7The versatility of the light-up aptamers is evidenced with their widespread usage in imaging and sensing.7,8 In in vivo imaging of RNA, monitoring the spatiotemporal dynamics of RNA molecules can be achieved by fusing the aptamers to the RNA molecules of interest.9,10 Light-up aptamers are able to activate fluorescence rapidly and are not restricted by photobleaching.11 Higher signal-to-background ratio and ease of modulation are actualizable with the application of light-up aptamers.12 Fluorogenic aptamers impart reversible fluorescence emission upon binding to their target, which is an added advantage to sensing applications as compared to ribozymes that undergo irreversible analyte-induced cleavage. In light of the attributes of light-up aptamers, in vitro diagnostics can also immensely benefit with their integration into these assays. One particular in vitro diagnostic format, which is the one-pot system, is a strategy formulated to contain all the ingredients entailed for a diagnostic system in one single reaction/tube, without the need for any extensive washing steps. The incorporation of the light-up aptamers into the assay permits the inference of the state of target-binding based on the fluorescence emission of the dye. An epiphany of the splendour of the light-up aptamers in revolutionizing the one-pot approach in diagnostic assays seems to set in among researchers, as there are several one-pot light-up aptamer-based diagnostic assays developed thus far. Equally incited by these endeavours, I provide an overview of the one-pot in vitro diagnostic assays designed based on light-up aptamers. Various configurations of the one-pot in vitro diagnostic assays developed based on light-up aptamers constitute the major core of this study, which will be explicated in detail. It is anticipated that this study will be one of the guidelines for any researcher who intend to parlay the remarkable properties of the light-up aptamers into one-pot in vitro diagnostic assays.
2. Incorporating light-up aptamers into in vitro diagnostic assays
Light-up aptamers are so versatile and one straightforward application is to fuse them to any aptamer of interest. For example, an aptamer specific to an analyte can be connected to the light-up aptamer Spinach via a transducer domain. The third stem of the Spinach aptamer (from the 5′-end) is able to be connected to any aptamer sequence without losing its the binding affinity to DFHBI.13 In the absence of a target, the aptamer is in an unfolded state. In the presence of the target, the interaction of the aptamer with the target subsequently induces the conformational change of Spinach, causing it to bind to DFHBI, resulting in the emission of fluorescence. The direct modulation of a light-up aptamer that engenders an analyte-specific aptamer-light-up aptamer chimera is readily applicable in any diagnostic assay for a targeted detection. A light-up aptamer can also be fragmented into independent modules that would be able to assemble in the presence of the target, in a strategy known as the split aptamer approach.14 A high specificity of detection is ascertained via this configuration as the assembly of the discrete modules into a functional unit only occurs in the presence of a specific target. Another configuration that also ensures high specificity is the strand-displacement strategy, which depends on the displacement of the light-up aptamer from the target in the presence of the complementary sequence. The non-activate dye does not emit any fluorescence.15 As light-up aptamers are the ‘inciter’ of fluorescence of their specific cognate dyes, they can be directly used per se as the ‘substrates’ for signal production, instead of coalescing them with any other aptamer or molecules. Any strategies that can produce multiple copies of the light-up aptamers, either via enzymatic action or by protein-mediated cleavage is able to generate fluorescence emission that ensures a highly sensitive detection. Approaches such as in vitro transcription reaction drives a high production of light-up RNA aptamer molecules, which can bind to the dyes and enhances their fluorescence.16 Protein-mediated cleavage by Cas nuclease can also cause the accumulation of light-up aptamer molecules, generating fluorescence upon interaction with their specific dye.17 One unique strategy is also to measure the concentration of ions required for the formation of G-quadruplex, based on the fluorescence emission of the light-up aptamer.18 Predicating upon these various formats, one-pot in vitro diagnostic assays were successfully developed, which will be expounded in the subsequent sections.3. Fusion of the light-up aptamer to the target-specific aptamer module
Direct modulation of the light-up aptamers involves the fusion of the aptamers to another aptamer module specific against one particular target. It is the simplest form of employing light-up aptamers for in vitro diagnostics that rely on the light-up aptamers themselves as the platform that can be modulated by the insertion of any aptamer of interest. In this assay, the binding of the target to the corresponding aptamer sequence fused to the light-up aptamer induces conformational change of the aptamer, affecting its ability to incite the fluorescence of its cognate dye. The very low cost of the fluorogenic dyes would make the overall cost of developing an in vitro diagnostic assay to be low. Kato et al., 2013 have successfully used a light-up aptamer selected against Dapoxyl dyes as a label-free turn-on sensor for the detection of thrombin. In their design, the thrombin-binding DNA aptamer was fused to the loop of the aptamer via a short stem. In the presence of the target thrombin, the binding of thrombin to the aptamer causes stem hybridization which promotes the binding of Dapoxyl dye to the aptamer, resulting in the fluorescence emission. However, in the absence of the Dapoxyl dye, the stem is unfolded, as such binding to the dye Daboxyl is affected (Fig. 1). In this study, the researchers have fused the thrombin-binding aptamer at two different positions at the loop structure of the light-up aptamer connected with different lengths of the connecting stem. One particular variant responded very well in the protein concentrations of thrombin between 100–500 nM. The tunability of the sensor is also evidenced when a different target was used, such as ATP, whereby the corresponding specific aptamer sequence was fused to the loop of the light-up aptamer. The detection limit of the ATP was recorded at 10 μM.19 An important feature of the study is the cruciality of the length of the stem, which could affect the degree of conformational change that the construct undergoes in the presence of the target. Extensive optimization of the stem length must be undertaken to ensure the optimal performance of the construct. The light-up aptamer-thrombin aptamer hybrid can be a ‘ready-to-be-applied’ structure that can be potentially extended to other aptamers, by simply replacing the thrombin sequence with the aptamer sequence of interest. As facile as it may imply, any sequence fused to the construct must be followed by conscientious secondary structural analysis of the whole hybrid by mfold prediction. Usually, aptamers that form simple secondary structures such as the basic stem-loop structures would impart minimal-to-no perturbation to the overall secondary structure of the hybrid. Other aptamers with complicated or extensive secondary structure such as the aptamer against PTTG120 must be carefully fused and subjected to secondary structural analysis. In the event of any perturbation of the whole secondary structure of the hybrid, the aptamer with a complex secondary structure can be ‘minimized’ by truncation to the core structure, prior to the fusion to the light-up aptamer. It requires an extensive optimization of both the length of the connecting module as well as the position of the analyte-specific aptamer at the loop of the light-up aptamer. An enhancement of the binding of the resulting new hybrid may also require the addition of monovalent or divalent cations in the incubation buffer, which also needs careful optimization to choose the best recipe for an optimal interaction and signal production.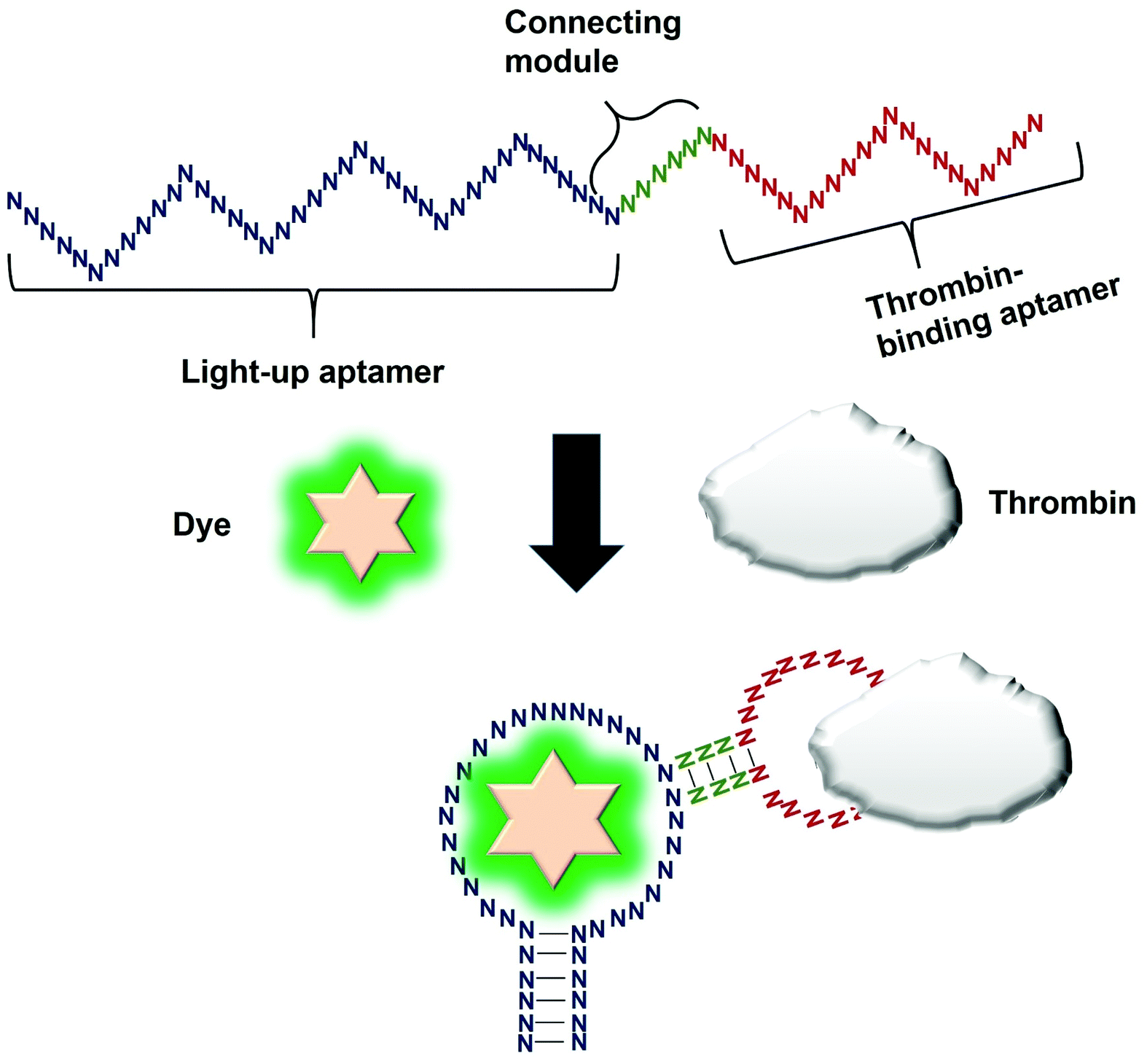 | ||
| Fig. 1 Direct modulation of the light-up aptamer with analyte-specific aptamer. In their design, the thrombin-binding DNA aptamer was connected to the loop of the aptamer via a short stem. Conformational change is induced upon binding of thrombin to the thrombin-specific aptamer. The conformational change is transduced to the light-up aptamer, causing its binding to the dye, resulting in fluorescence emission. | ||
4. Splitting light-up aptamers into discrete units that coalesce in the presence of a target
The most common structures of aptamers are hairpin and hairpin-like structures, whereby the binding of the target resides in the loop region. In the concept of split aptamers, aptamers are fragmented into several non-functional fragments. In the presence of a specific target, simultaneous interaction of the individual fragments with the target results in the assembly of the fragments into a complete functional aptamer. The hairpin and hairpin-like structures aptamers such as anti-thrombin21 and anti-cocaine aptamers22 are the most commonly used aptamers for split aptamer-based assays.23 The coalition of the discrete units into a functional aptamer strictly in the presence of the target is a sought-after property of any diagnostic assay to ensure high sensitivity and specificity. Previous studies have reported the fragmentation of the light-up aptamers into several discrete non-functional units for the purpose of RNA detection24–26 (Fig. 2a). In one split aptamer system invented by Kikuchi and Kolpashchikov, the Spinach aptamer was split into two fragments, whereby one fragment is complementary to one part of the target DNA or RNA. In the presence of the target, each of the fragments bind to one part of the target concurrently; these two juxtapositional fragments form a functional light-up aptamer, which then binds to DFHBI, resulting in fluorescence emission. However, in the absence of the target, the Spinach aptamer is not intact, as a result, it is not able to bind to DFHBI. By targeting a fragment of inhA gene from Mycobacterium tuberculosis (Mtb), two split-light-up aptamers (RNA and DNA probes) were designed. DNA probe was designed to target the DNA analyte of the gene while the RNA probe targeted the RNA counterpart. Linker was used to separate the analyte binding arms of the probes in order to prevent the potential interference with the binding of the probes to the target. These two probes are able to achieve an improvement in the fluorescence emission of up to 270 and 76 for the RNA and DNA probes, respectively upon binding to the fully matched analytes, achieving a detection limit in the low nanomolar range.14 One crucial aspect of their research is the ability of the probes to be selective and to differentiate single base-substituted analytes. The assay can be carried out at room temperature for a specific detection of a gene or RNA. In one unique approach known as the three-way junction, a sequence of destabilized Broccoli aptamer was hybridized to a sequence which is complementary to a target RNA. The stabilization of the aptamer is only restored in the presence of the target RNA, which enables the Broccoli aptamer to bind to DFHBI-1 T, causing the emission of fluorescence. The three-way junction strategy can be harboured for the diagnostic detection of a range of RNAs such as mRNA, miRNA or other non-protein coding RNAs.27 In tumor diagnosis, split aptamer system was also proven to be practical. In one split aptamer system, 2 aptamers were combined, one specific to human hepatocarcinoma cell line SMMC-7721 while another aptamer is specific against Thioflavin T,28 a water soluble fluorogenic dye with low fluorescence level in the unbound state. One probe was designed to contain both the recognition element of the tumor cell-specific aptamer and a partial fragment of the Thioflavin T-specific aptamer. The second probe was designed to contain the partial fragment of the tumor cell-specific aptamer and the recognition element of the Thioflavin T-specific aptamer. The recognition domain of the Thioflavin T-specific aptamer in the second probe is hybridized to one strand termed the helper strand. In the presence of the target cell SMMC-7721, the first probe which harbours the tumor cell recognition module undergoes a conformational change and the helper strand is released from the second probe. As a result, Thioflavin T is able to bind to the recognition element of the Thioflavin T in the second probe and the partial fragment of the Thioflavin T in the first probe, inducing fluorescence (Fig. 2b). The assay enables the detection of down to 125 cells in 100 μL buffer, with excellent specificity differentiating target cell SMMC-7721 from other cells including MCF-7 and HeLa.29 The researchers have also developed a diagnostic assay that is independent of any washing steps and is able to detect the targeted tumor cells in a single-step detection by a mere mixing of the split aptamer fragments, samples containing the target cells and the fluorescent dye. Deriving cues from this study, the split aptamer system should also be widely employed for the diagnostic detection of other tumor cells, whereby multiple cell-SELEX-derived aptamers30–32 can be employed in liquid biopsy-based diagnostics. For multiplex detection of tumor cells, different light-up aptamers can be fused to different cell-SELEX-derived aptamers and the specific interaction can be monitored via the distinct fluorescence emission profile for each pair.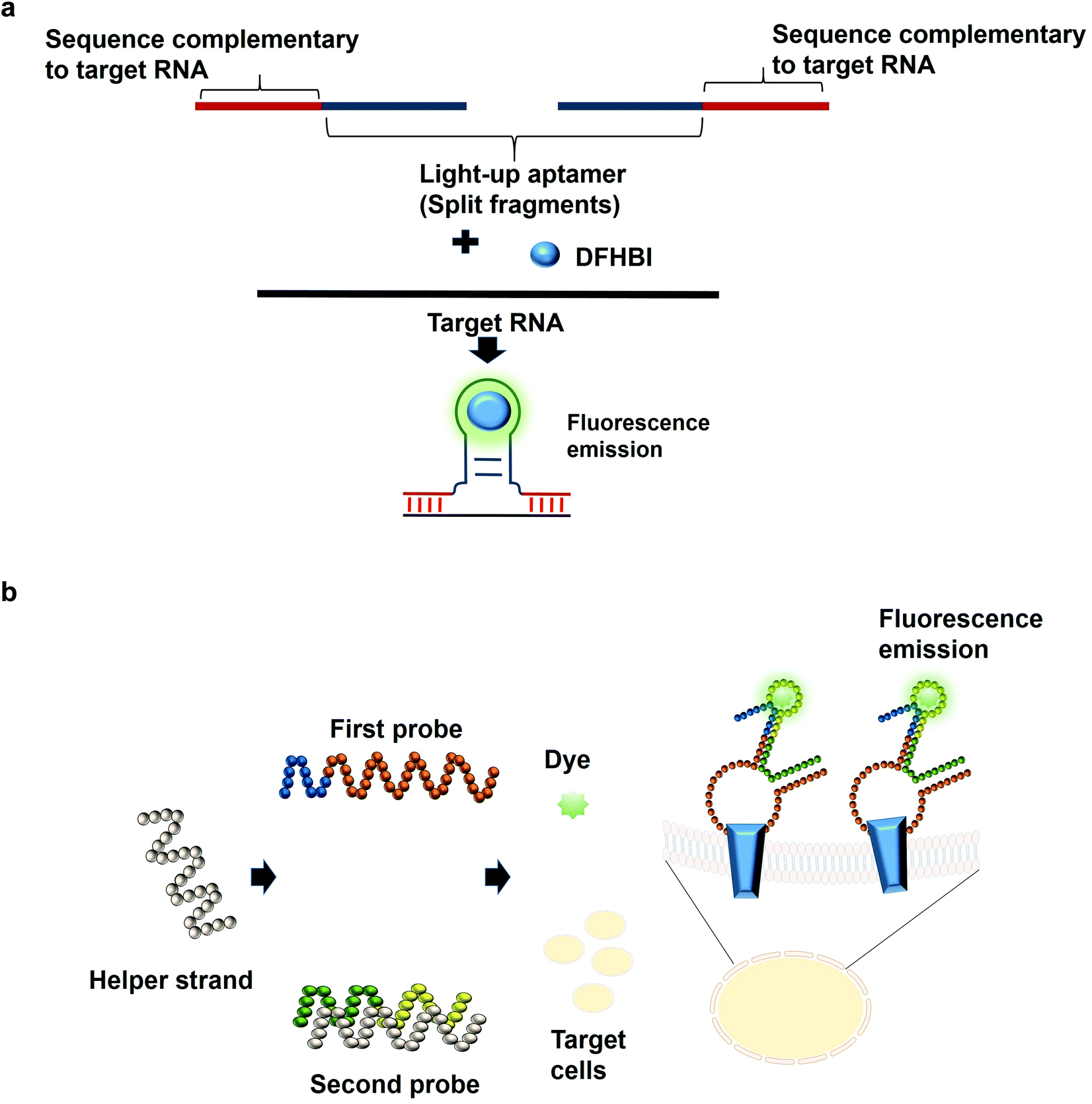 | ||
| Fig. 2 (a) A simple scheme of designing a split-aptamer. A light-up aptamer can be split into two discreet components, each of which carries a segment complementary to one section of a target RNA. In the presence of a target RNA, the binding of the individual components coalesces with each other to form a complete functional unit of a light-up aptamer. The assembled light-up aptamer then binds and activates the fluorescence emission of the dye. (b) Split aptamer-based configuration. The first probe was designed to contain both the recognition element of the tumor cell-specific aptamer and a partial fragment of the Thioflavin T-specific aptamer. The second probe comprise the partial fragment of the tumor cell-specific aptamer and the recognition element of the Thioflavin T-specific aptamer. The helper strand initially bound to the second probe is released in the presence of the specific tumor cell due to the conformational change. As a result, the first and the second probe combine to form a functional aptamer that is able to interact with the target protein on the surface of the cell while the light-up aptamer domain binds dye and incites its fluorescence emission. | ||
An imprecise fragmentation of the aptamers into discrete units would render the split aptamer assay futile, hence a precise monitoring of the fragment assembly is a requisite. In one study, the Spinach aptamer was split into two fragments and the stem of each fragment was extended using different short interfering RNA (siRNA) sequences. Complementary DNA blocker strands were used to block the individual RNA strands in order to preclude the formation of the fully functional split Spinach aptamer. To displace the blocking strand, strands complementary to the blocking strands can be added, freeing the split-Spinach aptamer fragments, which then coalesce to form a functional aptamer able to bind to DFHBI.33 This split aptamer approach can be feasible to monitor the assembly of either synthetic or natural RNA. Using a strategy known as the bottom-up approach, Torelli et al., 2018 have designed a light-up bio-orthogonal RNA origami nanoribbon. The biologically inert scaffold known as the De Bruijn scaffold sequences are able to form a three-way junction by binding to the complementary sequence fused with the split Broccoli aptamer. These complementary sequences act as the staple strands that promote the folding of the RNA scaffold sequence into a nanoribbon shaped structure by simple heating and cooling to room temperature. Only in the presence of the target RNA sequence, a complete assembly of the nanoribbon is promoted to restore the fully functional Broccoli aptamer that would be able to bind to DFHBI-1 T and emits fluorescence.34 They are the first group to demonstrate the formation of a nanostructure using scaffolded origami strategy and using Broccoli aptamer to monitor the self-assembly of the nanostructure. For the assembly of more than 2 fragments, one research study that can be based on is by a group of researchers that have constructed a structure known as the 6-stranded RNA nanoring. In this structure, the Spinach aptamer was truncated in order to fit into the interior of the RNA nanoring. The functional nanoring-split-light-up aptamer construct is able to bind and activate the fluorescence of DFHBI only in the presence of all the six strands of the nanoring.35 Although the researchers have developed the 6-stranded RNA nanoring for the purpose of RNA assembly, the ethos of the discovery can inspire researchers to extend the application of the RNA nanoring to the development of diagnostic assays for the detection of target RNA such as miRNA.
5. Strand displacement using the complementary strand of a light-up aptamer
Fast turnaround times is also one aspect desired in an early diagnosis to save time and for a high throughput analysis. The dynamicity of DNA nanotechnology is one possible attribute that can be largely deployed for rapid turnaround times in a diagnostic strategy. A great deal of focus can be given on the non-equilibrium state that exists during the DNA hybridization process.36 During this state, via a phenomenon coined as ‘base-pair breathing’, double-stranded DNAs are always on a constant switch between the ‘bound’ and ‘unbound states’, which occur primarily at the end of the helices.37 Exploiting this transitory moment, any complementary strands could be introduced that may disrupt the base-pairing between the double-stranded DNAs, displacing the other ‘less-competitive’ strand, depending on the overall kinetics of the reactions. Displacement of one strand from double helices by another equivalent or ‘better’ strands is the basis of DNA, RNA replication and genetic recombination in vivo. This principle was also based on for the solvation of the Hamiltonian path problem in the ground-breaking work by Adleman et al.38 As the displacement of one of the strands is governed by the kinetics and stoichiometry of the interaction, which is dictated by the inclusion of the competitor, nucleic acid-protein interaction-based kinetics could also be used as a disruptive force to dismantle the double helical structures. This is possible if the kinetics of the nucleic acid-protein interaction is way superior as compared to the hydrogen-bond kinetics that exist between the strands. Aptamers, as the nucleic acids that recognize protein, can be a source of the aforementioned kinetics.A simple strand displacement reaction can be used in developing a sensing application using light-up aptamers. In fact, a complementary sequence to the light-up aptamer can be utilized. Lloyd et al., 2018 incepted the concept kleptamers, which are anti-sense strands that are complementary to the aptamer, enabling the displacement of the aptamer from the its target. Kleptamers were used in conjunction with many light-up aptamers such as Spinach, Malachite Green and Mango aptamers.15 Islam et al., 2021 have developed an isothermal strand displacement assay using the aptamer that they have generated against Thioflavin T. One template was designed to contain the reverse complement sequence fragment of the target miR-215 at the 3′-end, the reverse complement sequence fragment of the nicking endonuclease recognition site (Nt·BstNBI) at the second domain while the reverse complement sequence fragment of the light-up aptamer at the 5′-end. Hybridization of the target miRNA to the complementary region in the template allows Polymerase to bind at the 3′-end of the miRNA to initiate extension, producing a DNA strand that consists both the Nt·BstNBI recognition site and the light-up aptamer. Nicking endonuclease nicks at the recognition site, causing a single-stranded DNA break or a nick. The resulting exposed 3′-end is bound by Bst Polymerase, which initiates the extension, producing another copy of the light-up aptamer while ‘knocks out’ the existing aptamer strand. This cycles continues and culminates in the accumulation of the light-up aptamer strands. The accumulated light-up aptamers then bind to the dye Thioflavin T, causing the emission of the fluorescence (Fig. 3). In an attempt to detect the target miR-215, a detection limit of 2.6 nM was achieved.39 Averting the need for any thermocyclers, the researchers have developed a cheap, simple, room temperature-operable assay for the diagnostic detection of miRNA, which can be further expanded into a point-of-care system. The system is also malleable by simply changing the 3-end sequence of the construct to be complementary to any target miRNA or non-protein coding RNAs such as bacterial small RNAs. Multiple other light-up aptamers available can also be explored for similar functionality and can be combined in a single-pot multiplex detection of miRNA. One particular concern of this experiment is the incumbency upon enzyme, which is the rate-limiting factor of the overall efficiency of the study as enzymes are proteinaceous, which makes them liable to temperature fluctuation-induced denaturation. As such, this study should also serve as an impetus to explore the potentiality of other enzyme-free and ‘all-DNA machine’-based strand displacement configurations to be used in concert with light-up aptamers, including toehold-mediated strand displacement.40,41 It is enzymeless and is even more highly sequence specific to ensure high sensitivity and specificity of detection. As an instance, Bhadra and Ellington. 2014 have showed the applicability of the toehold-initiated strand displacement using Spinach aptamer to discriminate sequences with single nucleotide mismatches.42
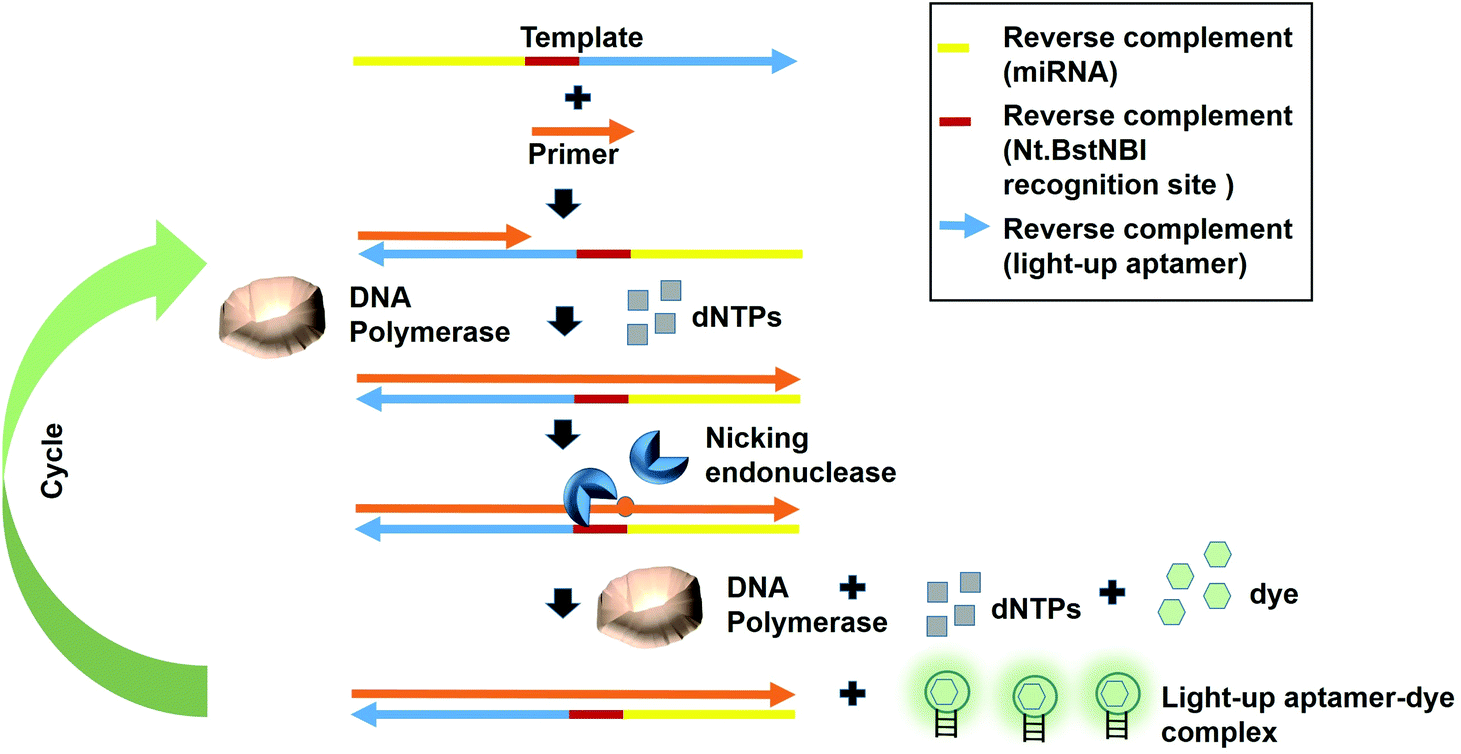 | ||
| Fig. 3 The template DNA was designed to contain the reverse complement sequence fragment of the target miR-215 at the 3′-end, the reverse complement sequence of the nicking endonuclease recognition site (Nt.BstNBI) in the middle and the reverse complement sequence fragment of the light-up aptamer at the 5′-end. Hybridization of the target miRNA at the 3′-end of the miRNA is followed by the extension mediated by DNA Polymerase to produce the second strand complementary to the template. Following the cleavage of the recognition site by the nicking endonuclease, the single-stranded DNA produced is bound by Bst Polymerase, which initiates the extension. The production of another light-up aptamer strand displaces the existing aptamer strand. The cycle starts all over again and more light-up aptamers are produced, which bind to the dye to generate fluorescence emission. | ||
6. In vitro transcription of the light-up aptamer
In vitro transcription-based diagnostic assay is based on the transcription of the multiple copies of RNA molecules from the starting DNA template, with the aid of RNA polymerase enzyme. From a minute amount of initial starting DNA template, microgram amounts of RNA can be generated, which can then be quantified by any means of detection, for example by fluorescence. The resulting RNA molecules can themselves act as a substrate for signal production and light-up RNA aptamers represent one class of perfect example. Sheng et al., have demonstrated a one-pot aptasensor based on transcription process. Using the aptamer Broccoli, they have segmented the Broccoli probe into 2 major modules. The first module is the template of the Broccoli aptamer, which has a promoter to drive the transcription that is able to produce the aptamer once the transcription is activated. The second module which is the recognition module contains a sequence termed binder. They have used an aptamer against S. aureus, and the module is hybridized to a sequence termed blocker. In the absence of the target, the sequence blocker is stably hybridized to the aptamer. However, in the presence of the target S. aureus, the binder domain in the second module would bind to the blocker, forming a duplex that acts as the template for in vitro transcription, producing multiple Broccoli RNA aptamers. The accumulated Broccoli RNA aptamers would be able to bind to DFHBI-1 T, resulting in fluorescence emission. The efficiency of the in vitro transcription and a high turn-on ratio of Broccoli-DFHBI-1 T pairs accounts for the highly sensitive label free-detection of S. aureus. The limit of detection achieved is 77.0 CFU mL−1. Moreover, the recovery rate for water samples (tap water and lake water) were also around 100%, with variation within 3.5%.16 Major milestone achieved in this study is that high sensitivity of detection can be still achieved even without the need for culture by broth or selective plating. Obviating the requirement for any temperature-control devices, detection can be achieved at a constant temperature (37 °C). This assay can be extended to the detection of other bacterial pathogens as a rapid, on-site diagnostic detection to facilitate an immediate decision making. In another study, light-up aptamer was evidenced for the detection of miRNA. In comparison to the study by Sheng et al., in this study, the strand was designed to also harbour a nicking site. Two different templates were designed. Specifically, the first template was designed to contain 3 modules, Region I, which is the miRNA complementary sequence, Region II, which is a recognition site for Nt.BstNBI nicking upon the formation of dsDNA and region 3, which is an amplification domain that is complementary to the product of the strand displacement process. On the other hand, template 2 consists of T7 promoter and the spinach aptamer. In the presence of the target miRNA, region I binds to the miRNA and is extended by the Bst DNA polymerase to form RNA/DNA duplex. Due to the repetitive cycles of strand displacement, nicking and polymerase activities, a large amount of sequences complementary to template 2 are produced, which then bind to template 2 forming double-stranded DNA. These double-stranded DNA molecules are then in vitro transcribed to produce RNA molecules that contains spinach aptamer domain, which binds to DFHBI to produce fluorescence. This assay allows a detection limit down to 5.12 × 10−18 M, allowing the differentiation of miRNAs from homologous family.43 The researchers have evinced that incorporating light-up aptamer allows the detection of low-abundance miRNA with high signal-to-background ratio with much lower noise signal as compared to the traditional SYBR Green I in a label-free manner. Moreover, two different approaches, which are the strand-displacement and in vitro transcription were conflated in a single system. As a whole, the resulting system ensures a high sensitivity of detection. Various other non-protein coding RNAs can also be tested using this system. However, this system contains more enzymes (not only RNA polymerase), which could make the overall detection assay much more expensive and care must be taken not to reversibly denature the enzymes involved.In one experiment, a novel proximity induced transcription assay (PITA) was exhibited for the detection of prostate specific antigen (PSA). A pair of PSA antibody-DNA oligonucleotide probes that interact at the different epitopes of PSA were used. The DNA of the first pair carries the T7 RNA Polymerase promoter sequence and a short region for ligation while the second DNA consists of the DNA template for Broccoli RNA aptamer production and also a short region for ligation. Upon fusion, both of these DNA fragments generate a DNA template for in vitro transcription. In the presence of the target PSA, as the two antibody-DNA pair that bind at different regions were brought in close proximity to each other, ligation is initiated by T4 DNA ligation enzyme and the connector oligonucleotide. The resulting ligation product is the DNA template for in vitro transcription reaction, which produces multiple molecules of Brocolli aptamer that bind to DFHBI-1 T to emit fluorescence.44 The limit of detection achieved was 0.15 ng mL−1, which is lower than the physiological PSA amount of 4.0 ng mL−1, exceeding of which is indicative of a high probability of prostate cancer. The researchers have also successfully analysed the applicability of the assay on 20 serum samples from prostate cancer patients. This assay is highly dependent on the binding affinity of the antibody pair against the target protein in ensuring a highly sensitive detection. Any researchers who intend to replicate this assay for their application must verify the non-overlapping nature of the antibody pair so that they concurrently recognize distinct epitopes on the surface of the target protein. Moreover, the DNA strands conjugated to the antibodies must also be ascertained not to be hindered by any steric hindrance effect, which could deter their ligation to each other. Future research can also involve the surrogation of the antibody pair with aptamers, which could impart less steric hindrance effect to the ligation of the DNA strands. A higher amount of DNA template is able to generate a much higher amount of RNA, which can amplify the signal production. One particular strategy is to use an immobilization platform that can harbour more DNA molecules. Dendrimer, which consists of multiple units of a monomer could increase the surface area to load more molecules. Capitalizing on this, a group of researchers have constructed a dendrimeric structure via layer-by-layer assembly strategy utilizing streptavidin and biotinylated BSA. The dendrimeric structure is able to bind to a higher amount of biotinylated DNA template, which can be transcribed to produce multiple copies of the RNA. The limit of detection achieved for the detection alpha-fetoprotein was 108 aM, which constitutes approximately 1500-fold of improvement as compared to that of conventional fluorescence ELISA, which is 160 fM.45
In a multiplex detection of miRNAs, two probes are designed, each of which is complementary to the region of miRNA. The first probe (probe A) which is complementary to the one region of the miRNA has the first T7 RNA polymerase promoter sequence for hybridization with the second T7 RNA polymerase promoter sequence. The second probe (probe B) also has a region complementary to one region of the miRNA. In the presence of the target, probe A is annealed to probe B with the aid of SplintR Ligase. SplintR Ligase relies on the RNA template that acts as a splint that can be bound juxtapositionally by two DNA strands, which are eventually ligated. As such, a high specificity of binding can be ensured by the enzyme. The ligated product acts as the DNA template for the in vitro transcription to produce RNA molecules that can then bind to the dyes and enhances fluorescence (Fig. 4a). To permit multiplex detection of miRNAs, the sequence of probe A or probe B can be varied to carry different DNA templates for different light-up RNA aptamers that are able to bind to various fluorophores. They have achieved a highly sensitive and selective detection of miR-21 and miR-141.46 Equivalently, using the same design, Woo et al., 2020, have acquired a detection of down to 0.1-attomolar of RNA with positive and negative predictive values of 95 and 100%, respectively using 40 nasopharyngeal SARS-CoV-2 samples.47 In this study, they have designed the probe that can be in vitro-transcribed to produce RNA aptamer that is specific against Malachite Green. Moreover, for a more sensitive detection, multiple light-up aptamer transcriptional units can be appended at the 3′-end of the aptamer to produce multiple copies of light-up aptamers that can bind to the dye and enhance the overall fluorescence emission (Fig. 4b). Collectively, in vitro transcription-based one-pot diagnostic assays are able to provide a fast turnaround time, simple, easy and are able to be conducted at room temperature, which are the ideal criteria for point-of-care diagnostics.
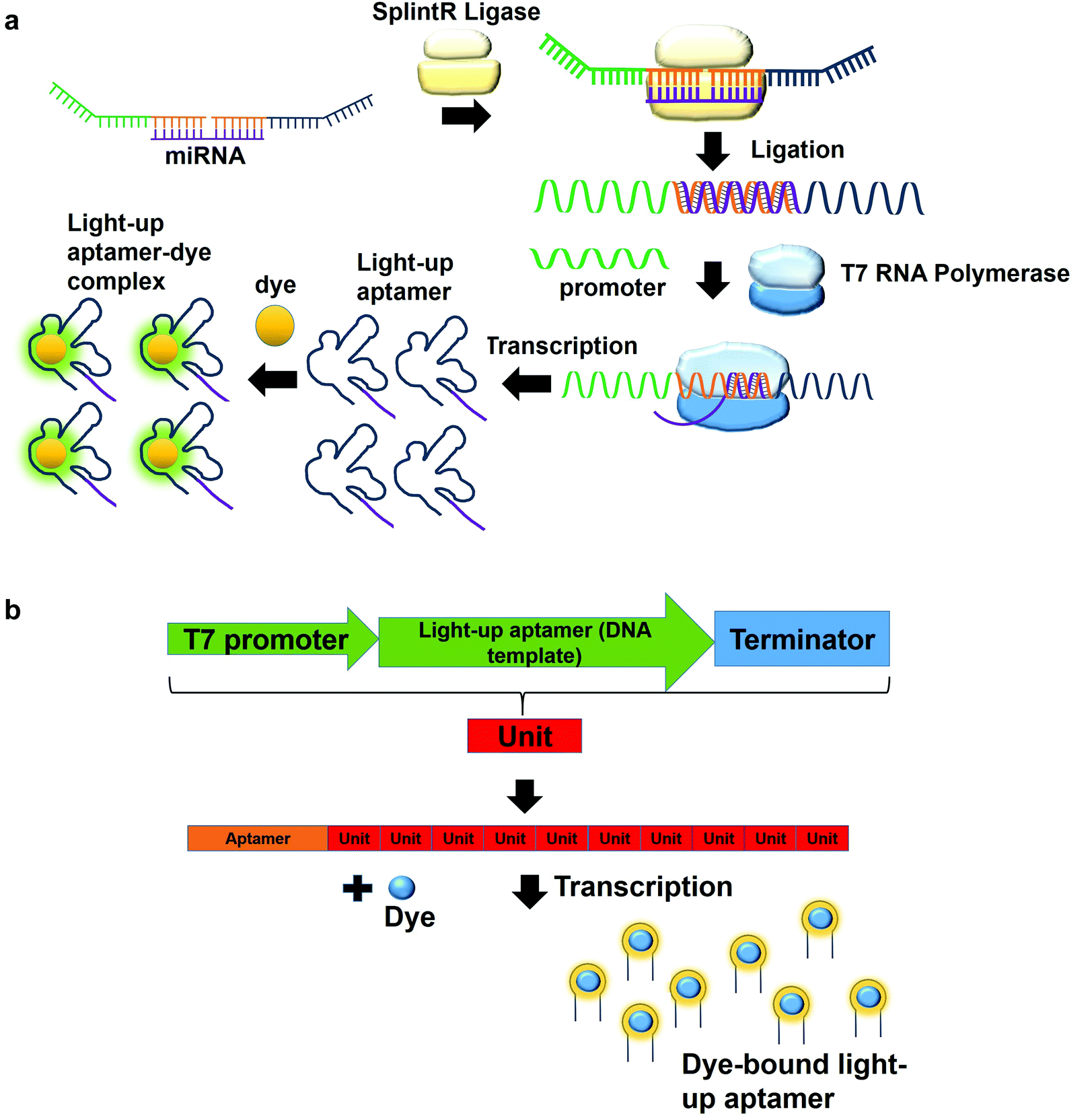 | ||
| Fig. 4 (a) Two probes are complementary to two regions of the miRNA. In the presence of the target miRNA, the two probes bind to the miRNA and is ligated by SplintR Ligase. The ligated product is used as the template for the production of RNA molecules by in vitro transcription, which then interact with the dye to produce fluorescence emission. (b) Aptamers can be appended with multiple transcriptional units at the 3′-end of the aptamer. In the presence of the T7 RNA Polymerase, the resulting light-up RNA aptamers can bind to the dye and amplifies the fluorescence. | ||
7. Collateral cleavage activity of CRISPR-Cas 13a on the light-up aptamers
The modification of the DNA sequence is a natural regulatory process and this property can also be deployed to perform the editing/changing of the targeted DNA region at will. CRISPR-based system is one such system, which comprise a CRISPR-associated protein (Cas) and a short guide RNA, which assists in homing the protein in on the targeted region.48,49 Two specific CRISPR-associated endonucleases Cas1250,51 and Cas1352,53 cleave DNA and RNA, respectively. Cas13 was used in a system known as specific high-sensitivity enzymatic reporter unlocking (SHERLOCK) while Cas12 was incorporated into a system known as the DNA Endonuclease-Targeted CRISPR Trans Reporter (DETECTR) for the diagnostic detection of virus. The collateral cleavage activity exhibited by these endonucleases, which refers to the cleavability of any DNA or RNA regardless of the sequences, allows for visual/fluorescence detection upon the addition of the reporter DNA or RNA.54,55Similarly, light-up RNA aptamers can be cleaved by Cas13a when its cleavage activity is activated by the interaction between the target RNA and crRNA. The cleaved light-up RNA aptamer would not be able to bind and activate the dye, hence no fluorescence is produced (Fig. 5a). In one study for the diagnostic detection of bacteria, Broccoli RNA aptamer is utilized. In the presence of the target RNA, crRNA binds and causes the activation of Cas13a to cleave the single-stranded RNA. The single-stranded Broccoli RNA aptamer is cleaved, as a result unable to bind and activate the fluorescence of the DFHBI-1 T. In the absence of the target RNA, there is no activation of Cas13a, causing the intact Broccoli aptamer to bind and activate the fluorescence of dye DFHBI-1 T (Fig. 5b).17 The researchers have developed nucleic acid amplification-free, reverse transcription-free, in vitro transcription-free and label-free method for the detection of bacteria as low as 10 CFU. The direct detection of RNA also allows for the measurement of the expression level of the gene, such as mRNA expression. In a recent work to detect SARS-CoV-2, Wang et al., 2021 have used similar method for the direct detection of the virus. A fascinating feature in this study as compared to the previous study is the ligation of two oligonucleotides to the target RNA, which collectively acts as the template for in vitro transcription. Single-stranded RNA from the virus serves as a template for the binding of two oligonucleotides which are placed in a juxtapositional manner to each other. T4 RNA ligase performs the ligation between these two oligonucleotides, producing the template for in vitro transcription to produce multiple copies of the transcripts. The ligation-mediated in vitro transcription increases the specificity of the detection. The transcripts are bound by crRNA that recruits and activates the ribonuclease activity of the Cas13a. As a result, Broccoli aptamers will be cleaved and thus fail to bind to the target DFHBI-1 T, resulting in no fluorescence emission. The assay developed enables the detection of various viruses including SARS-CoV-2, Middle East respiratory syndrome (MERS), SARS, H1N1, H7N9, and H9N2. The hallmark of the assay is the detection of as low as 82 copies of SARS-CoV-2 virus while able to distinguish D614G mutation of SARS-CoV-2 variant.56 Moreover, the assay is able to differentiate between throat swab samples of SARS-CoV-2-infected persons from that of healthy ones. The study can pave the way for the development of the point-of-care diagnostics. Studies based on CRISPR-Cas system should spark even more intense interest to widely use other light-up aptamers available in a singleplex or multiplex manner for the detection of virus.
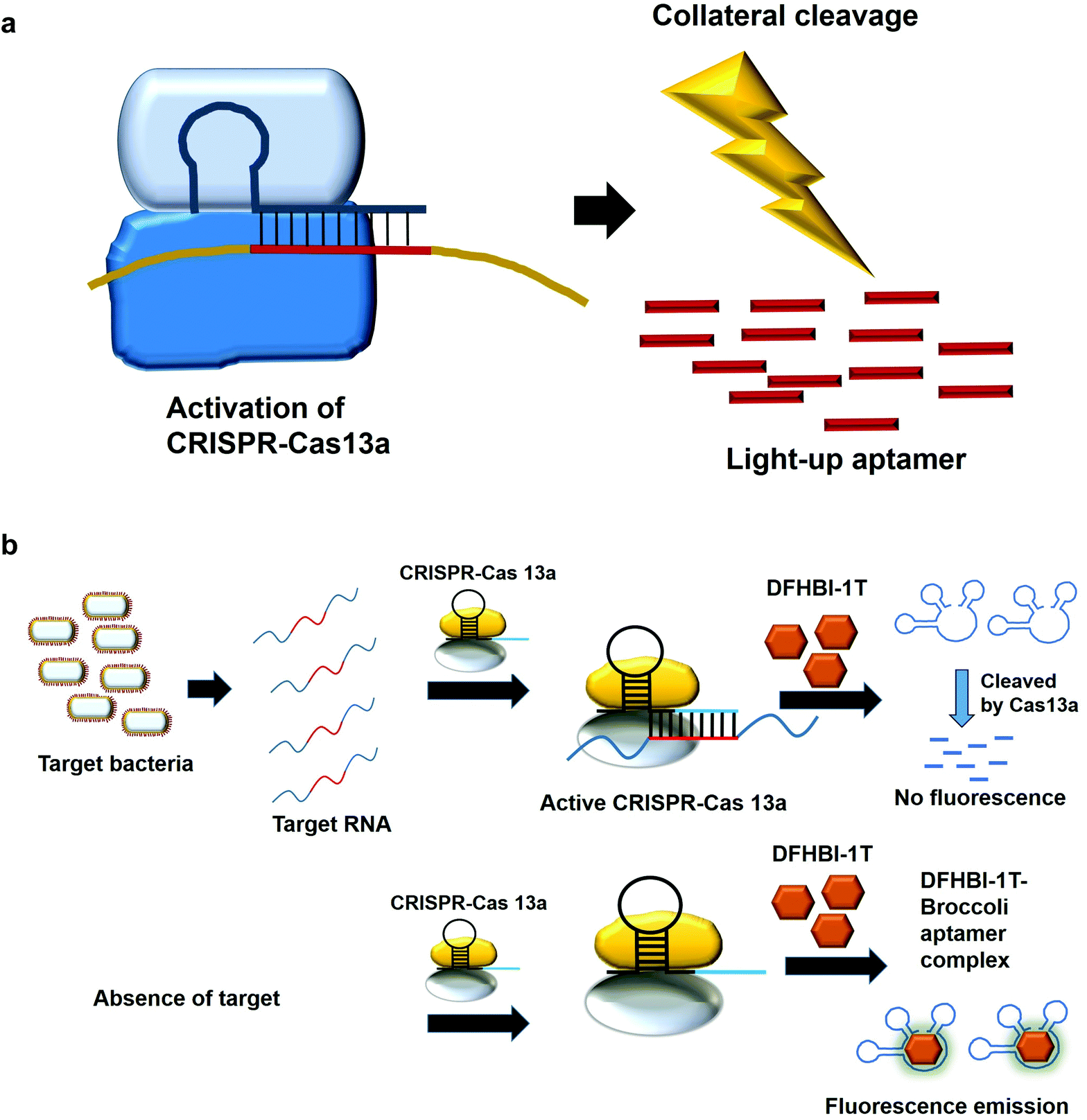 | ||
| Fig. 5 (a) CRISPR-Cas13a binds to the specific target RNA and this activates its non-specific cleavage ribonuclease activity, which cleaves the light-up RNA aptamers. The weak or absence of fluorescence is indicative of the presence of the target RNA. (b) In the presence of the target bacteria, the cRNA binds to one specific region of the RNA and activates Cas13a, which cleaves the light-up aptamer. No fluorescence emission is generated. In the absence of the target RNA, activation of the Cas13a failed, causing the intact Broccoli aptamer to bind and activate the fluorescence of dye DFHBI-1T. | ||
8. Measuring the fluorescence emission as the sensing strategy of the ion required for G-quadruplex formation
The detrimental effects of heavy metal ions on human health have raised much concern worldwide and also garnered attention from researchers to turn to aptamer-based sensing as the point-of-care monitoring strategy.57 Most of the sensors are architectured based on the fluorescence measurement of the certain aptamers at different concentrations of certain ions. For example, G quadruplex aptamers, which are G-rich non-canonical four stranded secondary structures, require ions such Na+ and K+ that can be located along the quartet plane or positioned in between the quartet planes for the stabilization of the structure. These ions can also aid in charge screening and forge the interaction with the loops and grooves of the nucleic acids.58 G-quadruplexes were also reported to be Pb2+-dependant, which sets out to be the basis for the development of Potassium-Lead-Switched G-Quadruplex-DNA-based logic gate.59 As Spinach aptamer was also discovered to form a G-quadruplex structure,60 this has galvanized DasGupta et al. 2015 to develop a sensor for the detection of Pb2+ using Spinach aptamer (Fig. 6). Spinach aptamer consists of two-layers of G-quadruplex motif that acts as the binding site for DFHBI.61 In the presence of Pb2+, G-quadruplex aptamers adopt a more compact structure than in the presence of monovalent cations due to the smaller size of Pb2+ and its high charge density.62 The sensor-based Spinach is quite selective against Pb2+, of up to more than 17000-fold as compared to against Ca2+. Specificity of the sensor was also tested in a metal soup that contains other ions such as Ag+, Ca2+,Co2+, Cr3+, Cd2+, K+, Na+, Cu2+, Fe2+, Mn2+, Ni2+, Zn2+, yet the sensor retains high selectivity towards Pb2+.18 In terms of stability, they have found that the sensor is stable to degradation between pH 3 and pH 11 during the period of detection, while is able to provide optimal detection between pH 6 and pH 8. The sensor is able to detect Pb2+ spiked into tap water in the range of 100 nM–5 μM. Instead of using light-up aptamers per se, aptamers selected against Hg2+,63 Zn2+,64 Ni2+,65 and K+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 66 can also be fused to the molecules. However, one concern is the fact that most of the metal ion-binding aptamers have poor dissociation constant, which may be translated into poorer diagnostic assay if they were used for the direct sensing of the ions.
66 can also be fused to the molecules. However, one concern is the fact that most of the metal ion-binding aptamers have poor dissociation constant, which may be translated into poorer diagnostic assay if they were used for the direct sensing of the ions.
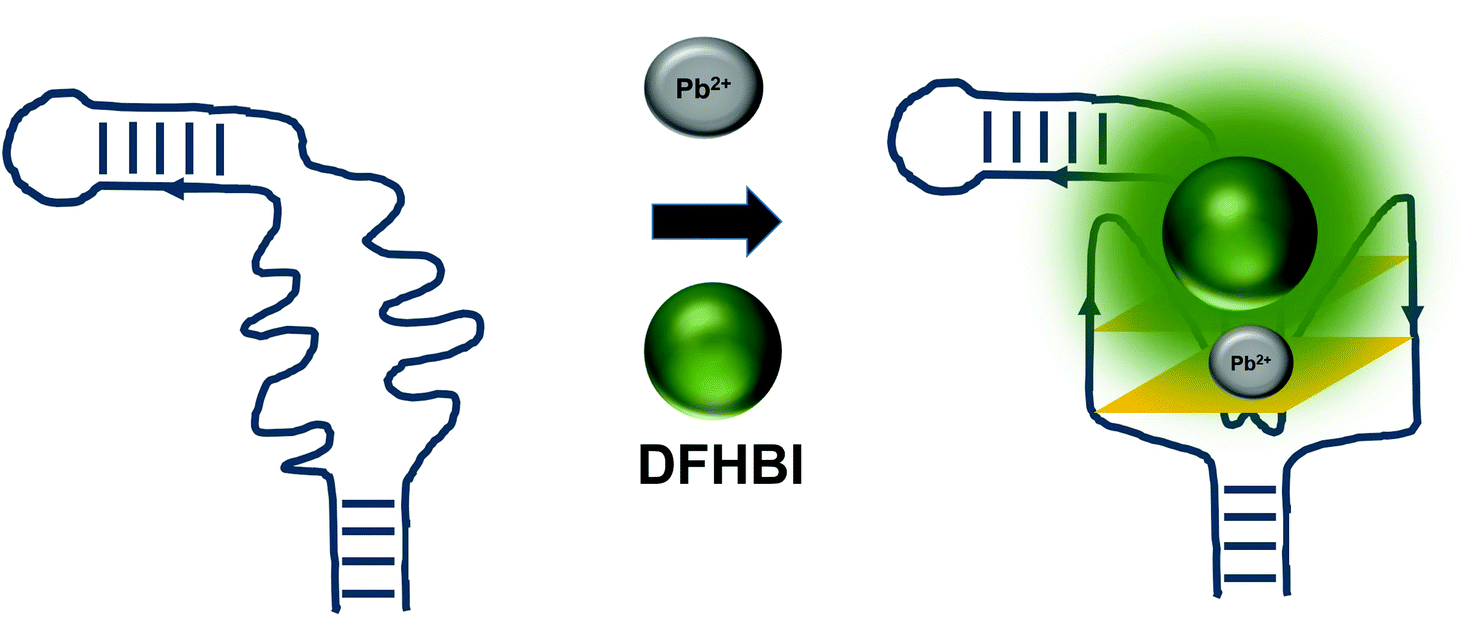 | ||
| Fig. 6 The presence of Pb2+ induces the formation of the G-quadruplex structure of the Spinach aptamer. The presence or absence of the ion can be used to measure the extent of G-quadruplex formation based on the fluorescence emission. | ||
9. Conclusion and future perspectives
The current pandemic of Covid-19 points out the pressing need for a fast turnaround time and mix-and-read diagnostic assay for an immediate decision making. To this end, it is perceptible that light-up aptamers have the potential to revolutionize in vitro diagnostic assays, facilitating the development of one-pot approach to detect various target molecules ranging from nucleic acids to protein. Considering the low cost of dyes and the fast kinetics of the light-up aptamer-based assays, researchers should ramp up the effort to integrate light-up aptamers into their diagnostic assays. Moreover, one-pot in vitro diagnostic assays empowered by light-up aptamers can also be built upon point-of-care-based framework by using portable paper-devices or handheld fluorometer. Researchers involved in generating aptamers should also expand the repertoire of the dyes used for in vitro selection so that more light-up aptamers are also available for the usage in diagnostic assays.Conflicts of interest
There are no conflicts to declare.References
- J. R. Babendure, S. R. Adams and R. Y. Tsien, J. Am. Chem. Soc., 2003, 125, 14716–14717 CrossRef CAS PubMed
.
- J. S. Paige, K. Y. Wu and S. R. Jaffrey, Science, 2011, 333, 642–646 CrossRef CAS PubMed
- R. L. Strack, M. D. Disney and S. R. Jaffrey, Nat. Methods, 2013, 10, 1219–1224 CrossRef CAS PubMed
- G. S. Filonov, J. D. Moon, N. Svensen and S. R. Jaffrey, J. Am. Chem. Soc., 2014, 136, 16299–16308 CrossRef CAS PubMed
- E. V. Dolgosheina, S. C. Y. Jeng, S. S. S. Panchapakesan, R. Cojocaru, P. S. K. Chen, P. D. Wilson, N. Hawkins, P. A. Wiggins and P. J. Unrau, ACS Chem. Biol., 2014, 9, 2412–2420 CrossRef CAS PubMed
- W. Song, G. S. Filonov, H. Kim, M. Hirsch, X. Li, J. D. Moon and S. R. Jaffrey, Nat. Chem. Biol., 2017, 13, 1187–1194 CrossRef CAS PubMed
- X. Tan, T. P. Constantin, K. L. Sloane, A. S. Waggoner, M. P. Bruchez and B. A. Armitage, J. Am. Chem. Soc., 2017, 139, 9001–9009 CrossRef CAS PubMed
- P. Swetha, Z. Fan, F. Wang and J. H. Jiang, J. Mater. Chem. B, 2020, 8, 3382–3392 RSC
- C. A. Kellenberger, S. C. Wilson, J. Sales-Lee and M. C. Hammond, J. Am. Chem. Soc., 2013, 135, 4906–4909 CrossRef CAS PubMed
- C. A. Kellenberger and M. C. Hammond, Methods Enzymol., 2015, 550, 147–172 CAS
- M. Sunbul, J. Lackner, A. Martin, D. Englert, B. Hacene, F. Grün, K. Nienhaus, G. U. Nienhaus and A. Jäschke, Nat. Biotechnol., 2021, 39, 686–690 CrossRef CAS PubMed
- J. Ouellet, Front. Chem., 2016, 4, 29 Search PubMed
- J. S. Paige, T. Nguyen-Duc, W. Song and S. R. Jaffrey, Science, 2012, 335, 1194 CrossRef CAS PubMed
- N. Kikuchi and D. M. Kolpashchikov, ChemBioChem, 2016, 17, 1589–1592 CrossRef CAS PubMed
- J. Lloyd, C. H. Tran, K. Wadhwani, C. Cuba Samaniego, H. K. K. Subramanian and E. Franco, ACS Synth. Biol., 2018, 7, 30–37 CrossRef CAS PubMed
- L. Sheng, Y. Lu, S. Deng, X. Liao, K. Zhang, T. Ding, H. Gao, D. Liu, R. Deng and J. Li, Chem. Commun., 2019, 55, 10096–10099 RSC
- T. Zhang, W. Zhou, X. Lin, M. R. Khan, S. Deng, M. Zhou, G. He, C. Wu, R. Deng and Q. He, Biosens. Bioelectron., 2021, 176, 112906 CrossRef CAS PubMed
- S. DasGupta, S. A. Shelke, N. S. Li and J. A. Piccirilli, Chem Commun., 2015, 51, 9034–9037 RSC
- T. Kato, I. Shimada, R. Kimura and M. Hyuga, Chem Commun., 2016, 52, 4041–4044 RSC
- S. S. Prabu, E. S. Ch'ng, P. Y. Woon, J. H. Chen, T. H. Tang and M. Citartan, Anal. Chim. Acta, 2020, 1138, 181–190 CrossRef CAS PubMed
- L. C. Bock, L. C. Griffin, J. A. Latham, E. H. Vermaas and J. J. Toole, Nature, 1992, 355, 564–566 CrossRef CAS PubMed
- M. N. Stojanovic, P. de Prada and D. W. Landry, J. Am. Chem. Soc., 2000, 122, 11547–11548 CrossRef CAS PubMed
- M. Debiais, A. Lelievre, M. Smietana and S. Müller, Nucleic Acids Res., 2020, 48, 3400–3422 CrossRef CAS PubMed
- K. K. Alam, K. D. Tawiah, M. F. Lichte, D. Porciani and D. H. Burke, ACS Synth. Biol., 2017, 6, 1710–1721 CrossRef CAS PubMed
- N. Kikuchi and D. M. Kolpashchikov, Chem. Commun., 2017, 53, 4977–4980 RSC
- D. M. Kolpashchikov, J. Am. Chem. Soc., 2005, 127, 12442–12443 CrossRef CAS PubMed
- Y. Furuhata, M. Kobayashi, R. Maruyama, Y. Sato, K. Makino, T. Michiue, H. Yui, S. Nishizawa and K. Yoshimoto, Rna, 2019, 25, 590–599 CrossRef CAS PubMed
- H. Wang, J. Wang, L. Xu, Y. Zhang, Y. Chen, H. Chen and R. Pei, Anal. Methods, 2016, 8, 8461–8465 RSC
- Y. Sun, B. Yuan, M. Deng, Q. Wang, J. Huang, Q. Guo, J. Liu, X. Yang and K. Wang, Analyst, 2018, 143, 3579–3585 RSC
- M. Sola, A. P. Menon, B. Moreno, D. Meraviglia-Crivelli, M. M. Soldevilla, F. Cartón-García and F. Pastor, Mol. Ther. – Nucleic Acids, 2020, 21, 192–204 CrossRef CAS PubMed
- N. Lin, L. Wu, X. Xu, Q. Wu, Y. Wang, H. Shen, Y. Song, H. Wang, Z. Zhu, D. Kang and C. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 9306–9315 CrossRef CAS PubMed
- K. Tsogtbaatar, D. A. Sousa, D. Ferreira, A. Tevlek, H. M. Aydın, E. Çelik and L. Rodrigues, Invest. New Drugs, 2021 DOI:10.1007/s10637-021-01161-y
- T. A. Rogers, G. E. Andrews, L. Jaeger and W. W. Grabow, ACS Synth. Biol., 2015, 4, 162–166 CrossRef CAS PubMed
- E. Torelli, J. W. Kozyra, J.-Y. Gu, U. Stimming, L. Piantanida, K. Voïtchovsky and N. Krasnogor, Sci. Rep., 2018, 8, 6989 CrossRef PubMed
- J. M. O'Hara, D. Marashi, S. Morton, L. Jaeger and W. W. Grabow, Nanomaterials, 2019, 9, 378 CrossRef PubMed
- T. Fu, Y. Lyu, H. Liu, R. Peng, X. Zhang, M. Ye and W. Tan, Trends Biochem. Sci., 2018, 43, 547–560 CrossRef CAS PubMed
- E. Cubero, E. C. Sherer, F. J. Luque, M. Orozco and C. A. Laughton, J. Am. Chem. Soc., 1999, 121, 8653–8654 CrossRef CAS
- L. M. Adleman, Science, 1994, 266, 1021–1024 CrossRef CAS PubMed
- M. M. Islam, V. M. Ghielmetti and P. B. Allen, Sci. Rep., 2021, 11, 4291 CrossRef CAS PubMed
- F. C. Simmel, B. Yurke and H. R. Singh, Chem. Rev., 2019, 119, 6326–6369 CrossRef CAS PubMed
- B. Yurke, A. J. Turberfield, A. P. Mills, Jr., F. C. Simmel and J. L. Neumann, Nature, 2000, 406, 605–608 CrossRef CAS PubMed
- S. Bhadra and A. D. Ellington, RNA, 2014, 20, 1183–1194 CrossRef CAS PubMed
- M. Zhou, X. Teng, Y. Li, R. Deng and J. Li, Anal. Chem., 2019, 91, 5295–5302 CrossRef CAS PubMed
- Z. M. Ying, H. Y. Xiao, H. Tang, R. Q. Yu and J. H. Jiang, Chem Commun., 2018, 54, 8877–8880 RSC
- J. Sim, J. Y. Byun and Y. B. Shin, Chem Commun., 2019, 55, 3618–3621 RSC
- Z. M. Ying, B. Tu, L. Liu, H. Tang, L. J. Tang and J. H. Jiang, Chem Commun., 2018, 54, 3010–3013 RSC
- C. H. Woo, S. Jang, G. Shin, G. Y. Jung and J. W. Lee, Nat. Biomed. Eng., 2020, 4, 1168–1179 CrossRef CAS PubMed
- M. M. Kaminski, O. O. Abudayyeh, J. S. Gootenberg, F. Zhang and J. J. Collins, Nat. Biomed. Eng., 2021, 5, 643–656 CrossRef CAS PubMed
- K. S. Makarova, Y. I. Wolf, J. Iranzo, S. A. Shmakov, O. S. Alkhnbashi, S. J. J. Brouns, E. Charpentier, D. Cheng, D. H. Haft, P. Horvath, S. Moineau, F. J. M. Mojica, D. Scott, S. A. Shah, V. Siksnys, M. P. Terns, Č. Venclovas, M. F. White, A. F. Yakunin, W. Yan, F. Zhang, R. A. Garrett, R. Backofen, J. van der Oost, R. Barrangou and E. V. Koonin, Nat. Rev. Microbiol., 2020, 18, 67–83 CrossRef CAS PubMed
- B. Zetsche, J. S. Gootenberg, O. O. Abudayyeh, I. M. Slaymaker, K. S. Makarova, P. Essletzbichler, S. E. Volz, J. Joung, J. van der Oost, A. Regev, E. V. Koonin and F. Zhang, Cell, 2015, 163, 759–771 CrossRef CAS PubMed
- S. Shmakov, O. O. Abudayyeh, K. S. Makarova, Y. I. Wolf, J. S. Gootenberg, E. Semenova, L. Minakhin, J. Joung, S. Konermann, K. Severinov, F. Zhang and E. V. Koonin, Mol. Cell, 2015, 60, 385–397 CrossRef CAS PubMed
- O. O. Abudayyeh, J. S. Gootenberg, S. Konermann, J. Joung, I. M. Slaymaker, D. B. Cox, S. Shmakov, K. S. Makarova, E. Semenova, L. Minakhin, K. Severinov, A. Regev, E. S. Lander, E. V. Koonin and F. Zhang, Science, 2016, 353, aaf5573 CrossRef PubMed
- A. East-Seletsky, M. R. O'Connell, S. C. Knight, D. Burstein, J. H. Cate, R. Tjian and J. A. Doudna, Nature, 2016, 538, 270–273 CrossRef CAS PubMed
- M. J. Kellner, J. G. Koob, J. S. Gootenberg, O. O. Abudayyeh and F. Zhang, Nat. Protoc., 2019, 14, 2986–3012 CrossRef CAS PubMed
- J. P. Broughton, X. Deng, G. Yu, C. L. Fasching, V. Servellita, J. Singh, X. Miao, J. A. Streithorst, A. Granados, A. Sotomayor-Gonzalez, K. Zorn, A. Gopez, E. Hsu, W. Gu, S. Miller, C.-Y. Pan, H. Guevara, D. A. Wadford, J. S. Chen and C. Y. Chiu, Nat. Biotechnol., 2020, 38, 870–874 CrossRef CAS PubMed
- Y. Wang, Y. Zhang, J. Chen, M. Wang, T. Zhang, W. Luo, Y. Li, Y. Wu, B. Zeng, K. Zhang, R. Deng and W. Li, Anal. Chem., 2021, 93, 3393–3402 CrossRef CAS PubMed
- W. Guo, C. Zhang, T. Ma, X. Liu, Z. Chen, S. Li and Y. Deng, J. Nanobiotechnol., 2021, 19, 166 CrossRef PubMed
- D. Bhattacharyya, G. Mirihana Arachchilage and S. Basu, Front. Chem., 2016, 4, 38–38 Search PubMed
- T. Li, E. Wang and S. Dong, J. Am. Chem. Soc., 2009, 131, 15082–15083 CrossRef CAS PubMed
- H. Huang, N. B. Suslov, N.-S. Li, S. A. Shelke, M. E. Evans, Y. Koldobskaya, P. A. Rice and J. A. Piccirilli, Nat. Chem. Biol., 2014, 10, 686–691 CrossRef CAS PubMed
- K. D. Warner, M. C. Chen, W. Song, R. L. Strack, A. Thorn, S. R. Jaffrey and A. R. Ferré-D'Amaré, Nat. Struct. Mol. Biol., 2014, 21, 658–663 CrossRef CAS PubMed
- H. Zhang, B. Jiang, Y. Xiang, J. Su, Y. Chai and R. Yuan, Biosens. Bioelectron., 2011, 28, 135–138 CrossRef CAS PubMed
- Y. Miyake, H. Togashi, M. Tashiro, H. Yamaguchi, S. Oda, M. Kudo, Y. Tanaka, Y. Kondo, R. Sawa, T. Fujimoto, T. Machinami and A. Ono, J. Am. Chem. Soc., 2006, 128, 2172–2173 CrossRef CAS PubMed
- J. Ciesiolka, J. Gorski and M. Yarus, Rna, 1995, 1, 538–550 CAS
- H. P. Hofmann, S. Limmer, V. Hornung and M. Sprinzl, Rna, 1997, 3, 1289–1300 CAS
- H. Chai, X. Ma, F. Meng, Q. Mei, Y. Tang and P. Miao, New J. Chem., 2019, 43, 7928–7931 RSC
| This journal is © The Royal Society of Chemistry 2022 |