
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Trimacrocyclic hexasubstituted benzene linked by labile octahedral [X(CHCl3)6]− clusters†
Zhenzhen
Lai
a,
Aimin
Li
a,
Sangshan
Peng
 a,
Jonathan L.
Sessler
b and
Qing
He
*a
a,
Jonathan L.
Sessler
b and
Qing
He
*a
aState Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, P. R. China. E-mail: heqing85@hnu.edu.cn
bDepartment of Chemistry, The University of Texas at Austin, 105 East 24th Street, Stop A5300, Austin, Texas 78712, USA
First published on 12th August 2021
Abstract
Crystalline supramolecular architectures mediated by cations, anions, ion pairs or neutral guest species are well established. However, the robust crystallization of a well-designed receptor mediated by labile anionic solvate clusters remains unexplored. Herein, we describe the synthesis and crystalline behaviors of a trimacrocyclic hexasubstituted benzene 2 in the presence of guanidium halide salts and chloroform. Halide hexasolvate clusters, viz. [Cl(CHCl3)6]−, [Br(CHCl3)6]−, and [I(CHCl3)6]−, were found to be critical to the crystallization process, as suggested by the single-crystal structures, X-ray powder diffraction (XRPD), thermogravimetric analysis (TGA), scanning electron microscopy with energy dispersive spectroscopy (SEM-EDS), and NMR spectroscopy. This study demonstrates the hitherto unexpected role that labile ionic solvate clusters can play in stabilizing supramolecular architectures.
Introduction
Crystallization or co-crystallization is ubiquitous in biology, chemistry, materials science, and manufacturing.1 It offers a very powerful synthetic strategy for fabricating interesting and useful ordered ensembles of molecules, e.g., nanoparticles, molecular crystals, colloids, semiconductor quantum dots, and phase-separated polymers.2–7 A particular subset of crystallization involves the formation of ensembles stabilized by non-covalent interactions. The resulting systems can be classified generally as those stabilized by (a) direct contact, wherein the individual components are connected through supramolecular interactions8 or mediated by intervening (b) neutral guests,9 (c) cations,10 (d) anions,11–13 or (e) ion-pairs (Fig. 1).14 Currently, we are unaware of any well-defined supramolecular constructs whose formation is mediated by labile anion solvate clusters, such as [X(CHCl3)6]− (X = Cl−, Br−, I−). Here we report what to the best of our knowledge are the first examples wherein [Cl(CHCl3)6]−, [Br(CHCl3)6]−, [I(CHCl3)6]− and [Br(CHBr3)6]− serve to promote the solid state crystallization of a trimacrocyclic hexasubstituted benzene (THB) 2 as inferred from single crystal X-ray diffraction analyses. Under conditions of rapid hexanes-induced precipitation, highly ordered cubic particles are formed. | ||
| Fig. 1 Cartoon illustration of supramolecular systems stabilized by (a) direct contact or mediated by intervening (b) neutral guests, (c) cations, (d) anions, (e) ion-pairs, or (f) octahedral anion solvate clusters (this work). Noncovalent interactions are indicated by red or blue dotted lines. | ||
Chloroform (CHCl3) is a common organic solvent, widely used both in the laboratory and in industry. Chloroform can exert a pronounced solvent effect, particularly in the presence of halide anions.15–20 Halide solvate clusters, such as [X(CHCl3)n]− where X = Cl−, Br−, I−, have attracted interest as models with which to explore these effects.21–27 On account of the weak X−⋯H–C interaction between the central halide anion and the chloroform molecules, [X(CHCl3)n]− clusters are inherently labile. As a result, most work to date has focused on theoretical investigations, as well as mass spectrometric and single-crystal X-ray crystallographic studies of clusters with low coordination numbers (n < 4).21,23,25 Although a few examples have been reported of [X(CHCl3)n]− clusters with high coordination number (e.g., [Cl(CHCl3)6]−), in certain lattices in the presence of transition metals.28,29 We are unaware of examples where [X(CHCl3)n]− clusters drive crystallization. As detailed below, we have now found that [X(CHCl3)6]− clusters can drive formation of well-organized, non-covalent crystalline structures.
Results and discussion
Design, synthesis and characterization of receptor 2
Hexasubstituted benzenes represent a versatile class of frameworks for constructing supramolecular receptors for either anions or cations.30–33 Usually, such architectures can access a variety of conformations due to the high degree of freedom of the substituents on the benzene core; this makes host–guest interactions with a putative substrate less entropically favorable. One way to overcome this latter energic penalty is to reduce the degrees of freedom by means of macrocyclization so as to generate, e.g., trimacrocyclic hexasubstituted benzene (THB) derivatives.33,34 Taking advantage of this latter strategy we have now prepared a trimacrocyclic hexasubstituted benzene 2 receptor wherein crown ethers are incorporated into the system (Fig. 2a). The synthesis of 2 is straightforward. Briefly, a key crown ether-like precursor 1 containing acetylene unit was prepared according to a reported approach (see Scheme S1†).35 A Co2(CO)8-catalyzed trimerization of 1 in dioxane at reflux overnight then gave receptor 2 in 80% yield after chromatographic purification over silica gel. The identity of 2 was confirmed by 1H/13C NMR spectroscopic and high-resolution mass spectrometric analyses (see the ESI†).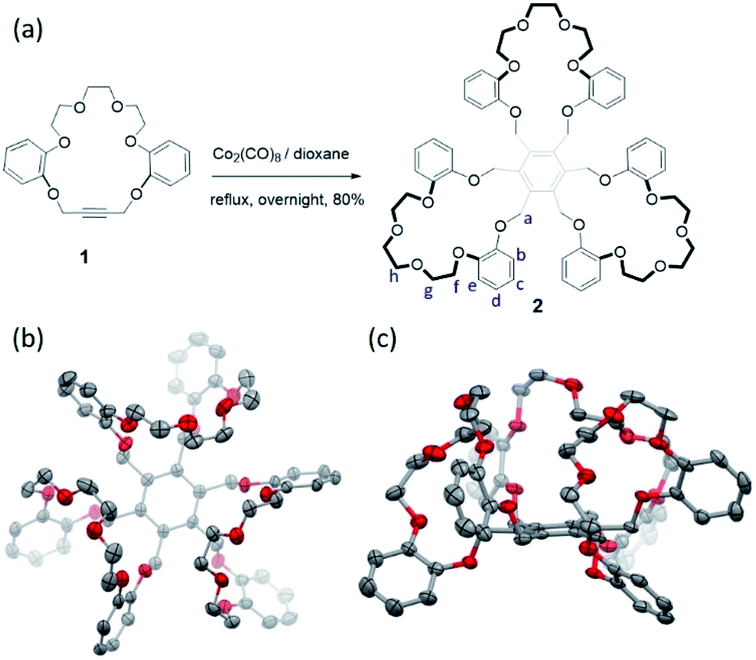 | ||
| Fig. 2 (a) Co2(CO)8-catalyzed synthesis of receptor 2; (b) top view and (c) front view of the solid-state structure of 2 as determined via single-crystal X-ray diffraction analysis. All solvent molecules and hydrogen atoms are omitted for clarity. | ||
Diffraction grade single crystals of receptor 2 were obtained by allowing a CHCl3/CH3OH solution of 2 to undergo slow evaporation over the course of two weeks. The resulting structure revealed a cis-like conformation, where all three crown ether moieties are oriented toward the same face of the benzene core (Fig. 2b and see ESI Fig. S1†). This finding led us to postulate that 2 might serve as a receptor for guanidium cations by virtue of its size, symmetry and presumed shape complementary (Fig. S2†).
Host–guest binding properties of receptor 2
To test the hypothesis that compound 2 could serve as a receptor for guanidium cation, we carried out 1H NMR spectroscopic experiments with receptor 2 and guanidium chloride in a mixture of CDCl3/CD3OD (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) at 298 K. Notably, only one set of resonances was seen for 2 under these conditions (Fig. S3†); this is as expected for a relatively flexible system that is in conformational equilibrium. Upon addition of excess guanidium chloride into a 2.0 mM solution of 2 in CDCl3/CD3OD (9:1, v/v), the Csp3–H peaks at 5.43 ppm (a), 3.94 ppm (f) were shifted upfield (Fig. S3†). In contrast, negligible changes were observed for any of the proton signals upon the addition of other larger and unsymmetrical guanidium chloride derivatives, viz. moroxydine hydrochloride (S6), and 1,1-dimethylbiguanide hydrochloride (S7), under identical conditions. Thus, we infer that receptor 2 favors guanidium chloride over other potential competing substrates. A 1H NMR spectroscopic titration yielded an association constant of K = (1.6 ± 0.4) × 104 M−1 for the interaction of guanidium chloride with 2 in a 1:1 binding model (Fig. S4 and S5†). The stability of the resulting complex was also evidenced by gas-phase molecular dynamics simulation studies (Fig. S6†).
:1, v/v) at 298 K. Notably, only one set of resonances was seen for 2 under these conditions (Fig. S3†); this is as expected for a relatively flexible system that is in conformational equilibrium. Upon addition of excess guanidium chloride into a 2.0 mM solution of 2 in CDCl3/CD3OD (9:1, v/v), the Csp3–H peaks at 5.43 ppm (a), 3.94 ppm (f) were shifted upfield (Fig. S3†). In contrast, negligible changes were observed for any of the proton signals upon the addition of other larger and unsymmetrical guanidium chloride derivatives, viz. moroxydine hydrochloride (S6), and 1,1-dimethylbiguanide hydrochloride (S7), under identical conditions. Thus, we infer that receptor 2 favors guanidium chloride over other potential competing substrates. A 1H NMR spectroscopic titration yielded an association constant of K = (1.6 ± 0.4) × 104 M−1 for the interaction of guanidium chloride with 2 in a 1:1 binding model (Fig. S4 and S5†). The stability of the resulting complex was also evidenced by gas-phase molecular dynamics simulation studies (Fig. S6†).
[X(CHCl3)6]− cluster-mediated single crystallization
Further evidence that compound 2 could act as a cation receptor for guanidium came from a single-crystal X-ray diffraction analysis of the guanidium chloride complex. Suitable crystals were obtained via the slow evaporation of a CHCl3/CH3OH solution of receptor 2 in the presence of excess guanidium chloride. The resulting structure revealed a 22·CN3H6+·CN3H5·[Cl(CHCl3)6]− complex (Fig. 3a). Due to the limitations of crystallography, positively charged guanidium and the charge-free guanidine species could not be distinguished from one another. From the relative number of chloride anions, we infer that the occupancies of guanidium and guanidine in each cavity of 2 are each 50%. As expected, the guest species was embedded within the cavity of 2 surrounded by three cyclic glycol chains via multiple C–H⋯O, N–H⋯O hydrogen bonding and cation-π interactions (Fig. 3b and S7†). A dimeric complex 22·CN3H6+·CN3H5 is found in the lattice (Fig. S8†). Much to our surprise, a careful inspection of the counter anions revealed that each chloride anion was surrounded by six chloroform molecules through multiple hydrogen bonding interactions within a [Cl(CHCl3)6]− cluster characterized by an average Cl−⋯C(HCl3) distance of 3.463 Å (Fig. 3c). Although [Cl(CHCl3)n]− clusters have been reported to be labile due to the weak Cl−⋯H–C(HCl3) bonds,23 in the current system, the octahedral [Cl(CHCl3)6]− clusters are found to mediate the formation of highly ordered 1D (one dimensional) supramolecular architectures (Fig. 3d) that are arranged in 2D networks (Fig. S8 and S9†).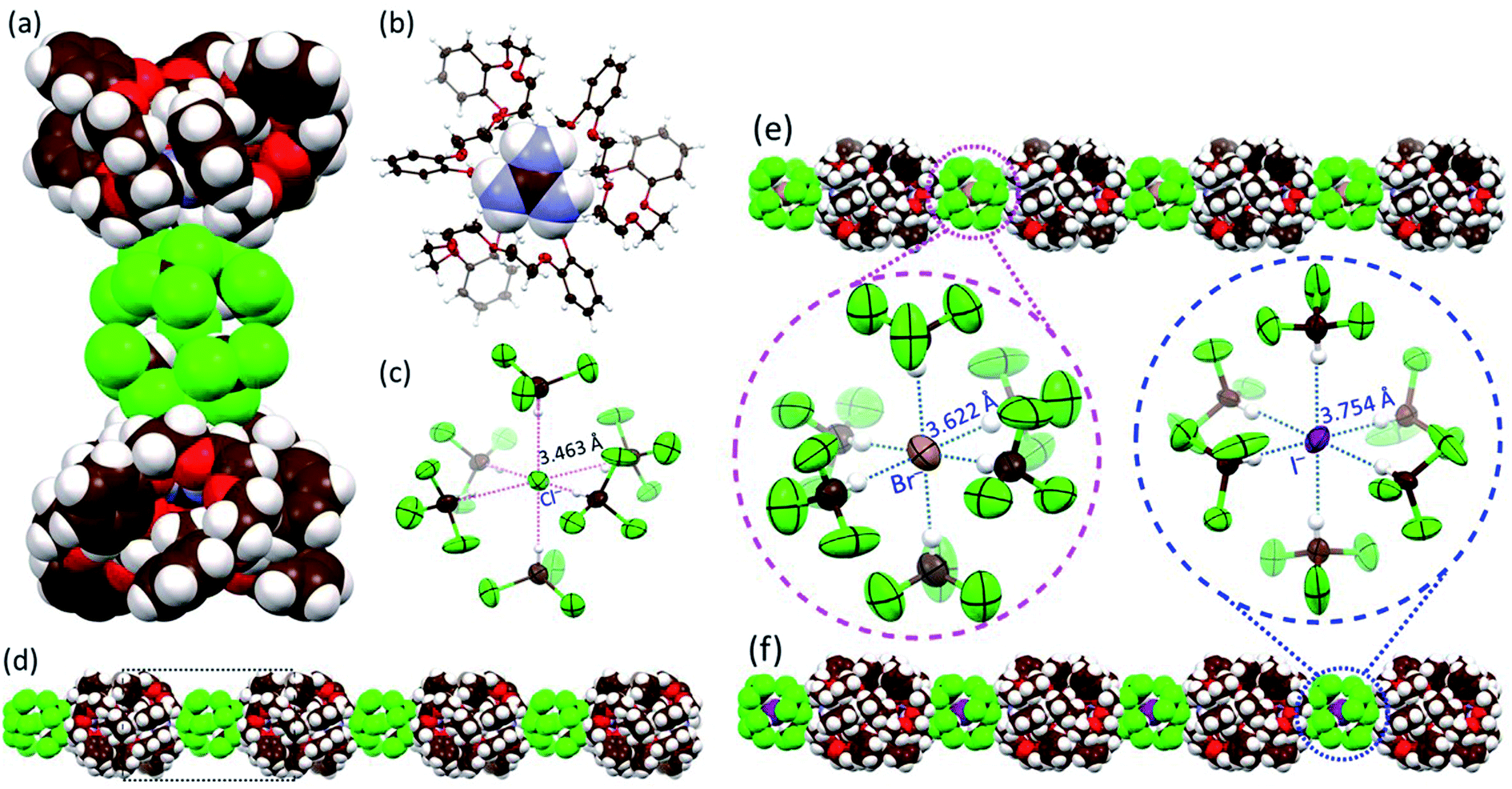 | ||
| Fig. 3 Different views and packing modes of single-crystal structures. (a) Complex 2·CN3H6+·CN3H5·[Cl(CHCl3)6]− shown in space-filling form; (b) host–guest complex 2·0.5CN3H6+·0.5CN3H5 with the receptor 2 shown in an ellipsoid model with the guanidium guest shown in space-filling form; (c) a [Cl(CHCl3)6]− cluster within the overall structure with hydrogen bonds indicated by red dotted lines for which a uniform Cl−⋯C(HCl3) distance of 3.463 Å is seen; views of the highly ordered linear architectures formed by dimeric 22·CN3H6+·CN3H5 complexes and (d) [Cl(CHCl3)6]− clusters; (e) [Br(CHCl3)6]− clusters; and (f) [I(CHCl3)6]− clusters shown in space-filling form. In (e and f), also shown are the [Br(CHCl3)6]− and [I(CHCl3)6]− clusters displayed as ellipsoid models with hydrogen bonds indicated by blue dotted lines. | ||
We next sought to explore whether other similar octahedral halide chloroform clusters, i.e., [Br(CHCl3)6]− and [I(CHCl3)6]−, could be stabilized in the solid state and mediate the generation of analogous supramolecular assembles as seen for the chloride anion. As above, diffraction-grade single crystals were obtained by slowly evaporating CHCl3/CH3OH solutions of 2 in the presence of guanidium bromide and guanidium iodide, respectively. The resulting structures revealed that, in analogy with what was seen in the presence of guanidium chloride, in both 22·CN3H6+·CN3H5·[Br(CHCl3)6]− (the complex formed from guanidium bromide) and 22·CN3H6+·CN3H5·[I(CHCl3)6]− (the complex formed from guanidium iodide) cluster-linked ensembles were observed (Fig. 3e and f and ESI Fig. S10 to S17†). Again, both bromide and iodide were found surrounded by six chloroform molecules with average anion–solvent distances of 3.622 Å and 3.754 Å for Br−⋯C(HCl3) and I−⋯C(HCl3), respectively, being found (Fig. 3e and f). The [Br(CHCl3)6]− and [I(CHCl3)6]− clusters were observed to bridge the individual 22·CN3H6+·CN3H5·[X(CHCl3)6]− complexes, resulting in the formation of 1D assemblies and 2D networks (Fig. 3e and f, see ESI Fig. S13 and S17†). Interestingly, upon replacement of CHCl3 with CHBr3, an analogous crystal structure of 22·CN3H6+·CN3H5·[Br(CHBr3)6]− complex was obtained in success and similar 1D assemblies and 2D networks were observed (Fig. S18 to S21†).
[X(CHCl3)6]− cluster-mediated rapid crystallization experiments carried out under more general conditions
All single crystals giving rise to the structurally characterized 22·CN3H6+·CN3H5·[X(CHCl3)6]− complexes (X = Cl−, Br−, I−) were grown over a period of several weeks. We were thus curious to explore whether labile [X(CHCl3)6]− clusters would support the robust co-crystallization of receptor 2 and guanidium halide in the presence of chloroform under more general conditions, such as those associated with rapid mixing and precipitation. To test this hypothesis, we sparged hexanes into a CHCl3/CH3OH (2:1, v/v) solution of 2 in the presence of 1 molar equivalent of guanidium chloride. This led to near immediate precipitation of what were found to be cubic crystalline particles as viewed under a polarizing microscope (Fig. S22 to S24†). These morphological features were further characterized by scanning electron microscopy (SEM) (Fig. 4a and ESI Fig. S25†). To gain greater insight into the nature of the cube-shaped microcrystalline material, SEM-EDS (energy dispersive spectrometry) experiments were performed. EDS elemental mapping associated with an SEM image revealed the occurrence of the expected elements (i.e., C, N, O, and Cl). This was taken as evidence that the microcrystals consisted of, at least, receptor 2 and guanidium chloride (Fig. S26†).
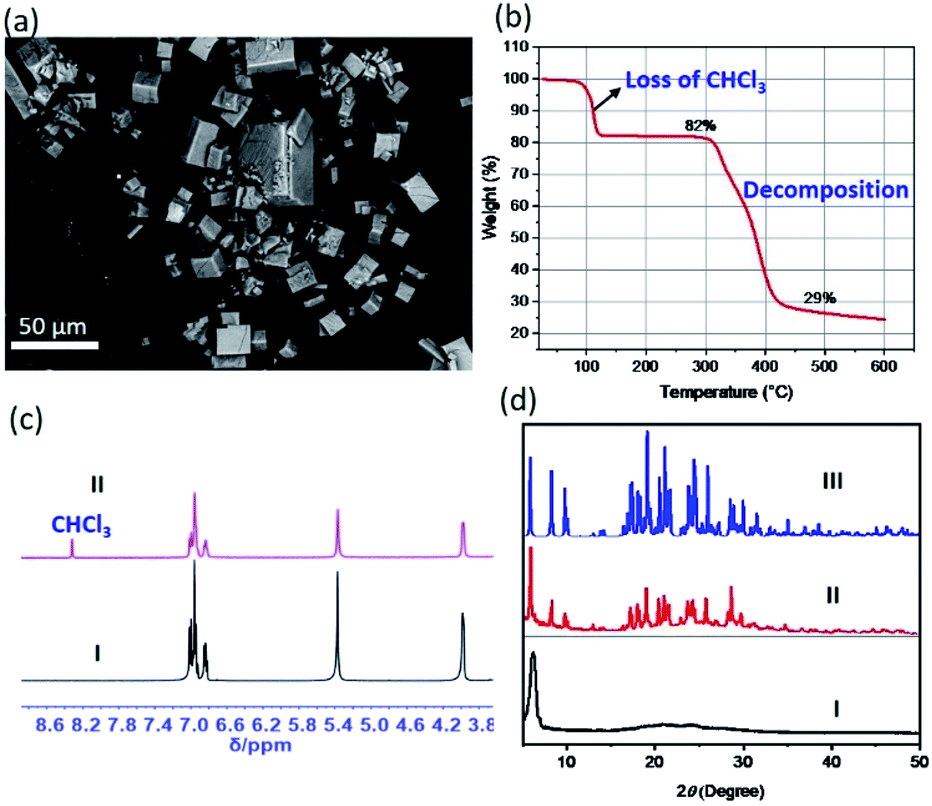 | ||
| Fig. 4 Characterization of [Cl(CHCl3)6]− cluster-mediated crystalline constructs under more general conditions. (a) SEM micrograph of cubic microcrystalline particles obtained by mixing 2 with guanidium chloride in the presence of CHCl3 under conditions of rapid precipitation (see text for details); (b) thermogravimetric decomposition trace for the particles in (a); (c) selected regions of the 1H NMR spectra of solutions of receptor 2 in DMSO-d6 (I) and the particles from (a) redissolved in DMSO-d6 (II); and (d) PXRD patterns of (I) solid obtained by drying 2 in the presence of CHCl3 but in the absence of a guest, (II) particles shown in (a), and (III) simulated PXRD pattern using the data from the single-crystal structure of complex 2·CN3H6+·CN3H5·[Cl(CHCl3)6]−. | ||
Thermogravimetric analyses (TGA), 1H NMR spectroscopic studies, and powder X-ray diffraction (PXRD) measurements were then carried out in an effort to confirm the inference that the microcrystalline material contained [Cl(CHCl3)6]− clusters. The above-mentioned precipitates were filtered off and dried naturally in a fume hood to allow evaporation of potential solvent residues on the surfaces. The resulting samples were subjected to TGA, NMR spectroscopic and PXRD analyses. Briefly, the TGA trace shows an initial mass loss of approximately 20% of the initial sample weight near 100 °C, a finding consistent with the release of CHCl3 from the crystalline materials (Fig. 4b). Additionally, a sharp CHCl3 peak was observed in the 1H NMR spectrum acquired by redissolving the microcrystals in DMSO-d6 (Fig. 4c). The observed content of CHCl3 in the precipitates were found slightly lower than that (22%) seen in the corresponding single crystal structures, which could be rationalized by the loss of CHCl3 during the sample preparation, drying and transfer due to the labile nature of the clusters (Fig. S27†). Finally, the PXRD pattern of the crystalline material was fully in accord with the simulated PXRD pattern using the diffraction data for the 2·CN3H6+·CN3H5·[Cl(CHCl3)6]− single crystals (Fig. 4d and S28†). In contrast, when chloroform was replaced by dichloromethane in the mixing of 2 with guanidium chloride, only column-like crystalline ensembles of 2 without guest species were observed (Fig. S29 and S30†). Taken together, these findings provide support for the notion that the acidic C–Hs of the CHCl3 serve as hydrogen bonding donors and play an essential role in holding together six chloroform molecules as labile octahedral halide anion solvate clusters (such as [Cl(CHCl3)6]−) that mediate formally the co-crystallization of 2 and guanidium chloride.
When mixtures of receptor 2 and guanidium bromide or guanidium iodide (in 1:1 ratios) were allowed to co-crystalize under otherwise identical conditions, cubic crystalline entities were also observed; again, the resulting constructs were fully characterized by microscopic techniques (Fig. S31–S33, S37 and S38†). The fact that [Br(CHCl3)6]− and [I(CHCl3)6]− clusters support the formation of cubic microcrystal materials was inferred from SEM-EDS experiments, thermogravimetric analysis, 1H NMR spectroscopy and powder X-ray diffraction studies as used for the [Cl(CHCl3)6]− cluster species described above (Fig. S33–S36 and S38–S41†).
Conclusions
In summary, a trimacrocyclic hexasubstituted benzene derivative 2 was synthesized via a Co2(CO)8-catalyzed [2 + 2 + 2] tricyclization of a monoyne-containing crown ether. This 3-fold symmetric system was found capable of trapping guanidium in a 1:1 ratio with an association constant of K = (1.6 ± 0.4) × 104 M−1 in a mixture of chloroform and methanol (9:1, v/v). A series of [X(CHCl3)6]− clusters, viz. [Cl(CHCl3)6]−, [Br(CHCl3)6]−, [I(CHCl3)6]−, and [Br(CHBr3)6]−, were observed to mediate the formation of 1D and 2D supramolecular entities as reflected in the corresponding single crystal X-ray diffraction-based structures. The octahedral halide chloroform clusters seen in the solid state proved key to the formation of cubic crystalline entities under conditions of rapid mixing and precipitation. To the best of our knowledge, this study represents the first example wherein labile halide solvate clusters serve to promote co-crystallization in the solid state. It may thus help advance our understanding how solvents affect organization at the molecular level while illustrating a new recognition motif that could prove useful in the creation of yet-more elaborate supramolecular architectures.
Data availability
All associated experimental and computational details are provided in the ESI.†Author contributions
Conceptualization and supervision: QH; synthesis, characterization, NMR, XRD, and TGA studies: ZL; single crystal growing, data collection and analysis: ZL and QH; theoretical calculations: AL; SEM-EDS experiments: SP; writing – original draft: ZL and QH; writing – review & editing, QH and JLS. All authors proofread, commented on, and approved the final version of this manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Science and Technology Plan Project of Hunan Province, China (Grant No. 2019RS1018 to Q. H.), the National Natural Science Foundation of China (22071050 and 21901069 to Q. H.), and Fundamental Research Funds for the Central Universities (Startup Funds to Q. H.). J. L. S. thanks the Robert A. Welch Foundation (F-0018) for financial support.Notes and references
- C. Q. Yuan, W. Ji, R. R. Xing, J. B. Li, E. Gazit and X. H. Yan, Nat. Rev. Chem., 2019, 3, 567–588 CrossRef CAS.
- D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, Nature, 2008, 451, 549–552 CrossRef CAS PubMed.
- D. V. Talapin, E. V. Shevchenko, A. Kornowski, N. Gaponik, M. Haase, A. L. Rogach and H. Weller, Adv. Mater., 2001, 13, 1868–1871 CrossRef CAS.
- Y. N. Xia, Y. J. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- X. B. Chen, A. C. S. Samia, Y. B. Lou and C. Burda, J. Am. Chem. Soc., 2005, 127, 4372–4375 CrossRef CAS PubMed.
- K. Margulis, X. Y. Zhang, L. M. Joubert, K. Bruening, C. J. Tassone, R. N. Zare and R. M. Waymouth, Angew. Chem., Int. Ed., 2017, 56, 16357–16362 CrossRef CAS PubMed.
- C. Knorowski, S. Burleigh and A. Travesset, Phys. Rev. Lett., 2011, 106, 215501 CrossRef CAS PubMed.
- M. Suzuki, J. F. K. Kotyk, S. I. Khan and Y. Rubin, J. Am. Chem. Soc., 2016, 138, 5939–5956 CrossRef CAS PubMed.
- R. Kaur, S. Sen, M. C. Larsen, L. Tavares, J. Kjelstrup-Hansen, M. Ishida, A. Zieleniewska, V. M. Lynch, S. Bahring, D. M. Guldi, J. L. Sessler and A. Jana, J. Am. Chem. Soc., 2020, 142, 11497–11505 CrossRef CAS PubMed.
- C. Caltagirone, N. L. Bill, D. E. Gross, M. E. Light, J. L. Sessler and P. A. Gale, Org. Biomol. Chem., 2010, 8, 96–99 RSC.
- H. Y. Gong, B. M. Rambo, E. Karnas, V. M. Lynch and J. L. Sessler, Nat. Chem., 2010, 2, 406–409 CrossRef CAS PubMed.
- H. Y. Gong, B. M. Rambo, V. M. Lynch, K. M. Keller and J. L. Sessler, J. Am. Chem. Soc., 2013, 135, 6330–6337 CrossRef CAS PubMed.
- A. B. Aletti, S. Blasco, S. J. Aramballi, P. E. Kruger and T. Gunnlaugsson, Chem, 2019, 5, 2617–2629 CAS.
- Q. He, G. I. Vargas-Zuniga, S. H. Kim, S. K. Kim and J. L. Sessler, Chem. Rev., 2019, 119, 9753–9835 CrossRef CAS PubMed.
- J. Li, K. Liu, Y. C. Han, B. Z. Tang, J. B. Huang and Y. Yan, ACS Appl. Mater. Inter., 2016, 8, 27987–27995 CrossRef CAS PubMed.
- S. C. Warren, L. C. Messina, L. S. Slaughter, M. Kamperman, Q. Zhou, S. M. Gruner, F. J. DiSalvo and U. Wiesner, Science, 2008, 320, 1748–1752 CrossRef CAS PubMed.
- S. Bai, C. Pappas, S. Debnath, P. W. J. M. Frederix, J. Leckie, S. Fleming and R. V. Ulijn, ACS Nano, 2014, 8, 7005–7013 CrossRef CAS PubMed.
- A. Uheida, Y. Zhang and M. Muhammed, Solvent Extr. Ion Exch., 2002, 20, 717–733 CrossRef CAS.
- M. Tokeshi, T. Minagawa and T. Kitamori, Anal. Chem., 2000, 72, 1711–1714 CrossRef CAS PubMed.
- H. Daud and R. W. Cattrall, Aust. J. Chem., 1982, 35, 1095–1103 CrossRef CAS.
- P. Botschwina, R. Oswald and V. Dyczmons, Int. J. Mass Spectrom., 2007, 267, 308–314 CrossRef CAS.
- S. Denifl, F. Zappa, I. Mahr, A. Mauracher, M. Probst, T. D. Mark and P. Scheier, J. Am. Chem. Soc., 2008, 130, 5065–5071 CrossRef CAS PubMed.
- P. V. Gushehin, G. L. Starova, M. Haukka, M. L. Kuznetsov, I. L. Eremenko and V. Y. Kukushkin, Cryst. Growth Des., 2010, 10, 4839–4846 CrossRef.
- D. M. Ivanov, A. S. Novikov, G. L. Starova, M. Haukka and V. Y. Kukushkin, Crystengcomm, 2016, 18, 5278–5286 RSC.
- T. V. Serebryanskaya, A. S. Novikov, P. V. Gushchin, M. Haukka, R. E. Asfin, P. M. Tolstoy and V. Y. Kukushkin, Phys. Chem. Chem. Phys., 2016, 18, 14104–14112 RSC.
- B. Watson, O. Grounds, W. Borley and S. V. Rosokha, Phys. Chem. Chem. Phys., 2018, 20, 21999–22007 RSC.
- K. Giles and E. P. Grimsrud, J. Phys. Chem., 1993, 97, 1318–1323 CrossRef CAS.
- X. Xie and R. E. McCarley, Inorg. Chem., 1997, 36, 4011–4016 CrossRef CAS.
- A. F. Hill, D. A. Tocher, A. J. P. White, D. J. Williams and J. D. E. T. Wilton-Ely, Organometallics, 2005, 24, 5342–5355 CrossRef CAS.
- M. Arunachalam, B. N. Ahamed and P. Ghosh, Org. Lett., 2010, 12, 2742–2745 CrossRef CAS PubMed.
- M. Arunachalam and P. Ghosh, Org. Lett., 2010, 12, 328–331 CrossRef CAS PubMed.
- S. Kondo, J. Masuda, T. Komiyama, N. Yasuda, H. Takaya and M. Yamanaka, Chem. –Eur. J., 2019, 25, 16201–16206 CrossRef CAS PubMed.
- R. Shukla, S. V. Lindeman and R. Rathore, J. Am. Chem. Soc., 2006, 128, 5328–5329 CrossRef CAS PubMed.
- S. Karabiyikoglu, B. A. Boon and C. A. Merlic, J. Org. Chem., 2017, 82, 7732–7744 CrossRef CAS PubMed.
- S. Kotha and G. T. Waghule, J. Org. Chem., 2012, 77, 6314–6318 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2068692–2068695 and 2088519. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03713g |
| This journal is © The Royal Society of Chemistry 2021 |