
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot fluorescent assay for sensitive detection of APOBEC3A activity†
Bingyao
Wang
a,
Xiong
Zhang
a,
Yafen
Wang
a,
Kun
Chen
 a,
Fang
Wang
b,
Xiaocheng
Weng
*a and
Xiang
Zhou
*a
a,
Fang
Wang
b,
Xiaocheng
Weng
*a and
Xiang
Zhou
*a
aThe Institute of Advanced Studies, College of Chemistry and Molecular Sciences, Key Laboratory of Biomedical Polymers of Ministry of Education, Hubei Province Key Laboratory of Allergy and Immunology, Wuhan University, Wuhan, Hubei 430072, China. E-mail: xzhou@whu.edu.cn; xcweng@whu.edu.cn; Fax: +86-27-68756663; Tel: +86-27-68756663
bWuhan University School of Pharmaceutical Sciences, Wuhan, 430071, China
First published on 30th April 2021
Abstract
We reported a one-pot fluorescence-based assay to quantitively detect A3A activity combined with cytosine deamination and uracil excision. After deamination by A3A and USER enzyme treatment, the fluorescent turn-on effect at 520 nm was observed, which can be used to evaluate the A3A activity and screen inhibitors.
Besides epigenetic modifications, gene expression can also be regulated by cytosine deamination by APOBECs (Apolipoprotein B mRNA-editing enzyme catalytic polypeptide-like).1 The APOBEC family, which consists of APOBEC1 (A1), APOBEC2 (A2), sub-family APOBEC3 (A3), APOBEC4 (A4) and activation-induced cytidine deaminase (AID), can catalyse the removal of an amino group from the cytosine (C) base to form uridine (U) in single strand DNA or RNA.2 The genetic alterations by APOBECs will change the transcription and mRNA processing, exhibiting diverse and important functions in human health.3,4
APOBECs were also found to be closely related to many diseases. Based on the deamination reaction of cytosine, APOBECs, in particular APOBEC3, can protect mammalian cells by providing defence against the replication of retroviruses and DNA viruses such as HIV, murine leukemia viruses (MLV), hepatitis B virus, herpesviruses, and human papillomavirus (HPV).5–10 However, this ability is a kind of double-edged sword where the overexpression of these proteins could lead to the development of cancer, for instance, breast cancer, clear cell ovarian cancer (CCOC), bladder cancer and so on.11–15 The misregulation of these proteins was shown to affect cellular localization, DNA targeting accuracy and failure to maintain genomic integrity. Among others, APOBEC3A (A3A) is known as the main source of APOBEC-signature mutations in tumors.16,17 Recent research found that a large proportion of the Asian, Amerindian, and Oceania population is associated with A3A overexpression, which may be responsible for their increased risk for cancer.18 Therefore, the detection of APOBEC activity is important to understand its relationship with malignancy.
Up to now, there are only a few ways to detect APOBEC activity. Almost all methods were designed based on the monitoring of the C to U deamination reaction on DNA oligos by APOBEC or followed uracil excision. Through a variety of measurement techniques, such as Nuclear Magnetic Resonance (NMR), gel electrophoresis or droplet digital PCR, the activity of APOBEC can be evaluated.17,19–22 The above methods are somewhat complicated, laborious and require specific instruments, resulting in difficulty for inhibitor discovery.23 In contrast, a fluorescence deamination assay based on FRET (Förster resonance energy transfer) is cheap, sensitive and easy to operate, and has been exploited to develop many innovative and powerful methods for nucleic acid or protein detection,24–26 including APOBECs.27 However, the present FRET-based deamination assay still has sufficient space for improvement. Above all, after the conversion from cytidine to the AP site, NaOH was used to cleave the AP site, which is not compatible with most biological systems and is harmful to DNA, proteins and the relative buffer or fluorophores. Next, HCl was needed to neutralize the pH, which makes the assay more complicated, and may lead to bias for fluorescent detection. Thus, although the present FRET-based assay is effective, the employment of NaOH requires several steps to perform the APOBEC activity assay, which brings a barrier for high throughput screening of inhibitors.
To reduce the experimental steps, here we introduced a one-pot fluorescent detection method for APOBEC3A activity with low background noise using a commercially available USER (Uracil-Specific Excision Reagent) enzyme, which is a mixture of Uracil DNA glycosylase (UDG) and DNA glycosylase-lyase endonuclease VIII. This method can provide a gentle way to cleave DNA after the deamination of A3A. The one-pot assay, which can mix all ingredients together, is more convenient for the screening of a large number of inhibitors in a single assay. Without harsh reaction conditions such as the use of NaOH, we optimized the assay to develop a one-step detection system in one tube, which makes the manipulation easier. By adding all materials at one time, the fluorescence can be recorded after a period of time. Following this design, A3A will catalyse the deamination of cytidine to uridine in the ssDNA probe. Then the USER enzyme in the mixture will firstly generate abasic sites (AP), and then cleave the ssDNA probe into two fragments. Because the fluorophore and quencher molecule in the ssDNA probe are far apart from each other after cleavage of the ssDNA probe, the FRET effect between them disappears, resulting in an increase of the fluorescence signal. Altogether, this fluorescent turn-on system can be applied to detect the activity of A3A (Scheme 1).
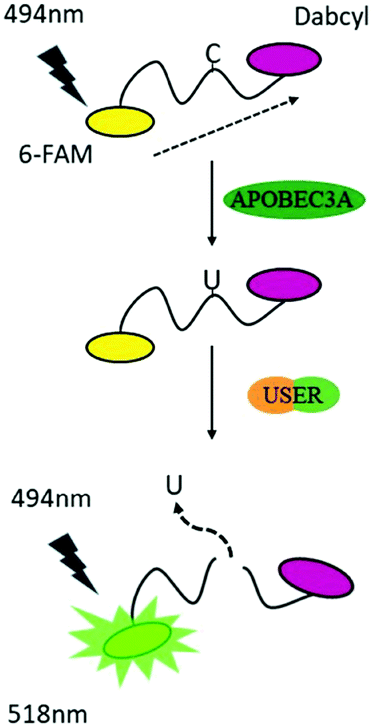 | ||
| Scheme 1 A strategy for detection of the activity of A3A. | ||
To obtain the best turn-on performance, the length of the ssDNA probe was firstly optimized. Different lengths of ssDNA probes with 6-FAM at the 5′-terminal and 3′-dabcyl at the 3′-terminal (DNA-C1) were designed. The sequence of TTCA which is the favoured sequence of A3A was included in all ssDNA probes.2 This indicated that the 6 nt DNA probe exhibited the maximum increase in fluorescence intensity (Fig. S1, ESI†). This is not surprising because the shorter distance between the fluorophore and quenching group has better FRET performance.28 The results of gel electrophoresis verified that the turn on effect was induced by cleavage of the ssDNA probe. Next, the reaction time was optimized. After the addition of the DNA probe and all enzymes together, the fluorescence spectra were recorded in different incubation times. The result showed that the highest fluorescence intensity was achieved at 8 h, which means the complete degradation of ssDNA (Fig. S2, ESI†). So, the 6 nt oligonucleotide DNA-C1 and 8 h incubation time were selected in further investigations.
Within our expectations, the fluorescent turn on phenomenon was only observed with the incubation of the DNA probe, A3A and USER together, while no change was seen with A3A or USER enzyme alone (Fig. 1a). These results further identify the feasibility of our design. All reaction products were analysed by denaturing polyacrylamide gel electrophoresis (PAGE) because cleaved ssDNA oligo showed faster migration. The result was in consensus with the fluorescence data while only the ssDNA probe in lane 4 was cleaved compared to the others (Fig. 1b). What's more, the fluorescence of the reaction system can be observed by the naked eye under UV irradiation (Fig. 1c).
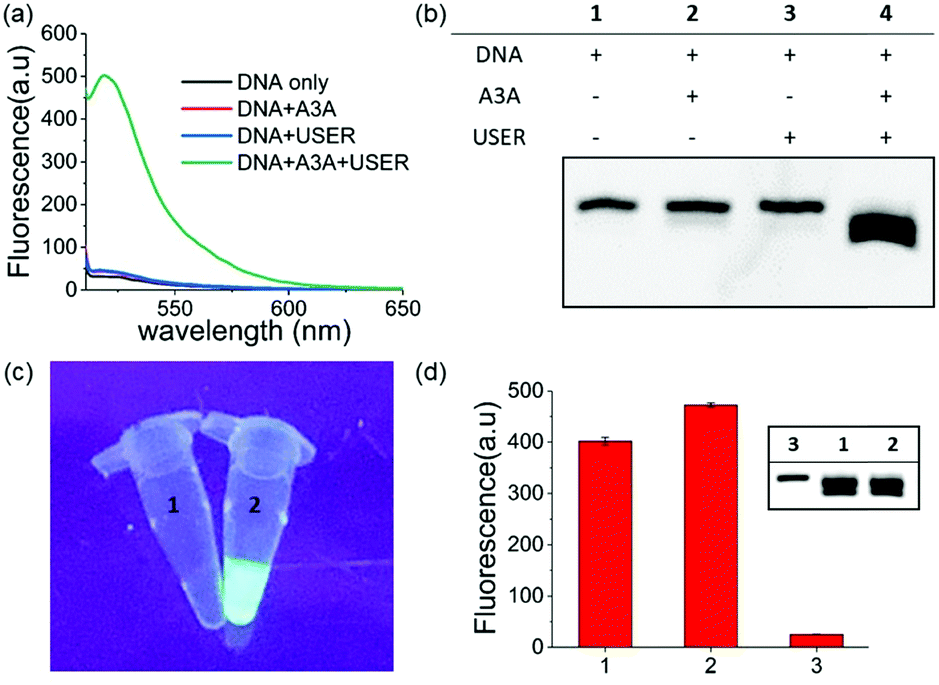 | ||
| Fig. 1 (a) Fluorescence spectra of different sensing systems. (b) PAGE analysis of the detection of A3A activity strategy. (c) Images of ssDNA untreated (1) and treated (2) by A3A/USER under UV light. (d) The fluorescence of the one step and multi-step detection strategy. Multi-step strategy (1 and lane 1); one step strategy (2 and lane 2); DNA without any treatment (3 and lane 3). | ||
As mentioned above, whether all steps of this assay can be fulfilled in one-pot and one tube is important to increase its application prospects, in particular for inhibitor screening. The mild reaction conditions in our assay provide the possibility to accomplish this goal. Then we added all reagents into the reaction buffer (25 mM HEPES buffer, pH 6.5, 1× CutSmart® buffer, pH 7.9) at one time and incubated it at 37 °C for 8 h. Compared with the experiments performed in multi-steps, even higher fluorescence intensity can be achieved in the one-pot experiment (Fig. 1d). This meant that the assay in this work is easier to be operated and showed wide application potential.
The reaction conditions of this one-pot assay were optimized in the above studies. The sensitivity and selectivity of this method were next investigated. As shown in Fig. 2a, the fluorescence was gradually enhanced as the concentration of A3A increased. In two A3A concentration ranges (from 0.005 μM to 0.03 μM and from 0.03 μM to 0.2 μM), good linear correlations between fluorescence and A3A concentration were observed (Fig. 2b). The detection limit of this method is as low as 5 nM of A3A in this system. The two linear ranges might be related to the feature of the enzyme. At the beginning of the enzymatic reaction, while most of A3A was in free status, the increase of A3A would lead to a fast enhancement of fluorescence. But when the A3A was almost saturated, the increase of A3A would lead to a slow increase of fluorescence. This kind of phenomenon was also reported in the detection of other enzymes such as polynucleotide kinase.29
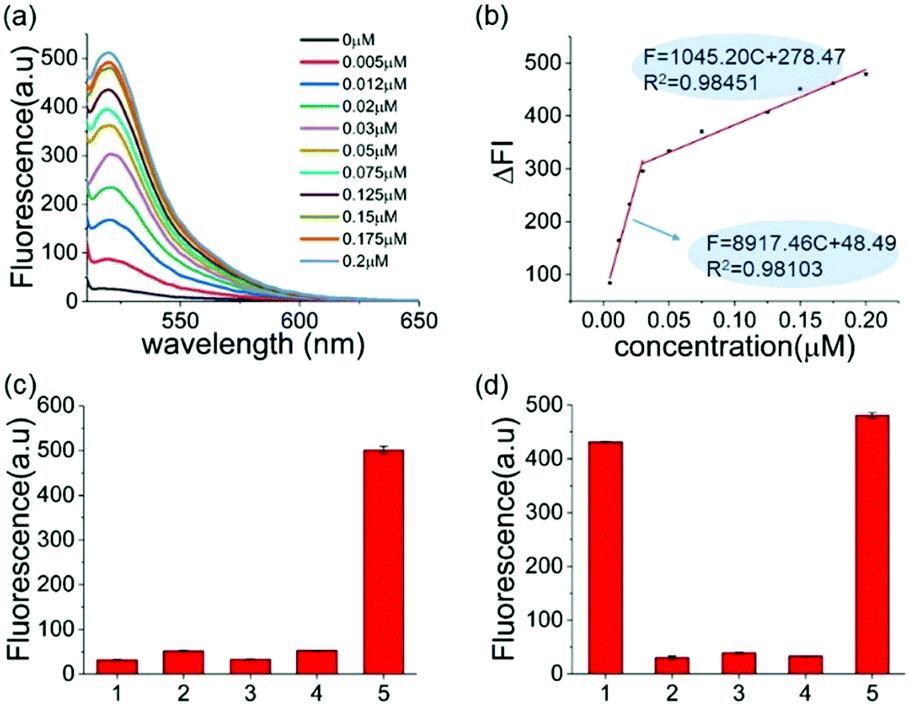 | ||
| Fig. 2 (a) Fluorescence spectra of the sensing system containing different amounts of A3A. (b) A linear relationship between the fluorescence intensity at 494 nm and the concentration of the A3A. (c) The selectivity of A3A to DNA-G. (1) DNA only; (2) DNA-G + A3A; (3) DNA-G + USER; (4) DNA-G + A3A + USER; (5) DNA-C1 + A3A + USER. (d) The selectivity of different proteins: (1) A3A; (2) BSA; (3) Klenow polymerase; (4) T4 PNK; (5) a mixture of the four proteins. (BSA) = 5 μM, the concentration of the other three enzymes was 50 U mL−1. | ||
To further prove that the fluorescence turn-on is induced by cytosine deamination, we changed the ssDNA probe without cytidines. DNA-G (GTTGAA) with guanine instead of cytosine was designed. Compared with the result with DNA-C1 within cytosine in the DNA probe, no obvious fluorescence enhancement can be observed for the DNA-G probe, whose fluorescence intensity was similar to the background. This indicated that the fluorescence turn-on was firstly induced by cytosine deamination, suggesting that this assay can indeed detect A3A (Fig. 2c). Then we investigated the selectivity of the assay and evaluated the influence by other enzymes or proteins such as BSA, Klenow polymerase and T4 PNK. No fluorescence turn-on can be obtained in these proteins unless in the mixture with A3A, indicating a good selectivity of this assay (Fig. 2d).
To further verify the feasibility of the strategy for A3A detection and the potential for inhibitor screening, we used aurintricarboxylic acid (MN1, Fig. 3a), a previously reported A3A inhibitor, to study the influence on this assay.27 We first incubated A3A with different concentrations of MN1 for 1 h. Then other component parts were added to the mixture and incubated for another 6 h at 37 °C. If A3A were inhibited by compound MN1, the cytosine deamination would be suppressed, which would result in less cleavage of the ssDNA probe and lower fluorescence intensity. The relative fluorescence intensity was in line with expectations, and decreased with the increase of the concentration of MN1 (Fig. 3b). The IC50 value, half-maximal inhibitory concentration, was calculated as 40.5 μM from the plot of the relative activity of A3A versus MN1 concentration, which is 0.03 μM for 0.0038 μM A3A in other literature.27 Then we chose five compounds (MN1, MN2, 17-AAG, PES, MN3, Scheme S1, ESI†) as the model to simulate the high-throughput screen (HTS) for small molecule inhibitors of A3A catalytic activity in 384-well plates. The solution of ssDNA probe, A3A and USER enzymes were separately mixed with each compound, and then the fluorescence was recorded by the fluorescence plate reader. As shown in Fig. 3c, different compounds exhibited various inhibition efficiencies. Among them, compound MN1 showed the best inhibition ability. The catalytic activity of A3A was almost totally inhibited by MN1 in this concentration (100 μM). Apart from MN3, no obvious fluorescence decline was obtained by the other three compounds compared with the control of sample 6, suggesting that only MN3 has a weak inhibitory effect. We also compared the activity of these compounds through a one-pot assay or multi-step assay to verify the reproducibility. All of them showed very similar inhibition results (Fig. 3d), which proved that our one-pot method could be used for the HTS of inhibitors.
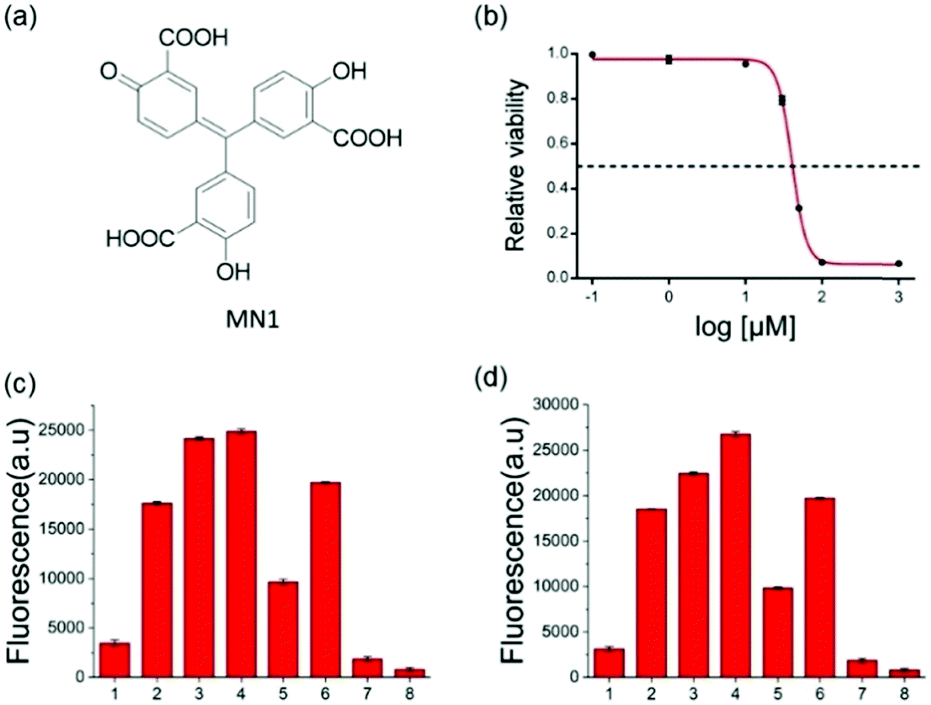 | ||
| Fig. 3 (a) The structure of MN1. (b) Representative A3A inhibitor dose–response assays. MN1 was chosen as the inhibitor of A3A (0.5 μM). The curve shows the relative activity of A3A in the presence of an increasing concentration of MN1. The concentrations of MN1 were 0.1 μM, 1 μM, 10 μM, 30 μM, 50 μM, 100 μM, and 1 mM. (c and d) The HTS assay to detect A3A activity through multi-steps (c) and one step (d). The meaning of the numbers in c and d are identical. 1 to 5 means the different compounds (100 μM): (1) MN1; (2) MN2; (3) 17-AAG; (4) PES; (5) MN3. (6) DNA-C1 + A3A + USER; (7) DNA-C1; (8) buffer. | ||
In addition to the in vitro assay and inhibitor screening, we extended this method to eukaryotic cell lysates. Two cell lines, HeLa and HEK 293T cells, were chosen in this study. After removing the culture media, the cells were washed and then lysed using a lysing buffer to prepare the lysates.17 The concentration of the cell lysates was determined by BCA protein assay. A normalized amount of the cell lysates was mixed and incubated with ssDNA probe or other constituents for 6 h at 37 °C, and then the fluorescence detection was performed. However, without USER Enzyme, the samples of ssDNA probe with cell lysates (samples 3 and 6, Fig. 4) also exhibited obvious fluorescence compared to the negative control (samples 1 and 2). This may be induced by the pre-existing uracil excision enzymes inside the cell. To identify whether the fluorescence enhancement was induced by cytosine deamination and uracil excision, we used a known uracil glycosylase inhibitor (UGI) to inhibit endogenous UDG in the cell lysates.30 After the addition of UGI, the fluorescence was decreased both in the HeLa and HEK 293T cells (samples 4 and 7). The reason why there was still some background might be due to the incomplete inhibition of UGI to UDG enzyme, or the degradation of DNA probe by other cellular components. Then extra USER Enzyme was added to compensate for the UDG inhibition by UGI, generating a recovery of the fluorescent signal (samples 5 and 8). The decline and restoration of fluorescence indicated that the fluorescent change was relative to the uracil excision and cytosine deamination, which can be used to preliminarily evaluate the catalytic ability of APOBECs. From the results, it was demonstrated that the content of APOBECs in HeLa cells is higher than in HEK 293T cells.
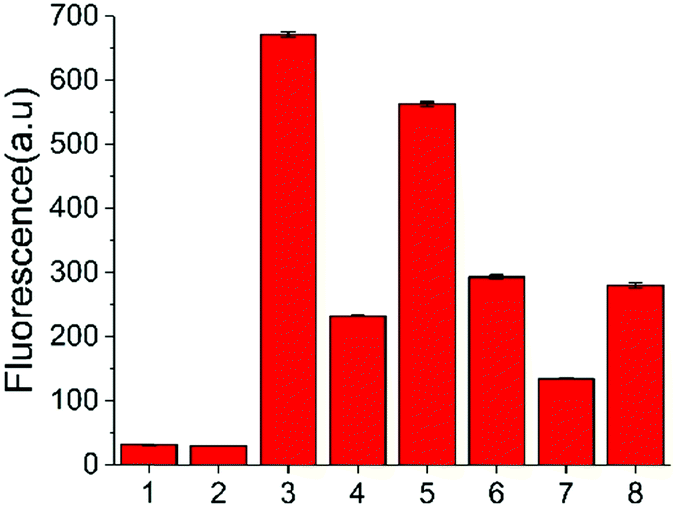 | ||
| Fig. 4 Fluorescence intensity of the sensing system containing cell lysates extracted from HeLa and HEK 297T cells (2 μg of protein). (1) DNA-C1; (2) DNA-C1 + USER; 3 to 5 used the cell lysates from HeLa cells: (3) DNA-C + cell lysates; (4) DNA-C + cell lysates + UGI; (5) DNA-C + cell lysates + UGI + USER; 6 to 8 used the cell lysates from HEK 293T: (6) DNA-C + cell lysates; (7) DNA-C + cell lysates + UGI; (8) DNA-C + cell lysates + UGI + USER. | ||
In conclusion, we have presented a convenient and effective strategy to detect the activity of APOBEC3A. In this strategy, we use the USER enzyme instead of NaOH/HCl to cleave DNA, which can avoid the side-effects of NaOH/HCl and allow the assay to be performed in a one-pot manner. In addition, the length of the ssDNA reporter was optimized and shortened to only 6 nt, exhibiting a stronger turn on performance compared to the present method. Next, we proved the feasibility of this strategy using the known A3A inhibitor MN1 and simply simulating this strategy for HTS. Based on the advantages of the one-step assay and enhanced turn on effect, this method is more convenient than the existing assay for APOBECs and competent for high-throughput inhibitor screening. Finally, the rough evaluation of APOBEC activity in cell lysates was preliminarily investigated. With increasing attention towards the deamination events in recent years, we hope that this new assay will provide a simple route for activity detection of APOBECs and the screening of new inhibitors.
This work was financially supported by the National Natural Science Foundation of China (NSFC) projects (21822704, 21778040 and 91940304 to X. W.; 22037004, 91753201 and 21721005 to X. Z.; 21778041 to F. W.).
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. A. Carpenter, M. Li, A. Rathore, L. Lackey, E. K. Law, A. M. Land, B. Leonard, S. M. Shandilya, M. F. Bohn, C. A. Schiffer, W. L. Brown and R. S. Harris, J. Biol. Chem., 2012, 287, 34801 Search PubMed.
- T. V. Silvas and C. A. Schiffer, Protein Sci., 2019, 28, 1552 Search PubMed.
- B. D. Anderson and R. S. Harris, Sci. Adv., 2015, 1, e1500296 Search PubMed.
- M. J. Wichroski, G. B. Robb and T. M. Rana, PLoS Pathog., 2006, 2, e41 Search PubMed.
- R. S. Harris, K. N. Bishop, A. M. Sheehy, H. M. Craig, S. K. Petersen-Mahrt, I. N. Watt, M. S. Neuberger and M. H. Malim, Cell, 2003, 113, 803 Search PubMed.
- E. M. Janahi and M. J. McGarvey, J. Viral Hepatitis, 2013, 20, 821 Search PubMed.
- A. M. Sheehy, N. C. Gaddis, J. D. Choi and M. H. Malim, Nature, 2002, 418, 646 Search PubMed.
- R. Suspene, M. M. Aynaud, S. Koch, D. Pasdeloup, M. Labetoulle, B. Gaertner, J. P. Vartanian, A. Meyerhans and S. Wain-Hobson, J. Virol., 2011, 85, 7594 Search PubMed.
- V. C. Vieira, B. Leonard, E. A. White, G. J. Starrett, N. A. Temiz, L. D. Lorenz, D. Lee, M. A. Soares, P. F. Lambert, P. M. Howley and R. S. Harris, mBio, 2014, 5, e02234 Search PubMed.
- Z. Wang, K. Wakae, K. Kitamura, S. Aoyama, G. Liu, M. Koura, A. M. Monjurul, I. Kukimoto and M. Muramatsu, J. Virol., 2014, 88, 1308 Search PubMed.
- M. B. Burns, L. Lackey, M. A. Carpenter, A. Rathore, A. M. Land, B. Leonard, E. W. Refsland, D. Kotandeniya, N. Tretyakova, J. B. Nikas, D. Yee, N. A. Temiz, D. E. Donohue, R. M. McDougle, W. L. Brown, E. K. Law and R. S. Harris, Nature, 2013, 494, 366 Search PubMed.
- A. A. Serebrenik, P. P. Argyris, M. C. Jarvis, W. L. Brown, M. Bazzaro, R. I. Vogel, B. K. Erickson, S.-H. Lee, K. M. Goergen, M. J. Maurer, E. P. Heinzen, A. L. Oberg, Y. Huang, X. Hou, S. J. Weroha, S. H. Kaufmann and R. S. Harris, Clin. Cancer Res., 2020, 26, 3397 Search PubMed.
- M.-J. Shi, X.-Y. Meng, P. Lamy, A. R. Banday, J. Yang, A. Moreno-Vega, C.-L. Chen, L. Dyrskjøt, I. Bernard-Pierrot, L. Prokunina-Olsson and F. Radvanyi, Eur. Urol., 2019, 76, 9 Search PubMed.
- M. B. Burns, N. A. Temiz and R. S. Harris, Nat. Genet., 2013, 45, 977 Search PubMed.
- S. Nik-Zainal, L. B. Alexandrov, D. C. Wedge, P. Van Loo, C. D. Greenman, K. Raine, D. Jones, J. Hinton, J. Marshall, L. A. Stebbings, A. Menzies, S. Martin, K. Leung, L. Chen, C. Leroy, M. Ramakrishna, R. Rance, K. W. Lau, L. J. Mudie, I. Varela, D. J. McBride, G. R. Bignell, S. L. Cooke, A. Shlien, J. Gamble, I. Whitmore, M. Maddison, P. S. Tarpey, H. R. Davies, E. Papaemmanuil, P. J. Stephens, S. McLaren, A. P. Butler, J. W. Teague, G. Jönsson, J. E. Garber, D. Silver, P. Miron, A. Fatima, S. Boyault, A. Langerød, A. Tutt, J. W. Martens, S. A. Aparicio, Å. Borg, A. V. Salomon, G. Thomas, A.-L. Børresen-Dale, A. L. Richardson, M. S. Neuberger, P. Andrew, P. J. Campbell and M. R. Stratton, Cell, 2012, 149, 979 Search PubMed.
- L. B. Alexandrov, S. Nik-Zainal, D. C. Wedge, S. A. J. R. Aparicio, S. Behjati, A. V. Biankin, G. R. Bignell, N. Bolli, A. Borg, A.-L. Børresen-Dale, S. Boyault, B. Burkhardt, A. P. Butler, C. Caldas, H. R. Davies, C. Desmedt, R. Eils, J. E. Eyfjörd, J. A. Foekens, M. Greaves, F. Hosoda, B. Hutter, T. Ilicic, S. Imbeaud, M. Imielinski, N. Jäger, D. T. W. Jones, D. Jones, S. Knappskog, M. Kool, S. R. Lakhani, C. López-Otín, S. Martin, N. C. Munshi, H. Nakamura, P. A. Northcott, M. Pajic, E. Papaemmanuil, A. Paradiso, J. V. Pearson, X. S. Puente, K. Raine, M. Ramakrishna, A. L. Richardson, J. Richter, P. Rosenstiel, M. Schlesner, T. N. Schumacher, P. N. Span, J. W. Teague, Y. Totoki, A. N. J. Tutt, R. Valdés-Mas, M. M. Van Buuren, L. Van, T. Veer, A. Vincent-Salomon, N. Waddell, L. R. Yates, J. Zucman-Rossi, P. Andrew Futreal, U. McDermott, P. Lichter, M. Meyerson, S. M. Grimmond, R. Siebert, E. Campo, T. Shibata, S. M. Pfister, P. J. Campbell and M. R. Stratton, Nature, 2013, 500, 415 Search PubMed.
- P. Jalili, D. Bowen, A. Langenbucher, S. Park, K. Aguirre, R. B. Corcoran, A. G. Fleischman, M. S. Lawrence, L. Zou and R. Buisson, Nat. Commun., 2020, 11, 1 Search PubMed.
- J. D. Salter, R. P. Bennett and H. C. Smith, Trends Biochem. Sci., 2016, 41, 578 Search PubMed.
- A. Furukawa, T. Nagata, A. Matsugami, Y. Habu, R. Sugiyama, F. Hayashi, N. Kobayashi, S. Yokoyama, H. Takaku and M. Katahira, EMBO J., 2009, 28, 440 Search PubMed.
- S. Harjes, W. C. Solomon, M. Li, K. M. Chen, E. Harjes, R. S. Harris and H. Matsuo, J. Virol., 2013, 87, 7008 Search PubMed.
- M. A. Carpenter, M. Li, A. Rathore, L. Lackey, E. K. Law, A. M. Land, B. Leonard, S. M. D. Shandilya, M.-F. Bohn, C. A. Schiffer, W. L. Brown and R. S. Harris, J. Biol. Chem., 2012, 287, 34801 Search PubMed.
- R. Buisson, A. Langenbucher, D. Bowen, E. E. Kwan, C. H. Benes, L. Zou and M. S. Lawrence, Science, 2019, 364, eaaw2872 Search PubMed.
- B. Zhang, B. Q. Liu, D. P. Tang, R. Niessner, G. N. Chen and D. Knopp, Anal. Chem., 2012, 84, 5392 Search PubMed.
- T. T. Huang, J. Wang, Q. S. Wang, D. P. Tang and Y. Lin, Microchim. Acta, 2020, 187, 563 Search PubMed.
- S. Z. Lv, Y. Tang, K. Y. Zhang and D. P. Tang, Anal. Chem., 2018, 90, 14121 Search PubMed.
- Q. Zhou, Y. X. Lin, M. D. Xu, Z. Q. Gao, H. H. Yang and D. P. Tang, Anal. Chem., 2016, 88, 8886 Search PubMed.
- M. Li, S. M. D. Shandilya, M. A. Carpenter, A. Rathore, W. L. Brown, A. L. Perkins, D. A. Harki, J. Solberg, D. J. Hook, K. K. Pandey, M. A. Parniak, J. R. Johnson, N. J. Krogan, M. Somasundaran, A. Ali, C. A. Schiffer and R. S. Harris, ACS Chem. Biol., 2012, 7, 506 Search PubMed.
- A. Plochowietz, R. Crawford and A. N. Kapanidis, Phys. Chem. Chem. Phys., 2014, 16, 12688 Search PubMed.
- Y.-P. Zhang, Y.-X. Cui, X.-Y. Li, Y.-C. Du, A.-N. Tang and D.-M. Kong, Chem. Commun., 2019, 55, 7611 Search PubMed.
- Y.-C. Du, Y.-X. Cui, X.-Y. Li, G.-Y. Sun, Y.-P. Zhang, A.-N. Tang, K. Kim and D.-M. Kong, Anal. Chem., 2018, 90, 8629 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cb00076d |
| This journal is © The Royal Society of Chemistry 2021 |