
Structural crystallisation of crosslinked 3D PEDOT:PSS anisotropic porous biomaterials to generate highly conductive platforms for tissue engineering applications†
Matteo
Solazzo
 ab and
Michael G.
Monaghan
*abcd
ab and
Michael G.
Monaghan
*abcd
aDepartment of Mechanical, Manufacturing and Biomedical Engineering, Trinity College Dublin, Dublin 2, Ireland. E-mail: monaghmi@tcd.ie
bTrinity Centre for Biomedical Engineering, Trinity College Dublin, Dublin 2, Ireland
cAdvance Materials and BioEngineering Research (AMBER) Centre at Trinity College Dublin and the Royal College of Surgeons in Ireland, Dublin 2, Ireland
dCÚRAM, Centre for Research in Medical Devices, National University of Ireland, Galway, Newcastle Road, H91 W2TY Galway, Ireland
First published on 19th February 2021
Abstract
An emerging class of materials finding applications in biomaterials science – conductive polymers (CPs) – enables the achievement of smarter electrode coatings, piezoresistive components within biosensors, and scaffolds for tissue engineering. Despite their advances in recent years, there exist still some challenges which have yet to be addressed, such as long-term stability under physiological conditions, adequate long-term conductivity and optimal biocompatibility. Additionally, another hurdle to the use of these materials is their adaptation towards three-dimensional (3D) scaffolds, a feature that is usually achieved by virtue of applying CPs as a functionalised coating on a bulk material. Poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) is by far one of the most promising CPs in terms of its stability and conductivity, with the latter capable of being enhanced via a crystallisation treatment using sulphuric acid. In this work, we present a new generation of 3D electroconductive porous biomaterial scaffolds based on PEDOT:PSS crosslinked via glycidoxypropyltrimethoxysilane (GOPS) and subjected to sulphuric acid crystallisation. The resultant isotropic and anisotropic crystallised porous scaffolds exhibited, on an average, a 1000-fold increase in conductivity when compared with the untreated scaffolds. Moreover, we also document a precise control over the pore microarchitecture, size and anisotropy with high repeatability to achieve both isotropic and aligned scaffolds with mechanical and electrical anisotropy, while exhibiting adequate biocompatibility. These findings herald a new approach towards generating anisotropic porous biomaterial scaffolds with superior conductivity through a safe and scalable post-treatment.
Introduction
The use of biomaterials in medical implants, sensors, devices and tissue engineered constructs is continuously evolving with more advanced materials (synthetic and natural) and novel manufacturing approaches emerging to meet new requirements and demands.1One such emerging group of biomaterials is that of intrinsically conductive polymers (CPs).2 These materials exist in many forms and can be fabricated into films,3 coatings,4 hydrogels5 and scaffolds.6,7 CP-based three-dimensional (3D) scaffolds can be challenging to fabricate as many CPs are thermoset materials, deeming them inappropriate for extrusion-based techniques, which primarily rely on polymer melting. One class of CPs of interest is polythiophenes, which can exist in a dispersed form in non-polar solvents. Poly(3,4-ethylenedioxythiophene) (PEDOT) is one such polythiophene conjugated polymer and it is the most widely investigated one because of its availability as a dispersion in aqueous solutions with polystyrene sulfonate (PSS) as a primary counterion. Despite this formulation impeding somewhat the original conductivity of pure PEDOT, the resulting material is relatively easy to be processed while offering exhibiting chemical stability.8,9 In addition to its use in electronic devices,10 it has emerged as a candidate in biomedical applications ranging from fabrication of scaffolds for tissue engineering10 to neural implants.11
The use of PEDOT:PSS in creating 3D scaffolds is limited by its inherent property of being dispersible in aqueous solutions, and for this reason, two main approaches have been adopted so far. First, the combination of the electroconductive compound within a bulk matrix, with one example, whereby EDOT was polymerised in the presence of alginate to achieve porous PEDOT alginate scaffolds with conductivity in the order of S m−1.12 Second, via chemically crosslinking of PEDOT:PSS slurries with crosslinking agents such as glycidoxypropyltrimethoxysilane (GOPS).10,13 The latter approach poses drawbacks in that a rather brittle structure is attained which at the same time compromises the overall conductivity of the scaffold. For instance, PEDOT:PSS films crosslinked with a content of GOPS greater than 1 v/v% have a 500-fold reduction in conductivity.14 Regardless, GOPS has demonstrated great potential in the fields of tissue engineering and biomaterials due to its adequate cytocompatibility and ease of processing.
Towards achieving improved electrical conductivities of PEDOT:PSS, several post-treatments have been reported,15 with the highest values to date resulting from the treatment of spin-coated PEDOT:PSS with sulphuric acid, a process that has adopted the nomenclature ‘crystallisation’ due to the stacked interwoven nanostructure imparted on the material.16 With this approach, it has been determined that the exposure of dried PEDOT:PSS with sulphuric acid removes PSS and generates nanofibrils of PEDOT from its characteristic grain-like morphology via a structural transition mechanism. Since this initial report, this crystallisation process has been applied in vitro17 and used in the generation of 3D hydrogels.18
In this study, we sought to establish if, in scaffolds previously crosslinked and annealed using GOPS, the use of sulphuric acid could achieve such crystallisation and circumnavigate the drawback imparted upon conductivity by GOPS. Therefore, we hypothesize that sulphuric crystallisation can enhance conductivity of GOPS crosslinked PEDOT:PSS scaffolds, while retaining their stability and cytocompatibility.
As PEDOT:PSS is water dispersible, it is an ideal candidate for the lyophilisation process when crosslinked, through which one can have control over pore size, quantity and directionality. Indeed, directional lyophilisation of GOPS crosslinked PEDOT:PSS scaffolds has been previously reported, yet with modest conductivities.6 Here, we document a new generation of sulphuric crystallised GOPS-crosslinked PEDOT:PSS porous scaffolds with a significant increase in conductivity and elasticity, while at the same time maintaining biocompatibility. We also present a detailed characterization of scaffold anisotropy not limited to the morphology but also extending to its mechanical and electroconductive properties.
Results and discussion
Crystallisation of 3D porous scaffolds increases their overall porosity and volume and can be fabricated into anisotropic porous scaffolds
High porosity, adequate pore size and fibre orientation are some of the topological requirements for tissue engineering scaffolds, together with suitable stiffness.2 With the ultimate goal of obtaining control over these features, several advanced manufacturing techniques have been implemented ranging from salt leaching, thermal induced phase separation, electrospinning and the more recent advent of additive manufacturing via exploitation of fused deposition modelling,19 and even more recently, melt electrospinning writing.20 In this context, based on clear and defined concepts of physics and thermodynamics, lyophilisation (also known as ice-templating or freeze-drying) has been demonstrated as an appropriate technique to achieve a high level of porosity and anisotropy within scaffolds. PEDOT:PSS is commonly dispersed in water, making it extremely suitable for freeze-drying. In this study, 3D GOPS-crosslinked PEDOT:PSS scaffolds were fabricated using a freeze-drying technique similar to that reported by others10,13 and were subjected to a sulphuric crystallisation treatment reported previously in two-dimensional coatings of PEDOT:PSS.16 The fabrication of these scaffolds enabled the creation of a broad range of sizes, shapes and anisotropy as shown in Fig. 1A and Fig. S1A.†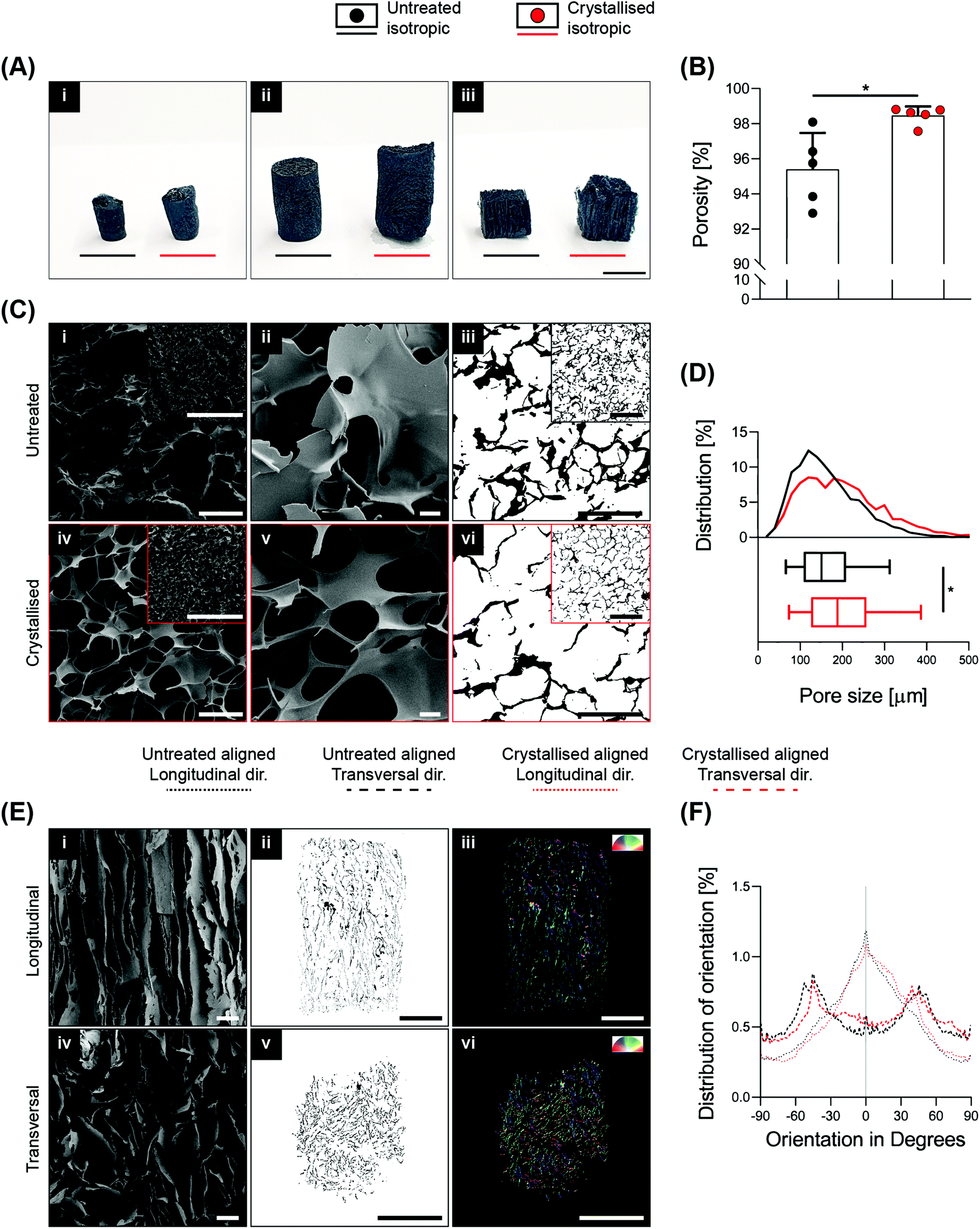 | ||
| Fig. 1 Processing effects on PEDOT:PSS 3D scaffolds. (A) Images of three pairs of freeze-dried scaffolds of different shapes, demonstrating both untreated and crystallised examples. i: “Isotropic small”, ii: “isotropic big” and iii: “aligned”. (B) Porosity of isotropic scaffolds evaluated by the ethanol intrusion method (n = 5). (C) Evaluation of the microarchitecture on isotropic scaffolds via SEM micrographs (C i/ii/iv/v) and sliced agarose embedded scaffolds (C iii/vi). (D) Pore size quantification from the analysis of sliced agarose embedded scaffolds (n = 3 per direction). Top: distribution of pore size as a percentage; bottom: cumulative mean and standard deviation. (E) Evaluation of the microarchitecture on aligned scaffolds via SEM micrographs (E i/iv) and sliced agarose embedded scaffolds (E ii/iii/v/vi). E iii/vi: colour survey representation of fibre orientation via OrientationJ (ImageJ plugin). (F) Distribution of orientation, with 0° representing the fibre direction in the longitudinal view (n = 3 per direction). Scale bars: A = 1 cm; C i/ii/iv/v = 100 μm; C i/iv inset = 1 mm; C iii/vi = 200 μm; C iii/vi inset = 500 μm; E i/iv = 200 μm; E ii/iii/v/vi = 2 mm. Bar graphs demonstrate the mean with error bars representing the standard deviation. Data values are presented as the associated points. * represents the statistical significance (p < 0.05) between the indicated groups using Student's unpaired t-test. | ||
Sulphuric crystallisation yielded a 10% decrease in mass together with a volumetric expansion of approximately 100% (Fig. S1B†), and an increase in porosity from 95.4 ± 2.1% to 98.5 ± 0.5% that was measured via ethanol intrusion (Fig. 1B). Scanning electron microscopy (SEM) validated a porous homogeneous structure for both untreated and crystallised PEDOT:PSS scaffolds, confirming that the sulphuric acid treatment did not lead to any major detrimental change in the porous microarchitecture (Fig. 1C). Morphological quantification of scaffold sections enabled an accurate understanding of pore size on isotropic scaffolds and detected a significant increase from 165 ± 78 μm to 202 ± 100 μm when the scaffolds were crystallised using sulphuric acid (Fig. 1C iii/vi and D).
In several tissue engineering applications, control over substrate topography and attainment of anisotropic constructs to mimic physiological substrates are important considerations. Here, freezing the PEDOT:PSS solutions along a preferential axis of heat transfer by virtue of a custom-made mould enabled the fabrication of highly aligned scaffolds (Fig. 1A iii). This preferential axis of heat transfer and subsequent sublimation of ice crystals generated a highly anisotropic structure consisting of parallel two-dimensional lamellae with a dominant directionality along the axis of heat transfer (i.e. freezing vertically), which is clearly demonstrated by the SEM micrographs in both longitudinal and transversal directions in Fig. 1E.
In a similar manner, aligned samples were sectioned for imaging analysis to quantify pore orientation within the internal structure (Fig. 1E ii/iii/v/vi). This quantification is summarised in Fig. 1F and testifies a high degree of pore alignment along the plane longitudinal to the fibers (i.e. identified at 0° in the plot) that results in a Gaussian-like distribution of orientation with a peak in proximity of the 0° direction, while a dispersed distribution with no prevalent directionality was found for the transversal plane.
SEM at higher magnifications revealed the presence of cracks along the surface of the material, which were not present on the smooth surface of the untreated porous scaffolds and that may represent crystal boundaries as previously reported16,18 (Fig. 2A i/iii). These boundary features have been previously reported as characteristic of the process of PEDOT:PSS crystallisation, attributed to the removal of excess PSS and to the stacking of PEDOT in a more lamellar fashion.16Fig. 2A ii/iv show a substantial thinning of the porous scaffold struts when crystallised, which is in agreement with the mass loss and the significant increase in volume, porosity and pore size caused by the process, ultimately suggesting a partial surface erosion of the porous scaffolds. From the same figures, it is possible to observe how the inner portion of the struts, despite being reduced in thickness, did not present any other major conformational changes after crystallisation, maintaining the same marble pattern, further evidencing that the crystallisation treatment mainly influences the outer part of the strut. Towards physicochemical verification of this treatment, XRD analysis was applied to dropcasted samples (of GOPS-crosslinked PEDOT:PSS) that underwent the same crystallisation protocol of the 3D scaffolds (Fig. 2B). GOPS-crosslinked PEDOT:PSS samples subjected to crystallisation lacked the characteristic peak of PSS at 2θ = 20.5°21 – an observation that has been reported in similar crystallisation treatments of PEDOT:PSS.16 Crystallised scaffolds demonstrated a significant increase in water intake when compared with uncrystallised scaffolds (up to 85 times their dry weight, Fig. 2C), which is remarkably higher when compared to 3D PEDO-alginate hydrogels currently reported12 and which we postulate as being a direct effect of the significant increase in porosity, volume and pore size. The efficacy of GOPS crosslinking was not compromised by the crystallisation treatment. Indeed, the scaffolds were able to maintain exceptional durability in a water environment, with no evidence of macroscopic deterioration after 3 months at room temperature.
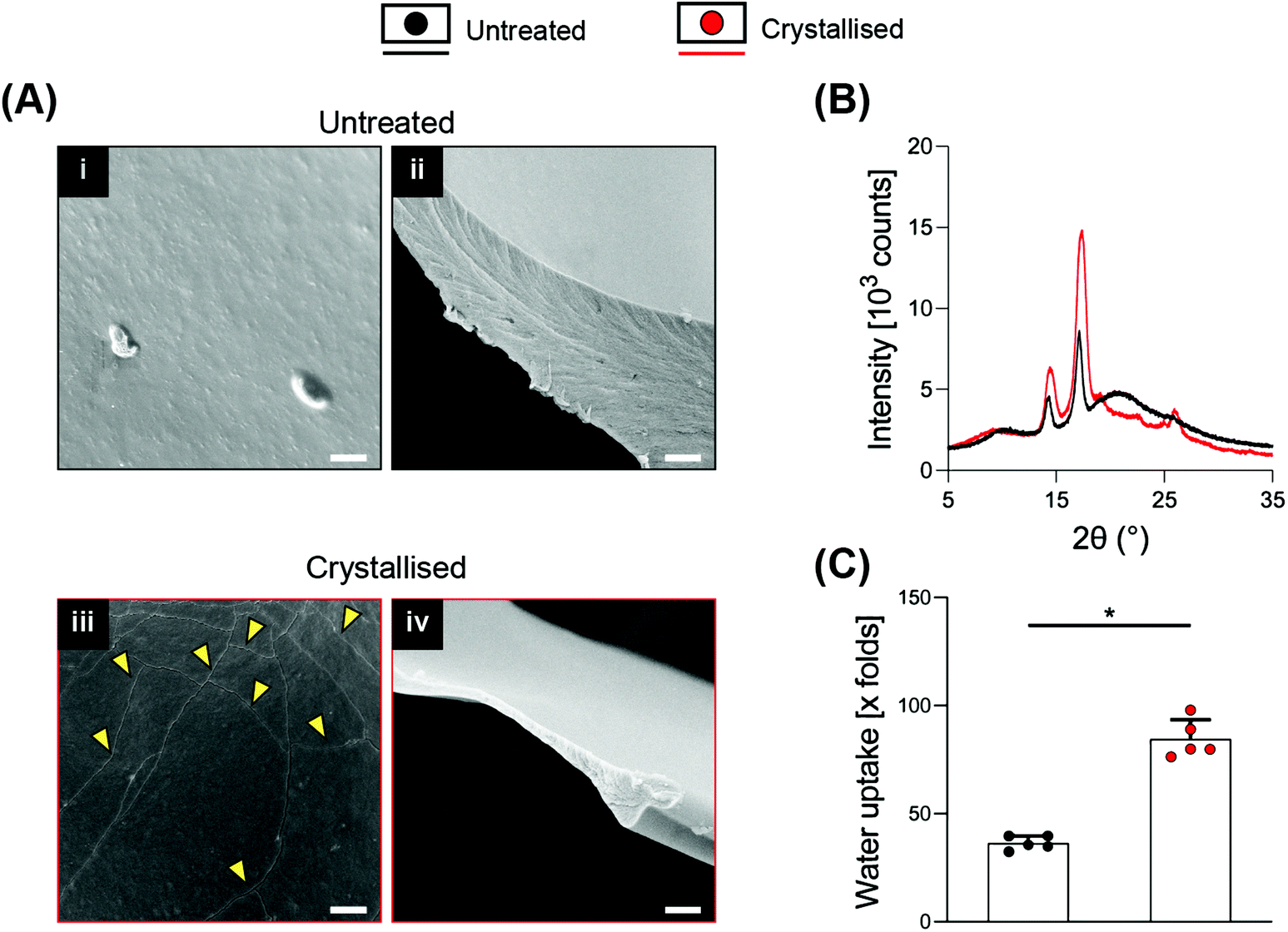 | ||
| Fig. 2 Assessment of crystallisation treatment on material microstructure, chemistry and swelling properties. (A) Micrographs from high magnification SEM showing details of the surface (i/iii) and of the section (ii/iv) of the scaffolds. Yellow arrows point to the cracks. (B) X-ray diffraction (XRD) patterns of the dropcasted thin sheets. (C) Swelling of the dry scaffolds soaked in deionized water, measured as water uptake (n = 5). Scale bars: A = 2 μm. Bar graphs demonstrate the mean with error bars representing the standard deviation. Data values are presented as the associated points. * represents the statistical significance (p < 0.05) between the indicated groups using Student's unpaired t-test. | ||
One previous study reports sulphuric crystallisation on thin PEDOT:PSS coatings (with a thickness in the range of hundreds of nm), whereby PEDOT:PSS was used in its pristine form with no additional molecules or crosslinkers. In order to achieve 3D conformations, a crosslinker such as poly(ethylene glycol) diglycidyl ether or GOPS can be adopted, but with the latter potentially lowering the conductivity of PEDOT:PSS.14 Whether the presence of a crosslinking agent may have hindered the efficacy of the sulphuric crystallisation was one point of query prior to beginning this study. However, crystallisation via sulphuric acid post-treatment in this study was successfully applied to 3D porous constructs, while at the same time enabling concise control over scaffold microarchitecture.
Crystallised aligned scaffolds exhibit anisotropic elastic behaviour
We performed a detailed series of mechanical tests to gain an overview of the mechanical performance of scaffolds under hydrated conditions, a typical environment of most biomedical applications. Indeed, the liquid present in the surrounding environment plays an important role, influencing the overall response of biomaterial scaffolds.22 This is particularly relevant for the constructs presented in this work, whereby a >95% porosity (Fig. 1B) and substantial water intake occurs (Fig. 2C), which may potentially impact their temporal viscoelastic properties. Therefore, a series of phases in mechanical testing allows one to decouple the different components and determine the extent to which the porous morphology and water uptake influence the overall mechanical outcome.23In Fig. 3, a combination of stress-relaxation and dynamic test is reported. Within the first phase of the test, the samples were compressed up to 15% strain with a relatively slow strain rate in order to derive the stiffness of the construct for a standard unconfined compression. The ramp modulus was derived as the slope of the curve in the range 5–10% of deformation, where the stress–strain relationship is linear, ultimately obtaining a stiffness value in the range of previous works on PEDOT:PSS.13 The 15% strain was then maintained for 45 minutes, to accommodate relaxation of the scaffold structure. The equilibrium modulus was derived as the ratio between stress and strain at the end of relaxation, providing a measure of stiffness without any hydraulic fluid pressurisation. The last cyclic loading phase enabled determination of the scaffold's responses in physiological regimes, where the viscous component of the fluid has a significant impact. Notably, the dynamic modulus reflects the permeability of the matrix and the ease at which the structure can expel fluids when subjected to a deformation. This aspect is an index of how a construct can generate fluid load support; all important considerations to be taken into account for anatomic structures that rely on a fluid component, for example, articular cartilage.22 The results are summarised in Table 1, demonstrating how the morphological anisotropy has a significant effect on the elasticity of the aligned scaffolds for all three types of the moduli investigated (ramp, equilibrium and dynamic), with a longitudinal/transversal ratio of 9.16, 5.24 and 17.74 for the untreated groups, and a diminished ratio of 3.54, 2.39 and 2.94, respectively, derived for the crystallized samples. Crystallisation of the scaffolds yielded a significant decrease in stiffness for the isotropic and the aligned scaffolds in the longitudinal direction, while no major effect was found in the transversal direction. This observation can be rationalized by the topographical alterations elicited by crystallisation; an increase in pore size and porosity accompanied by the thinning of the struts. As shown in Fig. S2,† significant trends in the peak stress to equilibrium stress ratio were observed between the different geometries, with a reduction in this ratio for isotropic crystallised scaffolds and in transversal direction for both untreated and crystallised aligned structures. These findings suggest that the sulphuric acid treatment and the direction of loading both contribute to a diminished ability to maintain the fluid load support.
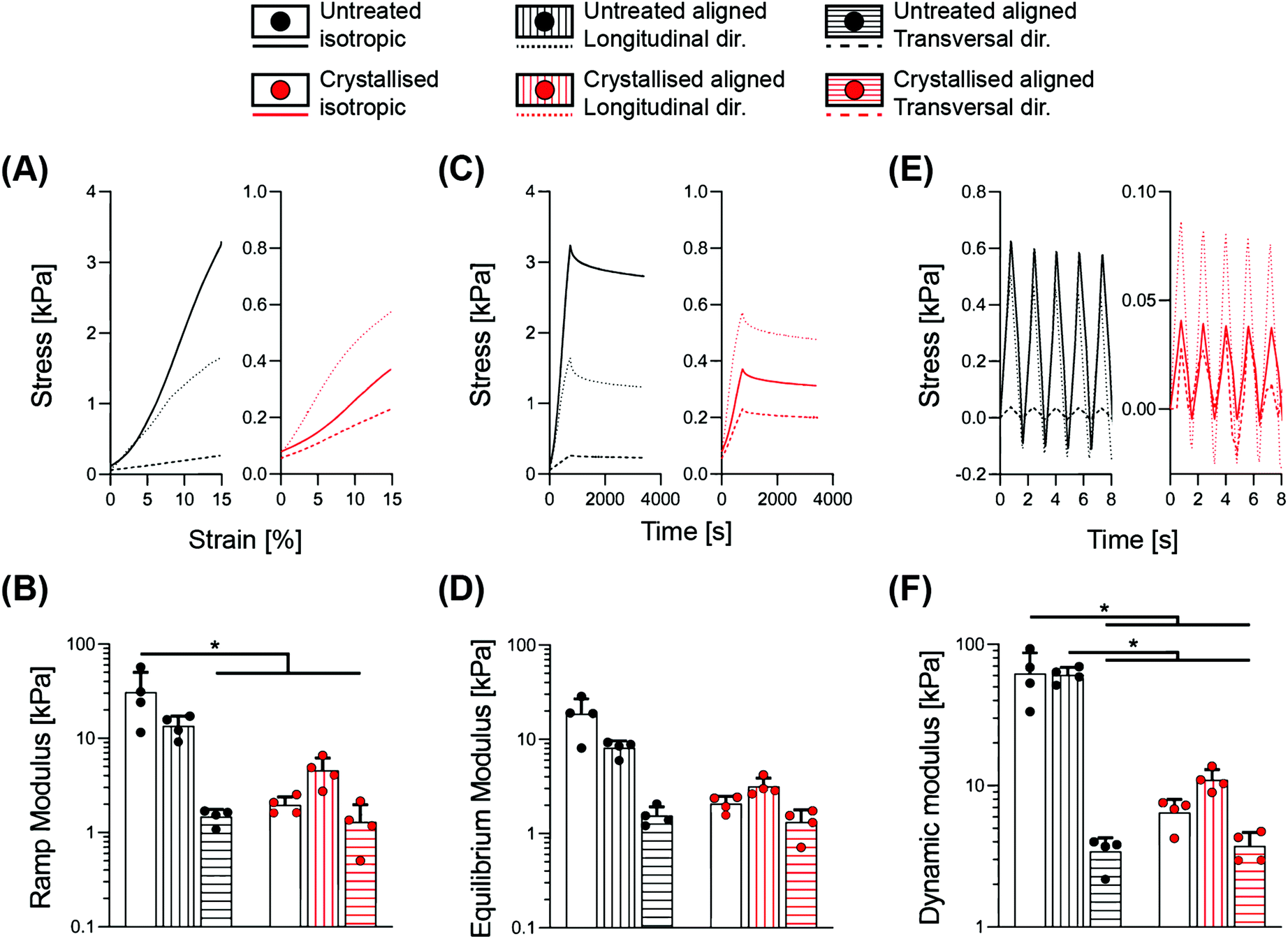 | ||
| Fig. 3 Stress relaxation followed by dynamic cyclic compression test. (A) Stress–strain curves representing the mean response during ramp phase. (B) Ramp modulus. (C) Stress–time curves representing the mean response during relaxation phase. (D) Equilibrium modulus. (E) Stress–time curves representing a standard response during the dynamic cyclic loading phase. (F) Dynamic modulus. Bar graphs demonstrate the mean of n = 4 replicates with error bars representing the standard deviation. Data values are presented as the associated points. * and # represent the statistical significance (p < 0.05) (* between the indicated groups and # with all other groups) using two-way ANOVA with Tukey's post-hoc test. | ||
| Material | Geometry | Ramp modulus [kPa] | Equilibrium modulus [kPa] | Dynamic modulus [kPa] | Conductivity [S m−1] |
|---|---|---|---|---|---|
| Untreated | Isotropic | 31.02 ± 19.23 | 18.66 ± 8.39 | 62.16 ± 25.26 | 7.01 ± 5.01 × 10−5 |
| Longitudinal | 13.61 ± 3.62 | 8.14 ± 1.49 | 60.82 ± 7.73 | 2.33 ± 2.05 × 10−5 | |
| Transversal | 1.49 ± 0.28 | 1.55 ± 0.37 | 3.43 ± 0.85 | 3.69 ± 3.51 × 10−6 | |
| Crystallised | Isotropic | 1.96 ± 0.43 | 2.08 ± 0.41 | 6.47 ± 1.52 | 3.45 ± 2.83 × 10−2 |
| Longitudinal | 4.58 ± 1.61 | 3.18 ± 0.71 | 11 ± 2.01 | 1.18 ± 0.53 × 10−1 | |
| Transversal | 1.30 ± 0.68 | 1.33 ± 0.45 | 3.75 ± 0.92 | 1.39 ± 1.42 × 10−2 |
Correlating the mechanical resilience of the structures with the dissipative energy of each deformation cycle, we performed a hysteresis analysis with cyclic loading constantly increasing the strain up to 60% (Fig. 4). The decrease of the ratio between the unloading area (recovered energy) and the loading area (applied energy) is characteristic of the transition between elastic to plastic deformation.24 This transition occurred within the range of 30–40% for both the isotropic and the aligned scaffolds when evaluated transversally, while an initial decrease was visible already at 15% strain for the longitudinally compressed aligned structures. This event is mainly based on the morphology of the aligned samples, as a compression parallel to the lamellae will first lead to a compaction between them, before beginning to deform permanently, and similarly for the isotropic structures where the pseudo-spherical shape of the pores allows for a higher range of elastic deformation. Instead, a compression perpendicular to the lamellae would directly operate on the struts, causing deformation starting from the lower strain levels.
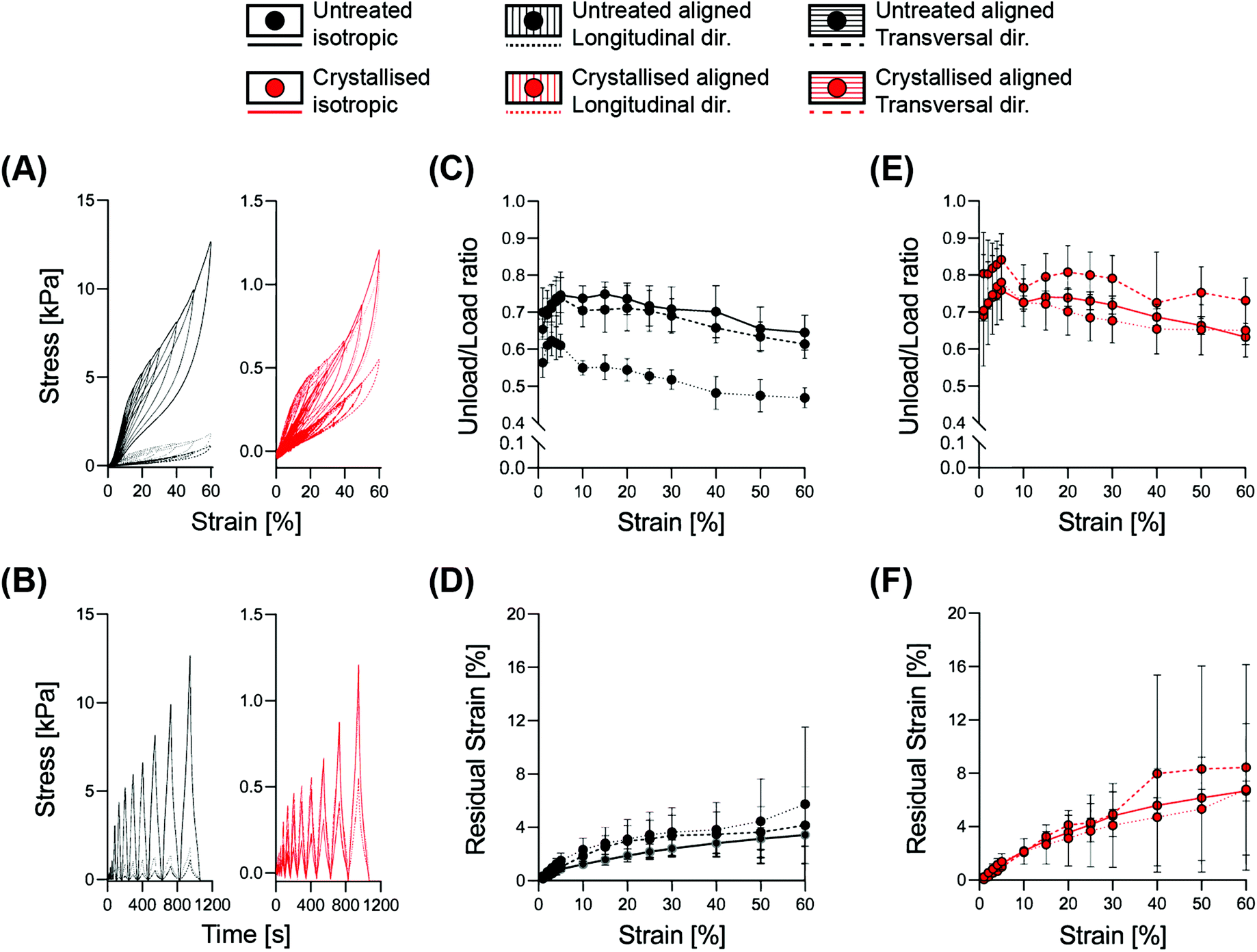 | ||
| Fig. 4 Elasto-plastic response from the analysis of hysteresis for the isotropic and aligned scaffolds. (A and B) Stress–strain and stress–time curves representing a standard response within the cycles. (C and E) Unload/load ratio response at different strain amplitudes. (D and F) Residual strain quantified as unrecovered deformation at zero force for each cycle. Data points demonstrate the mean of n = 4 replicates with error bars representing the standard deviation. | ||
Crystallisation of porous scaffolds yields over a thousand-fold increase in conductivity that can be tuned anisotropically
While it is often taken for granted that CPs are inherently easy to process in comparison with the standard metals – this is an oversimplification by nature of their classification as polymers. Achieving a construct based on CPs as the bulk and only material component has been mostly limited to two-dimensional films or thin sheets, or to 3D structures compromised by relatively low conductivity values. Most works report the fabrication of electrically conductive hydrogels or porous structures; however, all rely on the combination of the CPs with a supporting matrix.12,25Here we discovered that the sulphuric crystallisation yielded a profound increase in conductivity, of approximately 500-fold for the isotropic scaffolds, while the aligned scaffolds went even further with a conductivity increase in the order of 4000- and 5000-fold for the transversal and the longitudinal directions, respectively (Fig. 5C and Table 1). Furthermore, we also found a substantial difference in conductivity that was significantly influenced by the diverse geometries of the samples. The longitudinal direction, being constituted by parallel lamellae acting as a highway for the electrical conduction, leading to a conductivity of 6.3- and 8.4-fold higher when compared to the transversal direction, respectively, for both the untreated and the treated groups (Fig. 5C).
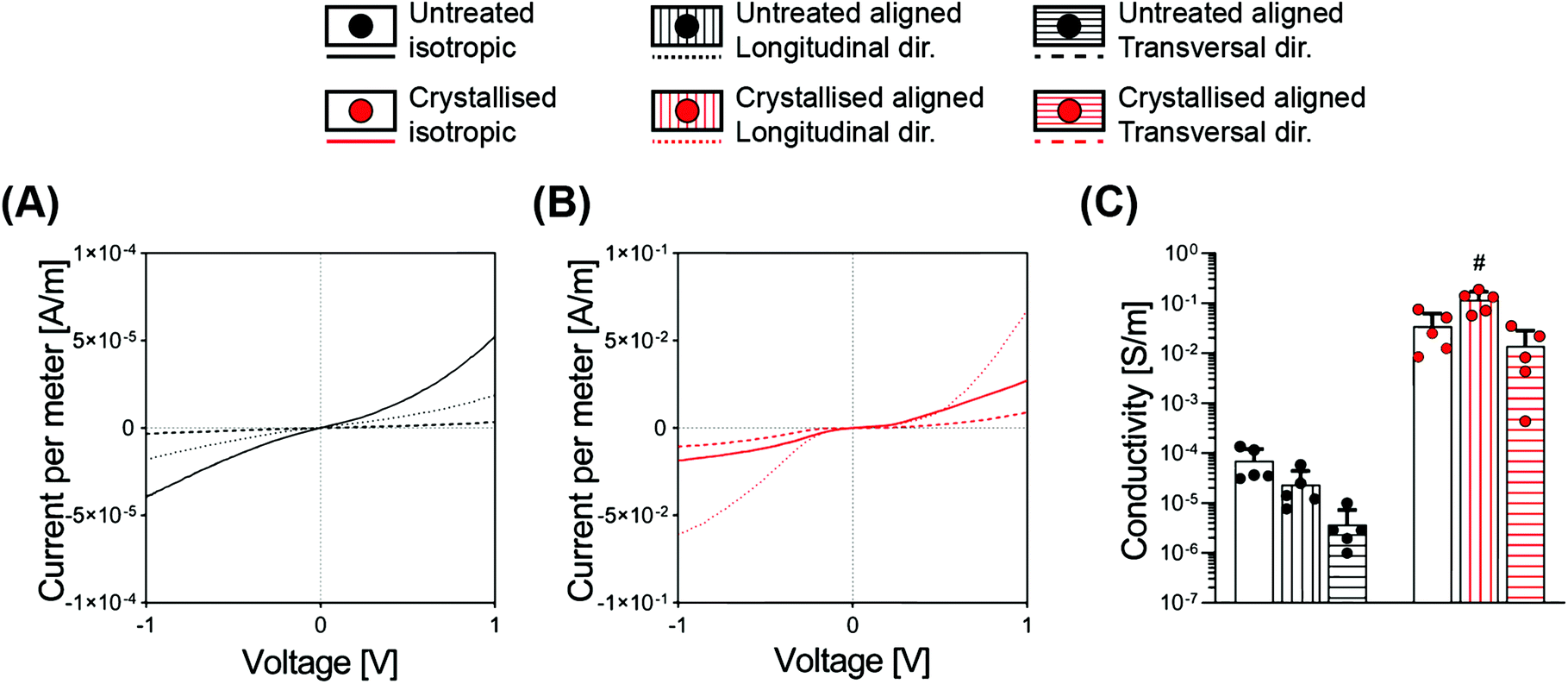 | ||
| Fig. 5 Electrical conductivity measurements via a 2-point probe. (A and B) I–V curves of untreated (A) and crystallised (B) PEDOT:PSS scaffolds. (C) Conductivity quantification from the linear regression of I–V curves in the linear region 0.5–1 V (n = 5). Bar graphs demonstrate the mean with error bars representing the standard deviation. Data values are presented as the associated points. # represent the statistical significance (p < 0.05) (with all other groups) using two-way ANOVA with Tukey's post-hoc test. | ||
When comparing electrical conductivity results reported from various studies, it is appropriate that the technique being adopted is considered with a 4-point probe being used for 2D substrates,3 a 2-point probe for 3D constructs10 and electrochemical impedance spectroscopy (EIS) as a valid method for both configurations.3,18 One example of a highly conductive material achieved via a solvent approach entailed the fabrication of thick 2D hydrogels and anisotropic drying without the addition of secondary molecules. Here the authors reported values in the range of 20 S cm−1 in PBS, with such conductivity acquired from 2D constructs using a 4-point probe setup, therefore not directly comparable to the scaffolds we report in this study.26
In this work, we adopted a 2-point probe measurement in order to successfully delineate a relationship between scaffold pore directionality and conductivity. Another point that one must consider is that the values we report here for all electrical characterization methods have been measured on samples that have been washed multiple times and freeze-dried again, a process that purifies the material from unreacted products, but that reduces conductivity. GOPS-crosslinked PEDOT:PSS porous constructs have been reported with electrical conductivities in the order of 1.5 ± 0.5 × 10−3 S m−1 when freshly prepared, which can deteriorate by one order of magnitude after 4 days in culture media.10 This mediocre conductivity can be somewhat overcome through the addition of secondary dopants (such as dodecylbenzenesulfonic acid, DBSA), which can boost the conductivity for the initial days of culture.
A stand-alone example of the PEDOT:PSS porous 3D structure with no presence of a supporting compound relied on the gelation process achieved by sulphuric acid; however, the 12-hour long post treatment potentially caused disadvantages in terms of biocompatibility that have not been assessed yet. Moreover, despite providing promising conductivity values up to 880 S m−1 (as measured by EIS), the gelation process adopted did not permit control over the microarchitecture.
To the best of our knowledge, this work is the first successful attempt to merge the precise optimization of the morphology via an ice-templating technique together with a crystallisation treatment on a PEDOT:PSS chemically crosslinked scaffold that had not been combined with a supporting material.
Crystallised PEDOT:PSS porous scaffolds are cytocompatible and serve as a 3D scaffold for 3D cell culture
An initial concern of using the sulphuric crystallisation approach was the risk of residual acid remaining on the scaffold. Phenol red-containing media were adopted for all the studies in this work, and no change was ever observed in the color of the media, an initial indication of the absence of acidic residue. Therefore, we proceeded with more thorough evaluations of cytotoxicity, via an indirect test and direct seeding of cells onto these scaffolds.The release of toxic by-products into media was assessed by applying the extraction media (whereby the scaffolds were placed in an extraction medium for 24 hours) onto the C3H10 cells seeded on the standard tissue culture plates. Cellular metabolism was evaluated using the alamarBlue™ assay (Fig. S3A†) and the results were normalized to the metabolism of cells grown in the standard media, which were conditioned in the same environment of the extraction media. This assay detected no statistically significant difference between the untreated and crystallised groups at the same time point. A qualitative observation of cell morphology via a nuclei (DAPI)/f-actin (phalloidin) fluorescence staining revealed no substantial difference in either cell adherence or spreading, suggesting that the crystallisation process did not have any residual toxic drawbacks on cell physiology.
Considering the promising results of this indirect evaluation of cytocompatibility, we proceeded with direct seeding of C3H10 cells onto these scaffolds with the scaffold dimensions being standardized to disks of 6 mm in diameter and 1 mm in thickness. 1-Ethyl-3-(-3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) crosslinked collagen porous scaffolds have been widely tested with positive outcomes in tissue engineering,27 hence they were adopted as a reference control in this work. The assessment of cell response was compared between day 1 and 7 after seeding, via live/dead fluorescence staining (Fig. 6A) that demonstrated a high viability at both time points, with the scaffolds exhibiting an adequate cell attachment at early time points and showing high proliferation for the untreated group at day 7. Despite the total number of live cells being significantly lower for the crystallised group at day 7 compared to the untreated one (Fig. 6B), an opposite trend was found once these values were normalized by the total number of cells and presented as viability % (Fig. S4†). This fact supports the claim that the diminished live cell density may be an effect of the reduced space offered by the struts, leaving cells further from each other and less capable of bridging the pores. This hypothesis was confirmed by fluorescence staining (Fig. 6C), where cells showed to be adherent to the substrates in all the groups and time points tested, excluding a detrimental effect due to the material chemistry. The combined increase in both porosity and mean pore size caused a reduced surface area available to the cells for attachment28 that can also explain their reduced proliferation compared to the untreated group and ultimately a lower value in DNA quantity (Fig. 6D) and reduction % in the alamarBlue™ assay(Fig. S5†). These findings support the claim of adequate biocompatibility of the crystallised GOPS-crosslinked PEDOT:PSS scaffolds, both with indirect test and with direct contact, paving the way for their use in the application of tissue regeneration, biosensors for in vivo studies or monitoring interfaces for in vitro platforms.
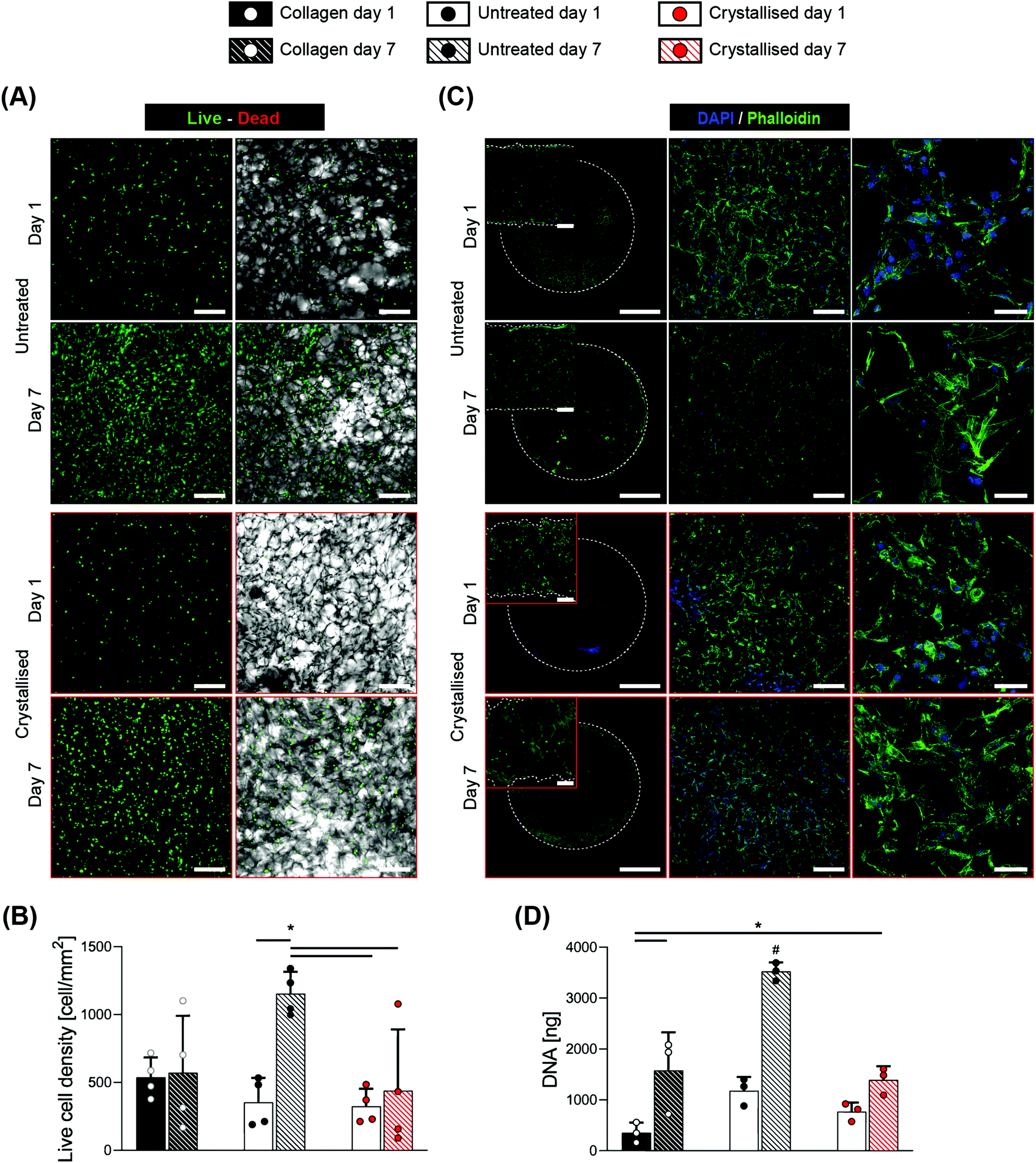 | ||
| Fig. 6 Cytocompatibility of C3H10 cells on scaffolds via a direct contact. (A) Micrographs reporting live/dead staining of CH310 cells at days 1 and 7, seeded on the scaffolds. Left: live/dead channels. Right: live/dead/bright-field channels. (B) Quantification of viability (extracted from live/dead staining) at days 1 and 7, quantified as the live cell density (n = 4). (C) Micrographs from confocal microscope fluorescence staining for nuclei/f-actin (DAPI and phalloidin, respectively), seeded on the scaffolds. Pictures refer to the top surface of the samples. Insets refer to the section view of the sample showing cells migrating through the thickness of the scaffolds. Dotted lines represent the borders of the samples. (D) Quantification of DNA via the Picogreen™ assay, expressed as ng per scaffold (n = 3). Scale bars: A = 200 μm; C left = 2 mm; C left insets = 200 μm; C middle = 200 μm; C right = 50 μm. Bar graphs demonstrate the mean with error bars representing the standard deviation. Data values are presented as the associated points. * and # represent the statistical significance (p < 0.05) (* between the indicated groups and # with all other groups) using two-way ANOVA with Tukey's post-hoc test. | ||
Experimental
Fabrication of 3D GOPS crosslinked porous scaffolds
PEDOT:PSS (1.3 wt%) (dispersion in water) and GOPS were purchased from Sigma-Aldrich (Sigma-Aldrich, Ireland). Solutions were prepared as previously described.3 Mixed solutions were placed into plastic or custom-made moulds. Highly porous scaffolds were achieved through lyophilisation using a freeze-dryer (FreeZone, Labconco Corporation, Kansas City, MO). The samples were kept at −40 °C for one hour, and afterwards, sublimation of ice crystals was induced by decreasing the pressure down to 0.2 mbar and increasing the temperature to −10 °C. After 18 hours, a secondary drying phase at 20 °C for 2 hours concluded the lyophilisation process. GOPS crosslinking of specimens required an additional annealing treatment in a vacuum oven at 140 °C for 1 hour. A custom-made mould containing a bottom stainless-steel layer and a top polydimethylsiloxane (PDMS, SYLGARD® 184) layer was fabricated to induce an ice crystal alignment by virtue of the different thermal conductivities of the two materials. To proceed with acidic crystallisation of the resultant porous constructs, we adopted a method previously reported16 that briefly entailed soaking the construct in 100% sulphuric acid. All samples, both untreated and crystallised, were subjected to multiple washings using deionized water and shaped to optimized thickness with a vibratome (VT 1200S, Leica, Germany). Finally, a second lyophilization process was carried out to fully dry the materials.For in vitro studies, collagen scaffolds were adopted as controls. Briefly, collagen type I was isolated from porcine tendons following previously developed protocols, and then collagen powder was solubilized in 0.1 M acetic acid to obtain a concentration of 15 mg ml−1. After being freeze dried with the same protocol of the PEDOT:PSS GOPS-crosslinked group, the collagen-based scaffolds underwent a standard EDC cross-linking.29
Scaffold morphology
Scaffold microarchitecture was qualitatively investigated by Scanning Electron Microscopy (SEM). Briefly, 3D porous scaffolds were mounted on aluminium stubs with a conductive carbon tape (Ted Pella, USA), and a gold–palladium layer of approximately 5 nm was sputter coated on the sample surface. Specimens were observed using a Zeiss SUPRA 40 field emission SEM (Zeiss, Germany) with a 5 kV electron beam.Quantitative analysis on pore size was performed similar to how it was reported by the O'Brien group.30 Samples were agarose-embedded and dehydrated using an automatic tissue processor (Leica ASP300), and then wax embedded and finally sliced. A total of n = 3 10 μm thick slices were sectioned using a rotary microtome (Leica Microtome RM2235) and sampled for imaging with a distance of 100 μm between the samples, and n = 3 replicates both on the transversal and longitudinal directions per condition. Micrographs were obtained using a ScanScope (Aperio Technologies Inc., USA) (Fig. 1C v–vi) and the image analysis was carried out using a custom-made Matlab® script capable of thresholding the image, skeletonizing the structure and finally deriving the average diameter pore size from the pore area approximated to a circle.
The micrographs from the aligned scaffolds were then post processed using ImageJ plugin OrientationJ31 in order to derive the lamellae alignment and the degree of anisotropy.
Porosity was evaluated using an ethanol intrusion technique. Briefly, the samples were immersed in 70% v/v ethanol solution and the volume of ethanol was derived from the global mass of the specimens and considered as the empty component of the structure as in eqn (1), with Pe = porosity by ethanol intrusion, Vs = volume of the scaffold, ds = density of the scaffold, Ms = mass of the scaffold, Ve = volume of ethanol solution, de = density of ethanol solution and Me = mass of ethanol solution.
 | (1) |
Similarly, the samples were weighed in their dry state and 1 hour after being soaked in distilled water. Water intake was estimated as a ratio between wet and dry mass.
X-ray diffraction (XRD)
XRD investigation was carried out on the two-dimensional drop-casted samples with a benchtop X-ray diffractometer (D2 Phaser 2nd generation, Bruker). The PEDOT:PSS-GOPS slurry was injected on PTFE substrates and left for drying for 2 hours at 60 °C. Afterwards, crystallisation and a series of washings were performed as for the 3D structures, finally followed by a second drying phase.Mechanical characterization
Two uniaxial unconfined compression tests were performed on n = 4 samples per group using a Zwick Roell® single column universal testing machine (Zwick/Roell. UK) and a 10 N load cell. Both tests were performed under hydrated conditions using PBS at room temperature. For isotropic samples, scaffolds with a cylindrical shape and approximately 6 mm diameter and 3 mm thickness were prepared, while aligned scaffolds were prepared into rectangular cuboids.In a similar manner to previous reports,23 the first test consisted of a stress relaxation phase, followed by a dynamic cyclic compression at a physiological frequency. Initially, the specimens were preloaded at 0.005 N, then a compression rate of 0.02% s−1 was applied up to 15% strain, and in this range, the slope of the curve was used to determine the ramp modulus of the material. Deformation was then kept constant for 45 minutes in order to understand the stress-relaxation response of the constructs and derive the equilibrium moduli. Finally, the dynamic modulus was obtained from the averaged slope of n = 5 cycles at 1 Hz between 15% and 16% strain.
Secondly, a cyclic test with increasing strain was performed to detect the measure of the viscoelastic response of the constructs.24 After the application of a preload of 0.005 N, the samples were compressed at 0.5% s−1 from 1% to 60% with variable steps of 1% strain per cycle between 1 and 5%, of 5% strain from 5% to 30%, and finally of 10% strain from 30% to 60%. The amount of hysteresis of each cycle were computed using a custom made Matlab® script as a ratio between the unloading/loading areas. The deformation at which an abrupt reduction of this ratio happened was identified as the elastic/plastic transition point. From the same dataset, it was also possible to derive the residual strain deformation at zero stress per every strain cycle.
Electroconductivity characterization
Electrical conductivity was determined using a custom-made two-point probe testing setup. The samples were compressed to approximately 5% strain between two parallel brass plates, and a sourcemeter Keithley 2400 (Tektronix, USA) was used to apply a sweep potential between −1 and +1 V and to measure the current with an increment of 5 mV per second, to derive the characteristic I–V curve. Resistance R was measured as the slope of the linear regression in the linear region, and afterward, the conductivity σ was calculated as in eqn (2), with ρ = resistivity, d = thickness of the sample and A = area of the cross section (derived by the diameter of the sample).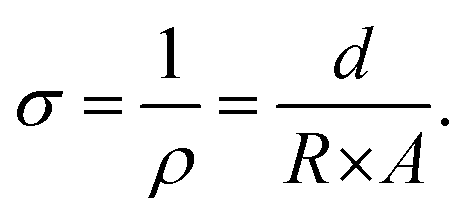 | (2) |
Biocompatibility studies
To assess any potential influence of the crystallisation process on material biocompatibility, indirect and direct cytotoxic tests were performed. C3H10 mouse embryonic fibroblasts (ATCC® CCL-226™) were cultured in growth media prepared using Dulbecco's Modified Eagle's Medium (DMEM) low glucose (Sigma-Aldrich) containing 10% v/v foetal bovine serum (FBS) (Gibco® by Life Technologies) and 2% v/v penicillin streptomycin (Pen–Strep) (Sigma-Aldrich) at 37 °C with 5% CO2. The cells between passages 12 and 15 were used.For the indirect contact assay, untreated and crystallised scaffolds were sterilised with multiple washings in 70% ethanol and exposed to UV light. Following rinsing with PBS, the specimens were let to macerate in growth media with a ratio of 10 mg of dry material per ml of media. We obtained the extraction media by substituting the liquid in contact with the material at day 1, 3, 5 and 7, in order to identify any potential effect of by-products release in the early stage phase. Cells were seeded on a standard tissue culture plate at a density of 10 × 104 cells per cm2 in standard growth media. At 24 hours, the standard media were replaced by extraction media that was left in contact with the cells for more 24 hours.
To evaluate the cell condition, we performed a standard alamarBlue™ metabolic assay, leaving cells for 3 hours in contact with the working solution, and we qualitatively observed the cell morphology by cytofluorescence staining. The samples were fixed in 4% w/v paraformaldehyde for 60 minutes at room temperature, and then they were incubated in a working dye solution of 4′,6-diamidine-2′-phenylindoledihydrochloride (DAPI, Sigma-Aldrich) (1 mg ml−1) and 1 μl ml−1 phalloidin (Santa Cruz Technology), to stain, respectively, the nuclei and the f-actin of the cell's cytoskeleton as in our previous work.3 Pictures were obtained using an Olympus IX83 epifluorescence microscope (Olympus, Germany).
For the direct contact in vitro study, scaffolds with 6 mm diameter and 1 mm thickness were adopted. The scaffolds were sterilised with multiple washes in 70% v/v ethanol solution and in sterile deionized water, and then they were incubated in the growth media for 24 hours. Following the pre-conditioning, a cell suspension of 20 μl growth media and 250k cells was injected on each scaffold, and after two hours, more culture medium was added. Growth media was replaced once every two days, up to day 7.
Cell viability was evaluated at day 1 and 7 with the live and dead assay adopting a solution of 2 μl ml−1 ethidium homodimer and 0.5 μl ml−1 calcein (Cambridge Bioscience) in PBS. The scaffolds were observed using a Leica SP8 scanning confocal microscope (Leica Microsystems, Germany). For quantitative analysis, three regions of each sample were imaged with each figure being the resultant z-projection of a z-stack; for this process, we used a constant z-step of 10 μm and penetration down to 100 μm from the surface. Pictures were later analysed using ImageJ and the live cell density was obtained as a ratio between live and total cells normalized by the ROI area. DNA was quantified using a Picogreen™ biochemical assay on papain digested specimens. As for the indirect contact test, both alamarBlue™ assay and staining for nuclei/f-actin were performed. For the alamarBlue™ assay, the culture media were replaced with fresh media containing 20% v/v alamarBlue™ at a specific time point. The media were gently mixed with pipette and incubated for 1 hour; afterwards, the media were mixed again and moved to a 96-well plate for analysis. For the fluorescence imaging, the same confocal microscope of the live and dead test was used.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 9 (GraphPad Software, USA). Where appropriate, one-way or two-way analysis of variance (ANOVA), followed by Tukey's multiple comparison test were performed. Unless otherwise specified, results are presented as mean ± standard deviation and differences were considered as statistically significant for p < 0.05.Conclusions
Several groups are currently working towards solutions to deliver highly conductive and tunable 3D constructs for tissue engineering and biomaterial applications. The platform we have described here has a significant advantage over similar materials reported to date and has significant potential within and outside the field of biomaterials and tissue engineering. Since its initial report and those following it, GOPS crosslinking of PEDOT:PSS has been associated with a compromise of conductivity properties. We have overcome this through a simple, safe and scalable sulphuric acid crystallisation step, which does not impact the stability of 3D porous scaffolds. The porous scaffolds reported in this study (both treated and crystallised) were capable of being fashioned into anisotropic porous configurations, which in turn enabled a morphological anisotropy (via pore orientation), which in itself imparted a mechanical and electrical anisotropy to the material. This platform could have significant impact in situations whereby topographical alignment of cellular growth and cell orientation, and in vitro electrical stimulation and/or sensing are warranted in tissue engineering and organoid approaches.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported through an SFI-HRB Wellcome Trust-ISSF Award Institutional Strategic Support Fund (Ref No. 204814/Z/16/Z), the Irish Research Council (Project No. GOIPG/2019/818) and Science Foundation Ireland (SFI), Ireland, through the Advanced Materials and Bioengineering Research (AMBER) Centre (SFI/12/RC/2278_P2, partly supported by the European Regional Development Fund). The SEM imaging for this project was carried out at the Advanced Microscopy Laboratory (AML), Trinity College Dublin, Ireland. The AML (http://www.tcd.ie/crann/aml) is an SFI supported imaging and analysis centre, part of the CRANN Institute and affiliated to the AMBER centre.Notes and references
- D. Olvera and M. G. Monaghan, Adv. Drug Delivery Rev., 2020 DOI:10.1016/j.addr.2020.09.011.
- M. Solazzo, F. J. O'Brien, V. Nicolosi and M. G. Monaghan, APL Bioeng., 2019, 3, 041501 CrossRef PubMed.
- M. Solazzo, K. Krukiewicz, A. Zhussupbekova, K. Fleischer, M. J. Biggs and M. G. Monaghan, J. Mater. Chem. B, 2019, 7, 4811–4820 RSC.
- D. Olvera, M. Sohrabi Molina, G. Hendy and M. G. Monaghan, Adv. Funct. Mater., 2020, 30, 1909880 CrossRef CAS.
- I. Del Agua, S. Marina, C. Pitsalidis, D. Mantione, M. Ferro, D. Iandolo, A. Sanchez-Sanchez, G. G. Malliaras, R. M. Owens and D. Mecerreyes, ACS Omega, 2018, 3, 7424–7431 CrossRef CAS PubMed.
- L. Groenendaal, F. Jonas, D. Freitag, H. Pielartzik and J. R. Reynolds, Adv. Mater., 2000, 12, 481–494 CrossRef CAS.
- M. N. Gueye, A. Carella, J. Faure-Vincent, R. Demadrille and J.-P. Simonato, Prog. Mater. Sci., 2020, 108, 100616 CrossRef CAS.
- P. Zhang, N. Aydemir, M. Alkaisi, D. E. Williams and J. Travas-Sejdic, ACS Appl. Mater. Interfaces, 2018, 10, 11888–11895 CrossRef CAS PubMed.
- I. Gualandi, M. Marzocchi, E. Scavetta, M. Calienni, A. Bonfiglio and B. Fraboni, J. Mater. Chem. B, 2015, 3, 6753–6762 RSC.
- A. G. Guex, J. L. Puetzer, A. Armgarth, E. Littmann, E. Stavrinidou, E. P. Giannelis, G. G. Malliaras and M. M. Stevens, Acta Biomater., 2017, 62, 91–101 CrossRef CAS PubMed.
- D. Khodagholy, T. Doublet, P. Quilichini, M. Gurfinkel, P. Leleux, A. Ghestem, E. Ismailova, T. Hervé, S. Sanaur, C. Bernard and G. G. Malliaras, Nat. Commun., 2013, 4, 1575 CrossRef PubMed.
- B. Yang, F. Yao, L. Ye, T. Hao, Y. Zhang, L. Zhang, D. Dong, W. Fang, Y. Wang, X. Zhang, C. Wang and J. Li, Biomater. Sci., 2020, 8, 3173–3185 RSC.
- S. Inal, A. Hama, M. Ferro, C. Pitsalidis, J. Oziat, D. Iandolo, A.-M. Pappa, M. Hadida, M. Huerta, D. Marchat, P. Mailley and R. M. Owens, Adv. Biosyst., 2017, 1, 1700052 CrossRef.
- A. Håkansson, S. Han, S. Wang, J. Lu, S. Braun, M. Fahlman, M. Berggren, X. Crispin and S. Fabiano, J. Polym. Sci., Part B: Polym. Phys., 2017, 55, 814–820 CrossRef.
- Z. Fan, P. Li, D. Du and J. Ouyang, Adv. Energy Mater., 2017, 7, 1602116 CrossRef.
- N. Kim, S. Kee, S. H. Lee, B. H. Lee, Y. H. Kahng, Y. R. Jo, B. J. Kim and K. Lee, Adv. Mater., 2014, 26, 2268–2272 CrossRef CAS PubMed , 2109.
- S.-M. Kim, N. Kim, Y. Kim, M.-S. Baik, M. Yoo, D. Kim, W.-J. Lee, D.-H. Kang, S. Kim, K. Lee and M.-H. Yoon, NPG Asia Mater., 2018, 10, 255–265 CrossRef CAS.
- B. Yao, H. Wang, Q. Zhou, M. Wu, M. Zhang, C. Li and G. Shi, Adv. Mater., 2017, 29, 1700974 CrossRef PubMed.
- U. Jammalamadaka and K. Tappa, J. Funct. Biomater., 2018, 9, 22 CrossRef PubMed.
- P. D. Dalton, Curr. Opin. Biomed. Eng., 2017, 2, 49–57 CrossRef.
- N. Zhang, X. Yu, J. Hu, F. Xue and E. Ding, RSC Adv., 2013, 3, 13740–13747 RSC.
- A. R. Gannon, T. Nagel and D. J. Kelly, Osteoarthritis Cartilage, 2012, 20, 1417–1425 CrossRef CAS PubMed.
- R. Schipani, S. Scheurer, R. Florentin, S. E. Critchley and D. J. Kelly, Biofabrication, 2020, 12, 035011 CrossRef CAS PubMed.
- N. T. Saidy, F. Wolf, O. Bas, H. Keijdener, D. W. Hutmacher, P. Mela and E. M. De-Juan-Pardo, Small, 2019, 15, e1900873 CrossRef PubMed.
- Y. Xu, M. Cui, P. A. Patsis, M. Gunther, X. Yang, K. Eckert and Y. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 7715–7724 CrossRef CAS PubMed.
- B. Lu, H. Yuk, S. Lin, N. Jian, K. Qu, J. Xu and X. Zhao, Nat. Commun., 2019, 10, 1043 CrossRef PubMed.
- B. Hafemann, K. Ghofrani, H. G. Gattner, H. Stieve and N. Pallua, J. Mater. Sci. Mater. Med., 2001, 12, 437–446 CrossRef CAS PubMed.
- F. J. O'Brien, B. A. Harley, I. V. Yannas and L. J. Gibson, Biomaterials, 2005, 26, 433–441 CrossRef.
- C. Yang, Bull. Mater. Sci., 2012, 35, 913–918 CrossRef CAS.
- M. G. Haugh, C. M. Murphy and F. J. O'Brien, Tissue Eng., Part C, 2010, 16, 887–894 CrossRef CAS PubMed.
- Z. Püspöki, M. Storath, D. Sage and M. Unser, Adv. Anat., Embryol. Cell Biol., 2016, 219, 69–93 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0bm02123g |
| This journal is © The Royal Society of Chemistry 2021 |