
Engineered drug-loaded cells and cell derivatives as a delivery platform for cancer immunotherapy
Lu
Wang
a and
Jinyao
Liu
 *ab
*ab
aShanghai Key Laboratory for Nucleic Acid Chemistry and Nanomedicine, Institute of Molecular Medicine, State Key Laboratory of Oncogenes and Related Genes, Shanghai Cancer Institute, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200127, China
bShanghai Key Laboratory of Gynecologic Oncology, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200127, China. E-mail: jyliu@sjtu.edu.cn
First published on 2nd November 2020
Abstract
Cancer immunotherapy, which provides durable clinical responses by restoring or boosting the patient's immune system to fight cancer, has become a promising strategy for cancer treatment. However, modest response rates and on-target off-tumor toxicity largely limit extensive implementation of this approach in clinical settings. Advances in drug delivery and combination with other cancer treatments are able to effectively promote the potency of cancer immunotherapy. Engineered natural particulates, such as cells and their derivatives, have been recently developed as prospective drug delivery systems that comprehensively combine genetic engineering, synthetic materials, and nanotechnology to enhance anticancer efficacy. Here, recent advances in improving cancer immunotherapy have been summarized with a focus on using functionalized intact cells and cell derivatives including cell membranes and extracellular vesicles as drug vehicles. The advantages and challenges of these unique systems have been further elucidated in terms of clinical translation. The results presented in this review would contribute to the development of advanced therapies for treating cancers.
 Lu Wang | Lu Wang received her bachelor's degree in biotechnology from Northwest A&F University in 2013 and Ph.D. in biology from the University of Science and Technology of China in 2019. Then she joined Prof. Jinyao Liu’ s group as a postdoctoral fellow at the School of Medicine, Shanghai Jiao Tong University. Her research focuses on cancer immunotherapy. |
 Jinyao Liu | Jinyao Liu is a full Professor in the Institute of Molecular Medicine, School of Medicine, Shanghai Jiao Tong University (SJTU), China. After receiving his PhD from SJTU in Materials Science and Engineering under the supervision of Prof. Deyue Yan, Jinyao joined Prof. Ashutosh Chilkoti’ group in the Department of Biomedical Engineering at Duke University (2013–2015) and Prof. Robert Langer's laboratory in the Koch Institute for Integrative Cancer Research at MIT (2015–2018) as a postdoctoral associate. His current research interests include oral delivery, bacteria-based bioagents, hydrogels and nanomedicine. Jinyao's publications have been featured in MIT News, Nature Communications Editors’ Highlights, Noteworthy Chemistry, Hunan TV, etc. |
1. Introduction
Cancer is a major public health problem worldwide and causes more deaths than AIDS, malaria and tuberculosis combined.1 Research in cancer therapeutics remains crucial to improve clinical outcomes. In addition to conventional chemotherapy, radiation and surgery, cancer immunotherapy that aims to activate or boost the response of the immune system through natural mechanisms has developed rapidly and revolutionized oncology in recent years.2 Due largely to durable clinical responses in a subset of patients who have failed in traditional drug treatment, immunotherapy is recognized as a new yet powerful treatment to fight many types of cancer in clinical settings.3 Immunotherapies approved for cancer treatment fall into several categories, including (1) cancer vaccines, which educate adaptive immune response via importing antigens and adjuvants, (2) agonistic or bispecific antibodies, which inhibit the proliferation of cancer cells or enhance T cell recognition and cytotoxicity against cancer cells, (3) immunomodulatory cytokines, which help to regulate the activation of immune cells, (4) immune-checkpoint inhibitors (ICI), which restore tumor-induced immune deficiency, (5) adoptive cell transfer, which directly provides enhanced cells to elevate immune response, and (6) oncolytic viruses or bacteria, which cause apoptosis in cancer cells and promote innate immune response.4 In particular, PD-1/PD-L1 blocking and CAR T cell therapies have demonstrated efficient immune responses and improved clinical outcomes. The unprecedented progress has led to immunotherapy take center stage in the cancer therapeutic arena.Despite these major advances, immunotherapies used in clinical settings encounter several limitations, such as unpredictable efficacy and on-target off-tumor toxicity. Poor immunogenicity, expression of targeted antigens on normal cells, insufficient local accumulation, and limited tumor infiltration are the main causes.5 To overcome these hurdles, drugs utilized for radiotherapy, chemotherapy, photodynamic therapy (PDT) and photothermal therapy (PTT) are often applied in combination therapy, which has been shown to enhance immunogenicity by inducing immunogenic cell death (ICD). Tumor cells undergoing ICD release more tumor antigens, danger-associated molecular patterns (DAMPs) and pro-inflammatory cytokines to facilitate the cancer immunity cycle.6 Furthermore, various synthetic platforms have been developed for the efficient delivery of immunoregulatory agents.7 With the ability to tune size, morphology, and surface groups, synthetic systems can be manipulated to increase accumulation at the tumor tissue, reduce systemic toxicity, and enhance drug solubility and stability. Nevertheless, most of these systems suffer from low efficiency of the enhanced permeability and retention (EPR) effect and nonspecific clearance caused by the foreign identity.8 Alternatively, as endogenous components, cells are able to avoid the induction of an adverse immune response and prolong the circulation of the loaded drugs.9 Moreover, the homing and chemotaxis properties of various cells enable the targeted delivery of various drugs.10 The targeted delivery of immunomodulators results in increasing their concentration in the target and inducing systemic specific anticancer immune response, which achieve a maximum therapeutic effect and lowered toxicity.11 Representatively, neutrophil and T cells, as sentinel and effector cells respectively, can be recruited by the corresponding disease sites.12 The capacity of cells to recognize and communicate with their surrounding microenvironments allows them to perform complicated assignments.13 Cell-based drug delivery has been explored to transport immunomodulatory proteins (cytokines, chemokines and proapoptotic proteins), toxins (chemotherapeutics, radiopharmaceuticals and oncolytic viruses), and tumor microenvironment activatable prodrugs.14–16 Currently, engineering of natural cells and their derivatives that combine genetic engineering, nanotechnology and materials science has evolved to achieve augmented therapeutic efficacy with minimal toxicity.17,18
In this review, we describe the recent progress in cell engineering aiming to improve the delivery of drugs for cancer immunotherapy. According to their structures, three main kinds of these systems, including intact cells, cell membranes and extracellular vesicles (EVs), will be focused upon and discussed (Fig. 1). We expect that elevating the efficacy and safety of immunotherapy by means of cell-based drug delivery could ultimately improve patient outcomes.
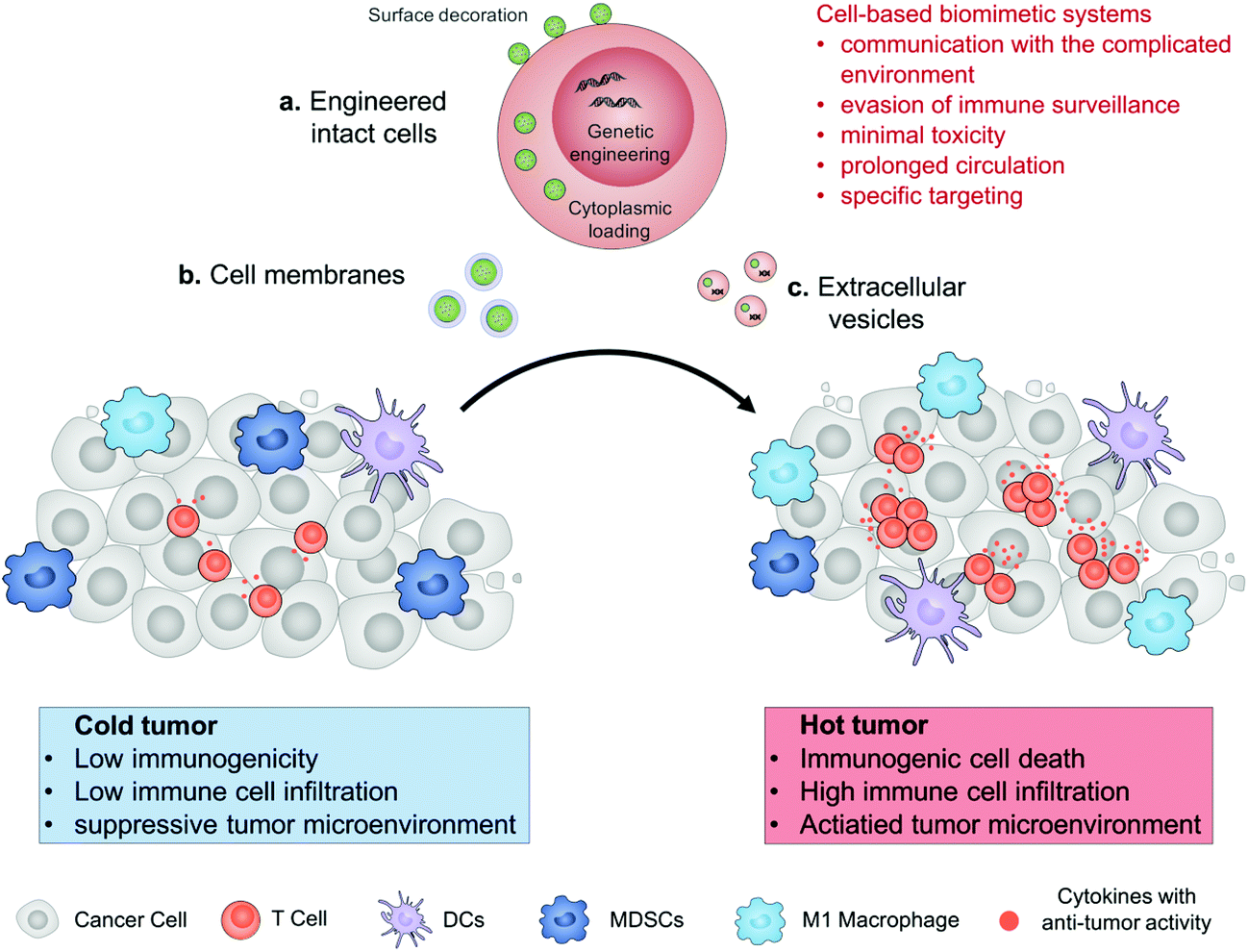 | ||
| Fig. 1 Schematic illustration of cell engineering aimed at improving the delivery of drugs for enhanced cancer immunotherapy. The cell-based drug delivery system including engineered intact cells, cell membranes or extracellular vesicles allows them to communicate with their surrounding microenvironments, evade from immune surveillance to reduce their toxicity, prolong their circulation, and enhance specific targeting. Drugs accumulated in the tumor tissue can convert an immunologically cold tumor to a hot one. | ||
2. Engineered intact cells
As drug vehicles, intact living cells retain their inherent characteristics that allow them to be regulated by dynamic environments. Abundant locations from the cell surface to the cytoplasm provide flexibility for drug carrying. Ihler and his colleagues kicked off the era of cell-based drug delivery with the first utilization of erythrocytes as a carrier model to deliver β-galactosidase in the 1970s.19 Banz et al. applied a semblable strategy for tumor immunotherapy in which antigen-loaded erythrocytes induced an effective and antigen-specific cytotoxic T lymphocyte (CTL) response.20 They demonstrated that dendritic cells (DCs) efficiently internalized erythrocytes loaded with ovalbumin (OVA) and produced durable OVA-specific T-cell response. Since then, a variety of cell-based drug loading methods, including surface decoration, cytoplasmic loading and gene editing, have been growing vigorously for tumor immunotherapy (Fig. 2).21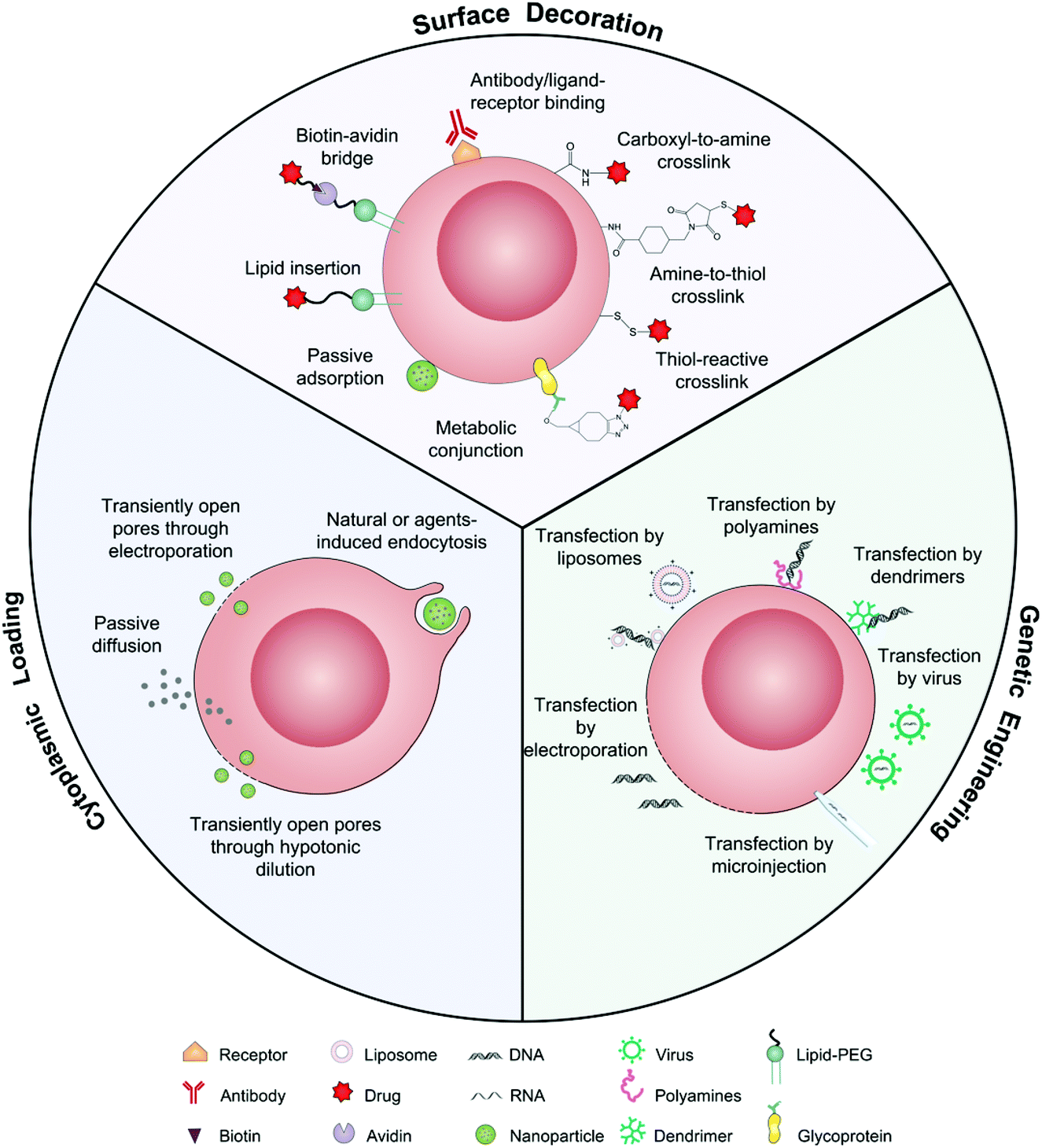 | ||
| Fig. 2 Schematic illustration of intact living cells as drug vehicles by means of surface decoration, cytoplasmic loading, and genetic engineering. | ||
2.1 Surface decoration
The presence of lipids and proteins on cell membranes provides facile decoration to anchor cargos by either covalent or non-covalent conjugation, including lipid insertion, biotin–avidin bridge, carboxyl-to-amine and amine-to-thiol crosslinks, antibody/ligand–receptor binding, metabolic labeling and passive adsorption.22 It is well known that macrophages can be largely recruited into tumors and influence tumor metastasis.23,24 Taking advantage of this feature, engineered macrophage-mediated drug delivery and immune modulation could further enhance therapeutic efficacy especially in metastatic tumors.25 For example, Cao et al. decorated both the legumain-specific propeptide of melittin (legM) and the cytotoxic soravtansine (DM4) prodrug onto the membrane of living macrophages.26 The attached drugs could be preferentially delivered to lung metastases for anti-metastasis therapy. The modified macrophages named LD-MDS responsively converted into DM4-loaded vesicles by legumain protease which was highly expressed in the metastatic niches. The damaged cancer cells induced by DM4-loaded vesicles released secondary vesicles and free drugs, which further destroyed neighboring cancer cells. This study provided a new opportunity to exploit macrophages to enhance targeted delivery and controlled release of drugs in metastatic lung tumors. Furthermore, Gu's group focused on the use of platelets as carriers for the delivery of immune checkpoint inhibitors.29 As cellular fragments released from megakaryocytes, platelets tend to accumulate at wounds and interact with circulating tumor cells (CTCs).27,28 Taking advantage of the intrinsic properties of platelets, they conjugated anti-PD-L1 (aPD-L1) to the surface of platelets, by which the aPD-L1 checkpoint inhibitor could target the residual cancer cells and CTCs in mice bearing B16 or triple-negative breast carcinoma that had been previously resected. The release of aPD-L1 was initiated by the generation of activated platelet-derived microparticles (PMPs) (Fig. 3). Given the characteristics, this approach was promptly extended to treat acute myeloid leukemia by systemically delivering anti-PD-1 (aPD-1) decorated platelets that were further conjugated with hematopoietic stem cells (HSCs), called the HSC-platelet-aPD-1 assembly.30 Relapse resulting from incomplete elimination of leukemia cells has become a predicament which impedes the cure of leukemia.31,32 HSC drove HSC-platelet-aPD-1 homing to the leukemia site, which released aPD-1 in situ by triggering the activation of platelets. This work pioneered a targeted release by HSC-platelet cellular combination. Compared to combination therapy with intravenous administration of free aPD-1, the cell-mediated delivery can enrich aPD-1 and the combined drugs simultaneously at the lesions to maximize the treatment outcome and minimize systemic toxicity. To achieve activation-responsive drug release, Jones et al. designed an approach in which cytolytic granules secreted by CTLs triggered the release of drugs from CTL-bound backpacks.33 CTLs killed target cells by secreting toxic proteases and perforin (a pore-forming protein) into the synapse which was initiated by the T-cell receptor (TCR) recognizing antigen.34,35 Perforin was concurrently used to lyse lipid nanoparticles conjugated to the surface of CTLs. IL-15 encapsulated within CTL-bound nanoparticles was released following co-culture of the CTLs with the target cells. Finally, they replaced lipid nanoparticles with protein nanogels as cell surface backpacks which selectively released protein drugs in response to CTL activation.36 The utilization of the characteristics of CTLs represents an elegant approach for achieving a targeting and controlled-release system simultaneously. In contrast to systemic administration of free IL-15, protein nanogels that carried an IL-15 super-agonist complex selectively expanded T cells 16-fold in tumors, which led to substantially enhanced therapeutic efficacy.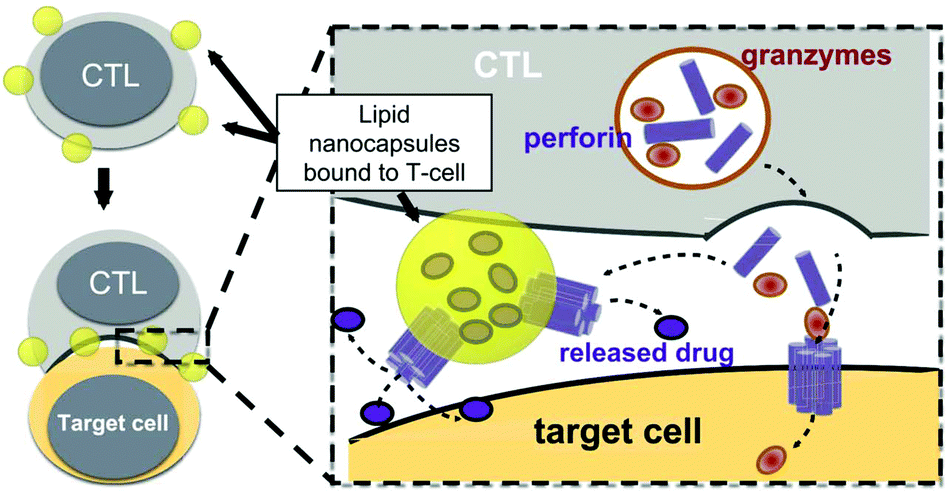 | ||
| Fig. 3 Strategy for CTL-triggered drug release from lipid nanocapsules. CTLs encountering target cells release perforin and granzymes into the immunological synapse formed between the CTL and target cell. Lipid nanocapsules (NCs) covalently anchored to CTL surface proteins traffic into the synapse, where they are exposed to perforin released by the CTL, enabling disruption of the NC and release of the encapsulated drug in tandem with the lysis of the target cell.33 Copyright © 2016 Elsevier Ltd. All rights reserved. | ||
Inevitably, there are potential challenges in drug delivery by cell surface modification. A constraint of surface modification is that drug loading may adversely affect cell functions including adhesion, migration, and signal transduction. As the loaded cargo on the cell surface can occupy the epitopes and partly hinder the interaction between surface ligands of the transferred cells and the relevant receptors on blood vessels or tumor sites, the limited drug loading also needs to be considered. In this case, myeloid cells such as neutrophils, macrophages, and monocytes may show advantages over lymphocytes due to their larger surface area. In another sense though, appropriate dosing prevents over-activation which can cause systemic toxicity. In addition, cell division, releasing vesicles and drug shedding can lead to dilution or premature release of the loaded cargoes.
2.2 Cytoplasmic loading
Different from surface decoration, loading inside the cytoplasm enables cells to serve as a “Trojan horse” that can deliver drugs covertly for cancer immunotherapy. Given that high doses of drugs can cause immediate cell death, nanoparticle capsules have been employed to load drugs before loading into live cells.37,38 Cytoplasmic loading can be achieved by natural or agent-induced endocytosis as well as transiently opening pores on cell membranes through electroporation or hypotonic dilution.39,40 Xue et al. reported that neutrophils internalized with paclitaxel (PTX)-loaded liposomes suppressed postoperative malignant glioma recurrence.41 Neutrophils can traverse the blood–brain barrier42 and migrate to inflamed tissues induced by surgical tumor removal.43,44 PTX-loaded liposomes were released from the neutrophils triggered by highly concentrated inflammatory signals and subsequently transported PTX into the tumor cells. While this neutrophil-mediated delivery of chemotherapeutics significantly improved the survival rates, recurrent growth of tumors could not be inhibited completely. Furthermore, DCs loaded with doxorubicin-polyglycerol-nanodiamond composites (Nano-DOX) improved the activation of mouse spleen-derived lymphocytes which suppressed the co-cultured glioblastoma cells (GCs) (Fig. 4).45 In patient-derived tumor xenograft (PDX) models, Nano-DOX-loaded DCs enhanced the infiltration and activation of lymphocytes. Since DCs possess the ability to migrate into the central nervous system (CNS) by receptor interactions between DCs and microvascular endothelial cells during injury,46 this strategy exhibits the potential to inhibit orthotopic glioblastoma (GBM). It is critical to avoid the degradation and release of drugs before reaching the targeted locations. Nanoparticles internalized by phagosomes especially in macrophages can fuse with lysosomes which contain an array of hydrolytic enzymes that can quickly digest drug carriers.47 To solve the issue, Zhang et al. created a nanocapsule platform using a solid silica sheath filled with a DOX–silica complex core, which was more resistant to degradation.48 Injecting nanocapsule-laden macrophages in an U87MG xenograft model showed efficient suppression of tumor growth and low systemic toxicity. Other than phagocytic cells, platelets have been often loaded with drugs by punching holes on cell membranes. For example, Rao et al. loaded photosensitive gold nanorods (AuNR) into platelets through an electroporation process.49 Photothermal material-mediated PTT is a promising way for cancer therapy through effectively ablating tumor tissues.50 AuNR-loaded platelets that combined the photothermal effect of AuNR with the prolonged circulation and tumor targeting ability of platelets could effectively inhibit the growth of head and neck squamous cell carcinoma (HNSCC) after local laser irradiation. In addition to these typical cell carriers, Wen et al. creatively developed adipocytes as drug depots which could modulate adipocyte–cancer cell interactions and coordinate an immunosuppressive tumor microenvironment (TME) to kill cancer cells.51 Adipose tissue supported tumor oncogenesis and metastasis by promoting angiogenesis, providing energy derived from fatty acid metabolism, and inducing PD-L1 expression.52 Hydrophobic rumenic acid (RA) and doxorubicin prodrug (pDox) were encapsulated through co-incubation as adipocytes naturally tend to accumulate lipids. The loaded RA and pDox were released by cancer cell-induced lipolysis and subsequently activated by intracellular reactive oxygen species (ROS).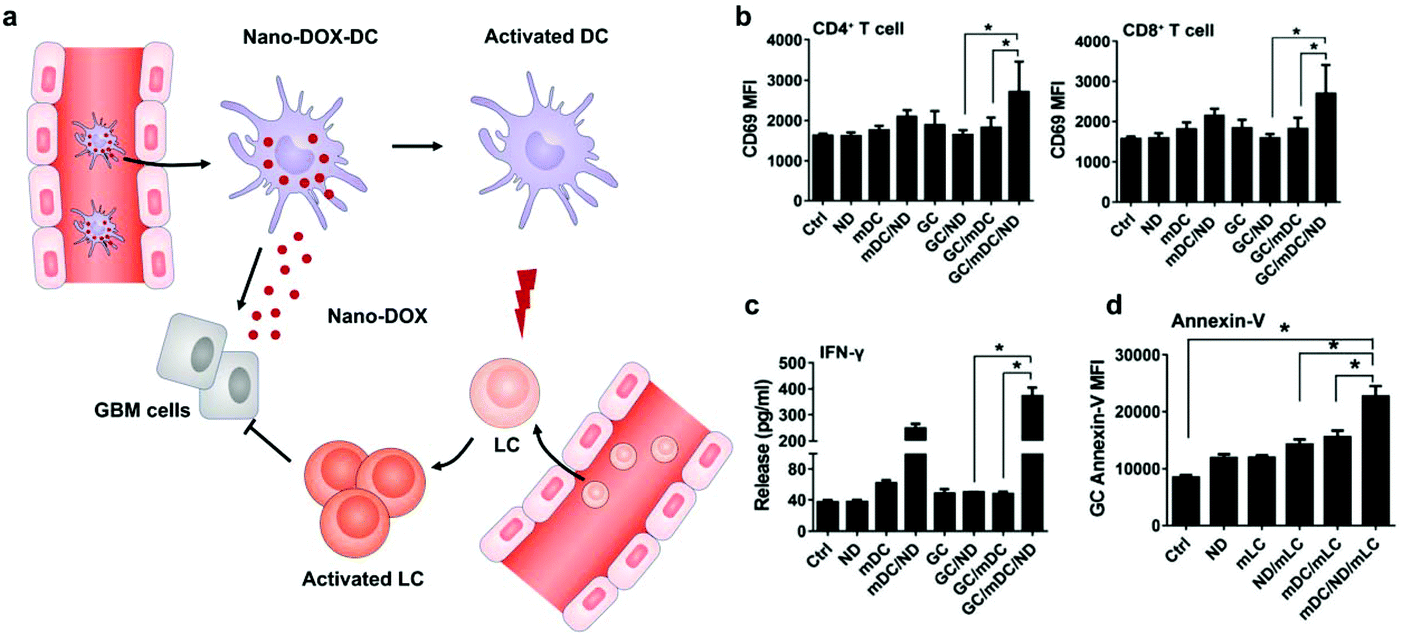 | ||
| Fig. 4 DC-mediated Nano-DOX delivery elicits potent anti-cancer immunity in GBM. (a) Schematic illustration of Nano-DOX-DCs enhancing the anti-cancer immune response in GBM by releasing Nano-DOX to induce the ICD of GCs and thereby enhancing GC immunogenicity. Subsequently, the activated and mature DCs present antigens and activate LCs (lymphocytes). (b) mDCs were cultured with GCs in the presence of Nano-DOX (2 mg mL−1) for 24 h and mLCs were added in the co-culture and harvested for CD69 staining and FACS analysis (gated on CD4+ and CD8+ subsets) after 24 h of incubation. (c) Cell culture media were collected and assayed for IFN-g by ELISA. (d) Apoptosis of GCs was detected by FACS analysis of surface-exposed annexin v immunofluorescent staining. Geometric means were used to quantify the fluorescence intensity. Values were means ± SD (n = 3, *p < 0.05).45 Copyright © 2018 Elsevier Ltd. All rights reserved. | ||
The above cell-based delivery systems mainly aim to improve the ICD of cancer cells and minimize nonspecific toxicity. It is worth noting that the balance between intracellular loading and biological activity of the cells needs to be further investigated. The integrity of cargos loaded into cells also needs to be considered.
2.3 Genetic engineering
Cells can be genetically encoded to secrete therapeutic agents, such as protein and peptide drugs.53 The methods capable of importing DNA or RNA molecules into cells mainly include chemical (lipids, polyamines and dendrimers), physical (electroporation and microinjection) and viral transfection.54 Genetic engineering has negligible influence on cells which can preserve their original functions. Among the cells that can be genetically engineered for cancer immunotherapy, the chimeric antigen receptor (CAR)-T cells are certainly the superstar which show the capacity to induce remission in patients associated with acute lymphoblastic leukemia and large B cell lymphoma.55 However, the implementation of this technology has been constrained by heterogeneous tumor antigen expression and single target-induced escape. To overcome this limitation, Choi et al. developed anti-EGFRvIII (a GBM-specific tumor antigen) CAR-T bispecific T-cell engager (BiTE) cells that could secrete BiTE to inhibit EGFR.56 The EGFR-specific BiTEs redirected CAR-T cells and recruited bystander T cells against wild-type EGFR to eliminate EGFRvIII-negative and EGFR-positive GBM. This approach therefore could relieve the effects of EGFRvIII antigen loss. The ingenious utilization of CAR-T cells to achieve the effective targeted delivery of BiTE-EGFR generated durable and specific antitumor responses against heterogeneous tumors. In view of the fact that signaling molecules derived from the tumor stroma mediate the recruitment and proliferation of mesenchymal stem cells (MSCs) for tissue construction,57 MSCs have been broadly explored to deliver gene-editing products for cancer immunotherapy, including interferons (IFNs), IL-2, IL-12, and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL).58–60 Advances in recent years have demonstrated the potential for producing functional platelets from genetically manipulated megakaryocytes (MKs) for clinical use.61 Gu and coauthors developed engineered platelets as blood-originated platelets cannot be expanded and genetically manipulated in vitro (Fig. 5). MKs were engineered to stably express murine PD-1 and produce mature PD-1-presenting platelets.62 Furthermore, genetically manipulated MKs could also carry cyclophosphamide into the cytoplasm, which is a chemotherapeutic drug that has been used to treat various cancers. Engineered platelets that combined with PD-1 blockade and chemotherapy further enhanced the antitumor effect within the surgical tumor microenvironment. Although as a fundamental tool they show plenty of scope for further development, the difficulty of operation limits the development of gene-editing cells.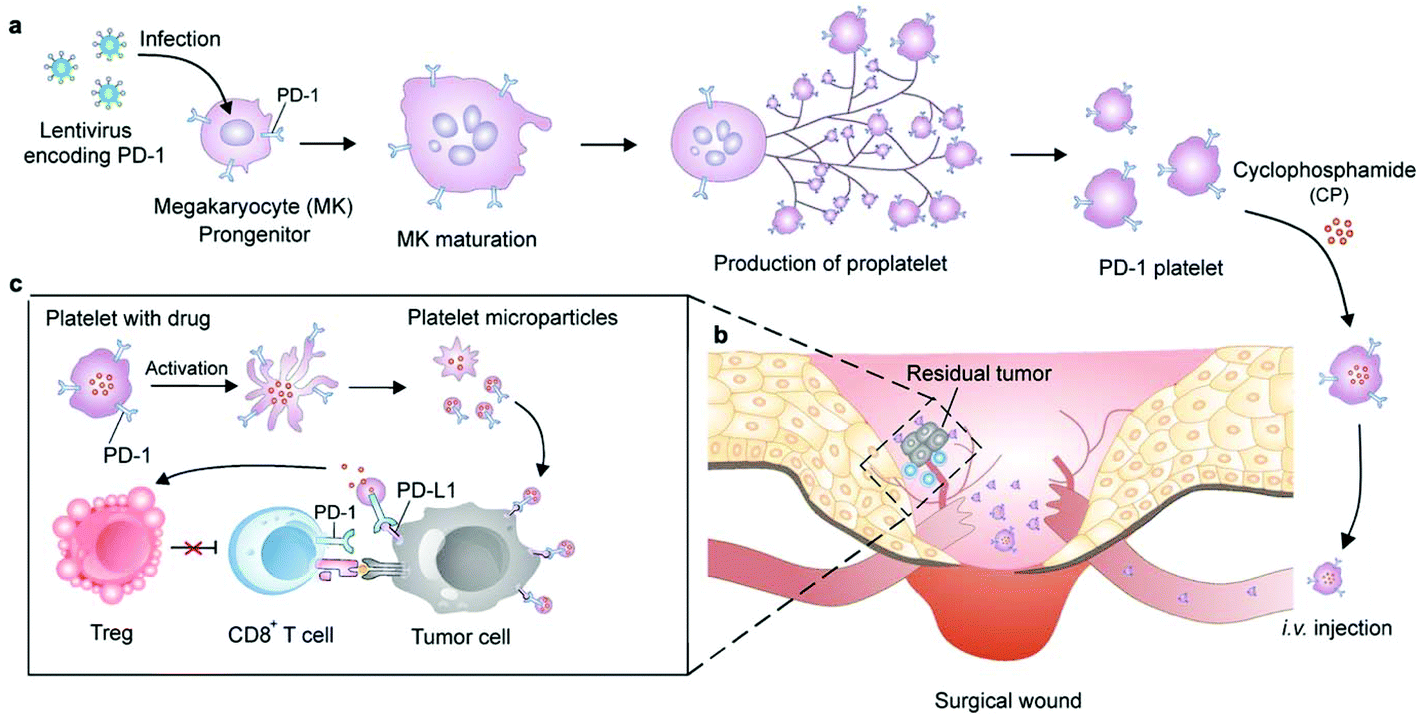 | ||
| Fig. 5 Schematic of the production of PD-1-expressing platelets and reinvigoration of CD8+ T cells. (a) The schematic shows the L8057 cell line stably expressing murine PD-1 and production of platelets. (b) PD-1-expressing platelets target tumor cells within the surgery wound. (c) PD-L1 blockade by PD-1-expressing platelets reverts exhausted CD8+ T cells to attack tumor cells.62 Copyright © 2018 American Chemical Society. | ||
3. Cell membranes
In comparison with intact cells, cell membranes are more widely applied for camouflaging cargoes because of the simplicity of operation and storage. Cell membrane-mediated drug delivery is usually characterized by a synthetic nanoparticle core that is coated with a layer of natural cell membrane (Fig. 6).63,64 The process of cell membrane extraction includes hypotonic lysis or repeated freeze/thaw cycles to achieve membrane lysis. Gradient centrifugation is sometimes needed to remove the cell nuclei and cytoplasm.65,66 There are three typical ways to wrap nanoparticle cores with cell membranes, including membrane extrusion through a nanoscale porous membrane, ultrasonic fusion leading to spontaneous formation of a core–shell nanostructure, and a microfluidic system combining rapid mixing with electroporation.65,67,68 Synthetic cores can be organic or inorganic, such as gold, silica, polymer, nanogel, protein/peptide, metal–organic frameworks (MOFs), iron oxide, upconversion nanoparticles (UCNPs) and black phosphorus. The composition of the cores can be modified to accommodate various payloads. Meanwhile, cell membranes bestow a wide range of functions on the nanoparticles according to source cells, such as immune cells, blood cells, stem cells and cancer cells.13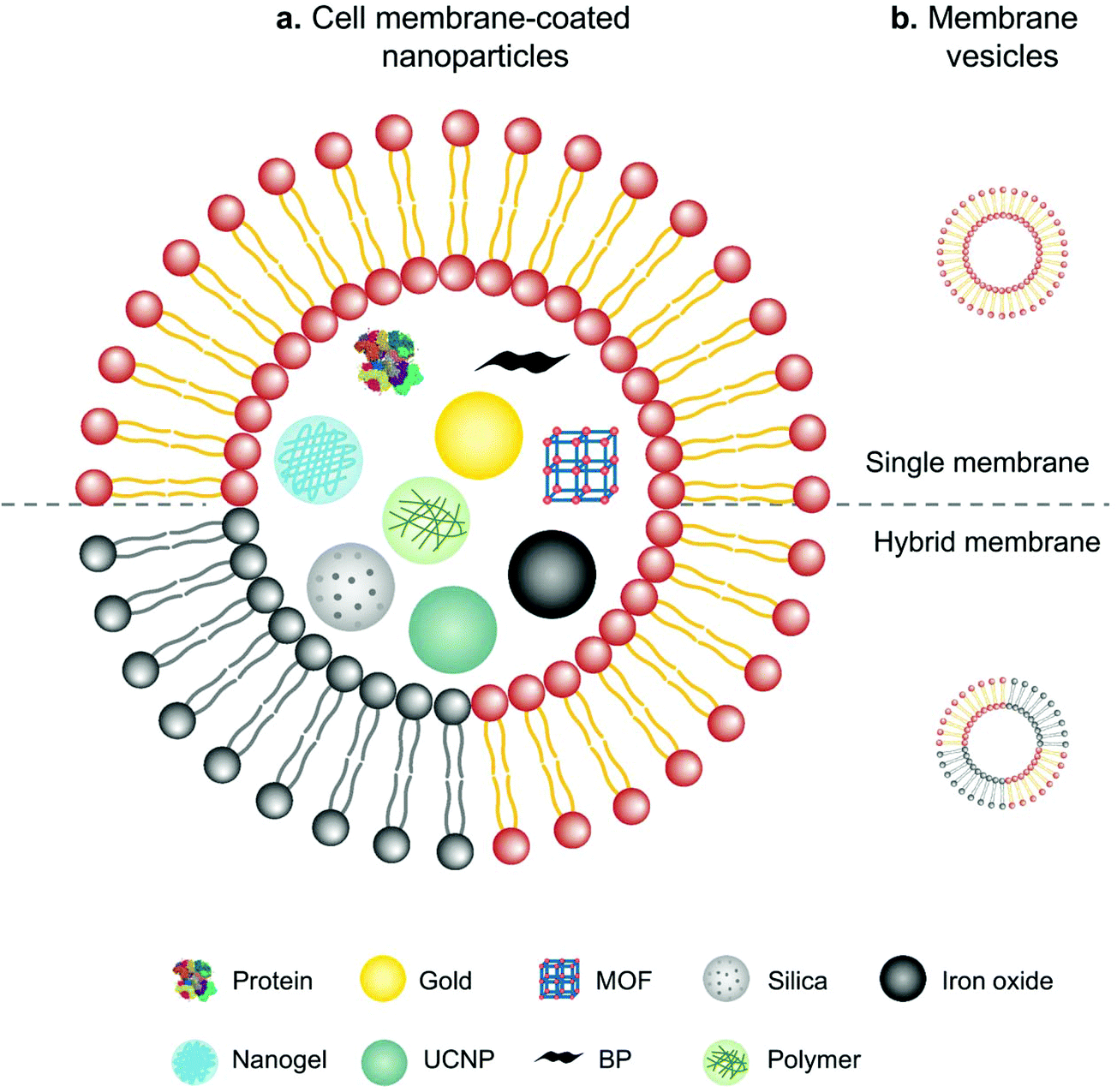 | ||
| Fig. 6 Schematic illustration of (a) cell membranes as drug vehicles by encapsulating various nanoparticles and (b) cell membrane-extruded vesicles. | ||
3.1 Natural cell membranes
Erythrocyte membranes are the first reported yet well-studied coating due to the simplicity of cell collection and membrane extraction.13 EM-coated black phosphorus quantum dot nanovesicles (BPQD) that rapidly converted near-infrared (NIR) light into thermal energy could induce triple-negative breast cancer cell apoptosis in situ with the help of irradiation.69,70 Furthermore, EM-coated BPQD-mediated PTT could combine with aPD-1 treatment to significantly inhibit tumor growth by increasing the infiltration and activity of CD8+ T cells in the tumor. Since the utilization of cancer cell membrane-coated nanoparticulates to elicit antitumor immunity by Kroll et al.,71 cancer cell membranes have been widely used to stimulate the immune system and deliver drugs into tumor sites. Therapeutic nanoparticles that combined starvation therapy and immunotherapy were coated with cancer cell membranes to achieve enhanced antitumor efficacy.72 Starvation therapy was based on mesoporous silica nanoparticles (MSN) that were loaded with glucose oxidase (GOx) to prevent energy supply and then encapsulated with cancer cell membranes to increase accumulation in the tumor tissue. This synthetic complex could induce the production of mature DCs, which efficiently enhanced the aPD-1 effect to eliminate the tumor. Different from the targeting behavior of cancer cell membranes, Li et al. exploited the ability of neutrophil membrane-coated nanoparticles (pCSs) to compete for myeloid-derived suppressor cells (MDSCs) which strongly inhibited anti-tumor immune responses and promoted tumor growth and metastasis.73,74 Similar to MDSCs in phenotype and morphology, neutrophils could neutralize MDSC-related cytokines, thereby inhibiting the function of MDSCs. As a result, pCS treatment dramatically increased the infiltration and activation of T cells in the tumor. In addition, combining with aPD-1 synergistically suppressed tumor progression and prolonged animal survival in murine breast cancer and melanoma models.74 As cancer vaccines, DCs pulsed with tumor-associated antigens (TAAs) in vitro have shown significant progress.75 The upregulated CD80/CD86 expression and the presence of MHC–peptide complexes on the cell surface endow DC membranes with the retained mature phenotype and ability to perform vaccine functions.76,77 Although the inefficiency of antigen presentation to induce potent antitumor responses is a common issue, Jiang et al. developed engineered artificial antigen presenting cells (aAPCs) to overcome the dilemma.78 Membranes from a model cancer cell line expressing the co-stimulatory marker CD80 were engineered and coated onto PLGA cores. These biomimetic nanoscale aAPCs contain two signals including various antigen peptide–MHC complexes from cancer cells and CD80, which are indispensable to promote T cell activation. The highlight of these aAPCs is that the intact membrane component enables the presentation of multiple tumor antigens.3.2 Modified cell membranes
Surface modification of cell membranes used for encapsulation can overcome the limitation of a single-targeted function. For example, a T-cell membrane coating increased the accumulation of a nano-photosensitizer (indocyanine green, ICG) in tumor sites by TCR-specific recognition. Metabolic labeling of glycans, such as the biorthogonal reaction between azide (N3) attached to T cell membranes and bicyclo[6.1.0]nonyne (BCN) incorporated in tumor cells, further enhanced the accumulation in tumors. The dual-targeting strategy facilitated the aggregation of ICG in the tumor and achieved highly efficient PTT outcomes such as prolonged survival rates (Fig. 7).79 In addition to cell membrane-coated nanoparticles, cell membrane extruded nanovesicles (MVs) can also deliver drugs efficiently. For instance, Ochyl et al. described that MVs derived from monophosphorylate lipid A (MPLA)-primed DC membranes showed the ability to load antigen peptides and promote antigen-specific T cell response.80 Moreover, Zhang et al. obtained PD-1 presented MVs from engineered HEK293 T cells that stably expressed mouse PD-1 on the cell membranes.81 PD-1 MVs capable of binding to PD-L1 on the surface of tumor cells remarkably increased the infiltration of CD8+ T cells in the tumor and enhanced antitumor responses.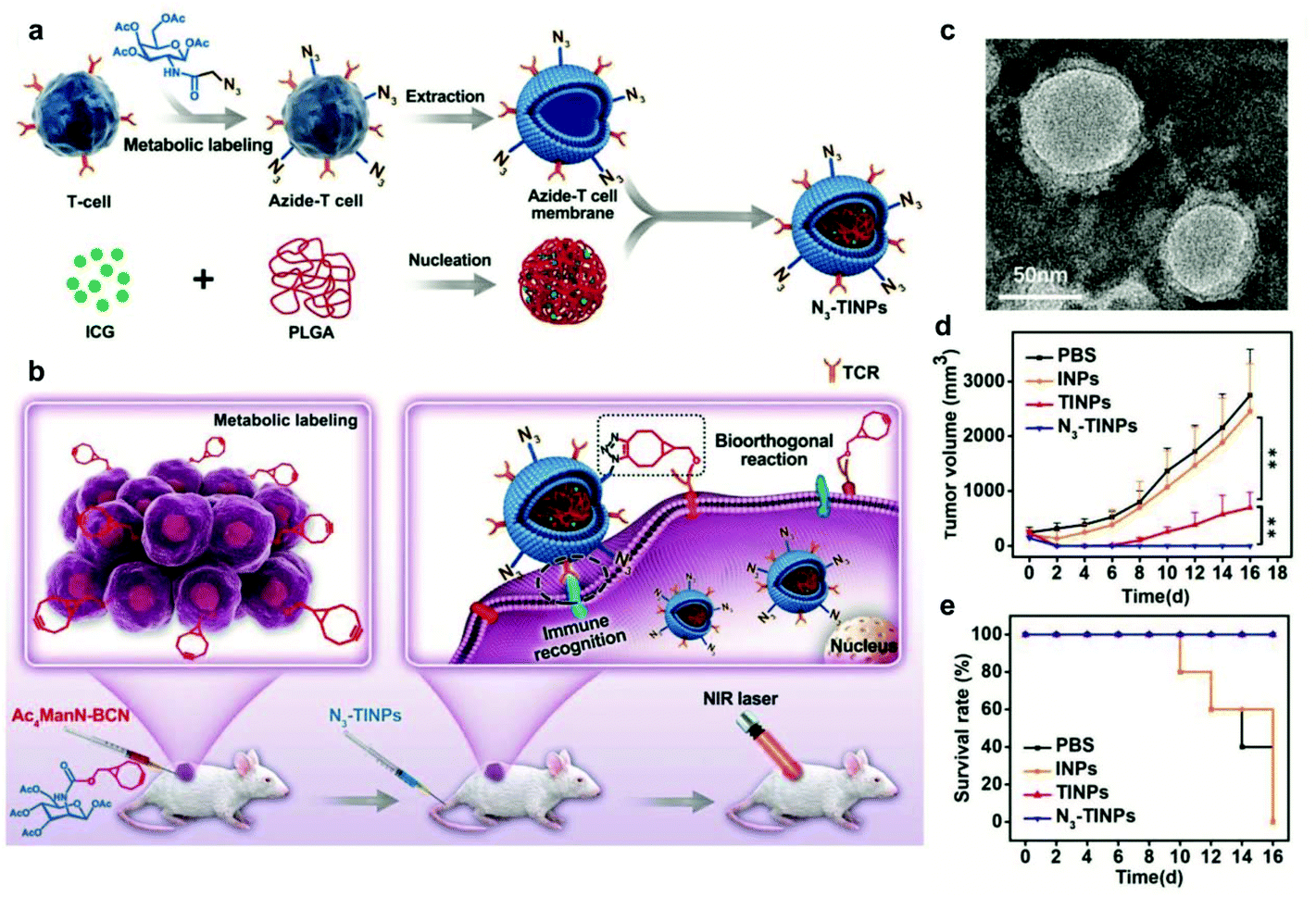 | ||
| Fig. 7 N3-Labeled T cell membrane-biomimetic nanoparticles with a dual-targeting mechanism for highly efficient PPT. (a) Schematic illustration of the synthesis of N3-TINPs. N3-labeling T cell membranes were coated on ICG-PLGA polymeric cores by extrusion. (b) Schematic illustration of tumor cells carrying the BCN group via natural glycometabolic labeling by pretreatment with Ac4ManN-BCN. N3-TINPs could target the tumor through immune recognition of the T cell membrane and the biorthogonal reaction between BCN and N3 groups, and effectively eliminate mouse tumors through ICG-mediated photothermal effects. (c) TEM image of N3-TINPs. (d) Raji tumor growth curves of different groups (n = 5). (e) Survival rates of the tumor-bearing mice.79 Copyright © 2019 the authors, published by WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
3.3 Fused cell membranes
Recently, to acquire multiple functions, multimodal coatings have emerged by fusing with different cell membranes.82–85 For example, to allow DCs to present more comprehensive tumor antigens, Liu et al. fused DCs and murine mammary carcinoma tumor (4T1) cells to form hybrid cells by polyethylene glycol (PEG)-mediated cell fusion (Fig. 8). Unlike the downregulated expression of tumor antigens in the immune escape mechanism, the hybrid cells could carry a whole array of highly expressed tumor antigens as well as DC-derived co-stimulatory molecules in the membranes.86 Consequently, the hybrid membrane-coated photosensitizer MOF (PCN224) resulted in a durable immune response to inhibit the recidivation of primary tumors as well as the growth of distant tumors.82 This work provided a general strategy as the fusion of DCs with different cancer cells can be expanded to the treatment of many types of cancer. Sonication and physical extrusion of the mixture of different cell membranes are alternatives for producing fused membranes. Han et al. leveraged erythrocytes to deliver TAAs to APCs in consideration of the fact that senescent or damaged erythrocytes were physiologically eliminated by scavenger cells, such as macrophages and DCs within the spleen.85,87 Erythrocyte and tumor cell membranes were fused through sonication and membrane extrusion to obtain Ag nanovesicles which effectively reached the spleen and activated T cell immune responses. The combination of Ag nanovesicles and aPD-1 blockade promoted tumor regression in murine B16F10 and 4T1 tumor models. Additionally, platelet–leukocyte hybrid membranes improved the efficiency and specificity of magnetic bead-mediated purification of CTCs.83 Iron oxide magnetic nanoparticles coated with cancer stem cell–platelet hybrid membranes could enhance the PTT of head and neck squamous cell carcinoma (HNSCC).84 In addition to synthetic nanoparticle cores, our group developed a unique strategy to camouflage living bacteria by wrapping with erythrocyte membranes using a simple extruding technique.88 This approach decreased the bodily clearance and side effects of bacteria, while enhancing their accumulation in tumor tissues. Given the intrinsic antitumor activities of bacteria,89 the application of this approach is expected to expand to cancer immunotherapy.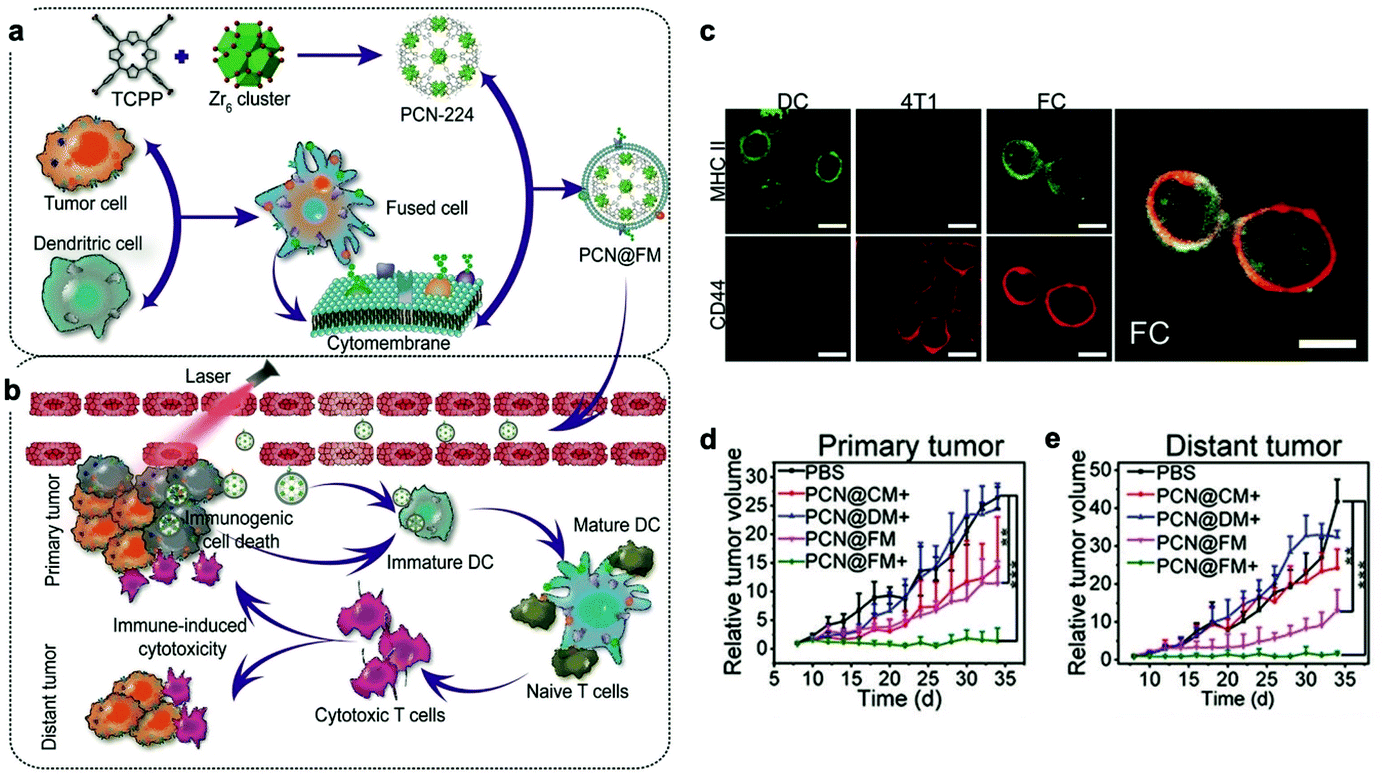 | ||
| Fig. 8 Cytomembranes of hybrid cells derived from cancer and DCs enhancing tumor-specific immunotherapy. (a) Schematic illustration of the preparation process of PCN@FM. (b) Combination between irradiation-mediated PDT and FM- and ICD-induced immunotherapy toward primary tumor and distant tumor therapy. (c) CLSM observation over anti-MHC II-FITC antibody (green) and anti-CD44-APC antibody (red) stained DC, 4T1, and FC cells. Scale bar: 16 μm. (d) Average volumes of the primary tumors in different groups (n = 6). *p < 0.05, **p < 0.01, ***p < 0.001. (e) Average volumes of the distant tumors in different groups (n = 6).82 Copyright © 2019, John Wiley and Sons. | ||
In brief, cell membrane coating is a top-down approach by which the natural functionality can be exploited directly without reversing biological processes and interactions. However, the process of extruding cell membranes may damage the structure of proteins on the surface and weaken their interaction with the tumor microenvironment. In addition, cell membrane-coated nanoparticles cannot penetrate deep into the tumor due to the elevated interstitial fluid pressure (IFP) and dense stroma in the tumor tissue. Finally, more efficient preparation, characterization, and preservation methods should be standardized for these coated nanoparticles.
4. Extracellular vesicles
As a mini cell clone that contains proteins, lipids and genetic materials, EVs are responsible for intercellular communications.90 Based on their origin and size, EVs can be divided into three groups, namely exosomes (30–150 nm), microvesicles (50 nm–1 μm) and apoptotic bodies (50 nm–5 μm).91 EVs play a crucial role in both primary tumor growth and metastatic evolution by orchestrating multiple pathophysiological processes to support pre-metastatic niche formation and subsequent metastasis.92 Furthermore, the intrinsic ability to protect the loaded cargoes from severe extracellular environments makes them ideal for drug delivery. A variety of cells, such as stem cells and cancer cells, have been investigated as sources for EV production.934.1 Production of EVs
Regardless of spontaneous EV release, approaches devoted to increase EV production by further stimulation mainly include (1) biological/chemical triggers induced by stress responses (serum deprivation and hypoxia), activated stimulations (TNF-α and IL-8) and chemical agents (cytochalasin B and ethanol) and (2) physical triggers based on the destruction of cell membranes (microfluidic device, cell-slicing system and centrifugal force combined with filters) and self-assembly into nanovesicles (Fig. 9).94 As a major cytokine that governs various biological characteristics of NK cells,95 IL-15 could improve the production of NK cell-derived EVs (NK-EVs) and enhance their cytolytic activity to significantly inhibit the growth of GBM xenografted in mice.96 Moreover, EVs derived from tumor-repopulating cells (TRCs) that were isolated from three-dimensional (3D) fibrin gels showed enhanced ability to deliver anticancer drugs as they were softer and more deformable.97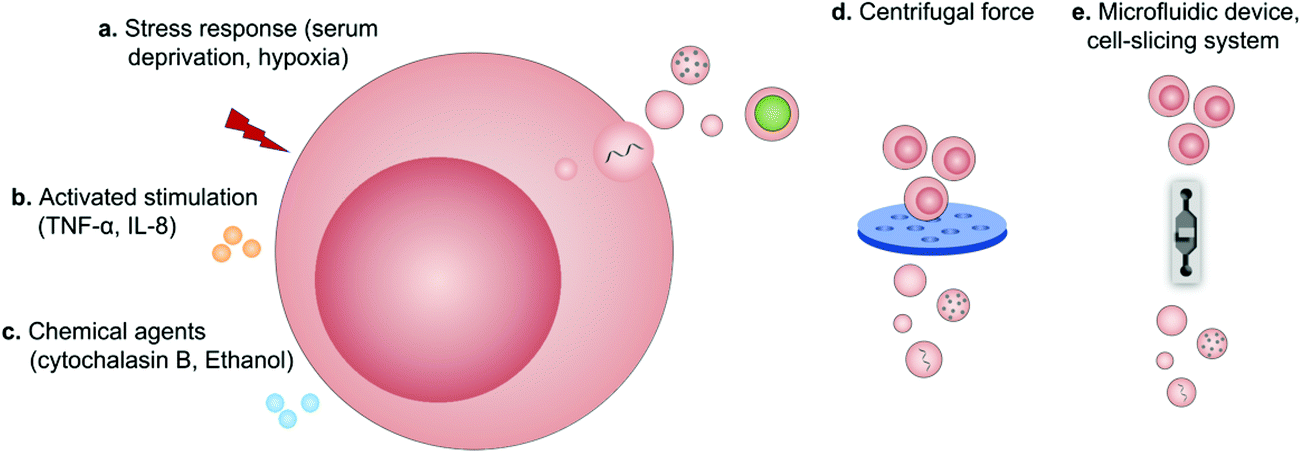 | ||
| Fig. 9 Schematic illustration of the production of extracellular vesicles by (a) stress responses, (b) activated stimulations, (c) chemical agents, (d) centrifugal force, and (e) microfluidic device-based cell-slicing systems. | ||
4.2 Pre-loaded EVs
An alternative for drug loading is to pre-load parental cells with cargoes so that the released EVs inherit both cell features and cytoplasmic solutes.98 Considering that EVs derived from cancer cells naturally contain microRNA (miRNA) and have the ability to target tumor sites, miRNA-based tumor therapies usually prefer EVs as delivery vehicles. Bose et al. transferred donor 4T1 cells to prepare anti-miRNA-21 loaded tumor cell derived EVs (TEVs) that could block the function of oncogenic miRNA-21 in cancer cells.99 The obtained TEV-anti-miRNA-21 could specifically accumulate in the tumor tissue and effectively kill the cancer cells. The therapeutic effect was further enhanced by combining with PTT using TEV-anti-miRNA-21 coated gold-iron oxide nanoparticles. Yong et al. developed exosome-sheathed Dox-loaded nanoparticles (Dox@E-NPs) by the exocytosis of cancer cells which were endocytosed with Dox-loaded nanoparticles (Dox@NPs).100 Dox@NPs were pre-endocytosed into cancer cells by incubation, then transported through autophagosomes and wrapped with exosomes via exocytosis. Dox@NPs were exocytosed from cancer cells in a time-dependent manner, and 96% of Dox@NPs were expelled out after culture in fresh medium for 18 h. Dox@E-NPs exhibited enhanced tumor accumulation and anti-tumor activity. This work provided a unique approach to prepare exosome-based carriers by concurrently using endocytosis and exocytosis of cancer cells. Similarly, Wang et al. exploited ultraviolet light irradiation-induced budding of parent cells for loading cargoes into EVs to produce Bi2Se3 nanodots and Dox co-embedded tumor cell-derived microparticles (Bi2Se3/Dox@MPs).101 Bi2Se3/Dox@MPs showed deepened tumor penetration and enhanced cellular internalization mediated through membrane fusion.4.3 Drug-loaded EVs
Additionally, cargoes can be loaded into EVs directly by electroporation or surface modification.98 For example, Taghikhani et al. successfully incorporated a miRNA mixture that can promote the differentiation and maturation of DCs into tumor-derived exosomes by electroporation.102 The surface of exosomes could be modified with immunostimulatory CpG DNA by streptavidin–biotin interaction.103 These results indicate that modified EVs represent a set of promising vaccines for cancer immunotherapy. Moreover, decoration on the surface of EVs can further improve tumor targeting of the encapsulated cargoes. To target αvβ3 integrin receptors on the surface of GBM, Zhu et al. decorated the surface of PTX-loaded EVs that were derived from embryonic stem cells by using lipid-inserted RGD (Arg-Gly-Asp) peptides for active tumor-targeting therapy.104 Tumor-specific aptamers could similarly direct EVs to deliver chemotherapeutics into tumors.105 Similar to the generation of hybrid cells, hybrid EVs could be obtained by fusion technology. Lin et al. successfully encapsulated the CRISPR/Cas9 expression vector into hybrid exosomes which were produced by incubating HEK293 cell-derived exosomes with expression vector-loaded liposomes.106 This work emerged as a new platform for delivering the CRISPR/Cas9 system for gene editing. These hybrid EVs could also be applied to the eukaryotic–prokaryotic vesicle (EPV) nanoplatform. Chen et al. integrated melanoma antigens with natural adjuvants by fusing melanoma cell membrane vesicles (CMVs) with bacterial outer membrane vesicles (OMVs) (Fig. 10). They demonstrated that the indocyanine green (ICG) implanted EPV combined with PTT could stimulate the immune system and enhance the antitumor effects as a therapeutic vaccine.107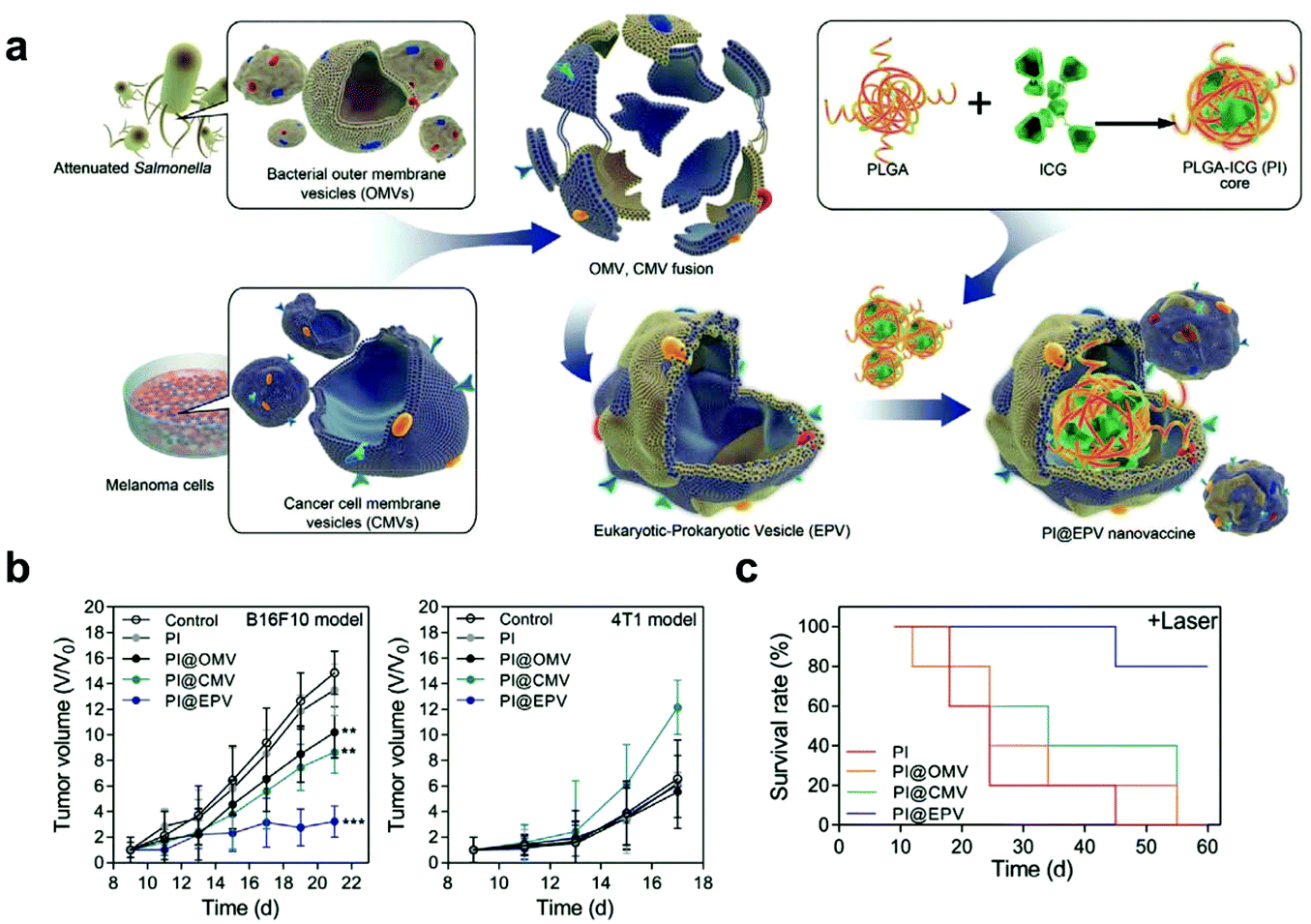 | ||
| Fig. 10 A hybrid OMV-CMV nanoplatform with PTT for enhanced antitumor vaccination. (a) Schematic illustration of the fabrication of the eukaryotic–prokaryotic vesicle coated PI@EPV nanovaccine. (b) Tumor growth curves of B16F10 or 4T1 after pretreatment with the nanovaccine. The error bars represent means ± SD. (c) Survival curves of mice after various treatments in the therapeutic assay.107 Copyright © 2020, WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
Taken together, EVs provide a promising platform for improving the delivery of therapeutic agents by virtue of their intrinsic properties. However, this technique has not been explored adequately yet. First, it remains unclear how EVs transport during cellular communication. The potential immunogenicity and the risk of promoting metastasis also need to be considered and verified. Second, drugs pre-loaded into parental cells may be affected by lysosomes and lose their integrity. Lastly, electroporation can potentially damage the component of EV membranes, which influences the recognition of EVs by recipient cells. The underlying mechanism of EVs regarding the delivery of immunotherapeutic drugs needs to be understood appropriately before the consideration of their clinical translation.
5. Conclusion
In this review, we have briefly summarized the advances in cell-mediated drug delivery for cancer immunotherapy. Engineered drug-loaded cells and cell derivatives intelligently exploit the unique delivery mechanisms of natural particulates. With endogenous characteristics, these delivery systems can communicate with complicated in vivo environments and achieve evasion of immune surveillance, minimal toxicity, prolonged circulation, and specific targeting. Given the great flexibility and versatile functions, engineered cells and their derivatives have been widely applied to guide cancer immunotherapy by combining with chemotherapy, radiotherapy, PDT and PTT. Although these combination therapies have achieved considerable progress, there are several challenges that need to be overcome for accelerating clinical translation. First, the source and quantity of cells used to prepare cell-based delivery systems limit scaling up of manufacture. Using cells that are easy to culture and expand in vitro could be a potential solution. Second, biomimetic systems cannot be completely comparable to natural cells in terms of bio-responsiveness and movement, which may result in delayed or unpredictable drug release. Lastly, the mechanism of cell-based delivery must be investigated comprehensively to ensure its safety in vivo. For example, the overall biodistribution, systemic immune response, effects on non-targeted tissues and long-term behaviors in biological systems need to be thoroughly assessed. With a deeper understanding of cell functions as well as the development of advanced synthetic techniques and multidisciplinary collaboration, it is promising that these biomimetic strategies could promote the rapid development of cancer immunotherapy both in fundamental research and clinical translation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21875135), the Recruitment Program of Global Youth Experts of China (D1410022), the Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant Support (20181704), and the Innovative Research Team of High-level Local Universities in Shanghai (SSMU-ZLCX20180701).References
- R. L. Siegel, K. D. Miller and A. Jemal, CA Cancer J. Clin., 2019, 69, 1 CrossRef.
- D. S. Chen and I. Mellman, Immunity, 2013, 39, 1 CrossRef CAS.
- J. D. Martin, H. Cabral, T. Stylianopoulos and R. K. Jain, Nat. Rev. Clin Oncol., 2020, 17, 4 Search PubMed.
- R. S. Riley, C. H. June, R. Langer and M. J. Mitchell, Nat. Rev. Drug Discovery, 2019, 18, 3 CrossRef.
- C. W. Shields, L. L. W. Wang, M. A. Evans and S. Mitragotri, Adv. Mater., 2020, 32, 13 CrossRef.
- J. Zhou, G. Wang, Y. Chen, H. Wang, Y. Hua and Z. Cai, J. Cell. Mol. Med., 2019, 23, 8 Search PubMed.
- Z. Zhao, L. Zheng, W. Chen, W. Weng, J. Song and J. Ji, J. Hematol. Oncol., 2019, 12, 1 CrossRef.
- Z. Chen, Q. Hu and Z. Gu, Acc. Chem. Res., 2018, 51, 3 CrossRef.
- C. H. Villa, A. C. Anselmo, S. Mitragotri and V. Muzykantov, Adv. Drug Delivery Rev., 2016, 106, 88 CrossRef CAS.
- B. Huang, W. D. Abraham, Y. Zheng, S. C. Bustamante López, S. S. Luo and D. J. Irvine, Sci. Transl. Med., 2015, 7, 291 Search PubMed.
- H. Dong, X. Xu, L. Wang and R. Mo, Biomater. Sci., 2020, 8, 9 Search PubMed.
- G. S. Selders, A. E. Fetz, M. Z. Radic and G. L. Bowlin, Regener. Biomater., 2017, 4, 1 CrossRef.
- R. H. Fang, A. V. Kroll, W. Gao and L. Zhang, Adv. Mater., 2018, 30, 23 Search PubMed.
- S. R. Denmeade, A. M. Mhaka, D. M. Rosen, W. N. Brennen, S. Dalrymple and I. Dach, et al. , Sci. Transl. Med., 2012, 4, 140 Search PubMed.
- S. H. Seo, K. S. Kim, S. H. Park, Y. S. Suh, S. J. Kim and S. S. Jeun, et al. , Gene Ther., 2011, 18, 5 CrossRef.
- A. Castleton, A. Dey, B. Beaton, B. Patel, A. Aucher and D. M. Davis, et al. , Blood, 2014, 123, 9 CrossRef.
- J.-W. Yoo, D. J. Irvine, D. E. Discher and S. Mitragotri, Nat. Rev. Drug Discovery, 2011, 10, 7 CrossRef.
- X. Xu, T. Li, S. Shen, J. Wang, P. Abdou and Z. Gu, et al. , Theranostics, 2019, 9, 25 Search PubMed.
- G. M. Ihler, R. H. Glew and F. W. Schnure, Proc. Natl. Acad. Sci. U. S. A., 1973, 70, 9 CrossRef.
- A. Banz, M. Cremel, A. Rembert and Y. Godfrin, Vaccine, 2010, 28, 17 CrossRef.
- S. Tan, T. Wu, D. Zhang and Z. Zhang, Theranostics, 2015, 5, 8 CrossRef.
- D. Sun, J. Chen, Y. Wang, H. Ji, R. Peng and L. Jin, et al. , Theranostics, 2019, 9, 23 Search PubMed.
- Q. Chen, X. H. F. Zhang and J. Massagué, Cancer Cell, 2011, 20, 4 Search PubMed.
- L. Wan, K. Pantel and Y. Kang, Nat. Med., 2013, 19, 11 CrossRef.
- Y. Xia, L. Rao, H. Yao, Z. Wang, P. Ning and X. Chen, Adv. Mater., 2020, 32, 40 Search PubMed.
- H. Cao, H. Wang, X. He, T. Tan, H. Hu and Z. Wang, et al. , Nano Lett., 2018, 18, 8 Search PubMed.
- L. J. Gay and B. Felding-Habermann, Nat. Rev. Cancer, 2011, 11, 2 CrossRef.
- J. W. Semple, J. E. Italiano and J. Freedman, Nat. Rev. Immunol., 2011, 11, 4 CrossRef.
- C. Wang, W. Sun, Y. Ye, Q. Hu, H. N. Bomba and Z. Gu, Nat. Biomed. Eng., 2017, 1, 2 CrossRef.
- Q. Hu, W. Sun, J. Wang, H. Ruan, X. Zhang and Y. Ye, et al. , Nat. Biomed. Eng., 2018, 2, 11 Search PubMed.
- M. M. Gottesman, T. Fojo and S. E. Bates, Nat. Rev. Cancer, 2002, 2, 1 CrossRef.
- L. Ding, T. J. Ley, D. E. Larson, C. A. Miller, D. C. Koboldt and J. S. Welch, et al. , Nature, 2012, 481, 7382 Search PubMed.
- R. B. Jones, S. Mueller, S. Kumari, V. Vrbanac, S. Genel and A. M. Tager, et al. , Biomaterials, 2017, 117, 44 CrossRef CAS.
- J. C. Stinchcombe, G. Bossi, S. Booth and G. M. Griffiths, Immunity, 2001, 15, 5 CrossRef.
- R. Basu, B. M. Whitlock, J. Husson, A. Le Floc'h, W. Jin and A. Oyler-Yaniv, et al. , Cell, 2016, 165, 1 CrossRef.
- L. Tang, Y. Zheng, M. B. Melo, L. Mabardi, A. P. Castaño and Y.-Q. Xie, et al. , Nat. Biotechnol., 2018, 36, 8 CrossRef.
- W.-C. Huang, W.-H. Chiang, Y.-H. Cheng, W.-C. Lin, C.-F. Yu and C.-Y. Yen, et al. , Biomaterials, 2015, 71, 71 CrossRef CAS.
- J. Choi, H.-Y. Kim, E. J. Ju, J. Jung, J. Park and H.-K. Chung, et al. , Biomaterials, 2012, 33, 16 Search PubMed.
- J. Nam, S. Son, K. S. Park, W. Zou, L. D. Shea and J. J. Moon, Nat. Rev. Mater., 2019, 4, 6 Search PubMed.
- C.-M. J. Hu, R. H. Fang and L. Zhang, Adv. Healthcare Mater., 2012, 1, 5 Search PubMed.
- J. Xue, Z. Zhao, L. Zhang, L. Xue, S. Shen and Y. Wen, et al. , Nat. Nanotechnol., 2017, 12, 7 CrossRef.
- D. Shlosberg, M. Benifla, D. Kaufer and A. Friedman, Nat. Rev. Neurol., 2010, 6, 7 Search PubMed.
- E. Kolaczkowska and P. Kubes, Nat. Rev. Immunol., 2013, 13, 3 CrossRef.
- A. D. Gregory and A. McGarry Houghton, Cancer Res., 2011, 71, 7 CrossRef.
- T.-F. Li, K. Li, Q. Zhang, C. Wang, Y. Yue and Z. Chen, et al. , Biomaterials, 2018, 181, 35 CrossRef CAS.
- D. Sagar, C. Foss, R. El Baz, M. G. Pomper, Z. K. Khan and P. Jain, J. Neuroimmune. Pharmacol., 2012, 7, 1 Search PubMed.
- P. Saftig and J. Klumperman, Nat. Rev. Mol. Cell Biol., 2009, 10, 9 CrossRef.
- W. Zhang, M. Wang, W. Tang, R. Wen, S. Zhou and C. Lee, et al. , Adv. Mater., 2018, 30, 50 Search PubMed.
- L. Rao, L.-L. Bu, L. Ma, W. Wang, H. Liu and D. Wan, et al. , Angew. Chem., Int. Ed., 2018, 57, 4 CrossRef.
- L. Cheng, C. Wang, L. Feng, K. Yang and Z. Liu, Chem. Rev., 2014, 114, 21 CrossRef.
- D. Wen, J. Wang, G. Van Den Driessche, Q. Chen, Y. Zhang and G. Chen, et al. , Matter, 2019, 1, 5 CrossRef.
- M. J. Khandekar, P. Cohen and B. M. Spiegelman, Nat. Rev. Cancer, 2011, 11, 12 CrossRef.
- X. Wang, N. Rivera-Bolanos, B. Jiang and G. A. Ameer, Adv. Funct. Mater., 2019, 29, 23 Search PubMed.
- T. K. Kim and J. H. Eberwine, Anal. Bioanal. Chem., 2010, 397, 8 Search PubMed.
- A. Yip and R. M. Webster, Nat. Rev. Drug Discovery, 2018, 17, 3 CrossRef.
- B. D. Choi, X. Yu, A. P. Castano, A. A. Bouffard, A. Schmidts and R. C. Larson, et al. , Nat. Biotechnol., 2019, 37, 9 CrossRef.
- Y. Shi, L. Du, L. Lin and Y. Wang, Nat. Rev. Drug Discovery, 2017, 16, 1 CrossRef.
- L. Elzaouk, K. Moelling and J. Pavlovic, Exp. Dermatol., 2006, 15, 11 CrossRef.
- M. R. Loebinger, A. Eddaoudi, D. Davies and S. M. Janes, Cancer Res., 2009, 69, 10 CrossRef.
- X. Yang, J. Du, X. Xu, C. Xu and W. Song, J. Immunol. Res., 2014, 2014, 318098 Search PubMed.
- X. Sim, M. Poncz, P. Gadue and D. L. French, Blood, 2016, 127, 10 CrossRef.
- X. Zhang, J. Wang, Z. Chen, Q. Hu, C. Wang and J. Yan, et al. , Nano Lett., 2018, 18, 9 CrossRef.
- C.-M. J. Hu, R. H. Fang, K.-C. Wang, B. T. Luk, S. Thamphiwatana and D. Dehaini, et al. , Nature, 2015, 526, 7571 CrossRef.
- C.-M. J. Hu, L. Zhang, S. Aryal, C. Cheung, R. H. Fang and L. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 27 CrossRef.
- T. Kang, Q. Zhu, D. Wei, J. Feng, J. Yao and T. Jiang, et al. , ACS Nano, 2017, 11, 2 Search PubMed.
- L. Rao, L.-L. Bu, J.-H. Xu, B. Cai, G.-T. Yu and X. Yu, et al. , Small, 2015, 11, 46 Search PubMed.
- L. Rao, B. Cai, L.-L. Bu, Q.-Q. Liao, S.-S. Guo and X.-Z. Zhao, et al. , ACS Nano, 2017, 11, 4 Search PubMed.
- J. A. Copp, R. H. Fang, B. T. Luk, C.-M. J. Hu, W. Gao and K. Zhang, et al. , Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 37 CrossRef.
- H. Liu, Y. Du, Y. Deng and P. D. Ye, Chem. Soc. Rev., 2015, 44, 9 RSC.
- X. Liang, X. Ye, C. Wang, C. Xing, Q. Miao and Z. Xie, et al. , J. Controlled Release, 2019, 296, 150 CrossRef CAS.
- A. V. Kroll, R. H. Fang, Y. Jiang, J. Zhou, X. Wei and C. L. Yu, et al. , Adv. Mater., 2017, 29, 47 CrossRef.
- W. Xie, W.-W. Deng, M. Zan, L. Rao, G.-T. Yu and D.-M. Zhu, et al. , ACS Nano, 2019, 13, 3 Search PubMed.
- J. E. Talmadge and D. I. Gabrilovich, Nat. Rev. Cancer, 2013, 13, 10 CrossRef.
- S. Li, Q. Wang, Y. Shen, M. Hassan, J. Shen and W. Jiang, et al. , Nano Lett., 2020, 20, 1 CrossRef.
- A. Gardner and B. Ruffell, Trends Immunol., 2016, 37, 12 CrossRef.
- S. K. Wculek, F. J. Cueto, A. M. Mujal, I. Melero, M. F. Krummel and D. Sancho, Nat. Rev. Immunol., 2020, 20, 1 CrossRef.
- K. Palucka and J. Banchereau, Immunity, 2013, 39, 1 CrossRef.
- Y. Jiang, N. Krishnan, J. Zhou, S. Chekuri, X. Wei and A. V. Kroll, et al. , Adv. Mater., 2020, 32, 30 Search PubMed.
- Y. Han, H. Pan, W. Li, Z. Chen, A. Ma and T. Yin, et al. , Adv. Sci., 2019, 6, 15 Search PubMed.
- L. J. Ochyl and J. J. Moon, Adv. Healthcare Mater., 2019, 8, 4 Search PubMed.
- X. Zhang, C. Wang, J. Wang, Q. Hu, B. Langworthy and Y. Ye, et al. , Adv. Mater., 2018, 30, 22 Search PubMed.
- W.-L. Liu, M.-Z. Zou, T. Liu, J.-Y. Zeng, X. Li and W.-Y. Yu, et al. , Adv. Mater., 2019, 31, 18 Search PubMed.
- L. Rao, Q.-F. Meng, Q. Huang, Z. Wang, G.-T. Yu and A. Li, et al. , Adv. Funct. Mater., 2018, 28, 34 Search PubMed.
- L.-L. Bu, L. Rao, G.-T. Yu, L. Chen, W.-W. Deng and J.-F. Liu, et al. , Adv. Funct. Mater., 2019, 29, 10 CrossRef.
- X. Han, S. Shen, Q. Fan, G. Chen, E. Archibong and G. Dotti, et al. , Sci. Adv., 2019, 5, 10 Search PubMed.
- J. Zhou, D. Weng, F. Zhou, K. Pan, H. Song and Q. Wang, et al. , Cancer Immunol., Immunother., 2009, 58, 10 Search PubMed.
- R. Ing, M. Segura, N. Thawani, M. Tam and M. Stevenson, J. Immunol., 2006, 176, 441 CrossRef CAS.
- Z. Cao, S. Cheng, X. Wang, Y. Pang and J. Liu, Nat. Commun., 2019, 10, 1 CrossRef.
- S. Zhou, C. Gravekamp, D. Bermudes and K. Liu, Nat. Rev. Cancer, 2018, 18, 12 CrossRef.
- G. Raposo and W. Stoorvogel, J. Cell Biol., 2013, 200, 4 CrossRef.
- R. Xu, A. Rai, M. Chen, W. Suwakulsiri, D. W. Greening and R. J. Simpson, Nat. Rev. Clin. Oncol., 2018, 15, 10 Search PubMed.
- A. Becker, B. K. Thakur, J. M. Weiss, H. S. Kim, H. Peinado and D. Lyden, Cancer Cell, 2016, 30, 6 CrossRef.
- A. Riches, E. Campbell, E. Borger and S. Powis, Eur. J. Cancer, 2014, 50, 5 CrossRef.
- M. Piffoux, A. Nicolás-Boluda, V. Mulens-Arias, S. Richard, G. Rahmi and F. Gazeau, et al. , Adv. Drug Delivery Rev., 2019, 138, 247 CrossRef CAS.
- N. Shimasaki, A. Jain and D. Campana, Nat. Rev. Drug Discovery, 2020, 19, 3 CrossRef.
- L. Zhu, S. Kalimuthu, J. M. Oh, P. Gangadaran, S. H. Baek and S. Y. Jeong, et al. , Biomaterials, 2019, 190–191 Search PubMed.
- Q. Liang, N. Bie, T. Yong, K. Tang, X. Shi and Z. Wei, et al. , Nat. Biomed. Eng., 2019, 3, 9 CrossRef.
- E. J. Bunggulawa, W. Wang, T. Yin, N. Wang, C. Durkan and Y. Wang, et al. , J. Nanobiotechnol., 2018, 16, 1 CrossRef.
- R. Jc Bose, S. Uday Kumar, Y. Zeng, R. Afjei, E. Robinson and K. Lau, et al. , ACS Nano, 2018, 12, 11 CrossRef.
- T. Yong, X. Zhang, N. Bie, H. Zhang, X. Zhang and F. Li, et al. , Nat. Commun., 2019, 10, 1 CrossRef CAS.
- D. Wang, Y. Yao, J. He, X. Zhong, B. Li and S. Rao, et al. , Adv. Sci., 2020, 7, 3 Search PubMed.
- A. Taghikhani, Z. M. Hassan, M. Ebrahimi and S. M. Moazzeni, J. Cell. Physiol., 2019, 234, 6 CrossRef.
- M. Morishita, Y. Takahashi, A. Matsumoto, M. Nishikawa and Y. Takakura, Biomaterials, 2016, 111, 55 CrossRef CAS.
- Q. Zhu, X. Ling, Y. Yang, J. Zhang, Q. Li and X. Niu, et al. , Adv. Sci., 2019, 6, 6 Search PubMed.
- J. Zou, M. Shi, X. Liu, C. Jin, X. Xing and L. Qiu, et al. , Anal. Chem., 2019, 91, 3 Search PubMed.
- Y. Lin, J. Wu, W. Gu, Y. Huang, Z. Tong and L. Huang, et al. , Adv. Sci., 2018, 5, 4 Search PubMed.
- Q. Chen, G. Huang, W. Wu, J. Wang, J. Hu and J. Mao, et al. , Adv. Mater., 2020, 32, 16 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |