
Realization of deep-blue TADF in sterically controlled naphthyridines for vacuum- and solution-processed OLEDs†
Gediminas
Kreiza
 a,
Dovydas
Banevičius
a,
Justina
Jovaišaitė
a,
Saulius
Juršėnas
a,
Tomas
Javorskis
b,
Vytenis
Vaitkevičius
b,
Edvinas
Orentas
b and
Karolis
Kazlauskas
*a
a,
Dovydas
Banevičius
a,
Justina
Jovaišaitė
a,
Saulius
Juršėnas
a,
Tomas
Javorskis
b,
Vytenis
Vaitkevičius
b,
Edvinas
Orentas
b and
Karolis
Kazlauskas
*a
aInstitute of Photonics and Nanotechnology, Vilnius University, Saulėtekio av. 3, LT-10257 Vilnius, Lithuania. E-mail: karolis.kazlauskas@ff.vu.lt
bDepartment of Organic Chemistry, Vilnius University, Faculty of Chemistry and Geosciences, Naugarduko 24, Vilnius LT-03225, Lithuania
First published on 11th May 2020
Abstract
Narrow-band deep-blue (emission peak < 460 nm) TADF emitters are in demand for commercial OLED display applications, yet the development of efficient emitters with low efficiency roll-off is very challenging. To address this issue, herein, we studied carbazole–naphthyridine (donor–acceptor)-based blue-emitting TADF compounds, which were designed by using both the H-bonding and sterically controlled charge-transfer (CT) interactions between D and A units. Methyl substitution employed at the first position of t-butyl-carbazole donors was found to affect CT strength and consequently the TADF properties of the studied compounds, enabling a significant reduction of delayed fluorescence lifetime (down to 3.1 μs) and enhancement of reverse intersystem crossing rate (up to 106 s−1). The naphthyridines were demonstrated to hold great potential as deep-blue TADF emitters suitable for both vacuum- and solution-processed TADF OLEDs. The optimized devices with 7 wt% naphthyridine emitter in a weakly polar mCP host delivered external quantum efficiencies (EQEs) of up to ∼17.6% and ∼13.5% for vacuum- and solution-processed OLEDs, respectively. Unsubstituted naphthyridine exhibited deep-blue (λmax < 460 nm) and narrow-band (FWHM = 66 nm) electroluminescence, whereas the more twisted methyl-substituted compound expressed broader band (FWHM > 80 nm) sky-blue (λmax ≈ 480 nm) emission. The demonstrated emitters are among the best-performing conventional D–A-type blue/deep-blue TADF emitters in terms of EQE and efficiency roll-off properties of their devices.
1. Introduction
The basic design principles of high performance thermally activated delayed fluorescence (TADF) compounds routinely rely on the combination of spatially twisted donor (D) and acceptor (A) moieties.1 This enables large separation of the highest occupied (HOMO) and lowest unoccupied (LUMO) molecular orbitals subsequently resulting in a small energy gap between the lowest excited singlet (S1) and triplet (T1) states. The twisted D–A compounds express charge-transfer (CT) character and are highly attractive for organic light emitting diode (OLED) applications due to the potentially fast and efficient reverse intersystem crossing (rISC).2 It allows the utilization of both singlet and triplet excitons formed in the emitting layer of a device after electron and hole injection.3 Although TADF-based OLEDs are frequently reported to reach 100% internal quantum efficiency,4 their application in commercial displays is limited by their broadened (FWHM ∼ 90 nm) and red-shifted fluorescence (FL) spectra due to the strong intramolecular CT and conformational disorder caused by variation in the twisting angle between D and A moieties.5 Therefore, the development of efficient narrow-band (FWHM < 60 nm) TADF emitters is considered to be a challenging task, especially for the blue/deep-blue OLEDs.6,7The reported approaches to achieve narrow blue TADF rely on rigid sterically hindered8 or donor-interlocked molecular structures,9 HOMO–LUMO separation by the multiple resonance effect10 and weak CT compounds.11 An additional involvement of H-bonding interactions between nitrogen heteroatoms in A and neighbouring C–H bonds in D units was also found to facilitate delivering narrow blue TADF emission.12 It seems that the combination of both H-bonding and CT interactions in a controlled manner could be an attractive strategy to achieve high-performance blue OLEDs, and more importantly, to gain understanding of the TADF mechanism of D–A systems containing nitrogen heteroatoms in their building blocks.
Recently, a nitrogen heteroatom-containing naphthyridine acceptor was successfully utilized in the construction of TADF emitters.13–16 The combination of naphthyridine with a variety of donors, e.g. those based on acridane, carbazole, phenoxazine and phenothiazine regularly employed for designing efficient TADF compounds, produced yellow/green/blue emitters with peak wavelength (λmax) above 460 nm. Meanwhile, implementing them in efficient OLEDs produced surprisingly low external quantum efficiency (EQE) roll-offs.15,16 Note, however, that for commercial applications like OLED displays, narrow-band deep-blue emitters with λmax < 460 nm are required.17 To reveal the potential of naphthyridine acceptors for the formation of deep-blue TADF emitters, we have designed a couple of 1,8-naphthyridine (ND)-based compounds with tert-butyl-carbazole (tCz) serving as the donor (Fig. 1). Although one of the compounds, tCz-ND, was recently reported,16 its most intriguing photophysical properties and its possibilities to be employed in deep-blue OLEDs remained overlooked. Herein, tCz-ND was also used for comparative analysis with the second compound MetCz-ND containing additional methyl (Me) moieties at the first position of the tCz donors. Me substitution was employed to alter steric hindrance and, consequently, twisting of the D and A units. The modification permitted assessing the impact of intramolecular D–A interaction and CT strength on TADF properties of the studied compounds and on their performance in both solution- and vacuum-processed OLEDs.
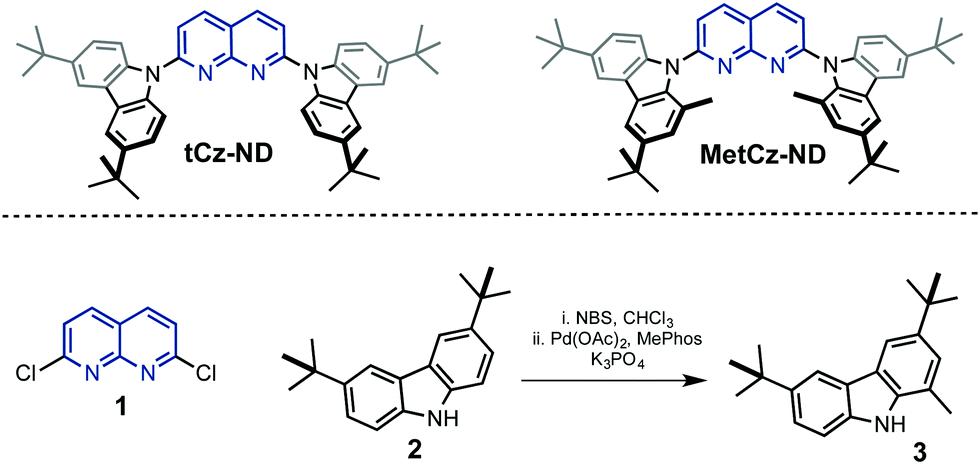 | ||
| Fig. 1 Chemical structures of the synthesized 1,8-naphthyridine (ND) derivatives and precursors 1–3. | ||
2. Experimental section
2.1. Sample preparation and instrumentation
Absorption spectra of dilute toluene solutions (10−5 M) of the investigated naphthyridine derivatives were recorded using a Lambda 950 UV-vis-NIR spectrophotometer (PerkinElmer). 1,3-Bis(N-carbazolyl)benzene (mCP) films doped with 7 wt% contents of the compounds were prepared by spin-coating chloroform solutions (20 mg mL−1) of mCP and compound mixtures on quartz substrates at 2000 rpm and annealing at 50 °C for 20 minutes to remove residual solvent. Steady state PL spectra were recorded using a PMA-12 back-thinned CCD spectrometer (Hamamatsu) by exciting samples with a xenon lamp coupled to a monochromator. Toluene solutions were degassed by the freeze–pump–thaw method. Fluorescence quantum yield (ΦFL) was estimated in oxygen-free media by using the integrating sphere (Sphere Optics) and comparative methods for solutions, where quinine sulfate in 0.1 M H2SO4 served as the reference.18 All samples were excited at 340 nm. FL transients were measured using an NT 242 nanosecond YAG:Nd3+ laser equipped with an optical parametric oscillator (Ekspla, excitation wavelength 340 nm, pulse duration 5 ns, and repetition rate 1 kHz) and an iStar DH340T time-gated intensified CCD camera (Andor) mounted on an SR-303i spectrograph (Shamrock). Measurements at low temperatures were performed in a 204N closed-cycle helium cryostat (Cryo Industries). Ionization potential (Ip) of the wet-casted films was estimated by photoelectron emission spectrometry in air.19 The films for the measurements were prepared by dissolving materials in THF and by casting the solutions on polyester film coated with an Al conductive layer and ∼0.5 μm-thick methylmethacrylate and methacrylic acid copolymer adhesive layers. Prior to experiments, the investigated compounds were purified using a DSU-20 vacuum sublimation system (CreaPhys).2.2. OLED fabrication and characterization
OLEDs were fabricated on pre-patterned ITO/glass substrates (purchased from Kintec company). ITO layer thickness was 100 nm with a sheet resistance of 15–20 Ω □−1. Prior to device fabrication, the substrates were cleaned by ultrasonication in a detergent (Hellmanex II) at 50 °C for 10 minutes; then, substrates were rinsed with running distilled water and blow-gunned ensuring no residual detergent is left. Afterwards, the substrates were sonicated consecutively in distilled water, acetone and isopropyl alcohol for 15 minutes (in each solvent). Immediately after the substrates were removed from the isopropanol bath, they were immersed in boiling hot distilled water and then dried. The substrates were then treated with O2 plasma and transferred into a nitrogen-filled glovebox with an integrated vacuum evaporation chamber. The vacuum-processed devices were fabricated in a vacuum chamber (at a base pressure of <10−6 Torr) by depositing multiple organic layers at a rate of 0.5–1.2 Å s−1. The doped emissive layer was obtained by co-evaporating materials from two different evaporation sources at different evaporation rates for the desired doping concentration.The solution-processed devices were fabricated by the following procedure. O2 plasma-treated ITO/glass substrates were coated with a PEDOT:PSS (poly(3,4-ethylenedioxythiophene)-poly(styrenesulfonate)) layer by spin-coating from a water solution at 5000 rpm. Before the coating, PEDOT:PSS (Al 4083, purchased from Ossila) was ultrasonicated and filtered through a 0.45 μm PES filter. The spin-coating resulted in a 40 nm-thick film, which was annealed at 200 °C for 10 minutes to remove residual H2O. Samples were then transferred into a nitrogen glovebox, where PVK (poly(9-vinylcarbazole)) was spin-coated at 2000 rpm from 6 mg mL−1 chlorobenzene solution, and annealed afterwards at 160 °C for 15 min resulting in a 15 nm-thick layer. Immediately after that, a 25 nm-thick emissive layer from a 2.5 mg mL−1 cyclohexane solution (emitter![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :host ratio of 7:93 by weight) was spin-coated on top at 1500 rpm. The samples were then left to dry for 30 minutes at room temperature before their transfer to an integrated vacuum chamber for further deposition of 5 nm-thick DPEPO (bis[2-(diphenylphosphino)phenyl] ether oxide) followed by a 50 nm-thick TmPyPB (1,3,5-tri(m-pyridin-3-ylphenyl)benzene) layer.
:host ratio of 7:93 by weight) was spin-coated on top at 1500 rpm. The samples were then left to dry for 30 minutes at room temperature before their transfer to an integrated vacuum chamber for further deposition of 5 nm-thick DPEPO (bis[2-(diphenylphosphino)phenyl] ether oxide) followed by a 50 nm-thick TmPyPB (1,3,5-tri(m-pyridin-3-ylphenyl)benzene) layer.
Finally, for both the solution- and vacuum-processed devices, the samples were transferred from the organic deposition chamber to a metal deposition chamber without breaking the vacuum, where (lithium fluoride) LiF and Al layers at the rates of 0.2 and 1.8 Å s−1, respectively, were successively deposited. The active area of the final devices was 1 mm2, 4 mm2 or 16 mm2 as defined by the cross-section of the patterned ITO anode and the shadow mask of cathode deposition. Before exposure to air, the devices were transferred from the vacuum chamber to a nitrogen atmosphere for encapsulation. The OLEDs were encapsulated using a UV-curable epoxy resin (DELO KATIOBOND LP655) and a cover glass on top.
A calibrated characterization set-up comprising an integrating sphere (Lab sphere), a photonic multichannel analyzer PMA-11 (Hamamatsu) and a source meter 2601A (Keithley) was employed to evaluate current–voltage–luminance characteristics and the EQE of the fabricated OLEDs.
3. Results and discussion
3.1. Synthesis
1,8-Naphthyridine derivatives tCz-ND and MetCz-ND were synthesized in one step from a common precursor, 2,7-dichloro-1,8-naphthyridine 1, and carbazole derivatives 2–3 possessing tert-butyl groups using Pd catalysed amination or nucleophilic aromatic substitution reactions, respectively (Fig. 1). The detailed information about the synthetic procedures and identification of molecular structures is provided in the ESI.† The methyl group-containing carbazole 3 was synthesized in high yield using a Suzuki coupling reaction between the corresponding 1-bromo-3,7-di-tert-butyl-9H-carbazole and methylboronic acid. The structures of tCz-ND and MetCz-ND were unambiguously confirmed by X-ray analysis (see ESI†). The ND compounds were purified by vacuum sublimation prior to experiments and device fabrication. Thermogravimetry analysis (TGA) of tCz-ND and MetCz-ND revealed high decomposition temperatures of 433 °C and 422 °C, respectively, implying high thermal stability of both compounds and thus suitability for practical applications (see Fig. S2, ESI†).3.2. DFT calculations
To gain insight into the photophysical properties of the synthesized compounds, their excitation energies, oscillator strengths, D–A dihedral angles and HOMO and LUMO distributions were calculated by using DFT at the B3LYP/6-31G(d) level (see Table S2 in ESI† and Fig. 2).20 The unmodified tCz-ND demonstrated rather weak CT character due to the small dihedral angle (31°) between tCz donor and ND acceptor. As expected, Me substituents in MetCz-ND imposed larger steric hindrance (dihedral angle 55°) resulting in considerably increased CT strength and thus reduced excitation energy, oscillator strength and ΔEST. Furthermore, analysis of molecular structures in optimized ground state geometries revealed proximity between the N atoms of the ND acceptor and the nearest H atoms of neighbouring D groups in both compounds, similarly to previously reported di(pyridinyl)methanone-based TADF emitters.12 C–H⋯N distances of 2.39 Å and 2.42 Å for tCz-ND and MetCz-ND, respectively, were determined, suggesting an involvement of intramolecular H-bonding interactions expected to increase the rigidity of the investigated compounds and thus impact their photophysical properties.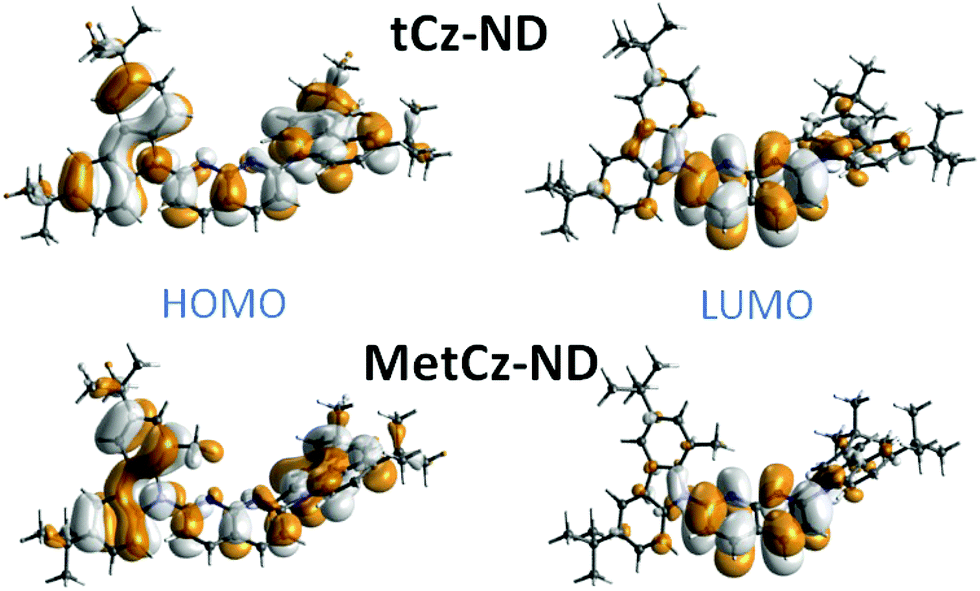 | ||
| Fig. 2 HOMO and LUMO distributions in 1,8-naphthyridine derivatives calculated by TD-DFT. | ||
3.3. Photophysical properties
Absorption spectra of the investigated ND derivatives in toluene solutions were found to be in good agreement with the DFT calculation results (see Fig. 3), i.e.tCz-ND expressed intense absorption with well-resolved vibronic structure, indicating weak CT character and a less-twisted molecular geometry, whereas MetCz-ND exhibited a less structured spectrum with 1.5-fold reduced oscillator strength likely due to enhanced CT and electron-vibronic coupling. Similarly, the FL spectrum of MetCz-ND in toluene was structureless and redshifted with respect to that of tCz-ND, signifying enhanced CT character (Fig. S3, ESI†). This was additionally verified by estimating the spectral responses of ND compounds with increasing solvent polarity (Fig. S4, ESI†). The change of the polarity from non-polar (cyclohexane) to highly polar (acetonitrile) resulted in a strong red shift and broadening of the FL spectra, which were obviously more pronounced for MetCz-ND than for tCz-ND. The photophysical properties of ND compounds are summarized in Table S3 (ESI†).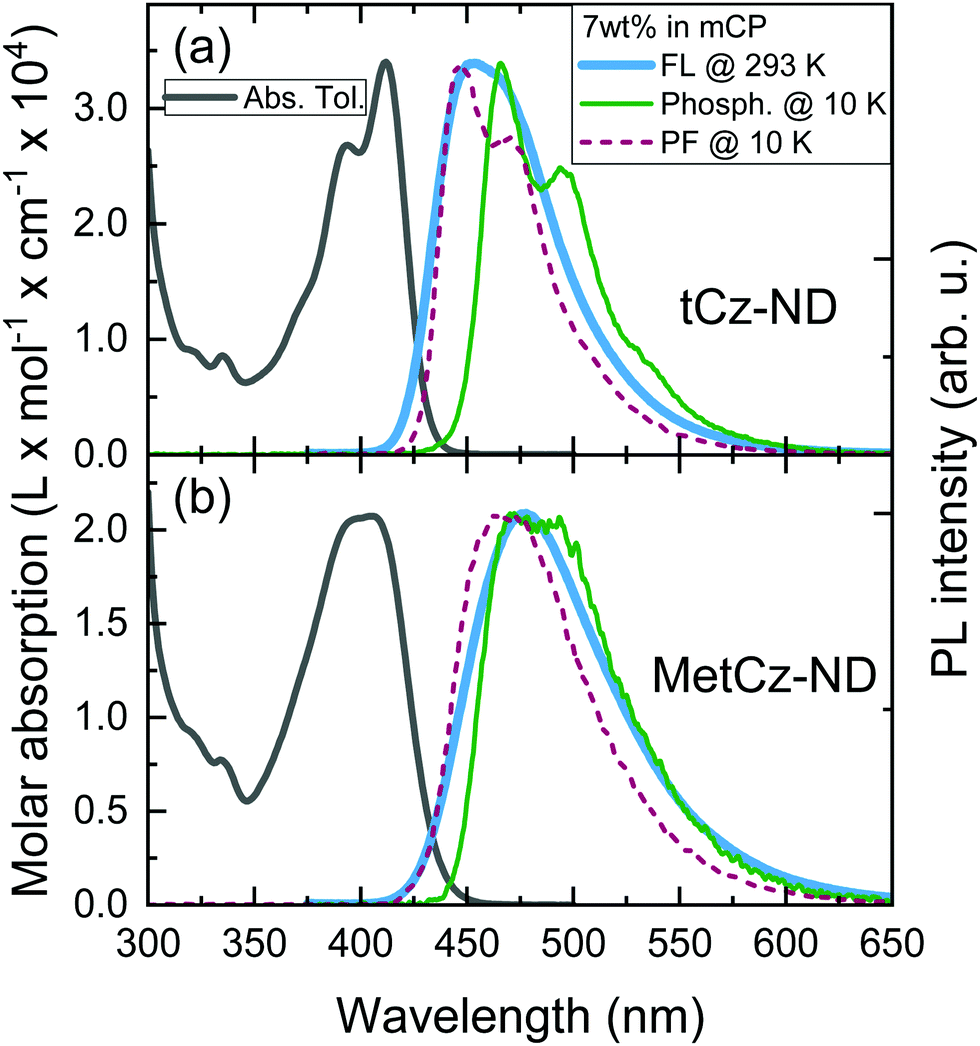 | ||
| Fig. 3 Absorption (in toluene), fluorescence and phosphorescence spectra (in mCP, and an oxygen-free environment) of tCz-ND (a) and MetCz-ND (b) at room temperature and 10 K. Prompt FL spectra at 10 K are also shown. | ||
All the studied compounds demonstrated oxygen-sensitive FL in solutions with distinct prompt (PF) and delayed (DF) FL components, signifying the presence of TADF (Fig. 4). Moderately high FL quantum yield (ΦFL) values of 0.53 and 0.64 obtained for tCz-ND and MetCz-ND, respectively, in solutions were increased up to 0.76 and 0.86, respectively, by dispersing ND derivatives in a rigid mCP host at 7 wt% concentration. This concentration was found to be optimal for the devices delivering maximal EQE. The ND-doped mCP film processing method, i.e. vacuum or solution process, was found to have no impact on the ΦFL values. The improved ΦFL values of the compounds in mCP were accompanied by notably enhanced contributions of the DF components (Table 1 and Table S3, ESI†), implying reduced non-radiative decay from the triplet states most likely due to suppressed vibronic coupling to the ground state.21 The vibronically-modulated prompt FL and phosphorescence spectra of tCz-ND in mCP obtained at 10 K again confirmed the origin of the emissive states to be of a more localized (and less CT) nature. Conversely, the analogous spectra of MetCz-ND with redshifted structureless emission clearly indicated the CT-like origin of the states (Fig. 3).
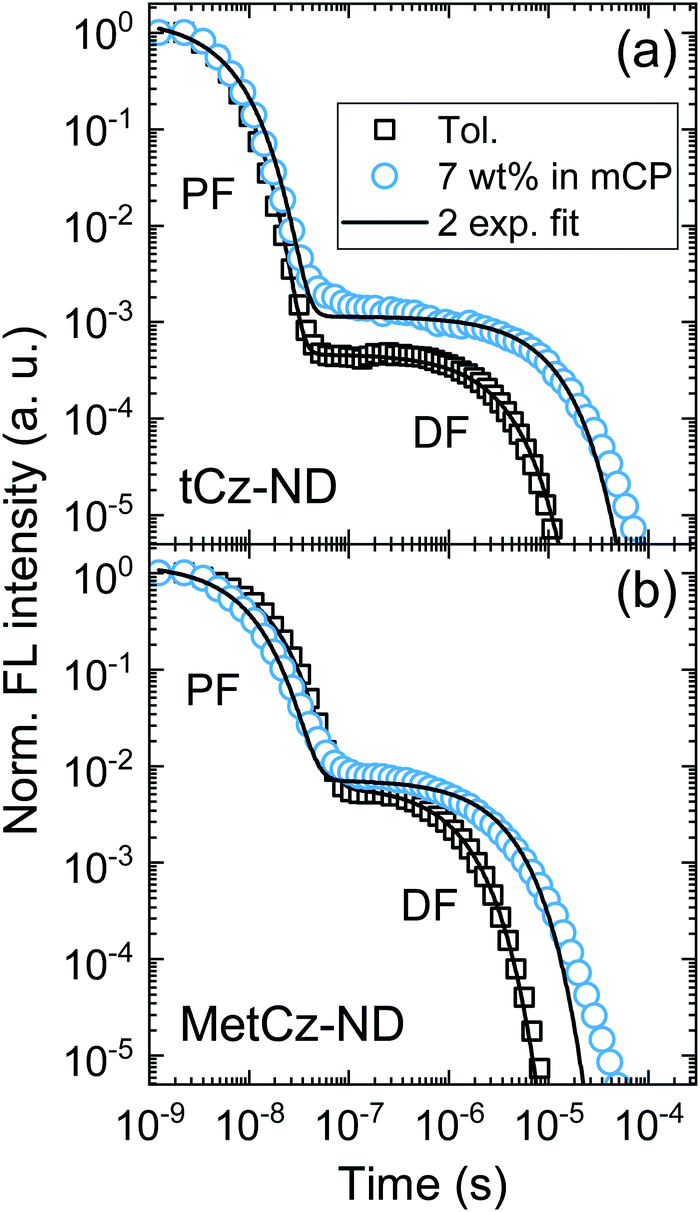 | ||
| Fig. 4 FL decay transients of tCz-ND (a) and MetCz-ND (b) in toluene (squares) and the mCP host at 7 wt% doping concentration (circles) in an oxygen-free environment. Solid lines represent double exponential fits. PF and DF components are indicated. | ||
| Compd. | λ max (nm) | Φ FL | Φ DF/ΦPFa | τ PF (ns) | τ DF (μs) | k r (107 s−1) | k ISC (107 s−1) | k rISC (106 s−1) | ΔESTb (eV) | HOMO/LUMOc (eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Estimated from the FL transient measurements. b ΔEST estimated from the onsets of FL (@293 K) and phosphorescence (@10 K) spectra. c HOMO obtained from ionization potential measured by photoelectron emission spectrometry; LUMO = HOMO − Eg. | ||||||||||
| tCz-ND | 452 | 0.76 | 0.53/0.23 | 5.2 | 8.8 | 4.4 | 14.8 | 0.34 | 0.18 | 5.6/2.7 |
| MetCz-ND | 478 | 0.86 | 0.61/0.25 | 8.3 | 3.1 | 3.0 | 9.0 | 1.06 | 0.09 | 5.6/2.7 |
DF and PF lifetimes (τDF and τPF) in different media were obtained by fitting the transients with double exponential decay profiles (Fig. 4). Slight deviation of the experimental DF points from the fits in the mCP host at the latest times could arise due to the small conformational disorder of the molecular geometry in the solid film.5 The determined lifetimes along with ΦFL and DF/PF ratios were further used to calculate the rISC rate (krISC) according to the previously described procedures assuming that non-radiative decay occurs mainly from the triplet states (as discussed above).21 The revealed krISC was found to be 3-fold larger for MetCz-ND (1.06 × 106 s−1 in mCP) as compared to that of the unmodified tCz-ND (0.34 × 106 s−1 in mCP), which could be a result of the enhanced coupling between S1 and T1 due to the small ΔEST (0.09 eV) and stronger vibronic coupling because of the more labile molecular structure. On the other hand, less structurally twisted tCz-ND showed the highest radiative decay rate (kr = 4.4 × 107), which, accompanied with a deep blue (λmax = 452 nm) and narrow (FWHM = 66 nm) FL spectrum, makes tCz-ND an excellent candidate for pure blue emitting TADF-OLEDs.
3.4. OLED performance
To evaluate the potential of naphthyridine compounds as blue TADF emitters for OLED applications, their electroluminescence (EL) properties were studied. The two ND compounds were compared in the same device architecture, where they were employed as dopants in the ND:mCP emissive layer at the same optimal doping concentration (7 wt%), identical to that used for FL studies (Fig. 3 and 4). Under such conditions, the obtained device characteristics including efficiency roll-off could be directly linked to the molecular properties of the ND compounds.The main characteristics of the vacuum-processed OLEDs are provided in Fig. 5, meanwhile the key parameters are summarized in Table 2. Vacuum-processed devices were fabricated using the following layer configuration: ITO(100 nm)/TAPC(30 nm)/TCTA(5 nm)/emitter(7 wt%): mCP(20 nm)/DPEPO(5 nm)/TmPyPB(50 nm)/LiF(0.8 nm)/Al(100 nm), where the emitter was either tCz-ND or MetCz-ND. An energy level diagram of the devices is displayed in Fig. 5b. 4,4′-Cyclohexylidenebis[N,N-bis(4-methylphenyl)benzenamine] (TAPC) and 4,4′,4′′-tris(carbazol-9-yl)triphenylamine (TCTA) acted as hole injection and transport layers, respectively, whereas LiF and TmPyPB were employed for electron injection and transport, respectively. A thin 5 nm layer of DPEPO possessing a large HOMO–LUMO gap and a high triplet energy (3.0 eV) was used to confine excitons within the emissive layer.
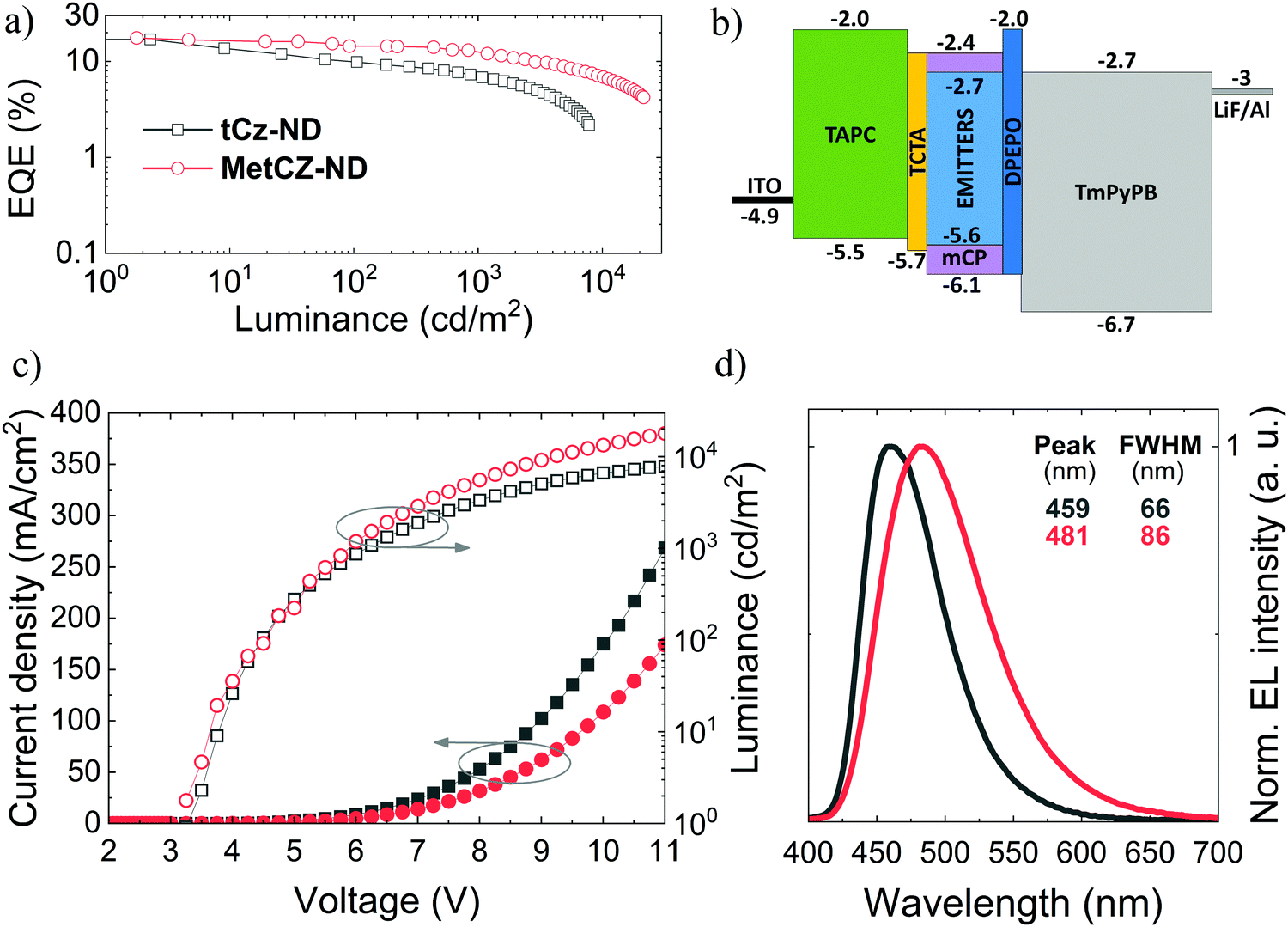 | ||
| Fig. 5 Characteristics of vacuum-processed OLEDs based on the naphthyridine TADF emitters (7 wt% in mCP): (a) EQE vs. luminance, (b) energy level diagram, (c) current density and luminance vs. applied voltage, and (d) normalized electroluminescence spectra. | ||
| Tech. | Emitter | V on (V) | EQEb (%) | L max (cd m−2) | CEmax (cd A−1) | LEmax (lm W−1) | λ max (nm) | FWHM (nm) | CIE 1931 (x, y) |
|---|---|---|---|---|---|---|---|---|---|
| a Turn-on voltage at 1 cd m−2. b Maximum EQE/EQE at 100 cd m−2/EQE at 1000 cd m−2. | |||||||||
| Vac. | tCz-ND | 3.25 | 17.0/9.9/7.0 | 8424 | 22.17 | 19.88 | 459 | 66 | (0.14, 0.16) |
| MetCz-ND | 3.25 | 17.6/14.4/12.5 | 21459 |
31.98 | 23.60 | 481 | 88 | (0.18, 0.32) | |
| Sol. | tCz-ND | 4.30 | 13.5/12.1/7.8 | 6840 | 17.26 | 10.24 | 452 | 66 | (0.15, 0.14) |
| MetCz-ND | 3.40 | 11.7/7.5/11.0 | 23028 |
23.84 | 11.76 | 479 | 80 | (0.16, 0.29) | |
OLEDs based on tCz-ND demonstrated a low turn-on voltage (Von) of 3.25 V, EL peak at 459 nm and FWHM of 66 nm rendering deep-blue narrow-band emission with Commission Internationale de L’Eclairage (CIE) coordinates of (0.14, 0.16). With these parameters, our device surpassed the recently reported vacuum-processed OLED device based on the same naphthyridine compound.16 Specifically, λmax and FWHM of the current device were 10 nm smaller as compared to those of the previously reported device. This was mainly attributed to the proper device optimization carried out at 3 times lower emitter concentration (7 wt%) in the less polar mCP host (versus strongly polar DPEPO host). Moreover, as will be shown below, the tCz-ND emitter was also confirmed to be suitable for fabrication of solution-processed deep-blue TADF OLEDs. The maximum EQE of tCz-ND-based devices was 17% at low brightness and low current density (Fig. 5a). At the practically useful brightness of 100 cd m−2, EQE decreased down to 9.9% with a further roll-off to 7% at 1000 cd m−2. Maximum brightness achieved in this device was 8424 cd m−2. A further increase of applied bias caused degradation of the device performance, most probably because of the exciton annihilation processes and loss of the current balance.
Although the MetCz-ND-based vacuum-processed OLEDs demonstrated similar turn-on characteristics with Von = 3.25 V, EL of the devices was considerably redshifted with the EL peak at 481 nm well matching the FL spectrum of the compound doped into mCP (Fig. 3). Additionally, the stronger CT character of MetCz-ND (as compared to that of tCz-ND) also implied much broader EL emission of the device (FWHM = 86 nm) typical of conventional TADF emitters. The CIE coordinates of this device (0.18, 0.32) corresponded to sky-blue emission. Similar to tCz-ND based OLEDs, the MetCz-ND based devices exhibited roughly the same maximum EQE of 17.6% at low brightness. However, in contrast to tCz-ND, the latter demonstrated reduced efficiency roll-off. Explicitly, EQE was reduced only down to 14.4% and 12.5% at the brightness of 100 cd m−2 and 1000 cd m−2, respectively. The device brightness maxed out at 21459 cd m−2 still maintaining EQE above 4%.
The more rapid efficiency roll-off in the OLED based on tCz-ND can be justified by the 3-fold longer τDF (8.8 μs) measured for this emitter as compared to that for MetCz-ND. Long triplet lifetimes are undesirable, since they increase the probability of triplet interaction causing detrimental annihilation effects. On the other hand, the fast rISC and short τDF (3.1 μs) of MetCz-ND significantly lowered the triplet population by rapid up-conversion to the singlet manifold resulting in only 29% loss of device efficiency at the brightness of 1000 cd m−2. For comparison, the device based on tCz-ND lost 60% of its initial efficiency at a similar brightness. It is worth noting that the maximum EQE values of ND-based OLEDs are in good agreement with the ΦFL values obtained for the ND-doped mCP films, if ∼20% light outcoupling efficiency of the devices (the case of random emitter orientation) is taken into account, i.e. EQE = 0.2 × ΦFL.22 This highlights the light outcoupling as the prime efficiency loss suggesting that the OLED structure is optimized at least for the low brightness regime. Compared to the performance of currently state-of-the-art narrow-band deep-blue (<460 nm) OLEDs based on conventional D–A-type TADF emitters, these ND-based (particularly tCz-ND-based) OLEDs are among the best devices in terms of the EQE and efficiency roll-off properties (see Table S5 in the ESI†).8,12,23–28 The results imply that sterically controlled CT interactions combined with H-bonding can indeed be promising in attaining narrow deep-blue TADF by employing ND acceptors.
Furthermore, both ND emitters were also tested in analogous solution-processed OLEDs in which TAPC and TCTA layers were replaced by solution-processable PEDOT:PSS and PVK (Fig. S5 in ESI† and Table 2). The performance of these OLEDs was found to be similar to that of the vacuum-processed devices at high current densities regardless of the inferior performance observed at low currents. The worsened performance in terms of the increased Von and the reduced EQE can be attributed to the reduced homogeneity of the solution processed layers as compared to the vacuum evaporated ones and rather poor hole injection through PVK causing unbalanced electron and hole currents. Evidently, increasing the current density raises the EQE of the solution processed OLEDs almost up to the point of the vacuum-processed devices indicating that the injection regime close to optimal is achieved. Similar to the vacuum-evaporated OLEDs, the solution-processed devices based on tCz-ND exhibited deep-blue (λmax = 452 nm) and narrow-band (FWHM = 66 nm) emission with an EQE of up to 13.5%, which could be of potential interest to display manufacturers.29 Although MetCz-ND showed a higher ΦFL (86%) as compared to that of tCz-ND (76%), the solution-processed device based on MetCz-ND exhibited a slightly lower maximum EQE (11.7%) than that of the tCz-ND-based device. This can be explained by the different EQE vs. luminance dependences observed for the two studied compounds (see Fig. S5a, ESI†), which suggest that the current imbalance is more pronounced for the MetCz-ND-based device causing a delayed rise in EQE and thus slightly reduced maximum EQE. Summing up, the demonstrated performance shows the suitability of the investigated naphthyridines to be employed also as solution-processable deep-blue TADF emitters.
4. Conclusion
In conclusion, relying on H-bonding and sterically controlled CT interactions, blue/deep-blue TADF emitters based on D–A-type carbazole–naphthyridine compounds have been developed. The ND compounds were found to express a small singlet–triplet energy gap (down to 0.09 eV), high fluorescence quantum yield (up to 0.86) and rather short delayed fluorescence lifetimes (down to 3.1 μs) resulting in a high rISC rate (up to 106 s−1) when dispersed in the mCP host. The TADF properties and emission wavelength and band-width of the ND derivatives were mainly governed by the CT strength of the D–A interaction, which could be controlled via methyl substituents introduced at the first linking position of t-butyl-carbazole donors. The less sterically hindered compound tCz-ND exhibited more narrow and shorter wavelength blue TADF as compared to that of the more twisted methyl-substituted compound MetCz-ND, implying a trade-off between the reduced rISC and improved emissive properties (λmax and FWHM). Importantly, the ND compounds were demonstrated to be suitable as TADF emitters for realization of vacuum- and solution-processed TADF OLEDs with low efficiency roll-off in sky-blue (CIExy: 0.18, 0.32) and deep-blue (CIExy: 0.14, 0.16) spectral ranges. The optimized devices with 7 wt% ND emitter in the weakly polar mCP host delivered up to ∼17.6% and ∼13.5% EQEs for the vacuum- and solution-processed OLEDs, respectively. The OLEDs based on unsubstituted tCz-ND exhibited deep-blue (λmax < 460 nm) and narrow-band (FWHM = 66 nm) electroluminescence, whereas those based on more twisted methyl-substituted MetCz-ND expressed broader band (FWHM > 80 nm) and sky-blue (λmax ≈ 480 nm) emission. The demonstrated ND-based TADF OLEDs are among the best blue/deep-blue-emitting devices in terms of EQE and efficiency roll-off properties. This highlights the potential of carbazole–naphthyridine-derived materials to become the basis in future engineering of deep-blue TADF emitters.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
The research was funded by the European Social Fund (project No 09.3.3-LMT-K-712-01-0084) under grant agreement with the Research Council of Lithuania (LMTLT). E. Kamarauskas is acknowledged for ionization potential measurements.References
- Y. Im, M. Kim, Y. J. Cho, J. A. Seo, K. S. Yook and J. Y. Lee, Chem. Mater., 2017, 29, 1946–1963 CrossRef CAS.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915–1016 RSC.
- T. Serevičius, R. Skaisgiris, J. Dodonova, K. Kazlauskas, S. Juršėnas and S. Tumkevičius, Phys. Chem. Chem. Phys., 2019, 22, 265–272 RSC.
- T. J. Penfold, F. B. Dias and A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935 RSC.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- L.-S. Cui, H. Nomura, Y. Geng, J. U. Kim, H. Nakanotani and C. Adachi, Angew. Chem., Int. Ed., 2017, 56, 1571–1575 CrossRef CAS PubMed.
- Y. J. Cho, S. K. Jeon, S. S. Lee, E. Yu and J. Y. Lee, Chem. Mater., 2016, 28, 5400–5405 CrossRef CAS.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- P. Pander, A. Swist, R. Motyka, J. Soloducho, F. B. Dias and P. Data, J. Mater. Chem. C, 2018, 6, 5434–5443 RSC.
- P. Rajamalli, N. Senthilkumar, P.-Y. Huang, C.-C. Ren-Wu, H.-W. Lin and C.-H. Cheng, J. Am. Chem. Soc., 2017, 139, 10948–10951 CrossRef CAS PubMed.
- X. Zhou, H. Yang, Z. Chen, S. Gong, Z. H. Lu and C. Yang, J. Mater. Chem. C, 2019, 7, 6607–6615 RSC.
- Y. Lee, S. J. Woo, J. J. Kim and J. I. Hong, Org. Electron., 2020, 78, 105600 CrossRef CAS.
- C. Chen, H. Y. Lu, Y. F. Wang, M. Li, Y. F. Shen and C. F. Chen, J. Mater. Chem. C, 2019, 7, 4673–4680 RSC.
- Y.-F. Shen, W.-L. Zhao, H.-Y. Lu, Y.-F. Wang, D.-W. Zhang, M. Li and C.-F. Chen, Dyes Pigm., 2020, 178, 108324 CrossRef CAS.
- M. Budzynski, T. Baumann and D. Ambrosek, in Organic Light Emitting Materials and Devices XXII, ed. F. So, C. Adachi and J.-J. Kim, SPIE, 2018, vol. 10736, p. 51 Search PubMed.
- M. J. Adams, J. G. Highfield and G. F. Kirkbright, Anal. Chem., 1977, 49, 1850–1852 CrossRef CAS.
- E. Miyamoto, Y. Yamaguchi and M. Yokoyama, Electrophotography, 1989, 28, 364–370 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- G. Kreiza, D. Banevičius, J. Jovaišaitė, K. Maleckaitė, D. Gudeika, D. Volyniuk, J. V. Gražulevičius, S. Juršėnas and K. Kazlauskas, J. Mater. Chem. C, 2019, 7, 11522–11531 RSC.
- N. C. Greenham, R. H. Friend and D. D. C. Bradley, Adv. Mater., 1994, 6, 491–494 CrossRef CAS.
- T. Serevičius, R. Skaisgiris, I. Fiodorova, V. Steckis, J. Dodonova, D. Banevičius, K. Kazlauskas, S. Juršėnas and S. Tumkevičius, Org. Electron., 2020, 105723 CrossRef.
- P. Rajamalli, D. Chen, W. Li, I. D. W. Samuel, D. B. Cordes, A. M. Z. Slawin and E. Zysman-Colman, J. Mater. Chem. C, 2019, 7, 6664–6671 RSC.
- Y. Im, S. H. Han and J. Y. Lee, J. Mater. Chem. C, 2018, 6, 5012–5017 RSC.
- M. Kim, S. K. Jeon, S.-H. Hwang and J. Y. Lee, Adv. Mater., 2016, 28, 603 CrossRef CAS PubMed.
- C. Chan, L. Cui, J. U. Kim, H. Nakanotani and C. Adachi, Adv. Funct. Mater., 2018, 28, 1706023 CrossRef.
- D. H. Ahn, S. W. Kim, H. Lee, I. J. Ko, D. Karthik, J. Y. Lee and J. H. Kwon, Nat. Photonics, 2019, 13, 540–546 CrossRef CAS.
- J. H. Lee, C. H. Chen, P. H. Lee, H. Y. Lin, M. K. Leung, T. L. Chiu and C. F. Lin, J. Mater. Chem. C, 2019, 7, 5874–5888 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1991494 and 1992697. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0tc01637c |
| This journal is © The Royal Society of Chemistry 2020 |