
Engineering the conductive carbon/PEO interface to stabilize solid polymer electrolytes for all-solid-state high voltage LiCoO2 batteries†
Jianneng
Liang
a,
Yipeng
Sun
a,
Yang
Zhao
a,
Qian
Sun
a,
Jing
Luo
a,
Feipeng
Zhao
a,
Xiaoting
Lin
a,
Xia
Li
a,
Ruying
Li
a,
Li
Zhang
b,
Shigang
Lu
b,
Huan
Huang
c and
Xueliang
Sun
 *a
*a
aDepartment of Mechanical and Materials Engineering, University of Western Ontario, Canada. E-mail: xsun9@uwo.ca
bChina Automotive Battery Research Institute Co., Ltd., China
cGlabat Solid-State Battery Inc., Canada
First published on 23rd November 2019
Abstract
All-solid-state lithium batteries (ASSLBs) are promising energy storage devices for application in electric transportation and large-scale energy storage systems. Polyethylene oxide (PEO)-based solid polymer electrolytes (SPEs) are attractive solid-state electrolytes for ASSLBs due to their high ionic conductivity, light weight, and low cost. However, the low electrochemical oxidation potential window of PEO seriously restricts its implementation with high voltage cathodes for high-energy-density ASSLBs. Effective interfacial engineering between high voltage cathodes and SPEs can be a solution. Most of the reported conventional cathode protection approaches have been focused on building coating layers on active material particles, which, however, can be insufficient because the conductive carbon is able to accelerate the decomposition of SPEs. In this work, atomic layer deposition (ALD) coating on the electrode instead of active material particles realizes a unique method to protect the cathode/SPE interface. As a successful example, a thin ALD-derived lithium tantalate coating on the high-voltage LiCoO2 electrode demonstrated good compatibility with PEO-based SPEs, significantly enhancing the cycling performance of the ASSLBs. The inner mechanism is attributed to the fact that the protection of the conductive carbon/SPE interface helps reduce the electrochemical oxidation of PEO-based SPEs. This work shall give new insights for the interfacial engineering of high voltage cathodes and solid polymer electrolytes.
Introduction
All solid-state lithium batteries (ASSLBs) are promising candidates for application in electric vehicles (EVs) and other large-scale energy storage systems due to their high energy density and enhanced safety. Feasible solid-state electrolytes (SSEs) are a key component to realize ASSLBs. Among all the solid-state lithium ion conductors, SSE systems including solid polymer electrolytes (SPEs), oxide-based SSEs, sulfide-based SSEs, and hybrid electrolytes received the most research interest.1–4 To realize the practical applications of ASSLBs, the following challenges of SSEs must be addressed: (i) high interfacial resistance, (ii) low ionic conductivity, (iii) narrow electrochemical window, and (iv) instability with electrodes.2,5,6SPEs based on polyethylene oxide (PEO) and lithium salt complexes are promising candidates for ASSLBs, owing to their high ionic conductivity at elevated temperature and low interfacial resistance toward electrodes. More importantly, PEO is commercially available, low cost, light weight, and environmentally friendly.6 However, (1) the low ionic conductivity at room temperature,6–8 (2) the susceptibility to lithium dendrites9 and (3) the instability at high voltage10 of PEO-based SPEs are the main issues that hinder their wide applications in ASSLBs.
Tremendous research efforts have been dedicated to address challenges (1) and (2) in recent years. For example, Cui's group enhanced the RT ionic conductivity of PEO-based SPEs from 10−7 S cm−1 to 4.4 × 10−5 S cm−1 by in situ formation of nanosized SiO2 particle fillers in the PEO matrix.8 Hu's group created a 3-dimensional garnet-type SSE nanofiber network for PEO to form a hybrid electrolyte that has an ionic conductivity of 2.5 × 10−4 S cm−1 at RT.11 Plenty of studies applying inorganic SSE fillers in the PEO matrix to enhance the RT ionic conductivity of PEO-based SPEs have also been reported.12,13 On the other hand, many studies were reported to address the lithium dendrite formation problem in ASSLBs with PEO-based SPEs.11–14 For example, Zhao et al. prepared a LLZTO–PEO hybrid electrolyte for ASSLBs and they proposed that anions were tethered in the hybrid electrolyte by the polymer matrix and ceramic fillers, which results in a uniform distribution of space charges and lithium ions, thus, resulting in dendrite-free lithium deposition.14
However, the issue of the instability of PEO-based SPEs at high voltage (challenge (3)) received little research attention. The instability of PEO at high voltage makes it challenging to couple with high voltage cathodes such as LiCoO2. Even though PEO-based SPEs show excellent stability with LiFePO4 cathode with charge voltage below 4 V (vs. Li/Li+),15 the limited theoretical energy density of LiFePO4 restrains its further application in future electric transportation. Alternatively, layer structured oxide cathodes such as LiCoO2 have a high specific capacity and high charge/discharge voltage, which makes them promising candidates for high-energy-density ASSLBs. Since the practical capacity of LiCoO2 is positively related to the charge cut-off voltage, a high charge cut-off voltage is required for high-energy-density applications.16 However, charge cut-off voltage over 4.5 V remains a challenge for PEO-based SPEs due to the decomposition of PEO-based SPEs at high voltage.17–19
One solution to enable PEO-based SPE coupling with high voltage LiCoO2 is interfacial engineering of SPEs and the LiCoO2 active material interface with coating layers. Coatings on LiCoO2 particles with Al2O3,19 Li3PO4,18 poly(ethylcyanoacrylate) (PECA),20 and Li1.4Al0.4Ti1.6(PO4)3 (LATP)21 have been reported to improve solid-state batteries performances. However, most of these previous studies only studied the coating effect on the active material/SPE interface while the conductive carbon/SPE interface is ignored. Unfortunately, conductive carbon has been proved to be able to accelerate the decomposition of SSEs including PEO-based SPEs, oxide-based SSEs and sulfide-based SSEs.10,22,23 Thus, the detrimental effect of conductive carbon on the PEO-based SPE cannot be neglected in high voltage ASSLBs. Therefore, systematic studies on the effects of coatings on active material particles versus on the whole electrodes (i.e. covering both active material and conductive carbon) are important to disclose the interfacial engineering mechanism and can provide us with helpful insights for future high-energy-density ASSLBs designs.
Herein, atomic layer deposition (ALD), as an emerging technique, which is capable of depositing uniform and conformal thin films with precise thickness control by self-limited chemical reactions, is chosen for realizing active material particle coating and whole electrode coating because the ALD process can be carried out at a low temperature compared to other chemical/physical deposition techniques such as chemical vapor deposition (CVD), physical vapor deposition (PVD), sol–gel methods, etc. Thus, ALD can be non-destructive to electrode components (cathode particles and conductive carbon). In this study, ALD derived lithium tantalate protective coatings are demonstrated to stabilize the interface between PEO-based SPEs and LiCoO2 electrodes at high voltage (4.5 V vs. Li/Li+). Effects of coatings on the LiCoO2 particles, the whole electrode, and conductive carbon particles are compared. The results show that the coating on LiCoO2 particles cannot improve the performance of ASSLBs, while the coating on electrode sheets (coating on both LiCoO2 particles and carbon particles) shows significant enhancement in cycling performance, the same as the coating on conductive carbon particles. The working mechanism of the ALD coating is illustrated in Fig. 1. These results highlight that a suitable coating approach is critical in preventing the electrochemical oxidation of PEO-based SPEs at high voltage for the stabilization of the high voltage performance of ASSLBs.
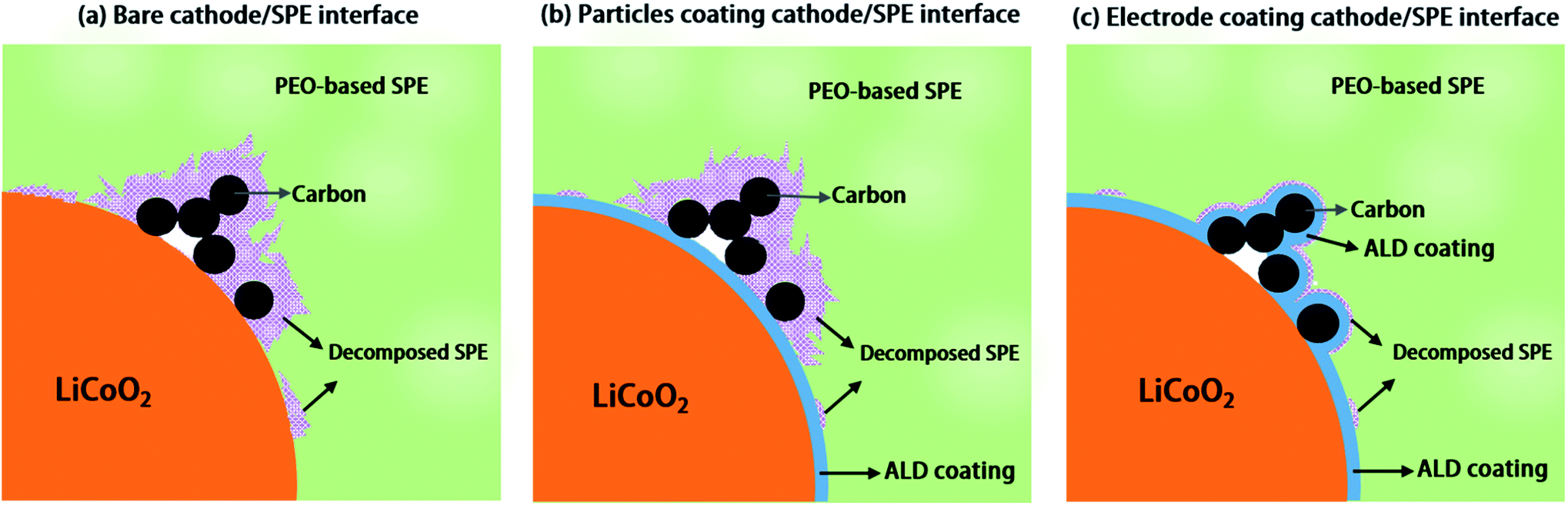 | ||
| Fig. 1 Schematic diagram showing the working mechanism of ALD coating for enabling stable, high voltage solid polymer electrolyte-based lithium ion batteries. The decomposition of SPEs on (a) unprotected LiCoO2 electrode and (b) electrode with protected LiCoO2 particles (LCO-coating), and the working mechanism of (c) protected electrode (LCO + CB-coating) in ASSLBs after extensive charge/discharge cycles. Conductive carbon can accelerate the decomposition of SPEs at high voltage, and thus, the protection of the carbon/SPE interface is very important for stabilizing high voltage solid-state lithium ion batteries. | ||
Experimental section
Preparation of PEO-based SPEs
PEO (M.W. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000), LiClO4 (purity, 99.9%) and garnet-type SSEs (Li6.4La3Zr1.4Ta0.6O12, LLZTO) were dried in a vacuum oven before using. 0.24 g of LLZTO was added into 50 mL of acetonitrile (AN) and mixed by ultrasonication for 5 h. Then, 1.2 g of PEO and 0.19 g of LiClO4 were added into the mixture and stirred overnight at room temperature. This homogeneous mixture was cast onto a Teflon substrate and dried at room temperature overnight to slowly evaporate the AN, followed by vacuum drying at 60 °C for 2 days. The obtained PEO–LiClO4–LLZTO SPE membrane was immediately transferred to an Ar-filled glovebox and rested over 3 days before use.
000000), LiClO4 (purity, 99.9%) and garnet-type SSEs (Li6.4La3Zr1.4Ta0.6O12, LLZTO) were dried in a vacuum oven before using. 0.24 g of LLZTO was added into 50 mL of acetonitrile (AN) and mixed by ultrasonication for 5 h. Then, 1.2 g of PEO and 0.19 g of LiClO4 were added into the mixture and stirred overnight at room temperature. This homogeneous mixture was cast onto a Teflon substrate and dried at room temperature overnight to slowly evaporate the AN, followed by vacuum drying at 60 °C for 2 days. The obtained PEO–LiClO4–LLZTO SPE membrane was immediately transferred to an Ar-filled glovebox and rested over 3 days before use.
LiCoO2 electrode preparation
Bare LiCoO2 electrodes were prepared by mixing 90 wt% of LiCoO2 particles, 6 wt% carbon-black (acetylene black) and 4 wt% poly(vinylidene) fluoride binder in the N-methylpyrrolidinone (NMP) solvent. The slurry was coated on Al foil by a doctor blade method. The electrode was obtained after drying in a vacuum oven at 120 °C overnight. The loading of LiCoO2 was around 1.5–2 mg cm−2. The obtained bare LiCoO2 electrode was used directly for electrode coating. ALD LTO coatings on LiCoO2 particles or electrodes followed a previously reported ALD procedure from our group.24 Briefly, the LTO coatings were deposited using an ALD reactor (Savannah 100, Cambridge Nanotechnology Inc., USA) by alternatively pulsing lithium tert-butoxide, tantalum(V) ethoxide and H2O at 235 °C.Electrochemical performance testing
All solid-state LiCoO2 batteries were assembled in 2032 type coin cells in an argon-filled glove box (Vacuum Atmosphere Company, moisture and oxygen level less than 1 ppm). The LiCoO2 electrodes and lithium foil were used as the working electrode and the counter electrode, respectively. The PEO–LiClO4–LLZTO SPE was used as both the separator and lithium ion conductor. No additional solvent or liquid electrolyte was applied into the LiCoO2 ASSLBs. Galvanostatic charge/discharge was performed between 2.7 and 4.5 V in a 60 °C oven using a LAND battery tester. The ASSLBs were rested at 60 °C over 30 h before testing. For LiCoO2 batteries with liquid electrolyte, 2032 type coin cells were assembled with a liquid electrolyte containing 1 M LiPF6 in ethylene carbonate (EC):ethylmethyl carbonate (EMC):diethyl carbonate (DEC) solvents with a volume ratio of 1:1:1, using a Celgard separator. Cyclic voltammetry of the LiCoO2 batteries was performed between 2.7 and 4.5 V (vs. Li/Li+) at 60 °C. Linear sweep voltammetry of Li/SPE/PEO–carbon cells was conducted at an open-circuit voltage of 4.5 V on a versatile multichannel potentiostat 3/Z (VMP3). PEO–carbon composite electrodes consisted of 70 wt% PEO and 26 wt% carbon-black (with/without LTO coating) and 4 wt% LiClO4 was used as the working electrode. Electrochemical impedance spectroscopy (EIS) was performed on a versatile multichannel potentiostat 3/Z (VMP3) by applying an AC voltage with 10 mV amplitude in the frequency range from 500 kHz to 0.01 Hz.
Materials characterization
The morphology of the samples was characterized using a Hitachi S-4800 field emission scanning electron microscope (FE-SEM) equipped with an energy dispersive X-ray spectrometer (EDX). FIB was conducted in a LEO (Zeiss) 1540XB FIB/SEM machine. Mass spectrometry was conducted on a Shimadzu GCMS-QP2010 gas chromatograph/mass spectrometer. X-ray absorption spectroscopy (XAS) measurements using total electron yield (TEY) and fluorescence yield (FLY) modes at the Co K-edge were performed at the Canadian light source (CLS) at the Soft X-ray Micro-characterization Beamline (SXRMB) with a photon energy range of 1.7–10 keV utilizing a Si (111) crystal monochromator.Results and discussion
Cathodes were prepared with LiCoO2 particles, carbon black (acetylene black), and a poly(vinylidene) fluoride binder at a weight ratio of 90:6:4. Using the advanced ALD technique, the lithium tantalate (LTO) coating was deposited on either the LiCoO2 particles before electrode preparation (coating on LiCoO2 active materials, referred to as LCO-coating) or on the electrode surface after casting and drying (coating on the electrode, referred to as LCO + CB-coating) or on the carbon black particles before electrode preparation (coating on CB particles, referred to as CB-coating). Fig. S1a–c† show the scanning electron microscopy (SEM) images of a pristine LiCoO2 electrode, an electrode with ALD LTO coated LiCoO2 particles (LCO-coating), and an ALD LTO coated electrode (LCO + CB-coating). The three electrodes with or without these two types of coatings show a very similar morphology, indicating that the ALD LTO coatings were conformal and non-destructive. The LCO-coating did not alter the distribution of the conductive carbon on the electrode surface, nor did LCO + CB-coating. In addition, from the SEM images, one can easily find that the electrode/SPE interface actually has a large proportion of the conductive carbon/SPE interface since carbon black is uniformly distributed/covered on the LiCoO2 particle surface.
Transmission electron microscopy (TEM), SEM and energy-dispersive X-ray spectroscopy (EDX) analyses were used to confirm the deposition of the ALD coatings on active materials and the electrode (Fig. 2 and S2–S5†). Fig. 2a shows the TEM image of the ALD coating on LiCoO2 particle and the schematic illustration of the LCO-coating electrode is presented in Fig. 2b. Differently, the ALD LTO coated electrode (LCO + CB-coating) shows a coating on both carbon black and LiCoO2 particles (Fig. 2c). The EDX mapping results in Fig. S3† confirm that ALD LTO is deposited on both the CB and LCO particle surface for the LCO + CB-coating electrode. This type of electrode structure is schematically illustrated in Fig. 2d. Hereby, two types of protective coverages are established: (I) partial protection between the LiCoO2/SPE interface only and (II) full protection on the entire electrode covering both LiCoO2 and conductive carbon. The electrochemical performance will be compared systematically.
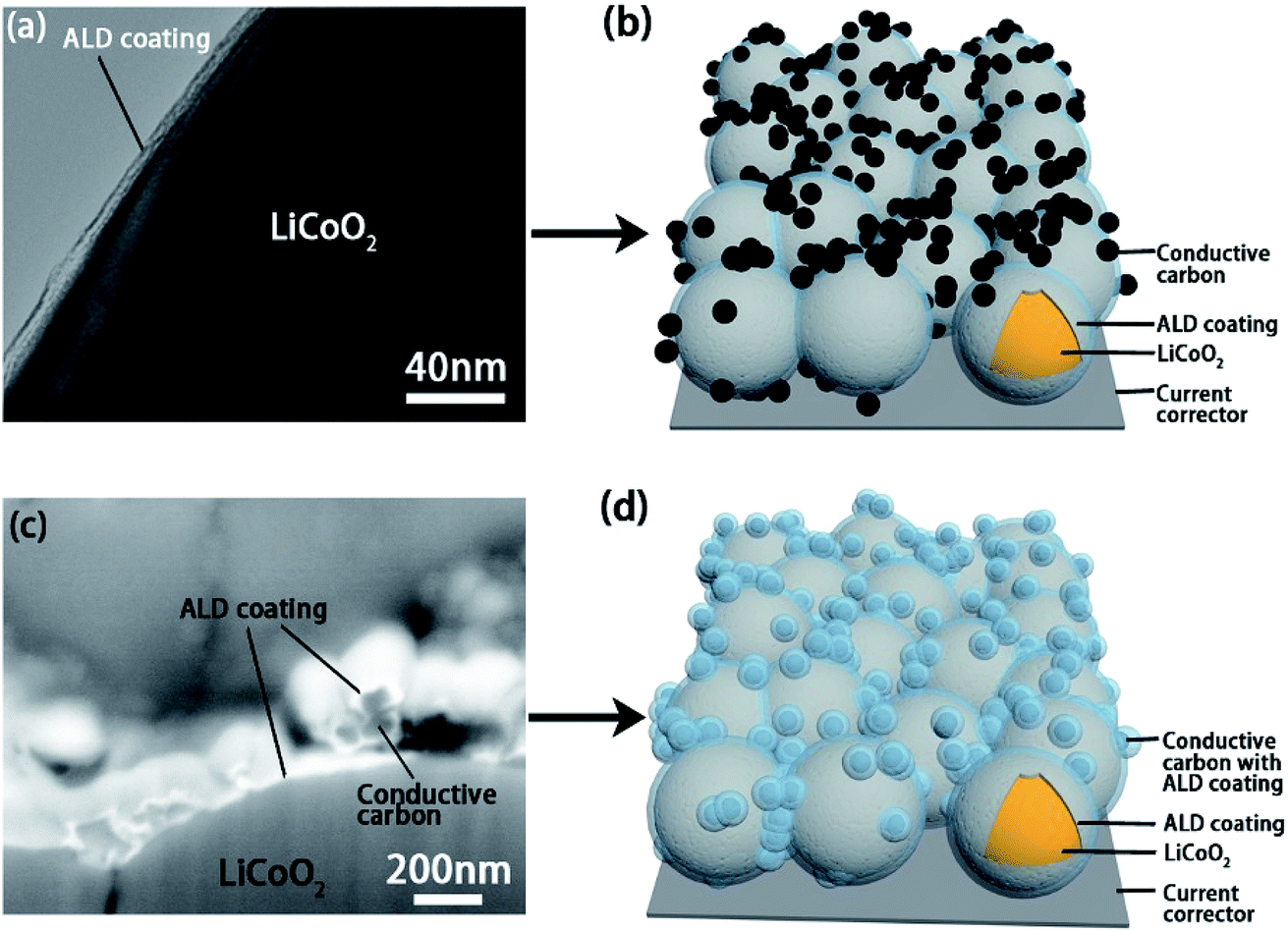 | ||
| Fig. 2 (a) A TEM image of the 10 cycles of ALD LTO (thickness is ∼5 nm) coating on LiCoO2 particles and its (b) schematic diagram of the LiCoO2 electrode with the LCO-coating where conductive carbon is not protected. (c) A SEM image in backscattered electron mode of the 20 cycles of ALD LTO (thickness is ∼10 nm) coating on both conductive carbon and LiCoO2 particles from the LCO + CB-coating sample after focused ion beam (FIB) cutting, and its (d) schematic diagram showing the LiCoO2 electrode where both LiCoO2 and conductive carbon are protected. The binder is omitted in the schematic diagrams. | ||
Cyclic voltammetry (CV) was performed to study the electrochemical process of LiCoO2 ASSLBs with different coatings. For comparison, CV of a regular LiCoO2 battery with a commercial carbonate-based liquid electrolyte was conducted for a comprehensive understanding. As shown in Fig. S6a,† the anodic peaks corresponded to the Li+ extraction process, and the cathodic peaks were related to the Li+ insertion process. There are three pairs of peaks which correspond to three oxidation/reduction couples. The redox pair at 4.07/3.74 V is the result of the conversion between Co3+ and Co4+ for the first-order phase transformation between two hexagonal phases.25 The other two pairs of anodic/cathodic peaks are related to the order–disorder phase transformation between hexagonal and monoclinic phases.26–28 In contrast, in ASSLBs, the three pairs of redox peaks are combined into a pair of broad peaks for all three cells with a bare LiCoO2 electrode, electrodes with LCO-coating protection and LCO + CB-coating protection (Fig. S6b–d†). This is probably because of the sluggish Li+ transport in SPEs and electrodes that broadens the peaks related to the first-order phase transformation reaction, overlapping the other weak peaks. Nevertheless, the ASSLBs with different cathodes still exhibit obvious difference in electrochemical behaviours. For the ASSLB with the bare LiCoO2 electrode, an anodic peak and a cathodic peak are observed with decreasing peak current intensities and increase in overpotentials over the three scans (Fig. S6b†). This is a typical indication of decay in electrochemical redox reversibility. The decrease in peak current intensities and increase in overpotentials also occur in the ASSLB with LCO-coating protection (Fig. S6c†). As a conclusion, LCO-coating protection is insufficient in preventing the reversibility decay. In great contrast, instead of a decrease in peak current intensities and increase in overpotentials, the slight increase in peak current intensities and the decrease in overpotentials indicate a more reversible electrochemical process of the LCO + CB-coating protected electrode (Fig. S6d†).
The electrochemical performances of ASSLBs with different protection coverages were characterized at 60 °C. First, the coating thickness is optimized. Different cycles of ALD LTO were conducted on LCO-coating and LCO + CB-coating electrodes and their results are presented in Fig. S7.† Clearly, the ALD LTO coating thicknesses were optimized to 10 ALD cycles for LCO-coating protection and 20 ALD cycles for LCO + CB-coating protection.
The ASSLBs performances with optimised ALD coating thickness for bare, LCO-coating, and LCO + CB coating are compared in Fig. 3. Consistent with the CV results, observable voltage polarization along with serious capacity fading is already observed during the first 20 cycles of galvanostatic testing at 0.2C for both ASSLBs with a bare LiCoO2 electrode (Fig. 3a) or LCO-coating protected electrode (Fig. 3b). This is possibly due to the severe decomposition of PEO-based SPEs at high voltage that increases the interfacial resistance. Even though the LCO-coating protection can protect the LiCoO2/SPE interface, the conductive carbon was still in direct contact with the SPE.
 | ||
| Fig. 3 Charge/discharge profiles of ASSLBs with (a) bare LiCoO2 electrode, (b) LCO-coating protected electrode, and (c) LCO + CB-coating protected electrode at 0.2C and 60 °C. Corresponding (d) cycling performance, (e) rate performance, and (f) long-term cycling performance after rate performance testing for ASSLBs with different LiCoO2 electrodes at 60 °C. | ||
In contrast, much smaller polarization increase is observed for the ASSLB with the LCO + CB-coating protected electrode (Fig. 3c). The discharge capacities are also relatively stable compared to the other two cells without protection or only with LCO-coating protection. Thus, the protection on the carbon/SPE interface is evidently very important.
The dramatically different effects between LCO-coating and LCO + CB-coating protections on the electrochemical performance can be further verified by cycling performance and rate performance. Under the optimized conditions, ASSLBs with LCO + CB-coating protection demonstrate substantially enhanced performance over the ASSLBs without protection or with LCO-coating protection (Fig. 3d–f). While all three ASSLBs deliver a similar initial discharge capacity around 170–177 mA h g−1, a retaining capacity of 110.4 mA h g−1 at 100th cycles for the ASSLB with the LCO + CB-coating protected electrode is significantly higher than the remaining capacity of ∼71 mA h g−1 for the other two ASSLBs (Fig. 3d). As shown in Fig. 3e, LCO + CB-coating protection also markedly enhanced the rate performance at lower current densities of up to 0.6C. However, at a higher current density, a higher capacity is achieved for the LCO-coating electrode. The reason can be possibly attributed to the higher lithium ion diffusion in the LCO-coating electrode compared to that of the bare and LCO + CB coating electrodes. In the LCO-coating electrode, LCO particles are totally coated with LTO, which is a good lithium ion conductor, while in the LCO + CB-coating protected electrode, only part of LCO particles are exposed for ALD LTO deposition. Therefore, the lithium ion diffusion of the LCO-coating protected electrode is better than that of the LCO + CB-coating protected electrode (Fig. S8†) and bare electrode. At high current density, lithium ion diffusion becomes a determining step for the redox reaction. Thus, the electrode with higher lithium ion diffusion can deliver a higher capacity.
LCO + CB-coating protection also can improve the initial coulombic efficiency and subsequent average coulombic efficiency. The initial coulombic efficiencies for the ASSLBs with a bare LiCoO2 electrode, LCO-coating protection, and LCO + CB-coating protection electrodes are 93.8%, 93.2%, and 94.9%, respectively; the subsequent average coulombic efficiencies are 99.0%, 98.6%, and 99.3% in order. The LCO-coating protection electrode actually shows barely any improvement over the bare LiCoO2 electrode, indicating the protection of only LiCoO2 particles is not enough in PEO-based ASSLBs. The higher coulombic efficiency by LCO + CB-coating protection indicates reduced decomposition of the PEO-based SPE during high voltage charge/discharge cycles in ASSLBs.
Electrochemical impedance spectroscopy (EIS) was conducted to study the evolution of interfacial resistance in the LiCoO2 ASSLBs upon cycling (Fig. 4). The three ASSLBs without coating or with LCO-coating or with LCO + CB-coating present EIS spectra with a similar shape but different evolution trends. There are two semicircles with one at high frequency and one at low frequency. Fig. 4d shows the equivalent circuit for fitting these EIS spectra. Re is the impedance from the solid polymer electrolyte, Ra is the interfacial impedance between the lithium anode and SPE, and CPE1 is its corresponding constant phase element. Rc is the interfacial impedance between the cathode and SPE, and CPE2 is its corresponding constant phase element.18 All ASSLBs show relatively stable anode/SPE interfacial resistance with similar values, while dramatic difference is present at the cathode/SPE interfaces. For the bare LiCoO2 ASSLB (Fig. 4a), a significant increase in the cathode/SPE interfacial resistance is observed over 30 cycles, enlarging from 1338 Ω at the 10th cycle to 1843 Ω at the 20th cycle to 2891 Ω at the 30th cycle. Similarly, consistent with the electrochemical performance, a significant increase in the cathode/SPE interfacial resistance is also observed in the ASSLB with LCO-coating protection (Fig. 4b). The increase in cathode/SPE interfacial resistance can be attributed to the accumulation of insulating SPE decomposition products, which can block the Li+ ion transportation at the interface. In contrast, the cathode/SPE interfacial impedance of the ASSLB with LCO + CB-coating protection is stabilized to ∼920 Ω after 30 cycles, confirming the stable SPE/cathode interface enabled by the ALD LTO coating on the electrode. The EIS results strongly support the superior effects of ALD LTO protection for high-voltage ASSLBs.
 | ||
| Fig. 4 EIS spectra of ASSLBs with (a) bare LiCoO2 electrode, (b) LCO-coating protected LiCoO2 electrode, and (c) LCO + CB-coating protected LiCoO2 electrode after 10, 20, and 30 cycles of charge/discharge. The inset tables show the critical frequency values on top of the high frequency semi-circle (Fa) and low frequency semi-circle (Fc) at different cycles of charge/discharge, respectively. (d) The equivalent circuit for fitting the EIS spectra. | ||
To confirm that the conductive carbon/SPE interface is important for the ASSLB performance enhancement, an ASSLB with a CB-coated electrode (an electrode with bare LiCoO2 and LTO coated CB) was assembled and studied. The results are presented in Fig. S10.† It shows that LTO coating on carbon particles can also enhance the cycling performance of the ASSLB, and its cycling performance is very similar to that of LCO + CB coated ASSLBs.
Why is the conductive carbon/SPE interface so critical? It is well known that the electrochemical oxidation potential of PEO-based SPEs is around 3.8 V vs. Li/Li+ according to the linear sweep voltammetry method using a PEO–carbon composite electrode.10 Conventionally, it has been reported that electronic conductive additives (usually carbon black) play an important role in realizing completely utilization of electrode active materials for batteries, because the occurrence of electrochemical reactions requires both electrons and Li+ ions. However, in ASSLBs with Li+ ion conductive SSEs, the presence of conductive carbons could also trigger irreversible decomposition of the SSEs,23 leading to low coulombic efficiency and performance decay.
Even conductive carbon black particles occupy a low weight content in the electrodes. The large volume of carbon black and resulted the large proportion of the carbon/SPE interface among the cathode/SPE interface cannot be neglected (Fig. S1a–c†). This could explain the similar performance of ASSLBs with the bare LiCoO2 electrode and LCO-coating electrode, whereas the carbon black particles are in direct contact with the SPE in either case. The linear sweep voltammetry results of carbon–SPE composite electrodes with or without ALD LTO coating also confirmed the importance of the carbon/SPE interface. As shown in Fig. 5a, the overshooting current approaching 4.5 V is observed for the bare carbon–PEO electrode, which indicates the serious decomposition of PEO at 4.5 V. In contract, this overshooting current is significantly reduced by the ALD LTO coated conductive carbon. As supported by the mass spectrometry results in Fig. S11b,† the significant signals from decomposed SPE products are presented in the bare carbon–PEO electrode. However, with the ALD LTO coating on carbon, the signals from the decomposed SPE are reduced (Fig. S11c†), which demonstrates the excellent capability of LTO in stabilizing the carbon/SPE interface at high voltage. This may be the reason why the protection of the conductive carbon/SPE interface can achieve such a significant enhancement in electrochemical performance.
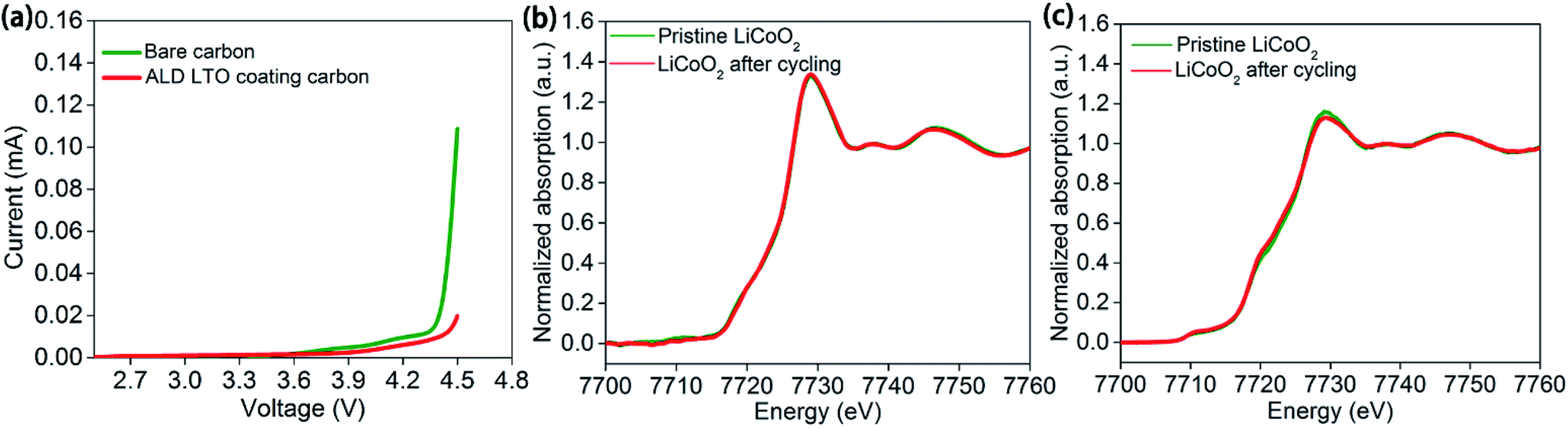 | ||
| Fig. 5 (a) Comparison of the linear sweep voltammogram of the Li/SPE/PEO–carbon composite cell and Li/SPE/PEO–LTO@carbon composite cell (scan rate = 0.3 mV s−1, from OCV to 4.5 V vs. Li/Li+). Comparison of the XAS Co K-edge spectra of unprotected LiCoO2 before and after 5 cycles of charge/discharge in ASSLBs in the discharge state in terms of (b) TEY mode and (c) FLY mode. | ||
Why does the LCO-coating electrode show almost no enhancement? It is possible that LiCoO2 active materials are stable and further ALD protection cannot enhance their stability in our study. This conclusion is supported by the X-ray absorption spectroscopy (XAS) of the unprotected LiCoO2 electrodes before and after cycling with the PEO-based SPE. The Co K-edge XAS results for LiCoO2 electrodes before and after 5 charge/discharge cycles at the discharge state in ASSLBs showed almost no difference under both total electron yield (TEY) mode (Fig. 5b) and fluorescence yield (FLY) mode (Fig. 5c). TEY mode provides surface chemical information up to several nanometers, and FLY mode reveals bulk chemical information. The XAS results indicated that the LiCoO2 active materials are relatively stable from the surface to bulk upon cycling in PEO-based ASSLBs. This is also supported by the stable cycling performance of the LiCoO2 battery with a carbonate-based liquid electrolyte (Fig. S12†). Since LiCoO2 is stable during change/discharge processes, it is reasonable that the ALD LTO coating on LiCoO2 particles (LCO-coating protection) showed little effect while the ALD LTO coating on the electrode (LCO + CB-coating protection) achieved significant enhancement.
Conclusions
The effects of ALD lithium tantalate coatings on cathode active material particles, carbon black particles and the electrode surface were studied systematically for enhancing the high-voltage performance of PEO-based ASSLBs. Using stable LiCoO2 particles as an example for a high voltage cathode up to 4.5 V, we demonstrated the importance of the protection on the conductive carbon/SPE interface. It is found that interfacial protection covering the carbon/SPE interface is very important to stabilize the PEO-based SPE at high voltage and enhance the cycling performance of ASSLBs. Significantly improved cycling performance and rate performance were demonstrated in ASSLBs with simple 20 ALD cycles of lithium tantalate coating on the electrode surface. This study sheds light on the rational design of protective coatings for the polymer electrolyte–electrode interface to enhance the high voltage performance of ASSLBs.Statement of contributions
J. Liang, Q. Sun and X. Sun conceived the idea and experiments; J. Liang conducted the electrode/SPE preparations, electrochemical performance testing, and SEM. Y. Zhao, F. Zhao and Y. Sun helped to conduct the ALD coating experiment. Y. Sun assisted in the synchrotron XAS measurement; J. Luo, X. Li, R. Li, X. Lin, L. Zhang, S. Lu and H. Huang participated in data analysis and discussion; J. Luo polished the manuscript; X. Sun supervised the overall project. All authors discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by China Automotive Battery Research Institute, the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canada Research Chair Program (CRC), the Canadian Light Source (CLS) and the University of Western Ontario. Jianneng Liang greatly appreciates the China Scholarship Council (CSC) for supporting this study. Qian Sun appreciates the support of MITACS Elevate postdoctoral program.Notes and references
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- C. Sun, J. Liu, Y. Gong, D. P. Wilkinson and J. Zhang, Nano Energy, 2017, 33, 363–386 CrossRef CAS.
- J. Liang, Q. Sun, Y. Zhao, Y. Sun, C. Wang, W. Li, M. Li, D. Wang, X. Li and Y. Liu, J. Mater. Chem. A, 2018, 6, 23712–23719 RSC.
- J. Liang, J. Luo, Q. Sun, X. Yang, R. Li and X. Sun, Energy Storage Materials, 2019, 21, 308–334 CrossRef.
- V. Thangadurai, S. Narayanan and D. Pinzaru, Chem. Soc. Rev., 2014, 43, 4714–4727 RSC.
- L. Long, S. Wang, M. Xiao and Y. Meng, J. Mater. Chem. A, 2016, 4, 10038–10069 RSC.
- F. Croce, G. Appetecchi, L. Persi and B. Scrosati, Nature, 1998, 394, 456 CrossRef CAS.
- D. Lin, W. Liu, Y. Liu, H. R. Lee, P.-C. Hsu, K. Liu and Y. Cui, Nano Lett., 2015, 16, 459–465 CrossRef PubMed.
- K. J. Harry, D. T. Hallinan, D. Y. Parkinson, A. A. MacDowell and N. P. Balsara, Nat. Mater., 2014, 13, 69 CrossRef CAS PubMed.
- Y. Xia, T. Fujieda, K. Tatsumi, P. P. Prosini and T. Sakai, J. Power Sources, 2001, 92, 234–243 CrossRef CAS.
- K. K. Fu, Y. Gong, J. Dai, A. Gong, X. Han, Y. Yao, C. Wang, Y. Wang, Y. Chen and C. Yan, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7094–7099 CrossRef CAS PubMed.
- J. Zhang, N. Zhao, M. Zhang, Y. Li, P. K. Chu, X. Guo, Z. Di, X. Wang and H. Li, Nano Energy, 2016, 28, 447–454 CrossRef CAS.
- C. Yang, B. Liu, F. Jiang, Y. Zhang, H. Xie, E. Hitz and L. Hu, Nano Res., 2017, 10, 4256–4265 CrossRef CAS.
- C.-Z. Zhao, X.-Q. Zhang, X.-B. Cheng, R. Zhang, R. Xu, P.-Y. Chen, H.-J. Peng, J.-Q. Huang and Q. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 11069–11074 CrossRef CAS.
- P. Hovington, M. Lagacé, A. Guerfi, P. Bouchard, A. Mauger, C. Julien, M. Armand and K. Zaghib, Nano Lett., 2015, 15, 2671–2678 CrossRef CAS PubMed.
- L. Wang, B. Chen, J. Ma, G. Cui and L. Chen, Chem. Soc. Rev., 2018, 47, 6505–6602 RSC.
- K. Nie, Y. Hong, J. Qiu, Q. Li, X. Yu, H. Li and L. Chen, Front. Chem., 2018, 6, 616 CrossRef CAS PubMed.
- S. Seki, Y. Kobayashi, H. Miyashiro, Y. Mita and T. Iwahori, Chem. Mater., 2005, 17, 2041–2045 CrossRef CAS.
- H. Miyashiro, Y. Kobayashi, S. Seki, Y. Mita, A. Usami, M. Nakayama and M. Wakihara, Chem. Mater., 2005, 17, 5603–5605 CrossRef CAS.
- J. Ma, Z. Liu, B. Chen, L. Wang, L. Yue, H. Liu, J. Zhang, Z. Liu and G. Cui, J. Electrochem. Soc., 2017, 164, A3454–A3461 CrossRef CAS.
- Q. Yang, J. Huang, Y. Li, Y. Wang, J. Qiu, J. Zhang, H. Yu, X. Yu, H. Li and L. Chen, J. Power Sources, 2018, 388, 65–70 CrossRef CAS.
- F. Han, Y. Zhu, X. He, Y. Mo and C. Wang, Adv. Energy Mater., 2016, 6, 1501590 CrossRef.
- W. Zhang, T. Leichtweiß, S. P. Culver, R. Koerver, D. Das, D. A. Weber, W. G. Zeier and J. r. Janek, ACS Appl. Mater. Interfaces, 2017, 9, 35888–35896 CrossRef CAS PubMed.
- J. Liu, M. N. Banis, X. Li, A. Lushington, M. Cai, R. Li, T.-K. Sham and X. Sun, J. Phys. Chem. C, 2013, 117, 20260–20267 CrossRef CAS.
- Y. Gu, D. Chen, X. Jiao and F. Liu, J. Mater. Chem., 2007, 17, 1769–1776 RSC.
- K. Dokko, S. Horikoshi, T. Itoh, M. Nishizawa, M. Mohamedi and I. Uchida, J. Power Sources, 2000, 90, 109–115 CrossRef CAS.
- J. N. Reimers and J. Dahn, J. Electrochem. Soc., 1992, 139, 2091–2097 CrossRef CAS.
- X. Li, J. Liu, X. Meng, Y. Tang, M. N. Banis, J. Yang, Y. Hu, R. Li, M. Cai and X. Sun, J. Power Sources, 2014, 247, 57–69 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta08607b |
| This journal is © The Royal Society of Chemistry 2020 |