
Micropores-in-macroporous gel polymer electrolytes for alkali metal batteries†
Hadi
Khani
 ,
Somayyeh
Kalami
and
John B.
Goodenough
*
,
Somayyeh
Kalami
and
John B.
Goodenough
*
Materials Science and Engineering Program and Texas Materials Institute, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: jgoodenough@mail.utexas.edu; Fax: +1 662 325 1618; Tel: +1 662 325 7608
First published on 2nd October 2019
Abstract
High-energy-density lithium and sodium batteries have the potential to meet the growing worldwide energy and power demands for all-electric vehicles. However, it is well known that alkali metal anodes impose a major safety issue when flammable organic-liquid electrolytes are used owing to the formation of anode dendrites during charge that can cause an internal short circuit, thermal runaway, and fire. Herein, a macroporous poly(vinylidene-fluoride-co-hexafluoropropylene) (PH) membrane is employed as a flexible three-dimensional macroporous polymer host that can incorporate additional microporous polymers. Introducing hyper-cross-linked microporous polymers within the PH host membrane creates a continuous polymeric network capable of trapping liquid electrolytes and creating a “liquid pathway” across the membrane. Compared to modern separators employed in liquid electrolyte systems, these quasi-solid polymer electrolytes offer superior safety and flexibility without sacrificing the high ionic conductivities of traditionally employed liquid electrolytes. Specifically, we synthesize macroporous PH films with hyper-cross-linked microporous polyfuran or polypyrrole incorporated within the 3D PH structure. These membranes are shown capable of immobilizing a liquid electrolyte within the microporous polymeric matrix that enables a quasi-solid electrolyte with high ionic conductivity, stability, and cycle life when employed in lithium and sodium-metal batteries. Full-cell lithium and sodium-batteries containing these micropores-in-macroporous polymer electrolyte membranes demonstrate rate and interface capabilities comparable to traditional liquid electrolytes, but with significantly improved cycling performance and coulombic efficiencies. Furthermore, these results indicate that the proposed micropores-in-macroporous polymer membranes containing an immobilized-liquid electrolyte can suppress dendrite growth and allow the safe implementation of metallic lithium or sodium anodes.
1. Introduction
Over the last few decades, the search for safe and economic power-storage devices with high enough energy and power densities to compete with fossil-fuel based internal combustion engines has been one of the greatest challenges in the field of materials science. While rechargeable lithium-ion batteries (LIBs) have achieved magnificent success in powering the wireless revolution, their future application in the fast-growing era of portable electronic devices and electric vehicles (EVs) is limited. Furthermore, the high raw-material demand of EVs and geographically isolated distribution of lithium resources urgently requires that alternative battery candidates (with similar working principle to LIBs but utilizing materials with higher resource abundancy compared to lithium) be developed. Sodium-ion batteries (SIBs) have been recognized as the most promising substitute to LIBs due to the lower cost and ubiquitous accessibility of sodium in nature.1 In this work, we address the inherent problem of poor electrode–electrolyte contact between a solid electrolyte and a solid metal electrode (caused by large volume fluctuations during charge and discharge) for the lithium and sodium batteries via a “trapped” liquid electrolyte approach.Replacing the low-capacity carbon-host anodes in LIBs and SIBs with metallic lithium or sodium anodes can increase the gravimetric energy densities of these devices by at least one order of magnitude. However, such metallic anodes are susceptible to dendrite growth and internal short-circuiting, which can be extremely hazardous where flammable organic liquid electrolytes are used. There is, therefore, a need to develop safe, high-energy-density lithium and sodium batteries without sacrificing rate performance, cycle life, or production costs. Although solid polymer electrolytes (SPEs) have been shown to address the safety challenges of alkali metal anodes by eliminating the flammable organic liquid electrolytes and dendrite growth,2 the poor room-temperature ionic conductivity (<10−5 S cm−1), high impedance of the polymer/electrode interface, and limited electrochemical stability of SPEs have largely hindered their commercial implementation.3
Alternatively, gel-polymer electrolytes (GPEs) are capable of providing key advantages of both SPEs and liquid electrolytes, including acceptable ionic conductivities (>10−4 at room temperature), good wettability by an alkali-metal anode, retention of intimate interfacial contact with both cathode and anode, and suppression of dendrite growth. However, homogeneous GPEs in which the polymer–salt system is dissolved in a plasticizer, an ionic liquid, and/or an organic solvent suffer from a poor mechanical integrity, thermal instability, and electrolyte leakage compared to SPEs. Furthermore, since most of the lithium/sodium solvation in homogeneous GPEs takes place at the polymer chains rather than in the carbonate solvents,4 and the room-temperature ionic conduction occurs in the swollen gelled phase,2 GPEs possess lower ionic conductivity compare to liquid electrolytes. In general, GPEs utilizing either a polymer host with a lower glass transition temperature or larger fraction of liquid phase can improve the ionic conductivity of GPEs; however, both changes are at the expense of mechanical integrity, flexibility, and thermal stability. Therefore, GPEs with poor mechanical strength are significantly more prone to internal short-circuiting (via dendrites) compared to traditional liquid electrolytes with a Celgard separator. There are ongoing attempts to overcome the trade-offs between the mechanical integrity and ionic conductivity of GPEs via “hard-soft” strategies that employ polymer–nanofiller composites, block copolymers, polymer blends, and cross-linked polymers.2,5,6
In heterogeneous GPEs, a porous, self-standing, and electrolyte-insoluble polymer host is used to immobilize a liquid electrolyte inside polymer cavities. These membranes benefit from the high mechanical strength and the flexibility of a porous polymer host and the high ionic conductivity of a liquid electrolyte within the interconnected pores. However, the performance of these GPEs is significantly impacted by their pore size, wettability, porosity, and overall interconnectivity of the pores. For example, membranes with large pore sizes tend to suffer from continuous electrolyte leakage at the electrode/electrolyte interface during cycling, bringing about all the issues associated with liquid-electrolyte/anode interfaces (e.g. continuous electrolyte decomposition) in addition to the possibility of dendrite penetration through the large pores. Alternatively, small pore sizes are more prone to be shut down at an elevated temperature and/or by a polymer swelling in liquid electrolytes, which results in high polarization of the cell potential.
Porous polymers are classified as microporous (pore size < 2 nm), mesoporous (2 nm < pore size < 50 nm), and macroporous (pore size > 50 nm) materials.7 Microporous polymer candidates having smaller pores and higher porosity can uptake a large quantity of liquid electrolyte, while providing minimum electrolyte leakage, and retain the ability to suppress dendrite formation and growth due to their small pore size. Recently, microporous hyper-crosslinked polymers (HCPs) have received significant attention (being employed for gas storage, separation, CO2 capture, catalysis, drug delivery, and sensing) thanks to their economic and facile synthesis that can utilize a variety of aromatic monomers.8,9 Nevertheless, the high degree of cross-linking in HCPs results in powder-based polymers that are inflexible and insoluble. Therefore, the intrinsic properties of HCPs hinder their application in systems where both mechanical integrity and polymer flexibility are desired.
Herein, we report a novel strategy to synthesize a micropores-in-macroporous polymer by incorporating a rigid microporous polymer into the macropores of a flexible polymer host through an in situ hyper-cross-linking polymerization. Macroporous poly(vinylidene-fluoride-co-hexafluoropropylene) (hereinafter abbreviated as PH), prepared via a phase inversion method, was chosen as the flexible polymer host due to its stability against alkali metal anodes (i.e. lithium and sodium) and high dielectric constant (εr = 8.4) which can promote the dissociation of the lithium and sodium salts. In this work, hyper-cross-linked microporous polyfuran (HCFu) and hyper-cross-linked microporous polypyrrole (HCPy) were synthesized in situ within the macropores of the PH membrane via a simple one-step Friedel–Crafts reaction; forming micropores-in-macroporous HCFu-PH and HCPy-PH membranes, respectively. The micropores of prepared HCFu-PH and HCPy-PH membranes were then filled with a liquid electrolyte (containing either lithium or sodium salts) to form a gel polymer electrolyte for lithium or sodium batteries. The Li-HCFu-PH and Na-HCPy-PH membrane electrolytes demonstrate high room-temperature ionic conductivities of 6.4 × 10−3 S cm−1 and 4.3 × 10−3 S cm−1, respectively, while maintaining excellent mechanical strength, flexibility, and thermal stability. The micropores of these micropores-in-macroporous polymer membranes are shown to retain an immobilized liquid electrolyte during cycling, which significantly reduces side reactions during cell operation. Long-term cycling performances at 1C rate of both Li/Li-HCFu-PH/LiFePO4 and Na/Na-HCPy-PH/Na3V2(PO4)3 full cells demonstrate remarkable capacity retentions and coulombic efficiencies, with no sign of short-circuiting, for over 1000 cycles.
2. Results and discussion
2.1. Electrolyte membrane preparation and characterization
The micropores-in-macroporous polymer strategy for the synthesis of Li-HCFu-PH and Na-HCPy-PH electrolyte membranes is shown schematically in Fig. 1. The initial step is the production of a macroporous PH film via a phase inversion method. In brief, a film (≈220 μm) of the gel polymer (consisting of a mixture of PH and DMF) was cast onto a glass plate and subsequently exposed to water vapor (a PH non-solvent) giving rise to displacement of DMF molecules with water molecules due to the strong hydrogen bonding of water molecules to the DMF molecules. Subsequent vacuum drying of the film results in a macroporous PH film with a thickness of ≈75 μm. The cross-sectional morphology characterization of the PH membrane by scanning electron microscopy (SEM) (Fig. 2a–c) shows a uniform distribution of interconnected honeycomb-like hollow macropores having widths in the range of 5–7 μm. The incorporation of microporous polymers within the PH macropores takes place through a three-step “impregnation–reaction–extraction” process including (i) monomer impregnation into the macropores, (ii) in situ Friedel–Crafts hyper-cross-linking of monomers forming a highly porous network, and (iii) Soxhlet extraction of the films to remove the FeCl3, impurities, and unreacted chemicals. Fig. 2e–h and i–l depict the cross-sectional SEM images of HCFu-PH and HCPy-PH membranes. As can be seen, the PH macropores are occupied with microporous HCFu or HCPy polymer particles. The incorporated HCFu and HCPy polymers are in the form of agglomerated spherical particles with an average size of ≈225 and 170 nm (Fig. S1†), respectively, that are in intimate contact with walls of the PH macropores. The particulate form of the microporous polymers, rather than being in the form of a single monolithic polymer, enables the PH polymer host to maintain its flexibility and mechanical strength. In addition, the macropores on the surface of the PH membrane (see Fig. 2d) are filled with incorporated HCFu or HCPy particles that are capable of suppressing dendrite formation while providing a continuous microporous pathway for lithium or sodium ions (see Fig. S2 and S3†). A uniform coating of HCFu or HCPy particles on the surface of PH membranes can be seen in Fig. S2 and S3† where the HCFu coating is in the form of particles, but that of HCPy coating is in the form of a continuous film well-adhered to the membrane surface. The SEM-EDX maps of oxygen and nitrogen, representing the existence of furan and pyrrole monomers, demonstrate that HCFu or HCPy are uniformly coated over the PH surface (Fig. S2 and S3†).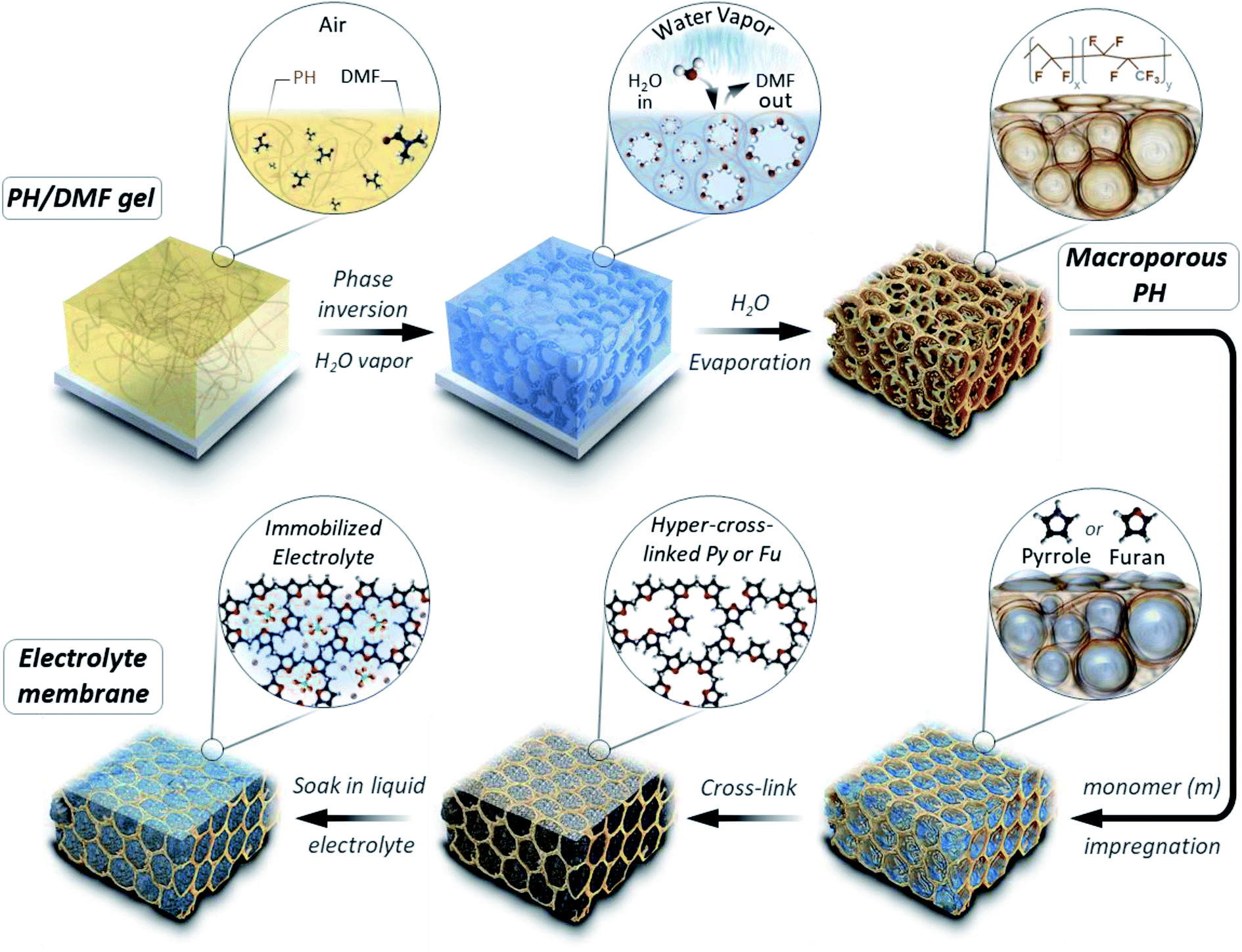 | ||
| Fig. 1 Schematic representation of the synthesis strategy for Li-HCFu-PH and Na-HCPy-PH micropores-in-macroporous polymer electrolyte membranes. | ||
 | ||
| Fig. 2 Cross-sectional and surface SEM images of (a–d) PH, (e–h) HCFu-PH, and (i–l) HCPy-PH membranes. | ||
The successful polymerization of HCFu and HCPy within the PH macropores is confirmed by FTIR spectroscopy characterization. The FTIR spectra of PH, HCFu-PH, and HCPy-PH are shown in Fig. S4.† In the FTIR spectra, broad peaks centered at 1584 cm−1 and 1620 cm−1 for the HCFu-PH and the peaks around 1495 cm−1, 1602 cm−1, and 1642 cm−1 in HCPy-PH can be assigned to aromatic ring skeleton vibrations (i.e. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration), indicating that the aromatic structure of furan and pyrrole have been retained after the polymerization.9–11 Following polymerization, the FTIR spectrum of HCPy-PH shows a new minor peak at 700 cm−1 and a broad peak around 3400 cm−1 that can be assigned to C–H deformation vibrations and N–H stretching vibrations of pyrrole, respectively.11 The peaks appearing at about 2912 cm−1 in the FTIR spectrum of both HCFu-PH and HCPy-PH can be attributed to the symmetrical and asymmetrical stretching vibrations of methylene (–CH2) linkers formed between the hetero-aromatic rings during the Friedel–Crafts reaction.12 These results confirm that both Py and Fu have been successfully polymerized within the PH host matrix via the Friedel–Crafts reaction.
C stretching vibration), indicating that the aromatic structure of furan and pyrrole have been retained after the polymerization.9–11 Following polymerization, the FTIR spectrum of HCPy-PH shows a new minor peak at 700 cm−1 and a broad peak around 3400 cm−1 that can be assigned to C–H deformation vibrations and N–H stretching vibrations of pyrrole, respectively.11 The peaks appearing at about 2912 cm−1 in the FTIR spectrum of both HCFu-PH and HCPy-PH can be attributed to the symmetrical and asymmetrical stretching vibrations of methylene (–CH2) linkers formed between the hetero-aromatic rings during the Friedel–Crafts reaction.12 These results confirm that both Py and Fu have been successfully polymerized within the PH host matrix via the Friedel–Crafts reaction.
Fig. 3a shows the nitrogen adsorption/desorption isotherms for the HCFu and HCPy samples. The BET surface areas of HCFu and HCPy samples were found to be 517 m2 g−1 and 576 m2 g−1, respectively. The micropore analysis of HCFu and HCPy samples indicated a pore-size distribution over the range 0.6–2.5 nm with dominant pore widths of 1.22 nm and 1.65 nm, respectively (Fig. 3b). The microporosity of the samples indicates a high degree of cross-linking in the polymer network. The observed hysteresis loop at low pressures (P/P0 ≈ 0.45) provides evidence of some mesoporosity in the samples due to pore network effects (e.g. mesopores that are accessible only via micropores).13 Overall, the high surface areas and tuned pore sizes of the HCFu and HCPy materials make them ideal candidates for lithium and sodium electrolyte membranes (which require high electrolyte uptake and retention).
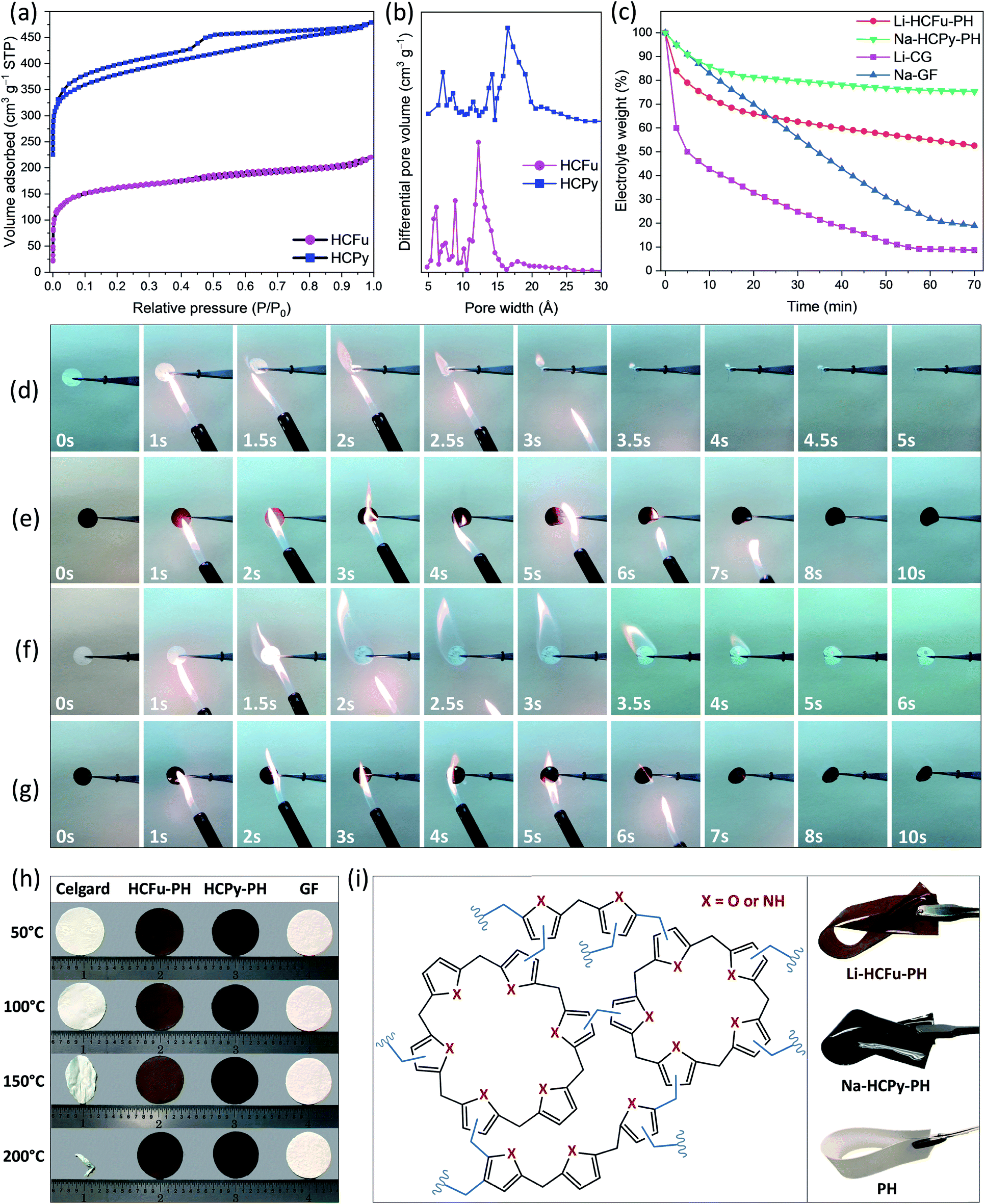 | ||
| Fig. 3 (a) N2 adsorption and desorption isotherms and (b) pore-size distribution for HCFu and HCPy; (c) electrolyte retention rate of Li-CG, Li-HCFu-PH, Na-GF, and Na-HCPy-PH membranes at 100 °C; photographs of time-frame flammability behavior of (d) Li-CG, (e) Li-HCFu-PH, (f) Na-GF, and (g) Na-HCPy-PH electrolyte membranes; (h) comparison photographs of the thermal shrinkage of CG, GF, HCFu-PH and HCPy-PH membranes at different temperatures each of which was held for 15 min; (i) schematic representation of the structure of HCFu-PH or HCPy-PH and the optical images of flexible Li-HCFu-PH, Na-HCPy-PH, and PH membranes. | ||
The electrolyte retention capability of GPEs at elevated temperatures, the thermal stability of the polymer backbone itself, and flame retarding ability of GPEs are critical factors in the safety of GPE-based lithium and sodium metal batteries. Fig. 3c demonstrates the electrolyte retention rate of the Li-CG, Li-HCFu-PH, Na-GF, and Na-HCPy-PH electrolyte membranes at 100 °C for a duration of 1 hour. As shown, the lithium liquid electrolyte in a commercial Celgard separator (Li-CG) readily drops to 50% of its initial weight within the first 5 minutes (loosing 89% of its initial weight after 1 hour) demonstrating that a poor retention of carbonate solvents (EC/DEC) within the Celgard pores. In contrast, Li-HCFu-PH demonstrated a superior electrolyte retention capability of 55% after 1 hour (due to the trapping of electrolyte solvents within the HCFu-PH micropores). Similarly, the Na-HCPy-PH membrane demonstrated an excellent electrolyte storage capability of 76% at 100 °C after 1 hour while the Na-GF membrane showed a 90% loss in its original electrolyte capacity under the same conditions. The sizes of PC, EC, and DEC solvents were calculated to be less than ≈6.5 Å (in their longest dimension); from the BET analysis, the micropores in HCFu and HCPy are both large enough to facilitate the absorption of electrolyte components within the pores and yet small enough to confine PC, EC, and DEC solvent molecules while allowing lithium or sodium ions to pass readily. Overall, the HCPy has demonstrated the highest electrolyte retention capability in carbonate-based electrolytes owing to the hydrogen-bonding interactions between –N–H functional groups of the polymer and the oxygen atoms of solvents.
The thermogravimetric analysis (TGA) results of HCFu-PH and HCPy-PH films in Fig. S5a† indicate their high thermal stability until 482 °C and 404 °C, respectively, where the thermal degradation of the polymers takes place. The high electrolyte retention capability and polymer thermal stability of Li-HCFu-PH and Na-HCPy-PH membranes (as discussed above) have enabled them to have a much lower flammability compared to conventional electrolyte/separator systems, which can remarkably improve the safety of batteries employing metallic lithium or sodium metal anodes. Fig. 3e and g show the time-frame flammability behavior of Li-HCFu-PH and Na-HCPy-PH membranes during a combustion test in which a flame (temperature ≈ 540 °C) was pointed at the membranes for 5 s and then moved away. As shown, both electrolyte membranes underwent a mild ignition after 5 s but were quickly self-extinguished and did not undergo significant change in their dimensions. The flame retarding ability of the electrolyte membranes originates from the low volatility of liquid electrolyte components trapped within the micropores, which decreases the overall vapor pressure of flammable species, as well as the high thermal stability (>400 °C) and incombustibility of HCFu-PH and HCPy-PH polymers. However, due to the intrinsic flammability of liquid electrolytes, a longer exposure of the films to the flame triggers a low combustion rate on the HCFu-PH and HCPy-PH disk membranes over a time period of ≈60 s with a tendency to produce char after combustion. The char formation indicates the absence of severe exothermic chain reactions during combustion (i.e. the amount of combustible and volatile polymer pyrolysis fragments is reduced); resulting in lower flame temperatures, reduction in the amount of heat released, and improved safety performance. In contrast, when the Li-CG or Li-PH membranes were exposed to the flame, they caught fire immediately and shrank drastically in less than 3 s (Fig. 3d and S6a†). High-flammability was also observed for the electrolyte membrane based on glass fiber separator (Na-GF) where the flammable electrolyte supplies enough energy during combustion to raise the temperature to the melting point of the glass fiber (≈1200 °C) within >6 s, as depicted in Fig. 3f.
The DSC result (Fig. S5b†) of HCFu-PH and HCPy-PH films shows an endothermic peak located around ≈160 °C, which belongs to the melting point of the PH polymer host. Interestingly, HCFu-PH and HCPy-PH membranes exhibited small thermal shrinkages (<5%) up to 200 °C (well above the melting point of the PH polymer host ≈160 °C); this remarkable thermal dimensional stability indicates that thermally stable HCFu and HCPy particles incorporated within the PH macropores help to maintain the 3D skeleton and porosity of the membranes by preventing the pores from “shutting down” when the PH polymer is melted or softened. These results are superior to that of PP-based Celgard and PE-based Celgard as the former decomposes around 280 °C and the latter “shuts down” at ≈130 °C (i.e. its melting point) when the CG pores collapse (causing deterioration of the membrane porosity, potential internal short-circuits, and thermal runaway).14,15 These processes can be clearly seen in Fig. 3h where the CG begins to curl when the temperature reaches 120 °C, undergoes a significant shrinkage at 150 °C, and completely shrivels up at 200 °C. The thermal-shrinkage test of the PH polymer host (Fig. S6b†) also demonstrates poor thermal dimensional stability at temperatures above 150 °C and eventually melts to transparency (at 200 °C) due to a collapse of the macropores; suggesting that thermally induced internal short circuiting/thermal runaway are possible at temperatures above ≈150 °C.
Fig. S7† displays the stress–strain curves for PH, HCFu-PH, and HCPy-PH films. The PH film demonstrates a tensile stress of 4.0 MPa with a maximum strain of 23.7%. For HCFu-PH and HCPy-PH films, the tensile strains at break are 48.3 and 35.9% while their corresponding tensile stresses at break are 4.6 and 4.1 MPa, respectively. These results indicate that the incorporation of HCFu and HCPy polymers into the macropores of PH improves the mechanical properties of the PH films without sacrificing their flexibility; as is shown in Fig. 3i. Therefore, the incorporated HCFu and HCPy solid polymers can be also considered as fillers (which could be the reason for the improved mechanical properties of HCFu-PH and HCPy-PH films) in the PH polymer host.
2.2. Electrochemical and battery characterization
Increased electrolyte uptake and salt dissociation capabilities are important factors affecting the ionic conductivities of membrane electrolytes. The electrolyte uptake in Li-HCFu-PH, Li-PH, Na-HCPy-PH, and Na-PH membranes were found to be 173%, 285%, 195%, and 265%, respectively. The lower electrolyte uptake for HCFu-PH and HCPy-PH membranes was expected since the PH macropores were occupied with HCPs. According to EIS measurements, the Li-HCFu-PH and Na-HCPy-PH membranes demonstrate ionic conductivities of 6.4 × 10−3 S cm−1 and 4.3 × 10−3 S cm−1 at 25 °C, respectively. These results were slightly higher than those of Li-PH (3.5 × 10−3 S cm−1) and Na-PH (9.0 × 10−4 S cm−1) GPEs, respectively. The higher conductivity of the membranes can be attributed to a high dissociation of LiPF6 and NaClO4 salts via electrostatic (electronic absorption effect) interactions with furan and pyrrole groups.16 Therefore, we anticipate that the high number of cross-linked monomers in close proximity to each other within the micropores of the HCFu and HCPy allow Li+ and Na+ cations to hop from pore to pore while remaining solvated by the solvent molecules. The outstanding hydrophilicity of HCFu-PH and HCPy-PH membranes (due to the existence of polar C–O–C and C–NH–C bonds) along with their remarkable electrolyte uptake (due to the incorporation of highly porous HCFu and HCPy polymers in the interconnected PH channels) results in a continuous liquid pathway across the membrane that brings about excellent cation conductivities and potential applications for use in batteries. The result of initial screening showed that the HCFu is not very stable against a sodium anode, and lithium cells based on HCPy produce a large charge–discharge polarization, apparently due to trapping of LiPF6 by pyrrole groups.The CV and LSV experiments were conducted on Li/Li-HCFu-PH/SS and Na/Na-HCPy-PH/SS cells to evaluate their plating/stripping behaviour and electrochemical stability window (Fig. 4a and b). From the CV experiments of both GPEs (the inset of Fig. 4a and b), the difference between the anodic peak potential, Epa, and the cathodic peak potential, Epc, was found to be ≈57 mV (ΔEp = Epa − Epc), indicating a reversible plating and stripping of metallic anodes. The LSV results for Li/Li-HCFu-PH/SS and Na/Na-HCPy-PH/SS cells did not show a significant anodic current up to 4.7 V vs. Li/Li+ and 4.5 V vs. Na/Na+, which are known to be the onset potentials for the electrochemical oxidation of lithium (LiPF6 in EC/DEC) and sodium (NaClO4 in PC + FEC) liquid electrolytes, respectively. As a result, the electrochemical stability window of the proposed GPEs is limited by the electrochemical stability of the impregnated liquid electrolyte rather than that of the polymer host. However, the anodic current densities (due to electrolyte decomposition) are much lower in the proposed GPEs compared to those of liquid electrolytes (having the same amount of electrolyte uptake as GPEs) paired with commercial separators. This lower current density can be attributed to the trapping of electrolyte solvents and negatively charged ions (e.g. PF6− and ClO4−) in the micropores as well as the superior anode/GPE interfacial compatibility. The retention of solvents within the micropores leads to a slow diffusion of solvent molecules to the electrode surface during cell operation, which limits their anodic electrooxidation current flow and the rate of decomposition. Moreover, trace impurities and moisture in the liquid electrolytes can be scavenged by the HCPy and HCFu micropores, thus inhibiting the accumulation of impurities at an electrode/electrolyte interface; providing an extended potential window and superior anode/GPE interfacial stability. The operation potentials of, for example, layered- and olivine-type lithium cathodes and fluorophosphate-, layered-, and NASICAN-type sodium cathodes fall into the electrochemical working window of Li-HCFu-PH and Na-HCPy-PH membranes, respectively, indicating the feasibility of these electrolytes for use in today's battery technology.
 | ||
| Fig. 4 CVs and LSVs for (a) Li/Li-HCFu-PH/SS and (b) Na/Na-HCPy-PH/SS cells recorded at 1 mV s−1 (SS: stainless-steal electrode); the galvanostatic cycling performance of (c) Li/Li-HCFu-PH/Li and (d) Na/Na-HCPy-PH/Na at 1 mA cm−2 with a capacity of 1 mA h cm−2; (e–g) schematic representation of Li or Na plating/stripping at traditional liquid/separator-based (CG or GF) cells compared to cells based on advanced Li-HCFu-PH or Na-HCPy-PH electrolyte membranes. | ||
In order to investigate the dynamic interfacial stability of metallic anodes with the GPEs and their dendrite-suppressing behavior, symmetric Li/Li-HCFu-PH/Li and Na/Na-HCPy-PH/Na cells were assembled and tested under the galvanostatic-polarization method at a constant current density of 1 mA cm−2 with a capacity of 1 mA h cm−2. As a control, symmetric Li/CG/Li and Na/GF/Na cells were also fabricated and subjected to the same galvanostatic experiments. As shown in Fig. 4c, Li/Li-HCFu-PH/Li cells demonstrate a stable voltage profile over 500 hours of continuous plating/stripping cycles (250 cycles), while the Li/CG/Li cells undergo a continuous increase in the overpotential that eventually leads to an internal short-circuit (as evidenced by a sudden voltage drop at ≈233 h). For the Li/Li-HCFu-PH/Li cells, the charging voltage gradually increased in the first few cycles to a stable value as cycling proceeded, whereas the opposite occurred for the Li/CG/Li cells. For example, the potential of Li/CG/Li increased by 35% at cycle 115 compared to cycle 10, while there was only 2.2% potential increase at 500th cycle (compared to the 10th cycle) for Li/Li-HCFu-PH/Li cells. The galvanostatic cycling performance of the Na/GF/Na cells exhibited cell failure by dendrite-induced short circuiting at cycles <22 (Fig. 4d). In sharp contrast, Na/Na-HCPy-PH/Na cells demonstrated excellent voltage stability after 500 h. The Na/Na-HCPy-PH/Na cells demonstrated a slightly higher plating/stripping overpotential in the first few cycles, but the charging potential eventually reached a stable value (i.e. lower potential) as cycling continued. The relatively higher voltage polarization in the first few cycles for Li/Li-HCFu-PH/Li and Na/Na-HCPy-PH/Na cells is attributed to the formation of an SEI layer from the decomposition of “free” liquid electrolyte present on the surface and/or residing within unoccupied macropores of the GPEs. This limited “free” liquid electrolyte could help to wet the GPE/electrode interface quickly; promoting the formation of a stable and conductive SEI layer while the “trapped” liquid electrolyte remains inaccessible to the anode. Therefore, HCFu-PH and HCPy-PH membranes prevent the continuous decomposition of liquid electrolyte while remaining conductive to Li+ and Na+, thereby enabling stable cycling. Notably, after the symmetric Li/Li-HCFu-PH/Li and Na/Na-HCPy-PH/Na cells were disassembled, it was observed that the GPEs still retained a large amount of liquid electrolyte (which was observed by hand-pressing the membranes) while the liquid electrolyte in Li/CG/Li and Na/GF/Na cells were almost completely dry and had visible signs of dendrite-induced punch marks on their surfaces. This observation can be attributed to the continuous decomposition of electrolyte in Li/CG/Li and Na/GF/Na cells, that forms a thick SEI layer on the Li and Na anodes and deteriorates the electrode/electrolyte interfaces (Fig. 4e–g). Disassembly of the symmetrical cells (Li/CG/Li and Na/GF/Na) revealed a thick SEI layer on the Li anode, which likely caused the observed increase in overpotential during cycling, while sodium-SEI layers were observed to lose their physical contact with the Na anode altogether; this can generate a fresh contact between the liquid electrolyte and the Na anode during cycling and thereby enable a steady overpotential but will ultimately exacerbate electrolyte decomposition and lead to rapid cell failure.
It is worth mentioning that the furan-based compounds have been known to promote the stability and conductivity of a lithium SEI layer, which decreases the interfacial electrode/electrolyte resistance and improves the coulombic efficiencies of lithium during plating/stripping cycles.17,18 Pyrrole-based macromolecules have also been shown to form a strong hydrogen bonding between pyrroles (N–H group) and counter anions (ClO4− in this work), leading to a strong trapping of counter anions and, hence, restricting their role in the formation of an SEI layer, thus improving the electrolyte/anode interfacial properties.16,19,20 Moreover, dendrite formation and growth across the GPEs are suppressed by HCFu or HCPy particles due to their extremely small micropores, while the relatively larger pores in Celgard (≈210 × 50 nm) and glass-fiber (micron-range pore sizes) provide sufficient pathways for dendrites to form and penetrate across these separators.
In order to evaluate the versatility of the Li-HCFu-PH in full-cell lithium metal batteries, a LiFePO4/C cathode was used to assemble Li/Li-HCFu-PH/LiFePO4 full cells. As shown in Fig. 5a, the charge/discharge profile of Li/Li-HCFu-PH/LiFePO4 full cells shows plateaus at 3.46/3.40 V (vs. Li/Li+) when cycled in the potential range of 2.6–4.0 V. The charge/discharge plateaus correspond to the reversible Fe3+/Fe2+ redox conversion, which is the typical electrochemical behavior of a LiFePO4 cathode. The Li/Li-HCFu-PH/LiFePO4 cells deliver a discharge capacity of 149.2 mA h g−1 at 0.5C, corresponding to 87.8% of its theoretical value (170.0 mA h g−1). Fig. 5b demonstrates that at C-rates of 0.05, 0.1, 0.5, 1, and 2C, the Li/Li-HCFu-PH/LiFePO4 cells offer high-rate capability with discharge capacities of 159.6, 154.5, 149.2, 139.7, 130.5 mA h g−1, respectively. This result is higher than the discharge capacities for Li/Li-CG/LiFePO4 cells, indicating a fast transport of lithium ions through the microporous HCFu. Furthermore, the cycling performance of Li/Li-HCFu-PH/LiFePO4 cells and conventional Li/Li-CG/LiFePO4 cells were investigated at a 1C rate over the potential range of 2.6–4.0 V. As shown in Fig. 5c, the discharge capacity of the Li/Li-CG/LiFePO4 cells exhibited an initial discharge capacity of 137 mA h g−1 followed by a rapid 23% capacity loss by the 357th cycle where it undergoes an internal cell short circuit. In sharp contrast, the Li/Li-HCFu-PH/LiFePO4 retained 84.2% (117.6 mA h g−1) and 78.0% (108.9 mA h g−1) of its initial capacity along with a stable coulombic efficiency of ≈99.7% after 500 and 1000 cycles at 1C, respectively.
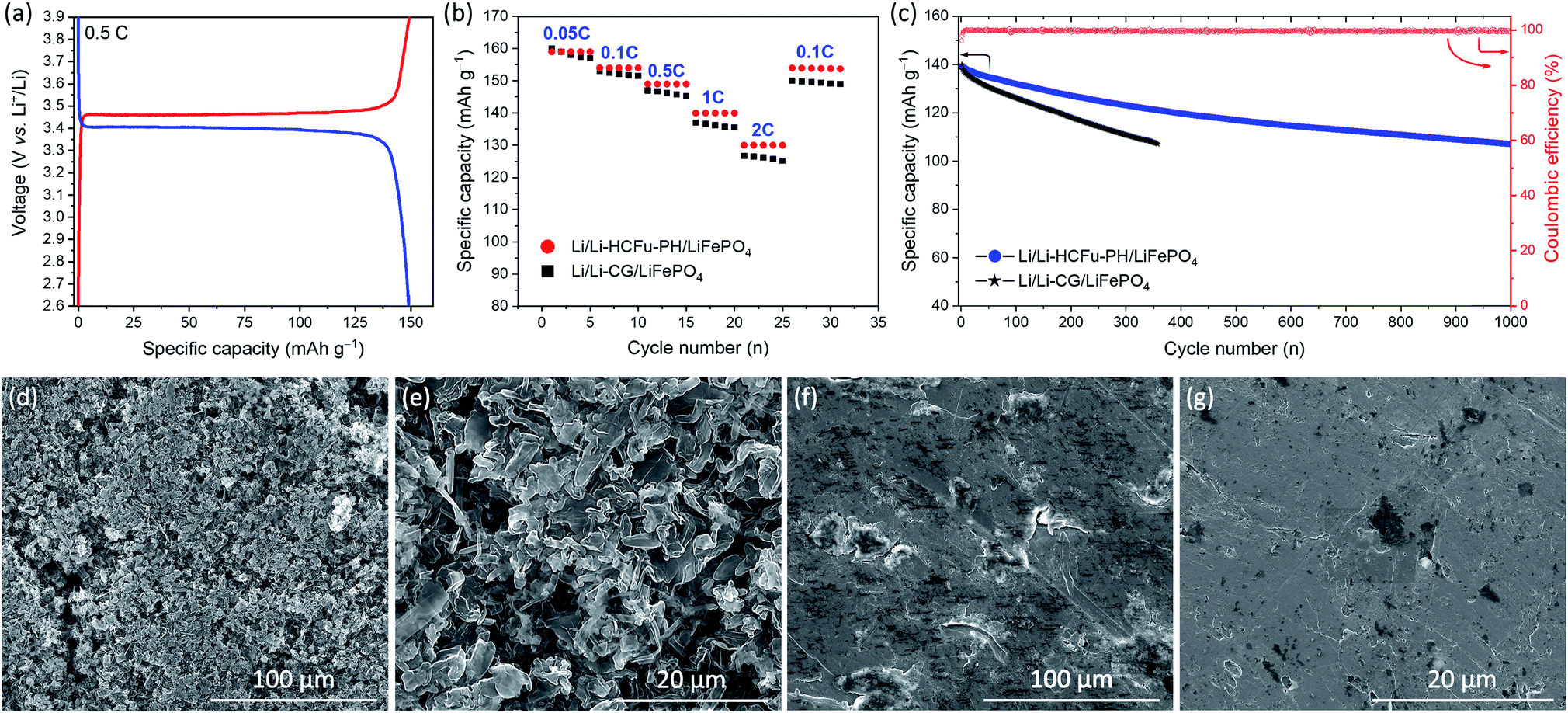 | ||
| Fig. 5 (a) Charge–discharge curve for Li/Li-HCFu-PH/LiFePO4 cell, (b) rate performances, and (c) cycle life and coulombic efficiency at 1C for Li/Li-HCFu-PH/LiFePO4 and Li/Li-CG/LiFePO4 cells; SEM surface morphologies of the lithium anode in (d and e) Li/Li-CG/LiFePO4 cell and (f and g) Li/Li-HCFu-PH/LiFePO4 cells after 200 cycles. | ||
SEM images of lithium metal anodes from the disassembled Li/Li-HCFu-PH/LiFePO4 and Li/Li-CG/LiFePO4 cells after 200 cycles at 1C rate are shown in Fig. 5d–g. As it is shown in Fig. 5d and e lithium anodes from disassembled cycled Li/Li-CG/LiFePO4 cells contained a porous and thick layer of filamentary- and particle-type dendrites on their surfaces. These dendrites are known to form during charging; as the newly plated metallic lithium (on the anode surface) is exposed to a pool of liquid electrolyte that quickly reacts with the fresh lithium to form a thick SEI layer. Such insulating SEI layers disrupt the uniform distribution of the electrical field on the metal anode leading to preferential plating of lithium on surface sites (i.e. thinner SEI layers and/or defects) which can facilitate a high flux of lithium ions, thus triggering the formation and growth of porous and/or fibrous Li deposits.21 The continuous growth of Li dendrites results in an increased surface area which exacerbates decomposition of the liquid electrolyte that is weekly retained in the CG separator, leading to low coulombic efficiencies, inferior cycling performance, and eventual internal cell short circuiting. In a sharp contrast, the morphology of lithium anode from a disassembled Li/Li-HCFu-PH/LiFePO4 cell (Fig. 5f and g) shows a smooth surface on the lithium anode surface with no apparent sign of dendrites. This result suggests that Li-HCFu-PH facilitates uniform lithium plating via a continuous network of lithium-ion pathways throughout the micropore channels (that are distributed across the electrolyte membrane as well as the surface of Li-HCFu-PH) contacting the lithium anode; this provides a uniform ionic flux to the anode surface leading to homogeneous Li plating/stripping. Furthermore, the ability of HCFu-PH to retain the electrolyte solvents within the micropores, while simultaneously allowing a fast transport of lithium ions during cycling, significantly reduces side reactions between freshly deposited lithium and solvent molecules; leading to the formation of a thin conductive SEI layer and uniform lithium deposition.
For the assessment of Na-HCPy-PH electrolyte membrane in a full-cell sodium battery, the NASICON-type structured Na3V2(PO4)3/C was chosen as the cathode due to its excellent electrochemical cycling in conventional sodium organic-based liquid electrolytes. As demonstrated in Fig. 6a, the fabricated Na/Na-HCPy-PH/Na3V2(PO4)3 full cells delivered a capacity of 101.0 mA h g−1 at 0.5C and showed charge/discharge plateaus at 3.40/3.36 V (vs. Na/Na+), indicating the reversible redox conversion of V3+/V4+ couples which is an electrochemical characteristic of the Na3V2(PO4)3 cathodes. As shown in Fig. 6b, when Na/Na-HCPy-PH/Na3V2(PO4)3 cells are subjected to a high charge/discharge rate of 2C, 89.2% (94.5 mA h g−1) of the discharge storage capacity is retained compared to 0.05C rate (106.0 mA h g−1), revealing that Na-HCPy-PH having a micropore structure can deliver a rate capability performance superior to that of a traditional Na/Na-GF/Na3V2(PO4)3 cell fabricated with a glass-fiber separator having large micron-size pores. Additionally, the long-term cycling performance of the Na/Na-HCPy-PH/Na3V2(PO4)3 full cells at a 1C rate after 500 and 1000 cycles revealed a remarkable capacity retention of 96.3% and 94.0%, respectively (Fig. 6d). The same cells also exhibited a high average coulombic efficiency of 99.8% after 1000 cycles, which indicates a superior reversible redox reaction at both Na/Na-HCPy-PH and Na-HCPy-PH/Na3V2(PO4)3 interfaces. In contrast, the Na/Na-GF/Na3V2(PO4)3 cells underwent a rapid short circuit at cycles <140 that, from the disassembled cells, was observed to be caused by fast growing sodium dendrites across the glass fiber separator.
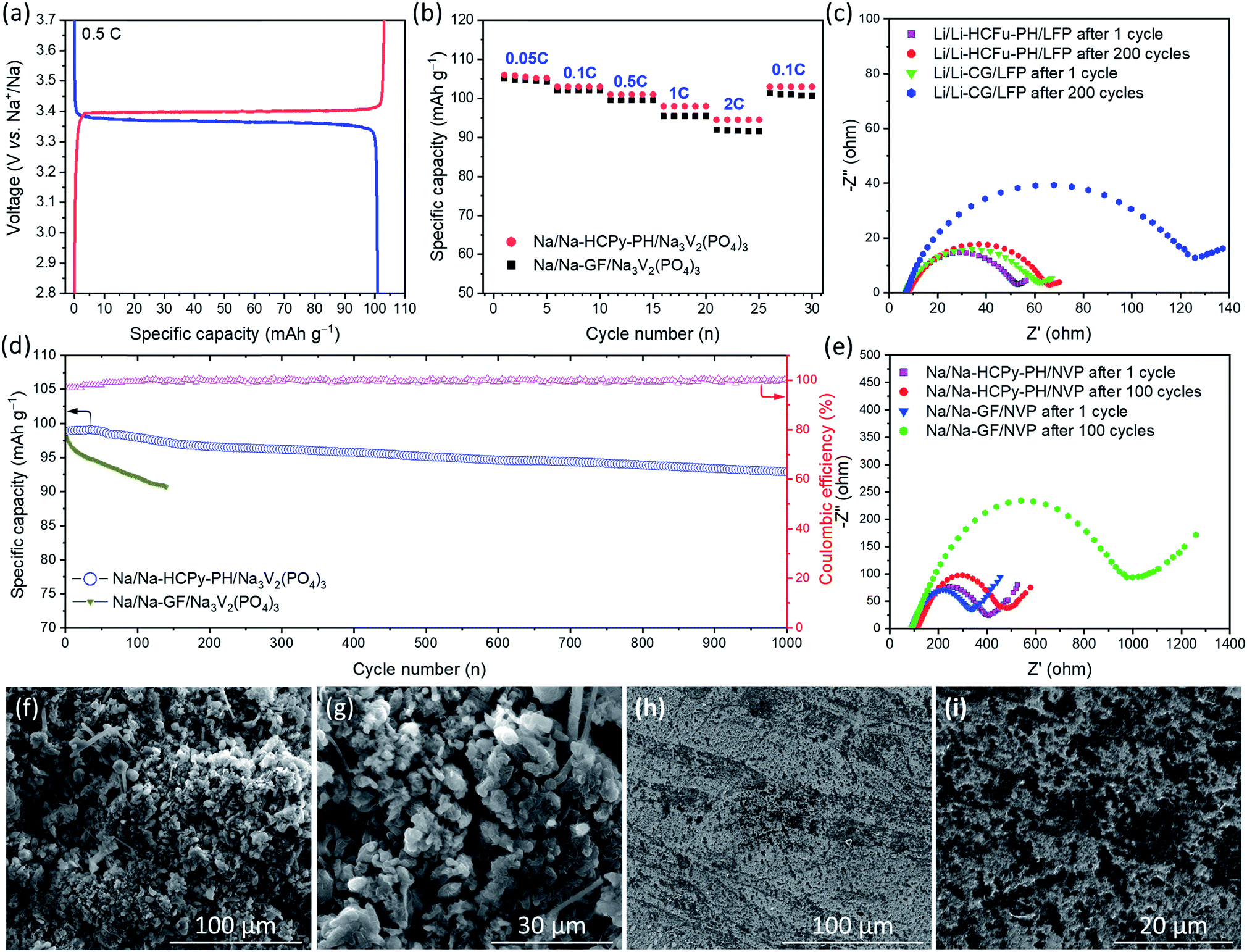 | ||
| Fig. 6 (a) Charge–discharge curve for Na/Na-HCPy-PH/Na3V2(PO4)3 cell, (b) rate performances, (c) EIS result of lithium batteries at different cycles, and (d) cycle life and coulombic efficiency at 1C for Na/Na-HCPy-PH/Na3V2(PO4)3 and Na/Na-GF/Na3V2(PO4)3 cells; (e) EIS result of sodium batteries at different cycles; SEM surface morphologies of the sodium anode in (f and g) Na/Na-GF/Na3V2(PO4)3 cell and (h and i) Na/Na-HCPy-PH/Na3V2(PO4)3 cells after 100 cycles. | ||
SEM images in Fig. 6f–i show the morphologies of the sodium anodes in Na/Na-GF/Na3V2(PO4)3 and Na/Na-HCPy-PH/Na3V2(PO4)3 cells after 100 cycles at 1C rate. As shown in Fig. 6f and g, the sodium anode (from a cycled Na/Na-GF/Na3V2(PO4)3) had a thick layer of sharp and “dead” dendrites on the surface. The severe side reaction of free liquid electrolyte (weekly retained in between the microfibers of GF separator) and the highly reactive sodium anode resulted in poor anode stability during plating/stripping; leading to continuous decomposition of electrolyte and formation of dead sodium-SEI dendrites. The low module properties GF separator provides sufficient pathways for Na dendrites to easily traverse through the separator and cause an internal short circuit. In contrast, as demonstrated in Fig. 6h and i, the Na-HCPy-PH electrolyte membrane (having a compact and robust architecture, high sodium-ion conductivity, superior electrolyte retention capabilities, and uniform ionic flux) enables homogeneous sodium plating/stripping over extended cycles. SEM images in Fig. 6h and i revealed black spots (residual HCPy particles) located on the surface of cycled Na anodes that remained after the membrane was peeled off; this observation demonstrates that the Na-HCPy-PH electrolyte membrane had excellent adherence (i.e. good interfacial anode/electrolyte contact) to the Na anode.
To further elucidate the underlying reasons for the improved cycling stability of the proposed GPE-based batteries, compared to batteries employing traditional separators, electrochemical impendence spectroscopy (EIS) measurements after deep cycles at 1C were performed to investigate the dynamics of the interfacial resistance. The observed semicircles in EIS spectra in Fig. 6c and e are assigned to the interfacial resistances (Ri) at the electrode–electrolyte interfaces which usually increase over extended cycling due to the progress of SEI formation. As seen in Fig. 6c, the EIS results of Li/Li-CG/LiFePO4 cells show a significant increase in Ri from ≈56 Ω at cycle 1 to 118 Ω after 200 cycles (ΔRi ≈ 62 Ω), whereas a much smaller change in Ri (ΔRi ≈ 10 Ω) for the Li/Li-HCFu-PH/LiFePO4 cells was observed after 200 cycles at 1C. Similarly, when Na/Na-HCPy-PH/Na3V2(PO4)3 cells were operated for 100 cycles at 1C, they exhibited a substantially smaller increase in the interfacial resistances (ΔRi ≈ 34 Ω) than those in Na/Na-GF/Na3V2(PO4)3 cells (ΔRi ≈ 698 Ω). The faster increase in interfacial resistance of Li/Li-CG/LiFePO4 and Na/Na-GF/Na3V2(PO4)3 cells during cycling likely stems from the poor capability of Celgard and glass-fiber separators to immobilize the liquid electrolyte. The continuous decomposition of liquid electrolyte in contact with the alkali metal anodes generates a thick insulating SEI layer during cycling; resulting in a larger increase in the interfacial resistance and eventual cell malfunction.
3. Conclusions
This work has presented a novel strategy to successfully incorporate rigid microporous polymers into the macropores of a flexible polymer host via an in situ polymerization technique. The PH polymer host provides the requisite mechanical strength and flexibility for the GPEs while the in situ incorporated microporous polymers enable the uptake and immobilization of a large fraction of liquid electrolyte within their matrix. In this work, furan and pyrrole monomers have been in situ hyper-cross-linked within the macropores of a flexible PH polymer and a gel-polymer electrolyte was prepared by impregnation with lithium or sodium liquid electrolytes. The prepared GPEs feature (i) high ionic conductivities rivaling those of liquid electrolytes, (ii) an excellent retention of electrolyte and fast transport of Li+ or Na+ ions, (iii) a flat charge/discharge plateau, indicating no side-reaction in the operating potential window of liquid electrolytes, (iv) a low and stable overpotential over extended cycles, indicating a high ionic conductivity and stability of the SEI layers, and (v) no evidence of any short-circuiting, implying the suppression of dendrite growth. The results indicate that the proposed micropores-in-macropores strategy allows the use of a metallic lithium or sodium anodes for large-scale applications. The performance of batteries based on these electrolyte membranes is comparable to that of the best in recent literature (see Table S1†).In addition, the proposed micropores-in-macroporous membrane approach can be extended to the preparation of micropores-in-mesoporous and mesopores-in-macroporous membranes. Therefore, this method allows the facile preparation of membranes that can be tailored based on the desired application. Alternative approaches can also be applied to this technique in order to in situ synthesize a variety of different types of porous organic and inorganic materials within the larger pores of a polymer host.
4. Experimental section
4.1. Electrolyte membrane preparation and characterization
The macroporous PVDF-HFP (PH) was prepared via a phase inversion technique as follows: PVDF-HFP (PH, 1 g, Sigma Aldrich, Mw = 455![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was completely dissolved in N,N-dimethylformamide (DMF, 4 mL, Sigma Aldrich, 99.8 wt%) under mechanical stirring at 40 °C for 5 h. The resulting viscous solution was cast onto a glass sheet by using a doctor blade with a gap of 220 μm and subsequently exposed to water vapor for 2 hours. The free-standing solidified film was then transferred into a tank of DI water and left undisturbed for 12 hours for a complete exchange of DMF with water to form a macroporous film. The macroporous PH membrane was rinsed with DI water and then vacuum dried at 70 °C for 12 hours. The dried film was punched into round disks with a diameter of 16 mm and stored in a glove box.
000) was completely dissolved in N,N-dimethylformamide (DMF, 4 mL, Sigma Aldrich, 99.8 wt%) under mechanical stirring at 40 °C for 5 h. The resulting viscous solution was cast onto a glass sheet by using a doctor blade with a gap of 220 μm and subsequently exposed to water vapor for 2 hours. The free-standing solidified film was then transferred into a tank of DI water and left undisturbed for 12 hours for a complete exchange of DMF with water to form a macroporous film. The macroporous PH membrane was rinsed with DI water and then vacuum dried at 70 °C for 12 hours. The dried film was punched into round disks with a diameter of 16 mm and stored in a glove box.
A few pieces of PH films were immersed in a furan (Sigma Aldrich, ≥99%) or pyrrole (Sigma Aldrich, 98%) monomer solution for 12 hours at 5 °C under Ar atmosphere to complete the impregnation of monomers within the PH macropores. The furan solution contains 5% (mol%) pyrrole monomers to prevent a ring-opening side reaction during the polymerization.22 To prepare the hyper-cross-linking solution, anhydrous ferric chloride (FeCl3, 1.00 g, Sigma Aldrich, ≥98.0%) and anhydrous 1,2-dichloroethane (DCE, 10 mL, Sigma Aldrich, 99.8%) were well-mixed in an oven-dried 25 mL round-bottom flask. After 5 min of stirring at 5 °C under Ar atmosphere, the external cross-linker formaldehyde dimethyl acetal (FDA, 0.47 g, Sigma Aldrich, ≥99.0%) was added to the mixture followed by another 10 min stirring. Afterwards, the Fu-soaked and Py-soaked PH films were slowly dipped into the cross-linking solution at 4 °C and kept there undisturbed for 1 hour. To complete the hyper-cross-linking process through the Friedel–Crafts reaction, a condenser was connected to the round-bottom flask and the mixture was heated to 80 °C for 3 h under Ar atmosphere with occasional stirrings. The obtained films were first washed with methanol several times followed by a Soxhlet extraction with methanol for 24 h. Finally, the films were vacuum dried at 80 °C for 24 h to get the hyper-cross-linked furan embedded in the macroporous PH (HCFu-PH) and hyper-cross-linked pyrrole embedded in the macroporous PH (HCPy-PH).
The lithium gel-polymer electrolyte based on the HCFu-PH film (Li-HCFu-PH) was achieved by immersing the dried HCFu-PH film into a commercial lithium electrolyte composed of 1 M LiPF6 in a mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) with the ratio of 1:1. The sodium gel-polymer electrolyte based on the HCPy-PH film (Na-HCPy-PH) was obtained by soaking the HCPy-PH film in 1 M NaClO4 in propylene carbonate (PC) solvent containing 5 wt% fluoroethylene carbonate (FEC). The soaking was continued for 72 hours in an argon-filled glovebox to ensure the complete permeation of the electrolyte ingredients into the micropores. Before a cell assembly, the surfaces of the electrolyte membranes were wiped with Kimwipe to remove any extra electrolyte that could add extra resistance to the cell upon cycling. The electrolyte uptake (η) was determined according to η = (Wt − W0)/W0 × 100% formula; where W0 and Wt represent the weight of the membranes before and after they absorbed the liquid electrolyte, respectively.
4.2. Cathode preparation
The LiFePO4 cathode (LFP) was prepared by slurry mixing of 80.0% LiFePO4 active material (MTI corporation), 6% acetylene black, 6% Timcal Super P graphite, and 8% polyvinylidene fluoride (PVDF) binder dissolved in an appropriate amount of N-methyl-2-pyrrolidone (NMP) solvent. The mixture was stirred at room temperature for 12 h until a homogeneous slurry was achieved. The slurry was coated onto a graphite-coated aluminum foil (18 μm thickness, MTI corporation). The cathode film was first dried in an air-circulating oven at 60 °C for 30 min followed by a vacuum oven drying at 110 °C for 12 hours. The mass loading of the cathode was ≈5 mg cm−2.The Na3V2(PO4)3/C sample was synthesized via a sol–gel method as follows: an aqueous solution of vanadium pentoxide (V2O5, 1.14 g, Sigma Aldrich, 99.99%), citric acid (C6H8O7, 1.81 g, Sigma Aldrich, ≥99.5%), ammonium dihydrogen phosphate (NH4H2PO4, 2.17 g, Sigma Aldrich, ≥99.999%), and sodium carbonate (Na2CO3, 1.00 g, Sigma Aldrich, ≥99.5%) with the molar ratio of 2/3:1:2:1 was stirred at 40 °C to obtain a homogeneous solution. The solution was continuously stirred at 90 °C to form a gel. Heating the gel in an oven for 12 h at 140 °C formed a porous foam product. The foam was first pyrolyzed at 350 °C for 4 h (at 1 °C min−1 heating rate) and then heated to 900 °C for 24 hours (at 1.67 °C min−1 heating rate) under Ar/H2 (95%/5%) followed by cooling down to room temperature at 3 °C min−1. The sodium cathode film was prepared with the same procedure as described for LFP films.
4.3. Material characterizations
The surface and cross-sectional morphology of the films were explored through SEM (FEI Quanta 650) after sputtering gold on the films. The thickness of the polymer films for SEM characterization were ≈100 μm. Thermogravimetric analysis (TGA, from 30 to 600 °C) and differential scanning calorimetry (DSC, from 30 to 200 °C) analysis of the membranes were conducted by a Mettler Toledo TGA/DSC 1 at a heating rate of 10 °C min−1. The Brunauer–Emmett–Teller (BET, Micromeritics ASAP 2020) test was conducted on the incorporated HCFu and HCPy after they were extracted from HCFu-PH and HCPy-PH membranes; to do so, the HCFu-PH and HCPy-PH membranes were soaked in hot DMF to completely dissolve the PH backbone. The remaining HCFu and HCPy were filtered and subjected to Soxhlet extraction for 24 h for the complete removal of PH. The obtained HCFu and HCPy particles were vacuum dried at 80 °C for 12 hours before the BET characterization. The spectroscopy characterizations included X-ray diffraction (XRD, MiniFlex 600 II) and Fourier transform infrared spectroscopy (FTIR, Thermo Scientific Nicolet iS5 FTIR Spectrometer, 4000–500 cm−1, resolution 2 cm−1). The dynamic mechanical analysis (DMA) of the films, which were cut into dog-bone-shaped specimens, were tested with an RSA-G2 Solids Analyzer according to the ASTM D882-18 standard method at room temperature.4.4. Electrochemical characterizations
To test the electrochemical properties of the GPEs, CR2032 coin cells were fabricated in an argon-filled glove box (H2O and O2 content <0.1 ppm), and all tests were carried out at room temperature. To assess the electrochemical stability window and the stripping/plating behavior of metallic anodes, the Li-HCFu-PH or Na-HCPy-PH were sandwiched between a metallic anode foil (Li or Na), as the reference and counter electrode, and a stainless-steel plate (SS) as a working electrode. The fabricated Li/Li-HCFu-PH/SS and Na/Na-HCPy-PH/SS cells were evaluated with linear-sweep voltammetry (LSV) and cyclic voltammetry (CV) tests at a scan rate of 1 mV s−1 with an electrochemical workstation (Solartron Analytical 1287A Cell Test System). To measure the ionic conductivities, the GPEs were sandwiched between two SS blocking electrodes, and the bulk resistance between these two SSs was measured by electrochemical impedance spectroscopy (EIS, Solartron 1260A Analyzer) with an AC perturbation of 10 mV over the frequency range of 100 kHz to 0.01 Hz. The ionic conductivities were then calculated by using the formula σ = d/ARb, where σ is the ionic conductivity (S cm−1), d is the thickness of the GPEs (cm), A represents the area of the GPEs (cm2), and Rb stands for the bulk resistance (Ω) obtained from the horizontal axis of the Nyquist plot. For galvanostatic plating/stripping cycling tests, symmetric cells were fabricated by placing the electrolyte membranes between two identical metallic foils in CR2032 cells. The galvanostatic cycling tests were carried out at a constant current density of 1 mA cm−1 with a capacity of 1 mA h cm−2 for 500 hours. For the full cell battery tests, the Li-HCFu-PH or Na-HCPy-PH electrolyte membrane was sandwiched between a metallic anode (Li or Na foil) and a cathode disk (LiFePO4/C or Na3V2(PO4)3/C) to fabricate Li/Li-HCFu-PH/LiFePO4 and Na/Na-HCPy-PH/Na3V2(PO4)3 full cells, respectively. For the sake of comparison, conventional Celgard (CG, 2500) and glass-fiber (GF, Whatman, 934-AH) separators were used to assemble lithium (Li/Li-CG/LiFePO4) and sodium (Na/Na-GF/Na3V2(PO4)3) full cells. For the full cell assembly, each LiFePO4/C and Na3V2(PO4)3/C cathode disk was arc-welded to the cap side of a CR2032 coin cell. A 3 μL of liquid electrolyte was dropped on the cathodes to ensure a complete wetting of the cathode matrix with the electrolyte. For the lithium cells, the galvanostatic charge–discharge tests were carried out at room temperature with an upper cut-off voltage of 4.0 V and a lower cut-off voltage of 2.6 V. The cutoff voltage limits for the sodium cells were 2.6–3.8 V. All full cells were cycled at a low C rate of C/10 for the first two cycles to produce a uniform SEI layer. A LANHE battery tester was employed for the galvanostatic cycling experiments.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Robert A. Welch Foundation, Houston, Texas (Grant No. F-1066). The authors would like to acknowledge Dr Nanshu Lu and Siyi Liu in the Department of Aerospace Engineering and Engineering Mechanics at the University of Texas at Austin for their help with mechanical testing of polymer membranes. The authors thank Timothy Dowell in the Department of Chemistry at Mississippi State University for his help with preparing the graphical abstracts.References
- J.-Y. Hwang, S.-T. Myung and Y.-K. Sun, Chem. Soc. Rev., 2017, 46, 3529–3614 RSC.
- L. Long, S. Wang, M. Xiao and Y. Meng, J. Mater. Chem. A, 2016, 4, 10038–10069 RSC.
- L. Yue, J. Ma, J. Zhang, J. Zhao, S. Dong, Z. Liu, G. Cui and L. Chen, Energy Storage Materials, 2016, 5, 139–164 CrossRef.
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- X. Cheng, J. Pan, Y. Zhao, M. Liao and H. Peng, Adv. Energy Mater., 2018, 8, 1702184 CrossRef.
- Q. Lu, Y.-B. He, Q. Yu, B. Li, Y. V. Kaneti, Y. Yao, F. Kang and Q.-H. Yang, Adv. Mater., 2017, 29, 1604460 CrossRef PubMed.
- W. Li, J. Liu and D. Zhao, Nat. Rev. Mater., 2016, 1, 16023 CrossRef CAS.
- L. Tan and B. Tan, Chem. Soc. Rev., 2017, 46, 3322–3356 RSC.
- Y. Luo, B. Li, W. Wang, K. Wu and B. Tan, Adv. Mater., 2012, 24, 5703–5707 CrossRef CAS PubMed.
- S. Glenis, M. Benz, E. LeGoff, J. L. Schindler, C. R. Kannewurf and M. G. Kanatzidis, J. Am. Chem. Soc., 1993, 115, 12519–12525 CrossRef CAS.
- B. A. Trofimov, N. I. Golovanova, A. I. Mikhaleva, S. E. Korostova, A. N. Vasil'ev and L. N. Balabanova, Chem. Heterocycl. Compd., 1977, 13, 734–738 CrossRef.
- B. Li, F. Su, H.-K. Luo, L. Liang and B. Tan, Microporous Mesoporous Mater., 2011, 138, 207–214 CrossRef CAS.
- P. M. Budd, E. S. Elabas, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib, C. E. Tattershall and D. Wang, Adv. Mater., 2004, 16, 456–459 CrossRef CAS.
- P. Arora and Z. Zhang, Chem. Rev., 2004, 104, 4419–4462 CrossRef CAS PubMed.
- C. T. Love, J. Power Sources, 2011, 196, 2905–2912 CrossRef CAS.
- A. Plewa, F. Chyliński, M. Kalita, M. Bukat, P. Parzuchowski, R. Borkowska, M. Siekierski, G. Z. Żukowska and W. Wieczorek, J. Power Sources, 2006, 159, 431–437 CrossRef CAS.
- M. Morita, S. Aoki and Y. Matsuda, Electrochim. Acta, 1992, 37, 119–123 CrossRef CAS.
- K. M. Abraham, J. S. Foos and J. L. Goldman, J. Electrochem. Soc., 1984, 131, 2197–2199 CrossRef CAS.
- A. M. Stephan, T. Prem Kumar, N. Angulakshmi, P. S. Salini, R. Sabarinathan, A. Srinivasan and S. Thomas, J. Appl. Polym. Sci., 2011, 120, 2215–2221 CrossRef CAS.
- M. Kalita, M. Bukat, M. Ciosek, M. Siekierski, S. H. Chung, T. Rodríguez, S. G. Greenbaum, R. Kovarsky, D. Golodnitsky, E. Peled, D. Zane, B. Scrosati and W. Wieczorek, Electrochim. Acta, 2005, 50, 3942–3948 CrossRef CAS.
- B. Wu, J. Lochala, T. Taverne and J. Xiao, Nano Energy, 2017, 40, 34–41 CrossRef CAS.
- R. M. McConnell, W. E. Godwin, S. E. Baker, K. Powell, M. Baskett and A. Morara, Int. J. Polym. Mater. Polym. Biomater., 2004, 53, 697–708 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9se00690g |
| This journal is © The Royal Society of Chemistry 2020 |