
Self-assembled binary multichromophore dendrimers with enhanced electro-optic coefficients and alignment stability†
Huajun
Xu
 *b,
Jianpeng
Liu
a,
Jun
Liu
*c,
Canwen
Yu
a,
Zhaofen
Zhai
a,
Gangzhi
Qin
a and
Fenggang
Liu
*a
*b,
Jianpeng
Liu
a,
Jun
Liu
*c,
Canwen
Yu
a,
Zhaofen
Zhai
a,
Gangzhi
Qin
a and
Fenggang
Liu
*a
aSchool of Chemistry and Chemical Engineering, Guangzhou University, Guangzhou 510006, P. R. China. E-mail: liufg6@gzhu.edu.cn
bDepartment of Chemistry, University of Washington, Seattle, WA 98195, USA. E-mail: fromhjx@foxmail.com
cSichuan Key Laboratory of Imaging & Department of Chemistry, School of Preclinical Medicine, North Sichuan Medical College, Nanchong, 637000, P. R. China. E-mail: ustbliujun@iccas.ac.cn
First published on 9th September 2019
Abstract
Novel binary self-assembled electro-optic (EO) dendrimers were developed to maximize the acentric order of chromophores with large hyperpolarizabilities and strong dipole–dipole interactions. To achieve high EO coefficient, a multichromophore dendritic structure and supramolecular self-assembly strategy through π–π stacking of fluoroaromatics and aromatics were adopted to minimize dipole–dipole interaction of the chromophores at high loading density. In these dendrimers, three tetraene chromophores were substituted with an aromatic dendron (HD) or a fluoroaromatic dendron (FD) on the donor ends as the peripheral groups. The neat dendrimer films show excellent film-forming ability and a higher refractive index (n = 1.81 for FDSD and n = 1.86 for HDSD at 1310 nm) compared with guest–host EO polymers (n ≈ 1.6, 1310 nm). Higher n is beneficial for reducing the driving voltage for Mach–Zehnder modulators. By doping a secondary chromophore (HJD, 25 wt%) into neat dendrimers and blending two dendrimers together with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (wt), three kinds of binary EO materials were prepared to afford better EO performance and larger index of refraction without phase separation. Due to supramolecular self-assembly through FD–HD π–π interactions, the poled binary film of 1:1 HDSD:FDSD exhibited a very large EO coefficient value up to 289 pm V−1 together with a large refractive index (n = 2.01) at 1310 nm, which represents a very high n3r33 figure-of-merit of 2347 pm V−1 at 1310 nm. Meanwhile, a non-covalently crosslinked network was formed by the HD–FD π–π interactions which afforded an improved long-term alignment stability. After annealing at room temperature for 1000 h, about 97% of the initial r33 values could be still maintained for the poled films of 1:1 HDSD:FDSD. The high poling efficiency and temporal EO stability combined with refractive index make them promising candidates for application in photonic devices.
:1 ratio (wt), three kinds of binary EO materials were prepared to afford better EO performance and larger index of refraction without phase separation. Due to supramolecular self-assembly through FD–HD π–π interactions, the poled binary film of 1:1 HDSD:FDSD exhibited a very large EO coefficient value up to 289 pm V−1 together with a large refractive index (n = 2.01) at 1310 nm, which represents a very high n3r33 figure-of-merit of 2347 pm V−1 at 1310 nm. Meanwhile, a non-covalently crosslinked network was formed by the HD–FD π–π interactions which afforded an improved long-term alignment stability. After annealing at room temperature for 1000 h, about 97% of the initial r33 values could be still maintained for the poled films of 1:1 HDSD:FDSD. The high poling efficiency and temporal EO stability combined with refractive index make them promising candidates for application in photonic devices.
1. Introduction
Organic electro-optic (OEO) materials are of great interest with increasing demand in applications related to telecommunication, optical computing, sensing, tetrahertz (THz) technology, and other categories of devices for high-speed data/signal processing.1–8 The OEO materials show great advantages in terms of EO coefficient (r33) and intrinsic response time (tens of femtoseconds), which comes from π-electron oscillations of nonlinear optical (NLO) chromophores, making it much faster than those of inorganic EO materials.2,9–15 Rational molecular design can be used to satisfy specific requirements of the EO material such as solubility, refractive index, and stability. The OEO materials also show excellent processability, which facilitates the fabrication of EO devices and integration with microelectronics and photonic circuits.16,17As the key component of OEO materials, the highly hyperpolarizable chromophores are generally covalently or non-covalently incorporated into NLO inactive hosts to fabricate photonic devices.18–20 The chromophores possess a push–pull electronic structure in which the π-conjugated bridge is end-capped by an electron-donor and an electron-acceptor.21 Due to the intramolecular charge transfer effect, the chromophores usually show large dipole moments which induce strong aggregations in an EO inactive manner at high chromophore loading.14,22–24 The dipolar chromophores need to be aligned by electric poling field to convert microscopic hyperpolarizability (β) to macroscopic EO activity.25–27 To achieve a large r33 value, the OEO materials should be rationally designed to get a combination of large chromophore β, high number density of chromophores (ρN), and high acentric order parameter 〈cos3θ〉 throughout the active region of the device. As chromophore number density increases, the unwanted chromophore aggregations become more severe which leads to the diminishment of the poling efficiency and increasing possibility of material inhomogeneity.28,29
Several strategies of molecular engineering for dipolar chromophores have been proposed in the last two decades, such as binary-chromophore systems,9,30,31 the site-isolation strategy,24,32–36 self-assembled molecular glass and dendritic materials.12,13,37–40 In recent years, multichromophore dendrimers were introduced as an effective molecular engineering strategy to combine the high poling efficiency, good film-forming ability and large refractive index (n > 1.70) together. In multi-chromophore dendrimers, three or more chromophores are combined into a core by flexible chains and functionalized with bulky groups. The chromophores are well isolated with each other to inhibit detrimental dipole–dipole interactions. This arrangement allows each individual chromophore to reorient more independently when an external poling field was applied.41,42 The multichromophore dendrimers can be cast into thin films with high optical quality, or used as a chromophore-containing matrix which can afford an increased chromophore loading without phase-separation and the loss of poling efficiency. According to the prediction of calculation modeling, the spherical molecular shape is ideal to maximize chromophore acentric order. Due to rational molecular engineering for molecular size, shape, and bulky groups, the multi-chromophore dendrimer can be controlled with a high degree of precision to form a spherical structure and isolate the dipolar unit from its surroundings.13,41,43 Another notable strategy is the concept of self-assembly active interaction, such as fluoroaromatic–aromatic,12 liquid crystalline,44–46 hydrogen bonding,47,48 and binary chromophore interactions,30,42 to drive spontaneous or enhanced poling-induced acentric ordering. These peripheral dendrons can reversibly self-assemble to build an extended supramolecular structure through active interactions.
Introducing fluoroaromatic–aromatic interactions through asymmetric synthesis methods has been proven effective for improving both the poling efficiency and long-term alignment stability in poled EO films.49 When the initially random binary material was heated to poling temperature, the fluoroaromatic–aromatic interactions can be dissociated by the poling field. After cooling down to room temperature, the fluoroaromatic–aromatic groups reassociated to improve acentric chromophore order and alignment stability. In this paper, we proposed two multi-chromophore dendrimers, HDSD and FDSD, and a tetraene chromophore HJD, as shown in Fig. 1. FDSD was reported in our previous work.42 In a dendrimer molecule, three tetraene chromophores are linked to the central core by covalent bonds, forming globular multi-chromophore dendrimers. Such attaching manner allows each individual chromophore to rotate more independently under the electric poling field. Additionally, the multi-chromophore structure can afford high chromophore loading and enhancement of refractive index without phase separation. HD and FD groups were introduced onto the donor end of the chromophores. As the bulky groups, HD and FD can afford large steric hindrance to isolate chromophore units and reversibly self-assemble to build an extended supramolecular structure through π–π interactions (Fig. 2). After electric field poling, the binary material (1:1 HDSD:FDSD, by weight) will form a non-covalently crosslinked network which contributes to locking the acentric order of chromophores. By doping a secondary chromophore HJD (25 wt%) into neat dendrimers and blending two dendrimers together with 1:1 ratio, three binary EO materials were prepared to afford higher chromophore loading better EO performance and larger index of refraction together with excellent film-forming ability. For EO activity, the poled 3:1 HDSD:HJD and 1:1 HDSD:FDSD films exhibited large r33 values up to 215 pm V−1 and 289 pm V−1, respectively. 1:1 HDSD:FDSD also showed a higher refractive index compared with other neat or binary materials. After 1000 hours at ambient temperature, ∼97% initial EO activity of 1:1 HDSD:FDSD can be maintained.
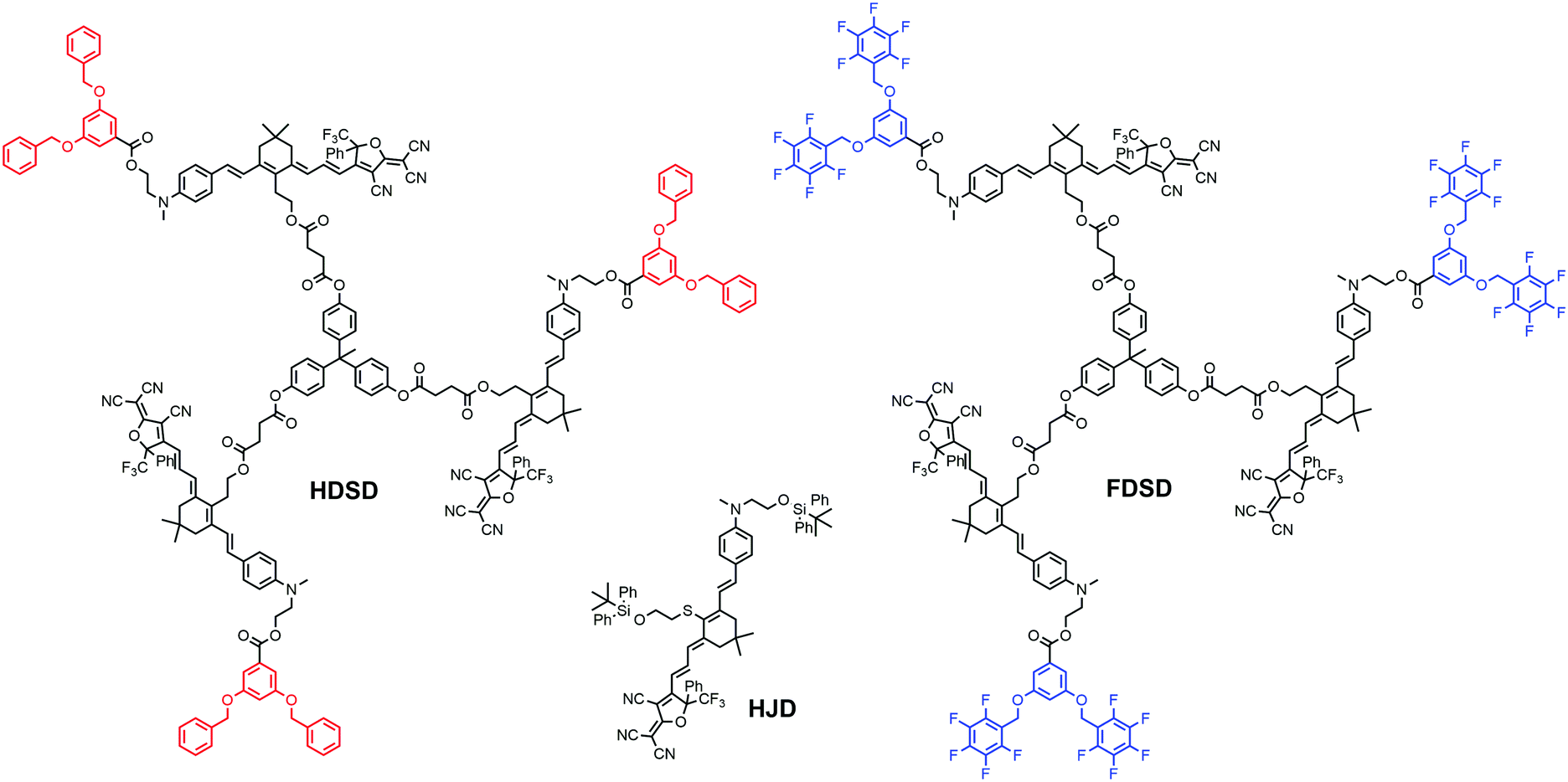 | ||
| Fig. 1 Chemical structure for chromophores HDSD, FDSD and HJD. | ||
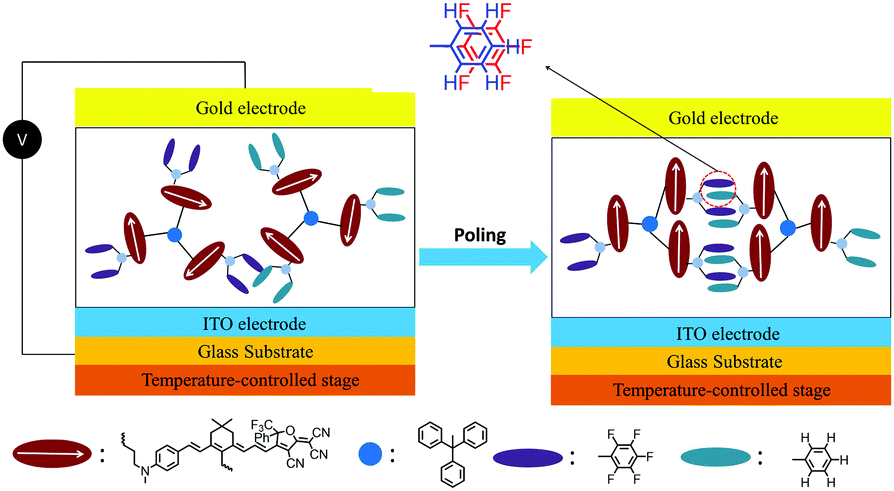 | ||
| Fig. 2 Graphical illustration of poling and alignment formation of self-assembled chromophores HDSD–FDSD by Ar–ArF interactions. | ||
2. Results and discussion
2.1 Synthesis and characterization of the chromophore
The synthesis of chromophore HDSD is depicted in Fig. 3. Starting from the aniline donor intermediate compounds 1a, chromophore HDSD was synthesized in good overall yield through the following reactions: in the presence of sodium ethoxide as the base, tetrahydropyranyl (THP) ether protected isophorone can be reacted directly with compound 1a in one-pot via Knoevenagel condensation to furnish compound 2 with a high overall yield. The free hydroxyl group on the aniline donor was protected by the bulky tert-butyldiphenylsilyl (TBDPS) group to afford compound 3. Compound 3 was then reacted with diethyl(cyanomethyl)phosphonate using the Wittig–Horner reaction to result in trienenitrile 4. The reduction with DIBAL-H followed by hydrolysis converted the nitrile group on 4 into the corresponding aldehyde 5. The TBDPS group on the donor was then removed under TBAF deprotection conditions. Aldehyde 6 was then reacted with 3,5-bis(benzyloxy)benzoic acid to introduce an aromatic dendron to it. Then the THP group on the bridge was then removed under mildly acidic conditions, followed by esterification to attach the 4-carbon diester linker producing compound 9.50,51 Then 3 equiv of the resulting compound 9 was then reacted with triphenol to furnish dendrimer 10. The aldehyde-containing donor-bridge 10 was condensed with CF3-Ph-TCF acceptor to furnish the chromophore HDSD as dark green solids. Synthetic material identity and purity was confirmed by 1H NMR, 13C NMR, mass spectrometry and HRMS. These chromophores possess good solubility in common organic solvents, such as acetone, chloromethane and dichloromethane.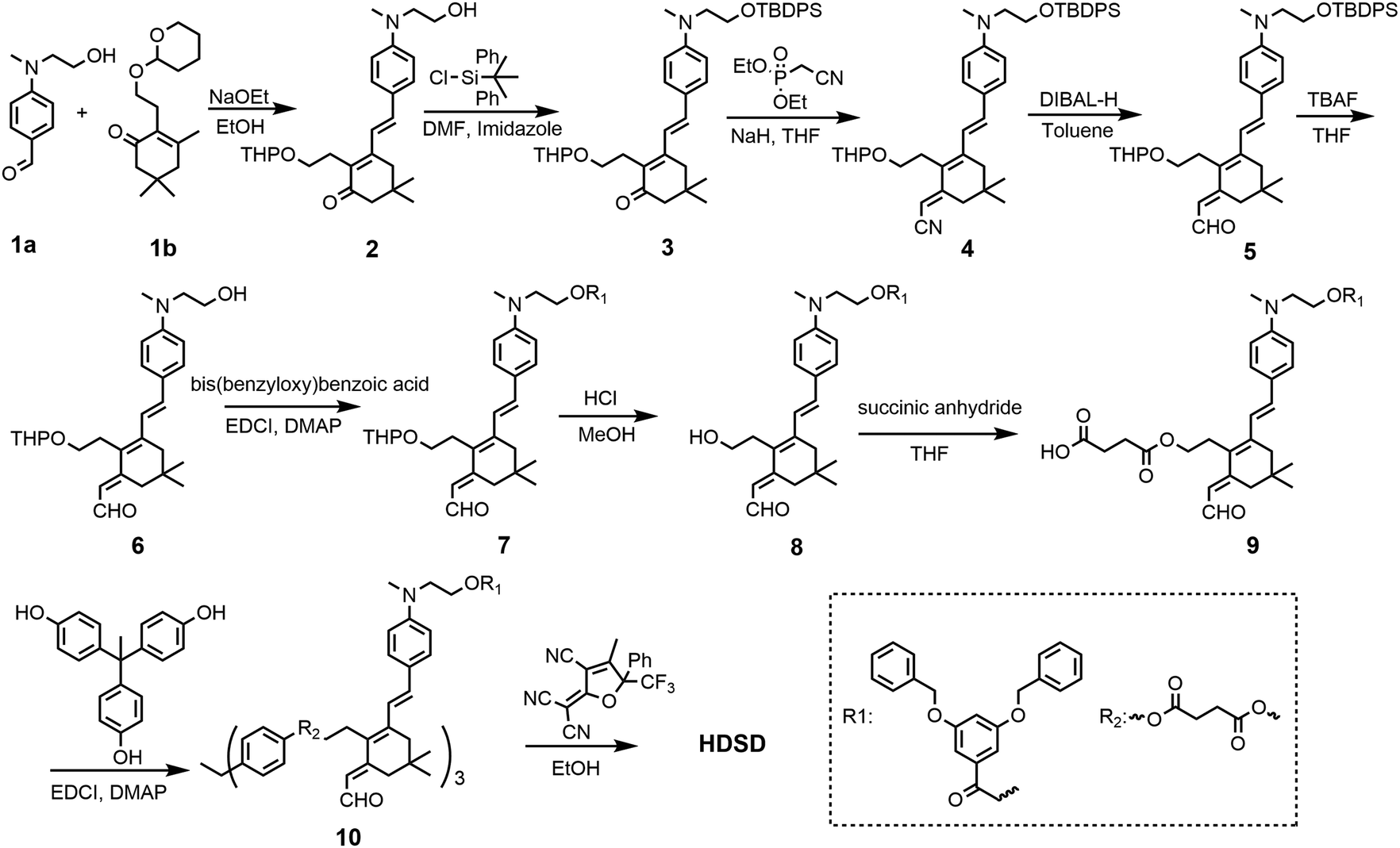 | ||
| Fig. 3 Synthetic routes for chromophore HDSD. | ||
2.2 Optical properties
UV-Vis absorption spectra were measured both in solution and film, as shown in Fig. 4, Fig. 5 and Table 1. The two dendrimers in chloroform solution exhibited similar charge-transfer bands, with a slight red shift in the maximum absorption wavelength (λmax) for HDSD (λmax = 751 nm) compared with FDSD (λmax = 735 nm). This was partially attributed to the electron-withdrawing effect of pentafluorinated aromatic rings by quadrupolar interactions with electron-rich parts of the chromophore π-system.52 The λmax of chromophores generally show redshifts with the increase of the solvent dielectric constant before reaching the cyanine limit. The two dendrimers exhibited similar redshifts (49 nm for HDSD and 43 nm for FDSD) when the solvents changed from 1,4-dioxane to chloroform. The absorption bandwidths of HDSD, FDSD and HJD in chloroform, defined as the full-width at half-maximum (FWHM), were 240 nm, 283 nm and 246 nm, respectively. In comparison with the absorption in solution, the thin films showed significant broadening of the FWHM bandwidth of 366 nm, 341 nm and 368 nm for HDSD, FDSD and HJD, respectively. These changes in the absorption bands when changing from solution to solid have been widely observed in NLO chromophore-doped guest–host materials, neat EO film materials and self-assembled dendrimers.52,53 This phenomenon may be attributed to the change of dielectric surrounding and solvatochromism of the electronic absorption characteristics associated with dye–dye and dye–polymer interactions.54 Compared with HDSD film, the blueshift of λmax for FDSD film was attributed to the easier formation of quadrupolar interactions between pentafluorinated aromatic rings and electron-rich parts of the chromophore π-system in the solid state. Meanwhile, the UV spectra of three binary materials, 3:1 HDSD:HJD, 3:1 FDSD:HJD and 1:1 HDSD:FDSD, were also tested as shown in Fig. 5. The binary material films exhibited redshifts of λmax compared with one component neat dendrimer films. The film of 1:1 HDSD:FDSD shows broad FWHM bandwidth o f 380 nm, and a large redshift to longer wavelengths of 812 nm, which might be due to the π–π interactions between HD and FD groups and their different nanoscale environment of dipole–dipole interactions.55
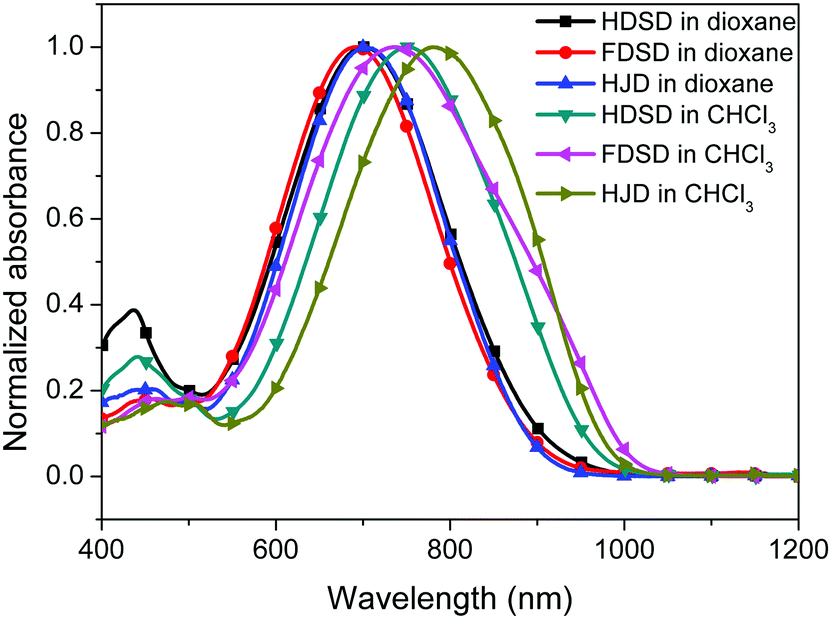 | ||
| Fig. 4 UV-Vis absorption spectra of chromophores HDSD, FDSD and HJD in chloroform and dioxane. | ||
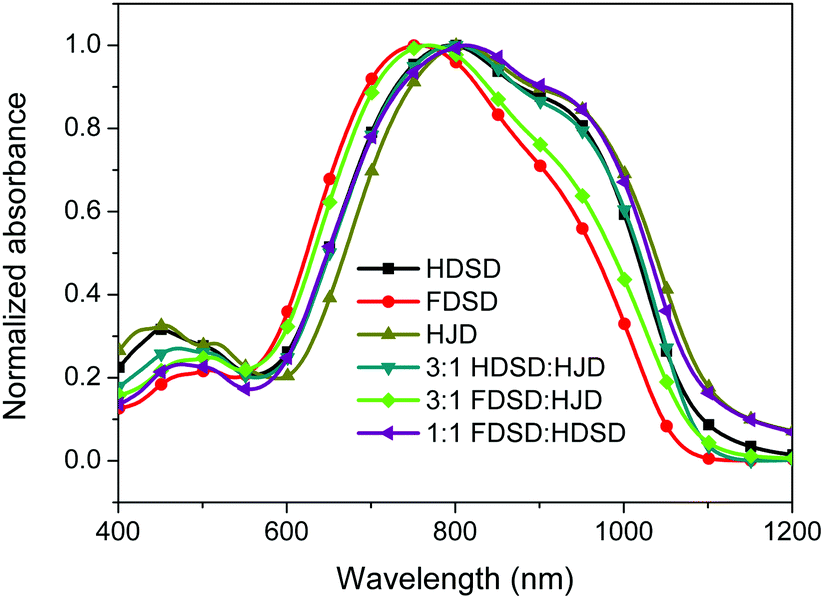 | ||
| Fig. 5 UV-Vis absorption spectra of chromophores HDSD, FDSD, HJD and their blends in film. | ||
2.3 Thermal property and VASE test
The glass transition temperature (Tg) of the neat and binary materials was measured by differential scanning calorimetry (DSC) with a heating rate of 10 °C min−1 under the protection of nitrogen as shown in Fig. S1 (ESI†) and Table 2. The neat HDSD exhibited a higher Tg (91.6 °C) than FDSD (81.1 °C) though the latter had a larger molecular weight. After blending with the chromophore HJD, the binary materials showed slightly decreased Tgs due to the relatively low Tg for HJD (64.6 °C). The thermal stability of the chromophores was also investigated using thermogravimetric analysis (TGA) with a heating rate of 10 °C min−1 under the protection of nitrogen as shown in Fig. S2 (ESI†). All the chromophores exhibited good thermal stabilities with the decomposition temperatures (Td) higher than 210 °C. All the thermal stabilities of these chromophores were high enough for application in EO device preparation.56| Chromophore | T g (°C) | λ max (nm) | n1c | n2d | T (°C) | ρ N-ave (×1020 cm−3) | r 33/Epg (nm2 V−2) | Max r33 (pm V−1) | Temp. stabh (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Glass transition temperature. b Neat films. c Index of refraction (n) tested at 1310 nm. d Index of refraction (n) tested at 1550 nm. e Poling temperature. f Number density (assumes mass density of 1 g cm−3). g Poling efficiency ± standard error. h Temporal alignment stability at room temperature after 1000 h. | |||||||||
| HDSD | 91.6 | 795 | 1.86 | 1.80 | 95 | 5.16 | 1.71 ± 0.09 | 179 | 83 |
| FDSD | 81.1 | 758 | 1.81 | 1.77 | 90 | 4.47 | 1.60 ± 0.08 | 147 | 72 |
| 3:1 HDSD:HJD |
85.5 | 803 | 1.87 | 1.81 | 90 | 5.15 | 2.13 ± 0.11 | 215 | 77 |
| 3:1 FDSD:HJD |
74.1 | 769 | 1.88 | 1.82 | 80 | 4.64 | 1.93 ± 0.14 | 203 | 65 |
| 1:1 HDSD:FDSD |
88.5 | 812 | 2.01 | 1.93 | 99 | 4.82 | 2.68 ± 0.13 | 289 | 97 |
The refractive index, n, of neat and binary films was measured using variable angle spectroscopic ellipsometry (VASE) as shown in Fig. 6 and Table 2.53 For Mach–Zehnder modulators, the high refractive index of EO materials at 1310 nm and 1550 nm (two important telecom wavelengths) is beneficial for reducing the operating voltage (Vπ) which is inversely proportional to the value of n3r33. The n values of neat and binary materials range from 1.81–2.01 at 1310 nm and 1.77–1.93 at 1550 nm, which are significantly higher than traditional guest–host polymeric materials (n ≈ 1.60).57 The refractive index of OEO materials has positive correlation with the number of chromophores per material unit volume. The neat HDSD shows a higher refractive index than FDSD due to the difference of ρN between two neat dendrimers. Meanwhile, introducing substituents with high molar refractions and reduced molar volumes can efficiently increase the refractive index. The binary material 1:1 HDSD:FDSD exhibits the largest n value (n = 2.01) though it has relatively low number of chromophores per material unit volume. This should be ascribed to the reduced molar volume of 1:1 HDSD:FDSD by the π–π interactions between HD and FD groups. More details for the optical constants of the single-component chromophores and chromophore blends are in Fig. 6 and Table 2.
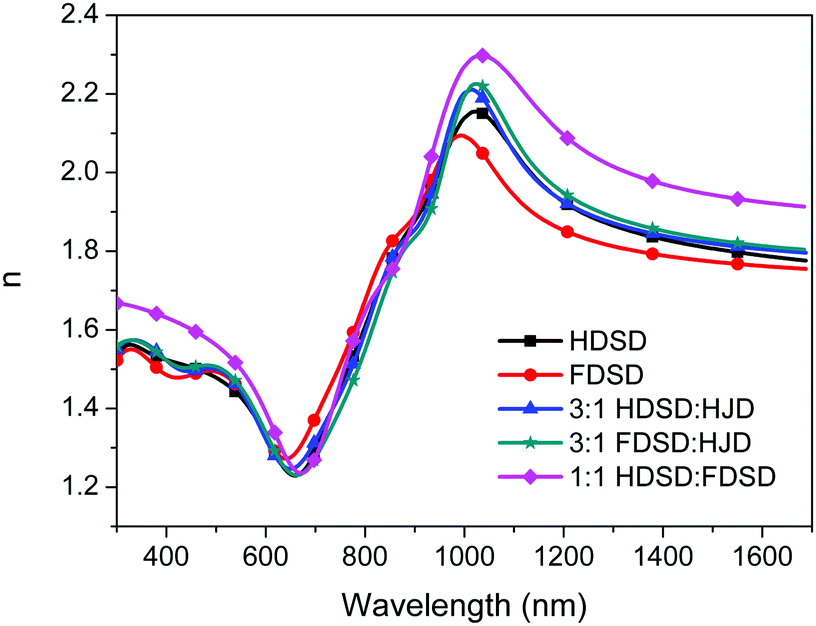 | ||
| Fig. 6 Index of refraction (n) of chromophores HDSD, FDSD, HJD and their blends. | ||
2.4 Electro-optic performance
Unlike traditional chromophores, which need to be doped in the EO inactive polymers to spin-coat films, the larger molecular weights (MW) and inherent amorphous nature of the multichromophore dendrimers HDSD and FDSD have allowed them to be cast into neat thin films with high optical quality, and used as an EO matrix. To evaluate the EO properties of molecular glassy films, the trichloroethane (TCE) solutions of HDSD, FDSD, HJD and their mixture (as shown in Table 2) at a concentration of about 9–11 wt% were filtered through a 0.2 mm PTFE-syringe filter and spin-coated onto indium tin oxide (ITO)/glass substrates to form neat thin films. The films were baked in a vacuum oven at 65 °C overnight to ensure the removal of any residual solvent. Then, a thin layer of gold (∼60 nm thick) was deposited onto the films by argon sputter coating as a top electrode for contact poling. The thickness of the EO film was measured to be around 1.2–1.8 μm. The barrier layer of benzocyclobutene (BCB) was fabricated from commercially available BCB polymer (Cyclotene 3022-46, from Dow Corporation). BCB solution was diluted to 20% w/w in mesitylene and filtered using a 0.45 μm PTFE filter and then applied directly to a clean ITO/glass substrate and spin cast at 4000 rpm for 60 seconds. The resulting films were dried on a hot plate at 120 °C for 15 minutes to remove residual solvent. The thickness of the BCB layer ranged from 60 to 100 nm. The EO activity (r33 values) of poled films was measured using the Teng–Man simple-reflection technique at a wavelength of 1310 nm using a carefully selected thin ITO electrode with low reflectivity and good transparency in order to minimize the measurement errors.58To achieve high EO coefficients, a large DC electric field was applied to induce acentric molecular order by heating the initially isotropic material to the poling temperature. At this temperature, the chromophores got enough thermal energy to rotate within the free volume created by the dendritic structure and peripheral group. Meanwhile, a large leakage current was observed which resulted from the low resistance of neat and binary EO materials at poling temperature, leading to a dramatic decrease in the poling field within the EO film. When using high poling field (>50 V μm−1), crosslinked benzocyclobutene (BCB) polymer was applied to serve as a charge barrier layer to inhibit leakage current during poling. Excessive voltage will cause damage to the gold electrode, so no more than 100 V voltage was applied. We totally poled and measured the r33 values of five kinds of neat and binary films. The measured r33 values and poling efficiencies (r33/Ep), defined as the contribution of the poling field to the EO coefficient, for neat and binary materials are summarized in Table 2, Fig. 7 and our previous work (FDSD).42 The average poling efficiencies of film HDSD and film FDSD were 1.71 ± 0.09 nm2 V−2 and 1.60 ± 0.08 nm2 V−2, respectively (Fig. 7), higher than the guest–host polymeric materials with similar chromophores (25 wt% YLD124 in PMMA, the poling efficiency is about 1.0 nm2 V−2).57 This proved that a dendritic structure can effectively prevent the dipole–dipole induced anti-parallel aggregations at high chromophore number density. In multichromophore dendrimers, the chromophore units are attached to a core by several covalent bonds. The chromophore units can freely rotate within the void created by the outer periphery and significantly be isolated from each other by the dendrimer core. The difference in poling efficiencies of HDSD and FDSD are mainly ascribed to the number of chromophores per material unit volume. The introduction of an HD group may be a better strategy in improving Tg, refractive index and poling efficiency compared with an FD group. It has been observed that the binary materials have an enhanced EO performance prepared by doping individual constituents to neat EO materials. Due to the larger molecular weights and inherent amorphous nature, HDSD and FDSD can serve as chromophore-containing hosts to prepare homogeneous binary films with high quality. According to previous reports,41 the EO coefficient of binary materials will be greatly enhanced by the increasing total concentrations of chromophores, and supermolecular interactions between the guest chromophore and host dendrimer. Here, the free chromophore HJD was chosen as the doping chromophore for the reason of good capability and large molecular hyperpolarizability. The average poling efficiencies of 3:1 HDSD:HJD and 3:1 FDSD:HJD were 2.13 ± 0.11 nm2 V−2 and 1.93 ± 0.14 nm2 V−2, respectively, higher than neat dendrimers. These enhancements of poling efficiency and the best r33 value for 3:1 HDSD:HJD are mainly resulted from the binary blend.59 The refractive index of 3:1 FDSD:HJD shows larger enhancement compared with that of 3:1 HDSD:HJD. The best r33 value for poled 3:1 HDSD:HJD is 215 pm V−1 which was 20% improvement compared with that of neat HDSD.
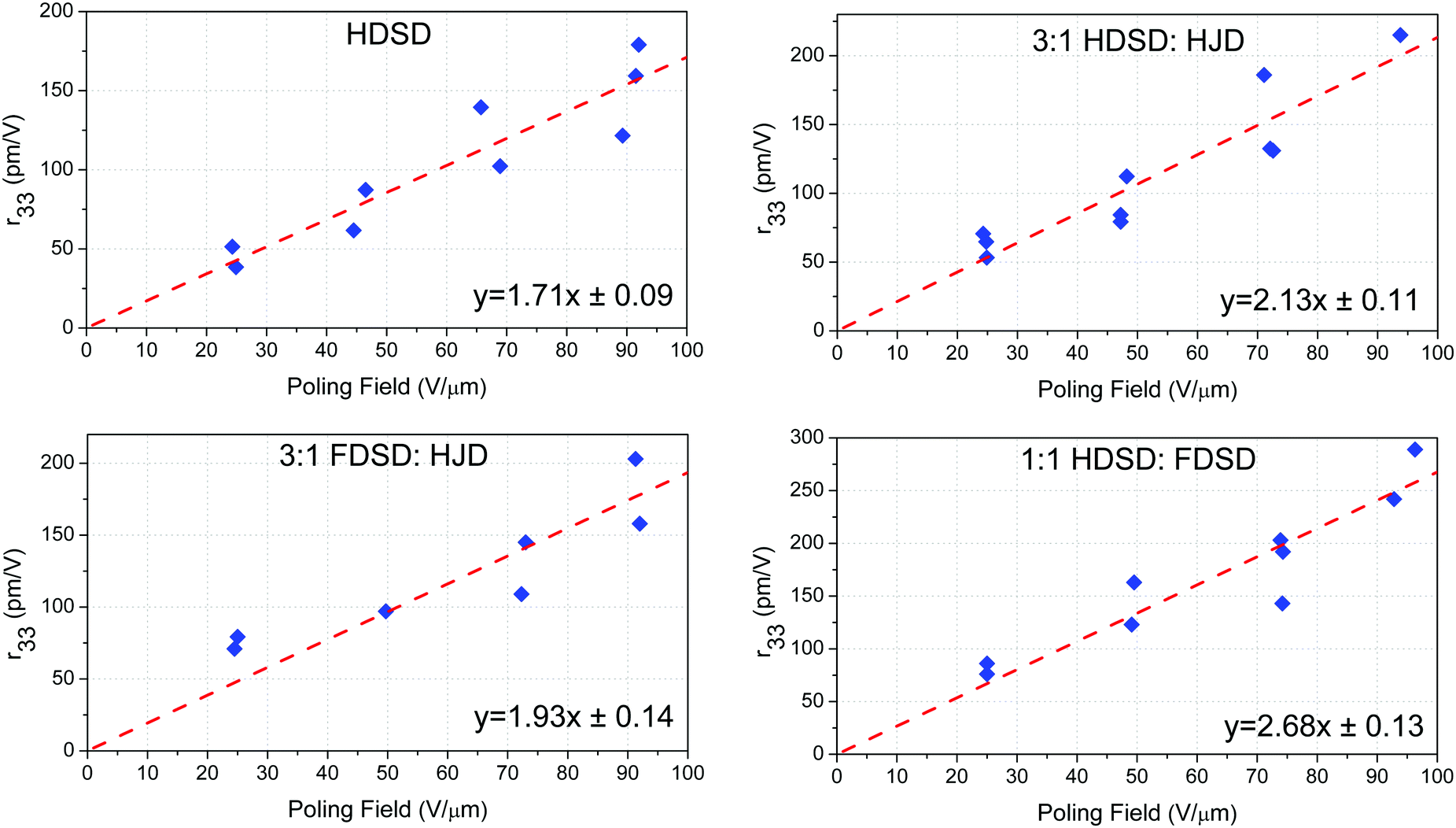 | ||
| Fig. 7 Poling curves of the EO film. Average r33/Ep ± standard errors are shown. | ||
To evaluate the self-assembly effect of HD–FD on EO performance, the basic devices of 1:1 HDSD:FDSD were fabricated and poled to calculate poling efficiency. The average poling efficiency of film HDSD–FDSD (1:1) was 2.68 ± 0.13 nm2 V−2, which is much higher than other neat and binary materials though with relatively low total chromophore number density compared with neat HDSD. The introduction of asymmetric interchromophore interactions by the electronegative HD groups and electropositive FD groups can help to obtain highly poling-induced order. The intermolecular interactions can be dissociated by the poling field when the initially random binary material was heated to poling temperature. Then the films were cooled with continued application of the poling field. As the films cooled to room temperature, the HD and FD groups reassociated to form a non-covalently crosslinking network which can enhance and stabilize the acentric chromophore order. One of the noteworthy results for 1:1 HDSD:FDSD is the high refractive index (n = 2.01) which can help to effectively reduce the energy consumption of the EO modulator. The best r33 value of 289 pm V−1 was achieved by 1:1 HDSD:FDSD at 94 V μm−1 poling field, which represents a very high n3r33 figure-of-merit of 2347 pm V−1 at 1310 nm.
In addition, good temporal stability can be achieved simultaneously with this self-assembled binary material. The poled film of 1:1 HDSD:FDSD showed very promising long-term alignment stability, about 97% of the initial r33 value could be still kept at ambient temperature for over 1000 h after an initial fast decay. While only 72% of the initial r33 values could be still kept for FDSD (Tg = 81.1 °C). This demonstrates that the self-assembled non-covalent crosslinking network plays an important role in improving both the poling efficiency and thermal stability in poled dendronized chromophore films. The self-assembled binary multichromophore dendrimers can improve both the EO activities and good temporal stability of the EO materials.
3. Conclusions
In conclusion, self-assembled multichromophore dendrimers HDSD and FDSD containing supramolecular self-assembly through HD–FD π–π interactions were successfully synthesized and demonstrated. The chromophore units are covalently attached to the core and functionalized with peripheral groups to form large branching structures. The chromophores are attached in such a manner that it can ensure that the chromophores can freely rotate and significantly decouple with other chromophore units. The inherent amorphous nature and large molecular weights of the resultant dendrimers mean that they can be cast into thin films with high optical quality, or used as a chromophore-containing matrix. The poled neat films exhibited enhanced best r33 values, poling efficiencies and refractive index compared with the guest–host polymeric material. Meanwhile, three binary materials were prepared by doping the secondary chromophore HJD into dendrimers and blending two dendrimers together. The enhanced EO performance was observed in poled films of binary materials. Due to the self-assembly effect of HD–FD active interactions, the poled films of 1:1 HDSD:FDSD exhibited large EO coefficients of 289 pm V−1 and refractive index (n = 2.01), which represents a very high n3r33 figure-of-merit of 2347 pm V−1 at 1310 nm. In the meantime, the long-term alignment stability of 1:1 HDSD:FDSD is greatly improved due to the non-covalent crosslinking network. After being annealed at room temperature for 1000 h, about 97% of the initial r33 values can be maintained for the poled film of 1:1 HDSD:FDSD. The ultrahigh electro-optic activity and high long-term alignment stability of these materials made them promising candidates for practical device exploration for photonic applications.
4. Experimental section
4.1 Materials and instruments
All chemicals are commercially available and are used without further purification unless otherwise stated. N,N-Dimethylformamide (DMF), dichloromethane and tetrahydrofuran (THF) were commercially available ultra-dry reagents. TLC analyses were carried out on 0.25 mm thick precoated silica plates and spots were visualized under UV light. Chromatography on silica gel was carried out on Kieselgel (200–300 mesh). Chromophores FDSD and HJD was synthesized referring to previous literature,42,60,61 respectively. Detailed synthesis procedures, spectrometric characterization, and official IUPAC names of compounds reported can be found in the accompanying Supporting Information.1H NMR and 13C NMR spectra were determined on an Advance Bruker 300M or 500 MHz NMR spectrometer (tetramethylsilane as internal reference). The MS spectra were obtained by MALDI-TOF (Matrix Assisted Laser Desorption/Ionization of Flight) on a BIFLEXIII (Broker Inc.,) spectrometer. The UV-Vis spectra were acquired on a Cary 5000 photo spectrometer. The Tg was determined by TA DSC Q10 with a heating rate of 10 °C min−1 under the protection of nitrogen. The TGA was determined by TA5000-2950TGA (TA co) with a heating rate of 10 °C min−1 under the protection of nitrogen.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Delwin L. Elder (University of Washington) for his help on refractive index measurement. We are grateful to the National Natural Science Foundation of China (No. 21805049) and the Science and Technology Program of Guangzhou, China (No. 201904010176) for the financial support.Notes and references
- M. Ayata, Y. Fedoryshyn, W. Heni, B. Baeuerle, A. Josten, M. Zahner, U. Koch, Y. Salamin, C. Hoessbacher, C. Haffner, D. L. Elder, L. R. Dalton and J. Leuthold, Science, 2017, 358, 630–632 CrossRef CAS.
- L. R. Dalton, P. A. Sullivan and D. H. Bale, Chem. Rev., 2010, 110, 25–55 CrossRef CAS.
- R. Tang, S. Zhou, Z. Cheng, G. Yu, Q. Peng, H. Zeng, G. Guo, Q. Li and Z. Li, Chem. Sci., 2017, 8, 340–347 RSC.
- S. R. Marder and J. W. Ferry, Science, 1994, 263, 1706–1707 CrossRef CAS.
- S. R. Marder, B. Kippelen, A. K.-Y. Jen and N. Peyghambarian, Nature, 1997, 388, 845–851 CrossRef CAS.
- L. R. Dalton, W. H. Steier, B. H. Robinson, C. Zhang, A. Ren, S. Garner, A. T. Chen, T. Londergan, L. Irwin, B. Carlson, L. Fifield, G. Phelan, C. Kincaid, J. Amend and A. K.-Y. Jen, J. Mater. Chem., 1999, 9, 1905–1920 RSC.
- L. Alloatti, R. Palmer, S. Diebold, K. P. Pahl, B. Chen, R. Dinu, M. Fournier, J.-M. Fedeli, T. Zwick and W. Freude, Light: Sci. Appl., 2014, 3, e173 CrossRef CAS.
- R. Palmer, S. Koeber, D. L. Elder, M. Woessner, W. Heni, D. Korn, M. Lauermann, W. Bogaerts, L. Dalton and W. Freude, J. Lightwave Technol., 2014, 32, 2726–2734 CAS.
- T. D. Kim, J. D. Luo, J. W. Ka, S. Hau, Y. Q. Tian, Z. W. Shi, N. M. Tucker, S. H. Jang, J. W. Kang and A. K.-Y. Jen, Adv. Mater., 2006, 18, 3038–3042 CrossRef CAS.
- J. Luo, Y.-J. Cheng, T.-D. Kim, S. Hau, S.-H. Jang, Z. Shi, X.-H. Zhou and A. K.-Y. Jen, Org. Lett., 2006, 8, 1387–1390 CrossRef CAS.
- Y. J. Cheng, J. D. Luo, S. Hau, D. H. Bale, T. D. Kim, Z. W. Shi, D. B. Lao, N. M. Tucker, Y. Q. Tian, L. R. Dalton, P. J. Reid and A. K.-Y. Jen, Chem. Mater., 2007, 19, 1154–1163 CrossRef CAS.
- T. D. Kim, J. W. Kang, J. D. Luo, S. H. Jang, J. W. Ka, N. Tucker, J. B. Benedict, L. R. Dalton, T. Gray, R. M. Overney, D. H. Park, W. N. Herman and A. K.-Y. Jen, J. Am. Chem. Soc., 2007, 129, 488–489 CrossRef CAS.
- P. A. Sullivan, H. Rommel, Y. Liao, B. C. Olbricht, A. J. Akelaitis, K. A. Firestone, J.-W. Kang, J. Luo, J. A. Davies and D. H. Choi, J. Am. Chem. Soc., 2007, 129, 7523–7530 CrossRef CAS.
- J. Wu, S. Bo, J. Liu, T. Zhou, H. Xiao, L. Qiu, Z. Zhen and X. Liu, Chem. Commun., 2012, 48, 9637–9639 RSC.
- W. Jin, P. V. Johnston, D. L. Elder, A. F. Tillack, B. C. Olbricht, J. Song, P. J. Reid, R. Xu, B. H. Robinson and L. R. Dalton, Appl. Phys. Lett., 2014, 104, 243304 CrossRef.
- W. Heni, Y. Kutuvantavida, C. Haffner, H. Zwickel, C. Kieninger, S. Wolf, M. Lauermann, Y. Fedoryshyn, A. F. Tillack, L. E. Johnson, D. L. Elder, B. H. Robinson, W. Freude, C. Koos, J. Leuthold and L. R. Dalton, ACS Photonics, 2017, 4, 1576–1590 CrossRef CAS.
- C. Wang, M. Zhang, X. Chen, M. Bertrand, A. Shams-Ansari, S. Chandrasekhar, P. Winzer and M. Lončar, Nature, 2018, 562, 101–104 CrossRef CAS.
- F. Meyers, S. R. Marder, B. M. Pierce and J. L. Bredas, J. Am. Chem. Soc., 1994, 116, 10703–10714 CrossRef CAS.
- M. Q. He, T. M. Leslie and J. A. Sinicropi, Chem. Mater., 2002, 14, 4662–4668 CrossRef CAS.
- C. Zhang, L. R. Dalton, M. C. Oh, H. Zhang and W. H. Steier, Chem. Mater., 2001, 13, 3043–3050 CrossRef CAS.
- S. R. Marder, C. B. Gorman, B. G. Tiemann, J. W. Perry, G. Bourhill and K. Mansour, Science, 1993, 261, 186–189 CrossRef CAS.
- H. Ma, B. Chen, T. Sassa, L. R. Dalton and A. K.-Y. Jen, J. Am. Chem. Soc., 2001, 123, 986–987 CrossRef CAS.
- L. R. Dalton, J. Phys.: Condens. Matter, 2003, 15, R897 CrossRef CAS.
- Z. a. Li, Z. Li, C. a. Di, Z. Zhu, Q. Li, Q. Zeng, K. Zhang, Y. Liu, C. Ye and J. Qin, Macromolecules, 2006, 39, 6951–6961 CrossRef CAS.
- B. Robinson and L. Dalton, J. Phys. Chem. A, 2000, 104, 4785–4795 CrossRef CAS.
- C. Zhang, C. Wang, J. Yang, L. R. Dalton, G. Sun, H. Zhang and W. H. Steier, Macromolecules, 2001, 34, 235–243 CrossRef CAS.
- B. C. Olbricht, P. A. Sullivan, G.-A. Wen, A. A. Mistry, J. A. Davies, T. R. Ewy, B. E. Eichinger, B. H. Robinson, P. J. Reid and L. R. Dalton, J. Phys. Chem. C, 2008, 112, 7983–7988 CrossRef CAS.
- H. Xu, N. Wang, X. Zhang, Z. Li and G. Deng, Dyes Pigm., 2018, 157, 230–237 CrossRef CAS.
- T. Baehr-Jones, B. Penkov, J. Q. Huang, P. Sullivan, J. Davies, J. Takayesu, J. D. Luo, T. D. Kim, L. Dalton, A. Jen, M. Hochberg and A. Scherer, Appl. Phys. Lett., 2008, 92, 3 CrossRef.
- T.-D. Kim, J. Luo, Y.-J. Cheng, Z. Shi, S. Hau, S.-H. Jang, X.-H. Zhou, Y. Tian, B. Polishak, S. Huang, H. Ma, L. R. Dalton and A. K. Y. Jen, J. Phys. Chem. C, 2008, 112, 8091–8098 CrossRef CAS.
- M. Li, S. Huang, X. H. Zhou, Y. Zang, J. Y. Wu, Z. C. Cui, J. D. Luo and A. K.-Y. Jen, J. Mater. Chem. C, 2015, 3, 6737–6744 RSC.
- Z. a. Li, G. Yu, P. Hu, C. Ye, Y. Liu, J. Qin and Z. Li, Macromolecules, 2009, 42, 1589–1596 CrossRef CAS.
- Z. Zhu, Z. a. Li, Y. Tan, Z. Li, Q. Li, Q. Zeng, C. Ye and J. Qin, Polymer, 2006, 47, 7881–7888 CrossRef CAS.
- Z. a. Li, W. Wu, Q. Li, G. Yu, L. Xiao, Y. Liu, C. Ye, J. Qin and Z. Li, Angew. Chem., Int. Ed., 2010, 49, 2763–2767 CrossRef CAS.
- W. Wu, C. Li, G. Yu, Y. Liu, C. Ye, J. Qin and Z. Li, Chem. – Eur. J., 2012, 18, 11019–11028 CrossRef CAS.
- W. Wu, J. Qin and Z. Li, Polymer, 2013, 54, 4351–4382 CrossRef CAS.
- H. Kang, G. Evmenenko, P. Dutta, K. Clays, K. Song and T. J. Marks, J. Am. Chem. Soc., 2006, 128, 6194–6205 CrossRef CAS PubMed.
- P. A. Sullivan, A. J. Akelaitis, S. K. Lee, G. McGrew, S. K. Lee, D. H. Choi and L. R. Dalton, Chem. Mater., 2006, 18, 344–351 CrossRef CAS.
- M. J. Cho, D. H. Choi, P. A. Sullivan, A. J. P. Akelaitis and L. R. Dalton, Prog. Polym. Sci., 2008, 33, 1013–1058 CrossRef CAS.
- H. Xu, M. Fu, S. Bo and X. Liu, RSC Adv., 2016, 6, 25023–25027 RSC.
- Y. V. Pereverzev, K. N. Gunnerson, O. V. Prezhdo, P. A. Sullivan, Y. Liao, B. C. Olbricht, A. J. P. Akelaitis, A. K.-Y. Jen and L. R. Dalton, J. Phys. Chem. C, 2008, 112, 4355–4363 CrossRef CAS.
- H. Xu, D. L. Elder, L. E. Johnson, B. H. Robinson and L. R. Dalton, ACS Appl. Mater. Interfaces, 2019, 11, 21058–21068 CrossRef CAS.
- W. Wu, C. Wang, Q. Li, C. Ye, J. Qin and Z. Li, Sci. Rep., 2014, 4, 6101 CrossRef CAS.
- S. J. Benight, D. H. Bale, B. C. Olbricht and L. R. Dalton, J. Mater. Chem., 2009, 19, 7466–7475 RSC.
- S. J. Benight, L. E. Johnson, R. Barnes, B. C. Olbricht, D. H. Bale, P. J. Reid, B. E. Eichinger, L. R. Dalton, P. A. Sullivan and B. H. Robinson, J. Phys. Chem. B, 2010, 114, 11949–11956 CrossRef CAS.
- S. J. Benight, D. B. Knorr Jr, L. E. Johnson, P. A. Sullivan, D. Lao, J. Sun, L. S. Kocherlakota, A. Elangovan, B. H. Robinson and R. M. Overney, Adv. Mater., 2012, 24, 3263–3268 CrossRef CAS.
- A. Facchetti, E. Annoni, L. Beverina, M. Morone, P. W. Zhu, T. J. Marks and G. A. Pagani, Nat. Mater., 2004, 3, 910–917 CrossRef CAS.
- O.-P. Kwon, M. Jazbinsek, H. Yun, J.-I. Seo, J.-Y. Seo, S.-J. Kwon, Y. S. Lee and P. Günter, CrystEngComm, 2009, 11, 1541–1544 RSC.
- P. A. Sullivan, A. J. P. Akelaitis, S. K. Lee, G. McGrew, S. K. Lee, D. H. Choi and L. R. Dalton, Chem. Mater., 2006, 18, 344–351 CrossRef CAS.
- M. Kotke and P. R. Schreiner, Synthesis, 2007, 779–790 CAS.
- M. T. Barros, C. D. Maycock and C. Thomassigny, Synlett, 2001, 1146–1148 CrossRef CAS.
- X. H. Zhou, J. Luo, S. Huang, T. D. Kim, Z. Shi, Y. J. Cheng, S. H. Jang, D. B. Knorr Jr, R. M. Overney and A. K. Y. Jen, Adv. Mater., 2009, 21, 1976–1981 CrossRef CAS.
- S. R. Hammond, J. Sinness, S. Dubbury, K. A. Firestone, J. B. Benedict, Z. Wawrzak, O. Clot, P. J. Reid and L. R. Dalton, J. Mater. Chem., 2012, 22, 6752–6764 RSC.
- R. R. Barto, C. W. Frank, P. V. Bedworth, S. Ermer and R. E. Taylor, J. Phys. Chem. B, 2004, 108, 8702–8715 CrossRef CAS.
- J. Wu, B. Wu, W. Wang, K. S. Chiang, A. K.-Y. Jen and J. Luo, Mater. Chem. Front., 2018, 2, 901–909 RSC.
- F. Liu, M. Zhang, H. Xiao, Y. Yang, H. Wang, J. Liu, S. Bo, Z. Zhen, X. Liu and L. Qiu, J. Mater. Chem. C, 2015, 3, 9283–9291 RSC.
- D. L. Elder, C. Haffner, W. Heni, Y. Fedoryshyn, K. E. Garrett, L. E. Johnson, R. A. Campbell, J. D. Avila, B. H. Robinson, J. Leuthold and L. R. Dalton, Chem. Mater., 2017, 29, 6457–6471 CrossRef CAS.
- C. C. Teng and H. T. Man, Appl. Phys. Lett., 1990, 56, 1734–1736 CrossRef CAS.
- T.-D. Kim, J. Luo, Y.-J. Cheng, Z. Shi, S. Hau, S.-H. Jang, X.-H. Zhou, Y. Tian, B. Polishak, S. Huang, H. Ma, L. R. Dalton and A. K. Y. Jen, J. Phys. Chem. C, 2008, 112, 8091–8098 CrossRef CAS.
- Y. J. Cheng, J. D. Luo, S. Huang, X. H. Zhou, Z. W. Shi, T. D. Kim, D. H. Bale, S. Takahashi, A. Yick, B. M. Polishak, S. H. Jang, L. R. Dalton, P. J. Reid, W. H. Steier and A. K.-Y. Jen, Chem. Mater., 2008, 20, 5047–5054 CrossRef CAS.
- F. G. Liu, S. J. Chen, S. M. Mo, G. Z. Qin, C. W. Yu, W. J. Zhang, W. J. Shi, P. L. Chen, H. J. Xu and M. K. Fu, J. Mater. Chem. C, 2019, 7, 8019–8028 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00508k |
| This journal is © the Partner Organisations 2020 |