
Electroactivation-induced IrNi nanoparticles under different pH conditions for neutral water oxidation†
Woong Hee
Lee‡
a,
Jaekyung
Yi‡
ab,
Hong Nhan
Nong
c,
Peter
Strasser
 c,
Keun Hwa
Chae
d,
Byoung Koun
Min
ae,
Yun Jeong
Hwang
*abf and
Hyung-Suk
Oh
*ab
c,
Keun Hwa
Chae
d,
Byoung Koun
Min
ae,
Yun Jeong
Hwang
*abf and
Hyung-Suk
Oh
*ab
aClean Energy Research Center, Korea Institute of Science and Technology (KIST), Hwarang-ro 14-gil 5, Seongbuk-gu, Seoul 02792, Republic of Korea. E-mail: yjhwang@kist.re.kr; hyung-suk.oh@kist.re.kr; Tel: +82 (0)2 952 5292
bDivision of Energy and Environmental Technology, KIST school, Korea University of Science and Technology, Seoul 02792, Republic of Korea
cThe Electrochemical Energy, Catalysis, and Materials Science Laboratory, Department of Chemistry, Chemical Engineering Division, Technical University Berlin, Berlin 10623, Germany
dAdvanced Analysis Center, Korea Institute of Science and Technology (KIST), Hwarang-ro 14-gil 5, Seongbuk-gu, Seoul 02792, Republic of Korea
eGreen School, Korea University, 145 Anam-ro, Seongbuk-gu, Seoul 02841, Republic of Korea
fDepartment of Chemical and Biomolecular Engineering, Yonsei-KIST Convergence Research Institute, Yonsei University, Seoul 03722, Republic of Korea
First published on 11th June 2020
Abstract
Electrochemical oxidation processes can affect the electronic structure and activate the catalytic performance of precious-metal and transition-metal based catalysts for the oxygen evolution reaction (OER). Also there are emerging requirements to develop OER electrocatalysts under various pH conditions in order to couple with different reduction reactions. Herein, we studied the effect of pH on the electroactivation of IrNi alloy nanoparticles supported on carbon (IrNi/C) and evaluated the electrocatalytic activities of the activated IrNiOx/C for water oxidation under neutral conditions. In addition, their electronic structures and atomic arrangement were analyzed by in situ/operando X-ray absorption spectroscopy (XAS) and identical location transmission electron microscopy techniques, showing the reconstruction of the metal elements during electroactivation due to their different stabilities depending on the electrolyte pH. IrNiOx/C activated under neutral pH conditions showed a mildly oxidized thin IrOx shell. Meanwhile, IrNiOx/C activated in acidic and alkaline electrolytes showed Ni-leached IrOx and Ni-rich IrNiOx surfaces, respectively. Particularly, the surface of IrNiOx/C activated under alkaline conditions shows IrOx with a high d-band hole and NiOx with a high oxidation state leading to excellent OER catalytic activity in neutral media (η = 384 mV at 10 mA cm−2) whereas much lower OER activity was reported under alkaline or acid conditions. Our results, which showed that electrochemically activated catalysts under different pH conditions exhibit a unique electronic structure by modifying the initial alloy catalyst, can be applied for the design of catalysts suitable for various electrochemical reactions.
1. Introduction
Because of the extensive combination of the oxidative half reaction for water splitting, the nitrogen reduction reaction and the carbon dioxide reduction reaction (CO2RR), the oxygen evolution reaction (OER) is an important step in the path toward developing a sustainable-energy-dependent and clean society.1–3 For instance, electrochemical water splitting has great significance in providing hydrogen, which is a key chemical fuel in regenerative energy cycles. Moreover, recently, the electrochemical conversion of CO2 to valuable chemicals has been considered as a promising technology to reduce greenhouse gases and create a clean hydrocarbon energy cycle simultaneously.4–6 Due to the low amount of CO2 gas molecules present in alkaline media or overwhelming hydrogen evolution reaction (HER) activity in acidic media, the CO2RR was typically conducted in neutral pH media such as a buffered bicarbonate electrolyte, which is not a popular condition for water splitting, and the reaction mechanism also varies depending on the pH of the electrolyte. The OER is a bottleneck reaction owing to the sluggish kinetics for the O–H destruction and the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond formation, leading to a high overpotential under neutral conditions as well.7–9 Therefore, considerable efforts have been focused on finding highly active OER catalysts in neutral media with different pH values to minimize the overpotential and increase the energy efficiency.
O bond formation, leading to a high overpotential under neutral conditions as well.7–9 Therefore, considerable efforts have been focused on finding highly active OER catalysts in neutral media with different pH values to minimize the overpotential and increase the energy efficiency.
In recent research, it has been emphasized that not only materials, but also electronic structures have been recently reported to have a strong influence on the catalytic activity for the OER by analyzing the surface states of the electrocatalysts. Even for Ir oxide, one of the best materials for the OER, major efforts have been devoted to transform its electrical structure to enhance its OER activity further.10–15 Heat treatment is a general method to produce metal oxides. However, rutile IrO2 synthesized by thermal treatment has a lower OER activity than amorphous IrOx, which is derived by electrochemical treatment.16 Jones et al. studied the electronic structure of these two different Ir oxides by X-ray photoemission spectroscopy (XPS).17 Rutile IrO2 is a composite of IrVI and OII− species, whereas amorphous IrOx additionally possesses IrIII and OI− species. Notably, OI− which is considered as an electrophilic oxygen is suitable for the nucleophilic attack of oxygen in water.18 These OI− species in the IrOx lattice can participate in water oxidation, leading to enhanced OER catalytic activity.19,20 Therefore, electrophilic oxygen (OI− species) should be introduced to improve the OER catalytic activity of Ir-based catalysts under neutral conditions.
Recently, alloys of Ir with other metals have been reported in efforts to improve the electrophilic oxidation properties of Ir oxides.21–30 To activate IrM alloy catalysts, electrochemical treatment was conducted in an acid electrolyte to leach out the non-noble metal components. The surface of the IrM alloy was converted to amorphous IrOx with increased d-band holes, resulting in the metal-leached IrM@IrOx metal oxide core–shell catalyst with abundant electrophilic oxygen. Jaramillo et al. reported an IrOx/SrIrO3 catalyst showing excellent OER activity with high durability.31 Sr is leached out during the initial electrochemical testing, leading to a highly active IrOx surface layer. Strasser et al. demonstrated nickel-leached IrNi@IrOx nanoparticles with a large number of d-band holes in the IrOx shell.32 It has been observed using operando X-ray absorption spectroscopy (XAS) that the Ni leached IrOx shell possesses a massive number of vacancies under oxygen evolution. This leads to the enhanced electrophilic character of oxygen in the catalyst, thus reducing the kinetic barriers for the OER. However, the alloyed metal can have a different chemical stability depending on the environmental conditions such as electrolyte, cation/anion conditions and potential, and the formation of an active layer would be affected by the electrolyte conditions. It has been reported that the unique electronic properties of the catalyst can be obtained by electrochemical treatment at different pH values.33–35
In this work, we have studied the effect of the electrochemical activation of IrNi nanoparticles at different pH values to improve the catalytic activity of the OER under neutral conditions. A carbon supported IrNi alloy nanocatalyst (denoted as IrNi/C) was synthesized by a modified polyol process. Electrochemical activations were carried out on IrNi/C by cyclic voltammetry (CV) under acidic, neutral, and alkaline conditions, which are denoted as IrNiOx/C–A, IrNiOx/C–N, and IrNiOx/C–Al, respectively. Changes in the electrocatalyst morphology before and after electrochemical activations were analyzed using identical location transmission electron microscopy (IL-TEM) and IL-HAADF-STEM elemental mapping analysis. IrNiOx/C–A exhibits a nickel-leached IrNi@IrOx structure, as reported previously.32 IrNiOx/C–Al shows a nickel-rich IrNiOx shell structure, while IrNiOx/C–N exhibits a softly oxidized nickel leached Ir oxide shell. IrNiOx/C–Al shows a highly improved OER catalytic activity in neutral media, and the cause of this result was identified by ex situ XPS and in situ/operando XAS measurements.
2. Experimental section
2.1. Synthesis of IrNi bimetallic alloy nanoparticles on carbon black
An IrNi bimetallic nanoparticle alloy supported on carbon black (IrNi/C) was synthesized using oleic acid and oleylamine.25 A total of 0.45 mmol of nickel acetate tetrahydrate (Sigma-Aldrich), 0.675 mL of oleic acid (Sigma-Aldrich, 70%), and 0.675 mL of oleylamine (Sigma-Aldrich) in 45 mL of dibenzyl ether (Sigma-Aldrich) were added to the reaction flask. The mixture was sonicated to produce a homogeneous solution. The solution was then stirred for 1 h under a nitrogen atmosphere at room temperature. Then, the mixture was heated to 80 °C and maintained at the same temperature for 30 min under a N2 atmosphere. Iridium acetate (60 mg) and 1,2-dichlorobenzene (5 mL) were added to the reaction mixture. The mixture was then heated to 240 °C for 1 h and subsequently cooled to room temperature. After this process, the mixture was dispersed in 10 mL of dichloromethane and 60 mL of absolute ethanol by adding to it a dispersion of 135 mg of carbon black (Ketjenblack EC 300J) in 30 mL of toluene. The resulting solution was stirred for 1 day at room temperature. The products were collected via centrifugation at 6000 rpm and then redispersed in absolute ethanol (Samchun, 100%). This rinsing process was then repeated three times. The resulting powder was dried under ambient conditions for several hours. For the thermal treatment of IrNi/C catalysts, the IrNi/C samples were annealed at 400 °C under 400 cc min−1 of N2 flow and 100 cc min−1 of H2 flow for 4 h.2.2. Electrochemical measurements
All the electrochemical tests were performed using a rotating disk electrode system (RDE, Pine Research instrument) and a potentiostat VSP (biologic) with a Pt wire as the counter electrode. Hg/Hg2SO4 (sat. K2SO4, acid, and neutral media) and Hg/HgO (1 M NaOH, alkaline media) were used as reference electrodes. Before the OER test, the electrochemical activation process was conducted by 50 cycles of cyclic voltammetry, between 0.05 and 1.5 V vs. RHE at a scan rate of 500 mV s−1 in acidic (0.05 M H2SO4 purged with N2), neutral (0.5 M KHCO3 purged with CO2) and alkaline media (0.1 M KOH purged with N2). The OER performance was measured in a CO2-saturated 0.5 M KHCO3 (99.7%, Sigma-Aldrich) aqueous solution (pH 7.4). For the working electrode, ink was prepared by homogeneously suspending 5 mg of the catalysts, 2.49 mL of isopropyl alcohol (Samchun, 99.5%), 2.49 mL of DI water, and 20 μL of 5 wt% Nafion solution (Sigma Aldrich). The synthesized inks were added dropwise onto a glassy carbon electrode (RDE tip, 0.196 cm2) using a micropipette. The total Ir loading on the RDE was fixed at 20.5 μg cm−2.2.3. Material characterization
For material characterization, an X-ray diffractometer (XRD, Malvern Panalytical, Empyrean) was used to identify the crystal phases of the synthesized catalysts. X-ray photoelectron spectroscopy (XPS, ULVAC PHI, VersaProbe PHI 5000) and X-ray absorption spectroscopy (XAS) were performed to analyze the oxidation state of the catalysts. For XPS analysis, the catalyst was loaded on carbon paper (SIGACET® GDL, 39 BC) by the spray-coating method. The electrode area was 1 cm2 and the iridium loading amount was 0.2 mg cm−2. After the electrochemical reaction, the electrode was washed with DI water and dried at room temperature. The morphological features were analyzed by transmission electron microscopy (TEM, FEI, Talos F200X) and energy dispersive spectroscopy (EDS). Changes in the same nanoparticle on electrochemical activation were observed by identical location transition electron microscopy (IL-TEM). The depth profile of Ir was determined using X-ray photoelectron spectroscopy (XPS) at the 4D beamline of the Pohang Accelerator Laboratory (PAL), Pohang, South Korea.2.4. In situ/operando X-ray analysis
In situ/operando X-ray absorption spectroscopy (XAS) measurements were performed in fluorescence mode at the 1D beamline of the Pohang Accelerator Laboratory (PAL), Pohang, South Korea. A custom-made in situ/operando electrochemical flow cell was used to measure the catalyst during oxygen evolution. Ag/AgCl (3.5 M KCl) and Pt wire were used as the reference electrode and counter electrode, respectively. To prepare the working electrode, the catalyst ink was sprayed onto a carbon-coated Kapton film. XAS data were collected at an angle of 45° with respect to the beamline and detector. The detailed setup of in situ/operando XAS was reported in a previous paper.283. Results and discussion
Carbon supported IrNi alloy nanoparticles (denoted as IrNi/C) were prepared using the modified polyol process and used as the initial material for electrochemical activation. The morphology of the synthesized IrNi/C and the distribution of the elements were confirmed by electron microscopy. Fig. 1 and S1† show the transmission electron microscopy (TEM) images of the prepared IrNi/C. The synthesized IrNi nanoparticles have a uniform spherical shape with a size range of 10–15 nm and they are homogeneously deposited on carbon black. Based on ICP-OES analysis, the mass ratios of the elements in IrNi/C were found to be 10.3 wt% Ir and 10.6 wt% Ni. The elemental distributions of Ir and Ni were also analyzed by energy dispersive X-ray (EDX) element mapping. As shown in Fig. 1b and c, Ir and Ni are homogeneously distributed, suggesting that Ir and Ni metals are well alloyed. Within one nanoparticle (Fig. 1d and e), the results of the EDS line scan of IrNi/C show that the Ir and Ni components are uniformly mixed as well. The formation of the IrNi alloy was further confirmed from X-ray diffraction (XRD) patterns (Fig. 1f). The three main diffraction patterns matched with the (111), (200), and (220) planes, showing an fcc structure.36 The XRD peak position of IrNi/C is located between Ir (JCPDS #06-0598) and Ni (JCPDS #04-0850), which clearly confirms that IrNi/C has formed a well alloyed bimetallic structure. Fig. 2 demonstrates a morphological transformation of IrNi/C on electrochemical activation under acidic, neutral, and alkaline conditions. The electronic structure of pristine IrNi/C was activated through 50 cycles of cyclic voltammetry (CV) between 0.05 and 1.5 V vs. RHE at a scan rate of 500 mV s−1. The activations were carried out in acid (0.05 M H2SO4 purged with N2, denoted as IrNiOx/C–A), neutral (0.5 M KHCO3 purged with CO2, denoted as IrNiOx/C–N), and alkaline media (0.1 M KOH purged with N2, denoted as IrNiOx/C–Al) to enhance the catalytic activity. Fig. 2a, b, and S2† show the morphology changes in IrNiOx/C–A by identical location TEM (IL-TEM). In previous research, it was observed that the dissolution and re-deposition of Ir occurred during fast CV scans in an acid solution, leading to the formation of amorphous IrOx.37,38 An Ir nanoparticle cluster was formed in the particles, whereas Ni was almost completely dissolved. This Ni-leached IrOx structure is formed because of the unstable nature of Ni in the acid electrolyte. According to the line-scan of IrNiOx/C–A (Fig. 2c), on the electrochemical activation under acid electrolyte conditions, Ni is detected only in the core of the particles, indicating that the surface Ni has dissolved, leaving behind the IrOx shell structure. To quantify Ni dissolution during activation, the atomic composition of Ir and Ni was determined using the EDX spectrum. After the electrochemical activation under acidic conditions, the atomic ratio of Ni/Ir was changed from 3.94 to 0.097, confirming the high dissolution of Ni in the acid electrolyte. This transformation of IrNiOx/C–A is consistent with the findings of a previous report.32 Upon the activation of IrNi/C under neutral conditions, the Ni dissolution rate is greatly reduced due to its low acidity and Ni leaching occurs only in the top layer of the IrNi nanoparticles, as shown in Fig. 1d, e, and S3.† Ir is also dissolved and redeposited during this electrochemical activation, so that some of the Ir nanoparticles are deposited in the form of small particles on the carbon support. These morphological transformations can be clearly observed in the EDS line scan. A large amount of Ni is located in the core of the particle, indicating a thin Ir oxide shell with an IrNi core structure. According to the EDX spectrum, the Ni/Ir atomic composition of IrNiOx/C–N is 3.46, suggesting that soft Ni leaching occurred during activation in neutral media. The electrochemical activation of IrNi/C under alkaline conditions tends to be different compared to that under neutral or acidic conditions. IrNiOx/C–Al formed a Ni oxide layer on the particle surface, because Ni forms a passivated oxide phase without dissolution in the alkaline electrolyte according to pH-potential stability (Fig. 2g, h, S4, and S5†). The Ni/Ir atomic ratio of IrNiOx/C measured using the EDX spectrum is 3.98, which is close to pristine IrNi/C (3.94), confirming no dissolution of Ni. Thus, the surface of IrNiOx–Al gets oxidized during the electrochemical activation in alkaline media, leading to the Ni-rich IrNiOx shell structure of the catalyst. The EDS line-scan result also confirms the Ni-rich shell structure of IrNiOx/C–Al (Fig. 2i).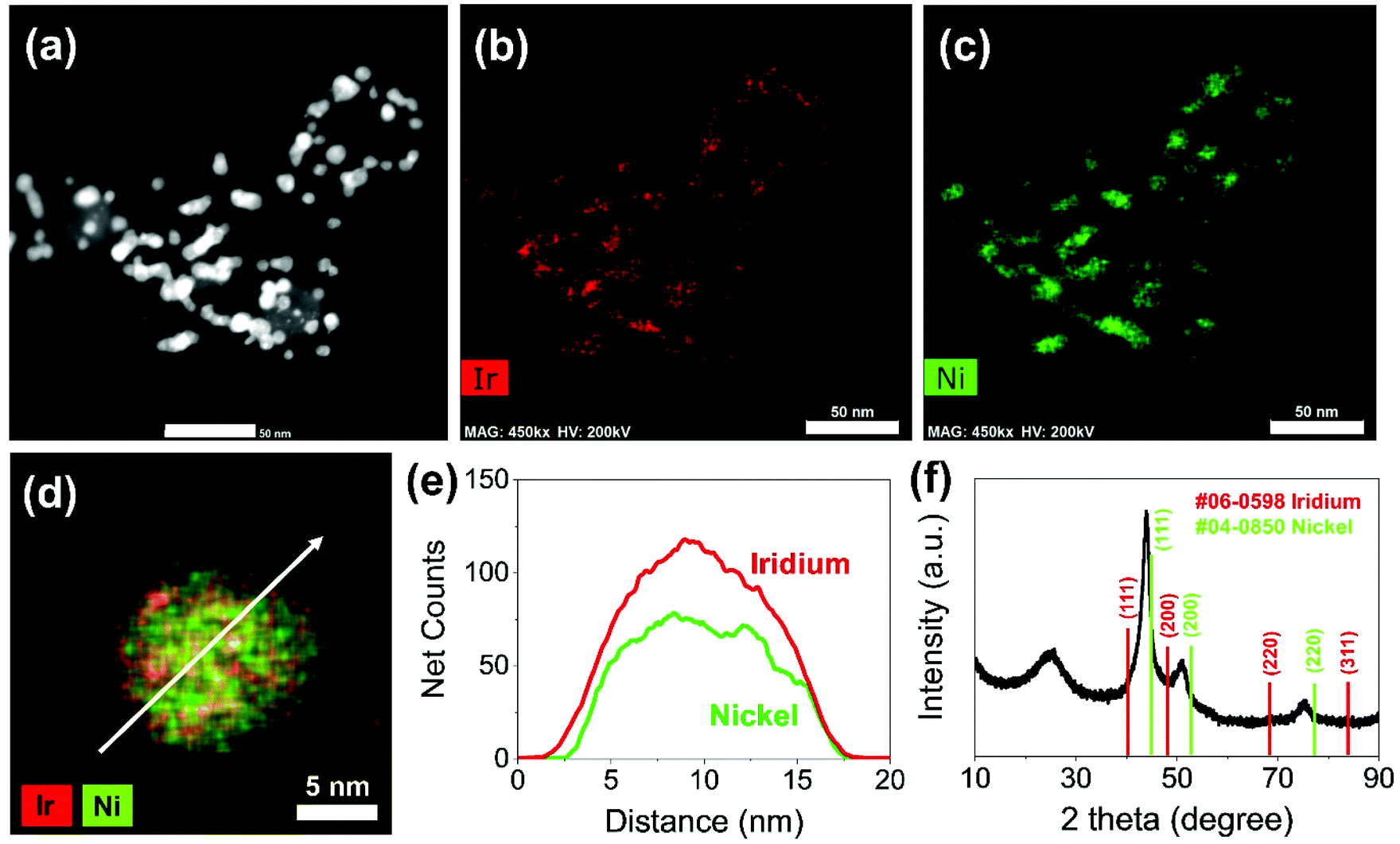 | ||
| Fig. 1 Morphology of carbon supported IrNi alloy nanoparticles (IrNi/C). (a) HAADF-STEM images of pristine IrNi/C (scale bar: 50 nm). HAADF-STEM elemental mapping analysis of IrNi/C (scale bar: 50 nm): (b) red and (c) green represent Ir and Ni, respectively. (d and e) Line-scan profiles of IrNi alloy nanoparticles. (f) X-ray diffraction pattern of IrNi/C. Reference patterns are given at the bottom: JCPDS card no. 06-0598 (Ir, red) and JCPDS card no. 04-0850 (Ni, green). | ||
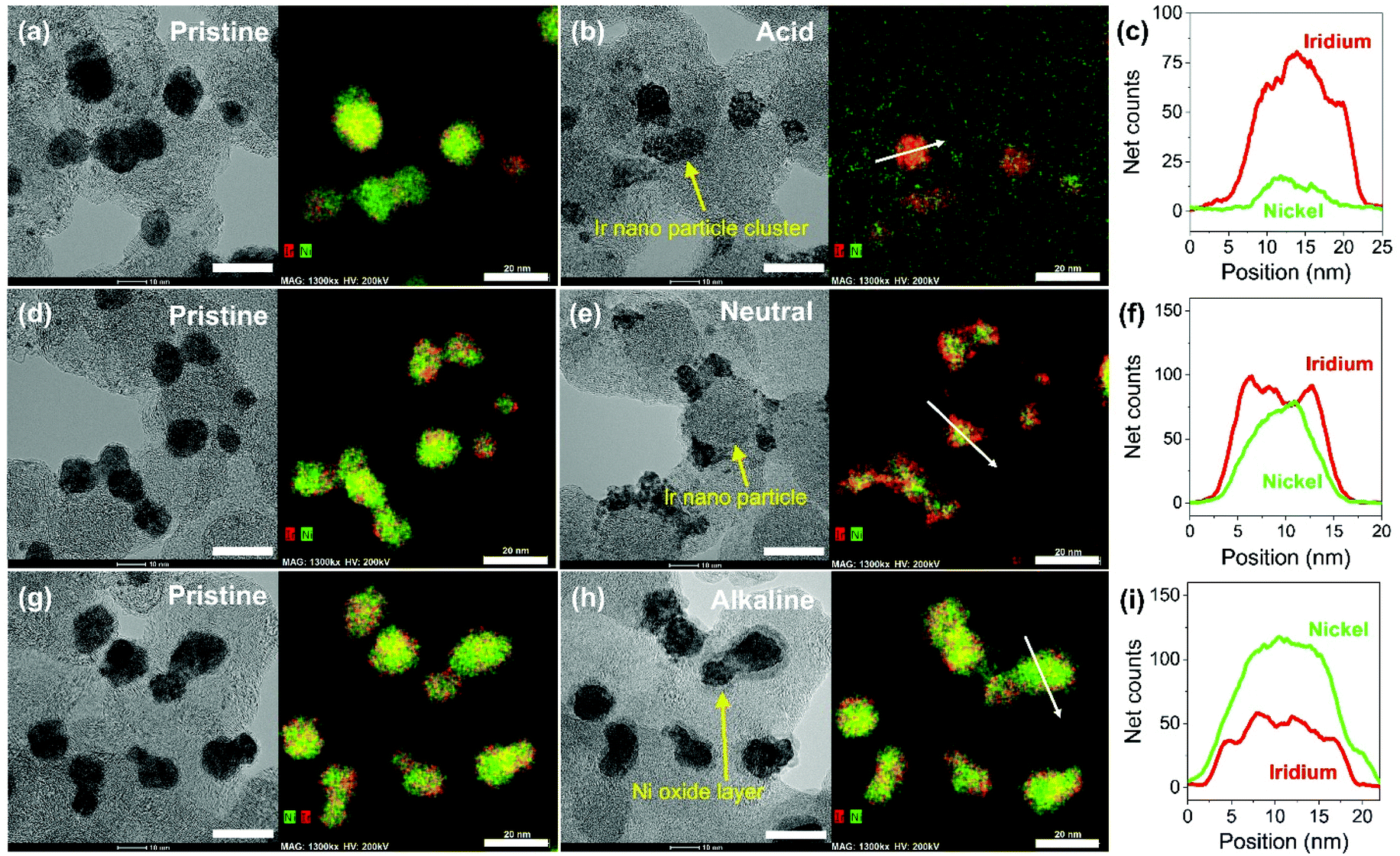 | ||
| Fig. 2 Morphological transformation of electrochemically activated IrNi/C under acidic, neutral, and alkaline conditions. Identical location transmission electron microscopy (IL-TEM) images and IL-HAADF-STEM elemental mapping analysis of pristine IrNi/C before electrochemical activation under (a) acid (d) neutral and (g) alkaline conditions (scale bar: 20 nm). Identical location transmission electron microscopy (IL-TEM) images and IL-HAADF-STEM elemental mapping analysis of electrochmically activated IrNi/C under (b) acid (e) neutral and (h) alkaline conditions, which are denoted as IrNiOx/C–A, IrNiOx/C–N, and IrNiOx/C–Al, respectively (scale bar: 20 nm). Line-scan profiles of (c) IrNiOx/C–A, (f) IrNiOx/C–N, and (i) IrNiOx/C–Al. | ||
To understand the changes in the electrochemical properties of IrNi/C by electrochemical activation under different pH conditions, the transformations of CV profiles (Fig. 3a–c) during electrochemical activation were observed. In all the samples, underpotentially deposited H (Hupd) peaks between 0.05 and 0.4 VRHE disappeared during activation, stipulating the oxidation of metallic IrNi/C in all media. In the case of acid activation, the redox peak associated with the Ir(III)/Ir(VI) redox couple increased, indicating the formation of an IrOx layer at the IrNiOx/C–A surface (Fig. 3a).39 IrNiOx/C–N possessed a smaller Ir(III)/Ir(VI) redox peak area of 0.499 mC cm−2 than both IrNiOx/C–A (0.823 mC cm−2) and IrNiOx/C–Al (0.773 mC cm−2), signaling only a small amount of the oxide species (Fig. 3b). Furthermore, the cathodic peak position of IrNiOx/C–N is located at 0.65 VRHE, which is lower than that of IrNiOx/C–A. In a previous report, it was expounded that the lower cathodic peak position of Ir represents the reduction of IrO2 and not IrOx with a high oxidation state.37,38 This implies that the electrochemical activation under neutral conditions slightly oxidizes the Ir surface to IrOx. IrNiOx/C–Al shows CV shapes different from both IrNiOx/C–A and IrNiOx/C–N. The redox peak associated with Ir(III)/Ir(VI) is located between 0.6 and 0.7 VRHE under alkaline conditions (Fig. 3c). The large Ni(II)/Ni(III) redox peak with an area of 2.086 mC cm−2 is observed at 1.2–1.3 VRHE, indicating the abundant Ni oxide of IrNiOx/C–Al.40,41 The above observations are in good agreement with the HR-TEM results.
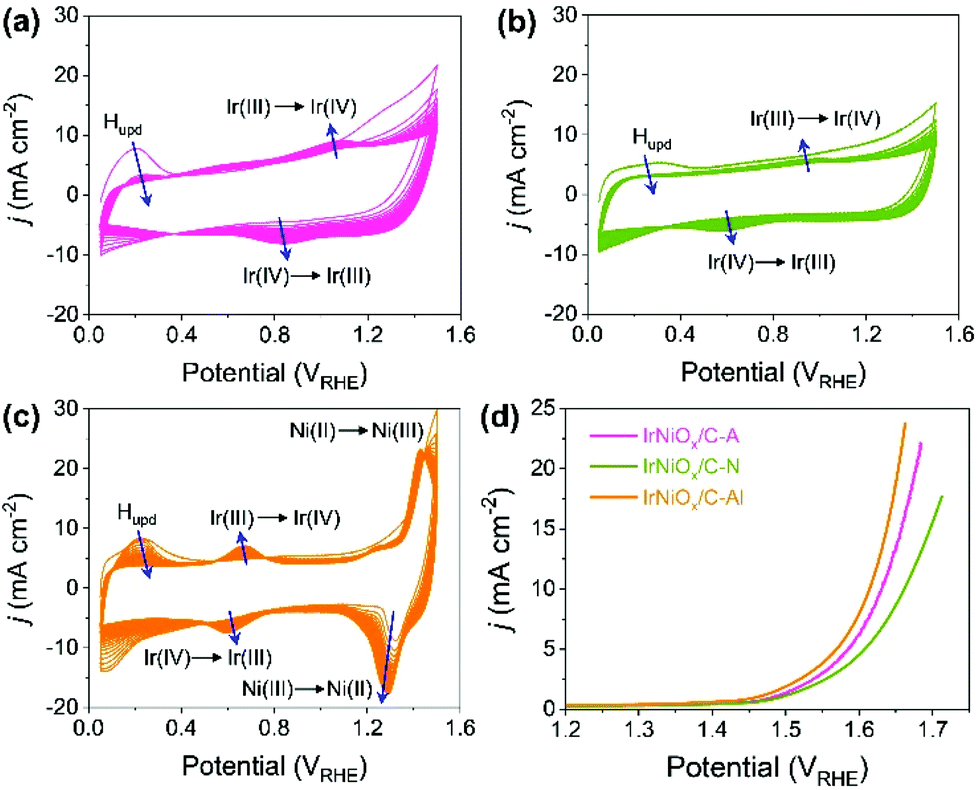 | ||
| Fig. 3 Electrochemical properties of activated IrNi/C under different pH conditions. Cyclic voltammogram (CV) profiles of IrNi/C between 0.05 and 1.5 V vs. RHE at a scan rate of 500 mV s−1 in (a) acid (0.05 M H2SO4 purged with N2), (b) neutral (0.5 M KHCO3 purged with CO2) and (c) alkaline media (0.1 M KOH purged with N2). (d) iR-corrected LSV curves of IrNiOx/C–A, IrNiOx/C–N and IrNiOx/C–Al in CO2-saturated 0.5 M KHCO3 at a scan rate 10 mV s−1. (e) Tafel plots of IrNiOx/C–A, IrNiOx/C–N, and IrNiOx/C–Al. | ||
The effects of the electrochemical activation in different media for OER catalytic activity were evaluated by linear sweep voltammetry (LSV) in CO2 saturated 0.5 M KHCO3 solution with a scan rate of 10 mV s−1 and a rotating speed of 1600 rpm. As presented in Fig. 3d, the LSV of IrNiOx/C–Al exhibited an anodic current onset at approximately 1.4 VRHE, which was 50 mV lower than that of IrNiOx/C–A and IrNiOx/C–N. Moreover, the overpotential at a current density of 10 mA cm−2 of IrNiOx/C–Al was only 384 mV, which was lower than that of IrNiOx/C–A (400 mV) and IrNiOx/C–N (431 mV). The enhanced OER activity in neutral media is compared with those previously reported as shown in Table S1.† These results suggest that IrNiOx/C–Al and IrNiOx/C–A possess higher OER catalytic activity than IrNiOx/C–N. Moreover, the observations reveal that despite using the same starting catalyst, different electrochemical activations can result in different catalytic activities. The electrochemical stability of the prepared catalysts was tested at 10 mA cm−2 constant water oxidation current density for 10 h in CO2 saturated 0.5 M KHCO3 solution and the results are shown in Fig. S6.† IrNiOx/C–N shows the gradually increased potentials, which indicate slow catalytic degradation. On the other hand, IrNiOx/C–A and IrNiOx/C–Al show stable behavior for 10 h in CO2 saturated 0.5 M KHCO3. These results indicate that IrNiOx/C–Al, which is electrochemically activated under alkaline conditions, is more efficient and durable than catalysts activated under acidic and neutral conditions.
To validate in detail the electronic structure of the electrochemically treated IrNi/C in different media, depth-profiled X-ray photoelectron spectroscopy (XPS) was performed with 210 eV and 550 eV kinetic energies (KE) at the 4D beamline of the Pohang Accelerator Laboratory (PAL). The deconvolution of the Ir 4f spectra yielded three components: Ir0 (metallic Ir, 60.9 and 63.9 eV), IrVI (rutile Ir oxide, 61.8 and 64.8 eV), and IrIII (amorphous Ir oxide, 62.3 and 65.3 eV). In Fig. 4a, the Ir 4f region of pristine IrNi/C indicates a mostly metallic state. The two dominant peaks for all electrochemically activated IrNi/C are located between the IrVI and IrIII species binding energy, denoting the oxidation states of the Ir species (Fig. 4b–d). The Ir 4f peak at 210 eV is more oxidized than that at KE 550 eV, confirming that IrNi/C was oxidized from the surface by electrochemical activation. Based on the XPS fitting at KE 210 eV, the IrIII species ratio was found to be 58.0% for IrNiOx/C–A and 66.6% for IrNiOx/C–Al. This shows that the surface of IrNi/C is converted to IrOx species by CV in acidic and alkaline media. The Ni peaks of IrNiOx/C–Al are located at 856 eV and are assigned to the binding energy of Ni(OH)2. This demonstrates that not only Ir, but also Ni is oxidized by the alkaline electrochemical activation (Fig. S7†). In contrast, IrNiOx/C–N exhibited 62.3% IrVI species component, signaling an Ir oxide layer with a small amount of IrOx. This observation implies that neutral conditions do not favor the oxidation of the catalyst surface, which is in agreement with the CV results. A similar observation is also found in the ex situ X-ray adsorption fine spectrum (XAFS). In Fig. S8,† the X-ray adsorption near edge structure (XANES) spectra and extended X-ray adsorption fine structure (EXAFS) spectra of the activated samples indicate that IrNiOx/C–A and IrNiOx/C–Al show higher oxidation states than IrNiOx/C–N.
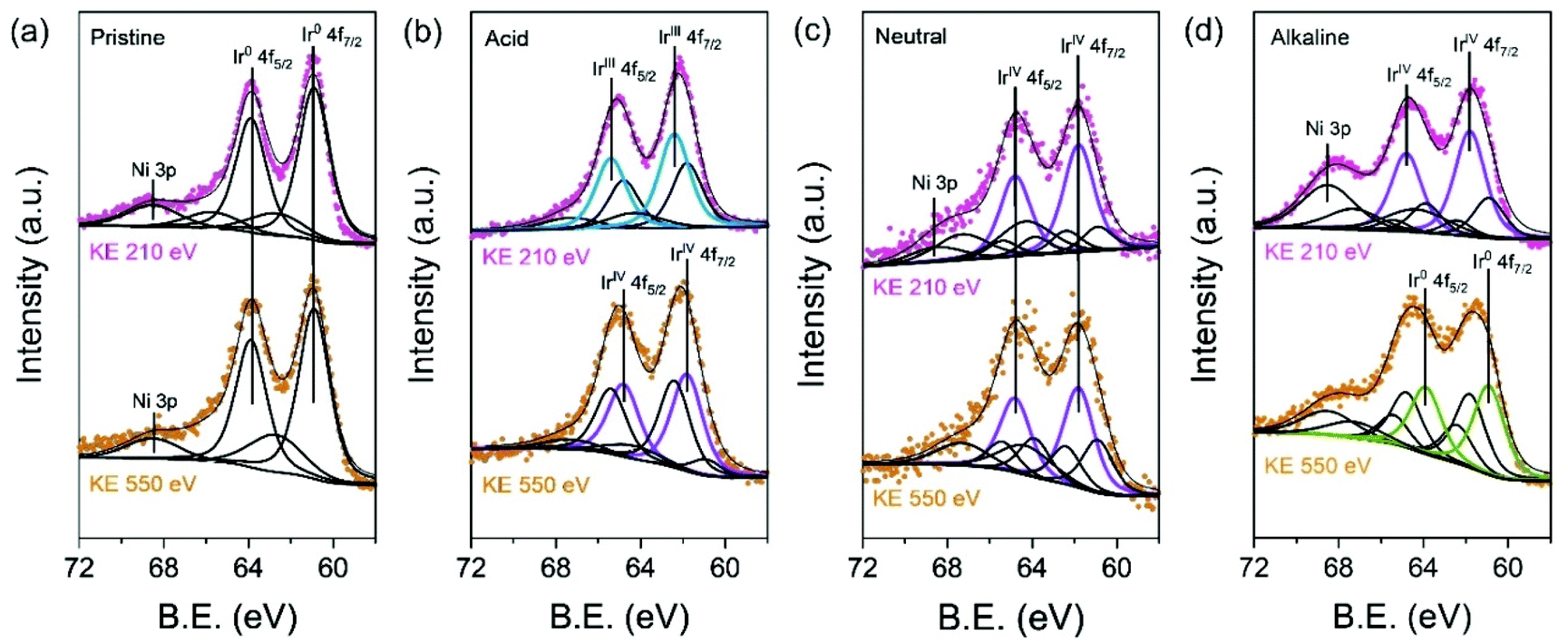 | ||
| Fig. 4 Electronic structures of electrochemically activated IrNi/C under acidic, neutral, and alkaline conditions. High-resolution XPS spectra for Ir 4f of (a) pristine IrNi/C, (b) IrNiOx/C–A, (c) IrNiOx/C–N, and (d) IrNiOx/C–Al at kinetic energies of 210 eV and 550 eV. | ||
To gain insight into the catalytic activity enhancement by electrochemical activation, the in situ/operando XAFS of the Ir L3-edge and Ni K-edge was carried out at the 1D KIST-PAL beamline of the Pohang Accelerator Laboratory. Customized electrochemical flow cells were used for the in situ/operando XAFS measurements. Fig. 5 shows that operando XAFS measurements were conducted during the OER under neutral conditions after electrochemical activation under different pH conditions. At the Ir L3-edge, the XANES spectra of IrNiOx/C–A and IrNiOx/C–Al showed a highly identical white line position, whereas that of IrNiOx/C–N exhibited a negatively shifted white line position, suggesting that IrNiOx/C–A and IrNiOx/C–Al possess a higher number of d-band holes than IrNiOx/C–N under OER conditions (Fig. 5a). The area of the white line peak of IrNiOx/C–A (21.03) was larger than that of IrNiOx/C–Al (19.80), indicating that the oxide thickness in the case of IrNiOx/C–A is more than that of IrNiOx/C–Al (Fig. S9†). This is further proved by the EXAFS results in Fig. 5b. The distance of Ir–M is reduced in the IrNi/C sample to be shorter than that of Ir black, confirming the existence of the IrNi alloyed structure. The Ir–O peaks of IrNiOx/C–A indicate fully oxidized IrNiOx nanoparticles, whereas the remaining Ir–M peaks of IrNiOx/C–Al represent the unoxidized core of the IrNiOx/C–Al nanoparticle during the OER. We expect that the thick Ni-rich IrNiOx layer of IrNiOx/C–Al observed by HR-TEM would inhibit the oxidation of the nanoparticle core. Strasser et al. reported that a higher number of d-band holes of Ir increase the hole character of the oxygen ligands, enhancing their electrophilic character.32 This promotes the nucleophilic attack of water molecules, thus reducing the kinetic barrier of the OER. IrNiOx/C–A and IrNiOx/C–Al possess more number of d-band holes under OER conditions than IrNiOx/C–N, indicating the greater OER activity of IrNiOx/C–A and IrNiOx/C–Al. In Fig. 5c, IrNiOx/C–Al reveals higher Ni XANES peak positions and areas than both IrNiOx/C–N and ex situ IrNiOx/C–Al in the OER under neutral conditions, indicating that the Ni in IrNiOx/C–Al has a higher oxidation state in the OER. This is also observed in the Ni-EXAFS peaks (Fig. 5d). An increased Ni–O bond peak is observed, indicating the existence of an oxide layer of IrNiOx/C–Al in the OER. In previous studies, high-valent Ni species of Ni-based catalysts were observed in the OER and are expected to act as active sites.42,43 The formation of higher oxidation states of nickel such as NiIV improves the OER catalytic activity under neutral conditions.44 Thus, Ni in IrNiOx/C–Al can be a good active site for the OER in neutral media. The above results reveal that IrNiOx/C–Al possesses a Ni-rich IrNiOx layer, with a configuration including IrOx active sites with a high number of d-band holes and NiOx active sites with a high oxidation state under OER conditions. This configuration of the two active species synergistically enhances the OER catalytic activity of IrNiOx/C–Al in neutral media.
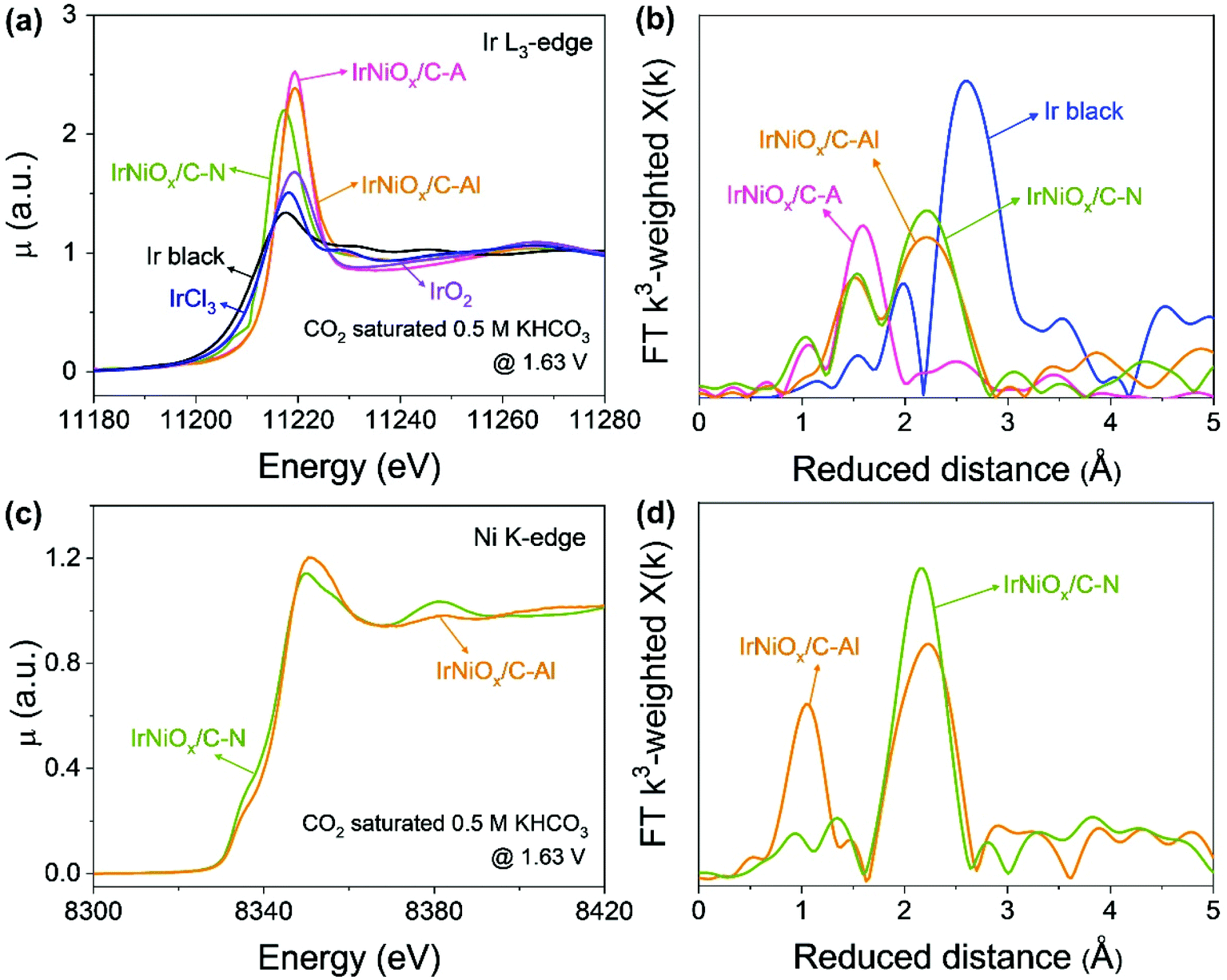 | ||
| Fig. 5 In situ/operando X-ray absorption spectroscopy (XAS) study under OER conditions in CO2-saturated 0.5 M KHCO3. (a) Ir L3-edge XANES region of IrNiOx/C–A, IrNiOx/C–N, and IrNiOx/C–Al at 1.63 VRHE in CO2 saturated 0.5 M KHCO3. Ir foil, IrCl3, and IrO2 as references were measured under ex situ conditions. (b) k3-Weighted Fourier transforms of EXAFS profiles collected at the Ir L3-edge of IrNiOx/C–A, IrNiOx/C–N and IrNiOx/C–Al. (c) Ni K-edge XANES region of IrNiOx/C–N and IrNiOx/C–Al at 1.63 VRHE in CO2 saturated 0.5 M KHCO3. (d) k3-Weighted Fourier transforms of EXAFS profiles collected at the Ni K-edge of IrNiOx/C–N and IrNiOx/C–Al. | ||
4. Conclusions
In summary, we have identified the effects of the electrochemical activation of IrNi nanoparticles under different pH conditions and evaluated the OER activity under neutral conditions applicable to CO2RR systems. IrNiOx/C–A activated under acidic conditions exhibits a Ni-leached IrNi@IrOx structure and IrNiOx/C–Al activated under alkaline conditions has a Ni-rich IrNi@IrNiOx structure. Meanwhile, IrNiOx/C–N activated under neutral conditions possesses a lower oxidized, thin Ir oxide shell structure. In situ/operando XAS demonstrates that the Ni-rich IrNiOx surface of IrNiOx/C–Al has an IrOx active site with a high number of d-band holes and a NiOx active site with a high oxidation state during the OER, leading to superior OER catalytic activity with an overpotential of 384 mV at 10 mA cm−2 under neutral conditions. These observations are also confirmed by synchrotron-based XPS depth profiling measurements. Our research suggests that the intrinsic properties of the catalyst change with the environment of the electrochemical activation, and that the catalyst structure suitable for the electrochemical reaction can be adjusted through the applied electricity and pH conditions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Korea Institute of Science and Technology (KIST) institutional program and partially supported by the National Research Foundation of Korea (NRF) grant funded by the Korea Government (MEST) (No. 2019R1A2C2005521). We acknowledge the Advanced Analysis Center at KIST for the TEM, XPS and ICP-MS measurements. We acknowledge 1D XRS KIST-PAL and 4D beamline of PAL for the XAS and XPS measurements, respectively.References
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- S. Ghosh and R. N. Basu, Nanoscale, 2018, 10, 11241–11280 RSC.
- S. Ma and P. J. Kenis, Curr. Opin. Chem. Eng., 2013, 2, 191–199 CrossRef.
- N. S. Spinner, J. A. Vega and W. E. Mustain, Catal. Sci. Technol., 2012, 2, 19–28 RSC.
- Y. Li and Q. Sun, Adv. Energy Mater., 2016, 6, 1600463 CrossRef.
- B. Zhang, Y. H. Lui, L. Zhou, X. Tang and S. Hu, J. Mater. Chem. A, 2017, 5, 13329–13335 RSC.
- Y. Dong, S. Komarneni, N. Wang, W. Hu and W. Huang, J. Mater. Chem. A, 2019, 7, 6995–7005 RSC.
- L. Cui, D. Liu, S. Hao, F. Qu, G. Du, J. Liu, A. M. Asiri and X. Sun, Nanoscale, 2017, 9, 3752–3756 RSC.
- L. Fu, X. Hu, Y. Li, G. Cheng and W. Luo, Nanoscale, 2019, 11, 8898–8905 RSC.
- M. Vuković, J. Appl. Electrochem., 1987, 17, 737–745 CrossRef.
- A. Grimaud, A. Demortière, M. Saubanère, W. Dachraoui, M. Duchamp, M.-L. Doublet and J.-M. Tarascon, Nat. Energy, 2016, 2, 1–10 Search PubMed.
- W. H. Lee and H. Kim, Catal. Commun., 2011, 12, 408–411 CrossRef CAS.
- Y. Zhao, M. Luo, S. Chu, M. Peng, B. Liu, Q. Wu, P. Liu, F. M. de Groot and Y. Tan, Nano Energy, 2019, 59, 146–153 CrossRef CAS.
- L. Fu, F. Yang, G. Cheng and W. Luo, Nanoscale, 2018, 10, 1892–1897 RSC.
- P. Jovanovič, N. Hodnik, F. Ruiz-Zepeda, I. Arčon, B. Jozinović, M. Zorko, M. Bele, M. Šala, V. S. Šelih and S. Hočevar, J. Am. Chem. Soc., 2017, 139, 12837–12846 CrossRef PubMed.
- V. Pfeifer, T. E. Jones, J. J. Velasco Vélez, C. Massué, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer, M. T. Greiner and J. Allan, Surf. Interface Anal., 2016, 48, 261–273 CrossRef CAS.
- V. Pfeifer, T. Jones, J. V. Vélez, C. Massué, M. Greiner, R. Arrigo, D. Teschner, F. Girgsdies, M. Scherzer and J. Allan, Phys. Chem. Chem. Phys., 2016, 18, 2292–2296 RSC.
- V. Pfeifer, T. E. Jones, J. J. V. Vélez, R. Arrigo, S. Piccinin, M. Hävecker, A. Knop-Gericke and R. Schlögl, Chem. Sci., 2017, 8, 2143–2149 RSC.
- C. Massué, V. Pfeifer, M. van Gastel, J. Noack, G. Algara-Siller, S. Cap and R. Schlögl, ChemSusChem, 2017, 10, 4786–4798 CrossRef PubMed.
- S.-A. Park, K.-S. Kim and Y.-T. Kim, ACS Energy Lett., 2018, 3, 1110–1115 CrossRef CAS.
- M. Tariq, W. Q. Zaman, W. Sun, Z. Zhou, Y. Wu, L.-m. Cao and J. Yang, ACS Sustainable Chem. Eng., 2018, 6, 4854–4862 CrossRef CAS.
- J. Feng, F. Lv, W. Zhang, P. Li, K. Wang, C. Yang, B. Wang, Y. Yang, J. Zhou and F. Lin, Adv. Mater., 2017, 29, 1703798 CrossRef PubMed.
- Y. Pi, Q. Shao, P. Wang, J. Guo and X. Huang, Adv. Funct. Mater., 2017, 27, 1700886 CrossRef.
- H. N. Nong, H. S. Oh, T. Reier, E. Willinger, M. G. Willinger, V. Petkov, D. Teschner and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 2975–2979 CrossRef CAS PubMed.
- C. Wang, Y. Sui, G. Xiao, X. Yang, Y. Wei, G. Zou and B. Zou, J. Mater. Chem. A, 2015, 3, 19669–19673 RSC.
- Q. Wang, X. Huang, Z. L. Zhao, M. Wang, B. Xiang, J. Li, Z. Feng, H. Xu and M. Gu, J. Am. Chem. Soc., 2020, 142(16), 7425–7433 CrossRef CAS PubMed.
- W. H. Lee, H. N. Nong, C. H. Choi, K. H. Chae, Y. J. Hwang, B. K. Min, P. Strasser and H.-S. Oh, Appl. Catal., B, 2020, 118820 CrossRef CAS.
- T. Reier, Z. Pawolek, S. Cherevko, M. Bruns, T. Jones, D. Teschner, S. r. Selve, A. Bergmann, H. N. Nong and R. Schlögl, J. Am. Chem. Soc., 2015, 137, 13031–13040 CrossRef CAS PubMed.
- G. Meng, W. Sun, A. A. Mon, X. Wu, L. Xia, A. Han, Y. Wang, Z. Zhuang, J. Liu and D. Wang, Adv. Mater., 2019, 31, 1903616 CrossRef PubMed.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang and J. K. Norskov, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- H. N. Nong, T. Reier, H.-S. Oh, M. Gliech, P. Paciok, T. H. T. Vu, D. Teschner, M. Heggen, V. Petkov and R. Schlögl, Nat. Catal., 2018, 1, 841–851 CrossRef CAS.
- S. Rudi, D. Teschner, V. Beermann, W. Hetaba, L. Gan, C. Cui, M. Gliech, R. Schlögl and P. Strasser, ACS Catal., 2017, 7, 6376–6384 CrossRef CAS.
- X. Chia, A. Ambrosi, D. Sedmidubský, Z. Sofer and M. Pumera, Chem. – Eur. J., 2014, 20, 17426–17432 CrossRef CAS PubMed.
- R. M. Arán-Ais, J. Solla-Gullón, M. Gocyla, M. Heggen, R. E. Dunin-Borkowski, P. Strasser, E. Herrero and J. M. Feliu, Nano Energy, 2016, 27, 390–401 CrossRef.
- W. Wu, Z. Chen, X. Lin, B. Li and X. Cong, Vacuum, 2011, 86, 429–437 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, A. Mingers and K. J. Mayrhofer, J. Electroanal. Chem., 2016, 773, 69–78 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, A. Mingers and K. J. Mayrhofer, J. Electroanal. Chem., 2016, 774, 102–110 CrossRef CAS.
- H.-S. Oh, H. N. Nong, T. Reier, M. Gliech and P. Strasser, Chem. Sci., 2015, 6, 3321–3328 RSC.
- C. Tang, H. S. Wang, H. F. Wang, Q. Zhang, G. L. Tian, J. Q. Nie and F. Wei, Adv. Mater., 2015, 27, 4516–4522 CrossRef CAS PubMed.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- M. Görlin, J. Ferreira de Araújo, H. Schmies, D. Bernsmeier, S. r. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert and H. Dau, J. Am. Chem. Soc., 2017, 139, 2070–2082 CrossRef PubMed.
- J. Yan, L. Kong, Y. Ji, J. White, Y. Li, J. Zhang, P. An, S. Liu, S.-T. Lee and T. Ma, Nat. Commun., 2019, 10, 1–10 CrossRef CAS PubMed.
- X. Zheng, B. Zhang, P. De Luna, Y. Liang, R. Comin, O. Voznyy, L. Han, F. P. G. de Arquer, M. Liu and C. T. Dinh, Nat. Chem., 2018, 10, 149 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr02951c |
| ‡ The authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2020 |