
A novel liquid biopsy-based approach for highly specific cancer diagnostics: mitigating false responses in assaying patient plasma-derived circulating microRNAs through combined SERS and plasmon-enhanced fluorescence analyses†
Adrianna N.
Masterson‡
a,
Thakshila
Liyanage‡
a,
Claire
Berman
a,
Hristos
Kaimakliotis
b,
Merrell
Johnson
 c and
Rajesh
Sardar§
*a
c and
Rajesh
Sardar§
*a
aDepartment of Chemistry & Chemical Biology, Indiana University-Purdue University Indianapolis, 402 N. Blackford Street, Indianapolis, Indiana 46202, USA. E-mail: rsardar@iupui.edu
bDepartment of Urology, Indiana University School of Medicine, 535 N. Barnhill Dr., Indianapolis, Indiana 46202, USA
cDepartment of Physics, Purdue University Fort Wayne, 2101 E. Coliseum Blvd., Fort Wayne, Indiana 46805, USA
First published on 25th May 2020
Abstract
Studies have shown that microRNAs, which are small noncoding RNAs, hold tremendous promise as next-generation circulating biomarkers for early cancer detection via liquid biopsies. A novel, solid-state nanoplasmonic sensor capable of assaying circulating microRNAs through a combined surface-enhanced Raman scattering (SERS) and plasmon-enhanced fluorescence (PEF) approach has been developed. Here, the unique localized surface plasmon resonance properties of chemically-synthesized gold triangular nanoprisms (Au TNPs) are utilized to create large SERS and PEF enhancements. With careful modification to the surface of Au TNPs, this sensing approach is capable of quantifying circulating microRNAs at femtogram/microliter concentrations. Uniquely, the multimodal analytical methods mitigate both false positive and false negative responses and demonstrate the high stability of our sensors within bodily fluids. As a proof of concept, microRNA-10b and microRNA-96 were directly assayed from the plasma of six bladder cancer patients. Results show potential for a highly specific liquid biopsy method that could be used in point-of-care clinical diagnostics to increase early cancer detection or any other diseases including SARS-CoV-2 in which RNAs can be used as biomarkers.
Introduction
Liquid biopsies involve analyzing circulating biomolecules in bodily fluids provide many advantages over traditional tissue biopsies for detecting cancers at an early stage as they are non-invasive, less expensive, and simpler.1 Small non-coding RNAs, known as microRNAs, have been found to regulate a large number of human genes and play a significant role in various cancer developments.2–7 Many studies have demonstrated that the detection of circulating microRNAs at an early stage, via liquid biopsies, could allow better treatment in an effort to prevent cancer progression and metastasis, increasing the chance of patient survival. The current “gold-standard” microRNA assays (i.e., quantitative reverse-transcription polymerase chain reaction (qRT-PCR)-based technologies) are capable of measuring microRNA levels reproducibly from bodily fluids in real-life clinical samples. However, this technology has several drawbacks including: (1) the treatment of biological fluid, (2) complicated labor-intensive RNA extraction, (3) required labeling and amplification prior to analysis, (4) complementary DNA conversion steps, and (5) the large volume of patient samples needed.8 Together, these traits hinder translation into clinical point-of-care (POC) diagnostics.In contrast, solid-state, label-free biosensing offers many unique advantages over qRT-PCR techniques, including simple sensor fabrication, high sensitivity, easy to miniaturization (of particular importance when adapting technologies to POC diagnostics), and elimination of receptor tagging with dyes or other specialized reagents (“label-free”). Among advantages, the latter is extremely important as it avoids undesirable interactions between labels and analytes, leading to more reliable and reproducible results. Current solid-state, label-free biosensing techniques such as microarray,9 electrochemical-,10,11 localized surface plasmon resonance (LSPR)-,12–14 surface-enhanced Raman scattering (SERS)-,15,16 and photonic microring resonator-based17 microRNA sensors provide good sensitivity. These single-mode detection methods rely on a specific sensing mechanism. For example, LSPR-based methods detect changes in local refractive index of the nanostructures while electrochemical-based measurements detect changes in current. Unfortunately, such changes can also result from non-specific adsorption of endogenous biomolecules from bodily fluids onto the sensor, as well as structural changes that can be induced following placement within bodily fluids during the measurements. As a result, these techniques suffer from low specificity, particularly when analyzing bodily fluids, and can in turn produce false test results. Further, aside from detecting/analyzing the specific signal change that occurs from the analyte, no additional confirmation of analyte attachment to the sensor can be obtained. Among these techniques, SERS-based biomolecular assays are capable of using Raman signal to provide rich structural information on the analytes. However, current solid-state SERS-based microRNA assays still show many problems, including low sensitivity, poor specificity, and inability to analyze real-life patient samples.15,16,18–21
High specificity is the most crucial factor in creating assays that can avoid false-positive and/or false-negative test results for the POC diagnosis.22,23 To overcome the current technological bottleneck in real-life patient sample analysis, we present, for the first time, the design of a solid-state nanoplasmonic sensor. This novel sensing approach is capable of assaying oncogenic microRNAs directly in human plasma with high sensitivity and specificity. Here, the unique LSPR properties24,25 of noble metal nanoparticles are utilized for the detection of microRNAs via a combined SERS26,27 and plasmon-enhanced fluorescence (PEF)28,29 approach. Importantly, the same nanoplasmonic sensor can be used in this multimodal detection method; no changes to structural parameters are required. To demonstrate the potential translational aspects of our dual detection technique in liquid biopsies for POC diagnosis, we successfully assayed two oncogenic microRNAs (microRNA-10b and microRNA-96). Analysis was performed directly in six bladder cancer patient plasma samples at sub-femtogram/microliter (fg μL−1) concentrations.
Experimental section
Materials
Chloro(triethylphosphine) gold(I) (Et3PAuCl, 97%) was purchased from Gelest Inc. Poly-(methylhydrosiloxane) (PMHS, Mn = 1700–3300), triethylamine (TEA, 98%), ACS grade acetonitrile (CH3CN, 99.9%), and 3-mercapto-1-propanol (3-MP, 95%) were purchased from Sigma-Aldrich. Thiol modified 5′-SH-(CH2)3-ssDNAs, and various microRNAs were purchased from Integrated DNA Technologies (IDT). RNase H enzyme and RBS detergent solution were purchased from Thermo Scientific. (3-Mercaptopropyl)-trimethoxysilane (MPTMS, 94%) was purchased from Alfa Aesar. All the chemicals were used without any further purifications. RNase free sterile water was obtained from Baxter Healthcare Corporation. The glass cover slips and the tris(2-carboxyethyl)phosphine (TCEP) solution were purchased from Fisher Scientific. Bladder cancer patient plasma samples were obtained from the Indiana University medical school and used as received. All water was purified using a Thermo Scientific Barnstead Nanopure system. Thiol modified -ssDNAs, microRNAs were stored at −80 °C. PBS buffer (pH = 7.2) was prepared using RNase-free sterile water. All experiments were performed in accordance with the Guidelines of the United States and approved by the ethics committee at Indiana University. Informed consents were obtained from human participants of this study.Fabrication of nanoplasmonic sensors
Gold triangular nanoprisms (Au TNPs) were synthesized using our well-established method.12,14,30 Et3PAu(I)Cl (10 mg) was dissolved in 20 mL of acetonitrile. After five minutes of stirring at room temperature, 0.019 mL (0.136 mmol) TEA was added, and the mixture was heated to 40 °C. Then, 0.3 mL (2.75 mmol) of PMHS was added, and the reaction ran until the desired LSPR peak of 800 nm was achieved. Au TNPs were then attached onto silanized glass coverslips as we reported previously.13 Separately, a mixture of 5 μM -C3-ssDNA-10b/96 with 0.1 M TCEP was prepared and allowed to react for 1 hour to break disulfide bonds. The solution was then diluted with PBS buffer, and the resulting solution was used to incubate Au TNPs for overnight. The addition of -ssDNA-10/96 solutions induced ligand exchange of Au TNPs by removing TEA and PMHS that were present on the surface during the colloidal synthesis. Next, -ssDNA-10b/96-functionalized Au TNPs were washed thoroughly with RNase free water, and both UV-Vis spectrum and Raman spectrum were acquired. Finally, 10 mL, 1.0 mM 3-MP solution was added to ssDNA-10b/96-functionalized Au TNPs to fabricate nanoplsmonic sensors. Various concentrations (100 nM–100 fM) of microRNA-10b or microRNA-96 in 10 mL PBS buffer were used to incubate nanoplasmonic sensors for 2 hours. MicroRNA bound nanoplasmonic sensors were washed thoroughly with RNase free water to remove any loosely bound microRNAs to the sensor. For the reversibility/regeneration tests, microRNA-bound nanoplasmonic sensors were incubated in15 units RNase H enzyme in RNase free water for 2 hours.UV-Vis and SERS measurements, and fluorescence confocal microscopy imaging
UV-Vis spectra were collected from 1100–300 nm on a Varian Cary 50 UV/Vis spectrophotometer, and all sensors were analyzed in RNase free water. SERS spectra were collected of the sensor on clean aluminum foil using a Foster-FORAM 785 HP Raman spectrometer with a 785 nm diode laser excitation source. Samples were placed on top of the aluminum foil when analyzed due to the requirement of a clean surface to analyze. The background scan was taken of a polystyrene bead to calibrate the laser. For all SERS measurements, two randomly selected spots were analyzed for each sensor and for three-separate sensors (a total of six measurements). Using a Fluoview FV1000 MPE laser scanning biological microscope, 100 nM Alexa-488 dye was used in the analysis of the sensors. A laser power of 40%, a 60× water objective, and gain of 1× were used for the image acquisition. The diameter of the laser spot was 5-μM.Scanning electron and atomic force microscopy analyses
The chemically synthesized Au TNPs attached onto the silanized glass coverslips were characterized by scanning electron microscopy (SEM) and atomic force microscopy (AFM). SEM micrographs were obtained using a Hitachi S-4700 Field Emission Scanning Electron Microscope (FESEM) at 20 kV accelerating voltage, and AFM images were obtained using a Bioscope AFM instrument.Construction of calibration curves
All SERS spectra were baseline corrected using OMNIC software. The Fit Peaks tool was used in Origin 2018b to find peak intensity with Lorentz fitting. Peak intensity at a specific wavenumber was graphed as a function of concentration. Linear fitting was used to find the R-squared value.Measuring false responses
In order to demonstrate the efficiency of our solid-state nanoplasmonic-based sensors mitigating false responses, we analyzed the patient samples that measured the highest and lowest concentrations. Nanoplasmonic-based sensors, which were used to quantify microRNA-10b/96 in patient plasma again re-incubated in 15 units RNase H enzyme in RNase free water for 2 h. Next, both SERS measurement fluorescence confocal measurements were performed. Again, the same sensors were incubated in a 100 nM solution of microRNA-FAM. After the sensors were thoroughly washed with RNase free water, SERS and fluorescence analyses were conducted.Results and discussion
To acquire both SERS and PEF signals from the same nanoplasmonic sensor, we used chemically-synthesized gold triangular nanoprisms (Au TNPs) to generate a high intensity LSPR response.31 This intensified LSPR arises due to strong electromagnetic field (EM) enhancement that occurs at the sharp tips and edges of Au TNPs.32–35 This leads to the formation of hot-spots, which are ideal for LSPR-based SERS and PEF mechanisms.26,29 For the first time, we have utilized PEF to unequivocally confirm microRNA attachment onto the nanoplasmonic sensor by utilizing fluorescence confocal imaging. Together, these methods fully eliminate the possibility of false assay responses. Further, in addition to their unique LSPR properties, the atomically flat Au TNPs allow -ssDNAs to form a tightly-packed self-assembled monolayer (SAM) on their surface, allowing for high reproducibly throughout the sensor. This is important in the context of making good interactions and forming a -ssDNA/microRNA duplex. Moreover, tightly-packed SAMs avoid non-specific adsorption of biomolecules onto the Au surface. Au is also extremely stable in human biofluids, therefore sensors are expected to maintain excellent structural integrity. Taking into consideration all their unique structural properties, we chose to Au TNPs with a 42 and 8 nm edge length and thickness, respectively, for the fabrication of our nanoplasmonic sensors, as shown in Fig. 1. | ||
| Fig. 1 Structural characterizations of Au TNPs. (A) A representative scanning electron microscopy image of Au TNPs attached onto a silanized glass substrate. Scale bar is 100 nm. (B) An atomic force micrograph of Au TNPs. | ||
The fabrication of nanoplasmonic sensors is illustrated in Fig. 2, where: (A) Au TNPs were chemically-attached onto silanized glass substrate; (B) TNPs were functionalized with -ssDNA-(10b/96) via the Au–S bond, and in order to increase their specificity in SERS, they were further functionalized with 3-mercapto-1-propanol (3-MP) (Fig. 2C) so that the -ssDNA-10b/96 would appear in a “standup” position. This standup formation creates an ideal distance between the fluorophore and Au TNPs to maximize PEF enhancement by reducing fluorescence quenching through the Förster resonance energy transfer (FRET) process. In additon, stand-up -ssDNAs provides adequate space for the microRNAs to form -ssDNA/microRNA duplexes. Finally, 3-MP also improves specificity, as reported in the literature.10 The functionalizations of Au TNPs with -ssDNA-(10b/96) and 3-MP represent our nanoplasmonic sensors. Fig. 2D shows attachment of a fully complimentary microRNA tagged with a 6-fluorescein (FAM), allowing complete -ssDNA/microRNA-FAM hybridization. Most importantly, as shown in Fig. 2D, the nanoplasmonic sensors are fully reversible and regenerative to allow use for multiple analyses. This is critical in the context of determining sensor stability and showing that they maintain complete structural integrity during assays.
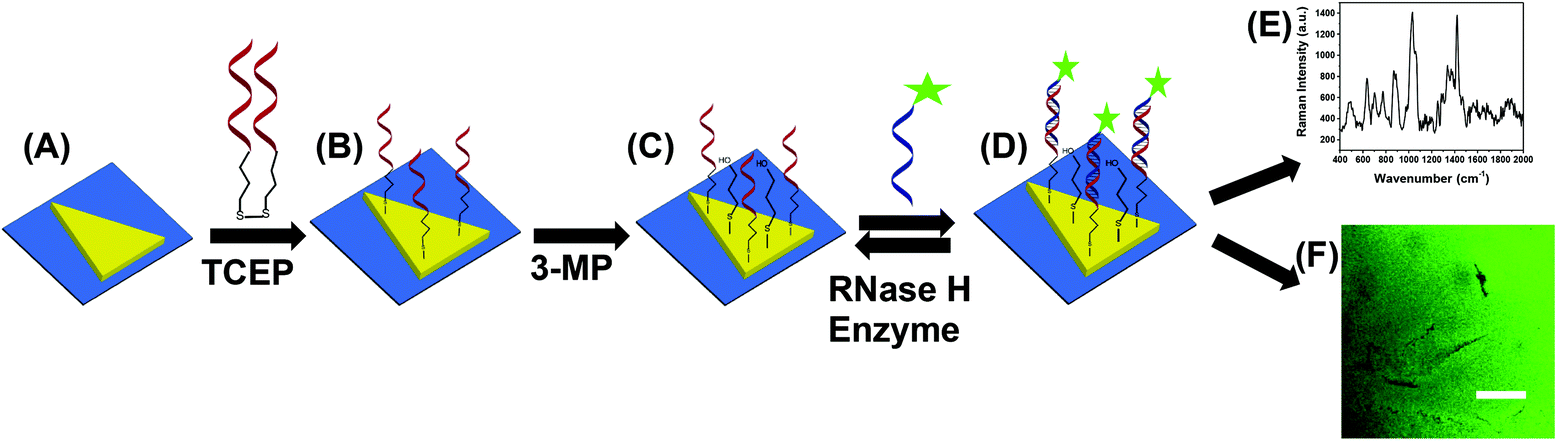 | ||
| Fig. 2 Design of a nanoplasmonic sensor for the detection of microRNAs using combined surface-enhanced Raman spectroscopy (SERS) and fluorescence confocal microscopy techniques. (A) Freshly synthesized Au TNPs were attached onto silanized glass coverslip. (B) Tris(2-carboxyethyl)phosphine (TCEP) addition to DNA-disulfide solution breaks disulfide bonds and subsequent incubation in the solution resulted in the attachment of -ssDNAs on the surface of TNPs. Generally, -ssDNAs were randomly organized on the flat metal surface (e.g., here TNPs).10 (C) -ssDNA-attached Au TNPs were further functionalized with 3-mercapto-1-propanol (3-MP) to allow -ssDNAs to “stand up” to achieve the most favorable -ssDNA/microRNA hybridization. The functionalization steps until (C) produced nanoplasmonic sensors, where microRNA-10b/96-FAM can selectively bind (D) to be analyzed by both SERS spectroscopy (E) and fluorescence confocal microscopy (F). Nanoplasmonic sensors can also be regenerated by incubating microRNA-bound sensors in a RNase H enzyme solution; image not to scale. | ||
Attachement of microRNA-FAM to the nanoplamionic sensor can be detected/confirmed utilizing both SERS (Fig. 2E) and fluorescence confocal microscopy (Fig. 2F) techniques. It is important to highlight the additional advantages of selecting this particular dimension Au TNP for a SERS-based microRNA assay: (1) As shown in Fig. S1,† the LSPR peak position of nanoplasmonic sensors is ∼860 nm. The LSPR peak of TNPs and the wavelength of incident laser light controls the intensity of hot-spots. Ideally, for the EM-field-driven SERS enhancement, the LSPR wavelength of nanostructures in the sensors should be longer than the wavelength of laser light; (2) Use of a low energy laser (e.g., 785 nm) that fits the excitation requirements for nanostructures is important for biosensing applications in order to avoid degradation of biomolecules, particularly microRNAs.
Each step in the fabrication of these nanoplasmonic sensors was characterized through SERS spectroscopy (see Fig. 3). Attachement of -ssDNA-(10b/96) via Au–S bonds results in the appearance of a C–S Raman stretch at 635 cm−1. Furthermore, several new Raman stretches that are characteristic of DNAs (see Fig. 3A and Fig. S2†) such as guanine (G) ring breathing at 688 cm−1, adenine ring breathing at 733 cm−1, cytosine ring breathing at 790 cm−1, and the phosphodiester group of the nucleic acids at 1289 cm−1 are also observed.18,21,36–38 The C–H Raman stretch at 1400 cm−1 further confirms the functionalization of Au TNPs with 3-MP (Fig. 3A-blue).39 Incubation of microRNA-(10b/96)-FAM to the nanoplasmonic sensor displays a prominent Raman peak at 1062 cm−1 of the additional PO2− symmetrical stretch.38 Furthermore, the Raman stretch at 1255 cm−1 corresponds to guanine C8–H bending18,21,36,37 and the low intensity Raman stretch at 1645 cm−1 is unique to the uracil C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch.18,21,36,37Fig. 3B illustrates SERS spectra of the microRNA-10b varying concentrations from 100 nanomolar (nM) to 100 femtomolar (fM).
O stretch.18,21,36,37Fig. 3B illustrates SERS spectra of the microRNA-10b varying concentrations from 100 nanomolar (nM) to 100 femtomolar (fM).
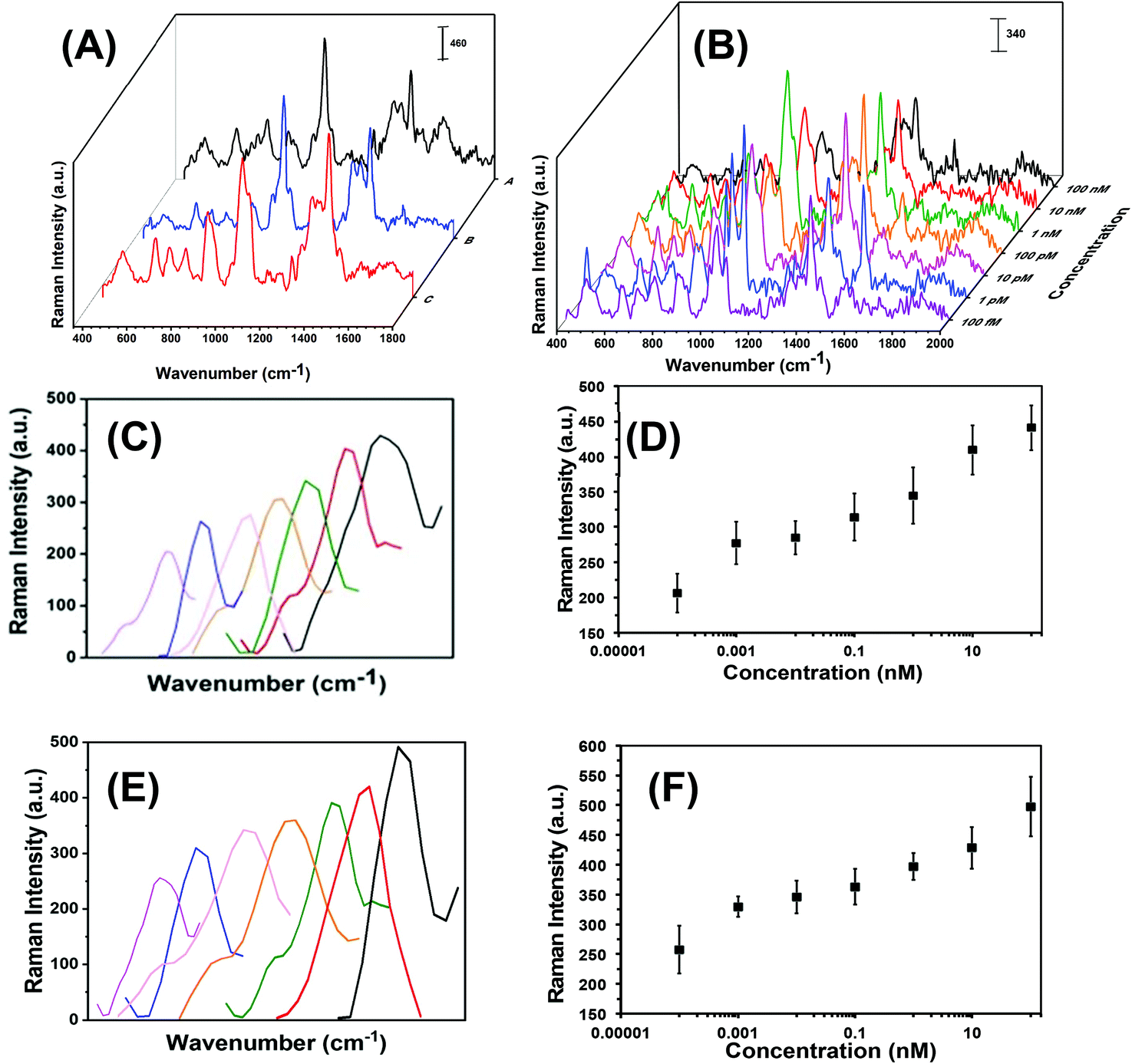 | ||
| Fig. 3 The SERS-based microRNA assay using nanoplasmonic sensors. (A) SERS spectra at different stages: Functionalization of AuNPs with -ssDNA-10b, A; after attachment of 3-MP on TNPs, B; and after incubation of nanoplasmonic sensors in a microRNA-10b solution, C. (B) SERS spectra of nanoplasmonic sensors upon incubation in different concentrations of microRNA-10b solution. (C) Expanded SERS spectra of the C8–H bending stretch of guanine at 1255 cm−1 in various microRNA-10b concentrations (light purple: 100 fM, blue: 1 pM, pink: 10 pM, orange: 100 pM, green: 1 nM, red: 10 nM, black: 100 nM). (D) Intensity of the C8–H bending stretch at 1255 cm−1 as a function of microRNA-10b concentration (logarithmic scale). Y = 15.94ln(X) + 361.88, R2 = 0.97. (E) Expanded SERS spectra of the C8–H bending stretch of guanine at 1255 cm−1 in various microRNA-96 concentrations (light purple: 100 fM, blue: 1 pM, pink: 10 pM, orange: 100 pM, green: 1 nM, red: 10 nM, black: 100 nM). (F) Intensity of the C8–H bending stretch at 1255 cm−1 as a function of microRNA-96 concentration (logarithmic scale). Y = 15.04ln(X) + 408.73, R2 = 0.95. In (C) and (E), the individual concentrations of the microRNA calibration curve are plotted in order to better visualize of the increase in intensity. Each spectrum has the same wavenumber of 1255 cm−1and does not shift in wavenumber as seen in the figure. | ||
The average edge-length of our chemically synthesized Au TNPs is 42 nm that provides a top surface area of 764 nm2. Considering all the -ssDNA probes were attached onto the top surface and thiolate has a 0.25 nm2 footprint, there would be approximately 3000 -ssDNA-(10b/96) attached per TNP. In the nanoplasmonic sensors fabrication, we used a high concentration of 3-MP in comparison to -ssDNA probes. Therefore, the -ssDNA grafting density is overestimated because a relatively large percentage of TNP surface would be occupied by 3-MP molecules. Further experimental characterization is required to quantitively determine the grafting density of -ssDNA probe that is currently under our investigation.
In literature, researchers have used a variety of SERS stretches to develop the calibration plots for microRNA assays, but have done so without providing any detailed rationale for their selections.16,19,40 Theoretical calculations show that the C8–H bending Raman stretch of guanine at 1255 cm−1 could be ideal to develop the calibration plot,41 thus we selected this SERS stretch for our studies. As shown in Fig. 3C and E for microRNA-10b and microRNA-96, respectively, a continuous increase in C8–H stretch intensity is displayed when corresponding nanoplasmonic sensors were incubated in various concentrations of microRNA solution. The SERS intensity of the C8–H bending mode versus the concentration of microRNAs is appeared to be linear over the entire concentration range (Fig. 3D and F, Tables S3 and S4†). The limit of detections (LODs) for microRNA-10b and microRNA-96 calibration was calculated using a published method that follows:40,42
| LOD = 103m/k fM |
Here, m is the relative standard deviation of the blank and k is the slope of the SERS intensity versus microRNA on the concentration curve. Our LODs for microRNA-10b and microRNA-96 are calculated at 1.13 pM and 0.030 pM, respectively. Importantly, this represents a 10-fold improved LODs in comparison to previously reported SERS-based microRNA assays.38 We believe that the high SERS sensitivity of our nanoplasmonic sensors is due the intensified hotspots at the sharp tips and edges of the Au TNPs, as previously shown through discrete dipole approximation calculations.32–34
Successful implementation of the developed biosensors in liquid biopsy-based cancer diagnostics requires unprecedentedly high specificity, specifically to avoid false test results.1 First, the specificity, which is the ability to unambiguously identify the analyte of our nanoplasmonic sensors, was investigated through fluorescence confocal imaging of labeled target microRNAs. We used microRNA-10b as a model system to study the specificity of the nanoplamonic sensors. Fig. 4A confirms that our nanoplasmonic sensors do not display any fluorescence (in the absence of microRNAs). Similarly, when Au TNPs were functionalized only with 3-MP (without -ssDNA-10b) and then incubated in a microRNA-10b-FAM solution, no fluorescence signals are detected (data not shown). Further, when the 3-MP functionalized Au TNPs were incubated in a -ssDNA-10b solution, no fluorescence is detected (Fig. 4B). However, when the nanoplasmonic sensors, which were constructed to detect microRNA-10b, were incubated in a microRNA-10b-FAM solution followed by copious rinsing, confocal imaging shows bright green fluorescence (Fig. 4C). Additionally, a control experiment was performed by incubating a MPTMS functionalized glass coverslip in -ssDNA and microRNA directly without the presence of Au TNPs. The obtained SERS and fluorescence signals showed no results (data not shown). These experimental data suggest the formation of a -ssDNA-10b/microRNA-10b-FAM duplex. Metallic nanostructures are capable of enhancing fluorescence signal through the PEF mechanism, in which LSPR properties of the nanostructure increase the radiative decay rate of the fluorophore.43 To reduce fluorescence quenching due to FRET processes between the nanostructure and fluorophore, it is important that the fluorophore resides within a particular distance from the metal surface. Recent studies showed that a distance of 7.0 to 16.2 nm, corresponding to 18 to 45 nucleotides, between Au nanostructures and fluorophores provides the strongest fluorescence enhancement.28 Importantly, microRNA-10b contains 23 nucleotides and thus the spacing between FAM molecules and Au TNPs is within the reported distance for highest fluorescence enhancement. To further confirm our confocal-based imaging results, which demonstrate that nanoplasmonic sensors unambiguously detect microRNAs with high specificity, we incubated microRNA-10b-FAM-attached nanoplasmonic sensors in a RNase H enzyme solution. This solution is expected to break the -ssDNA/microRNA duplex, removing the attached microRNA, to regenerate the original sensor. As predicted, following RNase H incubation, the fluorescence signal completely disappears, as shown in Fig. 4D. As illustrated in Fig. 4E, the fluorescence signal reappears when sensors were again incubated in then microRNA-10b-FAM solution. These results show the fully regenerative capabilities of the developed nanoplasmonic sensors. Sensor regeneration was further confirmed by the decrease or increase of the 1255 cm−1 peak of the C8–H bending mode in the SERS spectra after -ssDNA-10b attachment, 3-MP attachment, microRNA-10b-FAM attachment, incubation in RNase H enzyme, and re-incubation in microRNA-10b-FAM (Fig. S3†).
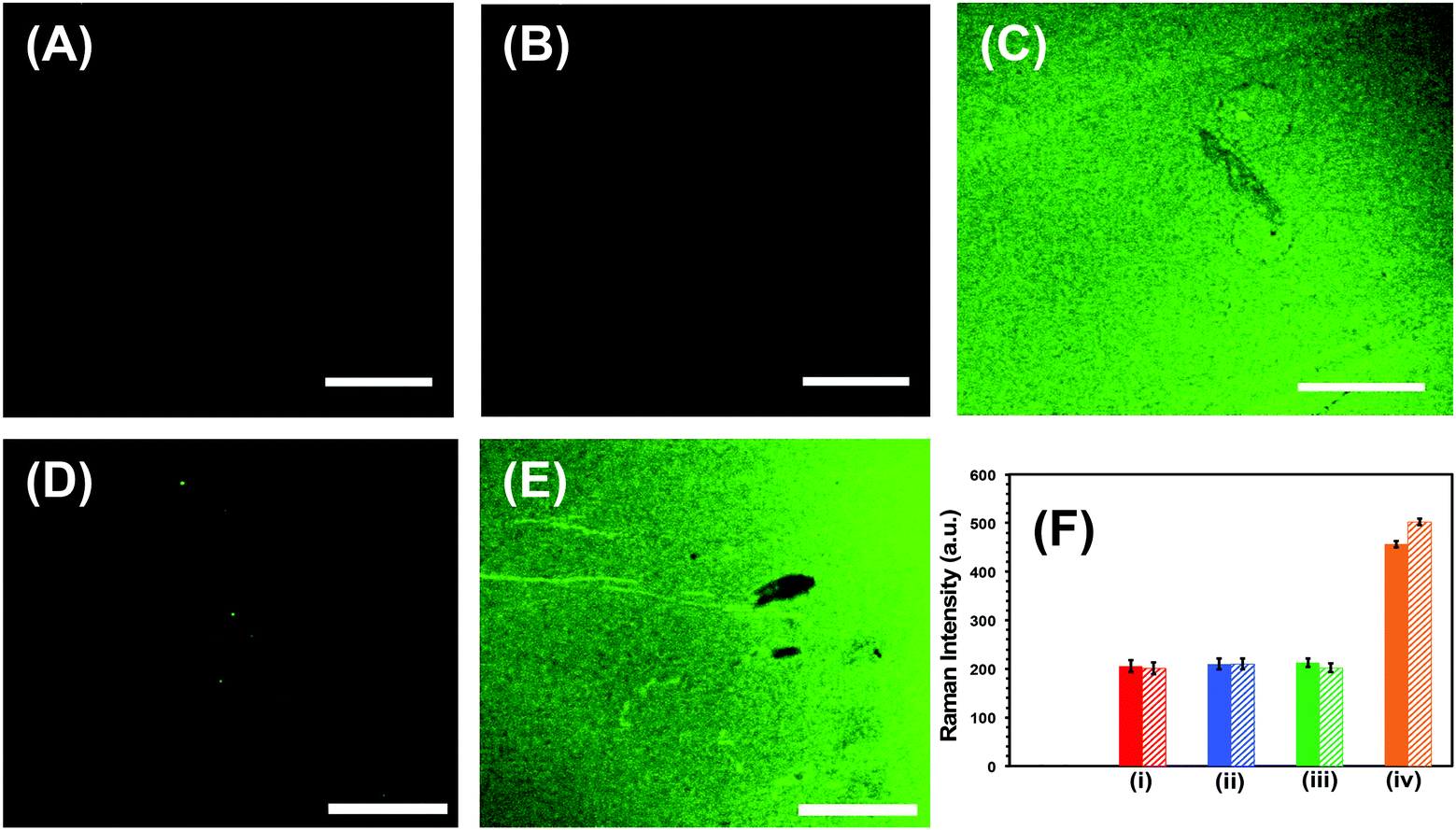 | ||
| Fig. 4 Specificity tests of nanoplasmonic sensors for the microRNA detection. Fluorescence confocal microscopy images of (A) glass substrate attached Au TNPs, (B) mixed -ssDNA-10b and 3-MP functionalized Au TNPs (nanoplasmonic sensor), (C) after incubation of nanoplasmonic sensors in a 100 nM microRNA-10b-FAM solution, (D) After treatment of nanoplasmonic sensors with RNase H enzyme to regenerate the sensor, (E) after re-incubation of nanoplasmonic sensors in a 100 nM microRNA-10b-FAM solution. Scale bars are 50 μm. (F) Normalized Intensity of the C8–H bending stretch at 1255 cm−1 for two different microRNAs (solid bars, microRNA-10b; dashed bards, microRNA-96). Black bars represent glass substrate attached AuNPs (not visible). (i) Red bars represent mixed -ssDNA-10b and 3-MP functionalized Au TNPs or -ssDNA-96 and 3-MP functionalized Au TNPs. (ii) Blue bars represent incubation of nanoplasmonic sensor in a control patient (without the history of cancer) plasma. Green bars represent incubation of nanoplasmonic sensors in a mixture of microRNA-96, 145, 143, and 490-5p solution for the -ssDNA-10b functionalized Au TNPs, or a mixture of microRNA-10b, 145, 143, 490-5p solution for the -ssDNA-96 functionalized Au TNPs. These microRNAs are non-complementary to either -ssDNA-10b or -ssDNA-96. Orange bars represent incubation of nanoplasmonic sensors either in 100 nM microRNA-10b or microRNA-96 solution. | ||
Next, we investigated the specificity of our label-free assay for microRNA-10b and microRNA-96 detection using the SERS technique, as shown in Fig. 4F and Fig. S4.† Here, the nanoplasmonic sensors, which are capable of detecting microRNA-10b, were incubated in a mixture of microRNA-96, microRNA-145, microRNA-143, and microRNA-490-5p. The levels of these microRNAs are commonly altered in patients with a history of bladder cancer.13 SERS analysis shows that the intensity of the C8–H bending mode at 1255 cm−1 does not increase when compared to the original nanoplasmonic sensors. This is because microRNA-96, microRNA-145, microRNA-143, and microRNA-490-5p are non-complementary to -ssDNA-10b. However, when the same nanoplasmonic sensor was incubated in the complimentary microRNA-10b solution, an increase in SERS intensity of the C8–H bending mode is observed. Thus, by measuring the Raman intensity in the presence of either non-complimentary/complementary microRNAs, we can unequivocally conclude the high specificity of our nanoplasmonic sensors towards targeted microRNAs. Taken together, our multimodal fluorescence and SERS analyses not only confirm the high specificity of the nanoplasmonic sensors towards the microRNA assay, but also demonstrate the reusability of the newly developed label-free technique. Sensor regeneration is a particularly an important aspect as the ability for repeated measurements not only provides unmatched accuracy, but also it is commercially important in low- and middle-income countries.
To demonstrate the translational aspects of our newly developed technology for clinical POC cancer diagnostics, we successfully assayed two oncogenic microRNAs (microRNA-10b and microRNA-96) directly in plasma from metastatic bladder cancer patients. Fig. 5A demonstrates microRNA-10b and microRNA-96 levels in six patients. It should be noted that all patients were categorized in the metastatic stage; however, the level of microRNA varies from one to another. To mitigate false positive and false negative test results, we re-analyzed the two nanoplasmonic sensors that measured the highest (patient 2 shows a high microRNA-10b level that could arise from false positive response) and the lowest (patient 5 shows a low microRNA-10b level that could lead to false negative response) microRNA-10b concentrations. It is expected that both sensors that were incubated in the patient plasma containing -ssDNA/microRNA-10b duplexes. As shown in Fig. 5B, SERS intensities decrease following RNase H enzyme treatment. Re-incubation of the regenerated sensors into microRNA-10b-FAM results in nearly the same SERS intensity values, as shown in Fig. 4F. Finally, we also characterized the nanoplasmonic sensor containing microRNA-10b-FAM by fluorescence confocal microscopy, which reveals excellent specificity (Fig. 5C). A similar SERS study was also conducted for microRNA-96 involving patient 3 and 6 (see Fig. 5D) as described for microRNA-10b.
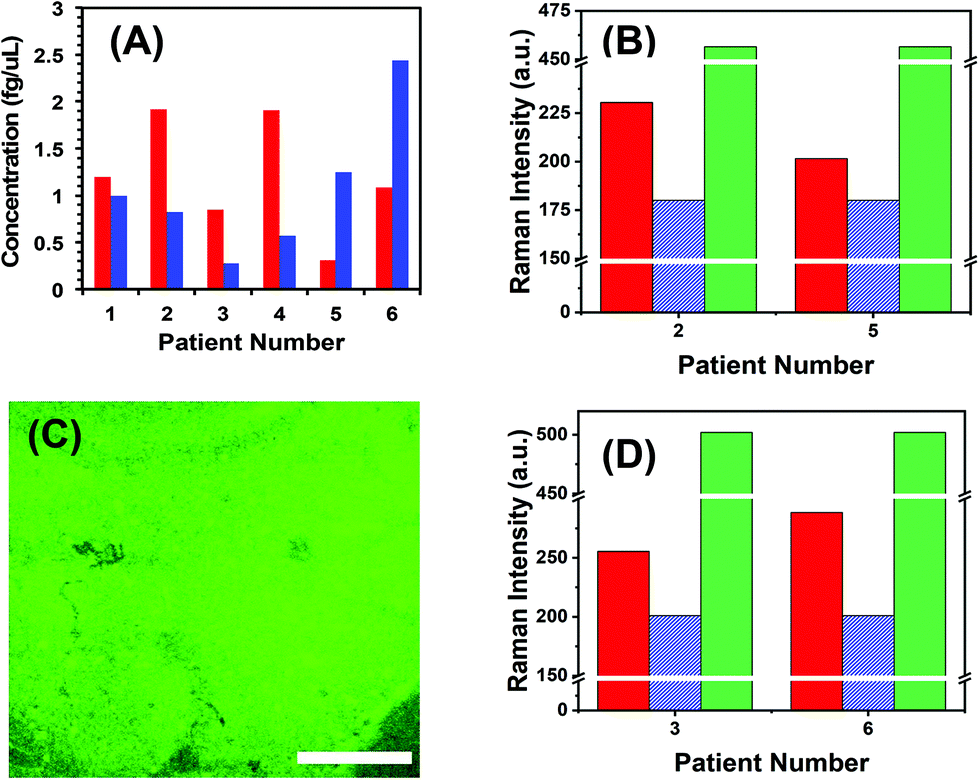 | ||
| Fig. 5 Nanoplasmonic sensor-based liquid biopsy for cancer detection. (A) SERS-based microRNA quantification in six different bladder cancer-related patient plasma samples. microRNA-10b and microRNA-96 are shown in red and blue bars, respectively. (B) Examination of false test resulting by measuring SERS intensities for two different patients containing the highest (patient 2) and the lowest (patient 5) microRNA-10b levels. Red solid bars represent the Raman intensity of C8–H bending stretch at 1255 cm−1, blue dashed bars after regeneration of nanoplasmonic sensors through RNase H treatment, and green dotted bars are after incubation of nanoplasmonic sensors in 100 nM microRNA-10b-FAM solution. (C) A representative fluorescence confocal image of the nanoplasmonic sensor after incubating in 100 nM microRNA-10b-FAM solution. (D) Examination of false test resulting by measuring SERS intensities for two different patients containing the lowest (patient 3) and the highest (patient 6) microRNA-96 levels. The bars are color coordinated as shown in panel (B). Scale bars is 50 μm. | ||
To the best of our knowledge, this is the first example where a SERS-based technique has been successfully implemented in the microRNA assay of clinically relevant samples without any sample processing. Experimental data confirm that the developed nanoplasmonic sensors did not compromise sensitivity or specificity aspects upon incubation in patient plasma, and the concentrations that were determined are true values.
Conclusion
In conclusion, we have designed and fabricated SERS-based, highly sensitive, specific, and regenerative nanoplasmonic sensors for the first time. These sensors were capable of quantifying oncogenic microRNAs at sub-fg μL−1 concentrations directly from patient plasma, thus obviating the complications associated with current gold-standard qRT-PCR-based technologies. The unique LSPR properties of Au TNPs were utilized to achieve a combined dual-detection SERS and fluorescence analysis that allows mitigation of false test results. The newly developed, label-free microRNA assay technique has unmatched potential to expand SERS-based biomarker quantification in the early detection of cancers through liquid biopsies. Importantly, this holds promise in advancing POC clinical diagnostics, which could be performed using a handheld/portable Raman instrument. Taken together, we believe our multimodal, innovative detection approach, which has been validated using plasma from bladder cancer patients samples, should also be applicable to other cancers (e.g., colon, breast, prostrate, and pancreatic) as well as various diseases (e.g., cardiovascular, Alzheimer, and infectious) that involve circulating nuclei acids (e.g., DNAs, long non-coding RNAs, and microRNAs) as biomarkers.8Conflicts of interest
There is no conflicts to declare.Acknowledgements
This work is supported by the National Science Foundation, award number CBET-1604617. We thank Dr Daniel Minner for valuable comments and suggestions. We would like to thank Indiana Clinical and Translational Sciences Institute, which was supported, in parts, by Award Number UL1TR002529 from the National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award.References
- J. Das and S. O. Kelley, Angew. Chem., 2020, 59, 2554–2564 CrossRef CAS PubMed.
- D. P. Bartel, Cell, 2004, 116, 281–297 CrossRef CAS PubMed.
- A. Esquela-Kerscher and F. J. Slack, Nat. Rev. Cancer, 2006, 6, 259–269 CrossRef CAS PubMed.
- D. J. Lockhart and E. A. Winzeler, Nature, 2000, 405, 827–836 CrossRef CAS PubMed.
- J. Lu, G. Getz, E. A. Miska, E. Alvarez-Saavedra, J. Lamb, D. Peck, A. Sweet-Cordero, B. L. Ebert, R. H. Mak, A. A. Ferrando, J. R. Downing, T. Jacks, H. R. Horvitz and T. R. Golub, Nature, 2005, 435, 834–838 CrossRef CAS PubMed.
- F. Petrocca and J. Lieberman, RNA Biol., 2009, 6, 335–340 CrossRef CAS PubMed.
- J. Wang, J. Chen and S. Sen, J. Cell. Physiol., 2016, 231, 25–30 CrossRef CAS PubMed.
- Z. Jin, D. Geißler, X. Qiu, K. D. Wegner and N. Hildebrandt, Angew. Chem., Int. Ed., 2015, 54, 10024–10029 CrossRef CAS PubMed.
- S. Fang, H. J. Lee, A. W. Wark and R. M. Corn, J. Am. Chem. Soc., 2006, 128, 14044–14046 CrossRef CAS PubMed.
- B. N. Johnson and R. Mutharasan, Anal. Chem., 2012, 84, 10426–10436 CrossRef CAS PubMed.
- M. Wanunu, T. Dadosh, V. Ray, J. Jin, L. McReynolds and M. Drndić, Nat. Nanotechnol., 2010, 5, 807–814 CrossRef CAS PubMed.
- G. K. Joshi, S. Deitz-McElyea, T. Liyanage, K. Lawrence, S. Mali, R. Sardar and M. Korc, ACS Nano, 2015, 9, 11075–11089 CrossRef CAS PubMed.
- T. Liyanage, A. N. Masterson, H. H. Oyem, H. Kaimakliotis, H. Nguyen and R. Sardar, Anal. Chem., 2019, 91, 1894–1903 CrossRef CAS PubMed.
- G. K. Joshi, S. Deitz-McElyea, M. Johnson, S. Mali, M. Korc and R. Sardar, Nano Lett., 2014, 14, 6955–6963 CrossRef CAS PubMed.
- D. Graham, B. J. Mallinder and W. E. Smith, Angew. Chem., Int. Ed., 2000, 39, 1061–1063 CrossRef CAS PubMed.
- Y. C. Cao, R. Jin and C. A. Mirkin, Science, 2002, 297, 1536–1540 CrossRef CAS PubMed.
- A. J. Qavi and R. C. Bailey, Angew. Chem., Int. Ed., 2010, 49, 4608–4611 CrossRef CAS PubMed.
- J. L. Abell, J. M. Garren, J. D. Driskell, R. A. Tripp and Y. Zhao, J. Am. Chem. Soc., 2012, 134, 12889–12892 CrossRef CAS PubMed.
- J. D. Driskell and R. A. Tripp, Chem. Commun., 2010, 46, 3298–3300 RSC.
- L. Qi, M. Xiao, X. Wang, C. Wang, L. Wang, S. Song, X. Qu, L. Li, J. Shi and H. Pei, Anal. Chem., 2017, 89, 9850–9856 CrossRef CAS PubMed.
- S. Tian, O. Neumann, M. J. McClain, X. Yang, L. Zhou, C. Zhang, P. Nordlander and N. J. Halas, Nano Lett., 2017, 17, 5071–5077 CrossRef CAS PubMed.
- P. S. Gaikwad and R. Banerjee, Analyst, 2018, 143, 1326–1348 RSC.
- S. K. Vashist, Biosensors, 2017, 7, 62 CrossRef PubMed.
- K. M. Mayer and J. H. Hafner, Chem. Rev., 2011, 111, 3828–3857 CrossRef CAS PubMed.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442–453 CrossRef CAS PubMed.
- P. L. Stiles, J. A. Dieringer, N. C. Shah and R. P. V. Duyne, Annu. Rev. Anal. Chem., 2008, 1, 601–626 CrossRef CAS PubMed.
- J. R. Lombardi and R. L. Birke, J. Chem. Phys., 2012, 136, 144704 CrossRef PubMed.
- Z. Mei and L. Tang, Anal. Chem., 2017, 89, 633–639 CrossRef CAS PubMed.
- J.-F. Li, C.-Y. Li and R. F. Aroca, Chem. Soc. Rev., 2017, 46, 3962–3979 RSC.
- G. K. Joshi, K. A. Smith, M. A. Johnson and R. Sardar, J. Phys. Chem. C, 2013, 117, 26228–26237 CrossRef CAS.
- T. Liyanage, M. Nagaraju, M. A. Johnson, B. B. Muhoberac and R. Sardar, Nano Lett., 2019, 20, 192–200 CrossRef PubMed.
- L. Scarabelli, M. Coronado-Puchau, J. J. Giner-Casares, J. Langer and L. M. Liz-Marzán, ACS Nano, 2014, 8, 5833–5842 CrossRef CAS PubMed.
- T. Liyanage, A. Rael, S. Shaffer, S. Zaidi, J. V. Goodpaster and R. Sardar, Analyst, 2018, 143, 2012–2022 RSC.
- G. K. Joshi, S. L. White, M. A. Johnson, R. Sardar and P. K. Jain, J. Phys. Chem. C, 2016, 120, 24973–24981 CrossRef CAS.
- G. K. Joshi, K. N. Blodgett, B. B. Muhoberac, M. A. Johnson, K. A. Smith and R. Sardar, Nano Lett., 2014, 14, 532–540 CrossRef CAS PubMed.
- J. D. Driskell and R. A. Tripp, Chem. Commun., 2010, 46, 3298–3300 RSC.
- F. Madzharova, Z. Heiner, M. Guehlke and J. Kneipp, J. Phys. Chem. C, 2016, 120, 15415–15423 CrossRef CAS PubMed.
- L. Qi, M. Xiao, X. Wang, C. Wang, L. Wang, S. Song, X. Qu, L. Li, J. Shi and H. Pei, Anal. Chem., 2017, 89, 9850–9856 CrossRef CAS PubMed.
- B. L. Lambert, S. Gronert, F. H. Shurvell and A. D. Lightner, Organic Structural Spectroscopy, 2011 Search PubMed.
- Z. Wang, S. Ye, N. Zhang, X. Liu and M. Wang, Anal. Chem., 2019, 91, 5043–5050 CrossRef CAS PubMed.
- B. Giese and D. McNaughton, Phys. Chem. Chem. Phys., 2002, 4, 5161–5170 RSC.
- Y. He, X. Yang, R. Yuan and Y. Chai, Anal. Chem., 2017, 89, 8538–8544 CrossRef CAS PubMed.
- Principles of fluorescence spectroscopy, ed. J. Lakowicz, Springer, 3rd edn, 2006 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures for Au TNPs synthesis, various spectroscopy and microscopy characterizations, and additional extinction and Raman spectra, and raw Raman data. See DOI: 10.1039/d0an00538j |
| ‡ These authors contributed equally to this work. |
| § Integrated Nanosystems Development Institute, Indiana University-Purdue University Indianapolis, 723 W. Michigan Street, Indianapolis, Indiana 46202, United States. |
| This journal is © The Royal Society of Chemistry 2020 |