
Fluorescent gels: a review of synthesis, properties, applications and challenges
Yuqiong
Li
a,
David James
Young
b and
Xian Jun
Loh
 *a
*a
aInstitute of Materials Research and Engineering, 2 Fusionopolis Way, Innovis, #08-03, 138634, Singapore. E-mail: liyqc@imre.a-star.edu.sg; lohxj@imre.a-star.edu.sg
bCollege of Engineering, IT & Environment, Charles Darwin University Darwin, Northern Territory 0909, Australia
First published on 30th May 2019
Abstract
Soft material gels are important biomaterials for tissue engineering and biomedicine. Fluorescent polymer gels possess the additional property of being light emitting, which endows this type of material with advanced applications in chemical and environmental sensing, photonics, bioimaging, and electrofluorochromic devices. In this review, we provide an overview of the recent development in the field of fluorescent polymer gels, with particular focus on hydrogels, inclusive of methods of synthesis of such gel materials, their mechanisms of fluorescence, physical and chemical properties, and their applications as demonstrated in laboratory studies. In recognition of the diverse chemical structures of fluorescent gel materials, examples of supramolecular fluorescent hydrogels and organogels are also included to provide a comprehensive survey of the development in the field. White-light emitting hydrogels are highlighted in this review due to their unique luminescence property, and their potential function in intelligent information displays. Challenges and future perspectives of this type of material are discussed, keeping in view continuing research efforts to engineer the material for macroscopic production and commercial usages.
 Yuqiong Li | Dr Yuqiong Li completed her Bachelor of Science at Cornell University (USA), Master of Engineering at Massachusetts Institute of Technology (USA) and PhD cum Diploma of Imperial College (DIC) from Imperial College London (UK) in 2016. A chemical engineer by training, she is currently a scientist at the Institute of Materials Research and Engineering, A*STAR Singapore. Her research focuses on polymers and photonic materials, and she is interested in developing these materials for biomedical applications or environmental remediation. She is a member of the Royal Society of Chemistry (MRSC) and an associate fellow of the Higher Education Academy (AFHEA, UK). |
 Xian Jun Loh | Xian Jun Loh is a polymer chemist working in the inter-disciplinary field of biomaterials. He is a senior scientist and currently the department head of the Soft Materials at the Institute of Materials Research and Engineering (IMRE). He is the current Vice President of the Singapore National Institute of Chemistry and a fellow of the Royal Society of Chemistry. He is a pioneering developer of biodegradable urethane-based thermogels. His main research interests are in the design of supramolecular and stimuli-responsive polymers and hydrogels for biomedical and personal care applications. |
Introduction
Fluorescent gels are soft materials that can be polymeric in nature or be made of small molecular gelators via the supramolecular assembly of a well-defined complex of molecules, held together by either covalent or noncovalent interactions that are typically dynamic and reversible.1 Such gels differ from conventional hydrogels or organogels in their light emitting properties, but retain their extended networks that can be extensively swollen with water (hydrogels) or organic solvent (organogels). Hydrogels, in particular, can potentially be a useful three-dimensional scaffold for tissue engineering2 due to their flexibility and water retention ability, similar to biological tissues.3–11 The prefix “hydro” indicates hydrophilicity although most gel structures do not dissolve in water.12 However, the hydrophilicity renders such gel structures capable of holding large amounts of water in their three dimensional networks.Light emitting materials that are embedded within these fluorescent gels can be used for sensing of chemical recognition events, such as for the detection of minute quantities of biochemical materials.13 Spectrofluorometers and related detection technologies are well established, and different assays have been designed for simultaneous high-throughput chemical screening in miniaturized systems.14 Incorporating fluorescent materials either into the gel skeleton, often polymer in nature, or as adorned fluorescent moieties, results in fluorescent polymer gels that possess several unique features which endow this type of material with potential applications as sensors,15–18 optical and electrical devices,19,20 imaging agents,21 in biomedicine22–25 or for memory and display devices.26,27
In this review, we discuss a myriad of luminescent mechanisms exhibited by the different fluorescent gels, with particular focus on fluorescent polymer hydrogels, which include ways of assembly of different luminescent entities (for example, small fluorescent dye molecules, quantum dots, carbon dots, lanthanide transition metals etc.) onto hydrogel networks (for example, supramolecular gel, polymer based matrix, gelatin hydrogels etc.). Sophisticated variations in structural architectures of complex gel materials are made possible by orthogonal metal coordination and host–guest interaction,28 incorporation of fluorophores into polymer matrices29 by infusion, self-assembly26 and co-assembly of gel hybrid system via bio-conjugation,30 or even co-assembly of peptide motifs and transition metal ions.31 Representative examples are selected here and their methods of synthesis, properties, applications, with future prospects are discussed. While the main topic of this review is on fluorescent polymer hydrogels, the readers are advised to read the following excellent reviews on fluorescent supramolecular hydrogels with low molecular weight gelators24,32 and fluorescent nanogels,33 both have slightly different focus and complement the scope of this work in terms of literature contributions to the field of fluorescent gels.
Mechanism of fluorescence
Fluorescence is the process of light emission by a material (e.g. quantum dots, fluorophore dyes, polymers with intrinsic fluorescence) that absorbs light at a lower wavelength (higher energy) and emits light of longer wavelength (lower energy). Fluorescence stops within a few hundred nanoseconds once the light source is removed. The ratio of the number of photons emitted to the number of photons absorbed is the fluorescence quantum yield, and the average time that a fluorophore remains in its excited state before the emission of photons is its fluorescence lifetime. The emission mechanisms of different fluorescent moieties associated with typical gel materials are discussed below.Fluorescence energy can be transferred between two chromophores within close physical proximity to each other via Förster (or fluorescence) resonance energy transfer (FRET).34 In this process, energy transfers from an excited donor chromophore to an acceptor chromophore through nonradiative dipole–dipole coupling, the mechanism of which is described by the Jablonski diagram (Scheme 1a). The efficiency of energy transfer is inversely proportional to the sixth power of the distance between donor and acceptor, which means FRET efficiency (E) is extremely sensitive to the distance of separation (r) between the two chromophores, with R0 being the Förster distance between the donor–acceptor pair at which the energy transfer efficiency is 50%.
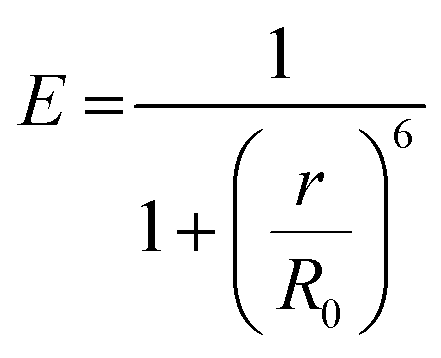
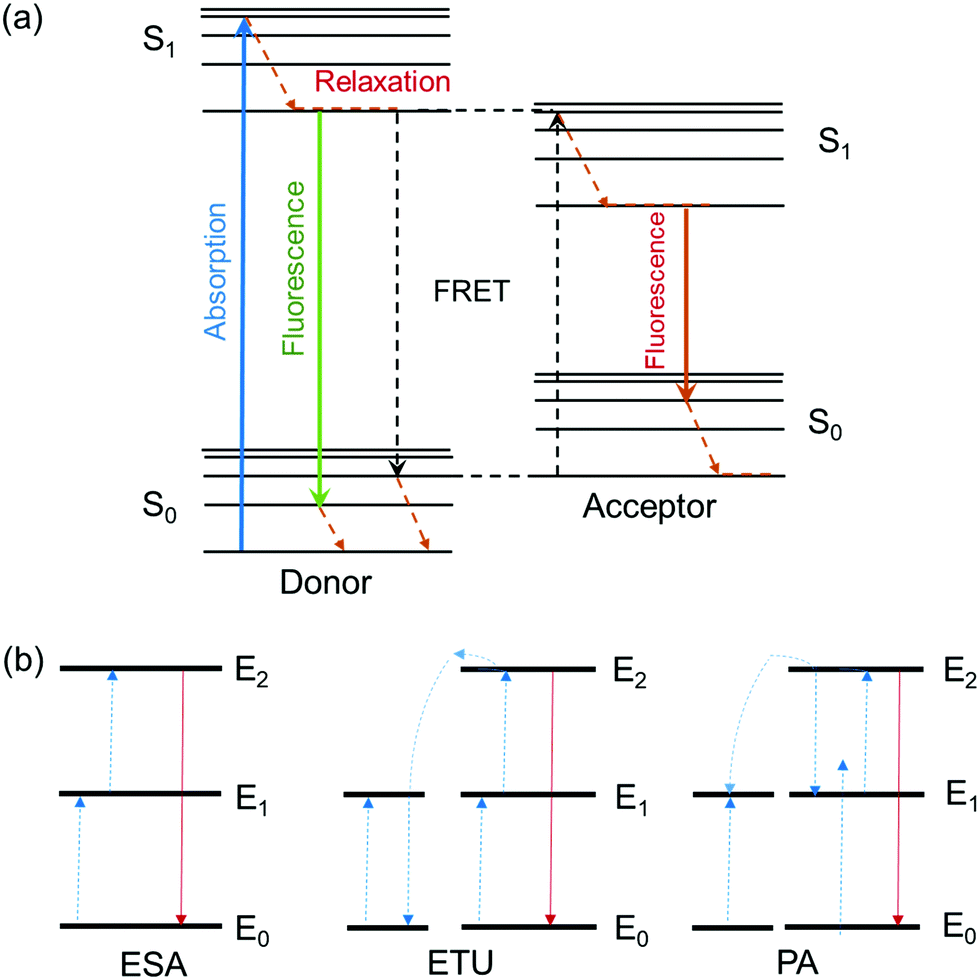 | ||
| Scheme 1 (a) The Jablonski diagram. (b) Principal upconversion processes for lanthanide-doped nanoparticles: (ESA) excited state absorption, (ETU) energy transfer upconversion, and (PA) photon avalanche. The dashed/dotted, dashed, and full arrows represent photon excitation, energy transfer, and emission processes respectively.35 | ||
Fluorescent moieties can be broadly classified as intrinsic or extrinsic fluorophores. Intrinsic fluorophores are naturally existing materials, such as green fluorescent proteins, while extrinsic fluorophores are usually synthetic nanomaterials, e.g. quantum dots. The latter are semi-conducting nanomaterial and the colour and efficiency of their fluorescence is strongly size-dependent with potentially high quantum yields (e.g. 0.65–0.85 for CdSe) and sharp emission bands.36,37 The nanometer-size of these dots enables their incorporation into supramolecular networks, such as polymer hydrogels.
Fluorescent quantum dots are generally heavy metal-based and are used in fluorescent gel materials as the light emitter.38,39 Their carbon counterpart used in gel materials, termed carbon quantum dots, are carbon-based fluorescent nanoparticles, that some have argued emit photons as a result of the quantum confinement effect resulting from their nanoparticle size.15–17,40–45 These fluorescent carbon dots were first extracted by electrophoretic purification of single walled carbon nanotube fragments in agarose gel.40 These materials fluoresced green-blue, yellow, and orange. The quantum yield of the yellow fluorescent dots was found to be 0.016, with a maximal lateral dimension less than 18 nm as determined by atomic force microscopy.40 Carbon dots have also been produced by laser ablation of a carbon target, followed by reflux treatment in an aqueous nitric acid solution.41 Upon surface passivation by organic capping agents such as poly-(ethylene glycol) in aqueous suspension, the quantum-size carbon analogues emit bright luminescence with quantum yields ranging from 0.04 to 0.10. Their carbon-based chemistry implies benign biocompatibility for biomedical imaging and therefore superiority relative to silicon-based quantum dots in this respect.43 Water soluble carbon dots also have better stability in hydrogels intended for biomedical applications, in comparison to semi-conductor quantum dots that are typically soluble in organic solvents and possess inherent bio toxicity due to their metallic nature.
A second, anti-Stokes type emission exists for photon upconversion materials. This process involves absorption of two or more photons followed by emission of light at a shorter wavelength than the absorption wavelength, for example absorption at infra-red wavelengths and emission of visible light.46,47 Typical upconversion processes can be divided into three broad categories: excited state absorption (ESA), energy transfer upconversion (ETU) and photon avalanche (PA). All three processes involve the sequential absorption of two or more photons.35 (Scheme 1b) One well-studied upconversion nanomaterial is lanthanide-doped nanoparticles that often consist of a transparent material (fluorides of ytterbium, e.g. NaYbF4, or oxides of gadolinium, e.g. Gd2O3) doped with lanthanide ions. The most common pairs of ions used in photon upconversion are erbium–ytterbium (Er3+, Yb3+) and thulium–ytterbium (Tm3+, Yb3+). The ytterbium ions are antennas that absorb light in the infrared region around 980 nm, and transfer it to the upconverter erbium ions or thulium ions. A characteristic green and red emission is obtained for an “activator-host” Er3+, Yb3+ pair, or blue and red emission for a Tm3+, Yb3+ pair.35 Such lanthanide transition ions are used in the synthesis of white light emitting gels due to their ability to fine-tune the emission color from the stoichiometry of different ions.29,48–51
Perhaps more intriguing is the surprising upconversion phenomenon also observed with some carbon dots materials.44 Li et al. have reported the facile, one-step, alkali-assisted electrochemical fabrication of carbon dots with sizes of 1.2–3.8 nm that exhibit size-dependent photoluminescence and excellent upconversion luminescence properties.44 The quantum yield of these carbon dots with yellow emission was about 0.12.44 Significantly, these carbon dots could be excited by long-wavelength light in the visible to infrared spectrum with upconverted emissions in the ultra-violet to blue light range.44 These upconversion characteristics may be attributed to multiphoton processes.43,44 and are also useful in the design of photo catalysts (TiO2/CQDs and SiO2/CQDs systems) to harness the full sunlight spectrum.44
Synthesis of fluorescent gels
A common fabrication strategy for fluorescent gels is incorporating fluorophore entities into the gel system.21,24,31,49,50 The gel matrix is formed via self-assembly of supramolecular gelators, while the photo luminescent tag molecules are introduced via either diffusion into the gel network or by co-assembly through orthogonal interaction with highly branched gel matrices.24 Alternatively, the gel body is constructed from fluorescent polymers that form the backbone of the gel network.26,52 Broadly classified, structures of gel materials are largely varied and those examples included in this section can be grouped into (1) conventional chemically crosslinked gels with covalent bonding; or (2) cross-linked polymer gels by non-covalent bond (coordination bonds, electrostatic bond, hydrogen bond, host–guest complexation, etc.); (3) low molecular weight gelators.Lu et al. have reported the synthesis of a multifunctional metallacage-core supramolecular organogel via metal-coordination self-assembly of cis-Pt(PEt3)2-(OTf)2, tetraphenylethene (TPE)-based sodium benzoate ligands and linear dipyridyl ligands28 (Fig. 1). Further host–guest interactions between the crown-ether moieties and bisammonium linkers form a final supramolecular polymer gel at high concentrations. The TPE derivatives act as fluorescent emitters. The higher-order of structure hierarchy and the inclusion of the dynamic metal-coordination and host–guest interactions means the gel network is capable of responding to multiple stimuli, and self-healing under conducive environments to restore the molecular bonds. The rigidity of the metallocages enhances the stiffness of the gel.
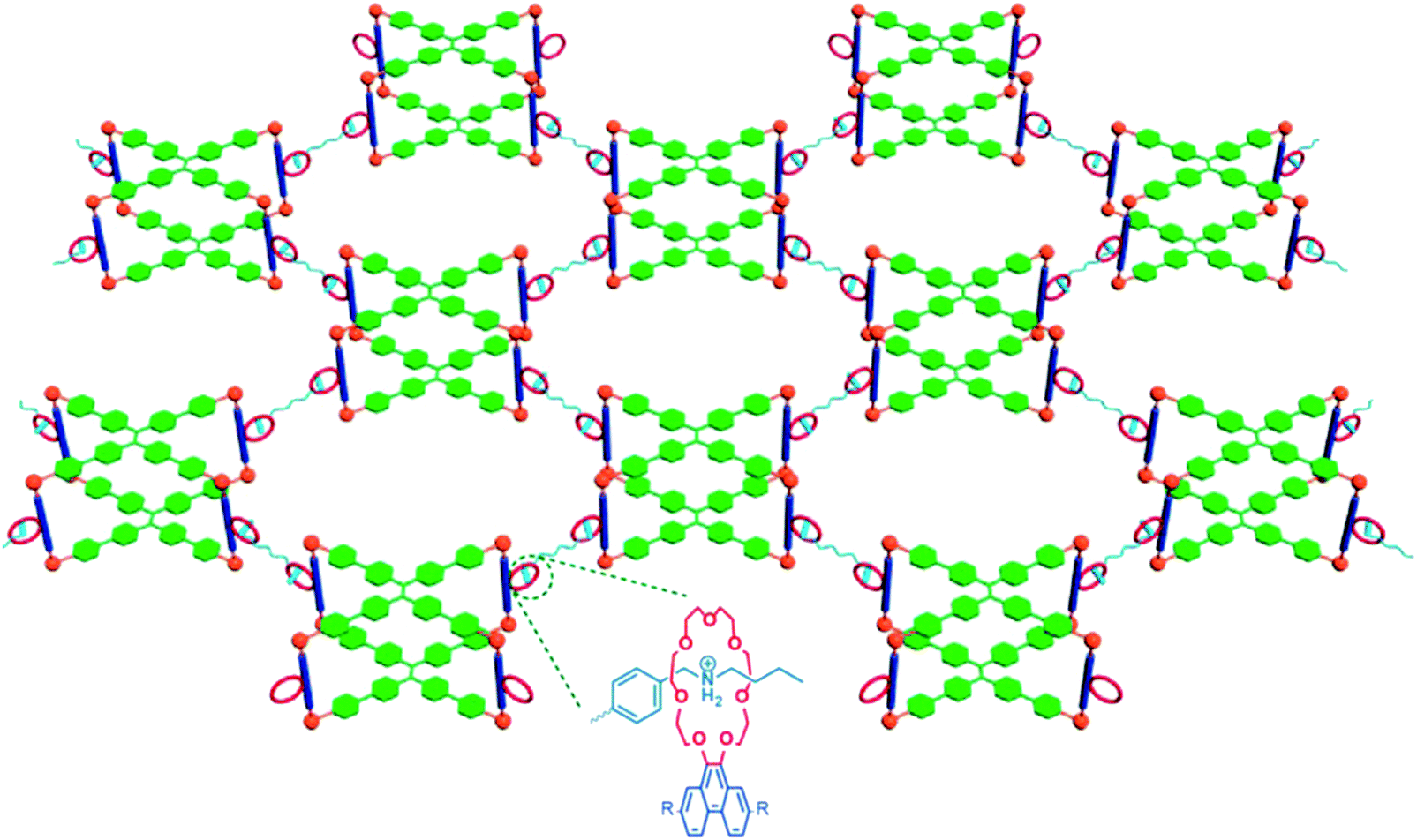 | ||
| Fig. 1 Metallocage-core supramolecular polymer gel network.28 21 C7-functionalized metallacages undergo self-assembly, and form cross-linked supramolecular polymer network with the addition of bisammonium salt cross-linkers. | ||
Ji et al. have reported a second example of host–guest mediated supramolecular polymer organogels with similar crown ether cum ammonium salt chemistry52 (Fig. 2). This network was constructed from a fluorescent conjugated polymer with dibenzo 24-crown-8 and secondary ammonium salts. The intensity of fluorescence of this gel network is enhanced by the presence of ionic salt species, pH change and heating. These stimuli cause disassembly of the conjugated gel network, and the fluorescence that was weakened due to aggregation of polymer chains is restored.
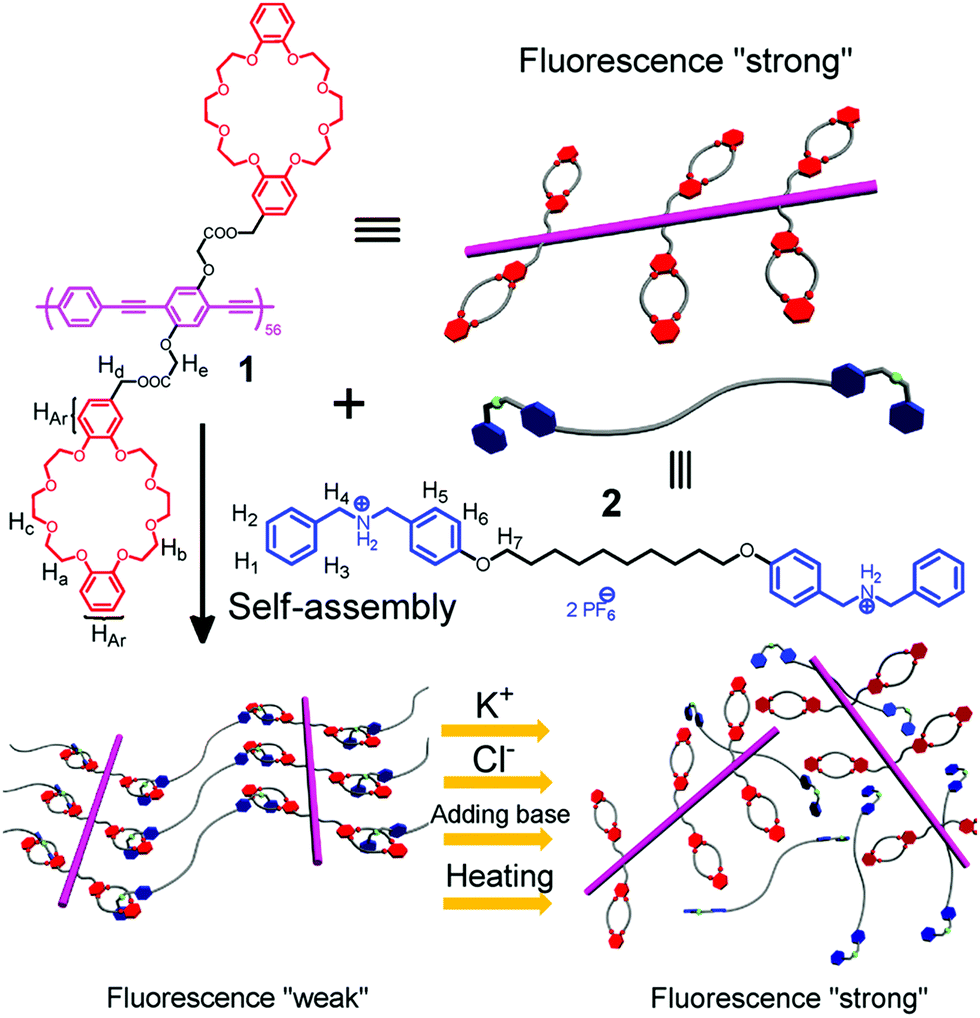 | ||
| Fig. 2 The formation of a supramolecular cross-linked conjugated polymer network and its disassembly induced by different ionic signals.52 | ||
Polymers with intrinsic fluorescence, e.g. fully π-conjugated polymer backbones, have been used by Elmalem et al. to develop mechanically strong luminescent hydrogels from just a single polymeric component.30 Specifically, a fully π-conjugated fluorine-phenylene backbone was chosen to form the gel framework. Flexible zwitterion-carrying side chains solubilize the system and act as cross-linkers. Cycling of temperature conditions converts gel–sol in a reversible manner, with minor hysteresis. This study reports a reproducible supramolecular assembly strategy that facilitates fabrication of gels from several semi-conducting conjugated polymers that can directly assemble into simple gel structures.
Other types of fluorescent supramolecular hydrogels are constructed by physically mixing the fluorophores with hydrogelators, or soaking as-formed hydrogels in dye solutions for diffusive incorporation.53,54 However, such non-covalent bonding of dye molecules with the gel network may later result in leaching of the fluorescent entities and/or aggregation of chromophores, resulting in loss of luminescence.31
Both peptide and DNA have been the biomaterials of choice for the construction of fluorescent hydrogels. One interesting study by Kim et al. reported a self-assembled peptide hydrogel consisting of Fmoc-diphenylalanine as a biosensing platform through the encapsulation of enzymes and quantum dots, which were physically immobilized within the gel matrix in situ by mixing with monomeric peptide solution.55 Xia et al. reports a hydrogel based co-assembly of peptides and transition metal ions.31 The as-designed system is mechanically stable enough for printing. The peptide hydrogelators encapsulate transition metal ions in a cage-like network through covalently linked ligands at specific positions. This effectively prevents dye aggregation and concentration-induced fluorescence quenching. Such metal–peptide ligand complexes also stabilize the gel network, making the gel even more stable. The hydrogel matrix is highly fluorescent and so a potentially versatile material for extrusion-based 3D printing of soft electronics.
DNA has also been an element of construction for hydrogels, with the incorporation of silver nanoparticles for both fluorescence and antibacterial activity.56,57 Guo et al. outlined a method to prepare silver nanocluster hydrogels.57 The nucleic acid containing systems self-assemble into Y-shape structures, where the loop region of the hybridized strands includes the specific sequence for the stabilization of red- or yellow-fluorescent Ag-nanoclusters. Geng et al. produced large, linear DNA chains by enzymatic polymerization that formed a distinctive network through physical entanglement.56 The entangled DNA chains were weaved into a 3D hydrogel with cytosine domains for high affinity with Ag nanoclusters.
Fluorescent moieties in fluorescent gels
The various fluorescent moieties, known as fluorophores, can be broadly classified as intrinsic fluorophores, for example, green fluorescent proteins;58 and extrinsic fluorophores, that include a list of small molecular fluorescent dyes, fluorescent conjugated polymers, Aggregation Induced Emissive dyes like tetraphenylethene (TPE),28 thienoviologen,26 lanthanides transition metal ions,29,48,50,51,59,60 quantum dots,16,38,39 and carbon dots15–17,29,48 are some examples of fluorescent moieties that have been attached to fluorescent gel matrices.Small molecule fluorescent dyes
Small molecular fluorescent dyes are excellent emitters with intense fluorescence and small molecular sizes that facilitate conjugation to the bulk gel network. Examples of such small fluorescent molecules include amino- or carboxyl-functionalized cyanides (e.g. cy3, cy5), pyridines (e.g. tripyridine, dipyridine), and fluorescein families.Kim et al. reported the synthesis of highly fluorescent blue-light emitting hydrogel consist of small fluorescent molecule tripyridine substituents, endowing the gel system with great potential for optical applications.61 The unique gelation capability of the hydrogel is due to the cooperative effects of the π–π stacking and the intermolecular hydrogen bonding interactions. Self-assembled supramolecular structure of the gelators further enhance the fluorescence emission at neutral pH 7. Such blue-light emissive hydrogels can be important materials in photochemistry and OLED fields.61
Aggregation induced emissive dyes
Aggregation induced emissive dyes exhibit enhanced fluorescence when they are in an aggregated state, e.g. high concentrations of dye molecules, within a confined polymer network, or in a sol–gel medium.62–65 Conventional luminogens lose the majority of their emission in such physical states due to photon quenching. Aggregation induced emissive dyes are particularly valuable materials with sensing applications. Confinement of these dyes within self-assembled polymer chains restricts intra-molecular rotation of, for instance, tetraphenylethene molecules and causes increased photon emissions from hydrogels made with TPE moieties29 (Fig. 3).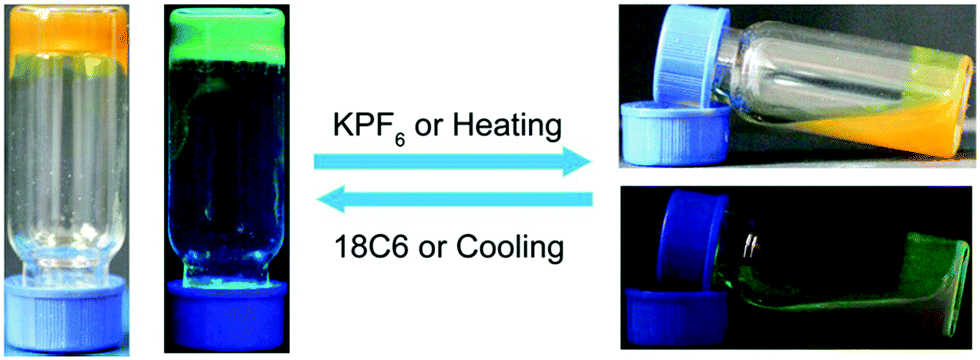 | ||
| Fig. 3 Optical and fluorescent photographs of the reversible gel–sol transition of TPE-based fluorescent polymer hydrogels.29 (left) Upon cooling or 18C6 addition, TPE-hydrogel in its solidified gel state with blue-green fluorescent emission image side by side; (right) Upon KPF6 addition or heating, the hydrogel turns liquid, and lost its fluorescence intensity as seen in the stacked images of the hydrogel in normal light and UV illumination. | ||
Lanthanide ions
Lanthanide-ligand metal coordination polymers have been much studied for the design of advanced supramolecular hydrogels because of their unique metal-coordinated fluorescence that can be easily modulated over a wide spectrum by varying the stoichiometry of the lanthanide ions.50,51,59,60,66 These dynamic bonds between the lanthanide ion and ligand also leads to reversible stimuli-responsive properties and self-healing structures. Such features improve overall hydrogel performance, including longer lifetimes.The lanthanide ions can be introduced as bridging links that form branched 2D and 3D coordination polymers and networks. Kotova et al. have prepared a fluorescent gel structure from Eu ions and a tripodal terpyridine-based ligand.59 Using a similar approach, Chen et al. incorporated red, green and blue fluorescent lanthanide complexes to give a white-luminescent fluid using appropriate lanthanide ions complexed by terpyridyl (Terpy)-end-capped poly(ethylene glycol) (4-Arm-PEG) ligands (Fig. 4).50 Titration of two lanthanide chromophores Eu (red):Tb (green) led to soft gels with a spectrum of color emission, with an intense white light emission at a Eu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Tb ratio of 4:96 (Fig. 5). Martínez-Calvo et al. showed that such a design strategy for lanthanide-based gels is feasible with a different ligand, using the pyridine-2,6-dicarboxylic acid derivative (4,4′-((pyridine-2,6-dicarbonyl)bis-(azanediyl))bis(methylene)-dibenzoic acid). Such ligands facilitate a 3D polymer gel network with the lanthanide ions as the chelating centers. The formulation was reproducible at achieving luminescent red (Eu) or green (Tb) hydrogels with remarkably self-healing characteristics as indicated by the dynamic host–guest interactions.66
:Tb ratio of 4:96 (Fig. 5). Martínez-Calvo et al. showed that such a design strategy for lanthanide-based gels is feasible with a different ligand, using the pyridine-2,6-dicarboxylic acid derivative (4,4′-((pyridine-2,6-dicarbonyl)bis-(azanediyl))bis(methylene)-dibenzoic acid). Such ligands facilitate a 3D polymer gel network with the lanthanide ions as the chelating centers. The formulation was reproducible at achieving luminescent red (Eu) or green (Tb) hydrogels with remarkably self-healing characteristics as indicated by the dynamic host–guest interactions.66
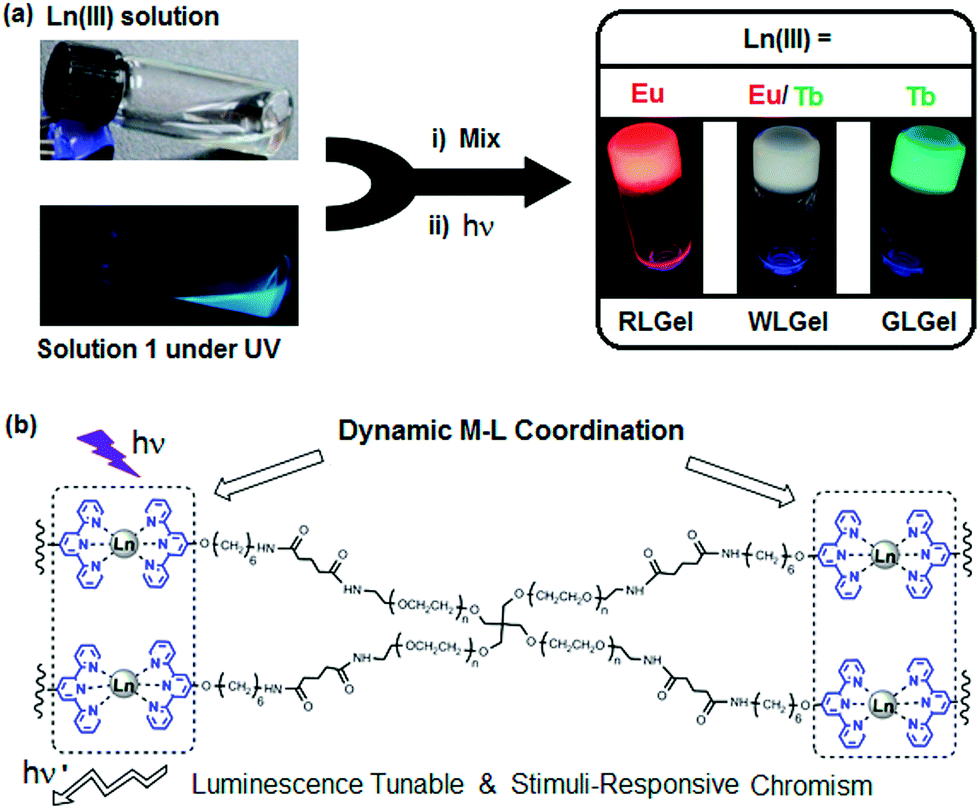 | ||
| Fig. 4 (a) Schematic preparation of Ln(III) coordination-based luminescent metallogels under UV light; (b) chemical structure of terpy-end-capped 4-arm PEG polymer cross-linked via Ln-terpy coordination.50 | ||
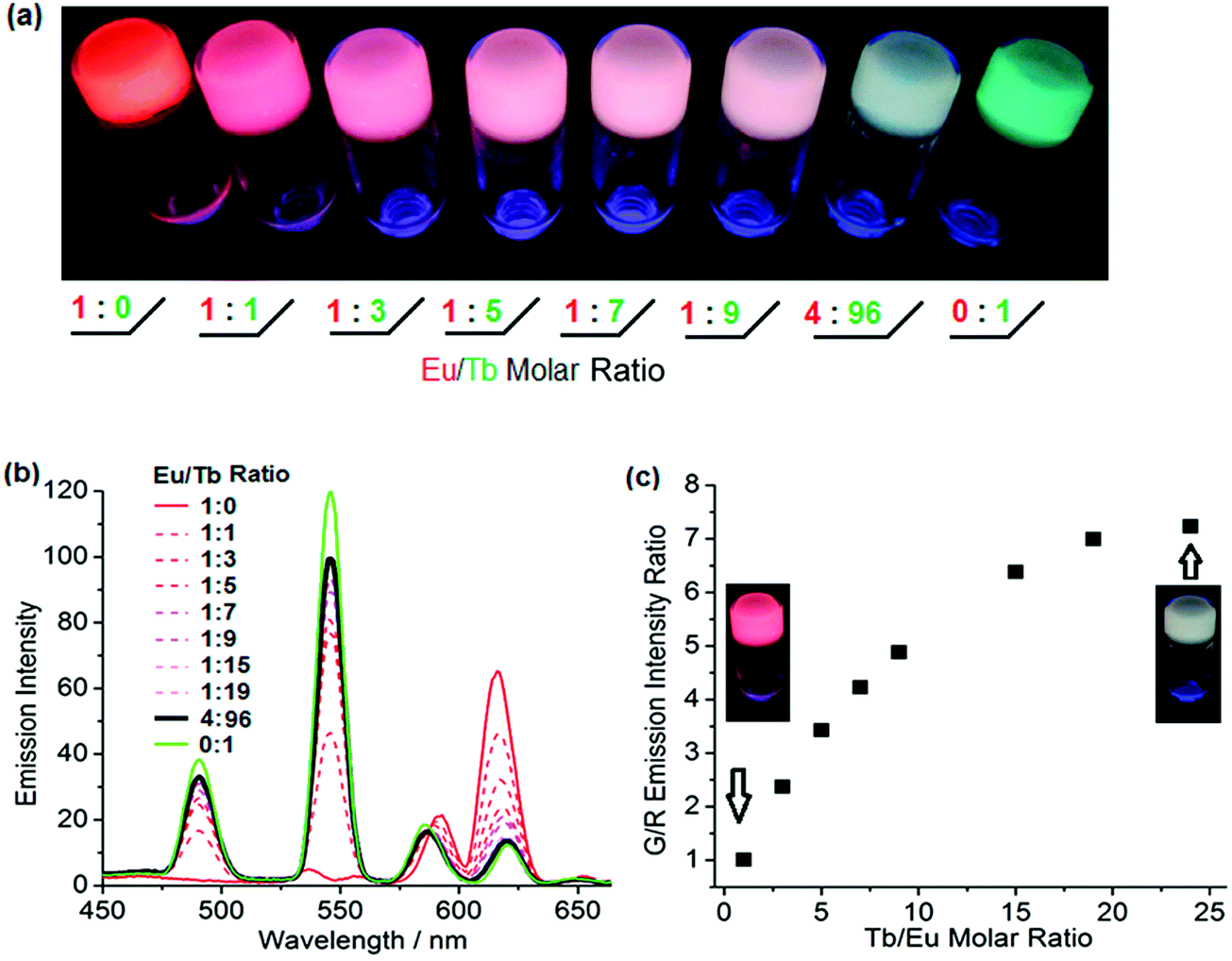 | ||
| Fig. 5 (a) Luminescence tuning of lanthanide based metallogels under UV irradiation; (b) emission spectra of metallogels of various Eu:Tb ratio; (c) Job's plot of green/red emission intensity ratio as a function of the Tb:Eu molar ratio.50 | ||
Quantum dots
Photo luminescent nanocrystals such as quantum and carbon dots are promising materials for coding elements because different wavelengths of emission and intensity can be encoded in to the design to achieve a high variability of fluorescence output. Both types of nanocrystals have sharp emission bands that correlate with the sizes of the nanoparticles; therefore both the color and the size information are discrete and distinct to each dot. This allows orthogonal tagging of the nanoparticles to gel matrices and discrete sensing mechanisms.15,17,38,45Zhao et al. have developed a hydrogel suspension array with quantum dot-encoding for full multiplexing label-free DNA detection.38 Hydrogel photonic beads are micro carriers in the suspension array. Specific hybridization of target DNA and the cross-linked, single stranded DNA in the gel induced hydrogel shrinking, and a corresponding blue shift in the Bragg diffraction peaks of the beads that correspond to the amount of target DNA detected.38
Carbon dots
Hybrid hydrogels with carbon dots have similarly been developed. Cayuela et al. synthesized a carbon dot–hydrogel hybrid nanomaterial from low molecular weight gelators with carbon dot–polymer gel conjugation.45 These functionalized carbon dots enhanced gelation and their inclusion in the hydrophobic gel resulted in a dramatic fluorescence enhancement. Gogoi et al. used chitosan based carbon dots in an agarose hydrogel as a hybrid sensing material with a color change for heavy metal detection within 5–10 seconds of exposure17 (Fig. 6).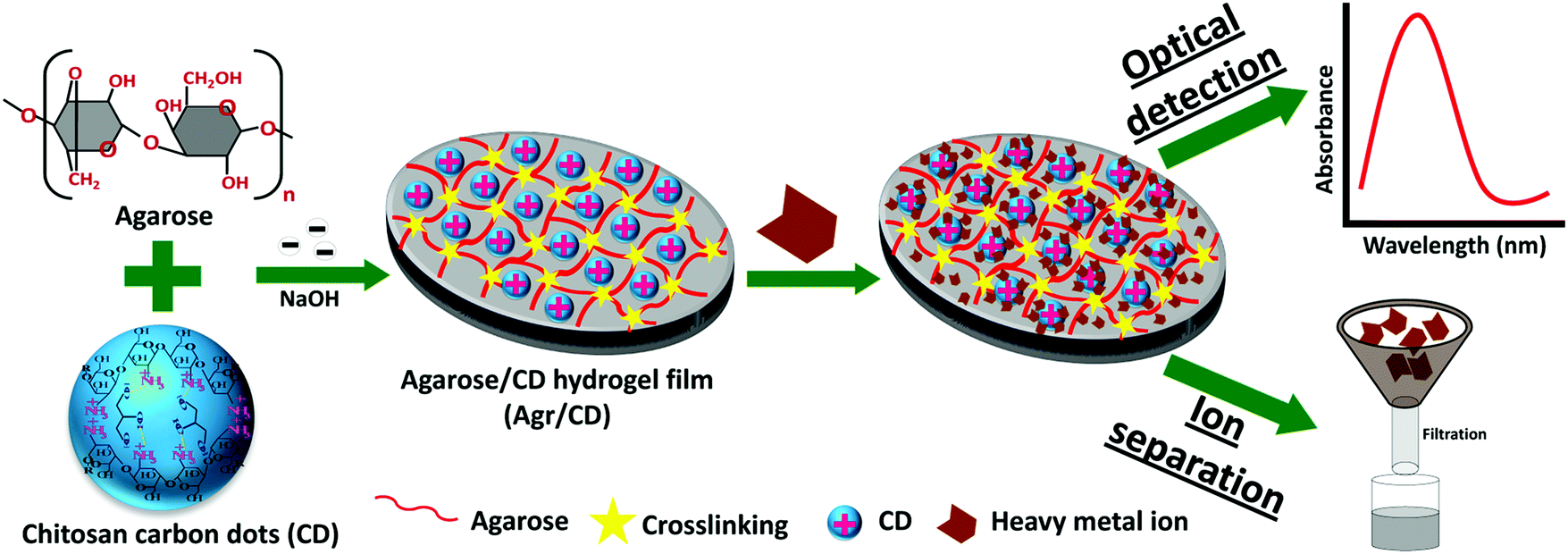 | ||
| Fig. 6 Fabrication of an agarose/carbon-dot/hydrogel film optical sensor for heavy metal ion detection.17 | ||
Another study, with a twist of engineering, by Bhattacharya et al. constructed a hybrid, carbon dot – hydrogel matrix for bacteria detection.15 Bacterially-secreted esterase cleaved ester bonds within the hydrogel scaffold, leading to fluidization of the gel network and aggregation of the embedded carbon dots with concomitant fluorescence quenching. This system was highly sensitive to the esterase secretions and so could distinguish between bacterial species by their fluorescence modulation.
A summary of selected types of gels and fluorophores referenced in this paper can be found in Table 1 below.
| Types of fluorophores | Types of gel structures | Ref.a |
|---|---|---|
| a Italic emphasis indicates hydrogels and bold emphasis indicates organogels. | ||
| Small molecular fluorescent dyes | Supramolecular structure | 24, 61 and 67 |
| Polymer | 22 | |
| Aggregation induced emissive dyes | Supramolecular structure | 28 |
| Polymer | 37 | |
| Lanthanide ions | Ligand-metal coordination polymer | 50, 51, 59, 60 and 66 |
| Peptide | 31 | |
| Quantum dots | Polymer | 15, 19, 38 and 39 |
| Peptide | 55 | |
| Carbon dots | Polymer | 17, 29 and 45 |
| Fluorescent conjugated polymers | Supramolecular structure | 68 |
| Polymer | 26, 30 and 52 | |
| Silver nanoparticles | DNA | 57 |
Properties of fluorescent gels
Fluorescent gels, in particular fluorescent polymer hydrogels, possess several properties that are intrinsic to the gel structure. Stimuli-responsiveness is the most important feature. Heat,39,52 pH,69 chemical sensing,14,16,17,52 mechanical agitation25 and electrofluorochromic stimulation26 are some examples of changes in the external environment that can induce fluorescence fluctuations in the gel.Ishiwari et al. have reported a solid-state fluorescent Ca2+ ion sensor for realizing extracellular Ca2+ imaging using a bioinspired design.21 Aggregation-induced emissive (AIE) luminogens were attached to a chemically-crosslinked polyacrylic acid gel, which underwent single-chain aggregation in the presence of Ca2+. Fluorescence emission was enhanced through the AIE effect when the polyacrylic acid chains aggregated, thereby successfully translating sub-millimolar concentrations of Ca2+ ions into fluorescence signals.
Most of the sensing mechanisms are rooted in non-covalent interactions between the gel network and external stimuli. Ji et al. selected crown ether macrocyclic hosts for preparing multi-responsive materials.52 Their gel body was constructed from a fluorescent conjugated polymer network and cross-linked by a bisammonium salt via host–guest interactions. At gelation state, the fluorescence of the conjugated polymer was quenched due to aggregation of single chains. Luminescence intensity could be restored when the polymer network was disassembled by multi-stimuli. The “host” dibenzo 24-crown-8 ether and the complementary “guest” ammonium salt formed a strong interaction that caused the aggregation of polymer chains. Treatment with K+, Cl−, pH or temperature change disrupted the host–guest bonding and collapsed the gel network. Hence, the system could be used as four types of sensors: a cationic K+ sensor, an anion Cl− sensor, a pH sensor and a temperature sensor.
The dynamic and reversible non-covalent bonds between the polymer chains in a gel structure constitute the basis for self-healing in fluorescent polymer hydrogels. The metallocage-core gel developed by Lu et al. shows macroscopic self-healing properties.28 A crack in the gel can be self-healed within several minutes (Fig. 7). The dynamic and reversible metal coordination and host–guest interactions are responsible for the gel healing process. Such non-covalent bonds are broken when the gel cracks. However, due to their dynamic and reversible nature, healing can be triggered to recover the mechanical damage. The presence of rigid metallocages not only enhances the stiffness of the gels, but also improves the self-healing process as these cages are less likely to be damaged in a crack, therefore preserving most of the gel shape.
 | ||
| Fig. 7 Photographic images of the self-healing process of the supramolecular hydrogel. The gel was cut and left standing to heal for (a) 0, (b) 0.5, (c) 1.0, (d) 1.5, (e) 2.0, (f) 2.5, (g) 3.0, (h) 4.0 min.28 | ||
Some fluorescent polymer hydrogels are mechanically strong. Elmalem et al. for instance, have presented a gel from a combination of a rigid, fully π-conjugated polymer backbone and zwitterionic polymer side-chains that self-assemble to form a mechanically robust and elastic gel network.30 The strength of the gel rigidity can be tuned via polymer loading, polymer chain length, temperature and ionic concentration variations.
White-light emitting hydrogels
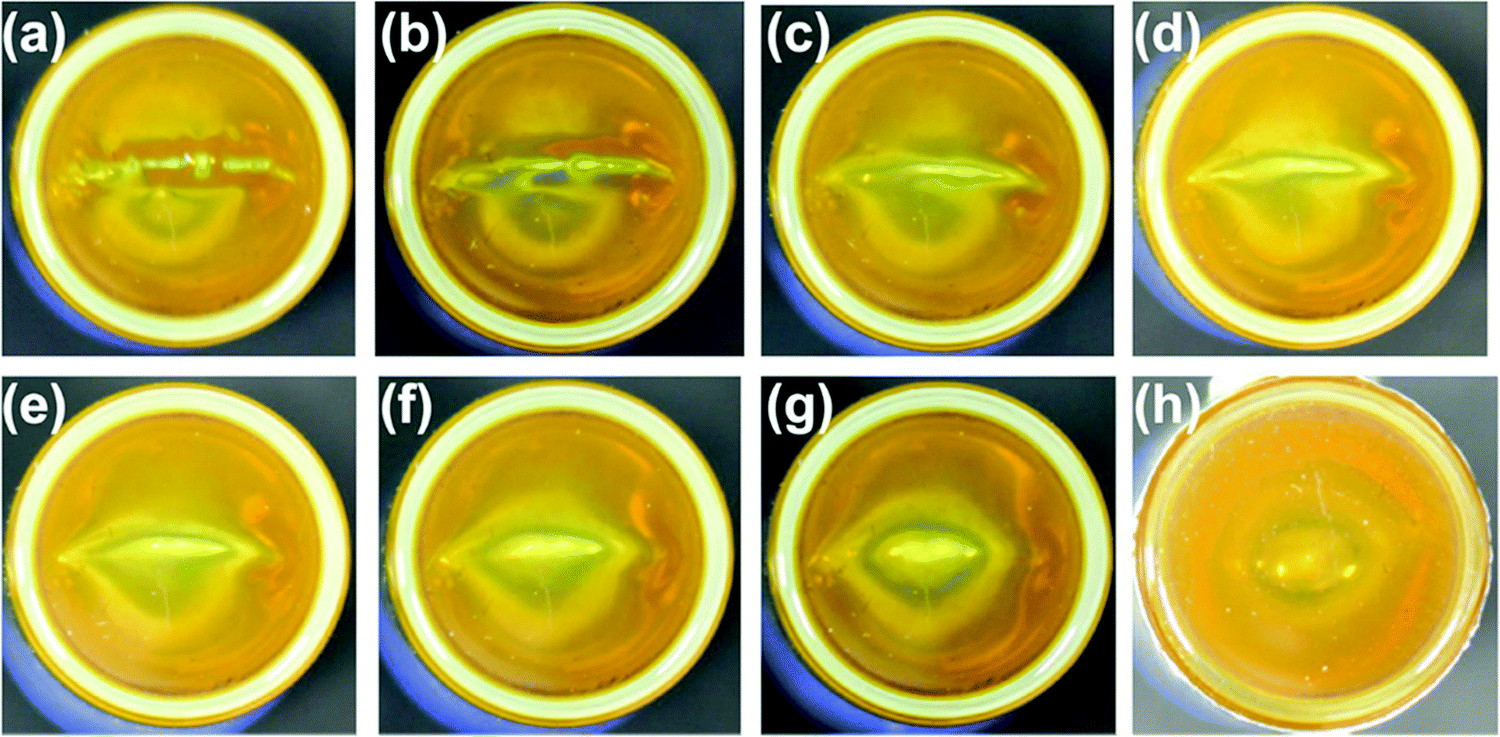
White-light emitting polymer hydrogels can potentially be used to construct intelligent information display and storage devices.68 An advantage of white luminescence is sensitivity to stimuli that results in chromic shifts.68 A common approach for preparation of white light emitting gels is by adjusting the molar proportions of fluorescent chromophores that emit the three primary colors (red, green and blue)48 or two complementary colored luminescent dyes.49Zhu et al. have prepared a white-light emitting hydrogel by incorporating lanthanide ions and carbon dots into a gel body of polyacrylamide and poly(acrylic acid) (Fig. 8).48 By tuning the stoichiometric ratio of blue-light emitting carbon dots, green- (Tb3+) and red-light (Eu3+) emitting lanthanide ions, white luminescence was obtained. Interestingly, this system also acted as a pH sensor (Fig. 9). Under acidic conditions, protonation of blue-light emitting terpyridine caused dissociation of the terpyridine–lanthanide complexes, leading to a decrease in the lanthanide-centered green-and-red emission that weakened the “antenna effect” from the blue light emitters. Under basic condition, dissociation of the terpyridine–lanthanide complex also caused fluorescence quenching, in addition to precipitation of lanthanide ions. Therefore, by exploiting the combined sensitivities of the various emitters, the polymer hydrogels could respond to pH conditions, including stimuli such as chemical vapors (HCl, and NH3) that induce changes in acidity or alkalinity in the environment.
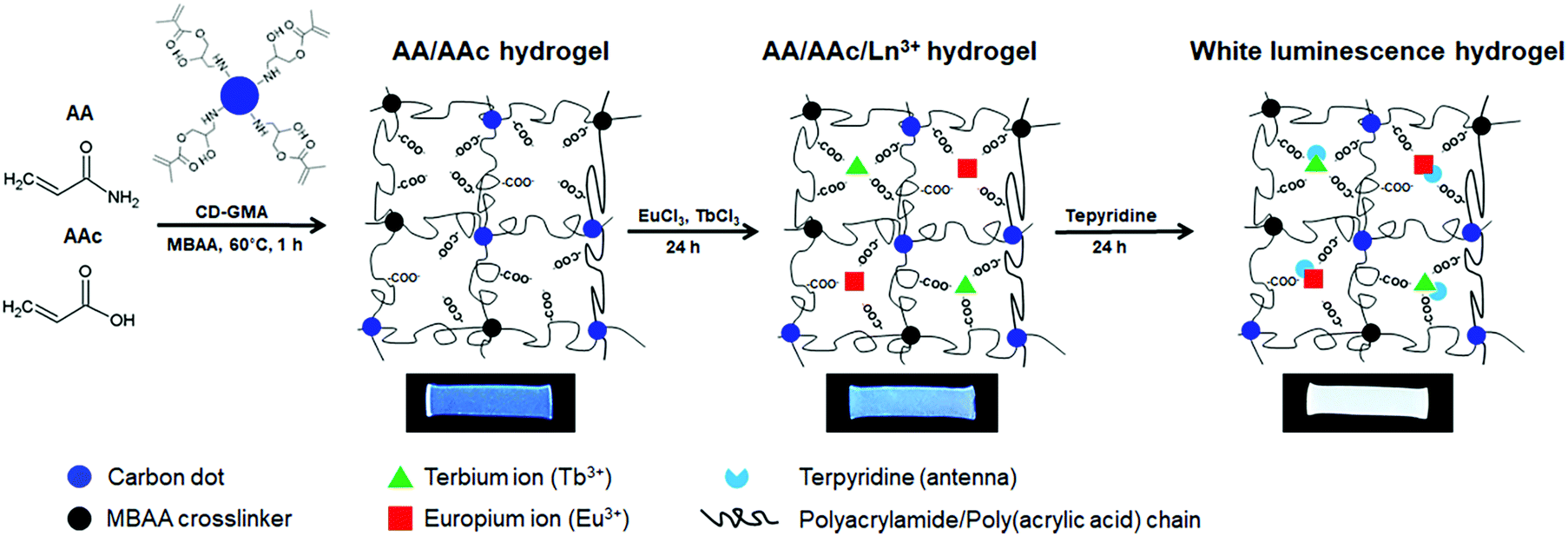 | ||
| Fig. 8 Fabrication process of white-light luminescent hydrogels.48 | ||
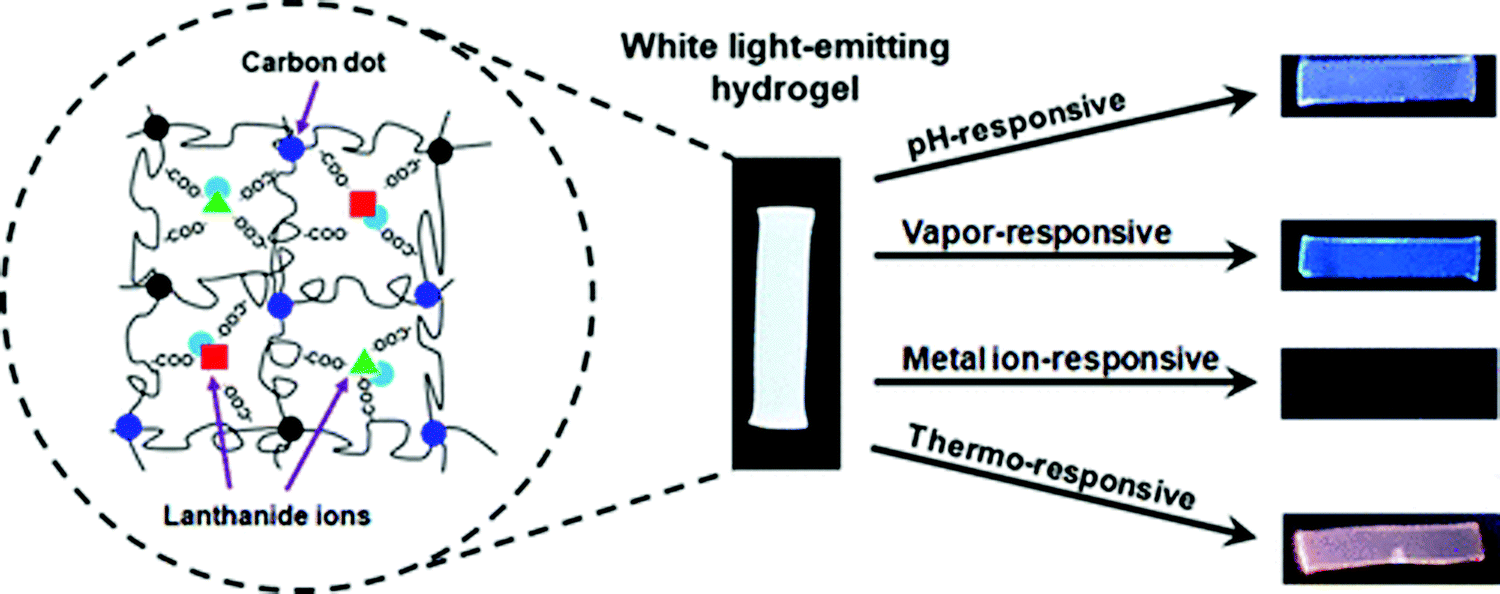 | ||
| Fig. 9 A white light emitting hydrogel sensor incorporating lanthanide ions and carbon dots and color-responsive to environmental cues including pH, chemical vapors, ions, and temperature.48 | ||
Zhao et al. have reported the design of a fluorescent tunable hydrogel that emits intense white light by swelling the hydrogel in solutions containing two complementary coloured dyes.49 This macrocycle-based, supramolecular hydrogel was prepared by a one-pot radical copolymerization reaction with the gel network containing binding sites for orthogonal supramolecular recognition of two dyes, viz. β-cyclodextrin-modified tetraphenylethene (TPECD) and 4-[4-(dimethylamino)styryl]-1-methylpyridinium iodide (DASPI). Tetraphenylethene is a typical blue aggregation induced emissive fluorophore, while DASPI is a twisted intramolecular charge transfer molecule that emits yellow light. When the gel is allowed to swell in the mixture of dye solutions, the self-sorting assembly of the fluorescent molecules exhibited fluorescence resonance energy transfer and color changes reflecting the different ratios of TPECD and DASPI.49
A different approach was taken by Wang et al. who have reported a novel and efficient white-light emitting gel design that involved aggregation of a single fluorescent chromophore.27 This aggregation by dynamic hydrogen bonding of ureidopyrimidinone (UPy) also endowed the fluorescent gel with self-healing properties that could be reversed by heating or change of solvent polarity, and recovered by cooling (or the removal of the polar solvent). Therefore, this fluorescent gel also acted as a sensor for reversible gel–sol transitions induced by environmental changes.
Applications of fluorescent gels
Fluorescent gels find major applications as bioimaging probes for disease diagnosis because of their 3-D structure, high water content, biocompatibility and smart responsivity to physiological stimuli.24 Supramolecular fluorescent hydrogelators that can be easily endocytosed into cells are favorable biological carriers to load imaging agents. Polymeric fluorescent nanofibers form the skeleton of the gel network and can be visualized by fluorescent microscopic channels. Cell–gel interactions can then be distinguished via differentiating the strong emission of the fluorescent nanofiber excitation from the stained cell excitation. A mapping of both the cell and the nanofibers can be accomplished. Overlaying the images of the cells and the fluorescent gel fibers make such luminescent hydrogels not just a tissue culture scaffold, but also an effective tool to reveal the interaction between polymer hydrogel substrates and cells.24,70–72Ikeda et al. have developed a photo-responsive supramolecular hydrogel consisting of 6-bromo-7-hydroxycoumarin-4-ylmethoxycarbonyl (Bhcmoc) tethered with a dipeptide (FF). The hydrogel was formed above a 0.4 wt% (20 mM) gelation concentration.72 An analogue structure consisting of a p-borono-phenylmethoxycarbonyl (BPmoc) unit with a similar dipeptide (FF) was redox-responsive and reacted with H2O2 to release the peptides.72 Glucose oxidase was encapsulated into the gel. H2O2 was produced in situ by the embedded glucose oxidase when glucose was present. The addition of glucose could thereby cause a gel–sol transition. This glucose-sensing response has been applied to control insulin release. FITC-(fluorescein isothiocyanate) labeled insulin was capped inside the glucose-oxidate/BPmoc-FF gel. When glucose was mixed into the solution, gel dissociation led to release of FITC-label insulin and therefore could be used as a diabetes diagnosis platform.
Takeuchi's group presents a long-term in vivo glucose monitoring sensor using glucose-responsive fluorescent hydrogel fibers (Fig. 10).73 The hydrogel was fabricated as a fibrous structure to anchor the sensor at the implantation site for a long-term application. In addition, the lengths of these fibers can be easily manipulated by cutting, to allow control of the amount of fluorescent sensors (Fig. 11) implanted, and facilitate the easy removal of these hydrogel fibers from the implantation site after use. Such polyethylene glycol-bonded polyacrylamide hydrogel fibers can continuously respond to blood glucose concentration variations for up to 140 days, allowing long-term application for in vivo continuous glucose sensing and monitoring.
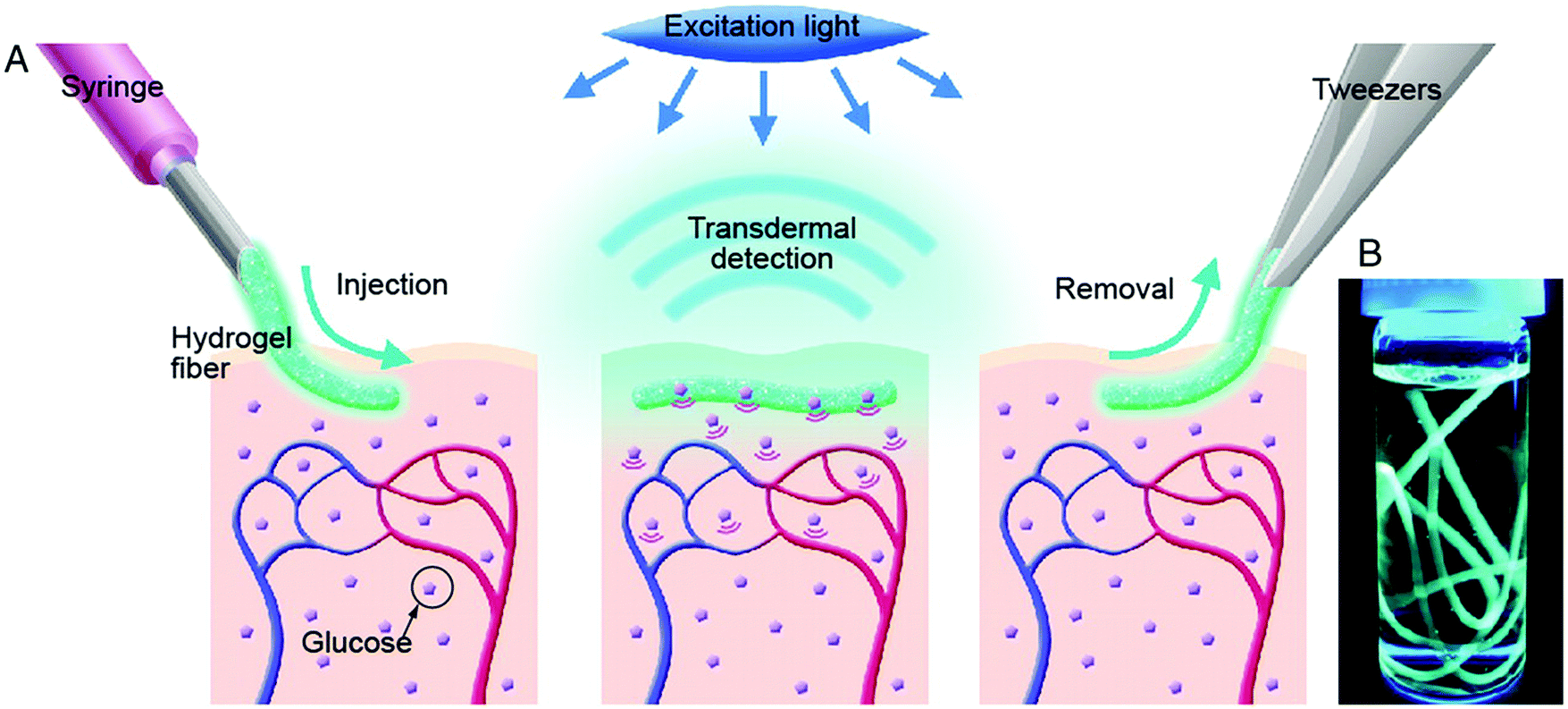 | ||
| Fig. 10 (A) Schematic illustration of the fluorescent hydrogel fiber for long-term in vivo glucose monitoring. The fiber is injectable to the sub-cutaneous tissues. The implant remains at the site for up to 140 days, and transmit fluorescent signals trans-dermally reflecting blood glucose concentration reading. The implant can be later removed. (B) Fluorescent hydrogel fibers in a glass vial with a 50% glucose solution. When the fiber is exposed to UV light, the fluorescence emission indicates glucose-responsive monomer is immobilized inside the hydrogel fibers.73 | ||
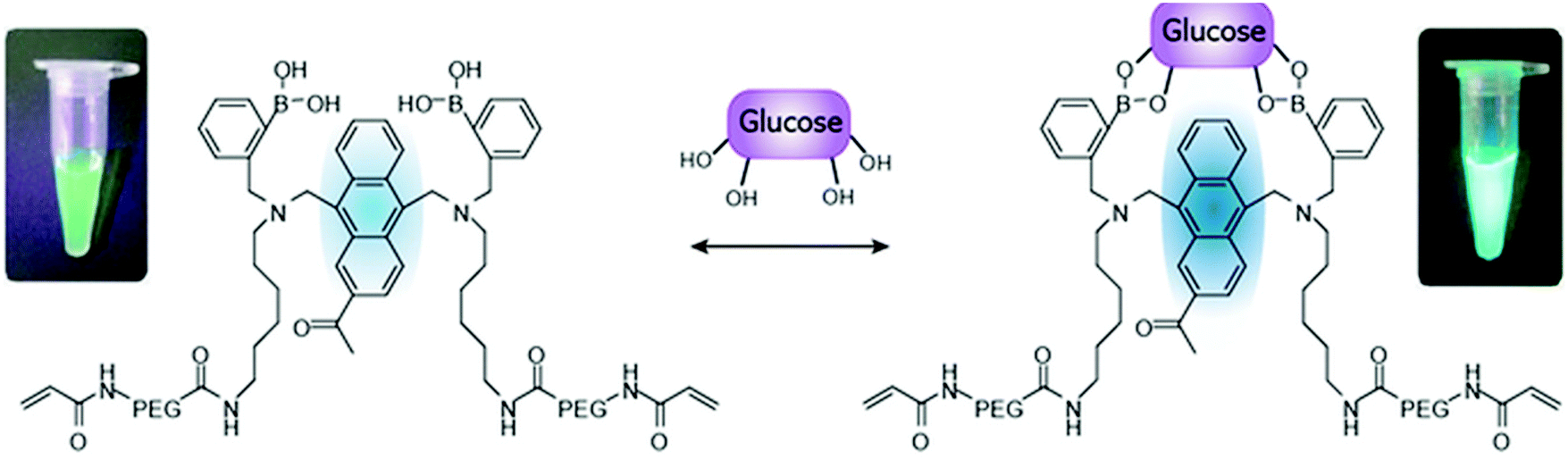 | ||
| Fig. 11 Schematic illustration of the glucose-recognition principle of the glucose-responsive monomer.73 The glucose-responsive monomer is composed of diboronic acids, anthracene acid, PEG, and a vinyl group; these components act as glucose-recognition sites, a fluorogenic site, spacers, and polymerization sites for PAM, respectively. In the absence of glucose molecules, the fluorescence of the anthracene is quenched by a photo-induced electron transfer (PET) that occurs from the unshared electron pair of the nitrogen atom to the anthracene. When glucose molecules bind to diboronic acid, a strong reaction between the nitrogen atom and a boron atom inhibits PET. As a result, the fluorescence of anthracene is higher than under glucose-free conditions.73 | ||
Such molecular recognition sensors based on hydrogels are not limited to glucose monitoring, but can be applied to enzymes and other biomolecules as well. Yoshimura et al. developed an artificial receptor and chemo-sensor that is based on semi-wet hydrogel (Fig. 12).74 Most of the guest-binding molecular recognition events happen in homogenous solution phase, whereas immobilization to solid supports often lead to loss of sensing functionality. Hydrogels provide a semi-wet environment for entrapping enzymes noncovalently, and provide a suitable medium for native peptide/protein binding with enzymes. Furthermore, the miniaturization of the functionalized hydrogel provide a new molecular array chip that enables fluorescent high-throughput assay of multiple analytes (Fig. 13).
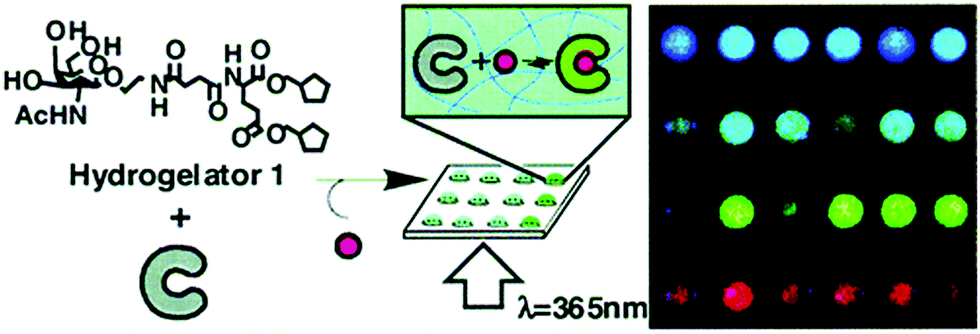 | ||
| Fig. 12 Schematic diagram showing the molecular structure of hydrogelator and the assembly of gel network and receptors into a semi-wet hydrogel array for fluorescent binding assay.74 | ||
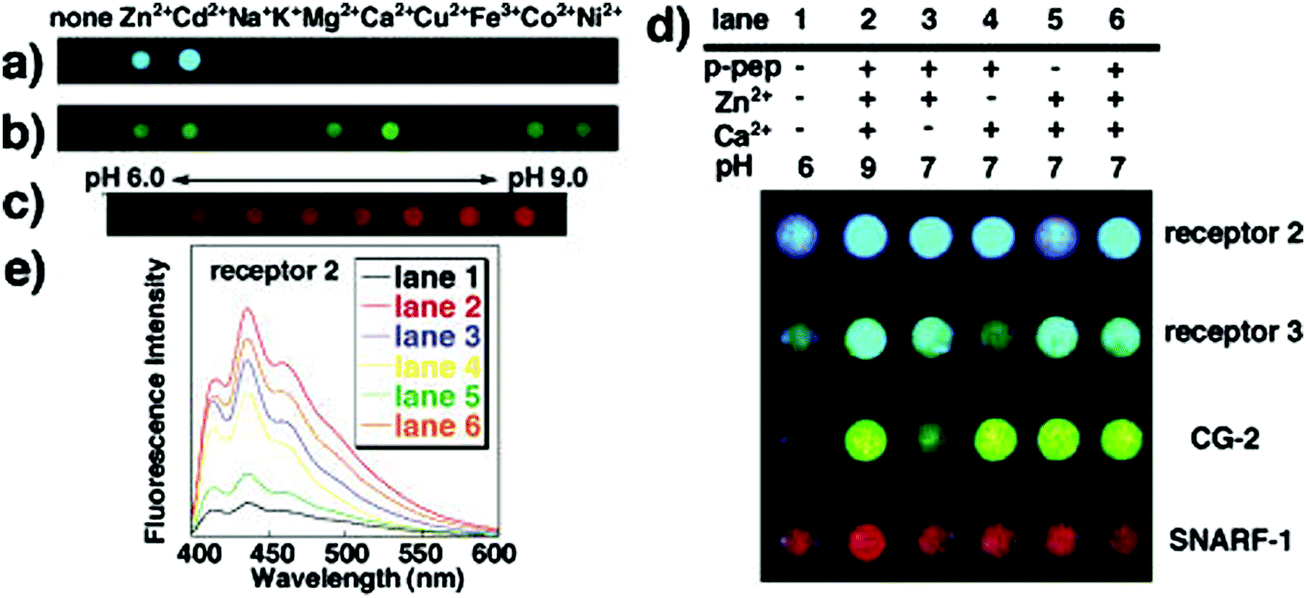 | ||
| Fig. 13 Photographs of sensing patterns of semi-wet chemo sensor chip made from hydrogels containing different receptors and the corresponding fluorescence emissive spots in reaction to mixed solutions containing the various analyte concentrations.74 | ||
Fluorescent hydrogels can also show promising applications in stem cell biology, specifically, embryonic body formation and osteogenic differentiation of embryonic stem cells.25 Zur Nieden et al. exploited substrate mechanics (e.g. stiffness) imposed by hydrogels to guide the lineage differentiation of stem cells. The gelatin gels synthesized were highly fluorescent and provided a soft, adhesive surface for the formation of embryonic bodies, potentially overcoming the challenge of imaging changes to the microenvironments of cells during 3-D cell culturing.
Fluorescent hydrogels also find novel applications in intelligent display and electrofluorochromic devices.26 Beneduci et al. fabricated a highly fluorescent (quantum yield 67%) electrofluorochromic gel incorporating thienoviologen fluorophores 4,4′-(2,2′-bithiophene-5,5′-diyl)bis(1-nonylpridinium)bistriflimide inside the polymer matrix.26 When the fluorophore aggregated, emission was observed at 630 nm. At low concentration of thienoviologen, emission was observed at 530 nm. The fluorescent state and the quenched state (with a contrast ratio of 337×) could be induced by direct electrochemical reduction of the thienoviologen fluorophore. A single layer electrofluorochromic device was assembled by sandwiching the polymer gels between two sheets of indium tin oxide (ITO) glass. The fluorescence intensity was then modulated between the emission and quenched states by electrochemical reduction of the thienoviologen fluorophore. The reversible interconversion between the two states had a short switching time of only a few seconds and a high reported contrast ratio.
Wang et al. have reported another fluorescent polymer hydrogels with white light emission based on the aggregation of a single fluorescent chromophore (ureidopyrimidinone uPy).27 The pyridinium salt was the monomer emitter with high-energy emission that emitted intense white light upon aggregation. Self-assembly of the polymer backbone by hydrogen bonding formed a gel that resulted in the pyridinium salt units becoming closer to each other and forming charge transfer complexes, giving rise to an additional low energy emission. A protected quick response (QR) code was fabricated by the group using this advanced material via laser carving of the photo luminescent gel. This material could hide information under natural light but displayed it under UV light illumination (Fig. 14). Additionally, due to the dynamic nature of the reversible hydrogen bonds, the structure was self-healing which is crucial for scratch-free display surfaces. The cut-QR code could be recovered via multiple hydrogen bonding, and self-reassembled to recover the damaged information.
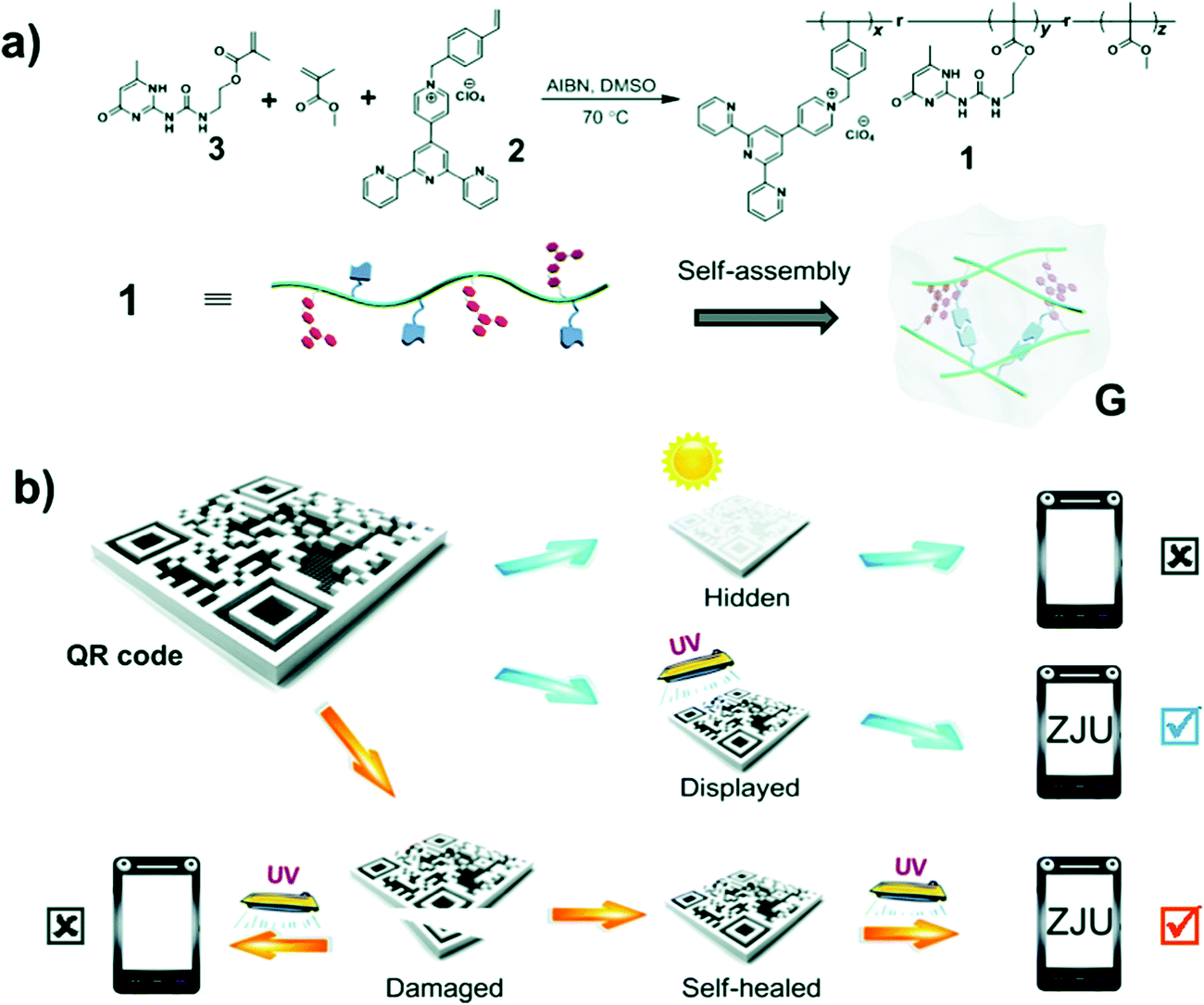 | ||
| Fig. 14 (a) Synthesis of ureidopyrimidinone uPy and its self-assembly in solution to form a white-light-emitting fluorescent supramolecular polymer gel via intermolecular hydrogen bonding. (b) Data recording, data security protection, and data recovery of a QR code constructed by using this gel.27 | ||
Future perspectives and challenges
Fluorescence polymer hydrogels have gained increasing interest recently due to their unique photoluminescence, tunable emission colors (including white light emission), self-healing capability and biocompatibility with high water content. Although still not commercially viable, there has been considerable effort towards the engineering of these gel systems for electronic displays, information storage, bioimaging and chemical sensing. Hydrogels have intrinsic hydrophilicity with high water content and biodegradability with a varying lifetime from gelator to gelator and (unfortunately) batch to batch.Synthesis of such supramolecular polymer gels are presently difficult, multi-step processes that involves complex chemistry, which limits large-scale production. These synthetic materials often lack reactive handles, making them difficult to functionalize. The many covalently conjugated or diffusion-adsorbed luminogens find sites of attachment at locations both on the gel surface and inside the gel body. When the luminogens are inside the gel network, fluorescence emission is shielded and weakened by the surrounding polymer gel material. For diffusion-infused dyes, the system may also have dye leaching issues and result in the loss of fluorescent moiety concentration in the gel body, with subsequent decrease in the emissive photon signals. Perhaps also intrinsic to the hydrogel material property, the polymer network must be stored hydrated. This restricts certain applications of the materials in places where a neat, dry environment is required, or when facile transfer of the material between places is necessary. On the occasions of the gel drying out, the fluorescence emission is usually quenched.
When used in macroscopic applications, such gel systems have an intrinsic thickness of at least hundreds of microns, if not of a centimeter length scale. This makes miniaturization of these hydrogels in thin film configurations difficult. Any embedding of the gel film into devices will need to account for the thickness of the gel and the requirement of maintaining it in a hydrated state by sealing or encasement.
Depending on the types of fluorescent moieties attached to the gel network, the typical brightness of the materials may vary. Using the brightest quantum dots, with a quantum yield between 0.65 to 0.85, the upper bound of quantum yield for hydrogels made from these luminophores may be near to these values or typically lower,36 due to quenching from aggregation or shielding effects for dots embedded in the deep interior of gel bodies.40 Aggregation induced emissive dyes can resist this type of quenching in the gel state. Hydrogels made with these dyes, such as tetraphenylethene, have the advantage of enhanced fluorescence quantum yields (0.02–0.045).28 Lanthanide-based fluorescence polymer gels usually have reported quantum yields of around 0.05 with the Eu(III) ion complex or 0.072 with the Tb(III) ion complex.66 Other high quantum yield hydrogels have been reported with thienoviologen fluorophores, at a quantum yield of 0.67.26
Another issue related to the biomedical applications of fluorescent polymer gels is the toxicity of the various fluorescent moieties in the gels over time, which have remained uncertain and largely un-investigated for such applications in medicine. There have been reports of cytotoxicity of semiconductor quantum dots for in vitro cellular studies and the renal clearance of nanoparticles of a particular cut-off size in vivo.75–77 While proper tuning of quantum dot dosage, surface passivation and delivery modality can help ensure in vitro cell delivery that exhibit minimal impact on cellular viability, cytotoxicity correlates with the release of metallic ions due to deterioration of the quantum dot lattice. Similarly, nanometer-sized particles of carbon dots or other fluorescent nanoparticles need to align with characteristics associated with optimal renal clearance from the host to avoid toxic accumulation inside the biological system, so as to improve the biocompatibility and biosafety of such gels for applications in medicine.78
Commercial-scale applications of such fluorescent polymer hydrogels require that the production cost of the polymeric matrices and the dye ingredients be as low as technically feasible. High durability of these gels and photostability in their hydrated state is also important. Similarly, gels need to be as non-toxic as possible, with high biodegradability. Although hydrogels have self-healing properties, re-wetting is needed for most biological applications. This often requires an enclosed system or a recycling process, that can be cumbersome.12
Challenges of these sorts are probably addressable with continued research into simpler gelator chemistry and more spontaneous self-assembly of such polymer chains to form gel bodies. With cross-linking or stronger chelating agents, the gain and loss moduli of the fluorescent hydrogels can be improved to yield mechanically robust gel networks with reproducible self-healing properties. Therefore, limitations of fluorescent supramolecular polymer hydrogels are also opportunities for future designs of these materials to enable their eventual applications beyond laboratory demonstrations.
Conflicts of interest
There are no conflicts to declare.References
- L. Gan, G. R. Deen, X. Loh and Y. Gan, Polymer, 2001, 42, 65–69 CrossRef CAS
.
- T. Billiet, M. Vandenhaute, J. Schelfhout, S. Van Vlierberghe and P. Dubruel, Biomaterials, 2012, 33, 6020–6041 CrossRef CAS PubMed
- V. P. N. Nguyen, N. Kuo and X. J. Loh, Soft Matter, 2011, 7, 2150–2159 RSC
- Z. X. Zhang, D. J. Young, Z. Li and X. J. Loh, Small Methods, 2019, 3, 1800270 CrossRef
- Z. W. K. Low, Z. Li, C. Owh, P. L. Chee, E. Ye, D. Kai, D. P. Yang and X. J. Loh, Small, 2019, 15, 1805453 CrossRef PubMed
- X. J. Loh, P. L. Chee and C. Owh, Small Methods, 2019, 3, 1800313 Search PubMed
- Z. Liu, S. S. Liow, S. L. Lai, A. Alli-Shaik, G. E. Holder, B. H. Parikh, S. Krishnakumar, Z. Li, M. J. Tan, J. Gunaratne, V. A. Barathi, W. Hunziker, R. Lakshminarayanan, C. W. T. Tan, C. K. Chee, P. Zhao, G. Lingam, X. J. Loh and X. Su, Nat. Biomed. Eng., 2019, 1, DOI:10.1038/s41551-019-0382-7
- H. Ye, K. Zhang, D. Kai, Z. Li and X. J. Loh, Chem. Soc. Rev., 2018, 47, 4545–4580 RSC
- K. Xue, S. S. Liow, A. A. Karim, Z. Li and X. J. Loh, Chem. Rec., 2018, 18, 1517–1529 CrossRef CAS PubMed
- X. Wang, D. J. Young, Y. L. Wu and X. J. Loh, Molecules, 2018, 23, 553 CrossRef PubMed
- Z. Liu, X. Su, S. S. Liow, M. J. Tan, Z. Li, X. J. Loh, C. Chee and G. Lingam, Invest. Ophthalmol. Visual Sci., 2018, 59, 5923 Search PubMed
- E. M. Ahmed, J. Adv. Res., 2015, 6, 105–121 CrossRef CAS PubMed
- L. Basabe-Desmonts, D. N. Reinhoudt and M. Crego-Calama, Chem. Soc. Rev., 2007, 36, 993–1017 RSC
- T. W. Bell and N. M. Hext, Chem. Soc. Rev., 2004, 33, 589–598 CAS
- S. Bhattacharya, S. Nandi and R. Jelinek, RSC Adv., 2017, 7, 588 RSC
- A. Cayuela, M. L. Soriano, S. R. Kennedy, J. W. Steed and M. Valcárcel, Talanta, 2016, 151, 100 CrossRef CAS PubMed
- N. Gogoi, M. Barooah, G. Majumdar and D. Chowdhury, ACS Appl. Mater. Interfaces, 2015, 7, 3058 CrossRef CAS PubMed
- M. Ikeda, T. Yoshii, T. Matsui, T. Tanida, H. Komatsu and I. Hamachi, J. Am. Chem. Soc., 2011, 133, 1670 CrossRef CAS PubMed
- E. Jang, S. Kim and W. G. Koh, Biosens. Bioelectron., 2012, 31, 529 CrossRef CAS PubMed
- A. R. Thete, T. Henkel, R. Göckeritz, M. Endlich, J. M. Köhler and G. A. Groß, Anal. Chim. Acta, 2009, 633, 81 CrossRef CAS PubMed
- F. Ishiwari, H. Hasebe, S. Matsumura, F. Hajjaj, N. Horii-Hayashi, M. Nishi, T. Someya and T. Fukushima, Sci. Rep., 2016, 6, 24275 CrossRef CAS PubMed
- J. F. Lovell, A. Roxin, K. K. Ng, Q. Qi, J. D. McMullen, R. S. Dacosta and G. Zheng, Biomacromolecules, 2011, 12, 3115 CrossRef CAS PubMed
- H. Shibata, Y. J. Heo, T. Okitsu, Y. Matsunaga, T. Kawanishi and S. Takeuchi, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17894 CrossRef CAS PubMed
- N. Mehwish, X. Dou, Y. Zhao and C.-L. Feng, Mater. Horiz., 2019, 6, 14–44 RSC
- N. I. zur Nieden, C. C. Turgman, X. Lang, J. M. Larsen, J. Granelli, Y.-J. Hwang and J. G. Lyubovitsky, ACS Appl. Mater. Interfaces, 2015, 7, 10599–10605 CrossRef CAS PubMed
- A. Beneduci, S. Cospito, M. L. Deda and G. Chidichimo, Adv. Funct. Mater., 2015, 25, 1240–1247 CrossRef CAS
- H. Wang, X. Ji, Z. Li, C. N. Zhu, X. Yang, T. Li, Z. L. Wu and F. Huang, Mater. Chem. Front., 2017, 1, 167–171 RSC
- C. Lu, M. Zhang, D. Tang, X. Yan, Z. Zhang, Z. Zhou, B. Song, H. Wang, X. Li, S. Yin, H. Sepehrpour and P. J. Stang, J. Am. Chem. Soc., 2018, 140, 7674–7680 CrossRef CAS PubMed
- B. Chen and J. Feng, J. Phys. Chem. C, 2015, 119, 7865 CrossRef CAS
- E. Elmalem, F. Biedermann, M. R. J. Scherer, A. Koutsioubas, C. Toprakcioglu, G. Biffi and W. T. S. Huck, Chem. Commun., 2014, 50, 8930–8933 RSC
- Y. Xia, B. Xue, M. Qin, Y. Cao, Y. Li and W. Wang, Sci. Rep., 2017, 7, 9691 CrossRef PubMed
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed
- W. Liu, W. Zhang, X. Yu, G. Zhang and Z. Su, Polym. Chem., 2016, 7, 5749–5762 RSC
- R. B. Sekar and A. Periasamy, J. Cell Biol., 2003, 160, 629–633 CrossRef CAS PubMed
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976–989 RSC
- A. P. Alivisatos, Science, 1996, 271, 933–937 CrossRef CAS
- M. Grabolle, M. Spieles, V. Lesnyak, N. Gaponik, A. Eychmüller and U. Resch-Genger, Anal. Chem., 2009, 81, 6285–6294 CrossRef CAS
- Y. Zhao, X. Zhao, B. Tang, W. Xu, J. Li, J. Hu and Z. Gu, Adv. Funct. Mater., 2010, 20, 976–982 CrossRef CAS
- J. Li, X. Hong, Y. Liu, D. Li, Y. W. Wang, J. H. Li, Y. B. Bai and T. J. Li, Adv. Mater., 2005, 17, 163 CrossRef CAS
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef CAS PubMed
- Y.-P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S.-Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., 2013, 125, 4045–4049 CrossRef
- L. Cao, X. Wang, M. J. Meziani, F. Lu, H. Wang, P. G. Luo, Y. Lin, B. A. Harruff, L. M. Veca, D. Murray, S.-Y. Xie and Y.-P. Sun, J. Am. Chem. Soc., 2007, 129, 11318–11319 CrossRef CAS PubMed
- H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. H. A. Tsang, X. Yang and S.-T. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430–4434 CrossRef CAS PubMed
- A. Cayuela, S. R. Kennedy, M. L. Soriano, C. D. Jones, M. Valcárcel and J. W. Steed, Chem. Sci., 2015, 6, 6139 RSC
- M. Pollnau, D. R. Gamelin, S. R. Lüthi, H. U. Güdel and M. P. Hehlen, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 3337–3346 CrossRef CAS
- X. Liu, C.-H. Yan and J. A. Capobianco, Chem. Soc. Rev., 2015, 44, 1299–1301 RSC
- Q. Zhu, L. Zhang, K. Van Vliet, A. Miserez and N. Holten-Andersen, ACS Appl. Mater. Interfaces, 2018, 10, 10409–10418 CrossRef CAS PubMed
- Q. Zhao, Y. Chen, S.-H. Li and Y. Liu, Chem. Commun., 2018, 54, 200–203 RSC
- P. Chen, Q. Li, S. Grindy and N. Holten-Andersen, J. Am. Chem. Soc., 2015, 137, 11590 CrossRef CAS PubMed
- H. Kim and J. Y. Chang, RSC Adv., 2013, 3, 1774 RSC
- X. Ji, Y. Yao, J. Li, X. Yan and F. Huang, J. Am. Chem. Soc., 2013, 135, 74–77 CrossRef CAS PubMed
- C. Y. Lee, O. K. Farha, B. J. Hong, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, J. Am. Chem. Soc., 2011, 133, 15858–15861 CrossRef CAS PubMed
- R. Haldar, K. V. Rao, S. J. George and T. K. Maji, Chem. – Eur. J., 2012, 18, 5848–5852 CrossRef CAS PubMed
- J. H. Kim, S. Y. Lim, D. H. Nam, J. Ryu, S. H. Ku and C. B. Park, Biosens. Bioelectron., 2011, 26, 1860 CrossRef CAS PubMed
- J. Geng, C. Yao, X. Kou, J. Tang, D. Luo and D. Yang, Adv. Healthcare Mater., 2018, 7, 1700998 CrossRef PubMed
- W. Guo, R. Orbach, I. Mironi-Harpaz, D. Seliktar and I. Willner, Small, 2013, 9, 3748–3752 CrossRef CAS PubMed
- A. P. Esser-Kahn and M. B. Francis, Angew. Chem., Int. Ed., 2008, 47, 3751–3754 CrossRef CAS PubMed
- O. Kotova, R. Daly, C. M. G. Dos Santos, M. Boese, P. E. Kruger, J. J. Boland and T. Gunnlaugsson, Angew. Chem., Int. Ed., 2012, 51, 7208 CrossRef CAS PubMed
- M. X. Wang, C. H. Yang, Z. Q. Liu, J. Zhou, F. Xu, Z. Suo, J. H. Yang and Y. M. Chen, Macromol. Rapid Commun., 2015, 36, 465 CrossRef CAS PubMed
- T. H. Kim, J. Seo, S. J. Lee, S. S. Lee, J. Kim and J. H. Jung, Chem. Mater., 2007, 19, 5815–5817 CrossRef CAS
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed
- R. Hu, Y. Kang and B. Z. Tang, Polym. J., 2016, 48, 359 CrossRef CAS
- S. S. Liow, Q. Dou, D. Kai, Z. Li, S. Sugiarto, C. Y. Y. Yu, R. T. K. Kwok, X. Chen, Y. L. Wu and S. T. Ong, Small, 2017, 13, 1603404 CrossRef PubMed
- S. S. Liow, H. Zhou, S. Sugiarto, S. Guo, M. L. S. Chalasani, N. K. Verma, J. Xu and X. J. Loh, Biomacromolecules, 2017, 18, 886–897 CrossRef CAS PubMed
- M. Martínez-Calvo, O. Kotova, M. E. Möbius, A. P. Bell, T. McCabe, J. J. Boland and T. Gunnlaugsson, J. Am. Chem. Soc., 2015, 137, 1983–1992 CrossRef PubMed
- P. Bairi, B. Roy, P. Chakraborty and A. K. Nandi, ACS Appl. Mater. Interfaces, 2013, 5, 5478 CrossRef CAS PubMed
- V. K. Praveen, C. Ranjith and N. Armaroli, Angew. Chem., Int. Ed., 2014, 53, 365 CrossRef CAS PubMed
- S. G. Liu, N. Li, Y. Ling, B. H. Kang, S. Geng, N. B. Li and H. Q. Luo, Langmuir, 2016, 32, 1881–1889 CrossRef CAS PubMed
- D. Grafahrend, K.-H. Heffels, M. V. Beer, P. Gasteier, M. Möller, G. Boehm, P. D. Dalton and J. Groll, Nat. Mater., 2010, 10, 67 CrossRef PubMed
- S.-M. Hsu, Y.-C. Lin, J.-W. Chang, Y.-H. Liu and H.-C. Lin, Angew. Chem., Int. Ed., 2014, 53, 1921–1927 CrossRef CAS PubMed
- M. Ikeda, T. Tanida, T. Yoshii and I. Hamachi, Adv. Mater., 2011, 23, 2819–2822 CrossRef CAS PubMed
- Y. J. Heo, H. Shibata, T. Okitsu, T. Kawanishi and S. Takeuchi, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13399–13403 CrossRef CAS PubMed
- I. Yoshimura, Y. Miyahara, N. Kasagi, H. Yamane, A. Ojida and I. Hamachi, J. Am. Chem. Soc., 2004, 126, 12204–12205 CrossRef CAS PubMed
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11–18 CrossRef CAS PubMed
- C. E. Bradburne, J. B. Delehanty, K. Boeneman Gemmill, B. C. Mei, H. Mattoussi, K. Susumu, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2013, 24, 1570–1583 CrossRef CAS PubMed
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicine, 2008, 3, 703–717 CrossRef CAS PubMed
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. Itty Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef CAS PubMed
| This journal is © the Partner Organisations 2019 |