 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInteractions of aggregating peptides probed by IR-UV action spectroscopy†
Sjors
Bakels
a,
Eline M.
Meijer
a,
Mart
Greuell
a,
Sebastiaan B. A.
Porskamp
a,
George
Rouwhorst
a,
Jerôme
Mahé
b,
Marie-Pierre
Gaigeot
 b and
Anouk M.
Rijs
*a
b and
Anouk M.
Rijs
*a
aRadboud University, Institute for Molecules and Materials, FELIX Laboratory, Toernooiveld 7-c, 6525 ED Nijmegen, The Netherlands. E-mail: a.rijs@science.ru.nl
bLAMBE CNRS UMR8587, Université d’Evry val d’Essonne, Blvd F. Mitterrand, Bât Maupertuis, 91025 Evry, France
First published on 8th January 2019
Abstract
Peptide aggregation, the self-assembly of peptides into structured beta-sheet fibril structures, is driven by a combination of intra- and intermolecular interactions. Here, the interplay between intramolecular and formed inter-sheet hydrogen bonds and the effect of dispersion interactions on the formation of neutral, isolated, peptide dimers is studied using infrared action spectroscopy. Therefore, four different homo- and heterogenous dimers resulting from three different alanine-based model peptides have been formed under controlled and isolated conditions. The peptides differ from one another by the presence and location of a UV chromophore containing end cap. The conformations of the monomers of the peptides direct the final dimer structure: strongly bonded or folded structures result in weakly bound dimers. Here, intramolecular hydrogen bonds are favored over new intermolecular hydrogen bond interactions. In contrast, linear monomers are the ideal template to form parallel beta-sheet type structures. The weak intramolecular hydrogen bonds present in the linear monomers are replaced by the stronger inter-sheet hydrogen bond interactions. The influence of π–π dispersion interactions on the structure of the dimers is minimal, and the phenyl rings have a tendency to fold away from the peptide backbone to favour intermolecular hydrogen bond interactions over dispersion interactions. Quantum chemical calculations confirm our experimental observations.
Introduction
The aggregation or self-assembly of peptides and proteins from their soluble, native structures into insoluble beta-sheet rich amyloid aggregates is directly related to both biological function and human disease.1–7 To date over 45 proteins and peptides are known to form disease related amyloid fibrils.5 This long list of amyloid diseases resulted in the toxic association of amyloid formation, with the most well-known examples being Alzheimer’s disease and Parkinson’s disease. The current general consensus that the early-stage formed intermediate oligomers are the actual malefactors8–10 has resulted in the demand to understand the structure and interactions of these oligomer intermediates.Most experiments aim to determine the structure of the full-grown fibrils or obtain an averaged picture of the ensemble of intermediate structures and morphologies.7,8,11–14 Extensive studies based on structural techniques such as X-ray diffraction, electron microscopy and solid-state NMR have revealed high-resolution structural information about these final states, but collecting information on earlier aggregation stages is hampered due to the non-crystalline nature and the broad heterogeneity of the pre-fibrils. Spectroscopic methods such as circular dichroism and FTIR are able to provide such structural information, but the information gained represents an average of all the aggregation states.15 It is thus impossible to obtain structural details of individual clustered peptide aggregates using these methods. Mass spectrometry combined with ion mobility has been very successful to study lowly populated species in heterogeneous mixtures and reveal how aggregates might evolve.4,16,17 More recently, spectroscopic experiments have been developed that allow detailed structural insights about low populated species.18–22 Electrospray ionization is generally used in combination with these advanced mass spectrometry methods to bring the peptide aggregates as charged species into the gas phase. It was shown that an increase in beta-sheet character, which is indicative of the regular aggregated structure, was observed in more extended oligomers where the charge states also played an important role.17 Studying peptide aggregation under neutral conditions might provide more insights into non-charge driven aggregation interactions, such as dispersion interactions, NH–π and π–π stacking, and intra- and intermolecular hydrogen bond interactions.23–26 For example, the π–π interactions are expected to play a directing role by stabilizing the hydrophobic domains crucial for the development of amyloid structures.24,27–31
Although cluster formation between (bio)molecules and solvent molecules, such as water or methanol, is rather straightforward by co-expanding the solvent molecules with the seed gas,32–35 the formation of aggregates of neutral peptides is not. Until recently, only a few examples were published where peptide dimer clusters were formed in the gas phase using laser desorption or thermal evaporation.27,31,36,37 Recently, we showed using laser desorption that the aggregation of overall neutral peptides is not limited to the formation of dimers anymore, but is brought into the regime of 10–15 peptides assembled together.38 This allows us to study the onset of peptide oligomerization in molecular-level detail.
Here, we aim to understand the intrinsic driving forces of peptide self-aggregation, i.e. probing the influence of intra- and inter-molecular interactions on the structures of formed peptide dimers. Therefore, we have studied three different aggregating model peptides, two where we incorporated a UV chromophore on either terminus of the peptide and a third one without a UV chromophore, see Fig. 1. By comparing the experimentally present conformers for the monomeric peptides with the formed homogeneous and heterogeneous dimers and theoretical quantum chemical calculations, we can elucidate whether the intramolecular hydrogen bonds of the bare monomeric conformer remain preserved or will be broken favouring new interactions upon dimerization. Moreover, the possible role of dispersion interactions can be probed by comparing the homogeneous and heterogeneous dimers, where the position of the phenyl ring is either altered or eliminated.
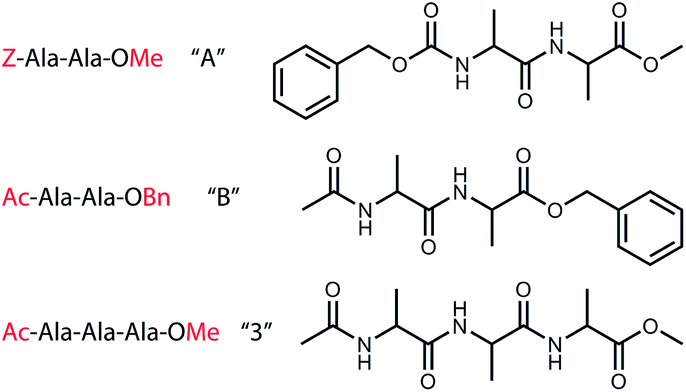 | ||
| Fig. 1 Chemical structures of the studied peptides (a) Z-Ala-Ala-OMe (peptide A), (b) Ac-Ala-Ala-OBn (peptide B), and (c) Ac-Ala-Ala-Ala-OMe (peptide 3) forming the homogeneous dimers “AA” and “BB” and the heterogeneous dimers “AB” and “B3”. | ||
Methods
(a) Experimental
A description of the laser desorption molecular beam set-up and experimental methods has been reported in great detail recently.39,40 Therefore, the experiment is described only briefly here by focusing on the important parameters for the experiments presented here.The peptide molecules studied in this work, Z-Ala-Ala-OMe, Ac-Ala-Ala-OBn, and Ac-Ala-Ala-Ala-OMe, were purchased from BioMatik (95% purity), and used without any further purification. These peptide molecules were mixed with carbon black and subsequently deposited on the surface of a graphite sample bar. This sample bar was placed via an airlock into a high vacuum (∼10−7 mbar) source chamber just in front of the nozzle of a pulsed valve (0.5 mm Ø, 10 Hz, Jordan). A low intensity (1 mJ per pulse) Nd:YAG laser (New Wave Research, Polaris II, 1064 nm, 10 Hz) was mildly focused on the surface of this sample bar, thereby desorbing the sample molecules from the graphite matrix. The sample bar was translated with a stepper motor to ensure that a new sample was provided at each laser shot. After desorption the neutral gas phase molecules were directly cooled by the molecular beam of argon with a backing pressure of 3 bar. The dimers discussed in this work and higher order clusters were obtained using a larger distance between the sample bar and nozzle opening than for monomeric peptides.38 About 10 cm downstream, the neutral molecular beam passes a skimmer and enters the differentially pumped reflector-based time-of-flight mass spectrometer (Jordan Co.). The molecular beam was subsequently perpendicularly crossed with a UV and IR laser beam to perform spectroscopy. Ions created by resonant enhanced [1 + 1] photoionization were accelerated into the reflector time-of-flight tube and detected with a dual microchannel plate detector (Jordan Co.), yielding mass spectra with a resolution of M/ΔM of about 2000.41
The phenyl groups in peptide A and B (see Fig. 1) have a ππ* transition around 266 nm and are used for resonant excitation. A second photon was used to subsequently ionize the molecules in our experiment. This technique is conformer and multimer specific, since every conformer and molecule has a different potential energy surface. IR absorption spectra were recorded using IR-UV ion dip spectroscopy (IR-IDS)32 by spatially and temporally overlapping the IR and UV lasers, where the IR laser arrives about 500 ns prior to the UV laser beam. The UV laser was fixed at a resonant wavelength, while the IR laser is scanned. When the IR laser is resonant with a vibrational transition of the studied peptide or peptide cluster, the population is transferred from the ground state to a vibrational level, resulting in a depletion of the ground state population. This will create a dip in the ion signal created by the UV laser as the number of produced ions is reduced. By measuring the ion yield of the mass of interest while scanning the IR laser wavelength, a mass- and conformer-selected IR ion-dip spectrum is recorded.
The IR spectra were obtained in the region between 1000 and 1800 cm−1. The infrared light was provided by the Free Electron Laser FELIX.42 FELIX produces pulses with a typical pulse duration of about 8–10 μs, pulse energies of about 100 mJ, and a spectral line width of about 0.5–1% of the IR frequency. IR on and off measurements were performed to correct for fluctuations in the signal, by running the experiment at 10 Hz (molecular beam, desorption and UV laser) and the IR laser at 5 Hz. Every measured point is an average of 30 shots with IR and 30 shots without IR. Absorbance spectra are obtained by taking the logarithm of the ratio between these two signals, dividing by the power of FELIX at each wavelength and multiplying by the photon energy in wavenumbers. For the monomeric peptides A and B the mid-IR spectra were also recorded between 3000 and 3650 cm−1, produced using a YAG-pumped (Innolas Spitlight 600) OPO/OPA system (LaserVision).
(b) Theoretical
Structural characterization relies on a comparison of the experimental IR spectra with the computed spectra resulting from quantum chemical calculations. First, a conformational search was performed using an amber force field.43 Here, a starting structure of the peptide or peptide cluster was heated up to 300 K for the dimers, and 1000 K for the monomers. The structures were subsequently cooled down to 0 K to find a minimum in the energy landscape and this structure was then stored. This was repeated 500 times, and from this about 50 unique structures were selected based on their relative energy, chemical intuition and structural family. To sample the conformational landscape as completely as possible, multiple searches were performed, with different starting structures. All selected conformations were further optimized at the B3LYP-D3/6-31+G* level of theory for the dimers and the B3LYP-D3/6-311+G** level of theory for the monomers and their vibrational frequencies were calculated using DFT in the Gaussian 09 environment.44,45 To correct for the anharmonicity a scaling factor of 0.976 was used for both the monomers in the infrared region between 1000 and 1800 cm−1, while for the amide A infrared region a scaling factor of 0.955 was used. For the dimers, the infrared region between 1000 and 1420 cm−1 and the amide I region were scaled by a factor of 0.976, while a mode dependent scaling factor of 0.962 was used in the dimers for the amide II modes (NH bending), the CH2 bending and CH3 umbrella, and in and out of plane deforming modes between 1420 and 1600 cm−1, as was discussed previously.38 The zero-point energy (ZPE)-corrected energies and Gibbs free energies at 300 K were evaluated at the B3LYP-D3/6-31+G* level of theory.Results
Peptide segments
In this work we focus on three alanine-based peptides. Previous work showed that alanine chains may favour the formation of beta-sheet dimers when they are clustered.24 In order to study the role of stabilizing dispersion interactions and intra- and intermolecular hydrogen bond preferences, the peptides differ in the position of a UV tag, i.e. (i) on the N terminal side for Z-Ala-Ala-OMe (peptide A) with a mass of 308 Da, (ii) on the C terminus for Ac-Ala-Ala-OBn (peptide B) with a mass of 292 Da, and (iii) no UV chromophore is present on the third peptide Ac-Ala-Ala-Ala-OMe (peptide 3) with a mass of 287 Da. For peptide A and B their monomeric conformers have been identified using IR-UV ion dip spectroscopy, which is not possible for peptide 3 due to the absence of a UV chromophore. Subsequently, the IR spectra have been recorded for the homogeneous dimers AA and BB, and heterogeneous dimers AB and B3.Monomeric peptides A and B
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 and 37700 cm−1, namely a slightly broadened peak around 37500 cm−1 and a set of sharp lines above 37550 cm−1. IR-UV hole-burning experiments confirmed that they originate from two separate conformations. Upon increasing the UV power to record the IR-UV ion dip spectra, a slight background appeared which originates from conformer A-II. In Fig. 2 the experimental IR spectra of the two conformations are shown. A-I and A-II are obtained at an excitation wavelength of 37523 cm−1 and 37558 cm−1, respectively.
300 and 37700 cm−1, namely a slightly broadened peak around 37500 cm−1 and a set of sharp lines above 37550 cm−1. IR-UV hole-burning experiments confirmed that they originate from two separate conformations. Upon increasing the UV power to record the IR-UV ion dip spectra, a slight background appeared which originates from conformer A-II. In Fig. 2 the experimental IR spectra of the two conformations are shown. A-I and A-II are obtained at an excitation wavelength of 37523 cm−1 and 37558 cm−1, respectively.
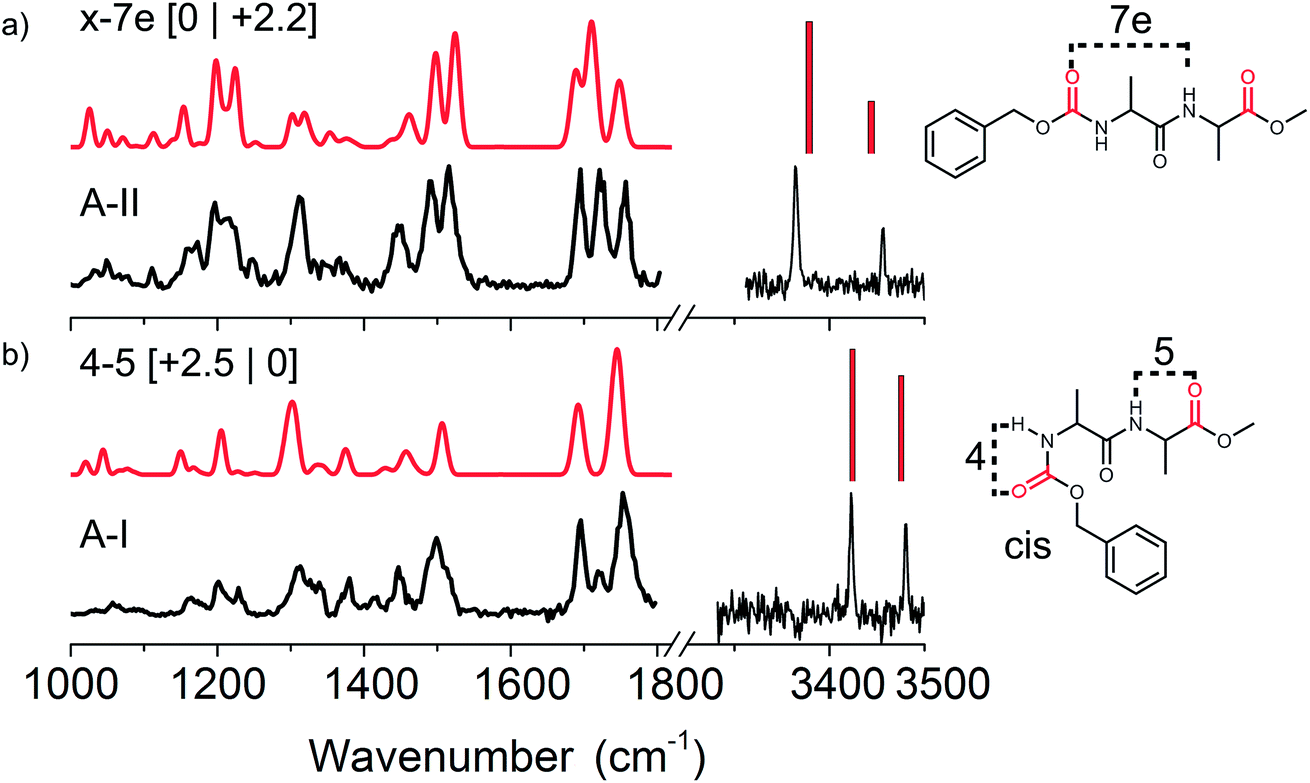 | ||
Fig. 2 Experimental (black) infrared spectra of the two conformations of Z-Ala-Ala-OMe with (a) conformer A-II and (b) conformer A-I both in the amide A (right panel) and mid-IR/fingerprint region (left panel). The red traces present the assigned calculated spectra with their respective structures given on the right, where ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups are indicated in red. The energetics are shown between brackets [zero-point energy | Gibbs free energy], both in kJ mol−1. O groups are indicated in red. The energetics are shown between brackets [zero-point energy | Gibbs free energy], both in kJ mol−1. | ||
The CO stretching region comprises 3 CO vibrations, two modes resulting from the ester group (O–CO) of both end caps, which typically absorbs between 1700 and 1770 cm−1. The third CO vibration results from a normal peptide bond CO and has its absorption between 1600 and 1700 cm−1. For these peptide bond CO vibrations it holds that a large red-shift coincides with a stronger hydrogen bond interaction. Two dominant peaks are observed in the experimental mid-IR spectrum of peptide A-I (left side of Fig. 2b) namely at 1696 and 1753 cm−1. A third small peak is present at 1719 cm−1. The most red located CO peak at 1696 cm−1 indicates that the peptide CO group is free, and thus not involved in any hydrogen bonding. The second peak at 1719 cm−1 is unexpected, since it suggests a very strong hydrogen bond of one of the ester CO groups, while both NH peaks in the amide A indicate only weak hydrogen bonds. Finally, the peak at 1753 cm−1 is significantly broader than the other peaks that were measured in this region (23 cm−1 instead of typically 15 cm−1 in this wavelength region). These findings indicate that this last peak actually consists of two CO vibrations, and that the peak at 1719 cm−1 originates from conformer A-II (see Fig. 2a). The UV peak of conformer A-I was originally located on a broad band resulting from conformer A-II, although upon improved cooling this contribution disappeared, see Fig. SI1.† Therefore, we have to keep in mind that strong IR signatures of conformer A-II might be present in the IR spectrum of A-I. The calculated IR spectrum of the assigned C4–C5 conformer also shows two peaks around 1753 cm−1 indicating that both ester CO groups of both end-caps at the C- and N-termini are similarly weakly involved in hydrogen bonding.
Additionally, the IR spectra show other structural diagnostic features such as the amide II region represented by the broad band around 1499 cm−1, which is most likely broadened on the blue side of the peak by the overlapping absorption of conformer A-II. The large peak at 1300 cm−1 originating from CH bending modes and the three backbone motion peaks around 1200 cm−1 confirm the assigned C4–C5 hydrogen bonded structure. The assigned structure has two weak hydrogen bonds making up a ring of 4 atoms and a second ring of 5 atoms, see the right panel of Fig. 2b. It is important to note that the N-terminal Z-cap CO is in a cis configuration with respect to the NH group of the first alanine. Natural peptide bonds are typically oriented in a trans configuration with the exception for proline.46 However, here the NH–CO peptide bond resulting from a bond between the Z-cap and the first alanine residue, appears to prefer to be in the cis configuration. As this involves a non-natural peptide bond, both cis and trans configurations were included in the computational search for the non-functional peptide moieties. The C4–C5 cis structure is only 2.5 kJ mol−1 higher in energy than the lowest energy conformer, but has the lowest energy when the Gibbs free energies are taken into account at 300 K.
O groups. The peak at 1695 cm−1 indicates that the CO in the centre of the peptide is not involved in hydrogen bonding, and the same holds for the ester CO group corresponding to the vibration at 1757 cm−1. However, the 1721 cm−1 peak is representative of a very strong hydrogen bond for one of the two ester CO groups at the termini. This is supported by the red shifted NH band in the amide II region. We have assigned this conformer to the x-7e structure, with the ester CO at the N-terminus involved in a C7 hydrogen bond with the alanine side group on an equatorial position. The NH located close to the N-terminus is not involved in hydrogen bonding. However, this peak is still slightly red shifted compared to the corresponding weakly hydrogen bonded NH (C4) of conformer A-I. This red-shift is known to appear when an adjacent CO group is involved in a strong hydrogen bond interaction, i.e. the so-called neighbouring effect.47 The doublet observed at 1200 cm−1 originating from amide III motions coupled with CH vibrations is also in agreement with the calculated spectrum, thereby confirming our assigned structure. This x-7e structure has the overall lowest zero-point corrected energy, and has a slightly elevated energy at 300 K with respect to the A-I conformer.
400 and 37750 cm−1 and shows three dominant peaks followed by a vibrational progression of the most intense peak at 37550 cm−1. Two conformers were identified using IR-UV hole-burning: the sharp peak at 37495 cm−1 was named conformer B-I, and a more intense, slightly broader band at 37551 cm−1 with a vibrational progression was assigned to conformer B-II. A third peak in between the two conformers at 37530 cm−1 was previously assigned to either a conformer similar to B-II kinetically trapped by argon, or a fragmented complex with argon.48 The experimental IR spectra presented in Fig. 3 were obtained at an excitation wavelength of 37495 cm−1 for conformer B-I (Fig. 3b) and 37551 cm−1 for conformer B-II (Fig. 3a).
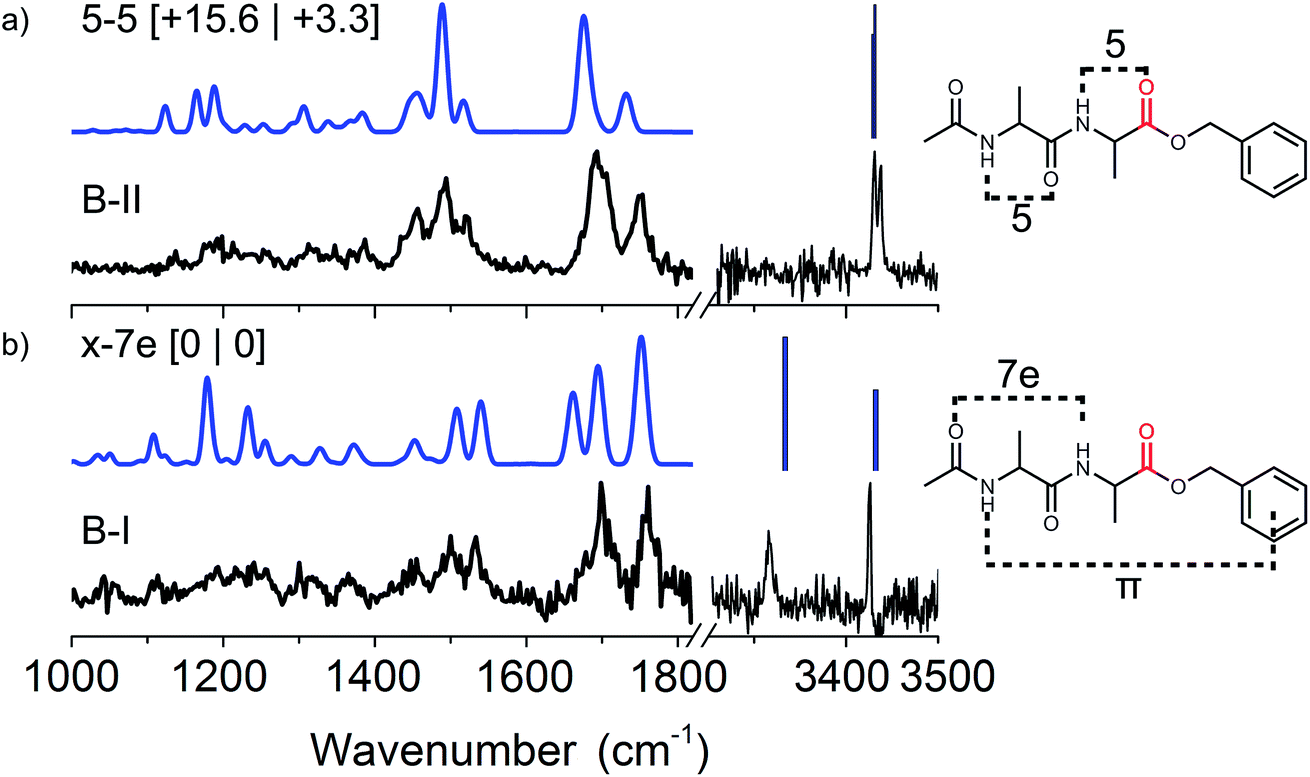 | ||
| Fig. 3 Experimental (black) IR spectra of conformer B-I and B-II of Ac-Ala-Ala-OBn (peptide B) measured in both the mid-IR/fingerprint region (left panel) and the amide A region (right panel). The theoretical IR spectra (blue) of the assigned conformers are plotted above the experimental spectra with their respective structures drawn on the right. The energetics are shown between brackets [zero-point energy | Gibbs free energy], both in kJ mol−1. | ||
O group of the acetyl-cap and the NH group of the second alanine residue. The 5–5 conformer (here conformer B-II) has two neighbouring C5 interactions, see the right panel of Fig. 3. The B-I conformer (5–5) is significantly higher in energy, however, when including Gibbs free energies at around 330 K this energy drops significantly. The higher energy conformer B-II is probably kinetically trapped, allowing us to observe it.49,50
Fig. 3 presents the experimental IR spectra of conformer B-I and B-II recorded in the mid-IR/fingerprint region from 1000–1800 cm−1. For completeness, the amide A region was measured as well from 3250 to 3500 cm−1. In this work, the functional B3LYP with the empirical dispersion D3 term from Grimme was used.44 This functional gives similar results to B-97D as used in the paper by Mons et al. In short, the assignments that were made by Mons et al. were confirmed by our work in the amide I, II and fingerprint infrared region.
Dimer aggregates
To probe the role of hydrogen bond and dispersion interactions on the formation of beta-sheet dimers, the IR spectra of two homogenous dimers (AA and BB) and two heterogeneous dimers (AB and B3) were measured. These dimers are expected to have different intermolecular and π–π interactions due to different ring and peptide moiety positions. The IR spectra and structure of both homogenous dimers of peptide A and peptide B are discussed in the next sections, followed by two heterogeneous dimers, one containing one A and one B peptide and a dimer containing peptide B and the non-chromophore containing peptide 3, respectively. Fig. 4 shows an overview of the infrared spectra of these four measured dimers, along with indications of the origin of the observed vibrational modes. The inset shows a zoom in of the diagnostic amide I region.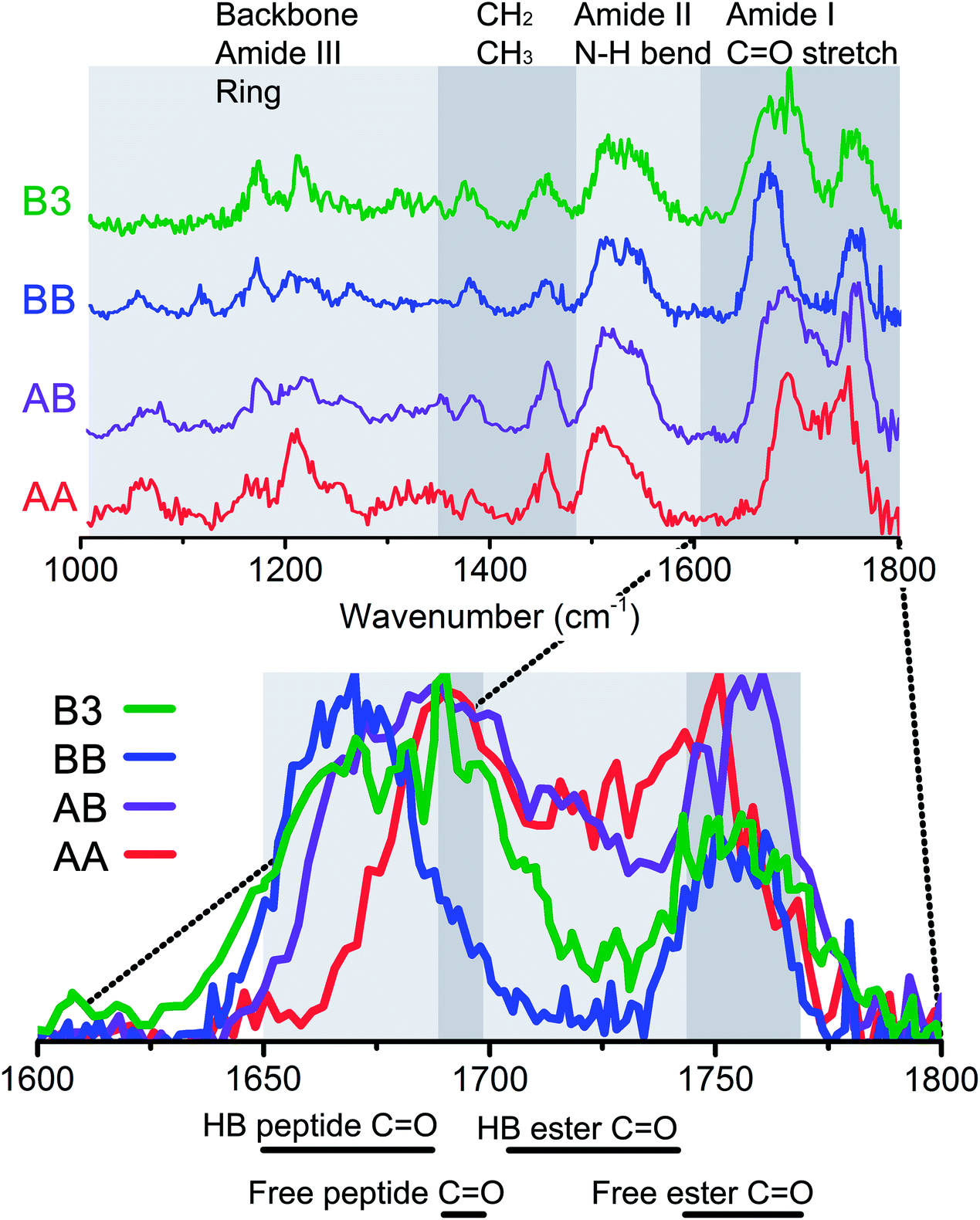 | ||
| Fig. 4 Top panel: Infrared spectra of all measured dimers, AA (red), AB (purple), BB (blue) and B3 (green) from bottom to top. The grey boxes indicate the vibrational modes in these regions. The bottom panel shows an enlargement of the amide I region, with the same colour coded spectra, but then on top of each other. Here, the spectrum is again divided into boxes, and below this figure a specification is given for the modes that can be found in these regions. | ||
430 and 37650 cm−1 (see red trace in Fig. SI3†). Infrared measurements were performed with the excitation laser fixed at 37480 cm−1, which coincides with the red-most feature in the REMPI spectrum. The obtained infrared spectrum is presented in Fig. 5c (red trace). The spectra of the two conformers of monomer A are displayed above the spectrum of the dimer to compare the IR absorption of both monomers to that of the dimer. An average of their combined experimental spectra (in black) is plotted on top of the experimental spectrum of the AA dimer.
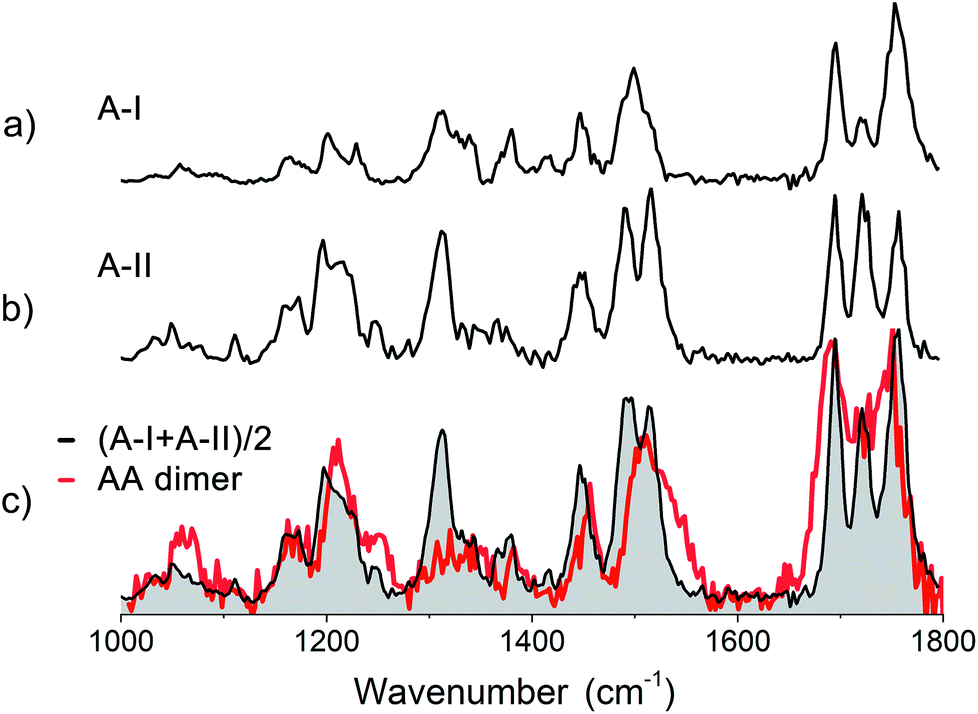 | ||
| Fig. 5 Experimental IR spectrum of (a) conformer A-I, (b) conformer A-II and (c) the dimer of Z-Ala-Ala-OMe (peptide AA) in red compared to the averaged experimental spectra (in black) of both assigned conformers of the peptide A monomer. | ||
The dimer AA has 6 CO groups. Although each monomer has two amino acid residues, due to the presence of the ester caps, four ester CO and only two peptide CO groups are present, see Fig. 1. The amide I region shows two dominant peaks for the AA dimer, with maxima at 1750 cm−1 and 1690 cm−1, respectively (see Fig. 5c). The latter peak is also the most red shifted amide I peak, suggesting that neither of the two peptide CO groups are involved in hydrogen bonding. Two ester CO moieties are likely weakly hydrogen bonded, while one ester CO appears to be strongly intramolecular hydrogen bonded and the last one is intermolecularly bonded. The amide II region shows a single broad peak at 1510 cm−1 with a shoulder slightly to the blue. No large shifts with respect to the individual monomer spectra are observed in both the amide I and the amide II regions. Whereas the monomers both show a large peak around 1300 cm−1, corresponding to Cα–H modes, the dimer shows a lower intensity, broader feature. The three peaks at 1169, 1211 and 1248 cm−1 in the dimer spectrum are similar to the peaks observed for monomer A-II, and were therefore assigned to peaks involving motions of Cα–H coupled to amide III modes. Based on a comparison of the dimer IR spectra with the two averaged spectra of both conformers of the monomer, it appears that the dimer of peptide A is only weakly bound and the monomer structures are largely retained.
Quantum chemical calculations were performed in order to confirm this preliminary conclusion. 90 structures were found in multiple rounds of molecular dynamics simulations starting from different structures. Both parallel, anti-parallel and random structures were used as input structures, see Fig. SI5b.† When a parallel type structure was used as a starting structure it always resulted in a range of different structures, except for a parallel type. For the other types, a family of structures similar to the original structure was present together with other structural families. The lowest energy structures were dominated by a mix of anti-parallel structures, a single hydrogen-bond parallel type and C8C8 structures (where a C16 ring was formed between the two monomers), see Fig. SI5a.† Above 10 kJ mol−1 with respect to the lowest energy structure, mainly structures were found where the two monomers were connected via a single hydrogen bond or via two hydrogen bonds in anti-parallel structures. Parallel structures almost exclusively appeared amongst the highest energetic structures.
Due to the large amount of calculated IR spectra resulting from these 90 structures without an obvious match with our experimental IR spectrum, we designed the following approach: each spectrum was evaluated by each diagnostic region (see Fig. 4), and discarded when no agreement between experiment and theory was present. Firstly, we evaluated the amide I region, where we discarded around 40% of all structures. Secondly, the amide II region allowed us to remove an additional 28% of the structures, and finally 7% of all the structures was eliminated based on the amide III region of the spectrum. However, almost all calculations predict the amide III peaks too far toward the higher energy side of the spectrum. Therefore, this 7% was based on relaxed criteria, and only structures were discarded when all the peaks were off for the amide III region.
The calculated IR spectra of the final 10 structures with energies up to 30 kJ mol−1 are presented in Fig. SI5c† together with the four overall lowest energy structures of each family and compared to the experimental spectrum in black. These calculated IR spectra include mostly low energy single hydrogen bonded structures, two anti-parallel structures with energies around +18 kJ mol−1 and a few high energy parallel structures (energies above 30 kJ mol−1). However, the average of the two monomers, as shown in Fig. 5c (red trace), still provides a better match to the experimental spectrum than any of the calculated structures.
The primary conclusions to be drawn from the above findings are that the structure of the homogeneous dimer of peptide A probably originates from both conformers of the monomer A which are at most only weakly bound to each other by a single hydrogen bond. This is reflected in the minimal shifts of the amide I and amide II frequencies, but also in the preservation of the high intensity peak in the conformationally important amide III region.
518 cm−1. The infrared spectra were predominantly recorded with a one photon energy of 37460 cm−1, in order to measure the trimer and other higher order clusters at the same time (not presented here). The infrared spectrum was also obtained at 37518 cm−1 which yielded the same IR spectrum indicating the presence of one dominant conformer, thereby justifying our chosen wavelength. Fig. 6 shows the experimental infrared spectrum in black, showing two clearly distinctive peaks in the amide I region which correspond to the 2 ester CO groups at 1753 cm−1 and the 4 peptide CO vibrations grouped together at 1670 cm−1. The experimental frequencies of the ester CO peaks indicate that they are not involved in intermolecular hydrogen bonding. This is confirmed if we compare the weakly bound (C5) and free ester CO groups in both monomers, which were observed at the same frequencies as those of the dimer.
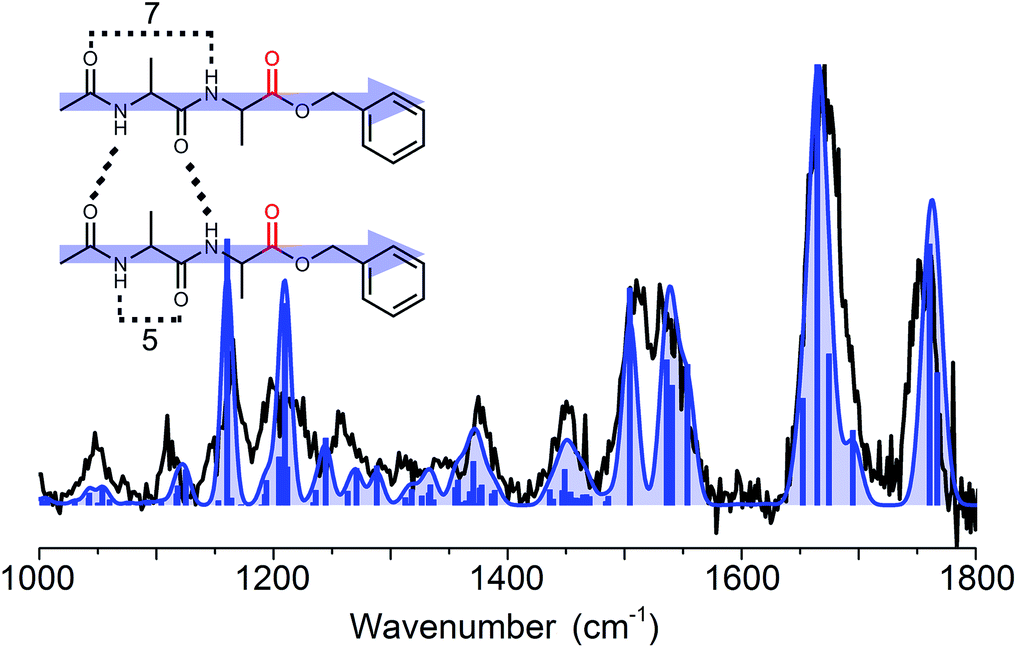 | ||
| Fig. 6 The experimental (black trace) spectrum of the dimer of Ac-Ala-Ala-OBn compared to the spectrum of the assigned calculated structure (blue trace). The inset shows the corresponding parallel type structure, the arrows pointing from the N-terminal side towards the C-terminus. | ||
To assign the structure, an extensive conformational search was performed resulting in 80 different structures which were categorized into several structural families. These families were based on their respective hydrogen bond patterns (e.g. number of hydrogen bonds, (anti-)parallel beta-sheet patterns, and positions of hydrogen bonds) as was similarly done for peptide dimer AA. Low lying energetic structures almost exclusively comprised beta-sheet structures with parallel type hydrogen bonds, whereas the first anti-parallel structure was over 20 kJ mol−1 higher in energy than the lowest energy structure (as shown in Fig. SI6†). The amide I region indicates that the two ester CO groups were not involved in hydrogen bonding, while the 4 peptide CO groups were all engaged in hydrogen bond interactions. This allowed us to exclude a high number of (higher energy) structural families and structures. Eight of the lowest energy structures including all families are plotted in Fig. SI6† for comparison. The remaining structural families were further examined, and based on the full spectrum between 1000 and 1800 cm−1 we assigned the structure to the overall lowest energy parallel beta-sheet type structure, see the inset of Fig. 6. The assigned structure is a combination of both monomer conformers, where the strong C7 intramolecular hydrogen bond of conformer B-II remained intact and only the weaker hydrogen bond (C5) in conformer B-I and the π-interaction in conformer B-II are broken to favour inter-strand hydrogen bonding.
:1 ratio and subsequently mixed with carbon black. The experiment was performed in a similar fashion as the homogeneous AA and BB dimer aggregates. Since both molecules have a slightly different mass (A: 308 Dalton, B: 292 Dalton), the dimers AA, AB and BB were distinguishable in the mass spectrometer, see Fig. SI4.† The REMPI spectrum of AB has a similar broad peak at the same UV excitation wavenumber as the AA and BB dimers, i.e. the rising edge is at the same position, but the peak ends in between that of the two other dimers (see Fig. SI3†). The excitation laser was fixed at 37490 cm−1 to obtain the IR-UV ion dip spectrum of the AB dimer that is shown in Fig. 4 in purple. The mass-selected dimer AB consists of one monomer of A and one monomer of B. This means that we have in total six CO groups of which there are three ester type CO groups and three peptide-like CO groups. The amide I region between 1600 and 1800 cm−1 shows two main bands: a main peak at 1757 cm−1, and a peak at 1687 cm−1, with a small shoulder at 1715 cm−1. The first peak is in accordance with the CO stretching vibration of a free ester CO group, and is slightly blue shifted with respect to the peak of the AA dimer. The width of the peak indicates that most likely two ester type CO groups are involved. The main peak has its maximum at 1688 cm−1, which is more or less the same as that for the AA dimer (1691 cm−1), and suggests free peptide CO groups as we observed for the AA dimer. However, the band extends at the red side with respect to the AA dimer and all conformers of the A and B monomers, and overlaps here with the BB dimer. This overlap with the BB dimer peptide CO vibrations indicates that at least one peptide CO group is involved in strong intermolecular hydrogen bonding.
The shoulder at 1715 cm−1 cannot be from a peptide CO absorption as it is shifted too much to the blue, and is therefore allocated to a strong hydrogen bonded ester CO moiety. This is confirmed by the NH bend vibrational modes in the amide II region, which show a peak at 1513 cm−1 and at 1539 cm−1. Moreover, the NH peak is more broadened towards the blue than was observed for both homogeneous dimer IR spectra indeed indicating strong intermolecular hydrogen bonds. The intensity of this peak suggests that it comprises two intermolecular hydrogen bonded NH bend vibrations. The peak at 1513 cm−1 has the same position as the C7 (intramolecular) hydrogen bonded NH bending mode as was observed for conformer A-II of the A monomer peptide. This suggests that we have both original intramolecular hydrogen bonds present preserved from the monomeric peptide structures involving NH/CO moieties in C7 or C5 intramolecular hydrogen bonds, while at the same time two other NH/CO pairs are involved in intermolecular hydrogen bonding.
We have performed an extensive conformational search, which resulted in about 125 structures. For this conformational search, we used both assigned conformers of peptide A, i.e. including structures with the CO and NH of the Z-cap in a cis position with respect to each other. The same approach was used as that described above in detail for dimer AA. By excluding all calculated structures which have poor agreement with the amide I region, 62% of all structures were discarded. Another 10% was rejected due to poor agreement in the amide II region. From the remaining structures, which were mainly singly hydrogen bonded structures, a handful of parallel and anti-parallel structures and a few low energy structures with different types of double hydrogen bonding (e.g. C7–C8, enclosing a ring of 15 atoms), the lowest energy structures were plotted in Fig. SI7.†
None of the calculated structures agree well enough with the experimental spectrum in order to make a conclusive assignment. However, considering the first 15 low energy structures (up to about 15 kJ mol−1) which are predominantly parallel type conformations, parallel structures are most likely to be present. This is confirmed when we compare the experimental IR spectra of the AA (red) and BB (blue) dimers with that of the AB dimer (purple), see Fig. 4. The IR signatures of the AB dimer are very similar to the signatures for the BB dimer throughout the full IR range, suggesting a similar structural arrangement to the peptide BB dimer, i.e. a parallel type beta-sheet. Zooming in on the amide I region, similarities with both the AA dimer and BB dimer are observed. The AB dimer shows a clear peak at 1715 cm−1 located in between the free ester-type CO modes centred at 1757 cm−1 and the peptide CO vibrations. This peak coincides with the strong intramolecular hydrogen bond interaction as was observed for conformer A-II as well. The peak of the AB dimer resulting from the peptide-type CO groups’ peaks at about 1687 cm−1, shows a combination of both AA dimer and BB dimer absorption where both CO moieties are engaged in intermolecular hydrogen bonds, see the left figure of Fig. SI6† (parallel C12 structure). The energies and spectral signatures indicate that conformer A-II and conformer B-II form this parallel beta-sheet. It is not surprising that conformer A-I is not observed within the AB dimer, due to its strong intramolecular hydrogen bond interactions and folded structure.
:1 ratio, showed a very similar broad peak to the homodimer BB, with its maximum around 37518 cm−1 (see Fig. SI3†). As explained previously for the BB dimer, we have measured the infrared spectra using UV photon energies of 37460 and 37518 cm−1. Both yielded the same IR spectrum, as was also observed for the BB dimer. The experimental infrared spectrum of the B3 dimer, presented by the black trace in Fig. 7 (and the green trace in Fig. 4), shows the same spectral features, although they appear to be slightly broadened and noisier with respect to the BB dimer due to experimental difficulties. Compared to the chemical structure of peptide B, the non-chromophore containing peptide 3 has one more peptide CO and NH group, but the same number of ester-type CO moieties, see Fig. 1. This is directly visible in the amide I region of the IR spectrum of the B3 dimer. The peak corresponding to the ester CO is similar in intensity, width and position as that for the BB dimer. This indicates that the ester type CO groups are free and not involved in any hydrogen bond interactions. The peak between 1600 and 1720 cm−1 shows the same behaviour as that for the BB dimer, however, there is an additional broadening to the blue side, with a maximum at 1692 cm−1, see the blue and purple traces in Fig. 4. This typically corresponds to a free peptide CO moiety or weakly hydrogen bond (C5, π) interaction. The amide II shows a similar peak shape to BB, but is slightly broadened and has more intensity at 1525 cm−1, which is around the same wavenumber as the C5 intramolecular hydrogen bond found in the monomers. The same holds for the diagnostic amide III and CH bending band between 1150 and 1250 cm−1. The peaks between 1000 and 1150 cm−1 originate from ring vibrations of the OBn-cap of peptide B. The weaker intensity of this group of peaks can be explained by the structure of the B3 dimer, which contains only one OBn group instead of two for the BB dimer. All these findings indicate a parallel structure for the B3 dimer, as was found for the BB dimer.
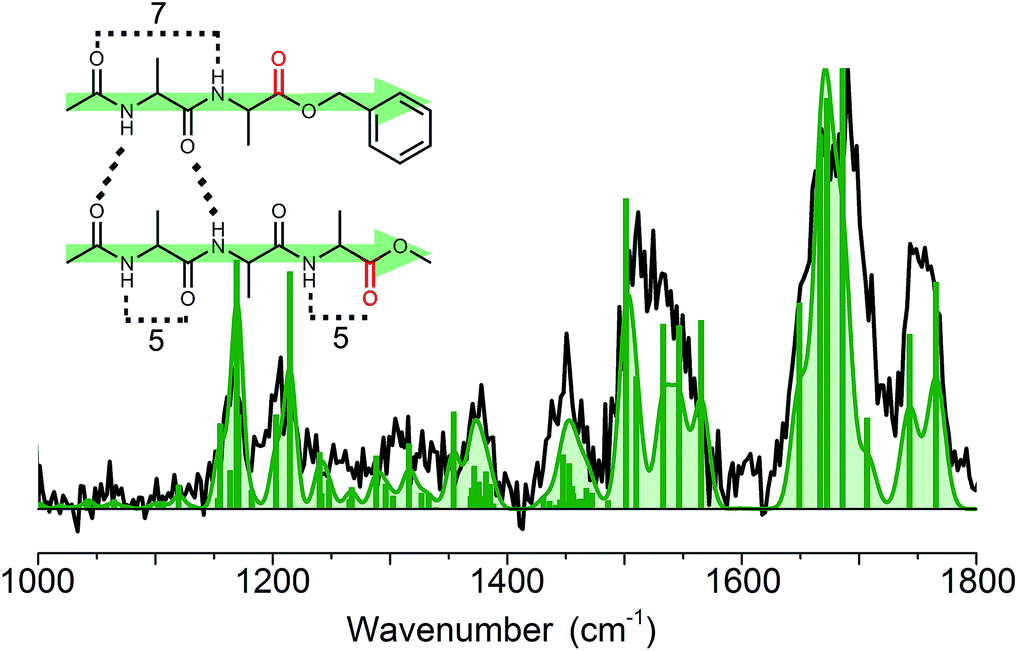 | ||
| Fig. 7 Experimental (black) and assigned calculated (green) infrared spectrum of the heterogeneous dimer B3. The inset shows the corresponding structure, which shows the same parallel intermolecular hydrogen bond pattern as the homogeneous dimer BB. | ||
Quantum chemical calculations were used to substantiate these findings. Therefore, the 30 structures found in molecular dynamics simulations were optimized and their IR frequencies were calculated. As was observed for the BB dimer, the lowest energy structures of the B3 dimer predominantly show parallel beta-sheet conformations, see Fig. SI8.† The first anti-parallel structure lies about 15 kJ mol−1 higher in energy than the lowest energy parallel structure. Other, more random structures, such as C6–C6 interactions or C7–C11, also have reasonable energies (<20 kJ mol−1). However, based on the amide I and amide II bands we could exclude the majority of the structures. The remaining conformers together with the lowest energy structures for each family resulted in eight structures, which are presented in Fig. SI8.† We assigned the structure to a parallel C12 intermolecular hydrogen bond as indicated in the inset of Fig. 7, although the other conformations with this similar parallel C12 interaction cannot be fully excluded. The assigned structure is analogous to the structure observed for the BB dimer.
Discussion
The structure of dimer BB is assigned to a parallel type beta-sheet structure, where the inter-strand hydrogen bonds form a C12 ring, see Fig. 8. The dimerization into parallel structures yields by far the most stable structures that are found for the BB dimer (see Fig. SI6a†). The two monomers from which this dimer is formed both originate from the original conformers found in our experiment. Conformer B-I forms a strong C7 (gamma turn) intramolecular hydrogen bond, which is preserved in the dimer structure. This C7 interaction slightly bends the backbone, thereby increasing the distance of the NH and CO moieties of the first alanine residue such that these are in an optimal position to form an inter-sheet hydrogen bond with conformer B-II. As can be seen from Fig. 8, the two UV chromophores are pointing in the same direction in this parallel beta-sheet BB dimer. However, the phenyl rings do not engage in any π–π interaction and are folded away from each other.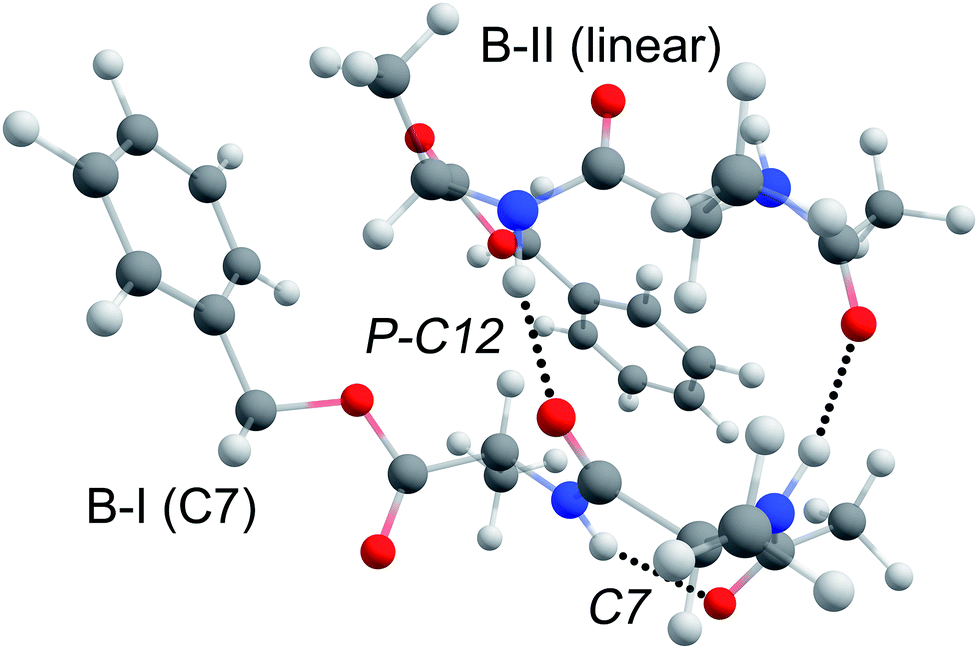 | ||
| Fig. 8 3D visualization of the assigned structure of peptide BB. The ring of the top peptide (B-II) is folded away such that it does not interact with the ring of the C7 intramolecularly hydrogen bonded B-I peptide. | ||
The heterogeneous B3 dimer shows the same parallel type structure as the BB dimer. The lowest energy structure has peptide B in the B-I conformation forming a C7 intramolecular hydrogen bond, while the linearly structured peptide 3 nucleates well on the template formed by the peptide B-I conformer, as was previously observed for the Ac-Val-Tyr-Me and Ac-Ala3-OMe dimer.24 However, the reversed parallel structure, with peptide 3 in the C7 conformation and peptide B in the B-II conformation, is only 2 kJ mol−1 higher in energy and shows only a slightly poorer agreement with the experimental spectrum. Therefore, this conformer cannot be excluded. Studying the infrared spectrum of peptide 3 using VUV ionization coupled to IRMPD would provide more insight into the structure of its monomers and could give an answer to this question.51 The parallel structures of the B3 dimer were also found to be the most stable structures (see Fig. SI8a†). Since the same structure is formed for dimer BB and B3, we can conclude that the intermolecular π–π interaction does not play a significant role in the aggregation of these peptides.
In contrast, the AA dimer does not seem to form any beta-sheet structures, i.e. the AA dimer appears to be only weakly bonded. The IR spectra of all dimers indicate that the monomeric conformers are retained in their dimers and dictate the observed structure. The observed monomers of peptide A both have strong intramolecular hydrogen bonds resulting in a compact cis conformer and a C7 gamma turn conformer. The latter would be an ideal platform to start self-assembly when linear-type conformers would be present with weak intramolecular hydrogen bonds as we observed for the B3 and BB dimers.
As for the other dimers containing at least one peptide B, the dimer of peptide AB was assigned to a parallel beta-sheet type structure as similar spectral signatures are found for the AB, B3 and BB dimer. Additionally, the IR spectrum of the AB dimer shows the diagnostic peak of the preserved intramolecular C7 hydrogen bond signature of conformer A-II of peptide A. These findings are substantiated by the large presence of parallel type structures in the low energy structures of the molecular dynamics search. However, where we observe that in the dimers of BB and B3 a linear conformer (either conformer B-II or a linear conformer of 3) is used as template that a C7 intramolecular hydrogen bonded structure nucleates onto, in peptide AB only the B-II conformer has a linear structure. This limits the possible combinations to a cluster of conformer B-II (linear with C5–C5 hydrogen bonds) with conformer A-II (C7 gamma turn) to form a parallel C12 structure. The dominance of this dimer structure is expected, as this linear conformation is required to form the higher order aggregates which we observed, as can be seen in Fig. SI4.†
Conclusions
The mass- and conformer-selective infrared spectra of four alanine-based dimers have been studied to probe (i) their propensity to form the well-known beta-sheet structures related to peptide aggregation and fibril formation, (ii) the key IR signatures for aggregation, (iii) the competition between intra- and intermolecular interactions, and (iv) the role of other actions such as dispersion. The studied peptide dimers consist of homogenous and heterogeneous combinations of Z-Ala-Ala-OMe (A), Ac-Ala-Ala-OBn (B), and Ac-Ala-Ala-Ala-OMe (3). The first two peptides have a benzyl group either on the N-terminus (A) or the C-terminus (B), which is used as a UV chromophore in our IR-UV ion-dip experiments. The monomers of both peptide A and B show 2 different conformations. For A the conformations are a γ-turn (making up a ring enclosing 7 atoms) and a surprising cis folded structure, while for peptide B a similar C7 γ-turn structure was found together with a linear C5–C5 conformation.Upon dimerization, the two homogenous dimers showed a different picture. For the dimer of peptide B the well-known beta sheet structure was observed in a parallel orientation. However, no beta-sheet structure was obtained for the AA dimer. Both monomer conformations of peptide A are engaged in either strong hydrogen bond interactions or multiple weaker interactions resulting in a fully folded structure. As a result, both structures do not provide a suitable template for peptide dimerization.
The IR spectra of both heterogenous dimers AB and B3 show similar spectral features to the BB dimer throughout the entire measured IR range, which is indicative of a beta-sheet structure with a parallel orientation. The linear conformation of at least one of the monomeric units in the dimer assists the beta-sheet formation. To conclude, the observed and assigned monomeric peptide conformers are retained upon dimerization, and thus direct their final structural arrangement. Currently, higher order peptide aggregates are being studied to evaluate how these conclusions can be extrapolated.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) for the support of the FELIX Laboratory. Calculations were carried out at the SurfSARA Cartesius cluster under NWO Rekentijd contract 16327.Notes and references
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2006, 75, 333–366 CrossRef CAS PubMed.
- C. M. Dobson, Nature, 2003, 426, 884–890 CrossRef CAS PubMed.
- T. P. J. Knowles, M. Vendruscolo and C. M. Dobson, Nat. Rev. Mol. Cell Biol., 2014, 15, 384–396 CrossRef CAS PubMed.
- A. E. Ashcroft, J. Am. Chem. Soc., 2010, 21, 1087–1096 CAS.
- D. Eisenberg, R. Nelson, M. R. Sawaya, M. Balbirnie, S. Sambashivan, M. I. Ivanova, A. O. Madsen and C. Riekel, Acc. Chem. Res., 2006, 39, 568–575 CrossRef CAS PubMed.
- K. A. Dill and J. L. MacCallum, Science, 2012, 338, 1042–1046 CrossRef CAS PubMed.
- J. Greenwald and R. Riek, Structure, 2010, 18, 1244–1260 CrossRef CAS PubMed.
- L. E. Buchanan, E. B. Dunkelberger, H. Q. Tran, P. N. Cheng, C. C. Chiu, P. Cao, D. P. Raleigh, J. J. de Pablo, J. S. Nowick and M. T. Zanni, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19285–19290 CrossRef CAS PubMed.
- A. Laganowsky, C. Liu, M. R. Sawaya, J. P. Whitelegge, J. Park, M. L. Zhao, A. Pensalfini, A. B. Soriaga, M. Landau, P. K. Teng, D. Cascio, C. Glabe and D. Eisenberg, Science, 2012, 335, 1228–1231 CrossRef CAS PubMed.
- P. Neudecker, P. Robustelli, A. Cavalli, P. Walsh, P. Lundstrom, A. Zarrine-Afsar, S. Sharpe, M. Vendruscolo and L. E. Kay, Science, 2012, 336, 362–366 CrossRef CAS PubMed.
- A. Bleem and V. Daggett, Biotechnol. Bioeng., 2017, 114, 7–20 CrossRef CAS PubMed.
- C. Seuring, K. Ayyer, E. Filippaki, M. Barthelmess, J. N. Longchamp, P. Ringler, T. Pardini, D. H. Wojtas, M. A. Coleman, K. Dorner, S. Fuglerud, G. Hammarin, B. Habenstein, A. E. Langkilde, A. Loquet, A. Meents, R. Riek, H. Stahlberg, S. Boutet, M. S. Hunter, J. Koglin, M. N. Liang, H. M. Ginn, R. P. Millane, M. Frank, A. Barty and H. N. Chapman, Nat. Commun., 2018, 9, 1836 CrossRef PubMed.
- Z. A. Levine and J. E. Shea, Curr. Opin. Struct. Biol., 2017, 43, 95–103 CrossRef CAS PubMed.
- C. Wasmer, A. Lange, H. Van Melckebeke, A. B. Siemer, R. Riek and B. H. Meier, Science, 2008, 319, 1523–1526 CrossRef CAS PubMed.
- P. I. Haris, Biochim. Biophys. Acta, Biomembr., 2013, 1828, 2265–2271 CrossRef CAS PubMed.
- S. L. Bernstein, N. F. Dupuis, N. D. Lazo, T. Wyttenbach, M. M. Condron, G. Bitan, D. B. Teplow, J. E. Shea, B. T. Ruotolo, C. V. Robinson and M. T. Bowers, Nat. Chem., 2009, 1, 326–331 CrossRef CAS PubMed.
- C. Bleiholder, N. F. Dupuis, T. Wyttenbach and M. T. Bowers, Nat. Chem., 2011, 3, 172–177 CrossRef CAS PubMed.
- W. Hoffmann, K. Folmert, J. Moschner, X. Huang, H. von Berlepsch, B. Koksch, M. T. Bowers, G. von Helden and K. Pagel, J. Am. Chem. Soc., 2018, 140, 244–249 CrossRef CAS PubMed.
- J. Seo, W. Hoffmann, S. Warnke, X. Huang, S. Gewinner, W. Schollkopf, M. T. Bowers, G. von Helden and K. Pagel, Nat. Chem., 2017, 9, 39–44 CrossRef CAS PubMed.
- M. Z. Kamrath and T. R. Rizzo, Acc. Chem. Res., 2018, 51, 1487–1495 CrossRef CAS PubMed.
- H. Elferink, M. E. Severijnen, J. Martens, R. A. Mensink, G. Berden, J. Oomens, F. Rutjes, A. M. Rijs and T. J. Boltje, J. Am. Chem. Soc., 2018, 140, 6034–6038 CrossRef CAS PubMed.
- J. Ujma, V. Kopysov, N. S. Nagornova, L. G. Migas, M. G. Lizio, E. W. Blanch, C. MacPhee, O. V. Boyarkin and P. E. Barran, Angew. Chem., Int. Ed., 2018, 57, 213–217 CrossRef CAS PubMed.
- N. Borho, M. A. Suhm, K. Le Barbu-Debus and A. Zehnacker, Phys. Chem. Chem. Phys., 2006, 8, 4449–4460 RSC.
- K. Schwing and M. Gerhards, Int. Rev. Phys. Chem., 2016, 35, 569–677 Search PubMed.
- E. Gloaguen and M. Mons, Top. Curr. Chem., 2015, 364, 225–270 CrossRef CAS PubMed.
- S. Habka, W. Y. Sohn, V. Vaquero-Vara, M. Geleoc, B. Tardivel, V. Brenner, E. Gloaguen and M. Mons, Phys. Chem. Chem. Phys., 2018, 20, 3411–3423 RSC.
- M. Gerhards and C. Unterberg, Phys. Chem. Chem. Phys., 2002, 4, 1760–1765 RSC.
- M. Gerhards, C. Unterberg and A. Gerlach, Phys. Chem. Chem. Phys., 2002, 4, 5563–5565 RSC.
- M. Gerhards, C. Unterberg, A. Gerlach and A. Jansen, Phys. Chem. Chem. Phys., 2004, 6, 2682–2690 RSC.
- A. Gerlach, C. Unterberg, H. Fricke and M. Gerhards, Mol. Phys., 2005, 103, 1521–1529 CrossRef CAS.
- T. D. Vaden, S. A. N. Gowers and L. C. Snoek, J. Am. Chem. Soc., 2009, 131, 2472–2474 CrossRef CAS PubMed.
- D. J. Bakker, A. Dey, D. P. Tabor, Q. Ong, J. Mahé, M. P. Gaigeot, E. L. Sibert and A. M. Rijs, Phys. Chem. Chem. Phys., 2017, 19, 20343–20356 RSC.
- M. Cirtog, A. M. Rijs, Y. Loquais, V. Brenner, B. Tardivel, E. Gloaguen and M. Mons, J. Phys. Chem. Lett., 2012, 3, 3307–3311 CrossRef CAS.
- A. M. Rijs, N. Sandig, M. N. Blom, J. Oomens, J. S. Hannam, D. A. Leigh, F. Zerbetto and W. J. Buma, Angew. Chem., Int. Ed., 2010, 49, 3896–3900 CrossRef CAS PubMed.
- H. Zhu, M. Blom, I. Compagnon, A. M. Rijs, S. Roy, G. von Helden and B. Schmidt, Phys. Chem. Chem. Phys., 2010, 12, 3415–3425 RSC.
- Y. J. Hu and E. R. Bernstein, J. Phys. Chem. A, 2009, 113, 8454–8461 CrossRef CAS PubMed.
- J. J. Lee, M. Albrecht, C. A. Rice and M. A. Suhm, J. Phys. Chem. A, 2013, 117, 7050–7063 CrossRef CAS PubMed.
- S. Bakels, S. B. A. Porskamp and A. M. Rijs, ChemRxiv, 2019 DOI:10.26434/chemrxiv.7833500.v1.
- A. M. Rijs, E. R. Kay, D. A. Leigh and W. J. Buma, J. Phys. Chem. A, 2011, 115, 9669–9675 CrossRef CAS PubMed.
- A. M. Rijs and J. Oomens, Top. Curr. Chem., 2015, 364, 1–42 CrossRef CAS PubMed.
- P. M. Johnson and C. E. Otis, Annu. Rev. Phys. Chem., 1981, 32, 139–157 CrossRef CAS.
- D. Oepts, A. F. G. van der Meer and P. W. van Amersfoort, Infrared Phys. Technol., 1995, 36, 297–308 CrossRef CAS.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerlin, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Götz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, 2012 Search PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104-1–154104-19 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. J. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision E.01, 2016 Search PubMed.
- I. Compagnon, J. Oomens, J. Bakker, G. Meijer and G. von Helden, Phys. Chem. Chem. Phys., 2005, 7, 13–15 RSC.
- A. M. Rijs, I. Compagnon, J. Oomens, J. S. Hannam, D. A. Leigh and W. J. Buma, J. Am. Chem. Soc., 2009, 131, 2428–2429 CrossRef CAS PubMed.
- E. Gloaguen, B. de Courcy, J. P. Piquemal, J. Pilme, O. Parisel, R. Pollet, H. S. Biswal, F. Piuzzi, B. Tardivel, M. Broquier and M. Mons, J. Am. Chem. Soc., 2010, 132, 11860–11863 CrossRef CAS PubMed.
- L. Voronina and T. R. Rizzo, Phys. Chem. Chem. Phys., 2015, 17, 25828–25836 RSC.
- J. A. Silveira, K. L. Fort, D. Kim, K. A. Servage, N. A. Pierson, D. E. Clemmer and D. H. Russell, J. Am. Chem. Soc., 2013, 135, 19147–19153 CrossRef CAS PubMed.
- V. Yatsyna, D. J. Bakker, P. Salen, R. Feifel, A. M. Rijs and V. Zhaunerchyk, Phys. Rev. Lett., 2016, 117, 118101 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8fd00208h |
| This journal is © The Royal Society of Chemistry 2019 |