
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTris(benzoimidazol)amine (L) complexes of pnictogen(III) and pnictogen(V) cations and assessment of the [LP]3+/[LPF2]3+ redox couple†
Ala'aeddeen
Swidan‡
 a,
Riccardo
Suter‡
*b,
Charles L. B.
Macdonald
*a and
Neil
Burford
*b
a,
Riccardo
Suter‡
*b,
Charles L. B.
Macdonald
*a and
Neil
Burford
*b
aDepartment of Chemistry and Biochemistry, University of Windsor, Windsor, Ontario N9B 3P4, Canada. E-mail: cmacd@uwindsor.ca
bDepartment of Chemistry, University of Victoria, Victoria, British Columbia V8W 3V6, Canada. E-mail: nburford@uvic.ca; risuter@uvic.ca
First published on 19th June 2018
Abstract
A series of cationic complexes involving a pnictogen(III) (Pn = P, As, Sb) centre and the tetradentate ligand tris((1-ethyl-benzoimidazol-2-yl)methyl)amine (BIMEt3) have been synthesized and comprehensively characterized. Oxidation of [P(BIMEt3)]3+ with XeF2 provides access to [PF2(BIMEt3)]3+ representing the first structurally characterized example of a phosphorus(V) centred trication.
Introduction
Phosphorus(III) centers can undergo reversible oxidative addition of N–H and O–H bonds and have potential application as catalysts in organic transformations.1 This traditional Lewis basic reactivity extends to pnictogen(III) centers in general, but has been challenged by the realization of the Lewis acid “umpolung” made possible by the introduction of a cationic charge.2 A variety of pnictogen(III) based cations have been synthesized by halide abstraction from PnX3 derivatives in the presence of various ligands and weakly coordinating anions.3 The cationic charge of such complexes not only lowers the energy of the pnictogen based LUMO but also lowers the energy of the HOMO, so that oxidation is impeded. Consequently, examples of redox couples of the type [Pn(III)L]3+/[Pn(V)R2L]3+ have not yet been reported. The fluorophilicity of ligand stabilized phosphenium cations4 and dications5 is well established and has led to the discovery of effective catalysts for hydrofluorination reactions.6 The Lewis acidity of [PRL]2+ with L = terpyridine renders it an active catalyst for dehydrofluorination of fluoroalkanes, and suggests it has a substantial fluoride ion affinity.7We have recently shown that the multidentate tris(benzoimidazol)amine ligand (BIMEt3) encapsulates a germanium dication that is readily oxidized to [GeF2BIMEt3][OTf]2,8 and we have now exploited the versatility of this ligand to synthesise derivatives of [Pn(BIMEt3)][OTf]3 for Pn = P, As and Sb. As an analogue of the Verkade superbases [P(PRZ)],9 [P(BIMEt3)]3+ adopts a proazaphosphatrane type cage structure. Oxidation of [P(BIMEt3)][OTf]3 by XeF2 gives [PF2(BIMEt3)][OTf]3, containing a rare example of a cationic complex of the high oxidation state Pn(V) center.10 Previous examples include derivatives of [PnPh3Lx][OTf]2 (L = pyridine N-oxide and Pn = As,11 Sb, Bi12) and [SbPh2Lx][OTf]3 (L = pyridine N-oxide13).
Results
The reaction of BIMH3 and PCl3 in the presence of a slight excess of NaH in THF at room temperature gives P(BIM).14 Quantitative formation of P(BIM) is evidenced by a single peak in the 31P NMR spectrum (31P δ = 44.7 ppm). Crystals of P(BIM) were obtained by slow evaporation of the solvent from a DCM/MeCN solution. In the solid-state structure (Fig. 1a), two enantiomers are present in the unit cell, with the benzoimidazole groups arranged either in an S or R configuration. The geometry at N1 is essentially planar (353.77 and 355.85°, respectively) and features a pre-ordered N1⋯P bond with an average distance of 2.925 Å. The P–N3 bonds (1.721 Å) are significantly longer than those observed in P(PZ) (e.g. 1.694 Å for N(CH2–CH2–NR)3P with R = CH(Me)Ph).15 The nucleophilic behaviour of P(BIM) was explored through reactions involving a variety of stoichiometric ratios of methyltriflate. In each case the 31P-NMR spectrum of the reaction mixture indicated the formation of several products that we speculate to result from simultaneous methylation of the nitrogen and phosphorus atoms. We have modelled the energetic profile of the reactions using DFT calculations at the PBEPBE/6-311+G(d,p) level of theory.16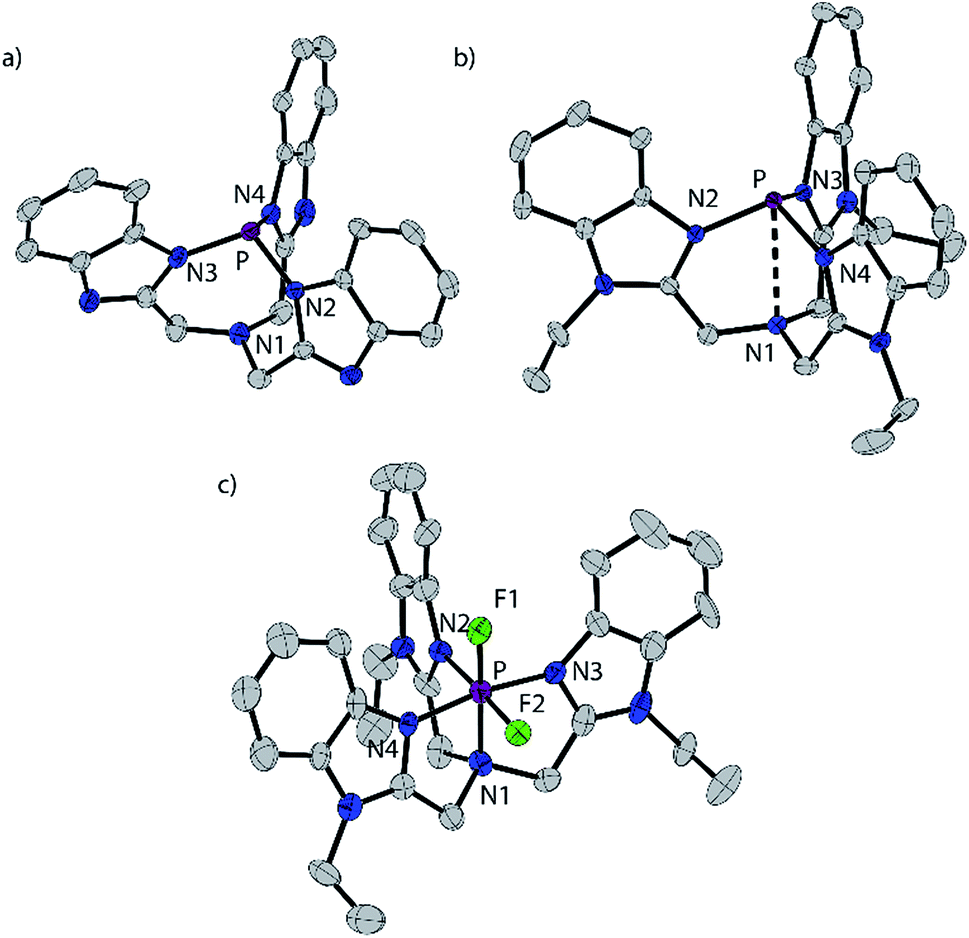 | ||
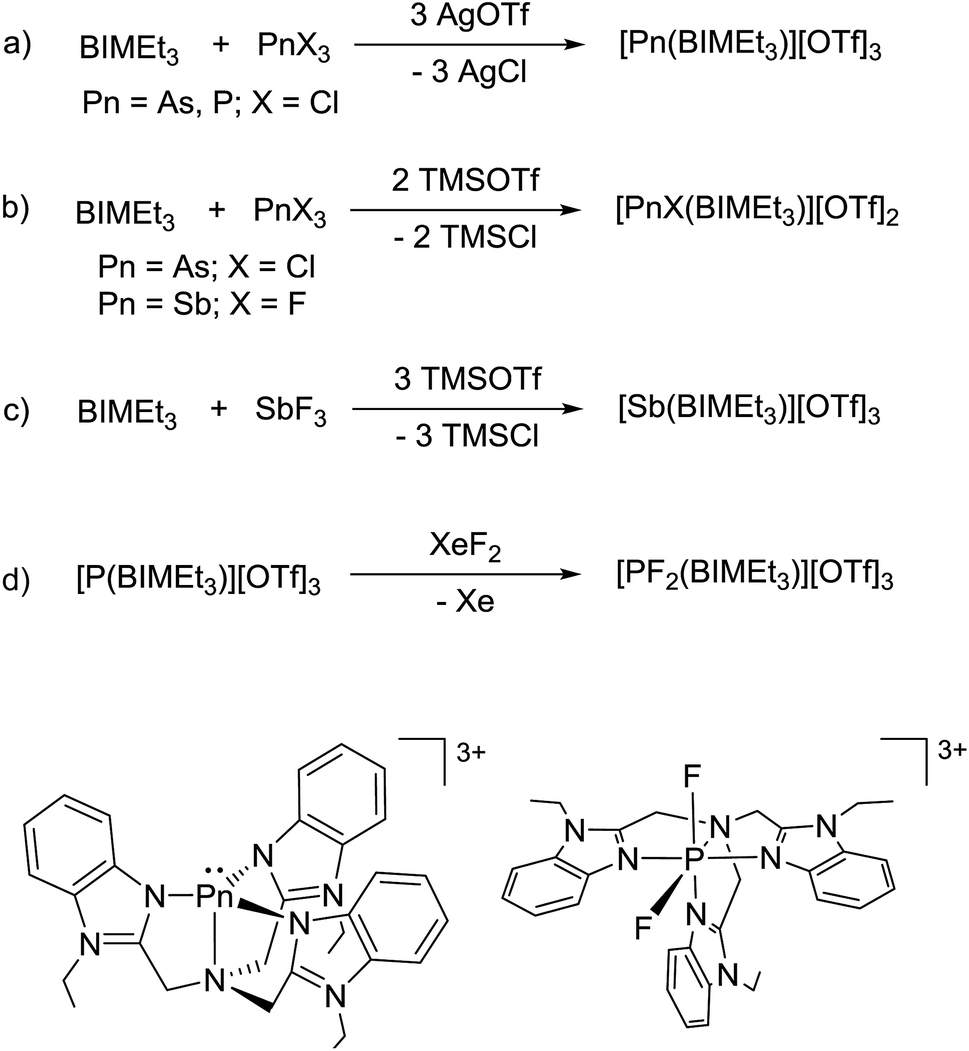
| Fig. 1 Solid state structures of one of two independent molecules of P(BIM) (a), the cation in [P(BIMEt3)][OTf]3·(MeCN)2 (b), and cation in [PF2(BIMEt3)][OTf]3·MeCN (c). Thermal ellipsoids are shown at a 50% probability level. Hydrogen atoms, solvent molecules and triflate anions are omitted for clarity. Inter-atomic distances and angles are summarized in Table 2. | ||
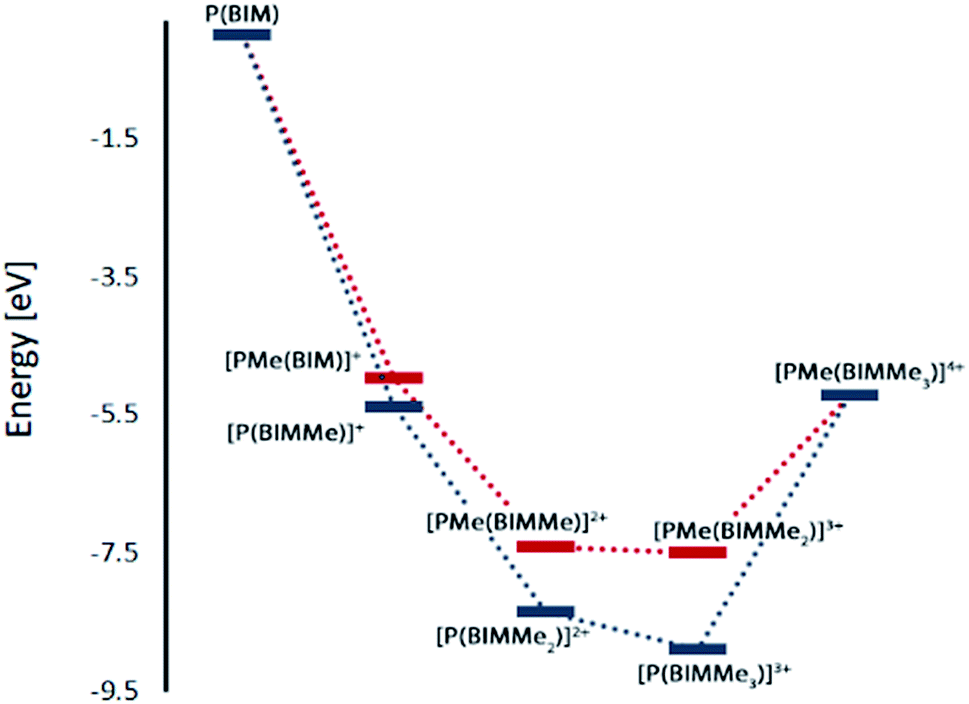
Geometry optimizations for each complex suggest that methylation is favoured at the imino nitrogen atoms (Table 1). In the gas phase each methylation of an imino centre is exergonic. Similar results were obtained with MeCN as a solvent model but an energetic minimum was not evident for [PMe(BIMMe3)]4+ (Table 1 and ESI†), and consistently, complicated reaction mixtures are observed for MeOTf and P(BIM). Nevertheless, addition of excess MeOTf resulted in a colourless precipitate (insoluble in common organic solvents) which suggests the potential formation of [PMe(BIMMe3)]4+. We have modelled the bonding in the potentially stable tricationic complex [BIMMe3P]3+ (at the PBEPBE/6-311+G(d,p) level of theory). NBO analysis on the optimized structure reveals a natural charge of +1.5 at phosphorus, and +0.5 on each benzoimidazole group, consistent with relatively strong P–N bonds (Wiberg bond index of 0.73).
|
|
|||
|---|---|---|---|
|
|
|||
| N-Methylation | eV | P-Methylation | eV |
| P(BIM) + 4 Me+ | 0 | P(BIM) + 4 Me+ | 0 |
| [P(BIMMe)]+ + 3 Me+ | −5.38 | [PMe(BIM)]+ + 3 Me+ | −4.96 |
| [P(BIMMe2)]2+ + 2 Me+ | −8.34 | [PMe(BIMMe)]2+ + 2 Me+ | −7.40 |
| [P(BIMMe3)]3+ + 1 Me+ | −8.90 | [PMe(BIMMe2)]3+ + 1 Me+ | −7.49 |
| [PMe(BIMMe3)]4+ | −5.21 | [PMe(BIMMe3)]4+ | −5.21 |
Based on the modelled stability of [P(BIMMe3)]3+, we examined the reaction of BIMEt3 (ref. 17) with “P(OTf)3” by mixing PCl3 and AgOTf acetonitrile in the presence of BIMEt3 (Scheme 1a). A singlet at δ = 56.1 ppm in the 31P NMR spectrum indicates the quantitative formation of [P(BIMEt3)][OTf]3 which has been separated from the AgCl by filtration, and crystallized by layering the reaction mixture with diethyl ether. The analogous reactions of AsCl3 or SbCl3 provided [As(BIMEt3)][OTf]3 and [Sb(BIMEt3)][OTf]3, respectively. In contrast, reaction of AsCl3 with BIMEt3 and excess TMSOTf formed [AsCl(BIMEt3)][OTf]2 as a primary product (Scheme 1b), which has been isolated in small quantities. The antimony fluoride derivative [SbF(BIMEt3)][OTf]2 is formed quantitatively in the reaction of SbF3 with BIMEt3 and two equivalents of TMSOTf (Scheme 1c). The derivatives of [MX(BIMEt3)][OTf]2 exhibit one set of signals for the benzoimidazole groups in the 1H-NMR spectrum, consistent with the triflate being labile in solution, as observed for analogous germanium complexes.8
 | ||
| Scheme 1 Synthetic procedure for derivatives of [PnX(BIMEt3)][OTf]2, [Pn(BIMEt3)][OTf]3 and [PF2(BIMEt3)][OTf]3. | ||
[P(BIMEt3)][OTf]3·(MeCN)2 crystallizes in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (see Fig. 1b). The three triflate anions are remote from the phosphorus centre (3.58–3.92 Å). The unique apex nitrogen atom (°ΣN1 = 350.5) is significantly bent out of plane towards the phosphorus centre consistent with a cross-ring bonding interaction. Nevertheless, N1⋯P(2.866(2) Å) is only slightly shorter than those in P(BIM) (2.912(2) and 2.938(2) Å). In the solid state [As(BIMEt3)][OTf]3 and [Sb(BIMEt3)][OTf]3 (Fig. 2c and d) exhibit a significantly different geometry around the pnictogen centre. The polygon described by the ligands is best described as a pentagonal bipyramid with three nitrogen atoms (N1, N2 and N3), one triflate oxygen atom and the lone pair in the plane. A second triflate oxygen atom and one nitrogen (N4) atom occupy the axial positions. This geometry enables significantly shorter Pn–N1 bond (As–N1 2.104(4) and Sb–N1 2.389(2) and 2.370(2) Å, respectively) compared to that in [P(BIMEt3)][OTf]3 (P–N1 2.879 Å). Two of the three triflate anions have a weak interaction with the pnictogen centre (Table 3). The mono-halide derivatives [SbF(BIMEt3)][OTf]2 and [AsCl(BIMEt3)][OTf]2 crystallise as dimeric structures. The antimony fluoride is linked by two triflate anions and the arsenic chloride is linked by chlorine substituents (As–Cl 2.3342(7) and 3.913(2) Å). The antimony centre adopts a tetragonal pyramidal geometry with two oxygen atoms from the triflate anions (Sb–OTf 2.551(2) and 2.793(2) Å) and two nitrogen atoms (Sb–N 2.214(2) and 2.165(2) Å) with the antimony centre 0.486 Å above this plane.
(see Fig. 1b). The three triflate anions are remote from the phosphorus centre (3.58–3.92 Å). The unique apex nitrogen atom (°ΣN1 = 350.5) is significantly bent out of plane towards the phosphorus centre consistent with a cross-ring bonding interaction. Nevertheless, N1⋯P(2.866(2) Å) is only slightly shorter than those in P(BIM) (2.912(2) and 2.938(2) Å). In the solid state [As(BIMEt3)][OTf]3 and [Sb(BIMEt3)][OTf]3 (Fig. 2c and d) exhibit a significantly different geometry around the pnictogen centre. The polygon described by the ligands is best described as a pentagonal bipyramid with three nitrogen atoms (N1, N2 and N3), one triflate oxygen atom and the lone pair in the plane. A second triflate oxygen atom and one nitrogen (N4) atom occupy the axial positions. This geometry enables significantly shorter Pn–N1 bond (As–N1 2.104(4) and Sb–N1 2.389(2) and 2.370(2) Å, respectively) compared to that in [P(BIMEt3)][OTf]3 (P–N1 2.879 Å). Two of the three triflate anions have a weak interaction with the pnictogen centre (Table 3). The mono-halide derivatives [SbF(BIMEt3)][OTf]2 and [AsCl(BIMEt3)][OTf]2 crystallise as dimeric structures. The antimony fluoride is linked by two triflate anions and the arsenic chloride is linked by chlorine substituents (As–Cl 2.3342(7) and 3.913(2) Å). The antimony centre adopts a tetragonal pyramidal geometry with two oxygen atoms from the triflate anions (Sb–OTf 2.551(2) and 2.793(2) Å) and two nitrogen atoms (Sb–N 2.214(2) and 2.165(2) Å) with the antimony centre 0.486 Å above this plane.
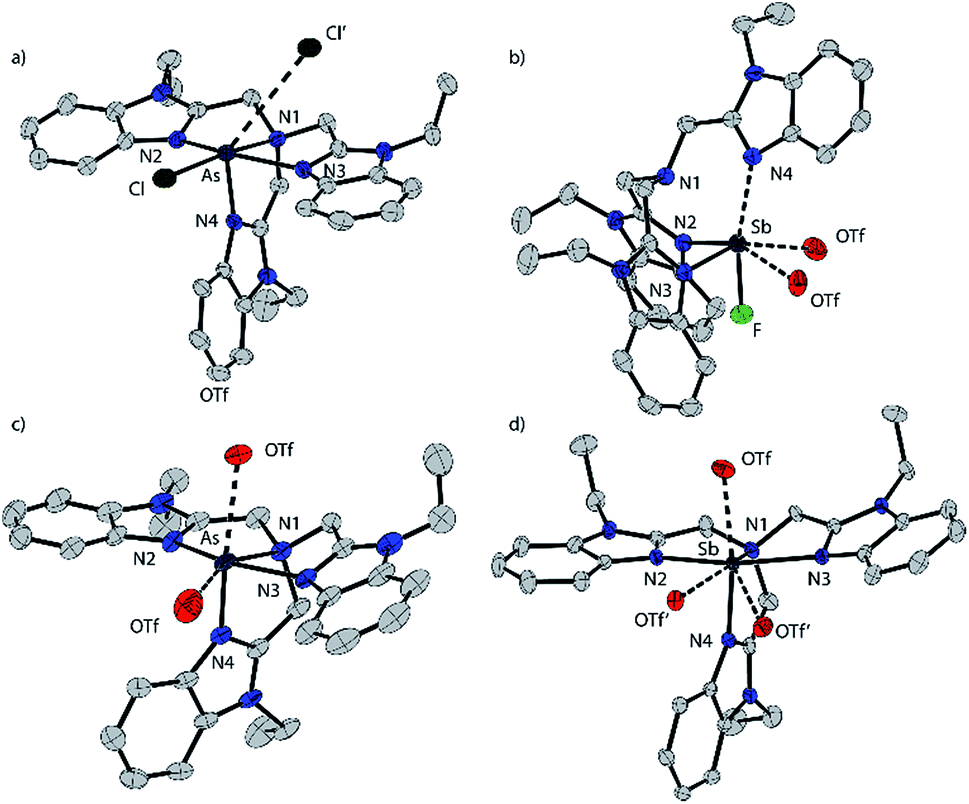 | ||
| Fig. 2 Solid state structure of the cations in [AsCl(BIMEt3)][OTf]2 (a), [SbF(BIMEt3)][OTf]2 (b), [As(BIMEt3)][OTf]3 (c), and [Sb(BIMEt3)][OTf]3 (d). Thermal ellipsoids are shown at a 50% probability level. Oxygen atoms of the triflate anions that interact with the pnictogen centres are shown, but the other atoms of the anions are omitted for clarity as well as the hydrogen atoms and solvent molecules. Interatomic distances and angles are summarized in Table 3. | ||
| P(BIM) | [P(BIMEt3)]3+ | [PF2(BIMEt3)]3+ | |
|---|---|---|---|
| P–N2 | 1.7281(11) | 1.7279(16) | 1.791(13) |
| 1.7159(11) | 1.7352(16) | 1.798(13) | |
| 1.7201(11) | 1.7364(16) | 1.773(13) | |
| 1.7246(12) | |||
| 1.7137(11) | |||
| 1.7242(12) | |||
| P–N1 | 2.912(2) | 2.866(2) | 1.893(13) |
| 2.938(2) | |||
| P–OTf | — | 3.591(2) | — |
| 3.922(2) | |||
| 3.584(2) | |||
| P–F | — | — | 1.604(9) |
| 1.577(9) | |||
| 31P δ | 44.7 (s) | 56.1 (s) | −127.8 (dd) |
| °ΣP | 319.48 | 320.2 | — |
| 317.75 | |||
| °ΣN1 | 353.77 | 350.5 | 328.0 |
| 355.85 |
| [AsCl(BIMEt3)]3+ | [As(BIMEt3)]3+ | [SbF(BIMEt3)]2+ | [Sb(BIMEt3)]3+ | ||
|---|---|---|---|---|---|
| Pn–N2 | 1.964(2) | 1.918(4) | 2.214(2) | 2.193(2) | 2.168(2) |
| 2.078(2) | 2.085(4) | 2.165(3) | 2.233(2) | 2.246(2) | |
| 2.251(2) | 2.195(4) | 2.335(2) | 2.346(2) | ||
| Pn–N1 | 2.268(2) | 2.104(4) | 2.635(3) | 2.389(2) | 2.370(2) |
| Pn–OTf | — | 2.646(4) | 2.551(2) | 2.421(2) | 2.485(2) |
| 2.796(4) | 2.793(2) | 3.181(2) | 3.171(2) | ||
| 2.833(2) | 2.820(2) | ||||
| 3.258(2) | 3.313(2) | ||||
| Pn–X | 2.3342(7) 3.913(2) X = Cl | 1.9344(19) X = F | |||
Equimolar mixtures of PCl5 and BIMEt3 with three equivalents of AgOTf give a mixture of products as evidenced by the 31P NMR spectrum of the reaction mixture. Isolation of [P(BIMEt3)][OTf]3 from this mixture implicates formation of [PCl2(BIMEt3)][OTf]3, which is subsequently reduced. In this context, we have studied the oxidation of [P(BIMEt3)][OTf]3 with the expectation to form cationic compounds analogous to [PPh3I][I3]18 or [Ph3P–I–I]19 However, reaction of addition of [P(BIMEt3)][OTf]3 with a large excess of I2 caused only a small chemical shift in the 31P NMR spectrum and a product could not be isolated. Nevertheless, Stephan's approach for oxidation of phosphenium cations4 was applied using equimolar mixtures of [P(BIMEt3)][OTf]3 and XeF2 (Scheme 1d) in acetonitrile to yield a mixture of products with a prominent doublet of doublets resonance in the 31P-NMR spectrum (31P δ = −127.8 ppm; 1JPF = 899.3 and 849.7 Hz) and consistently, two doublet of doublet resonances in the 19F NMR spectrum (δ = −65.5 and −33.9 ppm; 2JFF = 52.0 Hz). Attempts to scale up the reaction yielded several different fluorinated phosphorus species. Rapid crystallization from a saturated acetonitrile solution at −35 °C layered with diethyl ether yielded small amounts of fragile crystals, that have been characterized as [PF2(BIMEt3)][OTf]3. Consistent with the solution NMR spectra (Fig. 3), the solid state structure reveals, inequivalent fluorine substituents at phosphorus (Fig. 1c) and two different benzoimidazole environments.
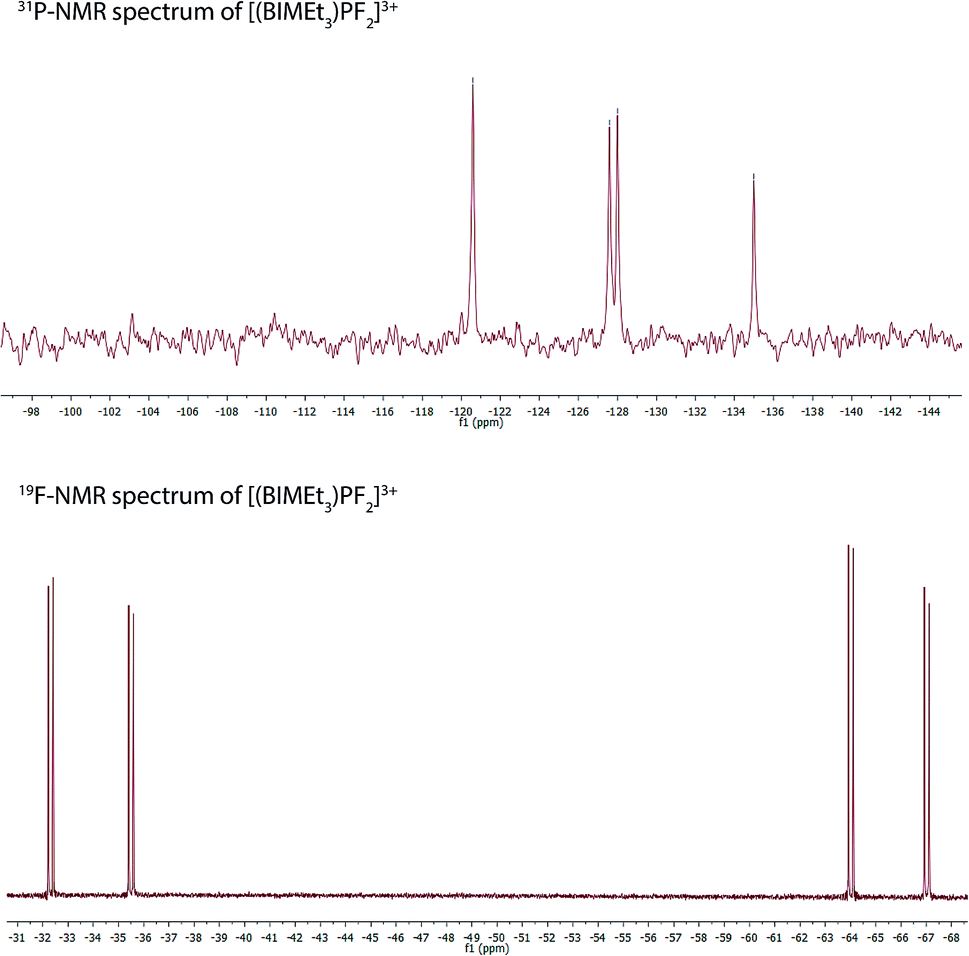 | ||
| Fig. 3 NMR spectra for [PF2(BIMEt3)][OTf]3 in CD3CN. | ||
The N1–P distance (1.893(13) Å) is significantly shorter than that in [P(BIMEt3)][OTf]3. The P–F1 bond trans to the tertiary amine is longer [1.577(9) Å] than P–F2 [1.604(9) Å]. Consequently, the pseudo C3V-geometry of the cation in [P(BIMEt3)][OTf]3 adjusts to Cs symmetry in the octahedral frame in [PF2(BIMEt3)][OTf]3. Moreover, P–N1 in [PF2(BIMEt3)][OTf]3 is shorter than that in [P(BIMEt3)][OTf]3 and the other P–N bonds are longer (Table 2). This substantive change in geometry is also evidenced by 31P NMR spectroscopy ([P(BIMEt3)]3+δ = −127.8 ppm) with a chemical shift difference of 184 ppm upon oxidation. There are no close interactions of the phosphorus centre with any of the triflate anions, so that the cation in [PF2(BIMEt3)][OTf]3 represents the first fully characterized tricationic P(V)3+ species.
Conclusions
We have presented a versatile and facile synthetic approach to a number of pnictogen(III) cations in the BIMEt3 ligand scaffold. The phosphorus derivative [P(BIMEt3)][OTf]3 can be readily oxidized to form the first tricationic phosphorus(V) complex, [LPVF2]3+. A fundamental study for general use of such pnictogen salts in Lewis acid catalysis, small molecule activation or as catalytic fluorinating agents is ongoing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The work was supported by the University of Victoria, University of Windsor and the Natural Sciences and Engineering Research Council of Canada (NSERC) 2016-05260 (NB) and 249809-2013 (CLBM). This research was enabled in part by support provided by WestGrid and Compute Canada (https://www.computecanada.ca). Robert Mac Donald from the University of Alberta is thanked for his assistance regarding X-ray crystallography and Hannah Sinclair from University of Victoria for preliminary synthetic work.Notes and references
- (a) A. J. Arduengo, C. A. Stewart, F. Davidson, D. A. Dixon, J. Y. Becker, S. A. Culley and M. B. Mizen, J. Am. Chem. Soc., 1987, 109, 627–647 CrossRef; (b) W. Zhao, S. M. McCarthy, T. Y. Lai, H. P. Yennawar and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 17634–17644 CrossRef PubMed; (c) D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC; (d) S. M. McCarthy, Y.-C. Lin, D. Devarajan, J. W. Chang, H. P. Yennawar, R. M. Rioux, D. H. Ess and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 4640–4650 CrossRef PubMed; (e) N. L. Dunn, M. Ha and A. T. Radosevich, J. Am. Chem. Soc., 2012, 134, 11330–11333 CrossRef PubMed; (f) J. Cui, Y. Li, R. Ganguly, A. Inthirarajah, H. Hirao and R. Kinjo, J. Am. Chem. Soc., 2014, 136, 16764–16767 CrossRef PubMed; (g) T. P. Robinson, D. M. De Rosa, S. Aldridge and J. M. Goicoechea, Angew. Chem., Int. Ed., 2015, 54, 13758–13763 CrossRef PubMed.
- (a) S. S. Chitnis, A. P. M. Robertson, N. Burford, B. O. Patrick, R. McDonald and M. J. Ferguson, Chem. Sci., 2015, 6, 6545–6555 RSC; (b) S. S. Chitnis, K. A. Vos, N. Burford, R. McDonald and M. J. Ferguson, Chem. Commun., 2016, 52, 685–688 RSC; (c) F. D. Henne, A. T. Dickschat, F. Hennersdorf, K. O. Feldmann and J. J. Weigand, Inorg. Chem., 2015, 54, 6849–6861 CrossRef PubMed; (d) J. Petuskova, M. Patil, S. Holle, C. W. Lehmann, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2011, 133, 20758–20760 CrossRef PubMed; (e) J. Carreras, M. Patil, W. Thiel and M. Alcarazo, J. Am. Chem. Soc., 2012, 134, 16753–16758 CrossRef PubMed; (f) J. J. Weigand, K.-O. Feldmann, A. K. C. Echterhoff, A. W. Ehlers and K. Lammertsma, Angew. Chem., Int. Ed., 2010, 49, 6178–6181 CrossRef PubMed.
- (a) T. A. Engesser, M. R. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789–899 RSC; (b) A. P. M. Robertson, P. A. Gray and N. Burford, Angew. Chem., Int. Ed., 2014, 53, 6050–6069 CrossRef PubMed; (c) H. Sinclair, R. Suter, N. Burford, R. McDonald and M. J. Ferguson, Can. J. Chem., 2018, 1–5 CrossRef; (d) R. Suter, P. A. Gray, N. Burford and R. McDonald, Chem.–Eur. J., 2018, 24, 4718–4723 CrossRef PubMed.
- (a) M. Mehta, M. H. Holthausen, I. Mallov, M. Pérez, Z.-W. Qu, S. Grimme and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 8250–8254 CrossRef PubMed; (b) M. H. Holthausen, M. Mehta and D. W. Stephan, Angew. Chem., Int. Ed., 2014, 53, 6538–6541 CrossRef PubMed.
- Á. Kozma, J. Rust and M. Alcarazo, Chem.–Eur. J., 2015, 21, 10829–10834 CrossRef PubMed.
- (a) C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374–1377 CrossRef PubMed; (b) J. M. Bayne and D. W. Stephan, Chem. Soc. Rev., 2016, 45, 765–774 RSC.
- S. Chitnis, F. Krischer and D. W. Stephan, Chem.–Eur. J., 2018, 24, 6543 CrossRef PubMed.
- R. Suter, A. Swidan, C. L. B. Macdonald and N. Burford, Chem. Commun., 2018, 54, 4140–4143 RSC.
- (a) H. Schmidt, C. Lensink, S. K. Xi and J. G. Verkade, Z. Anorg. Allg. Chem., 1989, 578, 75–80 CrossRef; (b) J. Tang, J. Dopke and J. G. Verkade, J. Am. Chem. Soc., 1993, 115, 5015–5020 CrossRef; (c) J. S. Tang and J. G. Verkade, J. Am. Chem. Soc., 1993, 115, 1660–1664 CrossRef; (d) P. B. Kisanga and J. G. Verkade, Tetrahedron, 2001, 57, 467–475 CrossRef; (e) S. Mummadi, D. Kenefake, R. Diaz, D. K. Unruh and C. Krempner, Inorg. Chem., 2017, 56, 10748–10759 CrossRef PubMed.
- P. A. Gray and N. Burford, Coord. Chem. Rev., 2016, 324, 1–16 CrossRef.
- M. Donath, M. Bodensteiner and J. J. Weigand, Chem.–Eur. J., 2014, 20, 17306–17310 CrossRef PubMed.
- A. P. M. Robertson, N. Burford, R. McDonald and M. J. Ferguson, Angew. Chem., Int. Ed., 2014, 53, 3480–3483 CrossRef PubMed.
- C. Frazee, N. Burford, R. McDonald, M. J. Ferguson, A. Decken and B. O. Patrick, Chem.–Eur. J., 2018, 24, 4011–4013 CrossRef PubMed.
- (a) S.-R. Zhang, D.-Y. Du, J.-S. Qin, S.-J. Bao, S.-L. Li, W.-W. He, Y.-Q. Lan, P. Shen and Z.-M. Su, Chem.–Eur. J., 2014, 20, 3589–3594 CrossRef PubMed; (b) X. Liu, Y. Bai and J. G. Verkade, J. Organomet. Chem., 1999, 582, 16–24 CrossRef; (c) P. L. Shutov, S. S. Karlov, K. Harms, D. A. Tyurin, A. V. Churakov, J. Lorberth and G. S. Zaitseva, Inorg. Chem., 2002, 41, 6147–6152 CrossRef PubMed.
- X. Liu, P. Ilankumaran, I. A. Guzei and J. G. Verkade, J. Org. Chem., 2000, 65, 701–706 CrossRef.
- M. J. Frisch, et al., Gaussian 16., Revision. A.03., Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- D. G. Lonnon, D. C. Craig and S. B. Colbran, Dalton Trans., 2006, 3785–3797 RSC.
- F. A. Cotton and P. A. Kibala, J. Am. Chem. Soc., 1987, 109, 3308–3312 CrossRef.
- (a) S. M. Godfrey, D. G. Kelly, C. A. McAuliffe, A. G. Mackie, R. G. Pritchard and S. M. Watson, J. Chem. Soc., Chem. Commun., 1991, 1163–1164 RSC; (b) T. P. Robinson, D. De Rosa, S. Aldridge and J. M. Goicoechea, Chemistry, 2017, 23, 15455–15465 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1578584–1578587, 1581022, 1581023 and 1819912. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01682h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |