
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Millifluidic culture improves human midbrain organoid vitality and differentiation†
Emanuel
Berger
a,
Chiara
Magliaro
 b,
Nicole
Paczia
c,
Anna S.
Monzel
a,
Paul
Antony
d,
Carole L.
Linster
c,
Silvia
Bolognin
a,
Arti
Ahluwalia
b and
Jens C.
Schwamborn
*a
b,
Nicole
Paczia
c,
Anna S.
Monzel
a,
Paul
Antony
d,
Carole L.
Linster
c,
Silvia
Bolognin
a,
Arti
Ahluwalia
b and
Jens C.
Schwamborn
*a
aUniversity of Luxembourg (UL), Centre for Systems Biomedicine (LCSB) – Developmental and Cellular Biology group, Luxembourg. E-mail: jens.schwamborn@uni.lu
bUniversità di Pisa, Centro di Ricerca “E. Piaggio”, Pisa, Italy
cUniversity of Luxembourg (UL), Centre for Systems Biomedicine (LCSB) – Enzymology & Metabolism group, Luxembourg
dUniversity of Luxembourg (UL), Centre for Systems Biomedicine (LCSB) – Experimental Neurobiology group, Luxembourg
First published on 11th September 2018
Abstract
Human midbrain-specific organoids (hMOs) serve as an experimental in vitro model for studying the pathogenesis of Parkinson's disease (PD). In hMOs, neuroepithelial stem cells (NESCs) give rise to functional midbrain dopaminergic (mDA) neurons that are selectively degenerating during PD. A limitation of the hMO model is an under-supply of oxygen and nutrients to the densely packed core region, which leads eventually to a “dead core”. To reduce this phenomenon, we applied a millifluidic culture system that ensures media supply by continuous laminar flow. We developed a computational model of oxygen transport and consumption in order to predict oxygen levels within the hMOs. The modelling predicts higher oxygen levels in the hMO core region under millifluidic conditions. In agreement with the computational model, a significantly smaller “dead core” was observed in hMOs cultured in a bioreactor system compared to those ones kept under conventional shaking conditions. Comparing the necrotic core regions in the organoids with those obtained from the model allowed an estimation of the critical oxygen concentration necessary for ensuring cell vitality. Besides the reduced “dead core” size, the differentiation efficiency from NESCs to mDA neurons was elevated in hMOs exposed to medium flow. Increased differentiation involved a metabolic maturation process that was further developed in the millifluidic culture. Overall, bioreactor conditions that improve hMO quality are worth considering in the context of advanced PD modelling.
Introduction
The marked difference between human (patho-)physiology and that of common laboratory animals as well as the difficulty of obtaining human tissue specimens suitable for experimental purposes underline the urgent need for high-quality human in vitro models.1,2 In general, the quality of an in vitro model is evaluated by the degree of accordance with the tissue or organ of interest on the one hand and its accessibility by biochemical analysis methods on the other. Over the last few years, the field of human stem cell technology has advanced from cultures containing single cell types to approaches that generate complete organ-like tissues containing multiple cell types in defined spatial orientations. These in vitro “organoids” are derived from stem cells that self-organize to form structures resembling the complexity of a layered tissue but are still well accessible by experimental tools.3 By definition, organoids contain several cell types in a spatial organization that closely reflects the organ of interest, including physiological cell–cell interactions. Several reports have shown that they are able to recapitulate characteristic functions of the modelled organ,4 such as neuronal activity in case of the human brain.5,6 Ideally, for applications such as in vitro disease modelling, drug testing and even cell replacement strategies, an organoid model should be stable for extended cultivation and manipulation, reproducible and offer the possibility for patient-specificity.4 Organoids modelling the human midbrain are of particular interest, since this region is specifically targeted by neuro-degeneration during Parkinson's disease (PD). PD, which is the second most common neurodegenerative disease after Alzheimer's disease, is an age associated progressive neurodegenerative disorder with yet unknown aetiology.7 It is characterised by the loss of dopamine-producing neurons in the substantia nigra located in the midbrain, which leads to a lack of the neurotransmitter dopamine in the striatum. Deprivation of dopamine leads to an impairment of basal ganglia function and causes the typical motor-symptoms such as rigidity, bradykinesia, akinesia and tremor.8 So far, owing to the complexity of the midbrain, very few organoids modelling brain structures relevant for PD have been described.5,9–11 Recently, we developed a protocol for deriving midbrain specific organoids (hMOs)5 valuable for PD disease modelling. For generating hMOs, human neuroepithelial stem cells (NESCs) derived from human induced pluripotent stem cells (hiPSCs) served as a starting population. hMOs contain all neural cell types comprising neurons, astrocytes and oligodendrocytes. A high proportion of the neuronal population are midbrain dopaminergic (mDA) neurons that show electrophysiological ion-channel activity including the typical “pace-maker” activity underlining the functional proximity of the model in comparison to the human midbrain.5 Nevertheless, the hMOs model shows intrinsic limitations. Most importantly, cells in the densely packed core region are unable to survive and accumulate to form a so-called “necrotic” or “dead” core.5 This has been described in cerebral brain organoids before.6 It was also observed in cardiac12 and liver organoids13 and is considered a limiting factor, hindering translational applications. Regarding brain organoids, different approaches have been applied to ensure proper medium supply, namely orbital shaking,11 spinning flasks6 and mini-bioreactors.10 However, applying fluidic systems (micro- or millifluidic) have not been considered, so far. In case of the hMOs, we hypothesize that keeping organoids under continuous orbital shaking as per published protocols5 might not be sufficient to ensure a proper supply of nutrients and oxygen. Therefore, to improve the quality of hMOs we investigated the effects of applying a continuous medium flow during culture. In this study we used the “Quasi Vivo®” (QV, Kirkstall, UK) millifluidic system rather than a microfluidic device14 for a number of reasons. First, following the concept of allometry, the size of an organoid should range between approximately 0.5 to 2 mm in order to exhibit physiologically scaled metabolism (oxygen consumption).15,16 In addition, millifluidic systems such as the QV allow the application of relatively high flow rates ensuring proper nutrient and oxygen supply without exposing the organoid to a high shear force due to the flow itself. This is due to a well-like design in which the medium inlet and outlet are located in the chamber lid whereas the organoid is placed a variable distance from the medium inlet.17,18 Finally, given their high volume to surface ratio, millifluidic systems do not require frequent media changes, thus organoid manipulation is reduced to a minimum during culture. Besides various applications in 2D cultures, the QV millifluidic system has been successfully used to culture liver organoids13,19 and 3D cardiac constructs20 derived from human (adult) stem cells.In this study, we established a stable midbrain organoid culture under millifluidic conditions and compared it to the state-of-the-art procedure of continuous orbital shaking using both a computational fluid dynamics (CFD) and an experimental approach. The CFD analysis was performed to determine if differences in calculated oxygen profiles in the two experimental set-ups could be used to explain any of the observed differences in organoids cultured in the two conditions. This comparison revealed improvements in the culture quality comprising a reduced “dead core”, corroborated by the models, and increased dopaminergic differentiation.
Material and methods
NESC maintenance, MO generation and cultivation
Human neuroepithelial stem cell (NESC) lines K7 WT (ID number 3.0.0.10.0), T12.9 WT (3.0.0.14.0) and 34769 WT (3.0.0.24.1) from three female healthy individuals (81, 53 and 68 years of age) were derived from human iPSC lines as described before.21 NESC lines were cultured on GelTrex (Thermo Fischer Scientific) coated plates. The culture medium (N2B27 maintenance medium) consisted of DMEM-F12 (Invitrogen) and Neurobasal (Invitrogen) media (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mixture) supplemented with 1:200 N2 supplement (Invitrogen), 1:100 B27 supplement lacking vitamin A (Invitrogen), 1% L-glutamine, 1% penicillin/streptomycin (Invitrogen), 3 μM CHIR-99021 (Axon Medchem), 0.75 μM purmorphamine (Enzo Life Science) and 150 μM ascorbic acid (Sigma).21
:50 mixture) supplemented with 1:200 N2 supplement (Invitrogen), 1:100 B27 supplement lacking vitamin A (Invitrogen), 1% L-glutamine, 1% penicillin/streptomycin (Invitrogen), 3 μM CHIR-99021 (Axon Medchem), 0.75 μM purmorphamine (Enzo Life Science) and 150 μM ascorbic acid (Sigma).21
As described in our original hMO generation protocol,5 on day 0 of the hMO culture, hNESCs at passage <24 were treated with accutase (Invitrogen) for 5 min at 37 °C, followed by gentle pipetting to generate single cells. 9000 cells were seeded into each well of an ultra-low attachment 96-well round bottom plate (Corning) and cultured in N2B27 maintenance media to form 3D NESC-colonies. The medium was changed every other day. On day 8 of culture, the 3D colonies were transferred to droplets of LDEV-free and growth factor reduced GelTrex (Thermo Fisher BD Bioscience) as previously described.22 Droplets were cultured in N2B27 maintenance media in ultra-low attachment 24-well plates (Corning) with one droplet per well for long-term cultures. On day 10, differentiation was initiated with N2B27 media supplemented with 10 ng ml−1 hBDNF (Peprotech), 10 ng ml−1 hGDNF (Peprotech), 500 μM dbcAMP (Peprotech), 200 μM ascorbic acid (Sigma), 1 ng ml−1 TGF-β3 (Peprotech) and 1 μM purmorphamine (Enzo Life Science). On this day, one half of the droplets were transferred into Quasi Vivo® 900 tray (Kirkstall, UK), with one droplet per chamber. Each chamber contained a nylon-plug at the bottom to reduce the chamber volume from 4 to 2 mL and to lift the hMO closer to the medium inlet. For more details regarding the experimental setup, see Fig. 2A. The medium flow was set to 240 μL min−1 using a peristaltic flow (PF) 600 cell culture tray pump (Parker, UK), which is high enough to allow nutrient turnover without generating turbulence – as per manufacturer's instructions. Trays and pump were placed into an incubator (5% CO2, 37 °C). The rest of the droplets stayed in 24-well ultra-low attachment 24-well plates with 600 μL of medium per well. To avoid a dropout of the hMOs from the matrix embedding due to agitation, the hMO containing 24-well plates were placed on an orbital shaker (IKA), rotating at 80 rpm, in an incubator (5% CO2, 37 °C) not before day 14. On day 16, the smoothened-agonist purmorphamine was removed by media change in both systems. Shaking organoids were kept in culture with media changes every second or third day. For QV cultures, after day 16, the medium in the reservoir bottle was changed every 7th day. Organoids were fixed on day 30 of differentiation in PBS buffered 4% paraformaldehyde overnight at RT and washed 3× with PBS for 1 h.
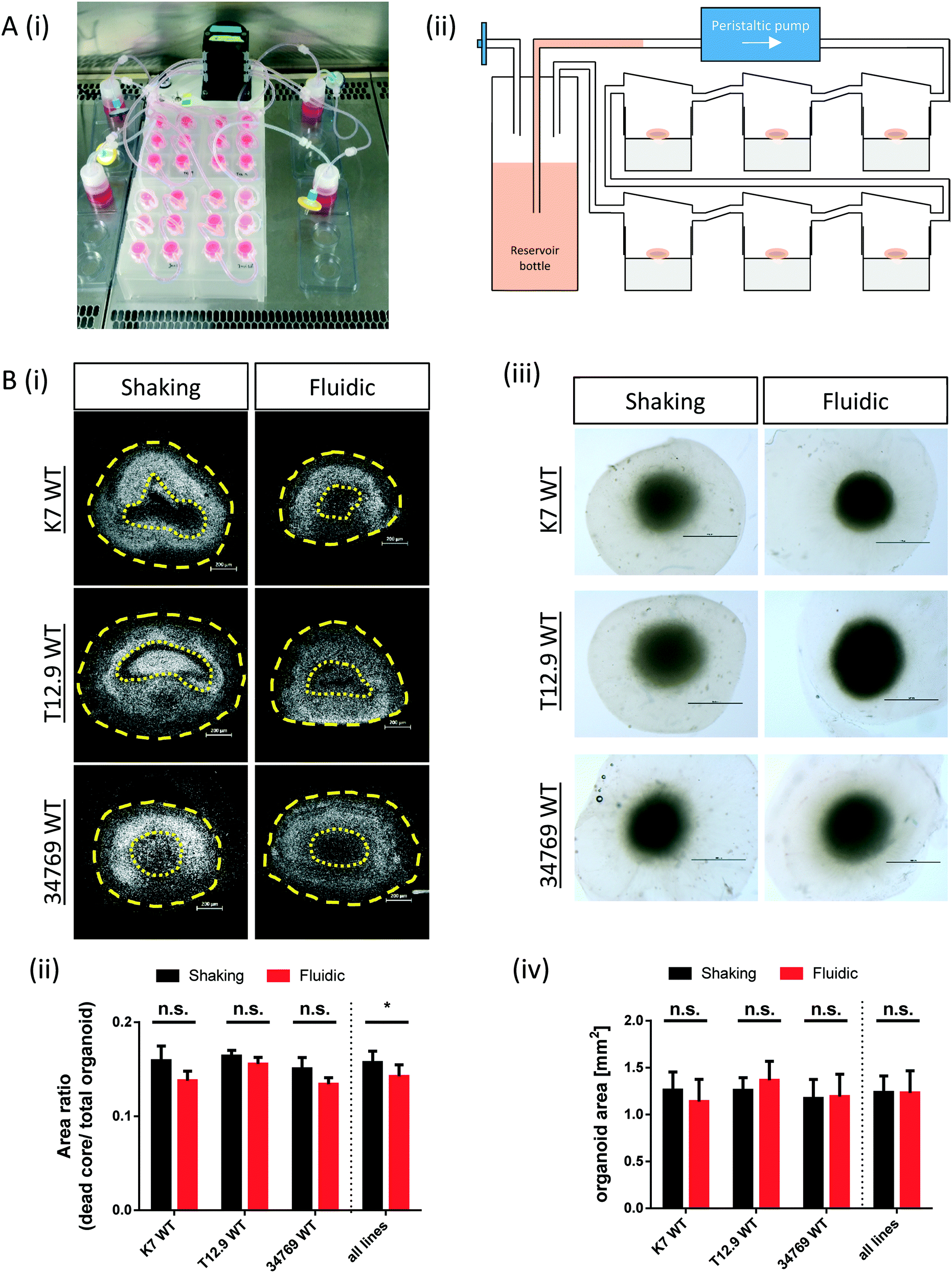 | ||
| Fig. 1 Reduced “dead core” size in hMO cultured in the millifluidic device. (Ai) Experimental setup for culturing hMO in the millifluidic device. The picture shows four QV900® circuits connected to one peristaltic pump. All chambers contain one hMO each. (Aii) Schematic drawing of one QV900® six-chamber circuit. (Bi) Hoechst staining of nuclei (white) of representative hMO sections derived from three different WT NESC lines and cultured under fluidic or shaking conditions. Yellow dotted lines indicate the area of the “dead core”, whereas the yellow dashed lines indicate the area of the entire hMOs (scale bar = 200 μm). (Bii) Quantification of area measurements as ratio of “dead core” area and total hMO area (n = 3 per line and culture condition, *p < 0.05). (Biii) Brightfield images of fixed (unsectioned) hMOs with surrounding halos of matrix (scale bar = 1 mm). (Biv) quantification of hMO area measurements (n = 6 per line and culture condition). | ||
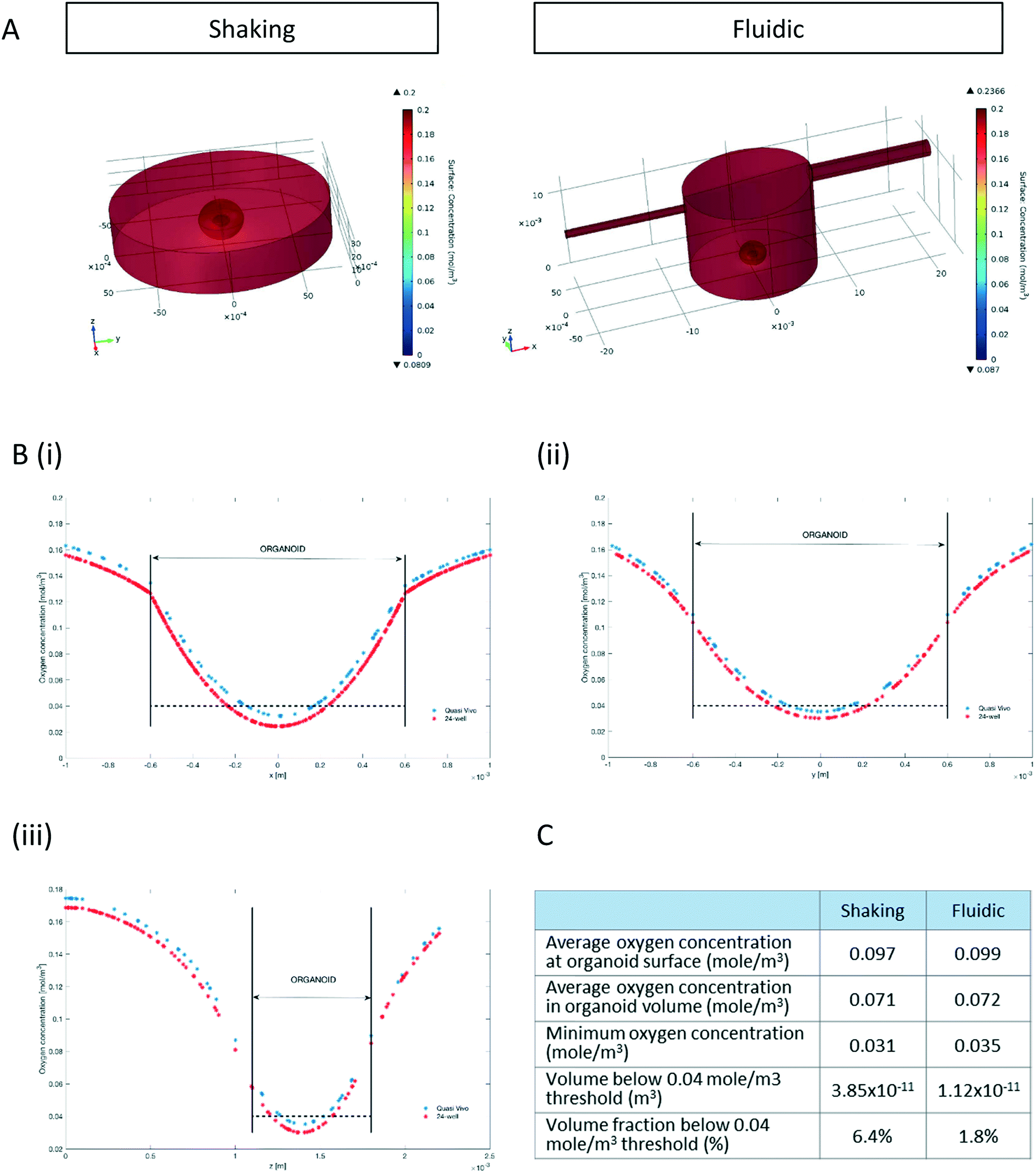 | ||
| Fig. 2 Increased predicted oxygenation of hMO under millifluidic culture conditions. (A) Surface plots of oxygen concentration resulting from the computational model of a hMO in a 24-well (left) or a millifluidic chamber (right). Oxygen concentration gradients are indicated by colours ranging from 0.2 mol m−3 (dark red) to 0 mol m−3 (dark blue). (Bi–iii) The computed oxygen concentration along the x- (i), y- (ii) and z- (iii) axis passing through the centre of a hMO cultured either in the millifluidic system (red line) or in the 24-well (blue line). Straight lines indicate the borders between the organoid and the surrounding medium. Dashed lines indicate the critical oxygen threshold of 0.04 mol m−3. (C) Tabular comparison of the modelling-based oxygenation status of hMOs cultured under fluidic or shaking conditions. | ||
All experiments with human stem cell lines were done only with lines were prior patient consent have been obtained. All these experiments were done in agreement with the relevant laws and followed the institutional guidelines. Approval for work with human stem cells has been obtained by the Ethics Review Panel (ERP) of the University of Luxembourg and by the Luxembourgish Comité National d'Ethique de Recherche (CNER).
Organoid sectioning, immunofluorescence staining and imaging
To measure the size of hMOS after fixation, brightfield images of six organoids per cell line and culture condition were taken with a stereomicroscope SMZ25 (Nikon) and the hMO area was measured using the NIS imaging software (Nikon).For immunofluorescence staining of organoid sections, fixed hMOs were embedded in 3% low-melting point agarose (Biozym) in PBS. hMOs were incubated in agarose for 15 min at 37 °C, followed by 30 min incubation at RT. Sections (50 μm thickness) was cut using a vibratome (Leica VT1000s) and centre-sections (middle position between first and last section) were used for assessing dead core size and TH/FOXA2/TUJ1 expression. For immune-stainings, sections were permeabilized with 0.5% Triton X-100 in PBS. Permeabilization times varied between 30 min and 2 h depending on the antibody used. Unspecific antigen blocking for 2 h in 2.5% normal donkey serum and 2.5% BSA, 0.1% Triton X-100 and 0.1% sodium azide was followed by primary antibody incubation for 48 h at 4 °C on a shaker. Antibodies were diluted in blocking buffer as follows: rabbit anti-TH (1:1000, Abcam), mouse anti-FOXA2 (1:250, Santa Cruz Biotechnology), chicken anti-TUJ1 (1:600 Millipore), goat anti-SOX2 (1:200, R&D Systems) and rabbit anti-CC3 (1:200, Cell Signalling). After incubation with the primary antibodies, sections were washed three times in PBS containing 0.01% Triton X-100 and blocked for 30 min at RT on a shaker, followed by incubation with the secondary antibodies diluted in PBS containing 0.01% Triton X-100 and Hoechst-33 342 nuclear dye (1:1000; Sigma-Aldrich). All secondary antibodies (Invitrogen) were conjugated to Alexa Fluor fluorochromes. Sections were mounted in Fluoromount-G mounting medium (Southern Biotech) and analysed with a confocal laser scanning microscope (Zeiss LSM 710).
Image quantification
Based on the Hoechst staining of nuclei in hMO centre sections, the area of the dead core and the area of the entire section was measured using the ZEN Software (Zeiss). Borders of the dead core area were assessed my means of zoomed-in views of the corresponding sections (see Fig. S1†).The immunofluorescence images of hMOs were analysed in Matlab (Version 2017a, Mathworks). The in-house developed image analysis algorithms automated the segmentation of nuclei (Hoechst), neurons (TUJ1), dopaminergic neurons (FOXA2 and TH) and proliferating cells (SOX2) with sub sequential feature extraction. The nuclear segmentation was computed by convolving a foreground image of the raw Hoechst channel with a Gaussian filter of size 21 and standard deviation 1. For the background image, a Gaussian filter of size 21 and standard deviation of size 3 was used. The difference was computed by subtracting the background from the foreground. For the segmentation of neurons, a median filter was applied to the raw TUJ1 and TH channels and the resulting images were convolved with a Gaussian filter of size 10 and 11 respectively, and standard deviation 1. TUJ1 and TH masks were defined by those pixels with values larger than 45 and 20 respectively. For the FOXA2 mask, a median filter was applied and all connected components smaller than 50 were removed. For the SOX2 mask, a median filter was applied and all connected components smaller than 40 were removed. The SOX2 and FOXA2 masks were identified within the nuclear mask of each image. For the dopaminergic neurons, a perinuclear region in FOXA2 positive neurons was identified, and the presence of TH staining was evaluated. A mask corresponding to the TH+ perinuclear area of FOXA2+ neuron was normalized to the perinuclear perimeter (TH/FOXA2 by nuclear perimeter). The SOX2 mask was normalized to the nuclear mask (SOX2 by nuclear mask).
Flow cytometry
For flow cytometry analysis, six hMOs of each cell line were dissociated to single cells by incubation in DMEM-F12 (Invitrogen) containing 0.18% Papain (Sigma), 0.04% EDTA (Sigma) and 0.04% L-cystein (Sigma) at 37 °C on an orbital shaker until any surrounding GelTrex disappeared (2–3 h). hMOs were then treated with accutase (Invitrogen) for further 2–3 h at 37 °C under continuous shaking. During accutase treatment, hMOs were firstly triturated with a 1000 μL pipette and eventually with a 200 μL pipette in order to support the dissociation into single cells. For intracellular staining, the transcription factor buffer set (BD Bioscience) was used according to the manufacturer's instructions. After fixation, cells were filtered through a 5 mL polystyrene round-bottom tube with cell-strainer cap (Corning). 500000 cells per condition were used and split into two samples. One was incubated with primary antibodies at the following dilutions: chicken anti-TH (1:50, Abcam) mouse anti-FOXA2 (1:300, Santa Cruz Biotechnology) and rabbit anti-LMX1A (1:100, Abcam). To set thresholds, the second sample was incubated with the following isotype control antibodies: normal chicken IgY Control (R&D Systems), normal rabbit IgG control (Santa Cruz Biotechnology), and purified Mouse IgG2a, κ isotype control (BioLegend). Isotype control antibodies were used at the same concentration as the detection antibody. All secondary antibodies (Invitrogen) were conjugated to Alexa Fluor fluorochromes. Flow cytometry was performed by using BD LSRFortessa cell analyser and data were analysed and represented by FlowJo software.
Modelling hMO oxygen transport and consumption
3D computational models for oxygen transport were developed using the COMSOL Multiphysics 4.3 software (COMSOL AB, Stockholm, Sweden). Firstly, we developed a reaction and diffusion multi-physics model of an organoid cultured in a 24 well-plate. In a second model, oxygen mass transport and consumption were coupled to fluid dynamics based on the geometry of the QV system. Given that oxygen consumption in mammalian cells is governed by Michaelis–Menten kinetics,15 we considered a mean literature value of Km = 3 μM (ref. 17 and 23) and an iPSC oxygen consumption rate (OCR) of 7 × 10−18 mol per cell per s), as estimated by.24Both models were based on the methods described in ref. 19. In particular, each configuration was represented using three sub-domains: i) a fluid domain, in which no oxygen consumption occurs and where both fluid-dynamics and the oxygen diffusion (D = 3 × 10−9 m2 s−1) and convective transport are solved, ii) the midbrain organoid, modelled as a solid ellipsoid (1200 × 1200 × 800 μm3), where only oxygen diffusion and consumption are solved (D = 1 × 10−9 m2 s−1), and iii) an ellipsoidal shell of GelTrex with a thickness of 1 mm and oxygen diffusion, D = 1 × 10−9 m2 s−1, surrounding the ellipsoid.25
Oxygen transport and consumption in both the 24-well- and the fluidic system were assumed to be governed by the generic advection and diffusion equation in its formulation for uncompressible fluid.30
The organoids cultured in wells are placed on an orbital shaker with radius of gyration 1 cm. At the gentle rotation rate used (80 rpm), the media was not observed to intersect the base of the well. In these conditions, the free surface level (h) in a cylindrical well of radius R undergoing rigid rotation at Ω radians per s as a function of the generic radius r can be described by:26

At the centre of the well, where the organoid is placed, since r = 0 the height h of fluid simplifies to:
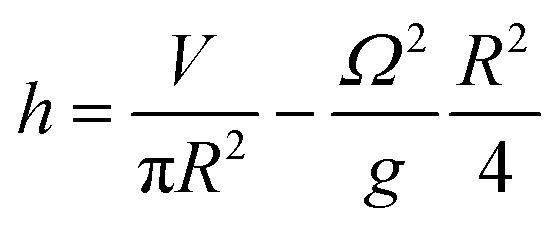
The height at the centre varies between 3.2 and 3.0 mm over each cycle. Given the small difference in height during a rotation, we assumed the fluid to be in quasi-static conditions with a mean height of 3.1 mm.
The velocity field resulting from fluid flow convection in the fluidic system was solved using the Navier–Stokes equation for Newtonian fluid, setting the inlet velocity to 5 × 10−4 m s−1 (corresponding to 240 μL min−1 inflow). In addition, since the QV fluidic system is made of PDMS, which is gas permeable,27 the inward oxygen flux through the bioreactor walls was modelled according to the expression:19

| Parameters | Values |
|---|---|
| Organoid width | 1200 μm |
| Organoid height | 800 μm |
| Mid-brain organoid volume (V) | 6.03 × 10−10 m3 |
| Number of cells within the organoid (#cells) | 1.8 × 105 cells |
| Cell density (ρ) |
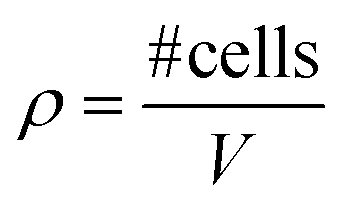
|
| Thickness of GelTrex coating around organoid | 1 mm |
| Initial and boundary oxygen concentration (CO2) | 0.2 mol m−3 or mM |
| Single cell maximum oxygen consumption rate (OCR) | 5.5 × 10−18 mol per cell s |
| Henry constant for oxygen (KH,O2) | 1.32 × 10−3 mol m−3 mmHg−1 |
| PDMS thickness (hPDMS) | 6 × 10−3 m |
| Ambient oxygen partial pressure (pO2) | 159 mmHg |
| Oxygen diffusion in water at 37 °C | 3 × 10−9 m2 s−1 |
| Oxygen diffusion in GelTrex and organoid at 37 °C | 10−9 m2 s−1 |
| Michaelis Menten constant (Km) | 5 × 10−3 mol m−3 |
Since the experimental set-up consists of six bioreactors in series, we also calculated the oxygen concentration at the outlet of the bioreactor so as to use the value as the input concentration for the second chamber, and so on. However, the outlet concentration was not significantly different from the inlet. All the boundary conditions are summarized in Table 2.
| Model | Surface | Boundary condition |
|---|---|---|
| Oxygen convection and diffusion | Quasi Vivo side walls | Inward oxygen flux through PDMS |
| Interface between the MO and the fluid sub-domain | Continuity | |
| Fluid domain inlet | Constant oxygen concentration | |
| Fluid domain outlet | Convective flux | |
| Navier–stokes | Solid–liquid interfaces | No slip |
| Fluid domain inlet | Normal inflow velocity | |
| Fluid domain outlet | Pressure, no viscous stress |
Estimation of metabolite consumption- and production rates within the midbrain organoid
For metabolite measurements, medium samples were taken from the QV reservoir bottles supplying either K7 WT, T12.9 WT or 34769 WT hMOs at day 23 and 30 of differentiation. For the shaking approach, medium samples were taken from individual wells and pooled for each hMO line at day 23 and 30 of differentiation (equals n = 3 medium samples per time point and culture condition). Extracellular glucose, glutamine, glutamate and lactate levels in cell culture media were measured using a YSI 2950D chemical analyser (Yellow Springs Instruments). Quantification was based on authentic standards. Each sample was analysed in technical triplicates. Quality controls were analysed after every sixth sample ensuring measurement quality. Sample size and reaction time was 40 μL and 30 seconds for glucose and lactate, 25 μL and 40 seconds for glutamine and glutamate, respectively.Based on the metabolite concentrations in the culture medium (Ct1), we calculated the metabolite consumption- and production rates for hMOs under flow or shaking conditions. According to the supplier's information, fresh organoid medium contains Ct0 = 20.4 mM glucose, 0 mM lactate, 2.0 mM glutamine and 0.024 mM glutamate. t1 equals the timespan between two medium refreshments (i.e. 2 days for the multi-well system and 7 days for QV). The metabolite consumption/production rates vmet were estimated as:
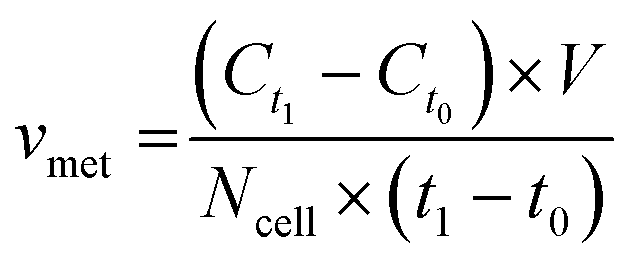
Statistics
The number of replicates “n” refers to the number of individual hMO per NESC line and culture condition. Statistical analysis was performed using the Prism 6 software (GraphPad). For comparison of shaking and millifluidic culture conditions in three different WT line-derived hMOs, t-tests (parametric data) or Mann–Whitney rank rum tests (non-parametric data) were performed. Regarding immunofluorescence image analysis, pixel ratio values for shaking conditions were set to 100% and the values for millifluidics were calculated correspondingly. In this case, statistics were performed applying a one-sample t-test.Results
Millifluidics reduce the size of the hMO dead core area
The commercially available millifluidic system QuasiVivo900® (Kirkstall, UK) was set up as follows: the chambers of a six-chamber tray were filled with one hMO each. The six lids of the chamber were connected in a row by tubing and each six-chamber-tray was connected to one reservoir bottle filled with 22 mL of differentiation medium (Fig. 1Ai+ii). This circuit was connected to a peristaltic pump to expose each organoid to a flow rate of 240 μm min−1. This value was chosen after various preliminary tests (data not shown) and in accordance with literature values using the QV system for liver organoid cultures.19 Up to four tray-units were connected to one poly-head peristaltic pump (Fig. 1A). Thanks to the flow and shared media, the number of media changes could be reduced from once every 2–3 days (shaking conditions) to once every 7 days (fluidic).After 30 days of differentiation, hMOs derived from three different WT NESC lines (K7 WT, T12.9 WT and 34769 WT) were fixed and sectioned. Measurements of the dead core area size in comparison to the overall size of the hMOs revealed a trend towards a reduced dead core size under millifluidic conditions in all three WT lines. This decrease in the fraction of the necrotic area under fluidic conditions reaches statistical significance upon averaging of the data for all three organoids (0.16 ± 0.012 vs. 0.14 ± 0.012, p = 0.017; Fig. 1Bi+ii; Fig. S1†). Of note, the overall size of the hMOs was not significantly different after millifluidic cultivation in comparison to shaking conditions in neither of the WT lines (Fig. 1Biii+iv). These data indicate that hMO vitality is superior in millifluidic culture conditions.
Millifluidics show increased oxygenation in a computational oxygen transport and consumption model
Since oxygen is considered as a limiting nutrient for 3D cell cultures and under-oxygenation of the organoid contributes to the appearance of the “dead core”, we assessed the O2 concentration profile in the culture medium and inside the organoids.Multi-physics 3D models that couple oxygen mass transport and consumption to fluid dynamics were used to predict oxygen concentration profiles during the 3D culture in both the 24-well and the QV chamber. Surface plots of steady state oxygen concentrations of both the 24-well and in the millifluidic chamber are shown in Fig. 2A. The streamline plot of the computed fluid velocity field shows laminar medium flow in the chamber (Fig. S2†). Having a closer look at the oxygen profiles around and inside the hMO along the x-, y- and z- axis (with origin fixed at the organoid centroid, Fig. 2B) reveals that the oxygen concentration is higher in the fluidic system than in the shaking 24-well plate. As shown in Fig. 2Biii, the oxygen profile along the vertical (z) axis is not symmetric with respect to the centroid because the ellipsoidal shape of organoids allows medium to flow and oxygen to diffuse underneath it. Overall, according to the computational model, the average oxygen concentrations in the hMO are similar in the two conditions (Fig. 2C). However, the minimal oxygen concentration inside the hMO is considerably higher under fluidic conditions. To determine the critical viable oxygen concentration, we compared the computed cross-sectional areas at the centre of the organoids with those obtained from image processing of Hoechst-stained slices. For both fluidic and shaking conditions, the results indicate that cells below 0.040 ± 0.011 mol m−3 are unlikely to survive in the core. The computed volume proportion of the hMO lying under this critical oxygen threshold of 0.04 mol m−3 is smaller in the Quasi-Vivo (1.8%) compared to shaking conditions (6.4%) indicating that the millifluidic system is more efficient for oxygen transport compared to the shaking in 24-well plates (Fig. 2C).
Millifluidics increase the proportion of dopaminergic neurons in a cell line specific manner
Convective transport not only increases oxygen supply but also exposes the organoids to higher amounts of growth- and differentiation factors. Since the differentiation process is directed towards mDA neurons, we checked for typical marker expression of this cell type by immunostaining of fixed hMO sections after 30 days of differentiation. The combined expression of tyrosine hydroxylase (TH) and neuron-specific class III tubulin (TUJ1) identifies the dopaminergic neuronal lineage and the presence of F-box protein A2 (FOXA2) indicates the midbrain fate (Fig. 3Ai). Computational image analysis revealed that hMOs kept under fluidic conditions reach higher proportions of TH+/FOXA2+/TUJ1+ neurons in comparison to shaking conditions. Interestingly, this effect was line dependent. The effect was significant for the 34769 WT line, whereas there was just a slight increase in mDA differentiation in the K7 WT line. For the T12.9 WT line, no difference could be detected (Fig. 3Ai+ii). Setting the staining signal of TH+/FOXA2+/TUJ1+ neurons under shaking conditions to 100% for each line makes the line specific increase in mDA differentiation under millifluidic conditions even more apparent (Fig. 3Aiii).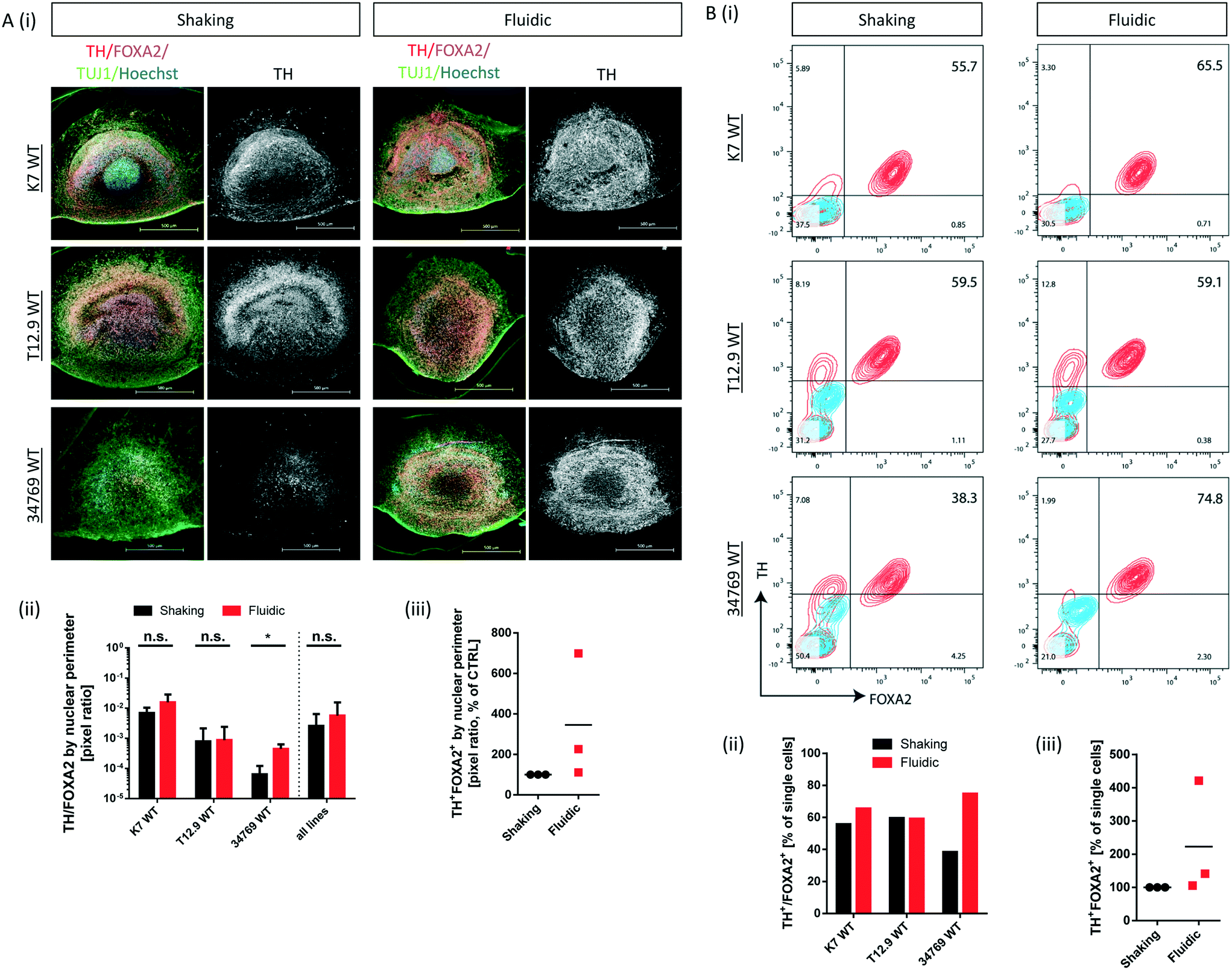 | ||
| Fig. 3 Line specific increase in mDA differentiation under millifluidic culture conditions. (Ai) Immunofluorescence stainings of representative hMO sections derived from three different WT NESC lines and cultured under shaking or fluidic conditions (merge of TH, FOXA2, TUJ1 and Hoechst staining; scale bar = 500 μm). For better comparability of TH quantities, the corresponding TH staining (white) is depicted next to the merge. (Aii) Mask-based image quantification (pixel ratios) of TH+/FOXA2+ cells in the corresponding immunofluorescence stainings (n = 3 per cell line and culture condition; *p < 0.05). (Aiii) mean pixel ratio values for TH+/FOXA2+ cells in hMO of three different WT lines under fluidic conditions relative to those under shaking conditions (set to 100%). (Bi) Flow cytometry analysis (contour plots) of single cells obtained from dissociated hMOs from three different WT NESC lines and cultured under shaking or fluidic conditions (blue lines = isotype control, red lines = target staining). Percentages of FOXA2+/TH+ cells are indicated in the upper right corner of the plots. (Bii) The bar chart summarizes the percentages of FOXA2+/TH+ cells presented above. (Biii) Percentage of FOXA2+/TH+ cells in MO under fluidic conditions relative to those ones under shaking conditions (set to 100%). | ||
Dopaminergic differentiation is fuelled by neuroepithelial stem cells that express the sex determining region Y-box 2 (SOX2) among other markers. Immunostaining for SOX2 revealed that a higher degree of mDA differentiation goes along with a reduced amount of SOX2+ NESCs and vice versa (Fig. S3i–iii†). This significant effect was still largely cell line dependent and was most apparent in the 34769 line-derived organoids. Here, hMOs cultured under shaking conditions maintained a bigger SOX2+ stem cell reservoir compared to the corresponding hMOs under millifluidic conditions.
To back up our observations based on imaging of immunostainings, we included a further level of evaluation. After 30 days of differentiation, hMOs were dissociated to singe cells, immunostained for TH and FOXA2 and subsequently analysed by flow cytometry. This analysis confirmed the significant increase in mDA neurons for the 34769 WT line, the trend towards higher proportions in the K7 WT line as well as the equal levels in the T12.9 WT line under millifluidic conditions (Fig. 3Bi–iii). Flow cytometry analysis of cells expressing TH and LIM homeobox transcription factor 1A (LMX1A), an additional marker of ventral midbrain specification, confirmed the quantitative differences between fluidic and shaking conditions (Fig. S4i–iii).
During differentiation towards mDA neurons, cells change their metabolic profile. While stem cells mainly convert glucose into lactate via anaerobic glycolysis to cover their energy demand, postmitotic neurons undergo a metabolic switch towards mitochondrial respiration.32 Therefore, we measured the concentration of specific metabolites in the culture medium of organoids after fluidic or shaking culture (Fig. 4A) at two different time points and calculated the consumption or production rate per cell. At both time points, hMOs cultured in millifluidic chambers consume less glucose und secrete less lactate into the culture medium compared to those ones under shaking conditions (Fig. 4i+ii). Furthermore, hMOs in the fluidic device take up significantly more glutamine and excrete significantly more glutamate (Fig. 4iii+iv). Secreted glutamate points to the presence of glutamatergic neurons, which can be considered as a by-product of mDA differentiation. Therefore, the metabolic profiles observed under fluidic culture conditions support the data obtained by marker expression indicating an increased differentiation efficiency under these conditions.
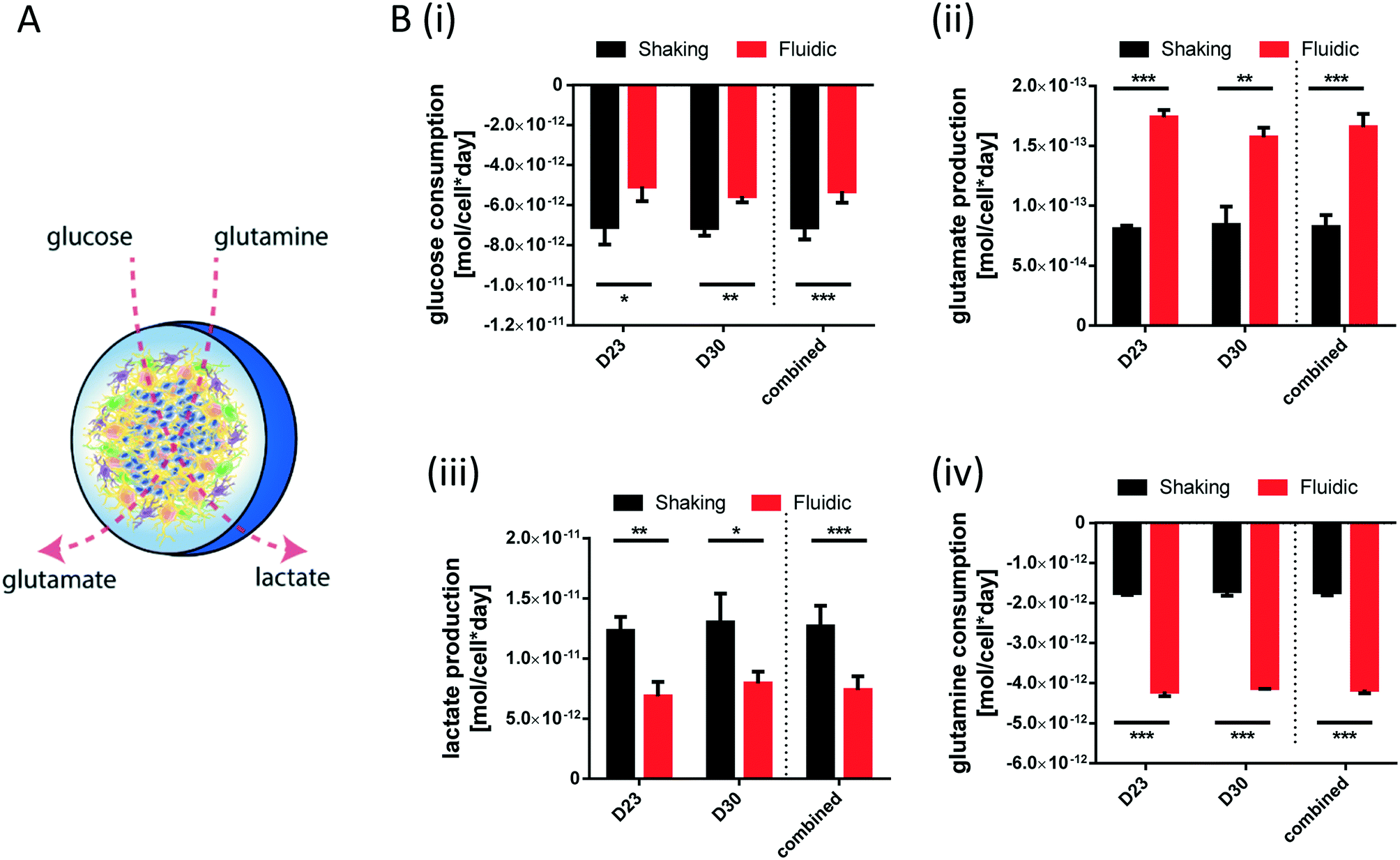 | ||
| Fig. 4 Shifted metabolite consumption and production in hMOs under fluidic conditions. (A) Schematic drawing of the experimental approach: measurement of glucose and glutamine uptake from the culture medium as well as lactate and glutamate excretion into the medium. Glucose (Bi) and glutamine (Bii) consumption as well as lactate (Biii) and glutamate (Biv) secretion rate in mole per cell per day by hMOs cultured in 24-wells or in QV chambers (n = 3 medium samples per line and culture condition; ***p < 0.001, **p < 0.01, *p < 0.05). | ||
Discussion
Over the past years, organoids have been derived from various stem cell sources to generate models of various brain regions. The properties of these brain organoid models have been reviewed recently.33 Apparently, orbital shaking or spinning bioreactors are the predominant culture systems for brain organoids, with little difference being reported between the two.34 Despite the fact that describing fluid dynamics accurately in shakers and spinners is extremely difficult due to the essentially turbulent nature of the flow, systems with a continuous medium flow have not been considered, so far. Our data show that human midbrain organoids can be cultured under continuous laminar medium flow and that these conditions improve the properties of hMO in comparison to orbital shaking.5 This improvement comprises a reduced size of the “dead core” area and an increased differentiation towards midbrain dopaminergic neurons.Whether the “dead core” is of necrotic or apoptotic nature is difficult to distinguish, since the core region usually disintegrated during sectioning. However, it should be noted that under ischemic conditions in the brain e.g. after stroke, necrotic and apoptotic mechanisms cannot be clearly separated.35 Experimental evidence indicates a time dependent convergence of both mechanisms.36,37 One possible explanation for the appearance of the “dead core” is persistent hypoxia in the core region due to a lack of vasculature. However, which oxygen concentration can be considered as hypoxic is a matter of debate. In the human brain, an average oxygen tension (partial pressure pO2) of above 35 mmHg (equals 0.05 mol m−3 at 37 °C according to Henry's law17) ensures normal oxygenation of the brain tissue,31 which is considerably less than the inhaled ambient oxygen tensions of 159 mmHg (21% of air). A value of 35 mmHg is considered as a “physiological hypoxia” or “in situ normoxia”.38 Our modelling approach reveals that oxygen concentrations in the centre of the organoid reach closely or even drop under 0.05 mol m−3. By overlapping the computed oxygen profiles across the centre of the organoids with images of necrotic nuclei, we identified a critical concentration threshold of 0.04 ± 0.011 mol m−3. In both the images and the models the zone lying under the threshold is smaller in hMOs cultured under millifluidic conditions. Besides oxygenation, other limiting factors could also contribute to the formation of a “dead core”. The diffusion of glucose and other nutrients to the core or the local accumulation of toxic compounds like acids (lactic acid, carbonic acid) and reactive oxygen species that cannot be removed properly due to the lack of vasculature have to be considered. Therefore, attempts towards a vascularization of the hMOs appear crucial for tackling the supply issues in this model.
Differences in oxygenation might also contribute to the increase in dopaminergic differentiation under millifluidic conditions. In the brain, neural stem cells (NSCs) and especially the embryonic neural tube are located in an environment that faces very low oxygen levels. For instance, in the dentate gyrus of the hippocampus, an area of adult neurogenesis, pO2 levels reach down to 6–8 mmHg.39 During embryonic development, the neural tube in both the hindbrain and midbrain regions is exposed to oxygen levels substantially below 7.6 mmHg.40 Obviously, NSCs seem to be very well adapted to this environment. In vitro studies have shown that hypoxia is able to promote an undifferentiated state in NSCs and neural crest stem cells.41 In case of our hMO model, the slightly reduced oxygenation under shaking conditions might maintain the stem cells longer in an undifferentiated state and diminish the differentiation to mDA neurons. Moreover, it is likely that the exposure of hMOs to a higher volume of medium and thereby a higher amount of neurotrophic factors in the millifluidic system contributes to the differentiation process. However, the effect of elevated dopaminergic differentiation is dependent on the individual NESC line used for hMO generation. Obviously, dopaminergic differentiation is following a saturation curve with a maximum at about 60–75% (at 30 days of differentiation), but the kinetics of this process are different between the respective NESC lines. This might be caused by the individual genetic background or differences in the culture history that the lines were facing. We have strong evidence, that the genetic background plays an important role for the dopaminergic differentiation efficiency in 2D cultures of NESC derived neurons (unpublished data).
Stem cells are adapted to low oxygen concentrations by conducting mainly glycolysis to cover their energy demand producing lactic acid to recycle their reduction equivalents. Postmitotic neurons, in contrast, undergo a metabolic switch towards mitochondrial respiration.32 In this context, the different metabolite profiles measured in the culture medium of shaking and fluidic hMO, adds further evidence for the increased differentiation efficiency that we observed under flow conditions. Since proliferation is an energy-demanding process and glycolysis is less energy efficient than oxidative phosphorylation, a higher proportion of NESC leads to higher glucose consumption and higher lactic acid levels in the medium as we see under shaking conditions. The switch from anaerobic glycolysis to oxidative phosphorylation also enables the conversion of glutamine into glutamate using the tricarboxylic acid cycle. Therefore, lower glutamine uptake and higher glutamate release can be a sign of increased differentiation as we see under millifluidic conditions. Recent data from our group show that besides the majority of mDA neurons also glutamatergic and GABAergic neurons arise from NESC in hMOs (unpublished data). Interestingly, the differences on metabolite levels are less cell-line dependent than the marker expression analysis mentioned before. This might be explained by the fact that the cell culture medium represents the average effect over a certain time period, whereas the marker analysis is a snapshot of the current cellular status within the hMO. Currently, we are working on a method to assess the release of the neurotransmitter dopamine by matrix-embedded hMOs as a direct measure of the differentiation efficiency.
Overall, our millifluidic approach delivered promising results regarding the quality of the organoid culture. Nevertheless, the throughput of organoids is rather low in the present system. The next step could be a size adaptation of the organoids, within the limitations of allometric scaling, to advance the system towards a midbrain-on-a-chip approach suitable for high-throughput and high-content assays using integrated sensor techniques. This would facilitate the use of organoids as a tool for Parkinson's disease modelling and personalized therapeutic screening.
Author contributions
EB and ASM performed the organoid experiments, CM and AA performed the computational modelling. NP and CL carried out the metabolic analyses. The automated image analysis was done by SB and PA. EB, CM, ASM, AA and JCS designed the project and wrote and revised the manuscript.Conflicts of interest
SB and JCS are co-founders of the biotech company Braingineering Technologies SARL. Currently, SB is an employee of Brainginerring Technologies SARL.Acknowledgements
The authors would like to thank Thea van Wuellen for excellent technical assistance. We thank Prof. Dr. Hans R. Schöler from the Max-Planck-Gesellschaft, Dr. Jared Sterneckert from the CRTD, Prof. Dr. Thomas Gasser from the Hertie Institute in Tuebingen and the Coriell Institute for providing us with cell lines. This project was supported by the LCSB pluripotent stem cell core facility. The JCS lab is supported by the Fonds National de la Recherché (FNR) (CORE, C13/BM/5791363 and Proof-of-Concept program PoC15/11180855 & PoC16/11559169). This is an EU Joint Programme – Neurodegenerative Disease Research (JPND) project (INTER/JPND/14/02; INTER/JPND/15/11092422). ASM is supported by a fellowship from the FNR (AFR, Aides à la Formation-Recherché). CM is supported by a fellowship from the Fondazione Umberto Veronesi. Finally, we also thank the private donors who support our work at the Luxembourg Centre for Systems Biomedicine.References
- W. E. Grizzle, W. C. Bell and K. C. Sexton, Issues in collecting, processing and storing human tissues and associated information to support biomedical research, Cancer Biomarkers, 2010, 9(1–6), 531–549 Search PubMed.
- A. Knight, Systematic reviews of animal experiments demonstrate poor human clinical and toxicological utility, ATLA, Altern. Lab. Anim., 2007, 35(6), 641–659 Search PubMed.
- X. Yin, et al., Engineering Stem Cell Organoids, Cell Stem Cell, 2016, 18(1), 25–38 CrossRef PubMed.
- M. A. Lancaster and J. A. Knoblich, Organogenesis in a dish: modeling development and disease using organoid technologies, Science, 2014, 345(6194), 1247125 CrossRef PubMed.
- A. S. Monzel, et al., Derivation of Human Midbrain-Specific Organoids from Neuroepithelial Stem Cells, Stem Cell Rep., 2017, 8(5), 1144–1154 CrossRef PubMed.
- M. A. Lancaster, et al., Cerebral organoids model human brain development and microcephaly, Nature, 2013, 501(7467), 373–379 CrossRef PubMed.
- J. M. Beitz, Parkinson's disease: a review, Front. Biosci., Scholar Ed., 2014, 6, 65–74 CrossRef.
- J. Jankovic, Parkinson's disease: clinical features and diagnosis, J. Neurol., Neurosurg. Psychiatry, 2008, 79(4), 368–376 CrossRef PubMed.
- V. Tieng, et al., Engineering of midbrain organoids containing long-lived dopaminergic neurons, Stem Cells Dev., 2014, 23(13), 1535–1547 CrossRef PubMed.
- X. Qian, et al., Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure, Cell, 2016, 165(5), 1238–1254 CrossRef PubMed.
- J. Jo, et al., Midbrain-like Organoids from Human Pluripotent Stem Cells Contain Functional Dopaminergic and Neuromelanin-Producing Neurons, Cell Stem Cell, 2016, 19(2), 248–257 CrossRef PubMed.
- P. Beauchamp, et al., Development and Characterization of a Scaffold-Free 3D Spheroid Model of Induced Pluripotent Stem Cell-Derived Human Cardiomyocytes, Tissue Eng., Part C, 2015, 21(8), 852–861 CrossRef PubMed.
- S. D. Ramachandran, et al., In Vitro Generation of Functional Liver Organoid-Like Structures Using Adult Human Cells, PLoS One, 2015, 10(10), e0139345 CrossRef PubMed.
- E. L. Moreno, et al., Differentiation of neuroepithelial stem cells into functional dopaminergic neurons in 3D microfluidic cell culture, Lab Chip, 2015, 15(11), 2419–2428 RSC.
- A. Ahluwalia, Allometric scaling in-vitro, Sci. Rep., 2017, 7, 42113 CrossRef PubMed.
- N. Ucciferri, T. Sbrana and A. Ahluwalia, Allometric Scaling and Cell Ratios in Multi-Organ in vitro Models of Human Metabolism, Front. Bioeng. Biotechnol., 2014, 2, 74 Search PubMed.
- G. Mattei, S. Giusti and A. Ahluwalia, Design Criteria for Generating Physiologically Relevant In Vitro Models in Bioreactors, Processes, 2014, 2(3), 548 CrossRef.
- D. Mazzei, et al., A low shear stress modular bioreactor for connected cell culture under high flow rates, Biotechnol. Bioeng., 2010, 106(1), 127–137 Search PubMed.
- G. Mattei, et al., On the adhesion-cohesion balance and oxygen consumption characteristics of liver organoids, PLoS One, 2017, 12(3), e0173206 CrossRef PubMed.
- S. Pagliari, et al., A multistep procedure to prepare pre-vascularized cardiac tissue constructs using adult stem sells, dynamic cell cultures, and porous scaffolds, Front. Physiol., 2014, 5, 210 Search PubMed.
- P. Reinhardt, et al., Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling, PLoS One, 2013, 8(3), e59252 CrossRef PubMed.
- M. A. Lancaster and J. A. Knoblich, Generation of cerebral organoids from human pluripotent stem cells, Nat. Protoc., 2014, 9(10), 2329–2340 CrossRef PubMed.
- P. Buchwald, FEM-based oxygen consumption and cell viability models for avascular pancreatic islets, Theor. Biol. Med. Modell., 2009, 6, 5 CrossRef PubMed.
- S. Varum, et al., Energy metabolism in human pluripotent stem cells and their differentiated counterparts, PLoS One, 2011, 6(6), e20914 CrossRef PubMed.
- R. J. McMurtrey, Analytic Models of Oxygen and Nutrient Diffusion, Metabolism Dynamics, and Architecture Optimization in Three-Dimensional Tissue Constructs with Applications and Insights in Cerebral Organoids, Tissue Eng., Part C, 2016, 22(3), 221–249 CrossRef PubMed.
- M. M. Salek, P. Sattari and R. J. Martinuzzi, Analysis of fluid flow and wall shear stress patterns inside partially filled agitated culture well plates, Ann. Biomed. Eng., 2012, 40(3), 707–728 CrossRef PubMed.
- K. J. Regehr, et al., Biological implications of polydimethylsiloxane-based microfluidic cell culture, Lab Chip, 2009, 9(15), 2132–2139 RSC.
- A. R. Frisancho, Functional adaptation to high altitude hypoxia, Science, 1975, 187(4174), 313–319 CrossRef PubMed.
- R. K. Kutty and M. D. Maines, Characterization of an NADH-dependent haem-degrading system in ox heart mitochondria, Biochem. J., 1987, 246(2), 467–474 CrossRef PubMed.
- G. Mattei and A. Ahluwalia, Sample, testing and analysis variables affecting liver mechanical properties: A review, Acta Biomater., 2016, 45, 60–71 CrossRef PubMed.
- A. Carreau, et al., Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia, J. Cell. Mol. Med., 2011, 15(6), 1239–1253 CrossRef PubMed.
- A. G. Jady, et al., Differentiation-Dependent Energy Production and Metabolite Utilization: A Comparative Study on Neural Stem Cells, Neurons, and Astrocytes, Stem Cells Dev., 2016, 25(13), 995–1005 CrossRef PubMed.
- E. Di Lullo and A. R. Kriegstein, The use of brain organoids to investigate neural development and disease, Nat. Rev. Neurosci., 2017, 18(10), 573–584 CrossRef PubMed.
- A. Przepiorski, et al., A simple bioreactor-based method to generate kidney organoids from pluripotent stem cells, bioRxivorg, 2017, 237644 Search PubMed.
- J. M. Lee, et al., Brain tissue responses to ischemia, J. Clin. Invest., 2000, 106(6), 723–731 CrossRef PubMed.
- I. Unal-Cevik, et al., Apoptotic and necrotic death mechanisms are concomitantly activated in the same cell after cerebral ischemia, Stroke, 2004, 35(9), 2189–2194 CrossRef PubMed.
- G. J. Muller, et al., Ischemia leads to apoptosis--and necrosis-like neuron death in the ischemic rat hippocampus, Brain Pathol., 2004, 14(4), 415–424 CrossRef.
- K. Zhang, L. Zhu and M. Fan, Oxygen, a Key Factor Regulating Cell Behavior during Neurogenesis and Cerebral Diseases, Front. Mol. Neurosci., 2011, 4, 5 Search PubMed.
- K. Zhang, et al., Reduced Cerebral Oxygen Content in the DG and SVZ In Situ Promotes Neurogenesis in the Adult Rat Brain In Vivo, PLoS One, 2015, 10(10), e0140035 CrossRef PubMed.
- E. M. Lord, L. Harwell and C. J. Koch, Detection of hypoxic cells by monoclonal antibody recognizing 2-nitroimidazole adducts, Cancer Res., 1993, 53(23), 5721–5726 Search PubMed.
- S. J. Morrison, et al., Culture in reduced levels of oxygen promotes clonogenic sympathoadrenal differentiation by isolated neural crest stem cells, J. Neurosci., 2000, 20(19), 7370–7376 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8lc00206a |
| This journal is © The Royal Society of Chemistry 2018 |