
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The integration of experiment and computational modelling in heterogeneously catalysed ammonia synthesis over metal nitrides
Constantinos D.
Zeinalipour-Yazdi
 ab,
Justin S. J.
Hargreaves
*c,
Said
Laassiri
c and
C. Richard A.
Catlow
abd
ab,
Justin S. J.
Hargreaves
*c,
Said
Laassiri
c and
C. Richard A.
Catlow
abd
aDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
bUK Catalysis Hub, Research Complex at Harwell, Rutherford Appleton Labs, Harwell Campus, OX11 0FA, UK
cWestCHEM, School of Chemistry, Joseph Black Building, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: Justin.Hargreaves@glasgow.ac.uk
dSchool of Chemistry, Cardiff University, Park Place, Cardiff CF10 1AD, UK
First published on 3rd August 2018
Abstract
In this perspective we present recent experimental and computational progress in catalytic ammonia synthesis research on metal nitrides involving a combined approach. On this basis, it suggested that the consideration of nitrogen vacancies in the synthesis of ammonia can offer new low energy pathways that were previously unknown. We have shown that metal nitrides that are also known to have high activity for ammonia synthesis can readily form nitrogen vacancies on their surfaces. These vacancies adsorb dinitrogen much more strongly than the defect-free surfaces and can efficiently activate the strong N–N triple bond. These fundamental studies suggest that heterogeneously catalysed ammonia synthesis over metal nitrides is strongly affected by bulk and surface defects and that further progress in the discovery of low temperature catalysts relies on more careful consideration of nitrogen vacancies. The potential occurrence of an associative pathway in the case of the Co3Mo3N catalytic system provides a possible link with enzymatic catalysis, which will be of importance in the design of heterogeneous catalytic systems operational under process conditions of reduced severity which are necessary for the development of localised facilities for the production of more sustainable “green” ammonia.
1. Introduction
One of the most studied reactions for the last 100 years has been ammonia synthesis. The significance of this reaction is evident by its association with three Nobel Prizes in chemistry, the first being awarded to Fritz Haber in 1918 for discovering that ammonia could be synthesised at high pressure and the next to Carl Bosch in 1931 for bringing the process to economical and technical industrial-scale. Haber in his Nobel lecture said that the main motivation for the discovery was the growing demand for food that requires fertilisers to be produced. He predicted that the amount of fixed N needed would increase from a few hundred thousand tons a year to millions of tons.1 More recently the fundamental surface science research approach of Gerhard Ertl led to the third Nobel Prize which was awarded in 2007 partially for his contribution to the enhanced understanding of the ammonia synthesis mechanism on the industrially relevant iron catalyst. He demonstrated how hydrogen dissociatively adsorbs on iron surfaces2 and that nitrogen dissociatively chemisorbs forming atomic N,3 leading to the production of ammonia according to a Langmuir–Hinshelwood mechanism (see Scheme 1).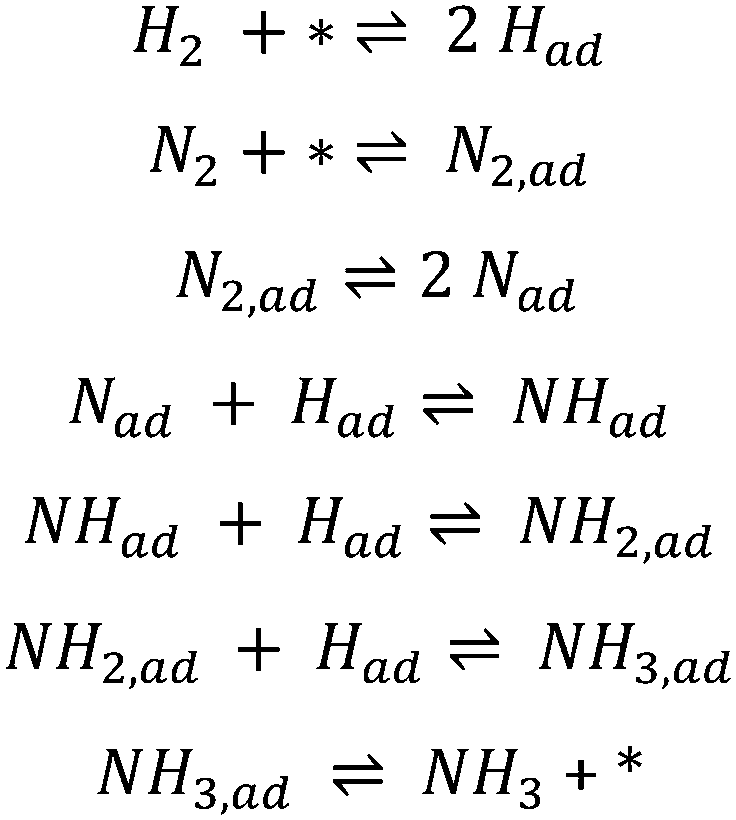 | ||
| Scheme 1 Elementary steps of the Langmuir–Hinshelwood mechanism in ammonia synthesis on iron.4 The asterisk (*) denotes a vacant adsorption site comprising an ensemble of surface atoms. | ||
Based on photoelectron spectroscopy, a structural model for the iron–nitrogen surface was deduced5 and an enhanced understanding of the kinetics of N2 adsorption on iron surfaces was obtained.6 The Langmuir–Hinshelwood mechanism has been previously modelled on Ru(0001) surfaces by Logadóttir and Nørskov7; and a simplified schematic of the reaction mechanism studied is presented in Scheme 2. Ru based catalysts are applied in industry and are more active, although less widespread, than their iron based catalyst counterparts.8
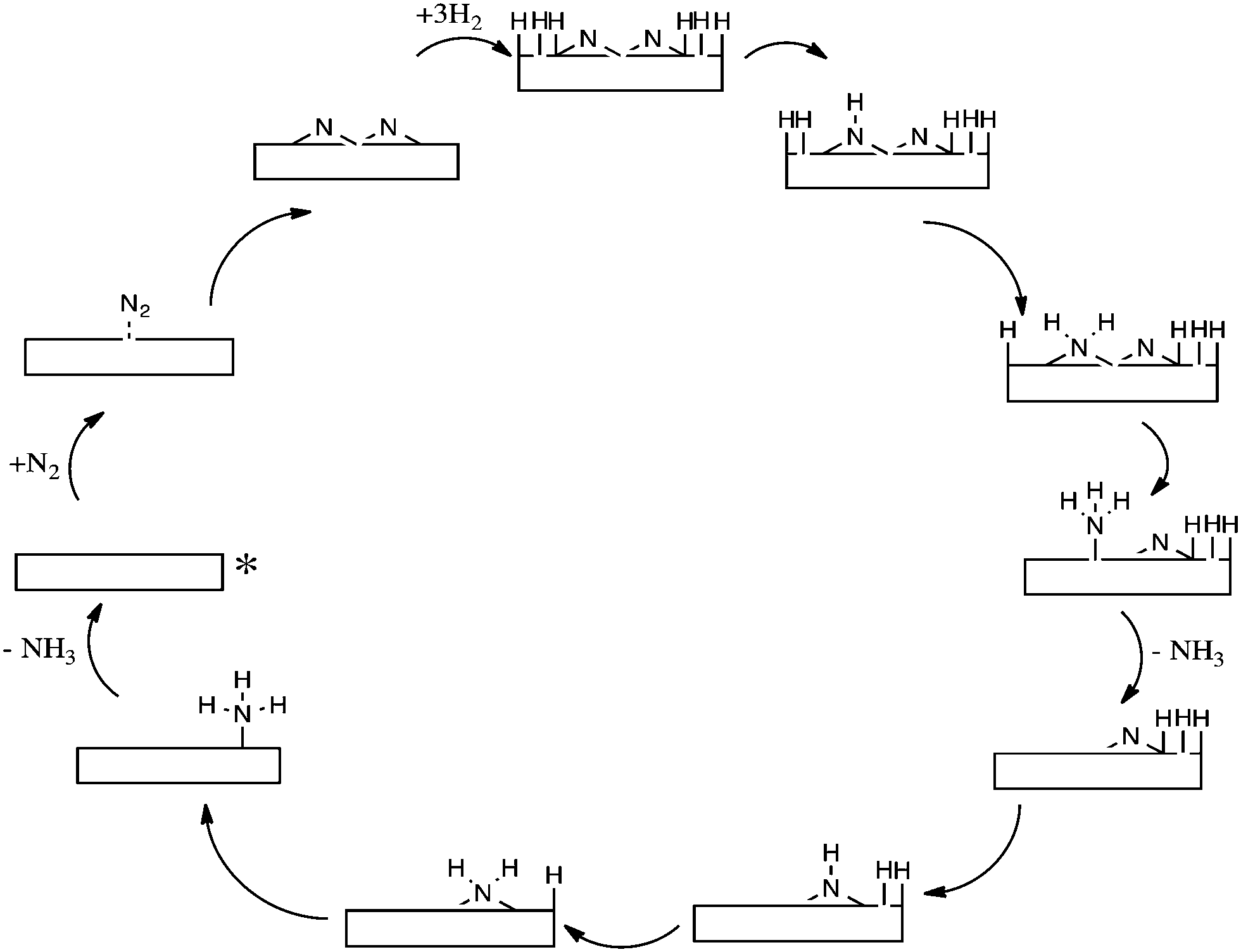 | ||
| Scheme 2 Simplified schematic of Langmuir–Hinshelwood mechanism on Ru(0001) surfaces. The starting point of the reaction is denoted by an asterisk. | ||
As currently operated on the industrial scale, ammonia synthesis via the Haber–Bosch process operates at a global annual production rate of ca. 174 million tonnes which is growing significantly.9 Around 85% of the ammonia produced is employed in synthetic fertilisers and accordingly the Haber–Bosch process can be directly credited with the sustenance of ca. 40% of the global population. The process is reliant upon the supply of high purity hydrogen and nitrogen feedstreams. The hydrogen required is generally produced from fossil based sources and thus the Haber–Bosch process as a whole accounts for around 2.5% of the worldwide fossil fuel based CO2 emissions, equating to around 670 million tonnes per year.9 Taken as a whole, the process is responsible for the consumption of around 2% of the global commercial energy requirement.9 In view of these considerations, it is not surprising that the production of more sustainable “green” ammonia is becoming a significant contemporary target. To this end, approaches which are being widely investigated include the development of photocatalytic10,11 and electrocatalytic12,13 routes as well as solar driven chemical looping approaches.14,15 An additional area of interest in this context relates to the development of more active ammonia synthesis catalysts, which can be used thermochemically. The discovery of such catalysts would lead to a reduction of the severity of operation of the traditional Haber–Bosch process which is operated at >100 atmospheres pressure and ca. 400 °C reaction temperature. Catalysts of enhanced activity, which allow operation in the more thermodynamically favourable lower reaction temperature regime, would facilitate operation at lower reaction pressure and on a more localised scale. Therefore, taking advantage of existing technology for the separation of nitrogen from air and the application of the ever increasing availability of renewably derived electricity for the generation of hydrogen from electrolysis of water, very localised smaller scale ammonia synthesis would be a realistic prospect. Indeed this approach would have additional benefits through further reduction of the carbon footprint associated with the transportation of ammonia based fertilisers over long distances and it could lead to a profitable use of the periodic over-supply of renewable electricity associated with wind or tidal derived sources. An interesting prospect also relates to the application of ammonia as a fuel and thus scenarios in which localised ammonia, and hence fertiliser, production coupled to its application in the agricultural field by tractors running on ammonia fuel become a tantalising, but realistic prospect; in relation to which it is argued that with the integration of ammonia as a fuel in new applications (e.g. within automotive industry) the global need for efficient ammonia production will be increasingly important.16 Ammonia can be used as a fuel in combustion engines with minimal modification of the engine. Currently there are also efforts to use it in gas turbines and ammonia fuel cells. The use of ammonia in automobiles was first demonstrated during 1943 in Belgium where it was used in buses. Ammonia combustion produces nitrogen and water in contrast to the combustion of fossil fuels that produce significant quantities of CO2. Therefore, the development of green non-fossil dependent routes to ammonia also offer the prospect of a substantial reduction in, or even elimination of, transportation related CO2 production.
2. Metal nitrides as ammonia synthesis catalysts
As described above, iron and ruthenium based catalysts which represent the most widely applied ammonia synthesis catalysts in the industrial context are generally accepted to operate via Langmuir–Hinshelwood based catalytic mechanisms. In recent years, we have become interested in the possibility that catalytically active nitride based materials may exhibit activity via Mars–van Krevelen based pathways in which their lattice nitrogen is hydrogenated to yield ammonia generating nitrogen vacancies which activate N2 closing the catalytic cycle. The role of metal nitrides in ammonia synthesis was recognised early on by Mittasch who reported 9% detectable N content during the catalytic reaction on molybdenum17 and we have drawn the analogy with the metal oxide catalysed oxidation which often involves participation of lattice oxygen species,18 thereby potentially allowing an element of design by directed tailoring of activity through variation of composition and structure. We have investigated such possibilities via a combination of experimental approaches and computational modelling and we suggest that the associated new insights provided might lead to the identification of the catalyst design parameters necessary for the step-change development novel catalysts active in temperature reaction regimes applicable to green ammonia production on a localised scale. To this end, amongst other systems, we have explored the origin of the catalytic activity of Co3Mo3N which is a system, particularly when promoted with appropriate levels of Cs+ dopant, reported to possess activities which exceed that of the conventional Haber–Bosch iron based system.19–23 Nørskov and co-workers have investigated this system and have rationalized its activity on the basis of a scaling relationship.24 Taking the enthalpy of N2 adsorption as an appropriate descriptor they have produced the volcano plot presented in Fig. 1. In this volcano plot, it can be seen that the combination of molybdenum which binds N2 too strongly with respect to Ru, the optimal catalyst, and cobalt with binds N2 too weakly, results in an alloy with a calculated turnover frequency higher than that of Ru, the most active material applied in an industrial context. It is proposed that the presence of interstitial N within the Co3Mo3N system leads to the expression of the active (111) surface plane in which both Co and Mo are present. The N itself was not proposed to participate in reaction. The rationale applied in which the (111) plane is active suggests the reaction to be strongly structure sensitive, although to the authors’ knowledge this has yet to be experimentally verified. In addition, applying the same principle, the combination of Mo and Ni would be predicted to result in an active catalyst and indeed Ni2Mo3N is known to be so,22,25 along with Fe3Mo3N.22 The high relative activities of Fe and Os are also evident in the plot. Indeed, the identification of the activity of osmium was made early in studies relating to ammonia synthesis although in an early example of the role of element scarcity, recognition of the fact that global reserve of Os only amounted to a small quantity at that time26 spurred the development of the now familiar iron based industrial catalyst.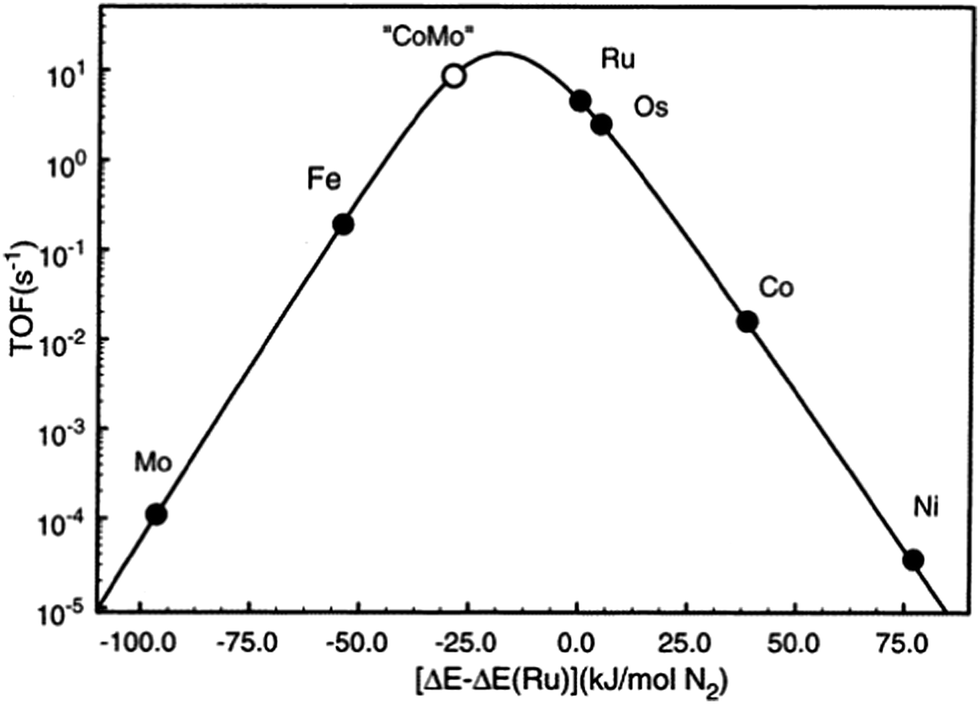 | ||
Fig. 1 Calculated turnover frequencies (TOFs) for ammonia synthesis as a function of the adsorption energy of nitrogen. The synthesis conditions are 400 °C, 50 bar, gas composition H2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N2 = 3:1 containing 5% NH3. Reprinted with permission from C. J. H. Jacobsen, S. Dahl, B. S. Clausen, S. Bahn, A. Logadóttir and J. K. Nørskov, J. Am. Chem. Soc., 2001, 123, 8404–8405. Copyright 2001 American Chemical Society. :N2 = 3:1 containing 5% NH3. Reprinted with permission from C. J. H. Jacobsen, S. Dahl, B. S. Clausen, S. Bahn, A. Logadóttir and J. K. Nørskov, J. Am. Chem. Soc., 2001, 123, 8404–8405. Copyright 2001 American Chemical Society. | ||
2.1 Experimental studies
Experimentally, the reactivity of lattice nitrogen via examination of the reduction properties of Co3Mo3N in an Ar/H2 (i.e. non-N2 containing) feed has been probed by us and the production of low levels of NH3 accomplished.27 Furthermore, it has been shown that reduction at elevated temperatures results in the loss of 50% of the lattice nitrogen originally present resulting in the unprecedented Co6Mo6N phase in which the remaining lattice N had relocated from its original 16c Wyckoff lattice site to the 8a site.28 The restoration of Co3Mo3N which can be achieved by direct reaction of Co6Mo6N with N2 at elevated temperature29 results in a potential N2 looping system for ammonia synthesis. Additional evidence for the participation of lattice N in the catalytic process has been supplied in studies of nitrogen isotopic exchange. Interestingly, studies undertaken relating to homo-molecular exchange in which the scrambling of a mixture of gas-phase 14N2 and 15N2 over Co3Mo3N indicate that the material is not particularly effective at activating, dissociating and recombining N2 at 400 °C, the reaction temperature commonly employed with this catalyst.30 In the context of Schemes 1 and 2 outlined earlier, this observation is somewhat surprising but, as shown later, computational modelling provides a potential explanation.Heterolytic isotopic exchange, in which the scrambling of gas-phase labelled N2 (15N2) with the lattice N of the nitride (the isotopic distribution of which is, of course, reflective of the natural isotopic distribution of N and is therefore overwhelmingly 14N) is monitored, provides a more direct means of probing the reactivity of the lattice N. Reactivity of lattice N is a requirement for the operation of the Mars–van Krevelen mechanism. In studies of heterolytic exchange undertaken at 600 °C, as presented in Fig. 2, exchange of a significant proportion of the total lattice N with gas-phase 15N2 occurs,30 as can be determined from the increase of gas-phase 14N containing species. It is proposed that the presence of hydrogen may facilitate this process via enhancement of lattice N vacancy concentration at lower reaction temperatures such as the 400 °C applied in ammonia synthesis. Also presented in this figure, is the influence of sample pre-treatment which is found to have a significant effect upon the exchange of gas-phase N2 and lattice N. Pre-treatment employing an Ar purging step inhibited exchange with the likely cause being related to surface segregation of zero valent Mo which acts as a diffusion barrier.
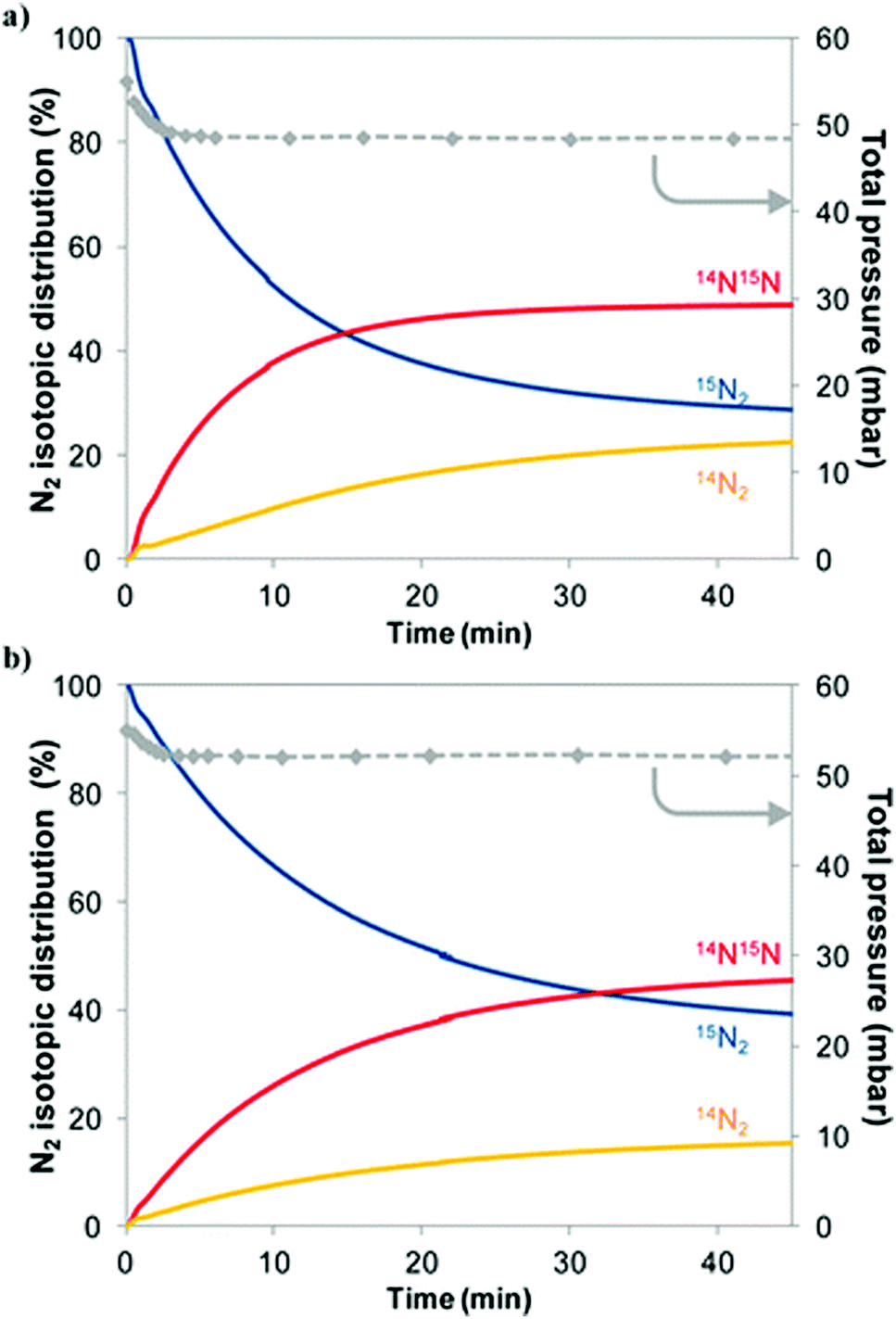 | ||
| Fig. 2 Evolution of the gas-phase N2 isotopic distribution and of the total pressure during a heterolytic isotopic nitrogen exchange experiment at 600 °C on Co3Mo3N material pretreated with 3:1 H2:N2 followed by (a) N2 purge for 30 minutes or (b) Ar purge and subsequent N2 purge for 30 minutes. Pre-treatment is undertaken at 700 °C. Reproduced from ref. 30. | ||
Additional experimental evidence for the role of lattice nitrogen in the ammonia synthesis reaction comes from our studies of the Co3Mo3C and Co6Mo6C systems which are isostructural with their nitride counterparts.31 Co3Mo3C is only active at temperatures above those required for Co3Mo3N and, following an initial lag period, the onset of activity is related the incorporation of lattice N in the 16c lattice site at 500 °C. Co6Mo6C, in which C is present in the 8a site, was not found to possess activity over prolonged testing periods.
2.2 Computational modelling
The computational modelling which we have undertaken supports and underpins our experimental observations. By modelling Co3Mo3N(111) surface slabs, calculated vacancy formation energies suggest that the surface nitrogen vacancy concentration will be significant in 3 fold hollow bound nitrogen surfaces at temperatures of interest for ammonia synthesis and that even at ambient temperature concentrations in excess of 1013 vacancies per cm2 are predicted.32 Furthermore, such vacancy sites have been shown to be effective for N2 activation. In relation to these results, the observed initial decline in pressure upon admission of the reactant gas as evident in the isotopic exchange studies presented in Fig. 2 becomes explicable based upon the replenishment of vacancies generated during the prior 700 °C pretreatment. It is also pertinent to note that the studies are consistent with recent experimental work which has been published by Hosono and co-workers.33,34 In this work, sodium naphthalenide driven reduction has been applied to prepare CoMo bimetallic particles supported upon CeO2 which possess enhanced activity with respect to Co3Mo3N. Characterisation suggested that the active phase comprises dispersed nanosized Co3Mo3N on the CeO2 support and interestingly, as dispersion increased, so did the ammonia synthesis turnover frequency which was related to the enhanced ease of formation of nitrogen vacancies.34Taking our work further, we have investigated in detail two mechanisms of ammonia synthesis on Co3Mo3N-(111) surfaces, which are depicted in Scheme 3.35 The first is a Langmuir–Hinshelwood based dissociative mechanism which happens at surface cavities. In this mechanism N2 adsorbs side-on at the catalytic site and dissociates into two –N species that reside at 3-fold hollows.36 These –N species subsequently hydrogenate with –H that originates from dissociatively chemisorbed molecular hydrogen, resulting in the successive formation of –NH, NH2 and NH3. Once NH3 is formed, it desorbs from the catalyst and hydrogenation of the second –N starts to occur. The second mechanism for ammonia synthesis that we explored was an Eley–Rideal/Mars–van Krevelen based associative mechanism which happens at surface nitrogen vacancies of 3-fold bound nitrogen. Gas phase nitrogen chemisorbs in an end-on configuration, which then further reacts with hydrogen, coming directly from the gas phase (hence the Eley–Rideal description applied). Interestingly, we have shown that this associative mechanism has smaller barriers than the Langmuir–Hinshelwood dissociative mechanism occurring at surface cavities, which has a large barrier for the reaction of surface bound –H and –N. This is a tantalizing suggestion, since it is generally acknowledged that in the drive to develop heterogeneous catalysts active at lower reaction temperatures, such as those necessary for localized ammonia synthesis, associative mechanistic pathways will assume a significantly greater degree of importance. In this context, it is interesting to bear in mind that enzymatic nitrogen activation which, of course, occurs at ambient temperature involves associative N2 activation.37
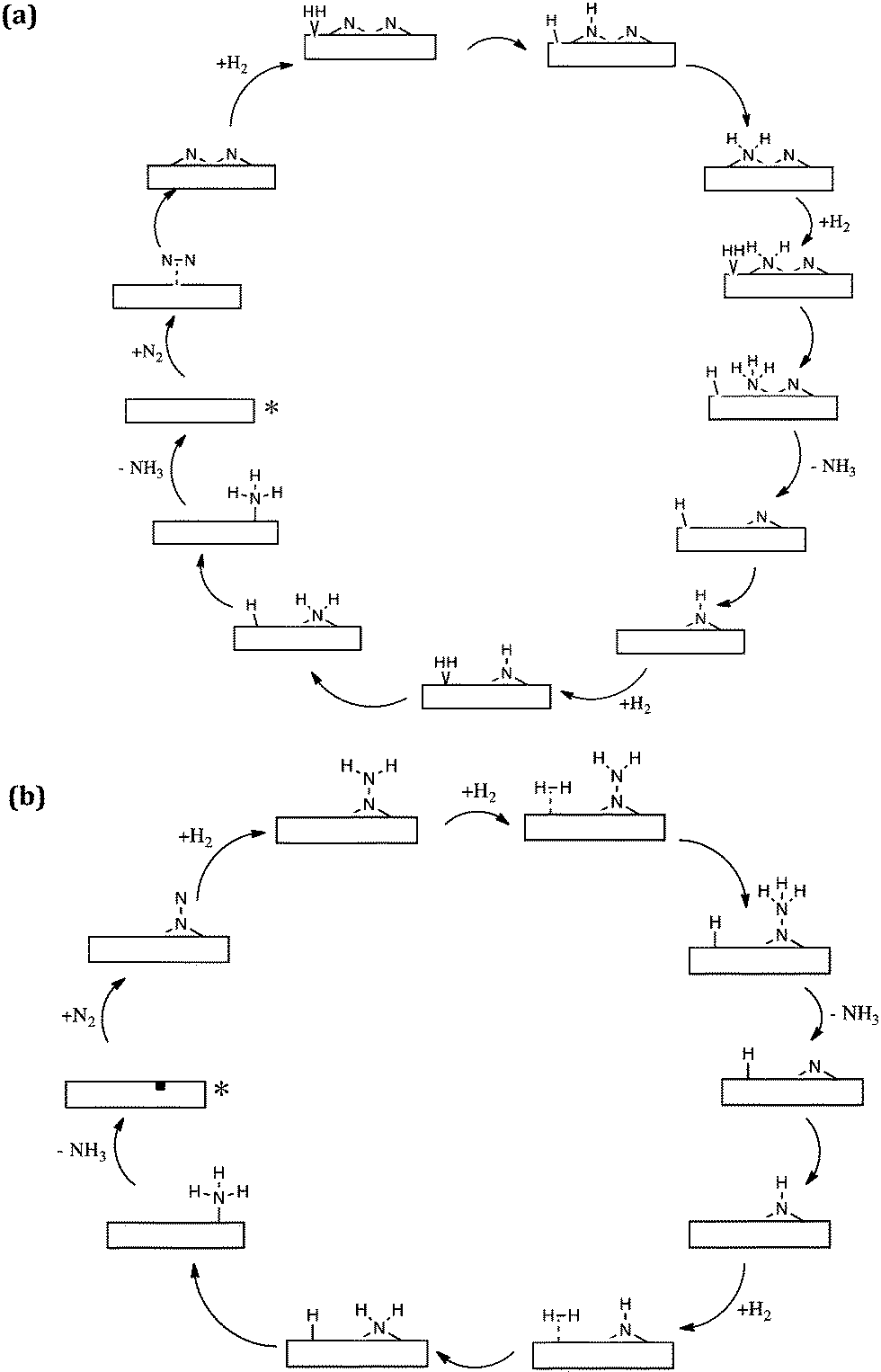 | ||
| Scheme 3 Simplified schematic of (a) the Langmuir–Hinshelwood based dissociative reaction mechanism of ammonia synthesis and (b) the Eley–Rideal/Mars–van Krevelen based associative mechanism for ammonia synthesis on Co3Mo3N according to ref. 35. The starting point of the reaction is denoted by an asterisk. | ||
The possible occurrence of the associative pathway, which has yet to be experimentally verified, is consistent with our observations in relation to the homomolecular exchange of 14N2/15N2 gas phase mixtures which indicate that, although an extremely good ammonia synthesis catalyst, Co3Mo3N is poor in relation to the dissociation and recombination of N2 – an observation which was perhaps surprising at first sight as discussed earlier. The novelty of the associative mechanism which we have modelled is firstly that H2 reacts directly from the gas phase with end-on activated nitrogen and secondly that the surface of the catalyst is considered to have a large number of nitrogen vacancies which can more effectively adsorb and activate N2 than the defect free surfaces. It would be extremely interesting and informative to see what other mechanisms, such as that involving the reaction of associatively activated N2 with dissociated H2, are available from a computational point of view and such studies may provide highly informative input into experimental work. Such experimental work should also involve detailed kinetic studies such that rate laws which can potentially discriminate against potential candidate mechanisms are obtained.
3. The role of promoters
In addition to defect structure and reaction mechanism, a further area which can profit from a combined experimental-computational modelling approach is to develop enhanced understanding of the effect of promoters (i.e. dopants) in ammonia synthesis. We have previously found that doping tantalum nitride (Ta3N5) with low levels of Co resulted in enhanced reactivity for ammonia production upon reduction.38 In a detailed DFT study we have shown that Co coordinates to the surface nitrogen preferably maximising its coordination number.39 These cobalt dopants chemisorb hydrogen dissociatively whereas the un-doped Ta3N5 surface chemisorbs H2 mostly molecularly, which is in agreement with H2 adsorption studies on cobalt dimers that showed that at low coverages cobalt atoms dissociatively chemisorb H2, whereas at coverages above 0.4 the adsorption starts to become molecular.40Another ammonia synthesis system where dopants were found to play a critical role in enhancing the activity of the catalyst is the Li–Mn–N system. We showed experimentally that doping manganese nitride with Li+ enhanced ammonia synthesis activity at low temperature.41 Furthermore, the Li–Mn–N presented high thermochemical stability after reduction with hydrogen. In related work, electronic structure calculations have been employed to show the role of doping in modifying the performance of manganese nitride for solar driven looping ammonia synthesis looping via destabilisation of lattice nitrogen.15,42 The free energy of the formation of bulk nitrogen vacancies has been determined from the difference in the total electronic energy of the metal nitride D0.25Mn1.75N (where D corresponds to the dopant) with 25 and 50% N vacancies with respect to stoichiometric Mn2N and the stoichiometric metal nitride plus the chemical potential of N2 in the gas-phase.42
The above case studies on various metal nitrides clearly show an important variation of the surface chemistry with the use of dopants which is a potential area for further development. In other work, dopants/promoters function by facilitating alternative mechanistic pathways. For example, lithium hydride has been reported to facilitate N2 activation in conjunction with transition metals and transition metal nitrides, by-passing limiting scaling and leading to enhanced activity at lower reaction temperatures for ammonia synthesis.43
4. Conclusion and outlook
The integration of experiment and computational modelling in ammonia synthesis can undoubtedly lead to a level of new insight. Whilst it is clear that computational modelling can play a significant role in facilitating enhanced interpretation and understanding of experimental observations, the next step to be taken is the computational design of the next generation of ammonia synthesis catalysts. Advances in theory make in silico design of new catalysts with tailored properties an increasingly realistic prospect. To date, many of the experimental advances in thermocatalytic ammonia synthesis relate to the identification of catalytic materials which possess enhanced activities within reaction regimes which are related to those operated in large scale practice. If the production of localised green ammonia by thermocatalytic routes is to be achieved it will be necessary to develop catalytic systems which possess enhanced activities under less severe operational conditions which will require a step-change advance. It is increasingly the case that the integration of computational design and experiment in a feedback loop may provide such a step-change.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to acknowledge the provision of financial support through EPSRC grants EP/L02537X/1, EP/L026317/1 and EP/K014714/1.References
- F. Haber, The Synthesis of Ammonia from its Elements, Nobel Lecture, 1920.
- F. Bozso, G. Ertl, M. Grunze and M. Weiss, Appl. Surf. Sci., 1977, 1, 103–119 CrossRef.
- G. Ertl, M. Huber, S. B. Lee, Z. Paál and M. Weiss, Appl. Surf. Sci., 1981, 8, 373–386 CrossRef.
- G. Ertl, in Catalytic Ammonia Synthesis, ed. J. R. Jennings, Plenum, New York, 1991, pp. 109–132 Search PubMed.
- R. Imbihl, R. J. Behm, G. Ertl and W. Moritz, Surf. Sci., 1982, 123, 129–140 CrossRef.
- G. Ertl, S. B. Lee and M. Weiss, Surf. Sci., 1982, 114, 515–526 CrossRef.
- A. Logadóttir and J. K. Nørskov, J. Catal., 2003, 220, 273–279 CrossRef.
- D. E. Brown, T. Edmonds, R. W. Joyner, J. J. McCarroll and S. R. Tennison, Catal. Lett., 2014, 144, 545–552 CrossRef.
- P. H. Pfromm, J. Renewable Sustainable Energy, 2017, 9, 034702 CrossRef.
- A. J. Medford and M. C. Hatzell, ACS Catal., 2017, 7, 2624–2643 CrossRef.
- L. Wang, M. Xia, H. Wang, K. Huang, C. Qian, C. T. Maravelias and G. A. Ozin, Joule, 2018, 2, 1–20 CrossRef.
- V. Kyriakou, I. Garagounis, E. Vasileiou, A. Vourros and M. Stoukides, Catal. Today, 2017, 286, 2–13 CrossRef.
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 CrossRef.
- R. Michalsky and A. Steinfeld, Catal. Today, 2017, 286, 124–130 CrossRef.
- R. Michaslky, A. M. Avram, B. A. Peterson, P. H. Pfromm and A. A. Peterson, Chem. Sci., 2015, 6, 3965–3974 RSC.
- C. Zamfirescu and I. Dincer, J. Power Sources, 2008, 185, 459–465 CrossRef.
- A. Mittasch and W. Frankenburg, Adv. Catal., 1950, 2, 81–104 Search PubMed.
- C. Doornkamp and V. Ponec, J. Mol. Catal. A: Chem., 2000, 162, 19–32 CrossRef.
- R. Kojima and K.-I. Aika, Appl. Catal., A, 2001, 215, 149–160 CrossRef.
- R. Kojima and K.-I. Aika, Appl. Catal., A, 2001, 218, 121–128 CrossRef.
- R. Kojima and K.-I. Aika, Appl. Catal., A, 2001, 219, 157–170 CrossRef.
- C. J. H. Jacobsen, Chem. Commun., 2000, 1057–1058 RSC.
- A. Boisen, S. Dahl and C. J. H. Jacobsen, J. Catal., 2002, 208, 180–186 CrossRef.
- C. J. H. Jacobsen, S. Dahl, B. S. Clausen, S. Bahn, A. Logadóttir and J. K. Nørskov, J. Am. Chem. Soc., 2001, 123, 8404–8405 CrossRef PubMed.
- N. Bion, F. Can, J. Cook, J. S. J. Hargreaves, A. L. Hector, W. Levason, A. R. McFarlane, M. Richard and K. Sardar, Appl. Catal., A, 2015, 504, 44–50 CrossRef.
- C. Flavell-While, Tce, 2010, 825, 54–55 Search PubMed.
- D. Mckay, D. H. Gregory, J. S. J. Hargreaves, S. M. Hunter and X.-L. Sun, Chem. Commun., 2007, 3051–3053 RSC.
- S. M. Hunter, D. Mckay, R. I. Smith, J. S. J. Hargreaves and D. H. Gregory, Chem. Mater., 2010, 22, 2898–2907 CrossRef.
- D. H. Gregory, J. S. J. Hargreaves and S. M. Hunter, Catal. Lett., 2011, 141, 22–26 CrossRef.
- S. M. Hunter, D. H. Gregory, J. S. J. Hargreaves, M. Richard, D. Duprez and N. Bion, ACS Catal., 2013, 3, 1719–1725 CrossRef PubMed.
- I. AlShibane, A. Daisley, J. S. J. Hargreaves, A. L. Hector, S. Laassiri, J. L. Rico and R. I. Smith, ACS Sustainable Chem. Eng., 2017, 5, 9214–9222 CrossRef.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves and C. R. A. Catlow, J. Phys. Chem. C, 2015, 119, 28368–28376 CrossRef.
- Y. Tsuji, M. Kitano, K. Kishida, M. Sasase, T. Yokoyama, M. Hara and H. Hosono, Chem. Commun., 2016, 52, 14369–14372 RSC.
- Y. Tsuji, K. Ogasawara, M. Kitano, K. Kishida, H. Abe, Y. Niwa, T. Yokoyama, M. Hara and H. Hosono, J. Catal., 2018, 364, 31–39 CrossRef.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves and C. R. A. Catlow, J. Phys. Chem. C, 2018, 122, 6078–6082 CrossRef.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves and C. R. A. Catlow, J. Phys. Chem. C, 2016, 120, 21390–21398 CrossRef.
- B. M. Hoffman, D. Lukoyanov, Z.-Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef PubMed.
- S. Laassiri, C. D. Zeinalipour-Yazdi, C. R. A. Catlow and J. S. J. Hargreaves, Catal. Today, 2017, 286, 147–154 CrossRef.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves, S. Laassiri and C. R. A. Catlow, Phys. Chem. Chem. Phys., 2017, 19, 11968–11974 RSC.
- C. D. Zeinalipour-Yazdi, Surf. Sci., 2017, 656, 54–59 CrossRef.
- S. Laassiri, C. D. Zeinalipour-Yazdi, C. R. A. Catlow and J. S. J. Hargreaves, Appl. Catal., B, 2018, 223, 60–66 CrossRef.
- R. Michalsky, P. H. Pfromm and A. Steinfeld, Interface Focus, 2015, 5, 20140084 CrossRef PubMed.
- P. K. Wang, F. Chang, W. B. Gao, J. P. Guo, G. T. Wu, T. He and P. Chen, Nat. Chem., 2017, 9, 64–70 Search PubMed.
| This journal is © the Owner Societies 2018 |