
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLaccase-catalyzed green synthesis and cytotoxic activity of novel pyrimidobenzothiazoles and catechol thioethers†‡
H. T. Abdel-Mohsen a,
J. Conradb,
K. Harmsc,
D. Nohrd and
U. Beifuss*b
a,
J. Conradb,
K. Harmsc,
D. Nohrd and
U. Beifuss*b
aChemistry of Natural and Microbial Products Department, Pharmaceutical Industries Research Division, National Research Centre, Cairo, Egypt
bBioorganische Chemie, Institut für Chemie, Universität Hohenheim, Garbenstr. 30, Stuttgart, D-70599, Germany. E-mail: ubeifuss@uni-hohenheim.de; Fax: +49 711 459 22951; Tel: +49 711 459 22171
cFachbereich Chemie, Universität Marburg, Hans-Meerwein-Str. 4, D-35032 Marburg, Germany
dInstitut für Biologische Chemie und Ernährungswissenschaft, Universität Hohenheim, Garbenstr. 30, Stuttgart, D-70599, Germany
First published on 20th March 2017
Abstract
The laccase-catalyzed reaction between unsubstituted catechol and 2-thioxopyrimidin-4(1H)-ones using aerial O2 as the oxidant delivers novel pyrimidobenzothiazoles with high yields in an aqueous solvent system under mild reaction conditions. With 4-substituted catechols, catechol thioethers are formed exclusively. The synthetic protocols developed provide a sustainable approach for these compound classes. In addition, the cytotoxicity of the products against HepG2 cell line is reported. Most compounds exhibit antiproliferative activities with IC50 values at the micromolar level. A structure–activity relationship study will facilitate the further development of these compounds as cytotoxic agents.
Introduction
Pyrimidines are the building blocks of pyrimidine nucleosides and thiamine (vitamin B1). Therefore, the pyrimidine skeleton is regarded as an interesting scaffold for the development of molecules with potential applications in medicine.1 Over the years, a wide range of pharmaceutical agents with a pyrimidine moiety have been developed.1–3 Zidovudine, stavudine and other potent anti-HIV agents are pyrimidines.2,3a Other important drugs with a pyrimidine ring are the antitumor agents fluorouracil (I), tegafur (II), nimustine (III), monastrol (IV) and pazopanib (V) (Fig. 1).2,3b,c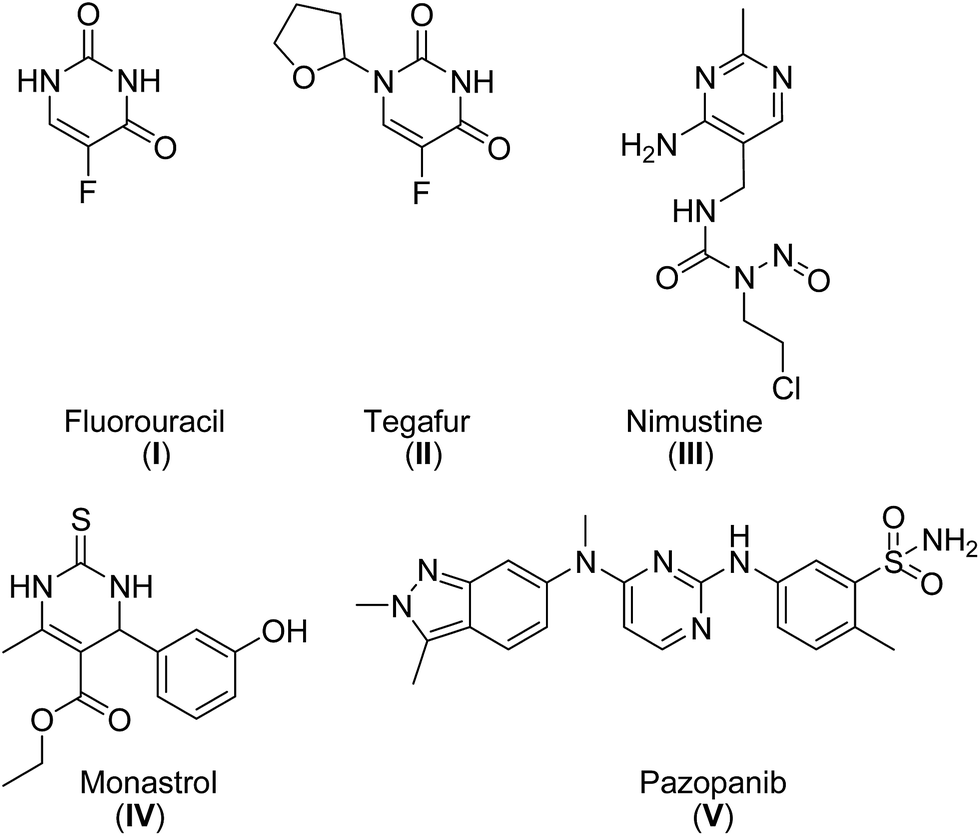 | ||
| Fig. 1 Antitumor agents with a pyrimidine moiety. | ||
Recently, the development of more environmentally friendly approaches for the synthesis of pharmaceutical active ingredients has increasingly come into focus of the pharmaceutical chemistry.4 Green chemistry is a valuable concept for the development of new, more effective, less toxic and cost efficient methods for the synthesis of bioactive molecules. This can be achieved, for example, by developing highly atom economic transformations,5 using nontoxic reagents and catalysts of natural origin (e.g. enzymes), the development of one-pot multi-step reactions, which reduce the amount of waste formed during reaction and work up as well as the development of environmentally benign reaction conditions.4,6
Hepatocellular carcinoma (HCC) is regarded as one of the leading causes of cancer related mortality in the world. Every year more than half a million new cases with HCC are diagnosed worldwide. Hepatitis B, hepatitis C viruses and non-alcoholic fatty liver disease are the main risk factors for the development of chronic liver disease and subsequent development of HCC.7a,b Despite the tremendous progress that has been achieved in cancer therapy over the last decades, the currently available drugs suffer from serious disadvantages, such as lack of selectivity, toxicity and resistance that limit their use.3b,7c For these reasons, the development of novel anti-tumor agents remains a challenge.
Against this background, the importance of enzyme-catalyzed transformations in organic synthesis is steadily growing.8 Laccases (benzenediol: O2 oxidoreductase E.C. 1.10.3.2.), which are multicopper oxidases, are among the most attractive enzymes in this respect.9 Over the last few years, laccases have proven their ability to catalyze a number of important oxidative transformations in aqueous solvent systems under mild reaction conditions (temperature, pH, pressure) using aerial oxygen as an oxidant. The oxidation of the substrates is linked to the reduction of oxygen to water which is the only byproduct of laccase-catalyzed reactions. Moreover, the application range of laccases can be broadened by employing mediators. Using laccase-mediator systems, the oxidation of substrates with higher oxidation potentials can also be achieved.10 Laccases have been successfully used to catalyze the oxidation of a number of functional groups,11 the dimerization of phenolic compounds by oxidative coupling12 and the oxidation of catechols and hydroquinones to the corresponding highly reactive quinoid systems, followed by reaction with different nucleophiles.13–16 With the latter approach, a range of simple 1,4-adducts13–15 as well as heterocyclic systems16 has been made available.
In contrast to the additions of C and N nucleophiles, only little is known about laccase-initiated 1,4-additions of S nucleophiles.15,16b,d Recently, we have studied the laccase-catalyzed generation of o-quinones and their reaction with different S nucleophiles for the synthesis of thioethers as well as pyrimidobenzothiazoles.15b,16d Ragauskas and coworkers have shown the successful application of laccases for the synthesis of 2,3-ethylenedithio-1,4-quinones.16b In 2016, Schauer et al. reported on the multiple C–S bond formation between p-hydroquinones and aromatic thiols using laccase as a catalyst.15a
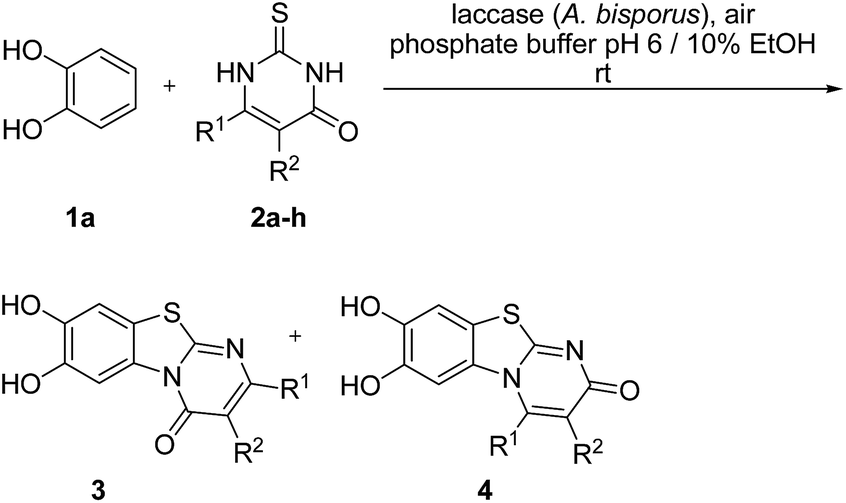
Here, we report on the laccase-catalyzed reaction between catechols 1 and 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2 for the green synthesis of novel biologically active compounds. Depending on the substitution pattern of the catechols 1, either pyrimidobenzothiazoles 3, 4 or catechol thioethers 5 are formed (Fig. 2). Moreover, the cytotoxic activity of selected reaction products against HepG2 cell line is reported.
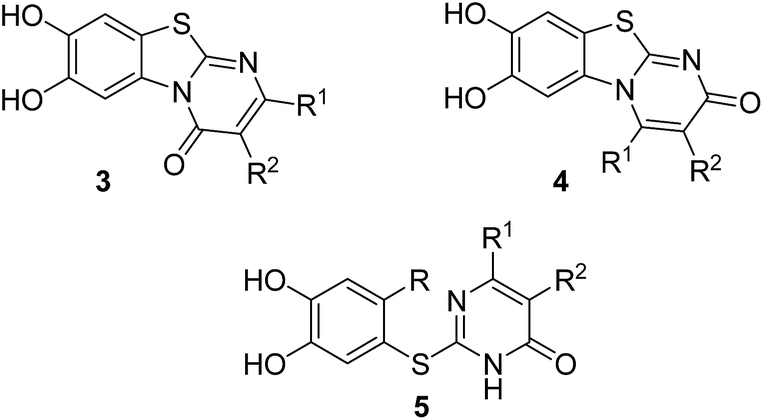 | ||
| Fig. 2 Structures of the regioisomeric pyrimidobenzothiazoles 3, 4 and the catechol thioethers 5. | ||
Results and discussion
Fig. 3 depicts the structures of the catechols 1 and the 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2 that were chosen as substrates for the enzyme-catalyzed oxidative transformations.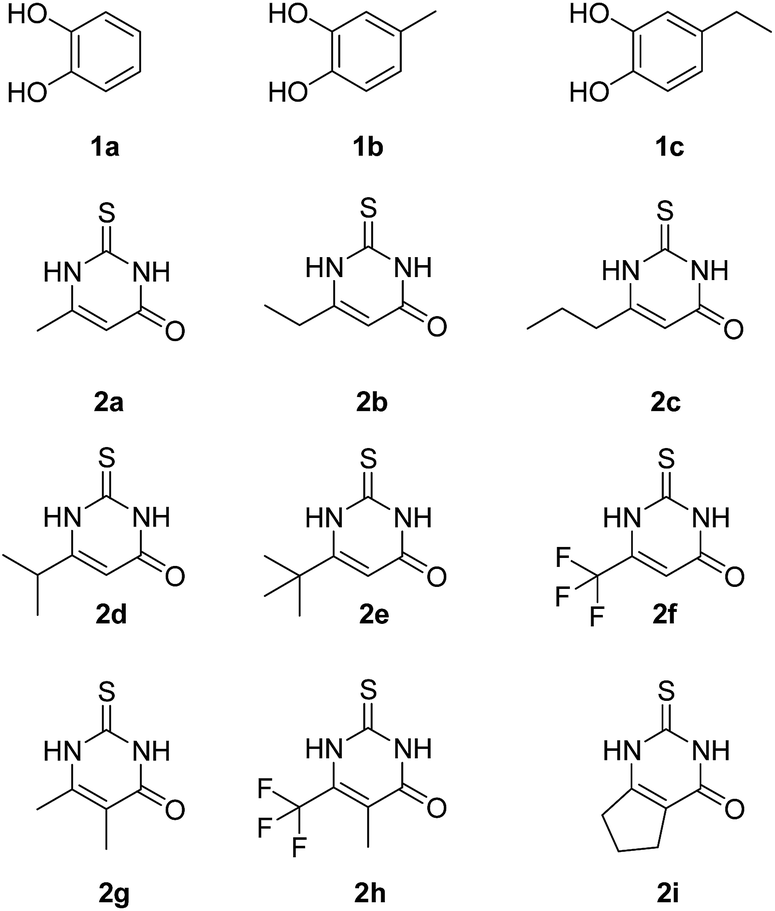 | ||
| Fig. 3 Substrates for the laccase-catalyzed transformations. | ||
The catechols 1a–c are commercially available, while the 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2a–h were synthesized by reaction of different acyclic β-ketoesters 6a–h with thiourea (7) under basic conditions in ethanol at 80 °C (general procedure I).17 2,3,6,7-Tetrahydro-2-thioxo-1H-cyclopenta[d]pyrimidin-4(5H)-one (2i) was obtained from cyclic β-ketoester 6i as substrate (Scheme 1).
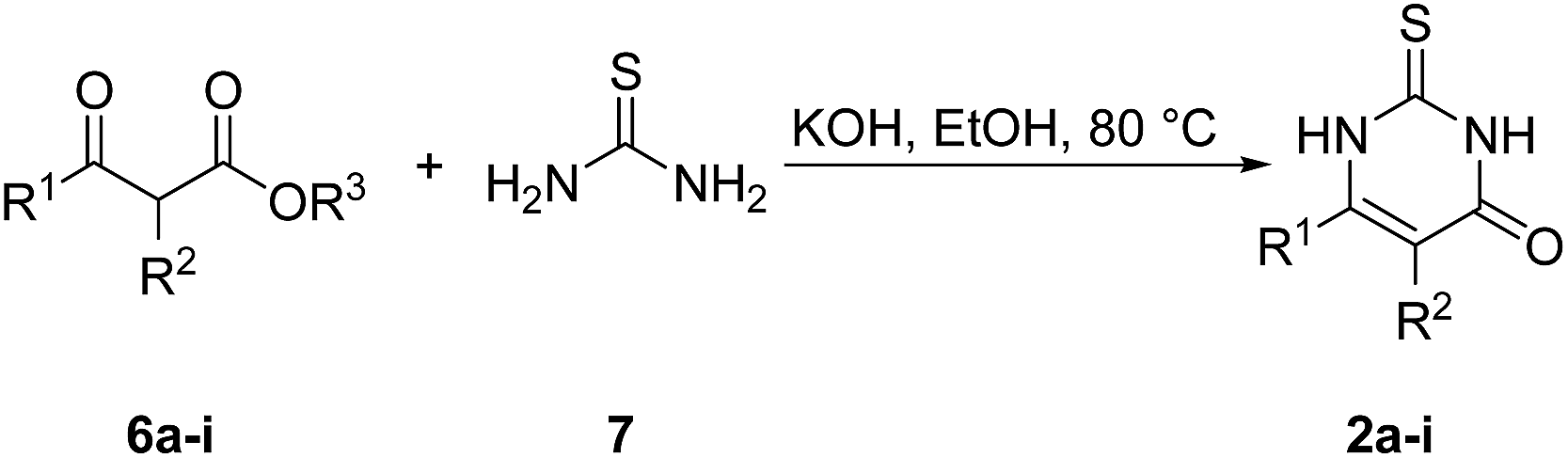 | ||
| Scheme 1 Synthesis of 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2a–i. | ||
First, the laccase-catalyzed reactions with the unsubstituted catechol (1a) were studied. In an initial experiment, 0.58 mmol (1.16 equiv.) catechol (1a) were reacted with 0.50 mmol (1 equiv.) 2,3-dihydro-6-methyl-2-thioxopyrimidin-4(1H)-one (2a) in the presence of 12 U laccase from Agaricus bisporus (1.2 U mg−1)18 as a catalyst in 20 mL of a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of phosphate buffer (pH 6) and ethanol under air (general procedure II). The reaction proceeded smoothly at room temperature and delivered 97% of a crude product (purity > 95%; 1H NMR) of a 37:63 mixture of the two regioisomers 7,8-dihydroxy-4H-2-methyl-pyrimido[2,1-b]benzothiazol-4-one (3a) and 7,8-dihydroxy-2H-4-methyl-pyrimido[2,1-b]benzothiazol-2-one (4a) after 12 h (Table 1, entry 1). A control experiment was conducted to show that the formation of the two regioisomers 3a and 4a does not proceed in the absence of the laccase (Table 1, entry 9). Subsequently, catechol (1a) was reacted with the other monosubstituted 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2b–f under similar conditions (Table 1, entries 2–6). With 2b–d, mixtures of the corresponding regioisomers 3b–d and 4b–d were obtained in excellent yields (purity of crude products >95%; 1H NMR) (Table 1, entries 2–4). When the 6-tert-butyl- and the 6-trifluoromethyl substituted 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2e, f were used as substrates, the 7,8-dihydroxy-4H-pyrimido[2,1-b]benzothiazol-4-ones 3e and 3f were isolated exclusively (Table 1, entries 5, 6). It seems that with increasing space demand of the R1 group in the 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2, the formation of regioisomers 3 is favoured over the formation of 4.
:1 mixture of phosphate buffer (pH 6) and ethanol under air (general procedure II). The reaction proceeded smoothly at room temperature and delivered 97% of a crude product (purity > 95%; 1H NMR) of a 37:63 mixture of the two regioisomers 7,8-dihydroxy-4H-2-methyl-pyrimido[2,1-b]benzothiazol-4-one (3a) and 7,8-dihydroxy-2H-4-methyl-pyrimido[2,1-b]benzothiazol-2-one (4a) after 12 h (Table 1, entry 1). A control experiment was conducted to show that the formation of the two regioisomers 3a and 4a does not proceed in the absence of the laccase (Table 1, entry 9). Subsequently, catechol (1a) was reacted with the other monosubstituted 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2b–f under similar conditions (Table 1, entries 2–6). With 2b–d, mixtures of the corresponding regioisomers 3b–d and 4b–d were obtained in excellent yields (purity of crude products >95%; 1H NMR) (Table 1, entries 2–4). When the 6-tert-butyl- and the 6-trifluoromethyl substituted 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2e, f were used as substrates, the 7,8-dihydroxy-4H-pyrimido[2,1-b]benzothiazol-4-ones 3e and 3f were isolated exclusively (Table 1, entries 5, 6). It seems that with increasing space demand of the R1 group in the 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2, the formation of regioisomers 3 is favoured over the formation of 4.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | 2 | R1 | R2 | t (h) | 3 + 4 | 3:4d |
Yield of 3 + 4e (%) |
| a General procedure II: reactions were carried out using 0.58 mmol catechol (1a), 0.50 mmol 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2a–f, laccase (A. bisporus, 10 mg, 12 U), phosphate buffer pH 6 (18 mL) and EtOH (2 mL).b General procedure III: reactions were carried out using 0.29 mmol catechol (1a), 0.25 mmol 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2g, h, laccase (A. bisporus, 10 mg, 12 U), phosphate buffer pH 6 (18 mL) and EtOH (2 mL).c Reaction was carried out using 0.58 mmol 1a, 0.50 mmol 2a, phosphate buffer pH 6 (18 mL) and EtOH (2 mL).d Ratios of regioisomers were determined by 1H NMR of the crude products.e Yields refer to crude products (purity > 95%, as determined by 1H NMR).f Crude product contain traces of unknown impurities.g No reaction. | |||||||
| 1a | a | CH3 | H | 12 | a | 37:63 |
97 |
| 2a | b | C2H5 | H | 12 | b | 54:46 |
95 |
| 3a | c | C3H7 | H | 18 | c | 44:56 |
94 |
| 4a | d | CH(CH3)2 | H | 13 | d | 76:24 |
95 |
| 5a | e | C(CH3)3 | H | 16 | e | 100:0 |
97 |
| 6a | f | CF3 | H | 16 | f | 100:0 |
78 |
| 7b | g | CH3 | CH3 | 28 | g | 36:64 |
89 |
| 8b | h | CF3 | CH3 | 17 | h | 100:0 |
95f |
| 9c | a | CH3 | H | 72 | — | — | —g |
When the disubstituted 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2g, h which carry an additional methyl group in 5-position were employed as substrates, the reactions were not complete even after 72 h. In addition to the cyclized products 3g/4g and 3h, uncyclized material could be detected by 1H NMR. Optimization experiments revealed that the reactions can be driven to completion by simply increasing the amount of laccase and solvent by a factor of 2. When 0.29 mmol catechol (1a) were reacted with 0.25 mmol of the 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2g, h in the presence of 12 U laccase from A. bisporus in 20 mL of a 9:1 mixture of phosphate buffer (pH 6) and ethanol under air (general procedure III) a mixture of regioisomers 3g and 4g in 89% yield (purity of crude product >95%; 1H NMR) and the regioisomer 3h in 95% yield were isolated (purity of crude product >95%; 1H NMR) (Table 1, entries 7, 8).
It can be expected that the reactions proceed via the laccase-catalyzed in situ oxidation of catechol (1a) to the highly reactive o-quinone (8a) (Scheme 2). The latter one is then attacked at C-4 by the S-atom of the 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2a–h to give the tautomers 9A and 10A. The intermolecular 1,4-addition is followed by a second laccase-catalyzed oxidation, namely the oxidation of the intermediates 9A and 10A to 9B and 10B, respectively. The final intramolecular 1,4-addition proceeds either by nucleophilic attack of N-3 or N-1 and delivers the corresponding regioisomeric cyclization products 3 and 4, respectively.
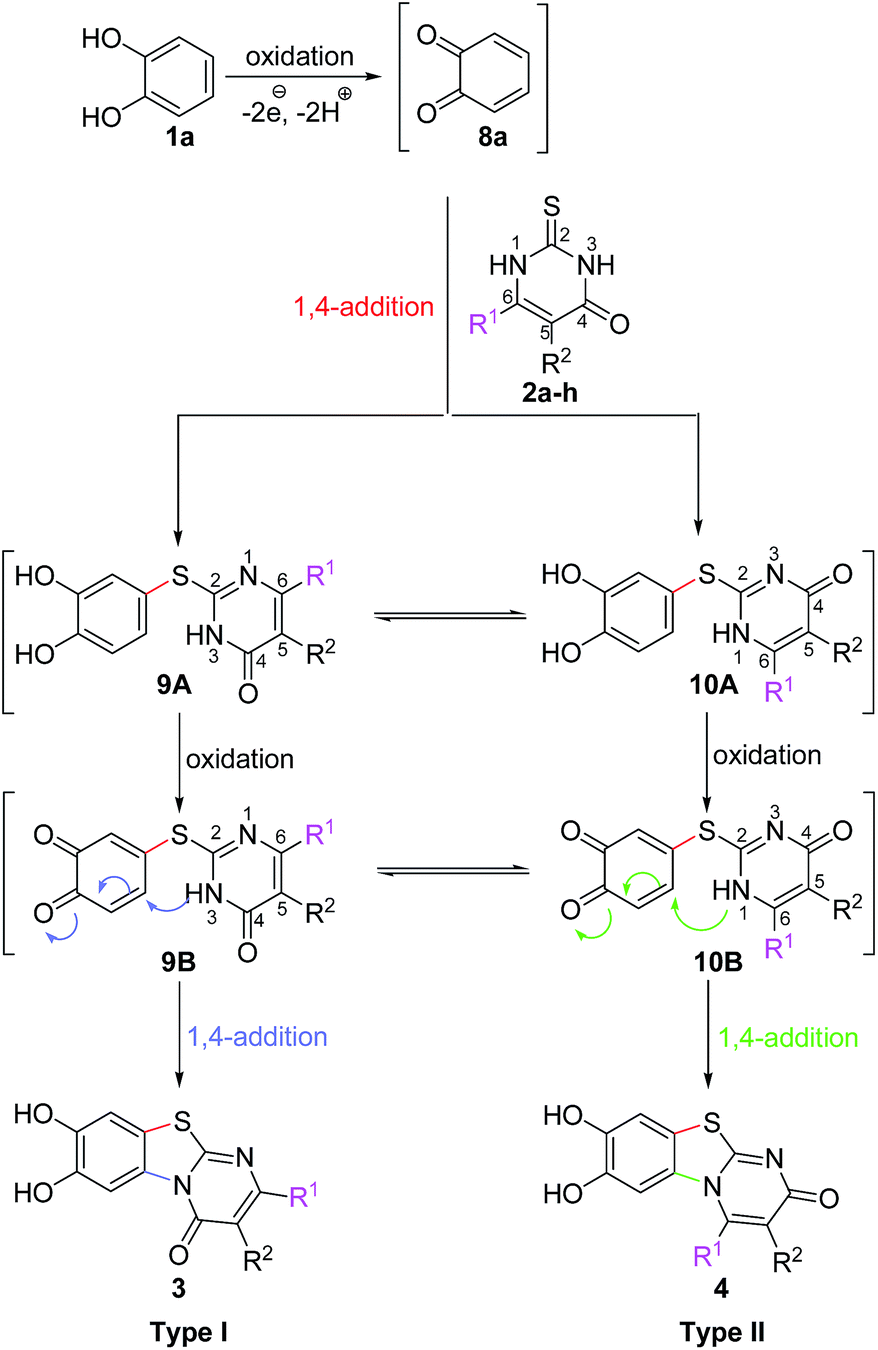 | ||
| Scheme 2 Mechanistic proposal for the laccase-catalyzed synthesis of pyrimidobenzothiazoles 3, 4. | ||
It is interesting to note that the substituent R1 at C-6 of the 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2a–h exerts a tremendous influence on the ratio of the regioisomeric 7,8-dihydroxy-4H-pyrimido[2,1-b]benzothiazol-4-ones 3 and 7,8-dihydroxy-2H-pyrimido[2,1-b]benzothiazol-2-ones 4. With unbranched alkyl groups at C-6, the formation of 7,8-dihydroxy-2H-pyrimido[2,1-b]benzothiazol-2-ones 4 is either favoured or roughly equal amounts of the two isomers 3 and 4 are observed (Table 1, entries 1–3, 7). With branched alkyl groups (i-propyl- or tert-butyl group) or a trifluoromethyl group at C-6, the 7,8-dihydroxy-4H-pyrimido[2,1-b]benzothiazol-4-ones 3 are strongly favoured (Table 1, entries 4–6, 8). These findings are in good agreement with the assumption that the intramolecular 1,4-addition via N-1 in 10B to regioisomers 4 (Scheme 2) is hindered by sterically demanding as well as strongly electronegative substituents.
The reactions between 1a and 2a–g delivered only either one major product (3e, 3f) or two major products, consisting of exclusively mixtures of the regioisomeric products 3a–d,g and 4a–d,g in remarkably high yields. No other products could be detected by 1H NMR. Only the reaction between 1a and 2h delivered in addition to 3h traces of an additional product of unknown structure (Table 1, entry 8).
Subsequently, the laccase-catalyzed reactions of 4-substituted catechols 1b, c with 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2a, c–e, g were studied. For this purpose, 4-methylcatechol (1b) and 4-ethylcatechol (1c) were reacted with selected 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2a, c–e, g under the conditions developed for the reactions of unsubstituted catechol (1a) (general procedure II). In all cases, the corresponding thioethers were formed exclusively with yields ranging between 76 and 92% (purity of crude products >95%; 1H NMR) (Table 2). Obviously, only the S atoms of the 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2 act as nucleophiles in the intermolecular 1,4-additions. This can be attributed to the higher nucleophilicity of 2-S in comparison to N-1 and N-3 (Scheme 3).
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entrya | 1 | R | 2 | R1 | R2 | Time (h) | 5 | Yieldb (%) |
| a General procedure II: reactions were carried out using 0.58 mmol 4-substituted catechols 1b, c, 0.50 mmol 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2a, c–e, g, laccase (A. bisporus, 10 mg, 12 U), phosphate buffer pH 6 (18 mL) and EtOH (2 mL).b Yields refer to crude products (purity > 95%, as determined by 1H NMR). | ||||||||
| 1 | b | CH3 | a | CH3 | H | 12 | a | 82 |
| 2 | b | CH3 | c | C3H7 | H | 13 | b | 88 |
| 3 | b | CH3 | d | CH(CH3)2 | H | 16 | c | 92 |
| 4 | b | CH3 | e | C(CH3)3 | H | 17 | d | 90 |
| 5 | b | CH3 | g | CH3 | CH3 | 20 | e | 86 |
| 6 | c | C2H5 | a | CH3 | H | 14 | f | 90 |
| 7 | c | C2H5 | d | CH(CH3)2 | H | 12 | g | 76 |
| 8 | c | C2H5 | e | C(CH3)3 | H | 16 | h | 81 |
| 9 | c | C2H5 | g | CH3 | CH3 | 16 | i | 91 |
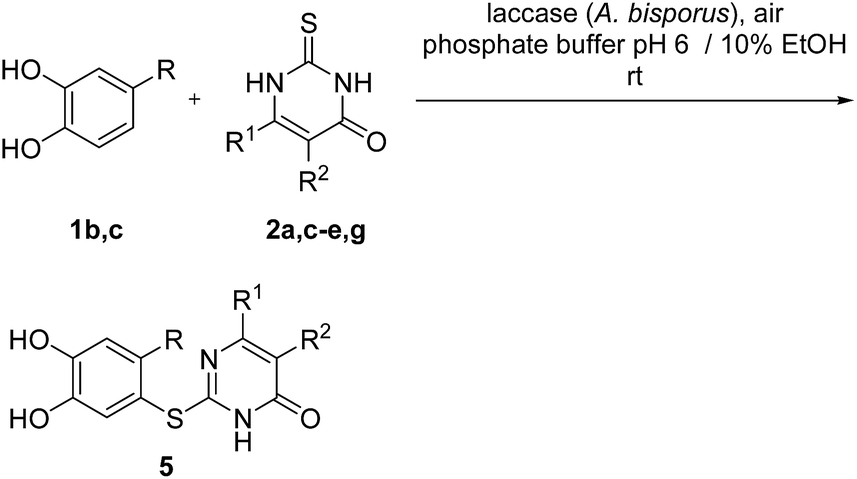
 | ||
| Scheme 3 Mechanistic proposal for the laccase-catalyzed synthesis of catechol thioethers 5a–i. | ||
Finally, the laccase-catalyzed reactions between the 4-substituted catechols 1b, c and 2,3,6,7-tetrahydro-2-thioxo-1H-cyclopenta[d]pyrimidin-4(5H)-one (2i) were studied (general procedure II) to give the corresponding catechol thioethers 5j, k in high yields (Table 3).
|
||||
|---|---|---|---|---|
| Entrya | 1 | R | 5 | Yieldb (%) |
| a General procedure II: reactions were carried out using 0.58 mmol 4-substituted catechols 1b, c, 0.50 mmol 2,3,6,7-tetrahydro-2-thioxo-1H-cyclopenta[d]pyrimidin-4(5H)-one (2i), laccase (A. bisporus, 10 mg, 12 U), phosphate buffer pH 6 (18 mL) and EtOH (2 mL).b Yields refer to crude products (purity > 95%, as determined by 1H NMR). | ||||
| 1 | b | CH3 | j | 90 |
| 2 | c | C2H5 | k | 76 |
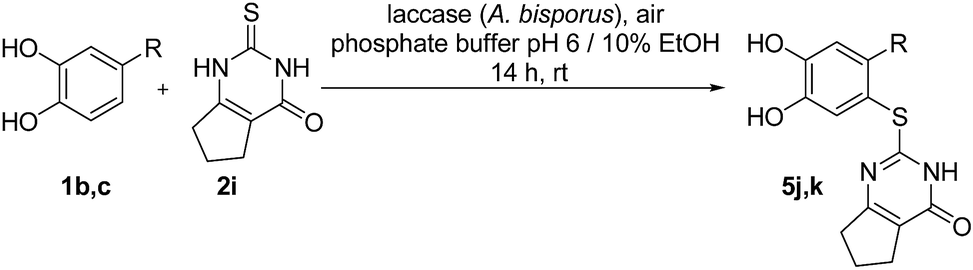
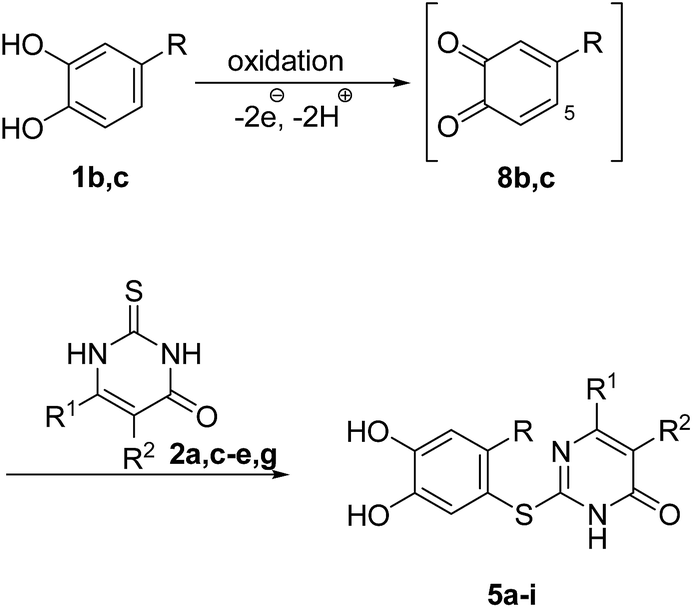
It can be taken for granted that the reactions start with the laccase-catalyzed oxidation of the 4-substituted catechols 1b, c to the corresponding o-benzoquinones 8b, c. This is followed by intermolecular 1,4-addition of the 2,3-dihydro-2-thioxopyrimidin-4(1H)-ones 2 via the S atom at C-5 to afford the corresponding catechol thioethers 5a–i as sole products. Due to the presence of a methyl or an ethyl substituent at C-4 of the catechol, the intramolecular 1,4-addition occurs exclusively at C-5 (Scheme 3).
It should be highlighted that the reactions presented are highly efficient and deliver the products 3, 4 and 5 in remarkably high yields and in a highly chemoselective manner. The reactions proceed without the formation of any relevant amounts of byproducts arising from competing reactions, such as (a) the homocoupling of the catechols 1; i.e., the reaction of the o-quinone intermediates 8 with the corresponding parent catechols 1, which is followed by formation of the benzofurans or (b) the formation of disulfides. We assume that this favourable outcome is the result of a combination of several factors. Among them are (a) the high nucleophilicity of the S-nucleophilic substrates 2, (b) the use of a laccase which is particularly suitable for this type of reactions and (c) the careful choice of reaction conditions, such as pH, reaction temperature and substrate concentrations. This view is supported by previous work done in our laboratory.16k The reactions between catechols and 1,3-dicarbonyls prove that the selection of laccase from A. bisporus as the catalyst as well as the choice of suitable reaction conditions exert a tremendous impact on product yields.
Structure elucidation
Structures of all pyrimidobenzothiazole regioisomers 3, 4 as well as catechol thioethers 5 were unambiguously elucidated by mass spectrometry and NMR spectroscopy including 2D NMR for the full assignment of the 1H and 13C chemical shifts. Analysis of the 1H NMR spectrum of the crude products obtained from the reaction between catechol (1a) and 2-thioxopyrimidin-4-ones 2a–h showed the appearance of either a mixture of 2 regioisomers 3 (type I) and 4 (type II) or a single regioisomer 3. 2D ROESY as well as super long range gHMBC experiments were carried out to differentiate between the 2 regioisomers. Taking the mixture of 3a and 4a as an example, in 4a a strong ROESY correlation between the methyl group at C-4 (δH 2.73 ppm) to both the aromatic proton 6-H (δH 7.52 ppm) and the aromatic proton 3-H (δH 6.07 ppm) confirms the type II regioisomer. However, in 3a only a single ROESY correlation between the methyl group at C-2 (δH 2.26 ppm) and the aromatic proton at 3-H (δH 6.17 ppm) can be observed (Fig. 4). 4J HMBC correlations (as observed in the super long range gHMBC spectrum) between 6-H (δH 8.44 ppm) and C-4 (δC 159.97 ppm) along with 9-H (δH 7.30 ppm) and C-10a (δC 161.71 ppm) established a type I regioisomer. Evaluation of the experimental 1H–13C long-range coupling constants (PIP-HSQMBC), for example in 4a, between 9-H (δH 7.25 ppm) to C-5a (3J = 9.5 Hz) and C-7 (3J = 7.6 Hz) allows the successful assignment of the quaternary carbons C-5a at δC 128.82 ppm and C-7 at δC 145.25 ppm. Similarly, two strong correlations can be seen from H-6 (δH 7.52 ppm) to C-9a (3J = 8.4 Hz) and C-8 (3J = 6.2 Hz), to be assigned at δC 112.65 ppm and δC 144.68 ppm, respectively. The quaternary carbons C-2, C-4, C-10a of ring C were assigned by standard gHMBC and super long range gHMBC at δC 166.32, 148.31, 164.60 ppm, respectively (Fig. 4).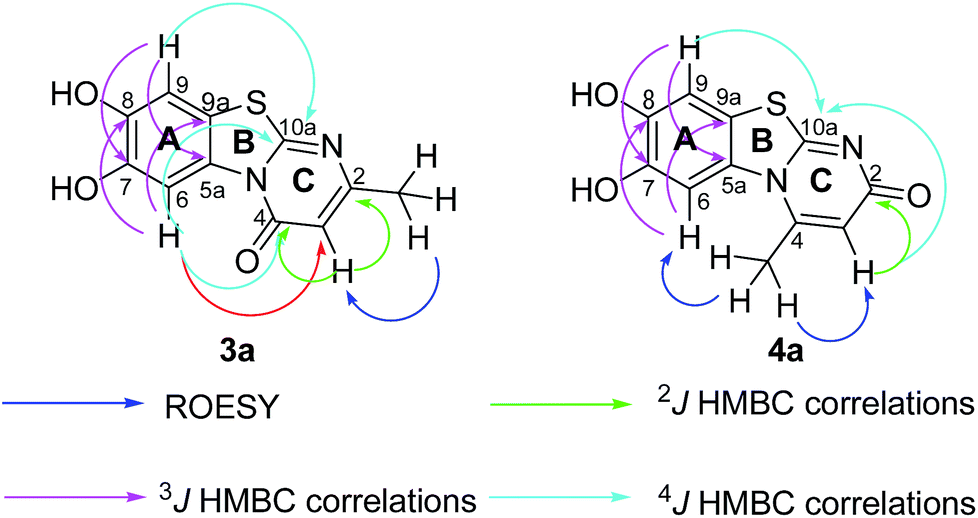 | ||
| Fig. 4 Important ROESY, 2J, 3J, and 4J HMBC correlations of 3a and 4a. | ||
In the reactions between 4-substituted catechols 1b, c and 2-thioxopyrimidin-4-ones 2a, c–e, g, i single products were formed exclusively. The products 5a–i consist of the 2 ring systems A and B. Taking 5d as an example, the complete assignment of ring A was carried out by PIP-HSQMBC 1H–13C correlations. The difficulty in assigning C-2, C-4, and C-6 was solved by 1H–13C super long gHMBC along with standard gHMBC correlations between 6′-H at δH 7.66 ppm and C-2 at δC 165.32 ppm (4J) as well as between 2′-CH3 at δH 2.42 ppm and C-2 at δC 165.32 ppm (5J), between 2′′-H at δH 1.14 and C-6 at δC 176.73 ppm (3J) and finally between 5-H at δH 6.43 ppm and C-4 at δC 167.61 (2J) as well as C-6 at δC 176.73 ppm (2J) (Fig. 5).
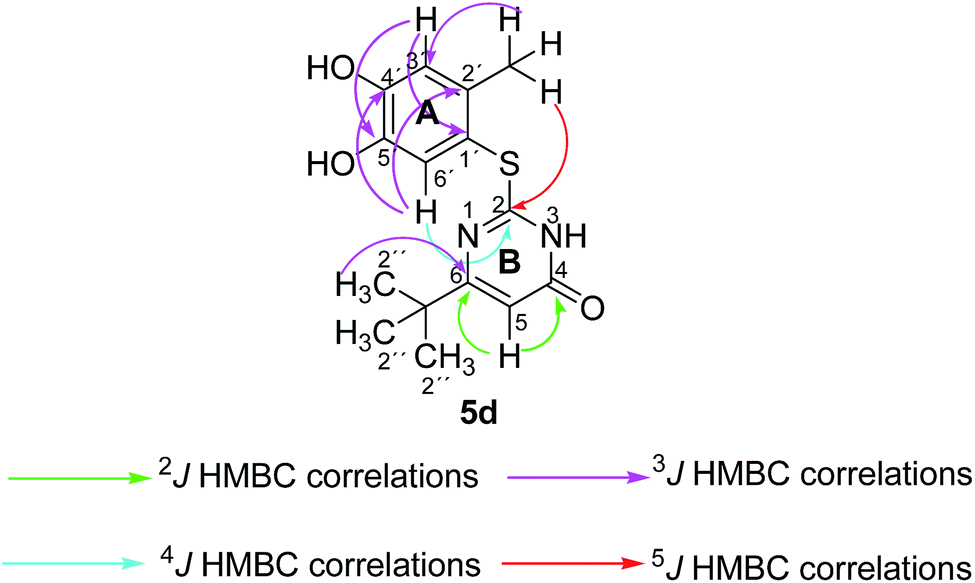 | ||
| Fig. 5 Important 2J, 3J, 4J and 5J HMBC correlations of 5d. | ||
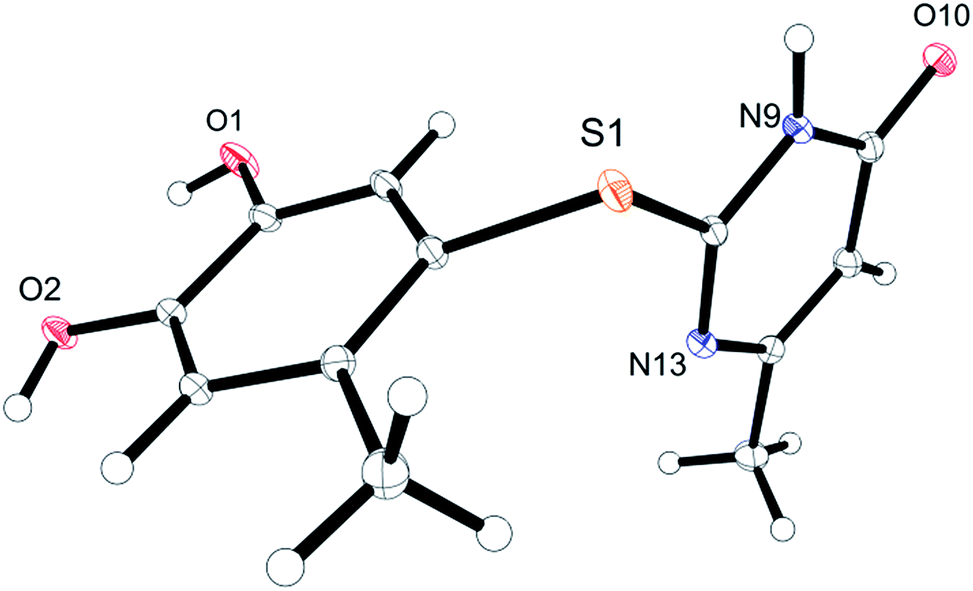
Unequivocal evidence for the structures of 3f and 5a was provided by X-ray crystal structure analysis.19 The molecular structures of 3f and 5a are depicted in Fig. 6 and 7.
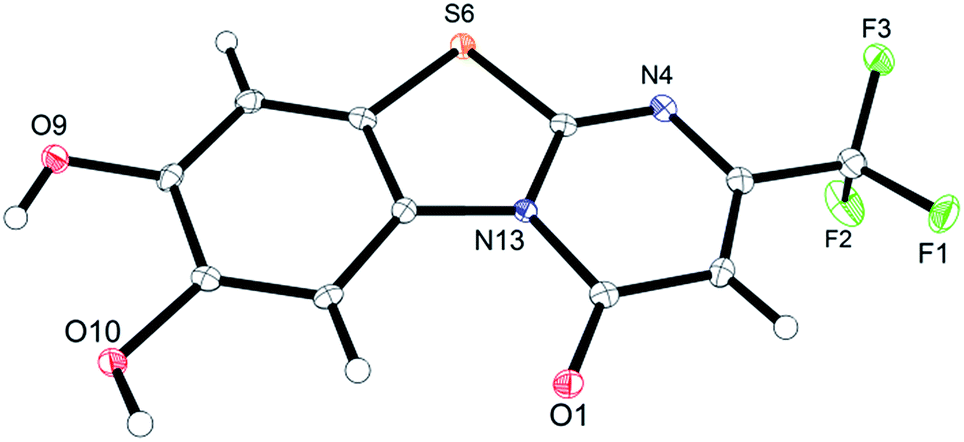 | ||
| Fig. 6 Molecular structure of 7,8-dihydroxy-4H-2-trifluoromethyl-pyrimido[2,1-b]benzothiazol-4-one (3f), derived from X-ray crystal structure analysis. | ||
 | ||
| Fig. 7 Molecular structure of 2-(4,5-dihydroxy-2-methylphenylthio)-6-methylpyrimidin-4(3H)-one (5a), derived from X-ray crystal structure analysis. | ||
Greenness of the reactions
The newly developed methods for the synthesis of pyrimidobenzothiazoles 3, 4 and catechol thioethers 5 address many of the principles of green chemistry.6,20 The reactions are enzyme-catalyzed transformations using completely safe and non-toxic aerial oxygen as the sole oxidant. The transformations deliver the pyrimidobenzothiazoles 3, 4 and the catechol thioethers 5 exclusively and with high yields. The only byproduct formed is water which stems from the reduction of oxygen. The laccase-catalyzed domino reactions presented combine several individual reactions in multistep processes. This allows the reduction of the amount of waste formed during reaction and work up and complies well with the first principle of green chemistry. In addition, environmentally benign reaction conditions have been developed. Taking the preparation of 3e as a typical example for the synthesis of pyrimidobenzothiazoles 3, the E-factor (kg waste per kg product)21 of the process is 4.57 kg kg−1. The atom economy5 of this transformation amounts to 94%. The reaction was carried out in a mixture of phosphate buffer (pH 6.0) and 10 vol% ethanol, which are completely safe and environmentally acceptable solvents.22 The method highlights the use of enzymes in catalytic rather than stoichiometric amounts. The turnover number of the laccase in this process is high, for the synthesis of 3e TON amounts to 4656; the turnover frequency of this transformation is good; in the example discussed TOF amounts to 291 h−1. Taking the preparation of 5j as a representative example for the synthesis of thioethers 5, the E-factor of this process was calculated to be 5.00 kg kg−1. The atom economy of this process, calculated as 94%, is also very good. The turnover number and the turnover frequency of the laccase for the synthesis of 5j are high. They amount to 4320 and 308.57 h−1, respectively.In vitro cytotoxic activity
Selected compounds and doxorubicin (positive control) were evaluated for their in vitro cytotoxic activity against HepG2 cancer cell line using SRB assay.23 The IC50 values of the tested compounds are summarized in Table 4. All compounds tested exhibited cytotoxic activities, but all of them were less potent than doxorubicin.| Entry | Product | IC50a,b (μM) |
|---|---|---|
| a IC50 are the mean of 2–5 independent experiments ± SE.b DMSO alone (2% final concentration) had no effect on the cell viability. | ||
| 1 | 3d | 23.28 ± 2.16 |
| 2 | 3e | 12.70 ± 0.53 |
| 3 | 3f | 12.01 ± 1.46 |
| 4 | 5a | >40 |
| 5 | 5b | 21.25 ± 0.46 |
| 6 | 5c | 7.77 ± 0.30 |
| 7 | 5d | 14.30 ± 0.11 |
| 8 | 5e | 30.57 ± 1.05 |
| 9 | 5f | 27.82 ± 2.36 |
| 10 | 5g | 2.74 ± 0.29 |
| 11 | 5h | 14.92 ± 1.16 |
| 12 | 5i | 31.98 ± 1.43 |
| 13 | 5j | 40.48 ± 3.52 |
| 14 | 5k | 31.34 ± 3.57 |
| 15 | Doxorubicin | 0.28 ± 0.04 |
In the catechol thioether series 5a–k, the substituents on C-5, C-6 and C-2′ have a great influence on the cytotoxic activity. In compounds 5a–e, it is assumed that increasing the chain length from methyl in 5a (IC50 > 40 μM, Table 4, entry 4) to n-propyl in 5b (IC50 = 21.25 μM, Table 4, entry 5) or introduction of a methyl group in the 5-position in 5e (IC50 = 30.57 μM, Table 4, entry 8) increases the potency. Moreover, the presence of a branched isopropyl group at C-6 in 5c results in a great increase in potency (IC50 = 7.77 μM, Table 4, entry 6) compared to 5a (IC50 > 40 μM, Table 4, entry 4). However, the presence of a bulky tert-butyl group in 5d results in a decrease of activity (IC50 = 14.30 μM, Table 4, entry 7) in comparison to 5c. In the 5f–i series, where an ethyl group is present at C-2′, the introduction of an isopropyl group in 5g increases the activity (IC50 = 2.74 μM, Table 4, entry 10) and the presence of a tert-butyl group in 5h decreases the potency (IC50 = 14.92 μM, Table 4, entry 11) in comparison to 5g. Introduction of a methyl group at C-5 of 5i results in a slight decrease of the cytotoxic activity (IC50 = 31.98 μM, Table 4, entry 12) in comparison to 5f (IC50 = 27.82 μM, Table 4, entry 9). The presence of a bicyclic system in 5j and 5k has no favourable effect on the cytotoxic activity compared to 5a and 5f. Comparison of the IC50 values of the 5a–e, j series with the 5f–i, k series reveals that the most potent compounds are 5c and 5g which have an isopropyl group at C-6. The presence of an ethyl group at C-2′ of 5g increases the cytotoxic potency in comparison to 5c. In the pyrimidobenzothiazole derivatives 3d–f, it was found that replacing the isopropyl group at C-2 of 3d (IC50 = 23.28 μM, Table 4, entry 1) with a tert-butyl group in 3e (IC50 = 12.70 μM, Table 4, entry 2) or a trifluoromethyl group in 3f (IC50 = 12.01 μM, Table 4, entry 3) results in an increase of the antiproliferative activity. The presented study provides a novel class of compounds, which will be further optimized to increase their potency. The mechanism of their cytotoxic activity is still under study.
Experimental
Chemistry
Synthesis and analytical data of 2,3-dihydro-6-methyl-2-thioxopyrimidin-4(1H)-one (2a).
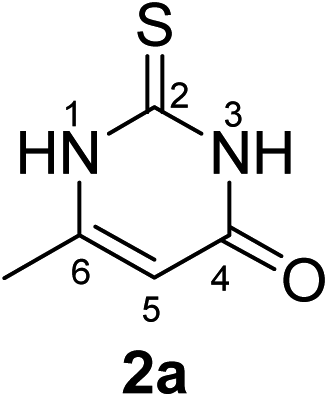
According to general procedure I, ethyl acetoacetate (6a) (6.50 g, 50 mmol), thiourea (7) (3.81 g, 50 mmol), KOH (2.81 g, 50 mmol), ethanol (80 mL) were reacted. Work up gave 2,3-dihydro-6-methyl-2-thioxopyrimidin-4(1H)-one (2a) as a white powder (5.00 g, 70%), mp > 300 °C (lit.24a > 300 °C); Rf = 0.54 (CH2Cl2/EtOAc = 2:1); δH (300 MHz; DMSO-d6) 2.05 (3H, s, 6-CH3), 5.66 (1H, s, 5-H), 12.22 (2H, br, 1-H and 3-H).
Synthesis and analytical data of 6-ethyl-2,3-dihydro-2-thioxopyrimidin-4(1H)-one (2b).
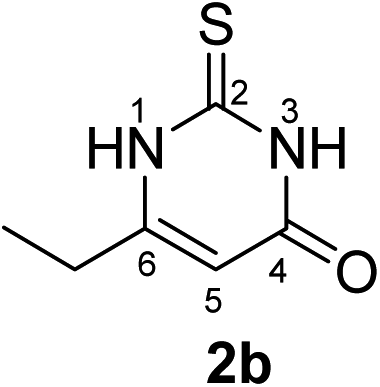
According to general procedure I, ethyl propionylacetate (6b) (1.44 g, 10 mmol), thiourea (7) (0.76 g, 10 mmol), KOH (0.56 g, 10 mmol), ethanol (20 mL) were reacted. Work up gave 6-ethyl-2,3-dihydro-2-thioxopyrimidin-4(1H)-one (2b) as a white powder (0.78 g, 50%), mp 227–229 °C (lit.24a 228.5–230.5 °C); Rf = 0.44 (CH2Cl2/EtOAc = 2:1); δH (300 MHz; DMSO-d6) 1.08 (3H, t, 3J = 7.5 Hz, CH3), 2.35 (2H, q, 3J = 7.5 Hz, CH2), 5.66 (1H, s, 5-H) and 12.24 (2H, s, 1-H and 3-H); δC (75 MHz; DMSO-d6) 11.60, 24.73, 102.04, 158.21, 161.19, 175.97.
Synthesis and analytical data of 2,3-dihydro-6-propyl-2-thioxopyrimidin-4(1H)-one (2c).
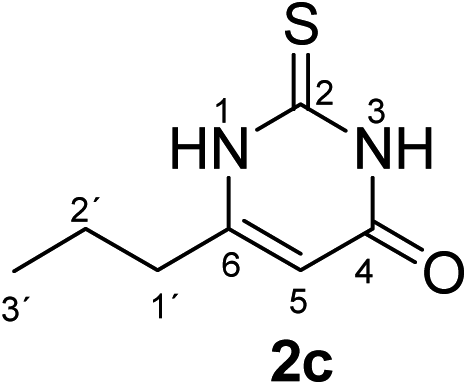
According to general procedure I, ethyl butyrylacetate (6c) (3.16 g, 20 mmol), thiourea (7) (1.52 g, 20 mmol), KOH (1.12 g, 20 mmol), ethanol (20 mL) were reacted. Work up gave 2,3-dihydro-6-propyl-2-thioxopyrimidin-4(1H)-one (2c) as a white powder (1.40 g, 41%), mp 216–218 °C (lit.24a 218–220 °C); Rf = 0.52 (CH2Cl2/EtOAc = 2:1); δH (300 MHz; DMSO-d6) 0.86 (3H, t, 3J = 7.4 Hz, 3′-H), 1.53 (2H, sex, 3J = 7.4 Hz, 2′-H), 2.30 (2H, t, 3J = 7.7 Hz, 1′-H), 5.65 (1H, s, 5-H) and 12.20 (2H, s, 1-H and 3-H); δC (75 MHz; DMSO-d6) 13.20, 20.50, 33.35, 102.90, 156.97, 161.17, 176.08.
Synthesis and analytical data of 2,3-dihydro-6-isopropyl-2-thioxopyrimidin-4(1H)-one (2d).
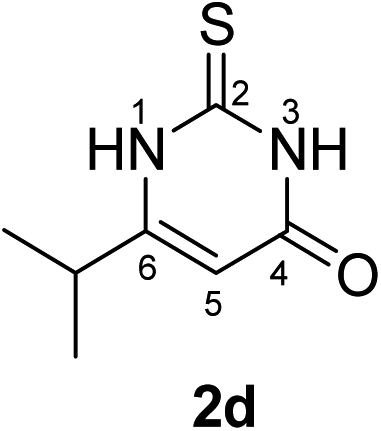
According to general procedure I, ethyl isobutyrylacetate (6d) (1.58 g, 10 mmol), thiourea (7) (0.76 g, 10 mmol), KOH (0.56 g, 10 mmol), ethanol (20 mL) were reacted. Work up gave 2,3-dihydro-6-isopropyl-2-thioxopyrimidin-4(1H)-one (2d) as a white powder (0.60 g, 35%), mp 176–178 °C (lit.24a 179–180 °C); Rf = 0.50 (CH2Cl2/EtOAc = 2:1); δH (300 MHz; DMSO-d6) 1.11 (6H, d, 3J = 6.9 Hz, CH(C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 3)2), 2.65 (1H, sep, 3J = 6.9 Hz, C(CH3)2), 5.65 (1H, s, 5-H) and 12.20 (2H, br s, 1-H and 3-H); δC (75 MHz; DMSO-d6) 20.39, 30.33, 100.33, 161.33, 162.22, 176.08.
3)2), 2.65 (1H, sep, 3J = 6.9 Hz, C(CH3)2), 5.65 (1H, s, 5-H) and 12.20 (2H, br s, 1-H and 3-H); δC (75 MHz; DMSO-d6) 20.39, 30.33, 100.33, 161.33, 162.22, 176.08.
Synthesis and analytical data of 6-tert-butyl-2,3-dihydro-2-thioxopyrimidin-4(1H)-one (2e).

According to general procedure I, ethyl 4,4-dimethyl-3-oxovalerate (6e) (1.72 g, 10 mmol), thiourea (7) (0.76 g, 10 mmol), KOH (0.56 g, 10 mmol), ethanol (20 mL) were reacted. Work up gave 6-tert-butyl-2,3-dihydro-2-thioxopyrimidin-4(1H)-one (2e) as a white powder (0.50 g, 27%), mp 179–181 °C (lit.24a 178–180 °C); Rf = 0.63 (CH2Cl2/EtOAc = 2:1); δH (300 MHz; DMSO-d6) 1.21 (9H, s, C(C3)3), 5.63 (1H, s, 5-H), 11.79 and 12.34 (2H, s, 1-H and 3-H); δC (75 MHz; DMSO-d6) 27.30, 34.49, 100.50, 161.16, 163.36, 176.49.
Synthesis and analytical data of 6-trifluoromethyl-2,3-dihydro-2-thioxopyrimidin-4(1H)-one (2f).
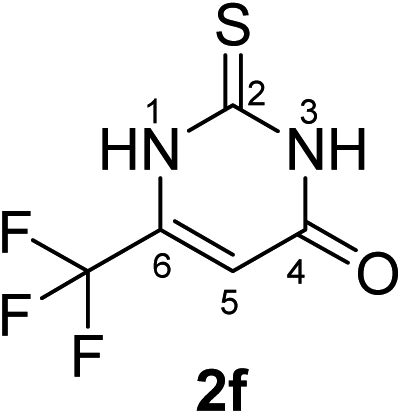
According to general procedure I, ethyl-4,4,4-trifluoroacetoacetate (6f) (1.84 g, 10 mmol), thiourea (7) (0.76 g, 10 mmol), KOH (0.56 g, 10 mmol), ethanol (20 mL) were reacted. Work up gave 6-trifluoromethyl-2,3-dihydro-2-thioxopyrimidin-4(1H)-one (2f) as a white powder (0.69 g, 35%), mp 246–248 °C (lit.24b 247–249 °C); Rf = 0.50 (CH2Cl2/EtOAc = 2:1); δH (300 MHz; DMSO-d6) 6.40 (1H, s, 5-H), 12.83 (1H, s, 1-H or 3-H) and 13.50 (1H, br, 1-H or 3-H); δC (75 MHz; DMSO-d6) 105.14 (q, 3JC,F = 3.9 Hz, C-5), 118.83 (q, 1JC,F = 273.7 Hz, CF3), 140.56 (q, 2JC,F = 36.1 Hz, C-6), 159.84, 176.82.
Synthesis and analytical data of 2,3-dihydro-5,6-dimethyl-2-thioxopyrimidin-4(1H)-one (2g).
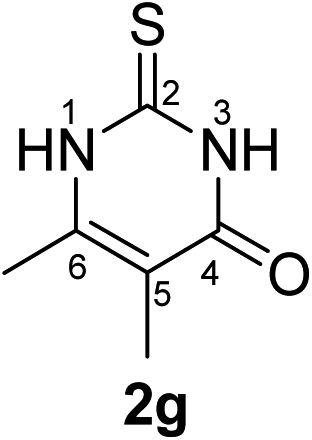
According to general procedure I, ethyl 2-methylacetoacetate (6g) (7.21 g, 50 mmol), thiourea (7) (3.81 g, 50 mmol), KOH (2.81 g, 50 mmol), ethanol (60 mL) were reacted. Work up gave 2,3-dihydro-5,6-dimethyl-2-thioxopyrimidin-4(1H)-one (2g) as a white powder (3.80 g, 49%), mp 283–285 °C (lit.24a 283–285 °C); Rf = 0.48 (CH2Cl2/EtOAc = 2:1); δH (300 MHz; DMSO-d6) 1.74 (3H, s, CH3), 2.09 (3H, s, CH3) and 12.19 (2H, br, 1-H and 3-H).
Synthesis and analytical data of 6-trifluoromethyl-2,3-dihydro-5-methyl-2-thioxopyrimidin-4(1H)-one (2h).
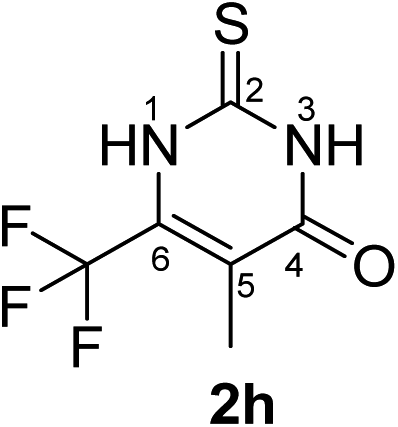
According to general procedure I, ethyl-4,4,4-trifluoro-2-methylacetoacetate (6h) (1.98 g, 10 mmol), thiourea (7) (0.76 g, 10 mmol), KOH (0.56 g, 10 mmol), ethanol (15 mL) were reacted. Work up gave 6-trifluoromethyl-2,3-dihydro-5-methyl-2-thioxopyrimidin-4(1H)-one (2h) as a white powder (0.60 g, 29%), mp 244–246 °C; Rf = 0.66 (CH2Cl2/EtOAc = 2:1); ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) max (atr)/cm−1 3150 (NH), 2874 (CH), 1664 (C
max (atr)/cm−1 3150 (NH), 2874 (CH), 1664 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1570 and 1220; δH (300 MHz; DMSO-d6) 1.93 (3H, q, 5JH,F = 3.3 Hz, 5-CH3) and 12.85 (2H, br, 1-H and 3-H); δC (75 MHz; DMSO-d6) 9.48 (q, 4JC,F = 2.5 Hz, 5-CH3), 115.57 (br, C-5), 119.89 (q, 1JC,F = 275.7 Hz, CF3), 135.16 (q, 2JC,F = 34.6 Hz, C-6), 161.18, 174.55.
O), 1570 and 1220; δH (300 MHz; DMSO-d6) 1.93 (3H, q, 5JH,F = 3.3 Hz, 5-CH3) and 12.85 (2H, br, 1-H and 3-H); δC (75 MHz; DMSO-d6) 9.48 (q, 4JC,F = 2.5 Hz, 5-CH3), 115.57 (br, C-5), 119.89 (q, 1JC,F = 275.7 Hz, CF3), 135.16 (q, 2JC,F = 34.6 Hz, C-6), 161.18, 174.55.
Synthesis and analytical data of 2,3,6,7-tetrahydro-2-thioxo-1H-cyclopenta[d]pyrimidin-4(5H)-one (2i).
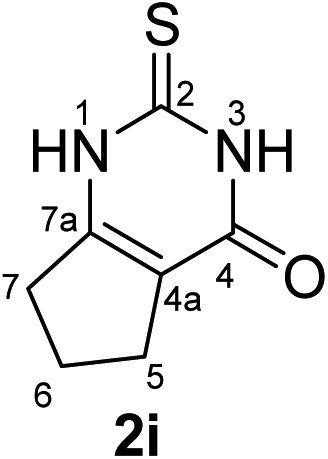
According to general procedure I, ethyl 2-oxocyclopentanecarboxylate (6i) (4.69 g, 30 mmol), thiourea (7) (2.28 g, 30 mmol), KOH (1.68 g, 30 mmol), ethanol (30 mL) were reacted. Work up gave 2,3,6,7-tetrahydro-2-thioxo-1H-cyclopenta[d]pyrimidin-4(5H)-one (2i) as a white powder (1.20 g, 24%), mp > 300 °C (lit.24a 336–337 °C); Rf = 0.38 (CH2Cl2/EtOAc = 2:1); δH (300 MHz; DMSO-d6) 1.94 (2H, quin, 3J = 7.3 Hz, 6-H), 2.47 (2H, ov. t like, 3J = 7.3 Hz, 7-H), 2.67 (2H, t, 3J = 7.6 Hz, 5-H), 12.17 and 12.18 (2H, br, 1-H and 3-H); δC (75 MHz; DMSO-d6) 20.78, 26.60, 31.00, 115.52, 156.51, 159.52, 175.57.
:1 to CH2Cl2/EtOAc/MeOH = 1:1:0.1) of the crude products.:1 to CH2Cl2/EtOAc/MeOH = 1:1:0.1) of the crude products.
Synthesis and analytical data of 7,8-dihydroxy-4H-2-methyl-pyrimido[2,1-b]benzothiazol-4-one (3a) and 7,8-dihydroxy-2H-4-methyl-pyrimido[2,1-b]benzothiazol-2-one (4a).

According to general procedure II, catechol (1a) (64 mg, 0.58 mmol), 2,3-dihydro-6-methyl-2-thioxopyrimidin-4(1H)-one (2a) (71 mg, 0.50 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 12 h. Workup gave a mixture of 7,8-dihydroxy-4H-2-methyl-pyrimido[2,1-b]benzothiazol-4-one (3a) and 7,8-dihydroxy-2H-4-methyl-pyrimido[2,1-b]benzothiazol-2-one (4a) as a brown powder (120 mg, 97%), mp > 300 °C; Rf = 0.27 (CH2Cl2/EtOAc/MeOH = 2:2:0.1); max (atr)/cm−1 3406 (OH), 3023 (CH), 1628 (CO), 1510 (CN) and 1185; δH (500 MHz; DMSO-d6) of 3a 2.26 (3H, s, 2-CH3), 6.17 (1H, s, 3-H), 7.30 (1H, s, 9-H), 8.44 (1H, s, 6-H) and 9.66 (2H, ov. br, 7,8-OH); δH (500 MHz; DMSO-d6) of 4a 2.73 (3H, s, 4-CH3), 6.07 (1H, s, 3-H), 7.25 (1H, s, 9-H), 7.52 (1H, s, 6-H) and 9.66 (2H, ov. br, 7,8-OH); δC (125 MHz; DMSO-d6) of 3a 23.10 (2-CH3), 105.68 (C-3), 106.38 (C-6), 108.29 (C-9), 113.21 (C-9a), 128.12 (C-5a), 144.96 (C-7), 145.65 (C-8), 159.97 (C-4), 161.71 (C-10a) and 161.99 (C-2); δC (125 MHz; DMSO-d6) of 4a 21.15 (4-CH3), 104.76 (C-6), 108.97 (C-9), 110.80 (C-3), 112.65 (C-9a), 128.82 (C-5a), 144.68 (C-8), 145.25 (C-7), 148.31 (C-4), 164.60 (C-10a) and 166.32 (C-2); MS (EI-70 eV) m/z 248 (M+, 100%), 220 (35) and 181 (15); HRMS calcd for C11H8N2O3S (248.0256), found 248.0253.
Synthesis and analytical data of 7,8-dihydroxy-4H-2-ethyl-pyrimido[2,1-b]benzothiazol-4-one (3b) and 7,8-dihydroxy-2H-4-ethyl-pyrimido[2,1-b]benzothiazol-2-one (4b).
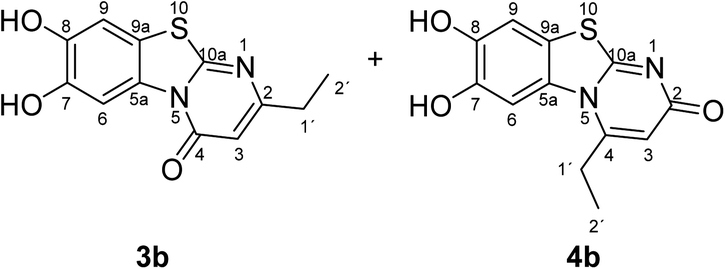
According to general procedure II, catechol (1a) (64 mg, 0.58 mmol), 6-ethyl-2,3-dihydro-2-thioxopyrimidin-4(1H)-one (2b) (78 mg, 0.50 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 12 h. Workup gave a mixture of 7,8-dihydroxy-4H-2-ethyl-pyrimido[2,1-b]benzothiazol-4-one (3b) and 7,8-dihydroxy-2H-4-ethyl-pyrimido[2,1-b]benzothiazol-2-one (4b) as a brown powder (124 mg, 95%), mp 298–300 °C; Rf = 0.27 (CH2Cl2/EtOAc = 2:1); max (atr)/cm−1 3400 (OH), 2979 (CH), 1625 (CO), 1512 and 1299; δH (500 MHz; DMSO-d6) of 3b 1.17 (3H, t, 3J = 7.5 Hz, 2′-H), 2.54 (2H, q, 3J = 7.5 Hz, 1′-H), 6.16 (1H, s, 3-H), 7.30 (1H, s, 9-H), 8.45 (1H, s, 6-H) and 9.65 (2H, ov. br, 7,8-OH); δH (500 MHz; DMSO-d6) of 4b 1.27 (3H, t, 3J = 7.1 Hz, 2′-H), 3.09 (2H, q, 3J = 7.1 Hz, 1′-H), 6.02 (1H, s, 3-H), 7.25 (1H, s, 9-H), 7.49 (1H, s, 6-H) and 9.65 (2H, br, 7,8-OH); δC (125 MHz; DMSO-d6) of 3b 12.25 (C-2′), 29.56 (C-1′), 104.36 (C-3), 106.37 (C-6), 108.30 (C-9), 113.24 (C-9a), 128.09 (C-5a), 144.94 (C-7), 145.65 (C-8), 160.25 (C-4), 161.82 (C-10a), 166.68 (C-2); δC (125 MHz; DMSO-d6) of 4b 11.57 (C-2′), 25.84 (C-1′), 104.93 (C-6), 108.52 (C-3), 108.96 (C-9), 112.73 (C-9a), 128.40 (C-5a), 144.63 (C-8), 145.33 (C-7), 153.26 (C-4), 164.91 (C-10a) and 166.35 (C-2); MS (EI-70 eV) m/z 262 (M+, 100%), 263 ([M + H]+, 37) and 219 (19); HRMS calcd for C12H10N2O3S (262.0412), found 262.0413.
Synthesis and analytical data of 7,8-dihydroxy-4H-2-propyl-pyrimido[2,1-b]benzothiazol-4-one (3c) and 7,8-dihydroxy-2H-4-propyl-pyrimido[2,1-b]benzothiazol-2-one (4c).
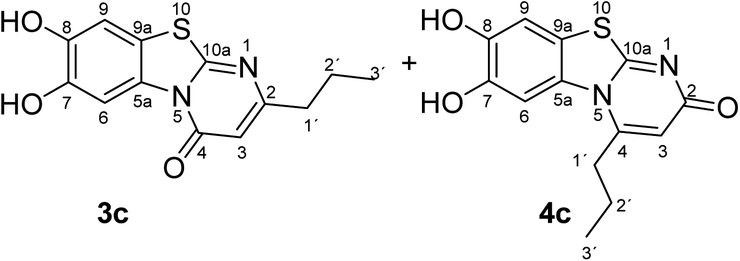
According to general procedure II, catechol (1a) (64 mg, 0.58 mmol), 2,3-dihydro-6-propyl-2-thioxopyrimidin-4(1H)-one (2c) (85 mg, 0.50 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 18 h. Workup gave a mixture of 7,8-dihydroxy-4H-2-propyl-pyrimido[2,1-b]benzothiazol-4-one (3c) and 7,8-dihydroxy-2H-4-propyl-pyrimido[2,1-b]benzothiazol-2-one (4c) as a brown powder (130 mg, 94%), mp 258–260 °C; Rf = 0.27 (CH2Cl2/EtOAc = 2:1); max (atr)/cm−1 3310 (OH), 2960 (CH), 1625 (CO), 1453 and 1307; δH (500 MHz; DMSO-d6) of 3c 0.89 (3H, t, 3J = 7.3 Hz, 3′-H), 1.65 (2H, ov., 2′-H), 2.49 (2H, ov., 1′-H), 6.15 (1H, br s, 3-H), 7.30 (1H, s, 9-H) and 8.45 (1H, s, 6-H); δH (500 MHz; DMSO-d6) of 4c 1.04 (3H, t, 3J = 7.1 Hz, 3′-H), 1.65 (2H, ov. sex, 3J = 7.7 Hz, 2′-H), 3.00 (2H, t, 3J = 7.5 Hz, 1′-H), 6.03 (1H, br s, 3-H), 7.25 (1H, s, 9-H) and 7.41 (1H, s, 6-H); δC (125 MHz; DMSO-d6) of 3c 13.50 (C-3′), 20.87 (C-2′), 38.26 (C-1′), 105.26 (C-3), 106.39 (C-6), 108.29 (C-9), 113.24 (C-9a), 128.09 (C-5a), 144.95 (C-7), 145.66 (C-8), 160.10 (C-4), 161.84 (C-10a), 165.30 (C-2); δC (125 MHz; DMSO-d6) of 4c 13.14 (C-3′), 20.09 (C-2′), 34.20 (C-1′), 104.63 (C-6), 109.00 (C-9), 109.73 (C-3), 112.73 (C-9a), 128.28 (C-5a), 144.72 (C-8), 145.38 (C-7), 151.48 (C-4), 164.98 (C-10a), 166.24 (C-2); MS (ESI) m/z 299 ([M + Na]+, 100%) and 277 ([M + H]+, 35); HRMS calcd for C13H12N2O3S + Na (299.0461), found 299.0457.
Synthesis and analytical data of 7,8-dihydroxy-4H-2-isopropyl-pyrimido[2,1-b]benzothiazol-4-one (3d) and 7,8-dihydroxy-2H-4-isopropyl-pyrimido[2,1-b]benzothiazol-2-one (4d).
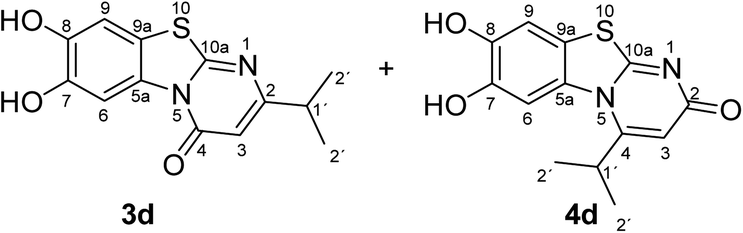
According to general procedure II, catechol (1a) (64 mg, 0.58 mmol), 2,3-dihydro-6-isopropyl-2-thioxopyrimidin-4(1H)-one (2d) (85 mg, 0.50 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 13 h. Workup gave a mixture of 7,8-dihydroxy-4H-2-isopropyl-pyrimido[2,1-b]benzothiazol-4-one (3d) and 7,8-dihydroxy-2H-4-isopropyl-pyrimido[2,1-b]benzothiazol-2-one (4d) as a brown powder (131 mg, 95%); mp 250–252 °C; Rf = 0.36 (CH2Cl2/EtOAc = 0.1:1); max (atr)/cm−1 3350 (OH), 2962 (CH), 1636 (CO), 1517 and 1234; δH (500 MHz; DMSO-d6) of 3d 1.17 (6H, d, 3J = 6.9 Hz, 2′-H), 2.77 (1H, sep, 3J = 6.8 Hz, 1′-H), 6.15 (1H, s, 3-H), 7.35 (1H, s, 9-H), 8.44 (1H, s, 6-H) and 9.77 (2H, ov. br, 7,8-OH); δH (500 MHz; DMSO-d6) of 4d 1.32 (6H, d, 3J = 6.3 Hz, 2′-H), 3.64 (1H, ov., 1′-H), 6.08 (1H, s, 3-H), 7.30 (1H, s, 9-H), 7.68 (1H, s, 6-H) and 9.77 (2H, ov. br, 7,8-OH); δC (125 MHz; DMSO-d6) of 3d 21.19 (C-2′), 34.65 (C-1′), 103.22 (C-3), 106.53 (C-6), 108.34 (C-9), 113.20 (C-9a), 128.02 (C-5a), 144.98 (C-7), 145.77 (C-8), 160.42 (C-4), 161.90 (C-10a) and 170.33 (C-2); δC (125 MHz; DMSO-d6) of 4d 21.48 (C-2′), 28.59 (C-1′), 105.33 (C-6), 106.43 (C-3), 109.02 (C-9), 112.65 (C-9a), 128.00 (C-5a), 144.81 (C-8), 145.64 (C-7), 158.10 (C-4), 165.25 (C-10a), 166.53 (C-2); MS (ESI) m/z 315 ([M + K]+, 20%), 277 ([M + H]+, 100), 249 (8); HRMS calcd for C13H12N2O3S + H (277.0641), found 277.0615. Column filtration using the eluent CH2Cl2/EtOAc = 1:1 gave 3d in pure form; mp 259–261 °C.
Synthesis and analytical data of 7,8-dihydroxy-4H-2-tert-butyl-pyrimido[2,1-b]benzothiazol-4-one (3e).
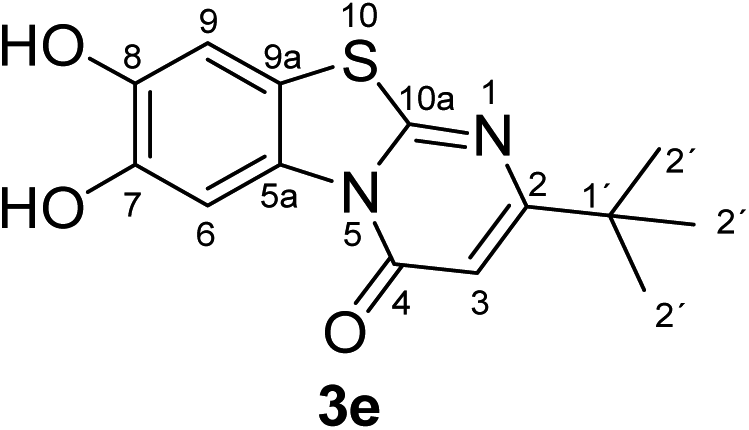
According to general procedure II, catechol (1a) (64 mg, 0.58 mmol), 6-tert-butyl-2,3-dihydro-2-thioxopyrimidin-4(1H)-one (2e) (92 mg, 0.50 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 16 h. Workup gave 7,8-dihydroxy-4H-2-tert-butyl-pyrimido[2,1-b]benzothiazol-4-one (3e) as a brown powder (140 mg, 97%), mp > 300 °C; Rf = 0.38 (CH2Cl2/EtOAc = 0.1:1); max (atr)/cm−1 3295 (OH), 2954 (CH), 1638 (CO), 1546 and 1185; δH (500 MHz; DMSO-d6) 1.23 (9H, s, 2′-H), 6.21 (1H, s, 3-H), 7.31 (1H, s, 9-H), 8.44 (1H, s, 6-H) and 9.75 (2H, br, 7,8-OH); δC (125 MHz; DMSO-d6) 28.67 (C-2′), 36.78 (C-1′), 102.08 (C-3), 106.42 (C-6), 108.32 (C-9), 113.22 (C-9a), 127.90 (C-5a), 144.94 (C-7), 145.73 (C-8), 160.52 (C-4), 161.36 (C-10a) and 172.33 (C-2); MS (ESI) m/z 329 ([M + K]+, 31%) and 291 ([M + H]+, 100); HRMS calcd for C14H14N2O3S + H (291.0798), found 291.0802.
Synthesis and analytical data of 7,8-dihydroxy-4H-2-trifluoromethyl-pyrimido[2,1-b]benzothiazol-4-one (3f).
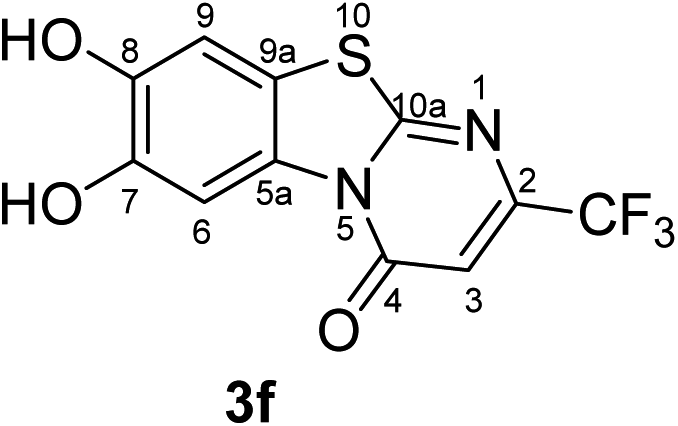
According to general procedure II, catechol (1a) (64 mg, 0.58 mmol), 6-trifluoromethyl-2,3-dihydro-2-thioxopyrimidin-4(1H)-one (2g) (98 mg, 0.50 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 16 h. Workup gave 7,8-dihydroxy-4H-2-trifluoromethyl-pyrimido[2,1-b]benzothiazol-4-one (3f) as a brown powder (118 mg, 78%), mp > 300 °C; Rf = 0.31 (cyclohexane/EtOAc = 1:1); max (atr)/cm−1 3408 (OH), 1654 (CO), 1526 (CN) and 1151; δH (500 MHz; DMSO-d6) 6.79 (1H, s, 3-H), 7.42 (1H, s, 9-H), 8.47 (1H, s, 6-H), 9.82 and 9.90 (2H, 2s, 7,8-OH); δC (125 MHz; DMSO-d6) 105.31 (q, 3JC,F = 3.1 Hz, C-3), 106.33 (C-6), 108.35 (C-9), 114.19 (C-9a), 120.78 (q, 1JC,F = 273.0 Hz, CF3), 127.82 (C-5a), 145.42 (C-7), 146.41 (C-8), 148.25 (q, 2JC,F = 34.4 Hz, C-2), 159.47 (C-4), 164.29 (C-10a); MS (ESI) m/z 341 ([M + K]+, 49%), 303 ([M + H]+, 100) and 283 (73); HRMS calcd for C11H5F3N2O3S + H (303.0046), found 303.0046.
Synthesis and analytical data of 7,8-dihydroxy-4H-2,3-dimethyl-pyrimido[2,1-b]benzothiazol-4-one (3g) and 7,8-dihydroxy-2H-3,4-dimethyl-pyrimido[2,1-b]benzothiazol-2-one (4g).
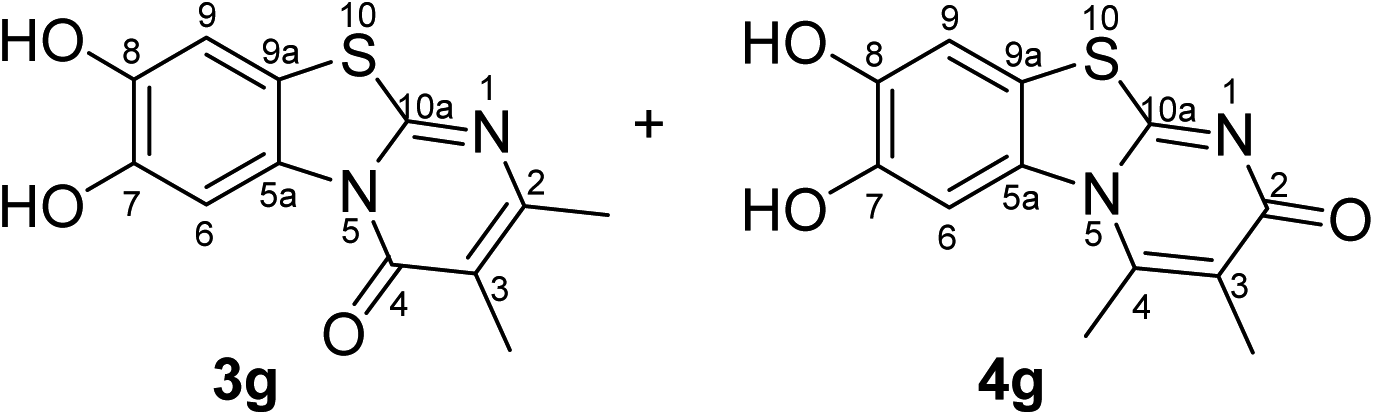
According to general procedure III, catechol (1a) (32 mg, 0.29 mmol), 2,3-dihydro-5,6-dimethyl-2-thioxopyrimidin-4(1H)-one (2g) (39 mg, 0.25 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 28 h. Workup gave a mixture of 7,8-dihydroxy-4H-2,3-dimethyl-pyrimido[2,1-b]benzothiazol-4-one (3g) and 7,8-dihydroxy-2H-3,4-dimethyl-pyrimido[2,1-b]benzothiazol-2-one (4g) as a brown powder (58 mg, 89%), mp > 300 °C; Rf = 0.46 (CH2Cl2/EtOAc = 1:1); max (atr)/cm−1 3352 (OH), 3023 (CH), 1631 (CO), 1488 and 1185; δH (500 MHz; DMSO-d6) of 3g 2.03 (3H, s, 3-CH3), 2.28 (3H, s, 2-CH3), 7.28 (1H, s, 9-H), 8.48 (1H, s, 6-H) and 9.61 (2H, ov. br, 7,8-OH); δH (500 MHz; DMSO-d6) of 4g 2.00 (3H, s, 3-CH3), 2.73 (3H, s, 4-CH3), 7.25 (1H, s, 9-H), 7.58 (1H, s, 6-H) and 9.61 (2H, ov. br, 7,8-OH); δC (125 MHz; DMSO-d6) of 3g 10.98 (3-CH3), 21.71 (2-CH3), 106.46 (C-6), 108.29 (C-9), 112.82 (C-3), 113.33 (C-9a), 128.12 (C-5a), 144.77 (C-7), 145.52 (C-8), 157.37 (C-2), 157.83 (C-10a) and 160.46 (C-4); δC (125 MHz; DMSO-d6) of 4g 12.01 (3-CH3), 17.97 (4-CH3), 105.06 (C-6), 108.99 (C-9), 112.76 (C-9a), 116.70 (C-3), 129.21 (C-5a), 144.00 (C-4), 144.49 (C-8), 145.02 (C-7), 162.86 (C-10a) and 166.21 (C-2); analytically pure product was obtained by acetylation; MS (ESI) m/z 369 ([M + Na]+, 93%), 347 ([M + H]+, 49), 305 (53) and 263 (100); HRMS calcd for C16H14N2O5S + Na (369.0516), found 369.0522.
Synthesis and analytical data of 7,8-dihydroxy-4H-3-methyl-2-trifluoromethyl-pyrimido[2,1-b]benzothiazol-4-one (3h).
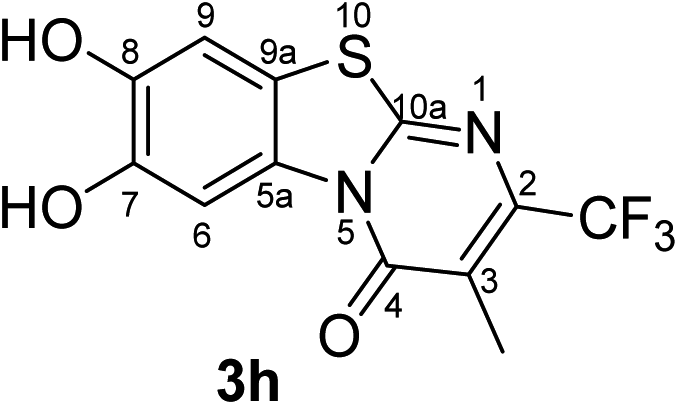
According to general procedure III, catechol (1a) (32 mg, 0.29 mmol), 6-trifluoromethyl-2,3-dihydro-5-methyl-2-thioxopyrimidin-4(1H)-one (2h) (52.5 mg, 0.25 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 17 h. Workup gave 7,8-dihydroxy-4H-2-trifluoromethyl-pyrimido[2,1-b]benzothiazol-4-one (3h) as a brown powder (75 mg, 95%), mp > 300 °C; Rf = 0.49 (CH2Cl2/EtOAc = 2:1); max (atr)/cm−1 3533 (OH), 3120 (CH), 1632 (CO), 1531 (CN) and 1224; δH (500 MHz; DMSO-d6) 2.20 (3H, q, 5JH,F = 2.2 Hz, 3-CH3), 7.39 (1H, s, 9-H), 8.49 (1H, s, 6-H) and 9.85 (2H, br, 7,8-OH); δC (125 MHz; DMSO-d6) 10.18 (q, 4JC,F = 2.2 Hz, 3-CH3), 106.40 (C-6), 108.36 (C-9), 114.29 (C-9a), 116.29 (br s, C-3), 121.78 (q, 1JC,F = 274.6 Hz, CF3), 127.59 (C-5a), 143.73 (q, 2JC,F = 33.0 Hz, C-2), 145.22 (C-7), 146.27 (C-8), 159.79 (C-10a) and 160.82 (C-4); MS (ESI) m/z 355 ([M + K]+, 72%), 317 ([M + H]+, 74), 297 (100) and 249 (60); HRMS calcd for C12H7F3N2O3S + H (317.0202), found 317.0193.
Synthesis and analytical data of 2-(4,5-dihydroxy-2-methylphenylthio)-6-methylpyrimidin-4(3H)-one (5a).
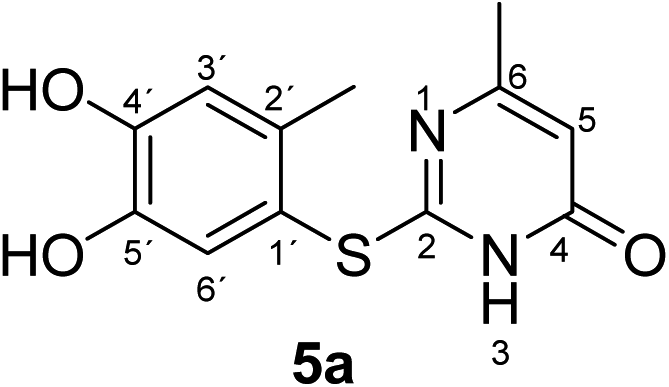
According to general procedure II, 4-methylcatechol (1b) (72 mg, 0.58 mmol), 2,3-dihydro-6-methyl-2-thioxopyrimidin-4(1H)-one (2a) (71 mg, 0.50 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 12 h. Workup gave 2-(4,5-dihydroxy-2-methylphenylthio)-6-methylpyrimidin-4(3H)-one (5a) as a pale yellow powder (108 mg, 82%), mp 213–215 °C; Rf = 0.27 (CH2Cl2/EtOAc/MeOH = 3:3:0.1); max (atr)/cm−1 3406 (OH), 2916 (CH), 1644 (CO), 1508 and 1264; δH (500 MHz; pyridine-d5) 2.13 (3H, s, 6-CH3), 2.43 (3H, s, 2′-CH3), 6.29 (1H, s, 5-H), 7.23 (1H, s, 3′-H) and 7.69 (1H, s, 6′-H); δC (125 MHz; pyridine-d5) 20.40 (2′-CH3), 23.67 (6-CH3), 105.59 (C-5), 116.82 (C-1′), 118.95 (C-3′), 125.04 (C-6′), 135.40 (ov., C-2′), 145.63 (C-5′), 149.64 (C-4′), 166.69 (C-6), 167.13 (C-2) and 167.87 (C-4); MS (ESI) m/z 287 ([M + Na]+, 100%), 265 ([M + H]+, 75) and 143 (60); HRMS calcd for C12H12N2O3S + H (265.0641), found 265.0643.
Synthesis and analytical data of 2-(4,5-dihydroxy-2-methylphenylthio)-6-propylpyrimidin-4(3H)-one (5b).
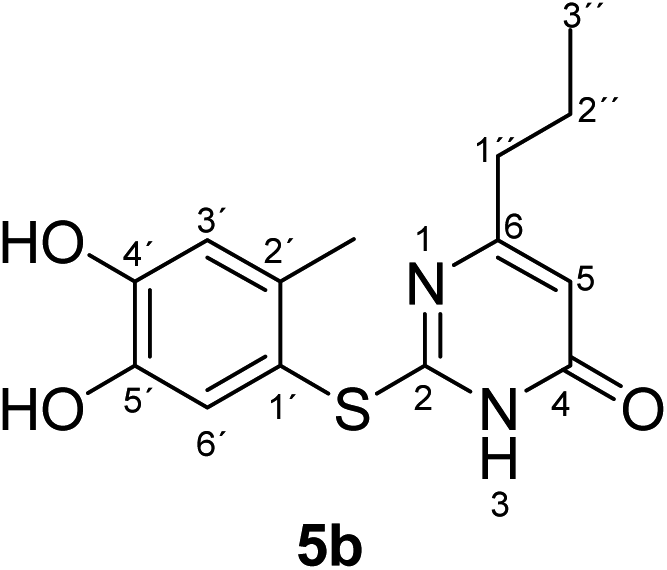
According to general procedure II, 4-methylcatechol (1b) (72 mg, 0.58 mmol), 2,3-dihydro-6-propyl-2-thioxopyrimidin-4(1H)-one (2c) (85 mg, 0.50 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 13 h. Workup gave 2-(4,5-dihydroxy-2-methylphenylthio)-6-propylpyrimidin-4(3H)-one (5b) as a pale yellow powder (128 mg, 88%), mp 189–191 °C; Rf = 0.40 (CH2Cl2/EtOAc/MeOH = 3:3:0.1); max (atr)/cm−1 3439 (OH), 3179 (NH), 2957 (CH), 1634 (CO), 1508 (CN) and 1227; δH (300 MHz; pyridine-d5) 0.75 (3H, t, 3J = 7.2 Hz, 3′′-H), 1.58 (2H, sex like, 3J = 7.5 Hz, 2′′-H), 2.39 (2H, t, 3J = 7.6 Hz, 1′′-H), 2.43 (3H, s, 2′-CH3), 6.31 (1H, s, 5-H), 7.25 (1H, s, 3′-H) and 7.67 (1H, s, 6′-H); δC (75 MHz; pyridine-d5) 13.64 (C-3′′), 20.34 (2′-CH3), 21.44 (C-2′′), 39.32 (C-1′′), 105.31 (C-5), 116.79 (C-1′), 118.85 (C-3′), 124.96 (C-6′), 135.39 (C-2′), 145.57 (C-5′), 149.60 (C-4′), 166.79 (C-2), 167.68 (C-4) and 170.09 (C-6); MS (ESI) m/z 331 ([M + K]+, 47%), 315 ([M + Na]+, 100), 293 ([M + H]+, 83) and 171 (53); HRMS calcd for C14H16N2O3S + Na (315.0774), found 315.0767.
Synthesis and analytical data of 2-(4,5-dihydroxy-2-methylphenylthio)-6-isopropylpyrimidin-4(3H)-one (5c).
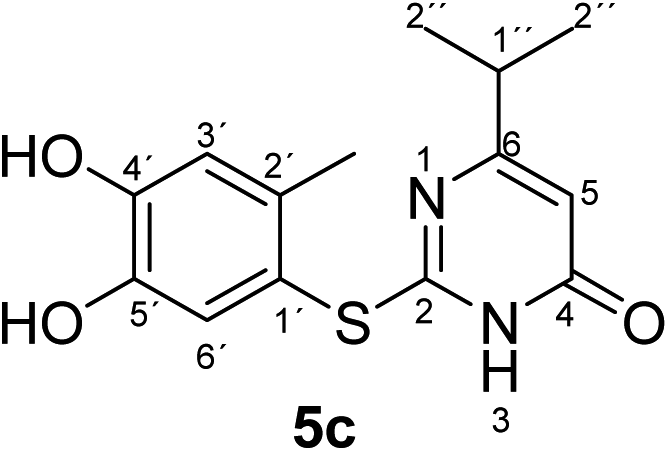
According to general procedure II, 4-methylcatechol (1b) (72 mg, 0.58 mmol), 2,3-dihydro-6-isopropyl-2-thioxopyrimidin-4(1H)-one (2d) (85 mg, 0.50 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 16 h. Workup gave 2-(4,5-dihydroxy-2-methylphenylthio)-6-isopropylpyrimidin-4(3H)-one (5c) as a pale yellow powder (134 mg, 92%), mp 204–206 °C; Rf = 0.27 (CH2Cl2/EtOAc/MeOH = 3:3:0.1); max (atr)/cm−1 3240 (OH), 2965 (CH), 1644 (CO), 1509 and 1281; δH (300 MHz; pyridine-d5) 1.09 (6H, d, 3J = 6.9 Hz, 2′′-H), 2.42 (3H, s, 2′-CH3), 2.65 (1H, sep, 3J = 6.9 Hz, 1′′-H), 6.32 (1H, s, 5-H), 7.26 (1H, s, 3′-H) and 7.67 (1H, s, 6′-H); δC (75 MHz; pyridine-d5) 20.32 (2′-CH3), 21.20 (C-2′′), 35.60 (C-1′′), 103.23 (C-5), 116.82 (C-1′), 118.77 (C-3′), 124.94 (C-6′), 135.40 (ov., C-2′), 145.51 (C-5′), 149.56 (C-4′), 166.51 (C-2), 167.84 (C-4) and 174.91 (C-6); MS (ESI) m/z 315 ([M + Na]+, 100%), 293 ([M + H]+, 44) and 171 (27); HRMS calcd for C14H16N2O3S + H (293.0954), found 293.0950.
Synthesis and analytical data of 2-(4,5-dihydroxy-2-methylphenylthio)-6-tert-butylpyrimidin-4(3H)-one (5d).
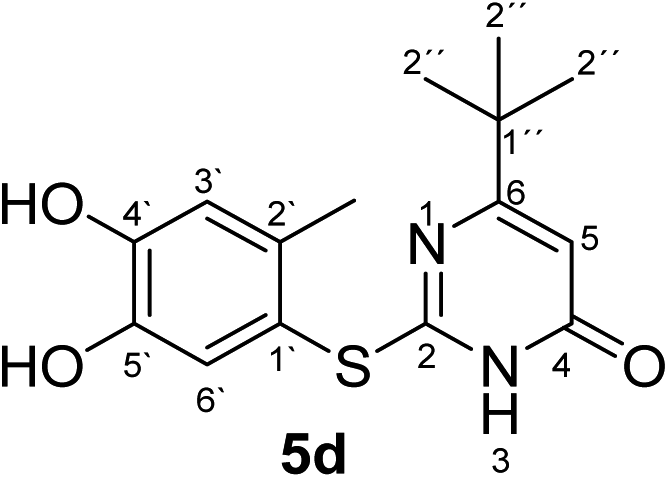
According to general procedure II, 4-methylcatechol (1b) (72 mg, 0.58 mmol), 6-tert-butyl-2,3-dihydro-2-thioxopyrimidin-4(1H)-one (2e) (92 mg, 0.50 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 17 h. Workup gave 2-(4,5-dihydroxy-2-methylphenylthio)-6-tert-butylpyrimidin-4(3H)-one (5d) as a pale yellow powder (138 mg, 90%), mp 205–207 °C; Rf = 0.45 (CH2Cl2/EtOAc/MeOH = 3:3:0.1); max (atr)/cm−1 3450 (OH), 2957 (CH), 1638 (CO), 1577 (CN) and 1279; δH (500 MHz; pyridine-d5) 1.14 (9H, s, 2′′-H), 2.42 (3H, s, 2′-CH3), 6.43 (1H, s, 5-H), 7.28 (1H, s, 3′-H) and 7.66 (1H, s, 6′-H); δC (125 MHz; pyridine-d5) 20.34 (2′-CH3), 28.73 (C-2′′), 37.20 (C-1′′), 102.37 (C-5), 116.75 (C-1′), 118.72 (C-3′), 124.98 (C-6′), 135.72 (C-2′), 145.49 (C-5′), 149.58 (ov., C-4′), 165.32 (C-2), 167.61 (C-4) and 176.73 (C-6); MS (ESI) m/z 345 ([M + K]+, 40%), 329 ([M + Na]+, 100), 307 ([M + H]+, 77), 229 (24) and 185 (54); HRMS calcd for C15H18N2O3S + H (307.1111), found 307.1111.
Synthesis and analytical data of 2-(4,5-dihydroxy-2-methylphenylthio)-5,6-dimethylpyrimidin-4(3H)-one (5e).
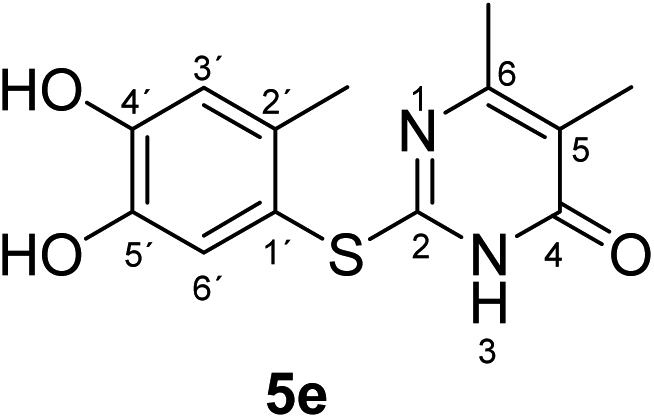
According to general procedure II, 4-methylcatechol (1b) (72 mg, 0.58 mmol), 2,3-dihydro-5,6-dimethyl-2-thioxopyrimidin-4(1H)-one (2g) (78 mg, 0.50 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 20 h. Workup gave 2-(4,5-dihydroxy-2-methylphenylthio)-5,6-dimethylpyrimidin-4(3H)-one (5e) as a pale yellow powder (120 mg, 86%), mp 229–331 °C; Rf = 0.45 (CH2Cl2/EtOAc/MeOH = 3:3:0.1); max (atr)/cm−1 3420 (OH), 2926 (CH), 1625 (CO), 1512 and 1264; δH (500 MHz; pyridine-d5) 2.06 (3H, s, 5-CH3), 2.13 (3H, s, 6-CH3), 2.46 (3H, s, 2′-CH3), 7.24 (1H, s, 3′-H) and 7.69 (1H, s, 6′-H); δC (125 MHz; pyridine-d5) 10.87 (5-CH3), 20.41 (2′-CH3), 21.61 (6-CH3), 113.56 (C-5), 116.85 (C-1′), 118.92 (C-3′), 125.00 (C-6′), 135.31 (C-2′), 145.61 (C-5′), 149.60 (C-4′), 161.78 (C-2), 162.20 (C-6) and 166.32 (C-4); MS (ESI) m/z 301 ([M + Na]+, 25%), 279 ([M + H]+, 100), 263 (25) and 157 (62); HRMS calcd for C13H14N2O3S + H (279.0798), found 279.0797.
Synthesis and analytical data of 2-(2-ethyl-4,5-dihydroxyphenylthio)-6-methylpyrimidin-4(3H)-one (5f).
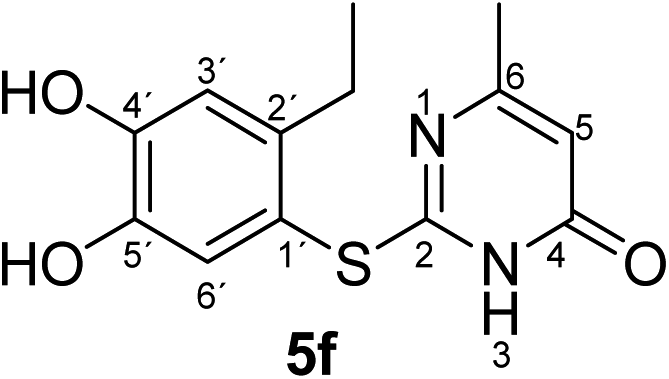
According to general procedure II, 4-ethylcatechol (1c) (80 mg, 0.58 mmol), 2,3-dihydro-6-methyl-2-thioxopyrimidin-4(1H)-one (2a) (71 mg, 0.50 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 14 h. Workup gave 2-(2-ethyl-4,5-dihydroxyphenylthio)-6-methylpyrimidin-4(3H)-one (5f) as a pale yellow powder (125 mg, 90%), mp 180–182 °C; Rf = 0.31 (CH2Cl2/EtOAc/MeOH = 3:3:0.1); max (atr)/cm−1 3400 (OH), 2964 (CH), 1645 (CO), 1503 (CN) and 1278; δH (500 MHz; pyridine-d5) 1.17 (3H, t, 3J = 7.5 Hz, 2′-CH2C3), 2.13 (3H, s, 6-CH3), 2.88 (2H, q, 3J = 7.5 Hz, 2′-C2CH3), 6.31 (1H, s, 5-H), 7.27 (1H, s, 3′-H) and 7.69 (1H, s, 6′-H); δC (125 MHz; pyridine-d5) 15.88 (2′-CH2![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H3), 23.64 (6-CH3), 27.30 (2′-H2CH3), 105.54 (C-5), 116.11 (C-1′), 117.44 (C-3′), 125.45 (C-6′), 141.24 (C-2′), 145.68 (C-5′), 149.92 (C-4′), 166.67 (C-6), 167.74 (C-2) and 167.93 (C-4); MS (ESI) m/z 301 ([M + Na]+, 100%), 279 ([M + H]+, 21) and 143 (18); HRMS calcd for C13H14N2O3S + H (279.0798), found 279.0790.
H3), 23.64 (6-CH3), 27.30 (2′-H2CH3), 105.54 (C-5), 116.11 (C-1′), 117.44 (C-3′), 125.45 (C-6′), 141.24 (C-2′), 145.68 (C-5′), 149.92 (C-4′), 166.67 (C-6), 167.74 (C-2) and 167.93 (C-4); MS (ESI) m/z 301 ([M + Na]+, 100%), 279 ([M + H]+, 21) and 143 (18); HRMS calcd for C13H14N2O3S + H (279.0798), found 279.0790.
Synthesis and analytical data of 2-(2-ethyl-4,5-dihydroxyphenylthio)-6-isopropylpyrimidin-4(3H)-one (5g).
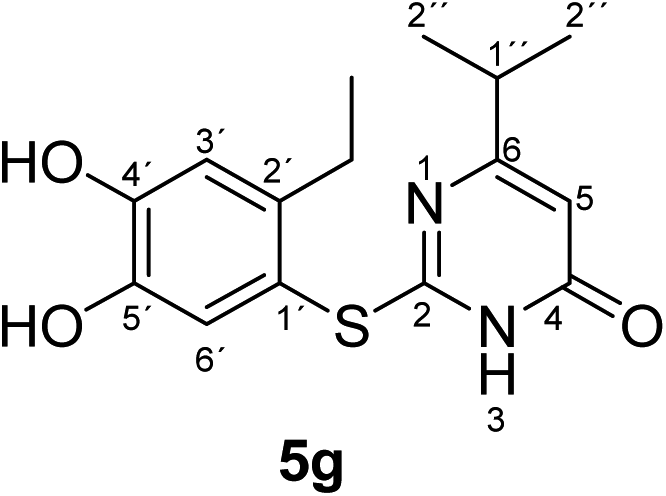
According to general procedure II, 4-ethylcatechol (1c) (80 mg, 0.58 mmol), 2,3-dihydro-6-isopropyl-2-thioxopyrimidin-4(1H)-one (2d) (85 mg, 0.50 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 12 h. Workup gave 2-(2-ethyl-4,5-dihydroxyphenylthio)-6-isopropylpyrimidin-4(3H)-one (5g) as a pale yellow powder (116 mg, 76%); mp 158–160 °C; Rf = 0.50 (CH2Cl2/EtOAc/MeOH = 3:3:0.1); max (atr)/cm−1 3323 (OH), 2962 (CH), 1630 (CO), 1514 and 1252; δH (300 MHz; pyridine-d5) 1.09 (6H, d, 3J = 7.0 Hz, 2′′-H), 1.16 (3H, t, 3J = 7.5 Hz, 2′-CH2C3), 2.66 (1H, sep, 3J = 7.0 Hz, 1′′-H), 2.86 (2H, q, 3J = 7.5 Hz, 2′-C2CH3), 6.32 (1H, s, 5-H), 7.28 (1H, s, 3′-H) and 7.67 (1H, s, 6′-H); δC (75 MHz; pyridine-d5) 15.80 (2′-CH2H3), 21.25 (C-2′′), 27.24 (2′-H2CH3), 35.63 (C-1′′), 103.06 (C-5), 116.20 (C-1′), 117.29 (C-3′), 125.44 (C-6′), 141.26 (C-2′), 145.57 (C-5′), 150.20 (ov., C-4′), 167.33 (C-2), 168.02 (C-4) and 175.01 (C-6); MS (ESI) m/z 345 ([M + K]+, 23%), 329 ([M + Na]+, 100), 307 ([M + H]+, 21) and 215 (14); HRMS calcd for C15H18N2O3S + H (307.1111), found 307.1103.
Synthesis and analytical data of 2-(2-ethyl-4,5-dihydroxyphenylthio)-6-tert-butylpyrimidin-4(3H)-one (5h).
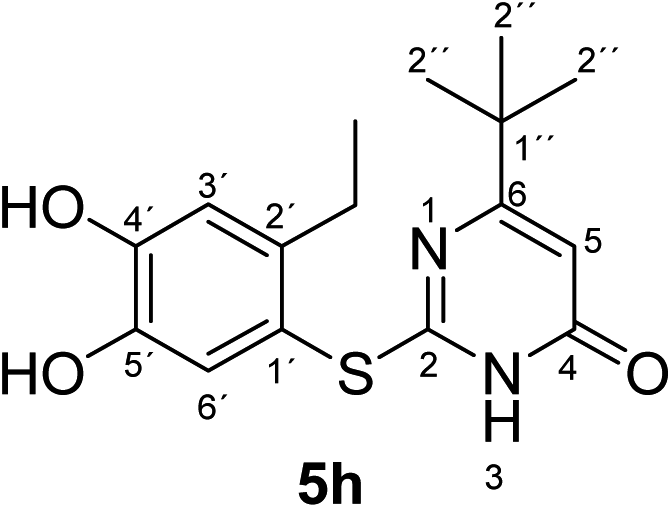
According to general procedure II, 4-ethylcatechol (1c) (80 mg, 0.58 mmol), 6-tert-butyl-2,3-dihydro-2-thioxopyrimidin-4(1H)-one (2e) (92 mg, 0.50 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 16 h. Workup gave 2-(2-ethyl-4,5-dihydroxyphenylthio)-6-tert-butylpyrimidin-4(3H)-one (5h) as a pale yellow powder (130 mg, 81%), mp 153–155 °C; Rf = 0.49 (CH2Cl2/EtOAc/MeOH = 3:3:0.1); max (atr)/cm−1 3410 (OH), 2962 (CH), 1632 (CO), 1512 (CN) and 1275; δH (300 MHz; pyridine-d5) 1.15 (9H, s, 2′′-H), 1.17 (3H, t, 3J = 7.5 Hz, 2′-CH2C3), 2.84 (2H, q, 3J = 7.4 Hz, 2′-C2CH3), 6.44 (1H, s, 5-H), 7.30 (1H, s, 3′-H) and 7.67 (1H, s, 6′-H); δC (75 MHz; pyridine-d5) 15.79 (2′-CH2H3), 27.24 (2′-H2CH3), 28.75 (C-2′′), 37.19 (C-1′′), 102.12 (C-5), 116.14 (C-1′), 117.20 (C-3′), 125.47 (C-6′), 141.28 (C-2′), 145.52 (C-5′), 149.8 (ov., C-4′), 166.20 (C-2 or C-4), 167.80 (C-2 or C-4) and 176.82 (C-6); MS (ESI) m/z 343 ([M + Na]+, 100%), 321 ([M + H]+, 38) and 185 (25); HRMS calcd for C16H20N2O3S + H (321.1267), found 321.1260.
Synthesis and analytical data of 2-(2-ethyl-4,5-dihydroxyphenylthio)-5,6-dimethylpyrimidin-4(3H)-one (5i).
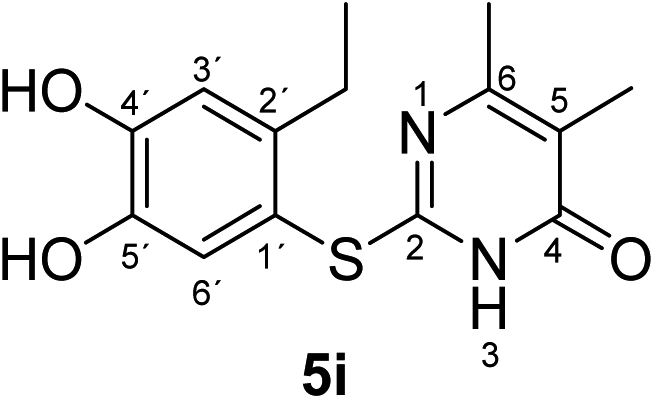
According to general procedure II, 4-ethylcatechol (1c) (80 mg, 0.58 mmol), 2,3-dihydro-5,6-dimethyl-2-thioxopyrimidin-4(1H)-one (2g) (78 mg, 0.50 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 16 h. Workup gave 2-(2-ethyl-4,5-dihydroxyphenylthio)-5,6-dimethylpyrimidin-4(3H)-one (5i) as a pale yellow powder (133 mg, 91%), mp 185–187 °C; Rf = 0.43 (CH2Cl2/EtOAc/MeOH = 3:3:0.1); max (atr)/cm−1 3403 (OH), 2927 (CH), 1629 (CO), 1523 (CN) and 1252; δH (500 MHz; pyridine-d5) 1.19 (3H, t, 3J = 7.5 Hz, 2′-CH2C3), 2.06 (3H, s, 5-CH3), 2.13 (3H, s, 6-CH3), 2.88 (2H, q, 3J = 7.5 Hz, 2′-C2CH3), 7.27 (1H, s, 3′-H) and 7.70 (1H, s, 6′-H); δC (125 MHz; pyridine-d5) 10.82 (5-CH3), 15.86 (2′-CH2H3), 21.55 (6-CH3), 27.27 (2′-H2CH3), 113.42 (C-5), 116.19 (C-1′), 117.38 (C-3′), 125.40 (C-6′), 141.15 (C-2′), 145.64 (C-5′), 149.80 (ov., C-4′), 162.20 (C-6), 162.43 (C-2) and 166.34 (C-4); MS (ESI) m/z 315 ([M + Na]+, 100%), 293 ([M + H]+, 69) and 157 (41); HRMS calcd for C14H16N2O3S + H (293.0954), found 293.0949.
Synthesis and analytical data of 2-(4,5-dihydroxy-2-methylphenylthio)-6,7-dihydro-3H-cyclopenta[d]pyrimidin-4(5H)-one (5j).
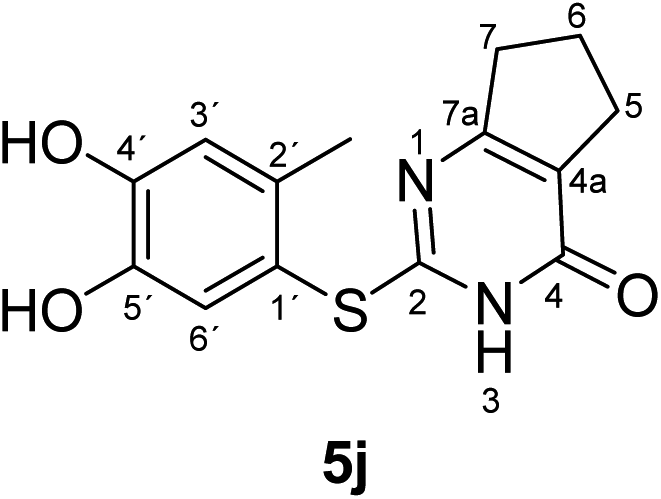
According to general procedure II, 4-methylcatechol (1b) (72 mg, 0.58 mmol), 2,3,6,7-tetrahydro-2-thioxo-1H-cyclopenta[d]pyrimidin-4(5H)-one (2i) (84 mg, 0.50 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 14 h. Workup gave 2-(4,5-dihydroxy-2-methylphenylthio)-6,7-dihydro-3H-cyclopenta[d]pyrimidin-4(5H)-one (5j) as a pale yellow powder (130 mg, 90%), mp 240–242 °C; Rf = 0.30 (CH2Cl2/EtOAc/MeOH = 3:3:0.1); max (atr)/cm−1 3480 (OH), 2924 (CH), 1639 (CO), 1512 and 1278; δH (500 MHz; pyridine-d5) 1.72 (2H, quin, 6-H), 2.47 (3H, s, 2′-CH3), 2.57 (2H, t, 3J = 7.7 Hz, 7-H), 2.75 (2H, t, 3J = 7.7 Hz, 5-H), 7.21 (1H, s, 3′-H) and 7.70 (1H, s, 6′-H); δC (125 MHz; pyridine-d5) 20.42 (2′-CH3), 21.37 (C-6), 27.41 (C-5), 34.89 (C-7), 115.94 (C-1′), 118.93 (C-3′), 119.48 (C-4a), 125.03 (C-6′), 135.35 (ov., C-2′), 145.71 (C-5′), 149.80 (ov., C-4′), 163.05 (C-4), 163.32 (C-2) and 171.02 (C-7a); MS (ESI) m/z 313 ([M + Na]+, 93%), 291 ([M + H]+, 100) and 169 (67); HRMS calcd for C14H14N2O3S + H (291.0798), found 291.0788.
Synthesis and analytical data of 2-(2-ethyl-4,5-dihydroxyphenylthio)-6,7-dihydro-3H-cyclopenta[d]pyrimidin-4(5H)-one (5k).
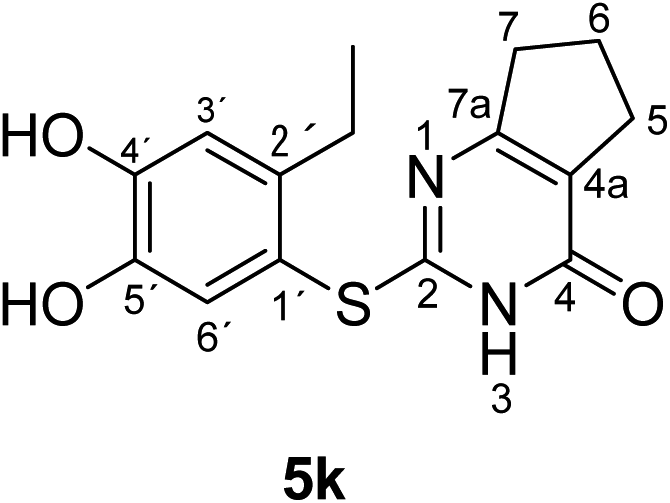
According to general procedure II, 4-ethylcatechol (1c) (80 mg, 0.58 mmol), 2,3,6,7-tetrahydro-2-thioxo-1H-cyclopenta[d]pyrimidin-4(5H)-one (2i) (84 mg, 0.50 mmol), ethanol (2 mL), phosphate buffer (0.2 M, pH 6.0, 18 mL) and laccase (12 U, 10 mg, A. bisporus) were reacted for 14 h. Workup gave 2-(2-ethyl-4,5-dihydroxyphenylthio)-6,7-dihydro-3H-cyclopenta[d]pyrimidin-4(5H)-one (5k) as a pale yellow powder (115 mg, 76%), mp 178–180 °C; Rf = 0.36 (CH2Cl2/EtOAc/MeOH = 3:3:0.1); max (atr)/cm−1 3500 (OH), 2962 (CH), 1639 (CO), 1534 (CN) and 1285; δH (500 MHz; pyridine-d5) 1.20 (3H, t, 3J = 7.6 Hz, 2′-CH2C3), 1.72 (2H, quin, 3J = 7.5 Hz, 6-H), 2.56 (2H, t, 3J = 7.5 Hz, 7-H), 2.75 (2H, t, 3J = 7.5 Hz, 5-H), 2.88 (2H, q, 3J = 7.6 Hz, 2′-C2CH3), 7.24 (1H, s, 3′-H) and 7.72 (1H, s, 6′-H); δC (125 MHz; pyridine-d5) 15.89 (2′-CH2H3), 21.36 (C-6), 27.30 (2′-H2CH3), 27.40 (C-5), 34.85 (C-7), 115.21 (C-1′), 117.41 (C-3′), 119.48 (C-4a), 125.41 (C-6′), 141.20 (C-2′), 145.76 (C-5′), 150.11 (C-4′), 163.06 (C-4), 163.85 (C-2) and 171.04 (C-7a); MS (ESI) m/z 327 ([M + Na]+, 100%), 305 ([M + H]+, 41) and 169 (14); HRMS calcd for C15H16N2O3S + H (305.0954), found 305.0946.
Biology
Conclusions
To summarize, simple-to-perform, efficient and sustainable methods for the synthesis of regioisomeric pyrimidobenzothiazoles 3, 4 as well as catechol thioethers 5 by laccase-catalyzed domino reactions between catechols 1 and 2,3-dihydro-2-thioxopyrimidin-4-(1H)-ones 2 have been developed. The transformations rely on the laccase-catalyzed oxidation of a catechol 1 to the corresponding o-benzoquinone 8 which in turn undergoes reaction with a 2,3-dihydro-2-thioxopyrimidin-4(1H)-one 2. Depending on the substitution pattern of the 2,3-dihydro-2-thioxopyrimidin-4(1H)-one 2, the laccase-catalyzed reactions with unsubstituted catechol (1a) deliver either 7,8-dihydroxy-4H-pyrimido[2,1-b]benzothiazol-4-ones 3 and/or 7,8-dihydroxy-2H-pyrimido[2,1-b]benzothiazol-2-ones 4. With 4-substituted catechols 1b, c, catechol thioethers 5 are formed exclusively. All reactions can be performed under mild reaction conditions with aerial O2 as the oxidant, and the products are formed with yields ranging between 76 and 97%. In addition, the cytotoxicity of selected pyrimidobenzothiazoles 3 and catechol thioethers 5 against HepG2 cell line is reported. A structure–activity relationship study reveals that the most potent compounds are 5c (IC50 = 7.77 μM) and 5g (IC50 = 2.74 μM) which carry an isopropyl group at C-6. The presence of an additional ethyl group at C-2′ of 5g increases the cytotoxic potency in comparison to 5c.Acknowledgements
We thank Mr M. Wolf (Institut für Chemie, Universität Hohenheim) for recording NMR spectra as well as Dipl.-Ing. (FH) J. Trinkner and D. Garnier (Institut für Organische Chemie, Universität Stuttgart) for recording mass spectra. Special thanks to Prof. Dr Lutz Graeve; (Institut für Biologische Chemie und Ernährungswissenschaft, Universität Hohenheim) for providing the HepG2 cell line. We acknowledge the help of A. Flaccus and D. Mvondo (Institut für Biologische Chemie und Ernährungswissenschaft, Universität Hohenheim) in carrying out the SRB assay. H. T. A.-M. is grateful to Science Technology Development Fund (STDF) for financial support (Project ID 11978).Notes and references
- (a) V. Sharma, N. Chitranshi and A. K. Agarwal, Int. J. Med. Chem., 2014, 202784 Search PubMed; (b) K. S. Jain, T. S. Chitre, P. B. Miniyar, M. K. Kathiravan, V. S. Bendre, V. S. Veer, S. R. Shahana and C. J. Shishoo, Curr. Sci., 2006, 90, 793 CAS.
- A. Kleemann, J. Engel, B. Kutscher and D. Reichert, Pharmaceutical Substances, Thieme, Stuttgart, 5th edn, 2000 Search PubMed.
- (a) E. Sherman, in Lippincott Illustrated Reviews: Pharmacology, ed. K. Whalen, Wolters Kluwer, Philadelphia, 6th edn, 2015, ch. 45, p. 567 Search PubMed; (b) K. LaPlant and P. Louzon, in Lippincott Illustrated Reviews: Pharmacology, ed. K. Whalen, Wolters Kluwer, Philadelphia, 6th edn, 2015, ch. 46, p. 587 Search PubMed; (c) R. Kaur, P. Kaur, S. Sharma, G. Singh, S. Mehndiratta, P. M. S. Bedi and K. Nepali, Recent Pat. Anti-Cancer Drug Discovery, 2015, 10, 23 CrossRef CAS PubMed.
- (a) R. Bandichhor, A. Bhattacharya, M. C. Bryan, A. Cosbie, A. Díaz-Rodríguez, L. Diorazio, Z. Fei, K. Fraunhoffer, J. Hayler, M. Hickey, L. Humphreys, P. Richardson, S. Roberts, T. White, S. Wuyts and J. Yin, Org. Process Res. Dev., 2016, 20, 1118 CrossRef CAS; (b) F. Roschangar, R. A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752 RSC; (c) E. A. Peterson and J. B. Manley, Green Chemistry Strategies for Drug Discovery, in RSC Drug Discovery Series, Royal Society of Chemistry, Thomas Graham House, Cambridge, 2015 Search PubMed; (d) P. Gupta and A. Mahajan, RSC Adv., 2015, 5, 26686 RSC; (e) H.-J. Federsel, Green Chem., 2013, 15, 3105 RSC.
- B. M. Trost, Angew. Chem., Int. Ed., 1995, 34, 259 CrossRef CAS.
- (a) R. A. Sheldon, I. W. C. E. Arends and U. Hanefeld, Green Chemistry and Catalysis, WILEY-VCH, Weinheim, 2007 Search PubMed; (b) P. T. Anastas and J. C. Warner, Green Chemistry Theory and Practice, Oxford University Press, New York, 2000 Search PubMed; (c) K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31 RSC; (d) D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer Jr, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. S. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC.
- (a) R. Dhanasekaran, A. Limaye and R. Cabrera, Hepatic Med., 2012, 4, 19 Search PubMed; (b) S. Mittal and H. B. El-Serag, J. Clin. Gastroenterol., 2013, 47, S2 CrossRef PubMed; (c) E. Carr and J. A. Killman, Basics of Cancer Chemotherapy (Clinical Nursing Series), Western Schools Press, Canada, 3rd edn, 1995 Search PubMed.
- (a) S.-I. Shoda, H. Uyama, J.-I. Kadokawa, S. Kimura and S. Kobayashi, Chem. Rev., 2016, 116, 2307 CrossRef CAS PubMed; (b) D. Monti, G. Ottolina, G. Carrea and S. Riva, Chem. Rev., 2011, 111, 4111 CrossRef CAS PubMed; (c) F. Hollmann, I. W. C. E. Arends, K. Buehler, A. Schallmey and B. Bühler, Green Chem., 2011, 13, 226 RSC.
- (a) O. V. Morozova, G. P. Shumakovich, M. A. Gorbacheva, S. V. Shleev and A. I. Yaropolov, Biochemistry (Moscow), 2007, 72, 1136 CrossRef CAS; (b) H. Claus, Micron, 2004, 35, 93 CrossRef CAS PubMed; (c) A. M. Mayer and R. C. Staples, Phytochemistry, 2002, 60, 551 CrossRef CAS PubMed; (d) C. F. Thurston, Microbiology, 1994, 140, 19 CrossRef CAS.
- (a) S. Witayakran and A. J. Ragauskas, Adv. Synth. Catal., 2009, 351, 1187 CrossRef CAS; (b) A. Mikolasch and F. Schauer, Appl. Microbiol. Biotechnol., 2009, 82, 605 CrossRef CAS PubMed; (c) A. Kunamneni, S. Camarero, C. García-Burgos, F. J. Plou, A. Ballesteros and M. Alcalde, Microb. Cell Fact., 2008, 7, 32 CrossRef PubMed.
- (a) J. González-Sabín, N. Ríos-Lombardía, I. García, N. M. Vior, A. F. Braña, C. Méndez, J. A. Salas and F. Morís, Green Chem., 2016, 18, 989 RSC; (b) K. Kędziora, A. Díaz-Rodríguez, I. Lavandera, V. Gotor-Fernández and V. Gotor, Green Chem., 2014, 16, 2448 RSC; (c) H. T. Abdel-Mohsen, K. Sudheendran, J. Conrad and U. Beifuss, Green Chem., 2013, 15, 1490 RSC; (d) H. T. Abdel-Mohsen, J. Conrad and U. Beifuss, Green Chem., 2012, 14, 2686 RSC; (e) H. Leutbecher, M.-A. Constantin, S. Mika, J. Conrad and U. Beifuss, Tetrahedron Lett., 2011, 52, 604 CrossRef; (f) A. Coniglio, C. Galli, P. Gentili and R. Vadalà, J. Mol. Catal. B: Enzym., 2008, 50, 40 CrossRef CAS; (g) F. d'Acunzo, P. Baiocco and C. Galli, New J. Chem., 2003, 27, 329 RSC; (h) M. Fabbrini, C. Galli, P. Gentili and D. Macchitella, Tetrahedron Lett., 2001, 42, 7551 CrossRef CAS; (i) A. Potthast, T. Rosenau, C.-L. Chen and J. S. Gratzl, J. Org. Chem., 1995, 60, 4320 CrossRef CAS.
- (a) A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, J. Mol. Catal. B: Enzym., 2016, 125, 34 CrossRef CAS; (b) C. Engelmann, S. Illner and U. Kragl, Process Biochem., 2015, 50, 1591 CrossRef CAS; (c) M.-A. Constantin, J. Conrad and U. Beifuss, Green Chem., 2012, 14, 2375 RSC; (d) M.-A. Constantin, J. Conrad, E. Merişor, K. Koschorreck, V. B. Urlacher and U. Beifuss, J. Org. Chem., 2012, 77, 4528 CrossRef CAS PubMed; (e) B. Pickel, M.-A. Constantin, J. Pfannstiel, J. Conrad, U. Beifuss and A. Schaller, Angew. Chem., Int. Ed., 2010, 49, 202 CrossRef CAS PubMed; (f) S. Ncanana, L. Baratto, L. Roncaglia, S. Riva and S. G. Burton, Adv. Synth. Catal., 2007, 349, 1507 CrossRef CAS; (g) C. Ponzoni, E. Beneventi, M. R. Cramarossa, S. Raimondi, G. Trevisi, U. M. Pagnoni, S. Riva and L. Forti, Adv. Synth. Catal., 2007, 349, 1497 CrossRef CAS; (h) S. Ciecholewski, E. Hammer, K. Manda, G. Bose, V. T. H. Nguyen, P. Langer and F. Schauer, Tetrahedron, 2005, 61, 4615 CrossRef CAS.
- (a) S. Emirdağ-Öztürk, S. Hajdok, J. Conrad and U. Beifuss, Tetrahedron, 2013, 69, 3664 CrossRef; (b) J. Pietruszka and C. Wang, Green Chem., 2012, 14, 2402 RSC; (c) J. Pietruszka and C. Wang, ChemCatChem, 2012, 4, 782 CrossRef CAS; (d) S. Hajdok, J. Conrad and U. Beifuss, J. Org. Chem., 2012, 77, 445 CrossRef CAS PubMed.
- (a) V. Hahn, A. Mikolasch, C. Kuhlisch and F. Schauer, J. Mol. Catal. B: Enzym., 2015, 122, 56 CrossRef CAS; (b) S. Herter, D. Michalik, A. Mikolasch, M. Schmidt, R. Wohlgemuth, U. Bornscheuer and F. Schauer, J. Mol. Catal. B: Enzym., 2013, 90, 91 CrossRef CAS; (c) K. W. Wellington and N. I. Kolesnikova, Bioorg. Med. Chem., 2012, 20, 4472 CrossRef CAS PubMed; (d) S. Herter, A. Mikolasch, D. Michalik, E. Hammer, F. Schauer, U. Bornscheuer and M. Schmidt, Tetrahedron, 2011, 67, 9311 CrossRef CAS; (e) V. Hahn, T. Davids, M. Lalk, F. Schauer and A. Mikolasch, Green Chem., 2010, 12, 879 RSC; (f) A. Mikolasch, A. Matthies, M. Lalk and F. Schauer, Appl. Microbiol. Biotechnol., 2008, 80, 389 CrossRef CAS PubMed.
- (a) M. Schlippert, A. Mikolasch, V. Hahn and F. Schauer, J. Mol. Catal. B: Enzym., 2016, 126, 106 CrossRef CAS; (b) H. T. Abdel-Mohsen, J. Conrad and U. Beifuss, Green Chem., 2014, 16, 90 RSC; (c) K. W. Wellington, G. E. R. Gordon, L. A. Ndlovu and P. Steenkamp, ChemCatChem, 2013, 5, 1570 CrossRef CAS; (d) K. W. Wellington, R. Bokako, N. Raseroka and P. Steenkamp, Green Chem., 2012, 14, 2567 RSC.
- (a) A. C. Sousa, M. F. M. M. Piedade, L. O. Martins and M. P. Robalo, Green Chem., 2015, 17, 1429 RSC; (b) M. D. Cannatelli and A. J. Ragauskas, J. Mol. Catal. B: Enzym., 2015, 119, 85 CrossRef CAS; (c) A. C. Sousa, M. C. Oliveira, L. O. Martins and M. P. Robalo, Green Chem., 2014, 16, 4127 RSC; (d) H. T. Abdel-Mohsen, J. Conrad and U. Beifuss, J. Org. Chem., 2013, 78, 7986 CrossRef CAS PubMed; (e) K. W. Wellington, G. E. R. Gordon, L. A. Ndlovu and P. Steenkamp, ChemCatChem, 2013, 5, 1570 CrossRef CAS; (f) M. Kidwai, A. Jain, A. Sharma and R. C. Kuhad, Catal. Sci. Technol., 2013, 3, 230 RSC; (g) H. Leutbecher, G. Greiner, R. Amann, A. Stolz, U. Beifuss and J. Conrad, Org. Biomol. Chem., 2011, 9, 2667 RSC; (h) S. Hajdok, J. Conrad, H. Leutbecher, S. Strobel, T. Schleid and U. Beifuss, J. Org. Chem., 2009, 74, 7230 CrossRef CAS PubMed; (i) H. Leutbecher, S. Hajdok, C. Braunberger, M. Neumann, S. Mika, J. Conrad and U. Beifuss, Green Chem., 2009, 11, 676 RSC; (j) S. Witayakran, L. Gelbaum and A. J. Ragauskas, Tetrahedron, 2007, 63, 10958 CrossRef CAS; (k) S. Hajdok, H. Leutbecher, G. Greiner, J. Conrad and U. Beifuss, Tetrahedron Lett., 2007, 48, 5073 CrossRef CAS; (l) S. Witayakran, A. Zettili and A. J. Ragauskas, Tetrahedron Lett., 2007, 48, 2983 CrossRef CAS; (m) S. Witayakran and A. J. Ragauskas, Green Chem., 2007, 9, 475 RSC; (n) H. Leutbecher, J. Conrad, I. Klaiber and U. Beifuss, Synlett, 2005, 3126 CAS.
- (a) M. Barmaki, G. Valiyeva, A. A. Maharramovm and M. M. Allaverdiyev, J. Chem., 2013, 176213 Search PubMed; (b) K.-P. Shao, X.-Y. Zhang, P.-J. Chen, D.-Q. Xue, P. He, L.-Y. Ma, J.-X. Zheng, Q.-R. Zhang and H.-M. Liu, Bioorg. Med. Chem. Lett., 2014, 24, 2741 CrossRef PubMed.
- S. Nicotra, A. Intra, G. Ottolina, S. Riva and B. Danieli, Tetrahedron: Asymmetry, 2004, 15, 2927 CrossRef CAS.
- Structure solution and refinement using programs SHELXT and SHELXL (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 CrossRef PubMed . CCDC 1514742 (3f) and 1514743 (5a) contain the supplementary crystallographic data for this paper.
- (a) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301 RSC; (b) S. L. Y. Tang, R. L. Smith and M. Poliakoff, Green Chem., 2005, 7, 761 RSC; (c) R. A. Sheldon, Chem. Soc. Rev., 2012, 41, 1437 RSC; (d) R. A. Sheldon, Chem. Commun., 2008, 3352 RSC.
- R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC.
- (a) R. K. Henderson, C. Jiménez-González, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854 RSC; (b) P. G. Jessop, Green Chem., 2011, 13, 1391 RSC; (c) C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927 RSC.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107 CrossRef CAS PubMed.
- (a) G. W. Anderson, I. F. Halverstadt, W. H. Miller and R. O. Roblin, J. Am. Chem. Soc., 1945, 67, 2197 CrossRef CAS PubMed; (b) W. H. Miller, A. M. Dessert and G. W. Anderson, J. Am. Chem. Soc., 1948, 70, 500 CrossRef CAS PubMed.
Footnotes |
| † This paper is dedicated to Professor Dr Dr hc Wolfgang Haubold on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Copies of the 1H NMR and 13C NMR spectra. CCDC 1514742 and 1514743. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra28102h |
| This journal is © The Royal Society of Chemistry 2017 |