
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the origins of selectivity in soluble epoxide hydrolase from Bacillus megaterium†
Eila
Serrano-Hervás
 a,
Marc
Garcia-Borràs
*b and
Sílvia
Osuna
*a
a,
Marc
Garcia-Borràs
*b and
Sílvia
Osuna
*a
aInstitut de Química Computacional i Catàlisi (IQCC) and Departament de Química, Universitat de Girona, Carrer Maria Aurèlia Capmany 69, 17003 Girona, Spain. E-mail: silvia.osuna@udg.edu
bDepartment of Chemistry and Biochemistry, University of California, Los Angeles, 607 Charles E. Young Drive East, Los Angeles, CA 90095, USA. E-mail: marcgbq@gmail.com
First published on 11th October 2017
Abstract
Epoxide hydrolase (EH) enzymes catalyze the hydration of racemic epoxides to yield their corresponding vicinal diols. These enzymes present different enantio- and regioselectivity depending upon either the substrate structure or the substitution pattern of the epoxide ring. In this study, we computationally investigate the Bacillus megaterium epoxide hydrolase (BmEH)-mediated hydrolysis of racemic styrene oxide (rac-SO) and its para-nitro styrene oxide (rac-p-NSO) derivative using density functional theory (DFT) and an active site cluster model consisting of 195 and 197 atoms, respectively. Full reaction mechanisms for epoxide ring opening were evaluated considering the attack at both oxirane carbons and considering two possible orientations of the substrate at the BmEH active site. Our results indicate that for both SO and p-NSO substrates the BmEH enantio- and regioselectivity is opposite to the inherent (R)-BmEH selectivity, the attack at the benzylic position (C1) of the (S)-enantiomer being the most favoured chemical outcome.
1. Introduction
Enantiomerically pure compounds are commonly identified as key synthons for the manufacturing of bioactive products in the pharmaceutical and agrochemical industries. Both epoxide-containing substrates and their corresponding 1,2-diol products are essential chiral building blocks of pharmaceuticals.1–5 For instance, aryl glycidyl and naphthyl ethers are potentially useful compounds for the production of chiral amino alcohols such as (S)-alprenolol and (S)-propranolol β-blocker drugs.6The resolution of epoxide racemic mixtures is an attractive synthetic strategy for obtaining optically pure bioactive compounds of pharmacological interest. To this end, many different strategies are available that include the use of metal or organo-based catalysts,7 but also biocatalysts such as monooxygenases.8 However, these (bio)chemical approaches are not very efficient, and usually offer moderate yields of pure enantio-enriched epoxides, thereby making them less useful for organic synthesis.9 In pursuit of more efficient methodologies, the use of epoxide hydrolases (EHs) has emerged as a potential synthetic route due to some of the EH key properties such as: (i) it is a cofactor independent enzyme; (ii) it is found in a huge number of organisms; (iii) it is capable of operating in organic solvents; and (iv) it can exhibit high enantio- and regioselectivity.10,11 In particular, Bacillus megaterium ECU1001 epoxide hydrolase (BmEH) has an inherently high (R) enantioselectivity towards phenyl glycidyl ethers (PGE, see Scheme 1),12–14 for which some other EHs have only shown modest enantioselectivity or are (S)-specific.15–17 The BmEH enzyme also exhibits an excellent enantiomeric ratio (E > 200) towards a para-nitro styrene oxide (p-NSO) compound.12 For the latter case, it was found that the nature of the para-substituent switches both BmEH enantio- and regioselectivity, with the hydrolysis of the (S)-enantiomer via the attack at the benzylic position being favoured (see Scheme 1).12,18
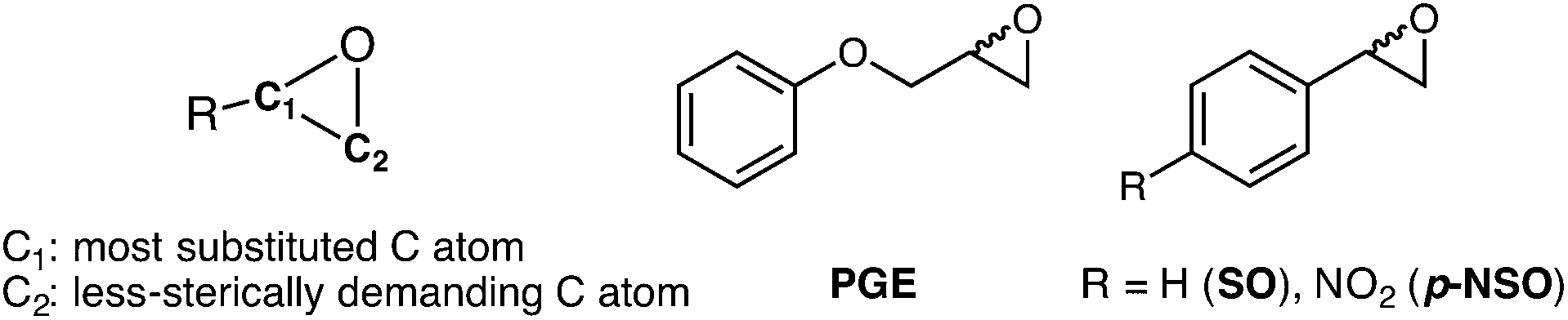 | ||
| Scheme 1 Left, general representation of epoxide (carbon atoms labelled). Right, phenyl glycidyl ether (PGE) and the substrates used in this study: styrene oxide (SO), and p-nitro styrene oxide (p-NSO). | ||
The BmEH enzyme belongs to the α,β-hydrolase fold, consisting of a three-dimensional (3D) structure that possesses an α,β sheet core domain and a lid domain that caps the active site.19–21 The EH members of this α,β-hydrolase superfamily are characterised for having a highly conserved catalytic triad (Asp–His–Asp/Glu), two tyrosine residues positioned on the lid domain responsible for substrate recognition and for assisting the epoxide ring opening, and a conserved oxyanion motif consisting of HGXP residues (X = F in BmEH, see Fig. 1).20,22,23
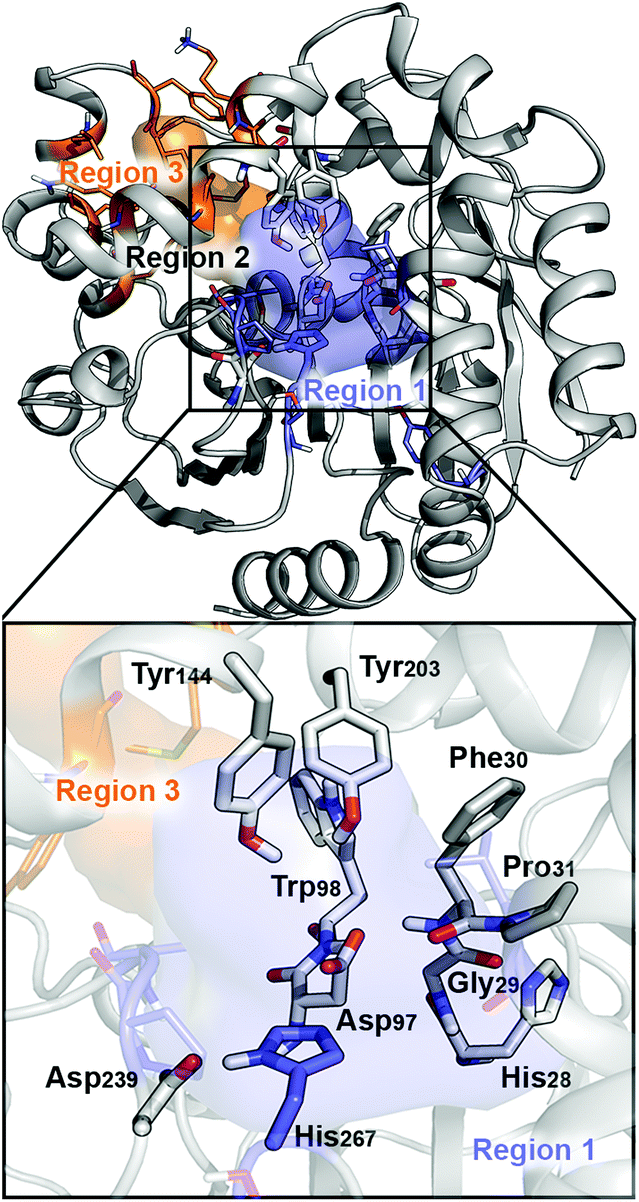 | ||
| Fig. 1 Representation of the active site (region 2) of the BmEH enzyme (PDB: 4NZZ). The most important residues for the reaction are represented in sticks, and non-polar hydrogens are omitted for clarity. The BmEH region 1 (substrate entrance) and region 3 (product release) surfaces are highlighted in blue and orange, respectively. The two possible binding modes are also indicated as “region 1”, i.e. substrate substituent pointing towards His267, and “region 3”, substituent pointing towards Trp98. | ||
EHs catalyse the addition of a water molecule to racemic epoxide-containing substrates to yield optically active 1,2-diols via biocatalytic hydrolytic kinetic resolution (HKR) and enantioconvergent hydrolysis of epoxides.24–28 The mechanism by which these enzymes operate has long been debated.29–33 However, it is generally accepted that EHs from the α,β-hydrolase fold share a common mechanism that takes place via a two-step mechanism through the formation of alkyl-enzyme intermediates (see Scheme 2, with BmEH labelling).34–36 Upon initial binding of the epoxide substrate with Tyr144 and Tyr203 (see Fig. 2), the first step involves the nucleophilic attack by the Asp97 side chain at one of the epoxide carbons to generate a covalently bound alkyl-enzyme intermediate (Int1 in Scheme 2). In the second step of the proposed mechanism, His267 acts as a general base to facilitate the nucleophilic activation of a water molecule to attack the Asp97 carbonyl, generating a new tetrahedral intermediate hereafter called Int2. The negatively charged tetrahedral intermediate is stabilized through hydrogen bond interactions between the backbone amide groups of the oxyanion hole residues Gly29–Phe30 and Asp97–Trp98. In the final step, the tetrahedral intermediate rearranges and dissociates to yield the corresponding 1,2-diol product (Prod in Scheme 2).
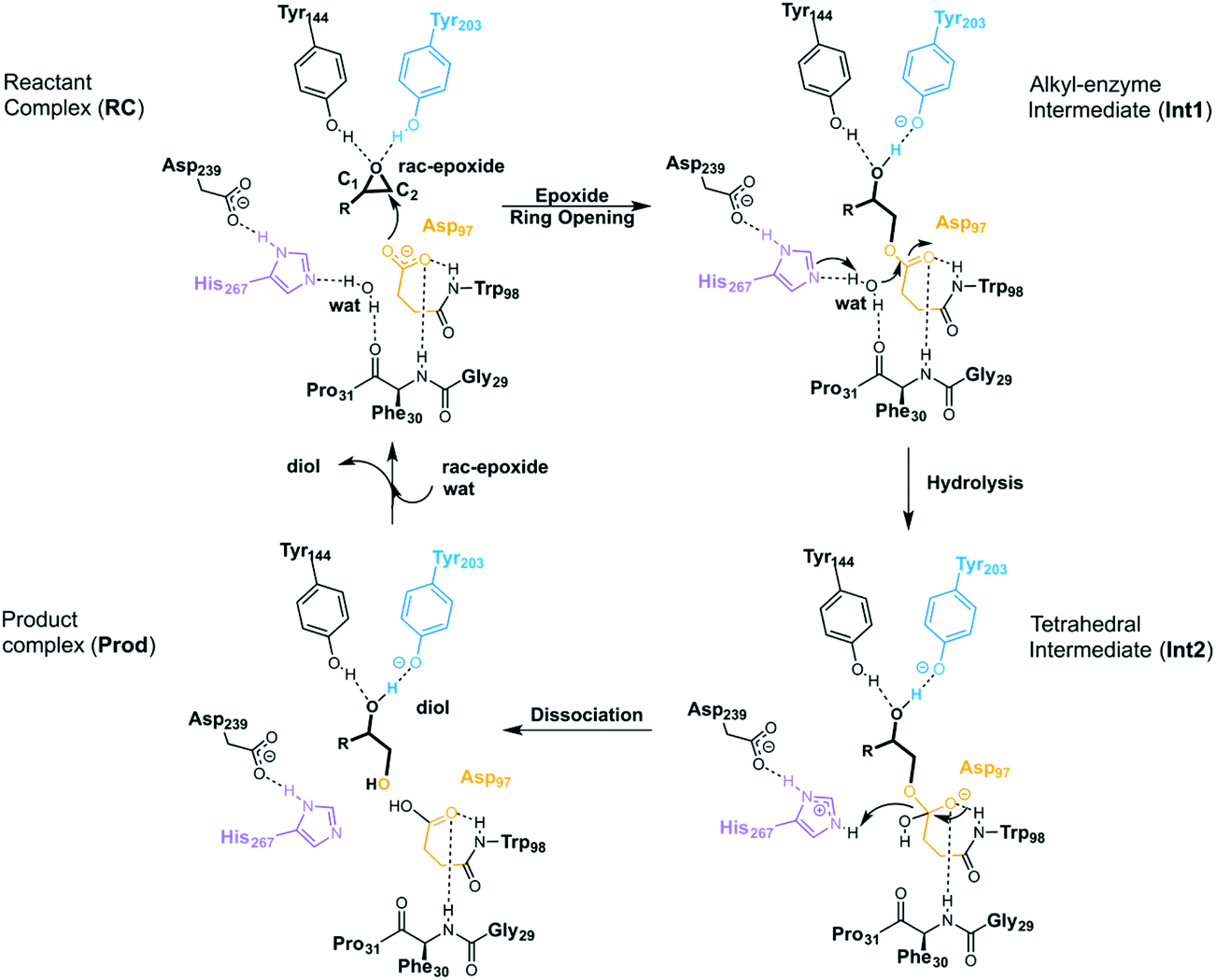 | ||
| Scheme 2 General reaction mechanism of soluble EH enzymes. BmEH wild-type (PDB: 4NZZ labelling). | ||
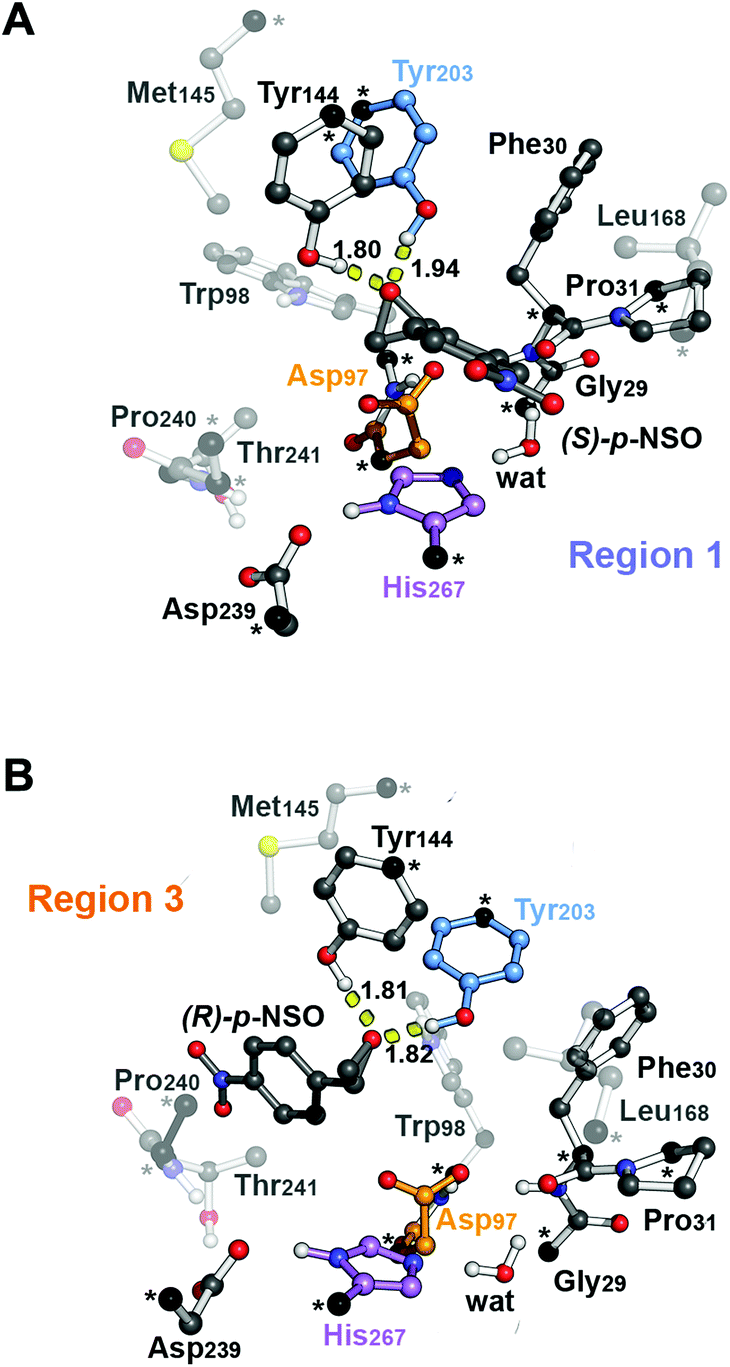 | ||
| Fig. 2 B3LYP-D3BJ/6-311+g(2d,2p)//B3LYP/6-31g(d,p) optimized geometries of the CM enzyme-structure complex (RC) in the active site model with (A) the phenyl substituent of (S)-p-NSO epoxide pointing towards the catalytic His267 residue (region 1, RC-S-r1) and (B) the phenyl substituent of (R)-p-NSO pointing towards Trp98 (region 3, RC-R-r3) (residue labelling as in BmEH). Active site pocket residues are shown as transparent sticks and spheres. Non-polar hydrogen atoms are omitted for clarity. The Asp97 nucleophile, the His267 base and the acid Tyr203 are highlighted in orange, violet, and blue, respectively. Atoms in black spheres and asterisks are kept fixed. | ||
Different computational approaches37 have been used to explore the overall mechanism and selectivity of soluble EHs (sEHs). Truncated theozyme and cluster model calculations have been successfully applied to study the role and impact of enzyme active site residues in catalysed epoxide ring opening reaction mechanisms and selectivities.38,39 Hopmann and Himo applied the cluster model (CM) approach on the X-ray structure of human soluble EH (sEH) to explore the role of the two conserved catalytic tyrosine residues.23 Their results showed that one Tyr is enough for the alkylation reaction to occur, although the barrier increases by ca. 6.7 kcal mol−1 as compared to the wild-type. A drastic change in the activation barriers was observed for the double tyrosine mutant (i.e. larger barriers of ca. 24.8 kcal mol−1 were found after mutating both Tyr to Phe) indicating that this enzyme variant must be inactive. These results demonstrate that the presence of H-bond donors to activate the epoxide ring and stabilize the negative charge developed during the course of the reaction is essential.40
Amrein et al. carried out empirical valence bond (EVB) simulations of the enantio- and regioselective hydrolysis of trans-stilbene oxide (TSO) catalysed by Solanum tuberosum epoxide hydrolase I (StEH1).30 EVB calculations suggested that the alkylation step for (S,S)-TSO at the benzylic C1 position is preferred by 1.7 kcal mol−1, whereas the attack at the terminal C2 is favoured by 3.6 kcal mol−1 for (R,R)-TSO. However, high-energy barriers were found for both enantiomers for the hydrolysis step of the alkyl-enzyme intermediate that is formed after the Asp nucleophilic attack at C1 (Int1 in Scheme 2). Therefore, the regioselectivity of the asymmetric epoxide ring opening of TSO by StEH1 is determined in the hydrolysis step, only the trajectories following the Asp attack at C2 for both enantiomers being productive. They also highlighted the relevance of considering a second active site histidine residue doubly protonated to properly describe the system. This histidine, which interacts with the nucleophilic aspartate, was thought to balance the negative charge developed during the catalysis.
In a very recent study, Lind and Himo applied the CM approach to explore the enantioconvergent resolution of racemic styrene oxide (SO) by using StEH1.41 To investigate the origins of StEH1 selectivity, a very large CM system (of 279 atoms) was designed based on the StEH1 X-ray crystal in complex with the competitive inhibitor valpromide (PDB: 2CJP). The authors investigated the reaction mechanism considering both enantiomers, and examined the two possible binding modes of each enantiomer considering both the shape of the active site pocket and the substitution pattern of the substrate. Their results showed that (S)-SO is preferably attacked at C1 by the catalytic Asp, whereas for the (R)-SO enantiomer the attack at C2 is favoured. Both cases lead to the formation of the (R)-diol, thus highlighting the enantioconvergent behaviour of StEH1 for the SO substrate. This is in contrast to previous results by Amrein et al. obtained for the TSO substrate, for which the reaction was favoured at the C2 position for both enantiomers. For TSO and (S)-SO substrates, the hydrolysis step was found to be the selectivity determining,30,41 whereas for the (R)-SO substrate the alkylation step is found to determine the regioselectivity of the process.
It should also be pointed out that Lind and Himo investigated the role of the protonation state of the histidine placed close to the catalytic Asp in the catalytic reaction. Their computed energy profiles suggested that including the doubly protonated His104 has a minor effect on their computed energy barriers. These studies show how despite all mechanistic studies carried out to date, the fine details of the sEHs mechanism still remain incompletely understood.
As most EHs preferentially accept the (S)-epoxide enantiomer, the comprehension of how (R)-selective BmEH operates, and the main factors that contribute to its selectivity and efficiency is of great interest. In this study, we computationally investigate the origins of the enantio- and regioselectivity in BmEH towards rac-SO and its derivative rac-p-NSO using density functional theory (DFT) calculations within the cluster model framework. We have computed the full reaction profile using a consciously designed quantum CM from the BmEH active site (PDB: 4NZZ) consisting of 195 and 197 atoms for rac-SO and rac-p-NSO, respectively (see Fig. 2 and Computational methods). Our results show that styrene oxide and its para-nitro derivative switch the inherent BmEH enantio- and regioselectivity, and demonstrate the minor effect of the nitro group on the enzyme selectivity.
2. Results and discussion
The BmEH enzyme presents different features in comparison with other EHs. The BmEH crystal structure, shown in Fig. 1, revealed that this enzyme has an independent product-release site (called region 3) that is not found in other EHs.6 The active site tunnel (region 2) connecting the substrate-entrance site (region 1) and the product-release site (region 3) was previously identified by an 80 ns Molecular Dynamics (MD) simulation.6 BmEH exhibits opposite (R)-selectivity in comparison with many other (S)-specific EHs, thereby providing an alternative approach to obtain those optically pure (S) aromatic epoxides, commonly identified as building blocks of active β-adrenergic agents.12 Many experimental studies reported on how the structure of the substrate or the presence of substituents on the aromatic ring moiety can switch the inherent selectivity of EHs.42 The inherent (R)-selectivity that BmEH exhibits for phenyl glycidyl ethers (PGE) is switched towards (S)-selectivity when the styrene oxide (SO) substrate and its p-NO2 derivative are considered.12 In order to shed some light on the role of the substrate structure and its substitution pattern, as well as to explore the origins of BmEH selectivity, we performed DFT-D3BJ calculations of both racemic styrene oxide (rac-SO) and racemic para-nitro styrene oxide (rac-p-NSO). Based on the X-ray BmEH wild-type structure and our MD simulations on the alkyl intermediate Int1, we have devised a truncated cluster model (CM) as done by Himo in previous studies (see Fig. 2 and Computational methods in the ESI†).29,43To investigate the origins of BmEH selectivity we have computed the EH mechanism described in Scheme 2, considering the first nucleophilic attack either at the epoxide-ring benzylic C1 or the terminal C2 positions for each enantiomer, but also accounting for the two possible substrate orientations regarding the BmEH active site tunnel regions (see Fig. 1 and 2).
Mechanism and selectivity of the BmEH enzyme with the rac-p-NSO substrate
The computed reaction energy profiles at the B3LYP-D3BJ/6-311+g(2d,2p)//B3LYP/6-31g(d,p) level of theory for all possible outcomes of rac-p-NSO BmEH hydrolysis are represented in Fig. 3. The DFT optimized structures of intermediates (Int), products (Prod), and transition states (TS) for the attack at the benzylic position (C1) of (S)-p-NSO oriented towards region 1 are shown in Fig. 4.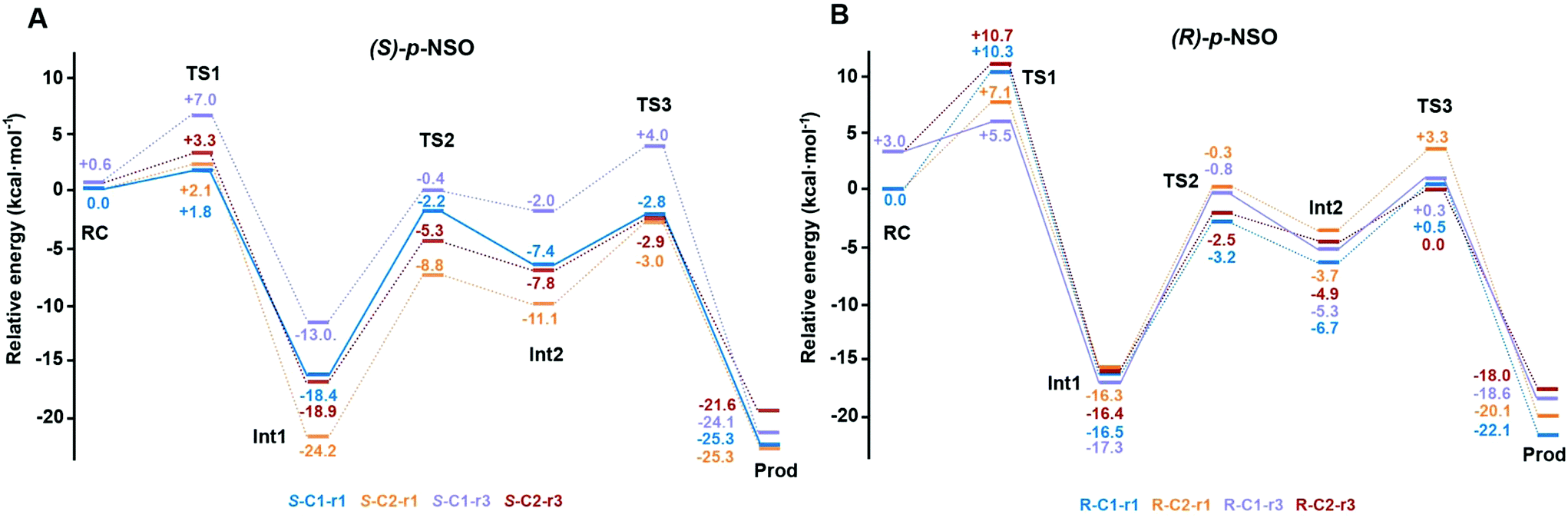 | ||
| Fig. 3 Energy profiles at the B3LYP-D3BJ/6-311+g(2d,2p)//B3LYP/6-31g(d,p) level of theory for all possible BmEH-mediated epoxide rac-p-NSO hydrolysis outcomes. All energies are in kcal mol−1 and referenced to the lowest energy RC for each enantiomer, RC-S-r1 and RC-R-r1, respectively. The kinetically favoured attack for each enantiomer is shown as solid lines (S-C1-r1 and R-C1-r3), the other possible pathways are shown as dashed lines. | ||
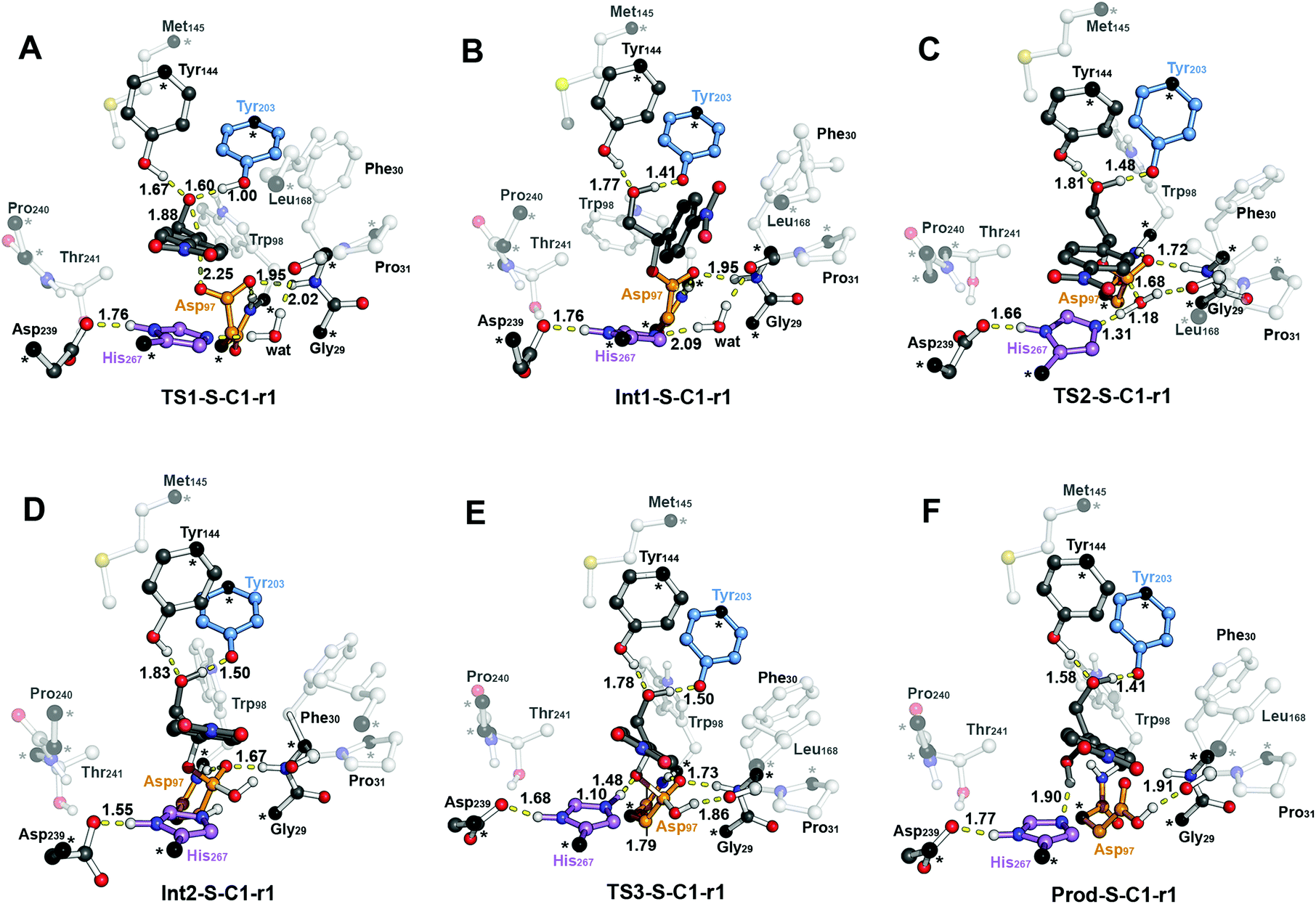 | ||
| Fig. 4 B3LYP-D3BJ/6-311+g(2d,2p)//B3LYP/6-31g(d,p) optimized geometries corresponding to the different steps of the BmEH mechanism of action for the (S)-p-NSO substrate oriented towards region 1 for the attack at the benzylic position (C1): (A) epoxide ring opening (TS1-S-C1-r1), (B) alkyl-enzyme intermediate (Int1-S-C1-r1), (C) hydrolysis of the alkyl-enzyme intermediate (TS2-S-C1-r1), (D) tetrahedral intermediate (Int2-S-C1-r1), (E) dissociation of tetrahedral (TS3-S-C1-r1) and (F) product complex (Prod-S-C1-r1). All distances are in Å. Active site pocket residues are shown as transparent sticks and spheres and non-polar hydrogen atoms are omitted for clarity. Atoms in black spheres and asterisks are kept fixed. | ||
From our computed energy profiles, small energy differences exist between both region 1 and 3 orientations of the substrate (ca. 0.6 kcal mol−1) in the enzyme–substrate complexes (RC) for the (S)-enantiomer, whereas a difference of ca. 3.0 kcal mol−1 is found for (R)-p-NSO RC. As shown in Fig. 2, in the DFT optimized RC geometries, the epoxide ring forms hydrogen bonds with both Tyr144 and Tyr203 residues, and C1 and C2 carbons are well pre-organized for the subsequent Asp97 nucleophilic attack. The carboxylate side-chain of Asp97 is well positioned for the catalysis thanks to hydrogen bond interactions with the amide backbone groups of Asp97–Trp98 and Gly29–Phe30. The ion-pair charge relay system consisting of Asp239–His267 residues properly interacts for assisting the water molecule activation. In addition, the position of the nucleophilic water is maintained fixed at the active site by hydrogen bonds to the general base His267 and to the backbone carbonyl group of Phe30–Pro31 residues.
The first step of the reaction mechanism after the enzyme–substrate complex (RC-S-r1) consists of the nucleophilic attack of Asp97 either at the most substituted carbon C1 (inverting its configuration) or at the less sterically hindered C2 of the epoxide ring to form an ester intermediate (retaining configuration, Int1-S-C1-r1, see Fig. 4B). The optimized TS for the Asp97 nucleophilic attack at the C1 position in (S)-p-NSO (TS1-S-C1-r1) exhibits a 2.25 Å distance between the O-Asp97 side-chain and the epoxide C1 atom. The distance between the epoxide oxygen and C1 atom is elongated up to 1.88 Å (see Fig. 4A), corresponding to an SN2-like concerted TS. The O–H distance of Tyr203 is slightly elongated (from 0.98 Å to 1.00 Å) in the alkylation TS, and the proton is completely transferred from Tyr203 to the epoxide O atom in the optimized covalently-bound enzyme intermediate Int1-S-C1-r1 (see Fig. 4B). The associated reaction barrier for TS1-S-C1-r1 is 1.8 kcal mol−1 relative to the reactant complex (RC-S-r1), and is 0.3 kcal mol−1 lower in energy than TS1-S-C2-r1 corresponding to the attack at the less hindered C2 position (see Fig. 3A). These results show that the intrinsic preference of the enzyme is to perform the nucleophilic attack at the C1 position of (S)-p-NSO. The resulting Int1-S-C1-r1 is stabilized by 18.4 kcal mol−1. Regarding the (R)-p-NSO energy profile diagram, the lowest alkylation TS (TS1-R-C1-r3) has an energy of 5.5 kcal mol−1 relative to RC-R-r1 (see Fig. 3B). Comparing both enantiomers, the lowest alkylation transition state (TS1) for each enantiomer differs by ca. 3.7 kcal mol−1. Note that the orientation of the phenyl ring of the epoxide is different for each case (see Fig. 4A and Fig. S2†). For the (S)-enantiomer, the phenyl substituent points towards His267, i.e. here called region 1, whereas the lowest alkylation energy barrier for the (R)-enantiomer is found when in the TS1 the phenyl substituent points towards Trp98 i.e. region 3 (see Fig. 2B). The analysis of both TS geometries indicates that the additional stabilization of TS1-S-C1-r1 might be due to the π-stacking interaction between (S)-p-NSO and the His267 residue, which is not possible in the R-C1-r3 case (see Fig. 4A and 5). Thus, at this first stage of the BmEH mechanism, our computed energy profiles indicate that the epoxide ring opening of (S)-p-NSO is more favoured than the (R)-enantiomer.
The next step in the reaction mechanism includes the nucleophilic attack by a water molecule (wat) to hydrolyse the previously formed ester bond, and the dissociation of the tetrahedral intermediate (Int2) generated at this half-hydrolytic reaction to yield the vicinal diol (Prod, see Scheme 2). In the DFT optimized TS2-S-C1-r1, the neutral His267 residue acts as a general base to activate the water molecule that attacks Asp97 generating the Int2-S-C1-r1 intermediate, as shown in Fig. 4C. The activation of the water molecule is possible because at TS2-S-C1-r1 the Asp239–His267 distance has been substantially shortened, allowing the stabilization of the protonated state of His267. Likewise, the observed distances in Int2-S-C1-r1 between the carbonyl oxygen atom of Asp97 and the amide bonds of Asp97–Trp98 and Gly29–Phe30 in the oxyanion hole indicate that the negative charge that arises at this stage is well stabilized (shorter distances of ca. 1.67 Å, see Fig. 4D). All these observed networks of interactions allow TS2 stabilization, thereby making it possible to overcome the computed Int1 → TS2 barriers of 16.2 kcal mol−1. Similar energy barriers were found for the most favoured (R)-enantiomer attack (TS2-R-C1-r3, ΔE‡ = 16.5 kcal mol−1), in which the covalent enzyme intermediate Int1-R-C1-r3 was found to be stabilized by 17.3 kcal mol−1.
To generate the final diol product, a final step is needed in which the C–O bond between the epoxide substrate and Asp97 is cleaved, followed by subsequent protonation (see Scheme 2). The corresponding transition state TS3-S-C1-r1 (ΔE‡ = 4.6 kcal mol−1 with respect to Int2) presents elongated C–O distances (ca. 1.79 Å), and the protonated His267 interacts with the (S)-p-NSO substrate delivering a proton for 1,2-diol formation (ca. 1.48 Å) as shown in Fig. 4E. Therefore, the catalytic His267 found to be important in the previous step for catalysing the hydrolysis acting as a general base has also a critical role at this final step for 1,2-diol generation. Note that this C–O bond breaking generates a diol product in which one of the oxygen atoms comes from the nucleophilic Asp97 residue (see Scheme 2). The activation barrier for the dissociative transition state TS3 for the (R)-enantiomer (TS3-R-C1-r3) is 5.6 kcal mol−1 with respect to Int2. As previously mentioned for the alkylation transition state (TS1), since for (R)-p-NSO the phenyl ring points towards region 3, this might lead to a less stabilized TS3 due to the lack of π-stacking interaction with the catalytic His267 residue (see Fig. S3†).
As shown in Fig. 4F, the new hydroxyl group in the product Prod-S-C1-r1 interacts with Tyr144-O–H (ca. 1.58 Å) and the deprotonated Tyr203-O− (ca. 1.41 Å). Prod-S-C1-r1 is −25.3 kcal mol−1 more stable than RC-S-r1, whereas for the (R)-enantiomer Prod-R-C1-r3 is −18.6 kcal mol−1 lower in energy than RC-R-r1. In all optimized Prod structures, Asp97 is in its protonated state whereas Tyr203 remains deprotonated. Thus, an additional acid–base step is needed, and the binding of a new epoxide and water molecule is required to regenerate the catalytic cycle (see Scheme 2). These steps are difficult to be accurately described with the current methodology.41 As proposed by Himo in previous studies,41 the overall energetics of the process can be roughly approximated by computing the free energy of the (S)-epoxide + water → (R)-1,2-diol reaction, which is exergonic by 8 kcal mol−1. This large energetic span between Prod-S-C1-r1 and RC (−8 + 25.3 = 17.3 kcal mol−1) should be added to the alkylation activation barrier of the next cycle, thus yielding an approximated barrier of ca. 19 kcal mol−1 for (S)-p-NSO. This estimation indicates that both alkylation and hydrolysis steps present similar barriers. Due to the large inaccuracies associated with the estimation of the Prod → React process, we focus our analysis on the computed reaction pathway displayed in Fig. 3, which shows that alkylation is selectivity-determining, whereas hydrolysis is rate-determining. Thus, to directly compare the reactivities of (S) and (R) substrates, we evaluate the activation barriers for the rate-determining hydrolysis step in the pathways that have the lowest alkylation barriers (TS1) for each enantiomer, i.e.S-C1-r1 and R-C1-r3, respectively.
Overall our computed reaction pathways indicate that the lowest energy intermediate corresponds to Int1, and the rate-determining transition state is TS2 for S-C1-r1 and TS3 for R-C1-r3 both corresponding to the dissociation of the covalently bound enzyme intermediate (Int1 in Scheme 2). The computed barriers are 16.2 kcal mol−1 for (S)-p-NSO (S-C1-r1 in Fig. 3A), whereas 17.6 kcal mol−1 for (R)-p-NSO (R-C1-r3 in Fig. 3B). Therefore, the hydrolysis of the (S)-enantiomer is kinetically favoured by ca. 1.4 kcal mol−1, which is in agreement with experimental observations for BmEH.12 It should also be noted that our computed activation barriers are in line with the experimental rate constants of 3–10 s−1 at 30 °C (ca. 16–17 kcal mol−1) for the related StEH1 enzyme.44,45
Substituent effect on the BmEH selectivity
Using DFT-D3BJ calculations we also examined the hydrolysis of the racemic styrene oxide (rac-SO) substrate to further explore how the substrate structure and its substitution pattern affect BmEH selectivity. To this aim, we also considered all possible outcomes for rac-SO, i.e. the nucleophilic attack either at benzylic C1 or terminal carbon C2 for each enantiomer oriented towards region 1 or 3 as described above (see Fig. 1 and Fig. S4†).The computed energy profiles for the rac-SO substrate follow a similar trend to those discussed in the previous section for its para-nitro derivative substrate rac-p-NSO (see Fig. 3 and Fig. S4†). As observed for rac-p-NSO energy profiles, the most favoured outcome for (S)-SO epoxide comes from the nucleophilic attack at the benzylic position (C1) when the phenyl moiety of the substrate is oriented towards His267 (ΔE‡ = 4.0 kcal mol−1 with respect to RC-S-SO-r1, and ΔE‡ = 15.3 kcal mol−1 computed from the lowest energy Int1, for TS3-S-SO-C1-r1).
For the (R)-enantiomer, the kinetically favored attack is found when the phenyl ring is oriented towards region 3. The alkylation barrier of TS1-R-SO-C1-r3 is 6.6 kcal mol−1 regarding the lowest energy RC for (R)-SO (RC-R-SO-r1, see Fig. S4†). The alkylation at position C2 is ca. 0.5 kcal mol−1 less favorable than the attack at C1 (TS1-R-SO-C2-r3). The hydrolysis step of Int1 is kinetically more favoured at the terminal carbon C2 as the computed activation barriers are ca. 19.8 and 17.4 kcal mol−1 for TS3-R-SO-C1-r3 and TS3-R-SO-C2-r3, respectively (computed from the lowest energy intermediate Int1).
As observed for p-NSO, the activation barriers obtained (shown in Fig. S4†) for (S)-SO at C1 are ca. 2.6 kcal mol−1 lower than those obtained for (R)-SO, thus suggesting that the nitro group has a very minor effect on the enantio- and regioselectivity of the process.
Overall our mechanistic studies also indicate that both rac-p-NSO and rac-SO switch the inherent (R)-BmEH selectivity observed towards PGE substrates, as described in previous experimental studies. A higher enantioselectivity of BmEH towards rac-p-NSO (ees ≥ 99%) than for rac-SO (ees = 53%) was observed experimentally.12 The energy differences between our computed activation barriers overestimate the ee for rac-SO. However, the relative stabilities of the RCs indicate that the binding of (R)-epoxides for both p-NSO and SO is more favourable than for (S), especially in region 1 due to the higher stabilization by non-covalent interactions with the active site residues, including π⋯π with His267 (see Fig. 5). Our computed energy differences between substrate bound RCs ((R) vs. (S)) for p-NSO are ca. 3.0 kcal mol−1, while it is only 1.4 kcal mol−1 for rac-SO. Thus, the higher enantioselectivity observed experimentally for rac-p-NSO can be attributed to the more favourable unproductive binding of (R)-p-NSO in region 1, as in this region the activation barriers for the alkylation and hydrolysis step are ca. 2 kcal mol−1 higher than in region 3.
 | ||
| Fig. 5 Non-covalent interaction representation of the lowest energy alkylation transition states (A) TS1-S-C1-r1 and (B) TS1-R-C1-r3 for the rac-p-NSO epoxide ring opening reaction using the NCIplot computational tool.44,45 NCI surfaces show intermolecular interactions between the epoxide substrate and the active site residues included in the CM model. All residues are represented as balls and sticks and the two possible orientations for each substrate are indicated for each case (region 1, His267 and region 3, Trp98). Only the most important catalytic residues are labelled regarding BmEH (PDB: 4NZZ numbering) and non-polar hydrogen are not shown for clarity. Grey arrows are used to highlight the most important noncovalent interactions. The most important residues are framed. | ||
Our calculations show that in the BmEH catalysed hydration of rac-p-NSO and rac-SO, TS3 energies are in general lower than TS1. This is in contrast to rac-SO hydrolysis by StEH1,41 where Himo and coworkers found that the hydrolysis step (TS3) is generally higher in energy than TS1. Although the energy differences are rather small (ca. ±3 kcal mol−1), our results show that the shape and conformation of the active site pocket, as well as the substrate orientation will modulate the selectivity-determining step, i.e. the alkylation (TS1) or the hydrolysis (TS3).
3. Conclusions
Our DFT-D3BJ calculations within the CM framework indicate that the substrate structure switches the usual BmEH (R)-selectivity towards (S) when racemic styrene oxide (rac-SO) and racemic para-nitro styrene oxide (rac-p-NSO) are considered. The computed energy profiles for the different substrates and enantiomers suggest that the first alkylation step (TS1) is responsible for the regioselectivity of the process, while the hydrolysis half reaction is the rate-limiting step. In both rac-SO and rac-p-NSO the (S)-enantiomer is preferentially attacked at the benzylic position (C1) when the phenyl ring moiety points towards region 1 (i.e. the substrate entrance). This orientation maximizes the π⋯π interactions between the substrate (both SO, and p-NSO) and the active site residues, in particular His267. Therefore, the catalytically competent poses of the (S)-enantiomer of the aromatic SO and p-NSO substrates are found in region 1. The least reactive (R)-epoxide has a preferential binding for region 1, however in this region the activation barriers are substantially higher as compared to those in region 3. This work demonstrates that the enantiopreference of BmEH towards aromatic epoxides can be directly assessed by the combination of DFT-D3BJ and the analysis of non-covalent interactions, and provides an alternative strategy for the engineering of EHs towards the resolution of synthetically useful epoxides.Computational methods
Full geometry optimizations were performed with the hybrid DFT B3LYP functional46,47 and the 6-31G(d) basis set48,49 using Gaussian 09,50 a widely accepted level of theory for studying mechanisms of organic reactions.51,52 The effects of the active site environment of the protein were implicitly included in all calculations using the Conductor-like Polarizable Continuum Model (CPCM)53,54 with diethyl ether as the solvent (ε = 4). The transition states (TS) were located by using a Bofill update TS search on a reduced potential energy surface.55 Analytical frequency calculations were performed at the same level of theory as the geometry optimizations to obtain the zero point energies (ZPE). The nature of the stationary points was determined in each case according to the Hessian matrix eigenvalues. We have also checked that imaginary frequencies exhibit the expected motion, while transition states have also been verified by intrinsic reaction coordinate (IRC) calculations. More accurate energies were obtained by single-point calculations including the DFT hybrid B3LYP-D3BJ dispersion correction with the larger 6-311+G(2d,2p) basis set. All energies are ZPE-corrected.The epoxide substrates used to model the mechanisms were the racemic styrene oxide (rac-SO) and its racemic para nitro-substituted derivative (rac-p-NSO, cluster models of 195 and 197 atoms, respectively). To explore the origins of the enantio- and regioselectivity of BmEH and the effect of the presence of the NO2 group, all plausible reaction mechanisms should be considered. Thus, a total of 16 reaction pathways need to be computed to take into account that the EH hydrolysis of rac-epoxides can take place via an attack on either carbon atom of each enantiomer, and also two possible binding orientations of the substrate on the basis of the proposed BmEH active site tunnel.6 The two possible binding poses arise from the orientation of the phenyl moiety of the substrate towards either region 1 (r1) placing the phenyl moiety close to His267, or region 3 (r3) when it points towards Trp98 (see Fig. 1 and 2).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
E. S.-H. thanks the Generalitat de Catalunya for a PhD fellowship (2017-FI-B-00118), M. G.-B. is grateful to the Ramón Areces Foundation for a Postdoctoral Fellowship. S. O. thanks the Spanish MINECO CTQ2014-59212-P, Ramón y Cajal contract (RYC-2014-16846), the European Community for CIG project (PCIG14-GA-2013-630978), and the funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (ERC-2015-StG-679001). We are grateful for the computer resources, technical expertise, and assistance provided by the Barcelona Supercomputing Center – Centro Nacional de Supercomputacion.Notes and references
- A. Archelas and R. Furstoss, Curr. Opin. Chem. Biol., 2001, 5, 112–119 CrossRef CAS PubMed.
- E. J. de Vries and D. B. Janssen, Curr. Opin. Biotechnol., 2003, 14, 414–420 CrossRef CAS PubMed.
- A. Archelas and R. Furstoss, Trends Biotechnol., 1998, 16, 108–116 CrossRef CAS PubMed.
- P. Besse and H. Veschambre, Tetrahedron, 1994, 50, 8885–8927 CrossRef CAS.
- K. Michael, A. Alain and W. Roland, Curr. Org. Chem., 2012, 16, 451–482 CrossRef.
- X.-D. Kong, S. Yuan, L. Li, S. Chen, J.-H. Xu and J. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15717–15722 CrossRef CAS PubMed.
- F. E. Held, S. Wei, K. Eder and S. B. Tsogoeva, RSC Adv., 2014, 4, 32796–32801 RSC.
- A. Schmid, K. Hofstetter, H. J. Feiten, F. Hollmann and B. Witholt, Adv. Synth. Catal., 2001, 343, 732–737 CrossRef CAS.
- P. Berglund, ChemBioChem, 2006, 7, 1280–1280 CrossRef CAS.
- M. Smit and M. Labuschagné, Curr. Org. Chem., 2006, 10, 1145–1161 CrossRef CAS.
- B. Van Loo, J. Kingma, M. Arand, M. G. Wubbolts and D. B. Janssen, Appl. Environ. Microbiol., 2006, 72, 2905–2917 CrossRef CAS PubMed.
- J. Zhao, Y.-Y. Chu, A.-T. Li, X. Ju, X.-D. Kong, J. Pan, Y. Tang and J.-H. Xu, Adv. Synth. Catal., 2011, 353, 1510–1518 CrossRef CAS.
- Y.-F. Tang, J.-H. Xu, Q. Ye and B. Schulze, J. Mol. Catal. B: Enzym., 2001, 13, 61–68 CrossRef CAS.
- P.-F. Gong and J.-H. Xu, Enzyme Microb. Technol., 2005, 36, 252–257 CrossRef CAS.
- Y. Xu, J.-H. Xu, J. Pan and Y.-F. Tang, Biotechnol. Lett., 2004, 26, 1217–1221 CrossRef CAS PubMed.
- M. T. Reetz, C. Torre, A. Eipper, R. Lohmer, M. Hermes, B. Brunner, A. Maichele, M. Bocola, M. Arand and A. Cronin, Org. Lett., 2004, 6, 177–180 CrossRef CAS PubMed.
- M. T. Reetz, L. W. Wang and M. Bocola, Angew. Chem., Int. Ed., 2006, 118, 1258–1263 CrossRef.
- P. Moussou, A. Archelas, J. Baratti and R. Furstoss, J. Org. Chem., 1998, 63, 3532–3537 CrossRef CAS.
- S. Barth, M. Fischer, R. D. Schmid and J. Pleiss, Proteins: Struct., Funct., Bioinf., 2004, 55, 846–855 CrossRef CAS PubMed.
- M. Nardini and B. W. Dijkstra, Curr. Opin. Struct. Biol., 1999, 9, 732–737 CrossRef CAS PubMed.
- D. Lindberg, S. Ahmad and M. Widersten, Arch. Biochem. Biophys., 2010, 495, 165–173 CrossRef CAS PubMed.
- R. N. Armstrong and C. S. Cassidy, Drug Metab. Rev., 2000, 32, 327–338 CrossRef CAS PubMed.
- K. H. Hopmann and F. Himo, J. Phys. Chem. B, 2006, 110, 21299–21310 CrossRef CAS PubMed.
- R. N. Patel, Green Biocatalysis, John Wiley & Sons, 2016 Search PubMed.
- W. J. Choi, Appl. Microbiol. Biotechnol., 2009, 84, 239–247 CrossRef CAS PubMed.
- P. Bauer, A. J. Carlsson, B. A. Amrein, D. Dobritzsch, M. Widersten and S. C. L. Kamerlin, Org. Biomol. Chem., 2016, 14, 5639–5651 CAS.
- D. Lindberg, A. Gogoll and M. Widersten, FEBS J., 2008, 275, 6309–6320 CrossRef CAS PubMed.
- E. Y. Lee and M. L. Shuler, Biotechnol. Bioeng., 2007, 98, 318–327 CrossRef CAS PubMed.
- K. H. Hopmann and F. Himo, Chem. – Eur. J., 2006, 12, 6898–6909 CrossRef CAS PubMed.
- B. A. Amrein, P. Bauer, F. Duarte, A. J. Carlsson, A. Naworyta, S. L. Mowbray, M. Widersten and S. C. L. Kamerlin, ACS Catal., 2015, 5, 5702–5713 CrossRef CAS PubMed.
- K. H. Hopmann and F. Himo, J. Phys. Chem. B, 2006, 110, 21299–21310 CrossRef CAS PubMed.
- B. Schiott and T. C. Bruice, J. Am. Chem. Soc., 2002, 124, 14558–14570 CrossRef CAS PubMed.
- M. T. Reetz, M. Bocola, L.-W. Wang, J. Sanchis, A. Cronin, M. Arand, J. Zou, A. Archelas, A.-L. Bottalla, A. Naworyta and S. L. Mowbray, J. Am. Chem. Soc., 2009, 131, 7334–7343 CrossRef CAS PubMed.
- R. N. Armstrong, Drug Metab. Rev., 1999, 31, 71–86 CrossRef CAS PubMed.
- G. M. Lacourciere and R. N. Armstrong, J. Am. Chem. Soc., 1993, 115, 10466–10467 CrossRef CAS.
- C. Morisseau and B. D. Hammock, Annu. Rev. Pharmacol. Toxicol., 2005, 45, 311–333 CrossRef CAS PubMed.
- A. Romero-Rivera, M. Garcia-Borràs and S. Osuna, Chem. Commun., 2017, 53, 284–297 RSC.
- K. Hotta, X. Chen, R. S. Paton, A. Minami, H. Li, K. Swaminathan, I. I. Mathews, K. Watanabe, H. Oikawa, K. N. Houk and C.-Y. Kim, Nature, 2012, 483, 355–358 CrossRef CAS PubMed.
- Y. Zou, M. Garcia-Borràs, M. C. Tang, Y. Hirayama, D. H. Li, L. Li, K. Watanabe, K. N. Houk and Y. Tang, Nat. Chem. Biol., 2017, 13, 325–332 CrossRef CAS PubMed.
- L. T. Elfström and M. Widersten, Biochem. J., 2005, 390, 633–640 CrossRef PubMed.
- M. E. S. Lind and F. Himo, ACS Catal., 2016, 8145–8155 CrossRef CAS.
- P. Moussou, A. Archelas, J. Baratti and R. Furstoss, Tetrahedron: Asymmetry, 1998, 9, 1539–1547 CrossRef CAS.
- K. H. Hopmann, B. M. Hallberg and F. Himo, J. Am. Chem. Soc., 2005, 127, 14339–14347 CrossRef CAS PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- G. Kiss, N. Celebi-Oelcuem, R. Moretti, D. Baker and K. N. Houk, Angew. Chem., Int. Ed., 2013, 52, 5700–5725 CrossRef CAS PubMed.
- F. Himo, Theor. Chem. Acc., 2006, 116, 232–240 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404–417 CrossRef CAS.
- S. K. Burger and P. W. Ayers, J. Chem. Phys., 2010, 132, 234110 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ob01847a |
| This journal is © The Royal Society of Chemistry 2017 |