
Supramolecular organization of a H-bonded perylene bisimide organogelator determined by transmission electron microscopy, grazing incidence X-ray diffraction and polarized infra-red spectroscopy†
Alexandru
Sarbu
a,
Patrick
Hermet
bc,
David
Maurin
d,
David
Djurado
e,
Laure
Biniek
a,
Morgane
Diebold
a,
Jean-Louis
Bantignies
*d,
Philippe
Mésini
*a and
Martin
Brinkmann
 *a
*a
aUniversité de Strasbourg, CNRS, ICS UPR22, F67000 Strasbourg, France. E-mail: martin.brinkmann@ics-cnrs.unistra.fr; Philippe.mesini@ics-cnrs.unistra.fr
bInstitut Charles Gerhardt Montpellier, UMR 5253 CNRS-UM-ENSCM, Place E. Bataillon, 34095 Montpellier, France
cInstitut Laue Langevin, 6 Rue Jules Horowitz, B.P. 156, 38042 Grenoble, France
dLaboratoire Charles Coulomb, UMR 5221 CNRS-Université de Montpellier, 34095 Montpellier, France. E-mail: Jean-Louis.Bantignies@univ-montp2.fr
eUniversité Grenoble Alpes, CNRS, CEA, INAC, SyMMES, F-38000 Grenoble, France
First published on 20th November 2017
Abstract
An organogelator based on a N,N′-substituted H-bonding perylenebisimide (PBI-C10) self-assembles to form either a green J-type (form I) or a red H-type (form II) aggregate structure. The molecular packing of both polymorphs was determined from a combination of Transmission Electron Microscopy (TEM) (low dose electron diffraction and high resolution), Grazing incidence X-ray diffraction and polarized infrared spectroscopy. To that aim, highly oriented films have been prepared by mechanical rubbing at controlled film temperature and DFT calculations were performed to identify representative vibrational IR bands and their associated polarizations. H-Bonding between amides generates either a rectangular columnar phase (form I) in the dried gel or a hexagonal packing of supramolecular 21/1 helices with a long period of 97 Å (form II) in annealed thin films. In aligned films of form I, polarized FTIR spectroscopy helps determine the orientation of both intermolecular H-bonds and the PBI core with respect to the substrate. In form II, PBI-C10 molecules assemble into pairs to form off-centered 21/1 helices whose helical axis is made of strongly H-bonded amides. TEM investigations show that three 21/1 helices are packed in a frustrated trigonal structure formed by H-bonding. The Form I → Form II transformation implies a redistribution of a single population of strong intra-columnar H-bonds between amides in form I to a mixture of strong and weak H-bonds in the supramolecular helices, the strong H-bonds forming the spine of the helices.
I. Introduction
Self-assembly of conjugated semi-conductors is a powerful bottom-up approach for the design of new materials of interest in electronic and optoelectronic applications.1–5 π-Conjugated organogelators are an original class of functional materials because their self-assembly in certain organic solvents leads usually to anisotropic 1D nanostructures such as nanofibrils while bearing interesting fluorescence properties and/or p- or n-type semi-conductivity.6–10 The gelation of these systems arises from the buildup of a 3D network of intermingled nanofibrils in given organic solvents. During the last decade, various organogelators based on conjugated oligomers of phenylenevinylene, thienylenevinylene or thiophene were synthesized and their charge transport or fluorescence properties were investigated.6–17 Among all conjugated systems, perylene-3,4:9,10-tetracarboxydiimides (PBI) has attracted much attention because of its pronounced 1D π-stacking order and because it is one of the few n-type conjugated semiconductors used in Organic Field Effect Transistors (OFETs), organic solar cells or stimuli-responsive thin films.18–29 Moreover, PBI is a widely-used building block for the design of alternated co-polymers, donor–acceptor co-oligomers or organogelators because the aromatic core can be substituted in various positions (imide, ortho or bay).30–37 Functionalization of PBIs with H-bonding groups such as amides is a common way to impart self-assembling properties that might result in organogelating properties while other substitutions can lead to the buildup of helical columnar mesophases.2–6,20,38Besides the control of electronic properties of PBIs via chemical engineering, mastering their structure and polymorphism is key since molecular stacking determines the electronic properties in the solid state. More specifically, PBI systems are characterized by a typical crystallochromism: they form either J- or H-type aggregates whose optical absorption or fluorescence depend on the overlap between PBIs in the crystal.39–42 The generated structures depend on the preparation conditions (solvent, temperature,…).43–47 The strong structure–properties correlation in PBIs is particularly interesting to design stimuli-responsive materials.26,27,36,47 As an example, N,N′-siloxane substituted PBIs can form mechanoresponsive materials since a mechanical stress on a thin film modifies substantially their fluorescence.36
Herein we focus on the compound PBI-C10 (Fig. 1a) which Würthner and coworkers47,48 originally described as forming J-type fibrillar aggregates in p-xylene. As recent studies have demonstrated, this molecule forms two polymorphs with typical J- and H-type aggregate properties that can be interconverted by two different stimuli i.e. temperature or the presence of H-bonding non-solvents.47 The green J-type form I obtained in thin films cast from a p-xylene solution transforms into the red H-type form II via annealing at 190 °C. The reverse transformation is induced when form II films are dipped in liquid H-bonding non-solvents such as linear alcohols or acetone. The underlying structural transformation rests on a redistribution of H-bonds as inferred by the analysis of the amide A bands as well as structural studies by AFM and TEM.47 Moreover, PBI-C10 molecules generate helical stacks in the red form II with a long period of 97 Å, contrary to the simple columnar phase of form I. However, in the absence of highly organized and oriented systems, no complete structural model could be obtained by TEM or GIXD for both polymorphs. This contribution describes a more detailed structural analysis of the polymorphs of PBI-C10. To this aim, means to prepare highly aligned films of both polymorphs have been investigated. In particular, it is shown that high-temperature rubbing yields highly aligned films of organogelating systems. While this method has been recently introduced to prepare highly oriented films of various p- and n-type semiconducting polymers, it was never used to align self-assembling molecules such as H-bonding PBIs investigated herein.49–53 A combination of GIXD, TEM and polarized FTIR spectroscopy combined with proper modeling (DFT calculations) helps uncover the packing of both forms. Whereas form I is poorly ordered, form II reveals supra-molecular long range order and an interesting trigonal structure that harbors three isochiral 21/1 helices. Despite low structural order in form I, linearly polarized FTIR spectroscopy evidences a high level of molecular orientation in the aligned thin films as inferred from the high dichroism of FTIR bands related to intermolecular H-bonding and to the PBI core. It is demonstrated that form II is an interesting example of a frustrated structure formed by H-bonding in a self-assembled conjugated system.
 | ||
| Fig. 1 (a) Molecular structure of PBI-C10. (b, c, e and f) Polarized optical microscopy using crossed polarizers and polarized UV-vis spectroscopy (d and g) of highly oriented PBI-C10 films of J-type aggregate i.e. form I (b–d) and H-type aggregates of form II (e–g). The UV-vis spectra were recorded for parallel (‖) and perpendicular (⊥) orientation of the incident polarized light with respect to the rubbing direction. | ||
II. Experimental section
(a) Sample preparation
The synthesis of PBI-C10 (chemical structure shown in Fig. 1a) has been described in the literature.47,48 Thin films of PBI-C10 were prepared with a home made doctor-blade apparatus on clean glass slides for UV-vis and TEM studies (the cleaning of the glass slides is described in ref. 54) and on Si(100) substrates (Silchem) for FTIR measurements. For each sample, 50 μL of a PBI-C10 solution in p-xylene (3 mg mL−1) were doctor bladed on the substrates maintained at 130 °C.A home-made rubbing machine was used to align the films following the procedure used in recent studies on conjugated polymers.49–53 The rubbing machine is composed of a rotating cylinder (4 cm diameter) covered with a microfiber cloth. Rubbing is performed by applying the rotating cylinder with a 1.5–2.0 bar pressure on the translating sample holder (5 cm s−1). The sample holder is heated between 100 °C and 175 °C and is allowed to equilibrate for 1–2 min before rubbing. A rubbing cycle is characterized by the so-called rubbing length i.e. the length of the rubbing tissue applied on a given point of the sample. In this study, it is approx. 50 cm. The rubbing machine is placed in a golvebox (Plaslab) under nitrogen atmosphere.
The form I of PBI-C10 was obtained by solvent vapor annealing in chloroform. The rubbed films were placed in a closed glass container filled with 5 mL of chloroform at room temperature for 30 min. The form II of PBI-C10 was obtained by thermal annealing in a Linkam hot stage (LTS420) under N2 atmosphere at 190 °C for 5 h and slow cooling to room temperature at a rate of 0.5 °C min−1.
For TEM investigations, the PBI-C10 films were coated with a thin amorphous carbon film and removed from the glass substrate by floating on a diluted aqueous HF solution (10 wt%) and subsequently recovered on TEM copper grids.
(b) Structural characterization
Polarized optical microscopy was performed on a Leica DMR-X microscope equipped with Nikon Coolpix 995 digital camera. Transmission electron microscopy was performed on a Philips CM 12 (120 keV) microscope equipped with a CCD MVIII digital camera. The samples were observed in bright field, high resolution and electron diffraction modes. The rotation-tilt experiments were carried out using a Phillips PW6594 sample holder. The image treatments were carried out using AnalySYS (Soft Imaging Systems) and ITEM (FEI) softwares. Filtering of the HR-TEM images was performed following the procedure described in the literature in order to avoid generation of ghost lattice images.55 The structural modeling and the calculation of electron diffraction patterns were performed using the proper modules of the Cerius2 Software on a SiliconGraphics Octane workstation.Grazing incidence X-ray diffraction (GIXD) experiments were performed at the ESRF on line BM02 using a monochromated X-ray beam at 23.14 keV. The oriented samples were prepared on Si(100) substrates by doctor blading, following the same alignment procedure as used for the samples on glass substrates.
(c) Spectroscopic investigations (UV-vis and FTIR) and DFT calculations
UV-visible spectroscopy was performed on an Agilent Cary 5000 spectrometer and a Shimadzu UV-2010PC spectrometer equipped with a goniometer.Linear dichroism infrared experiments are very sensitive to preferential molecular orientation on surfaces. Therefore, oriented PBI-C10 films of form I were prepared by following the previous preparation protocols on Si(100) substrates and IR spectra were recorded in transmission using a polarized infrared beam. Polarized micro infrared experiments in the middle infrared range (800–4000 cm−1) were carried out in transmittance on a Bruker IFS 66 V spectrometer coupled with a Hyperion 2000 microscope (Bruker Inc.). A mercury cadmium telluride detector, a KBr beam splitter, and a blackbody source were used for our experiments. The spectral resolution was 2 cm−1, and 256 scans were coadded for each spectrum. The samples used for polarized FTIR spectroscopy are prepared on Si(100) substrates with their native oxide.
The experimental polarization at 0° refers to the electric field (E) parallel to the substrate and parallel to the rubbing direction (R). The polarization at 90° refers to E parallel to the substrate and perpendicular to the rubbing direction.
DFT calculations were performed on a cluster of two molecules using the SIESTA package56 and the generalized gradient approximation to the exchange correlation functional as proposed by Perdew, Burke and Ernzerhof.57 Core electrons were replaced by nonlocal norm-conserving pseudopotentials. The valence electrons were described by a double-ζ singly polarized basis set. The localization of the basis is controlled by an energy shift of 50 meV. Real space integration is performed on a regular grid corresponding to a plane-wave cutoff around 350 Ry. Only the G-point is taken into account for the one-dimensional Brillouin zone integrations. Atomic positions were relaxed using a conjugate gradient until the maximum residual atomic force was smaller than 0.02 eV Å−1. The vacuum size was fixed at 11 Å. Zone-center phonons were calculated within the harmonic approximation by finite difference of the Hellmann–Feynman forces with an atomic displacement of 0.03 Å. Positive and negative displacements are used to minimize anharmonic effects. Born effective charge tensors were calculated using the Berry phase method as formulated by King-Smith and Vanderbilt.58 The polarized infrared absorption spectra were calculated following the methodology described in ref. 59.
III. Results and discussion
(1) Orientation of form I and II by high-temperature rubbing
Structural investigations by TEM on the two polymorphs of PBI-C10 were conducted on highly oriented films. The gel phase of PBI-C10 was tentatively oriented on friction-transferred poly(tetrafluorethylene) or by shearing, but only high-T rubbing after removal of the solvent afforded highly oriented films of form I suitable for TEM and GIXD analysis.As demonstrated by POM, high alignment of form I of PBI-C10 films is indeed observed upon high-T rubbing (see Fig. 1b and c). The optimum rubbing temperature was determined (in the range 25–175 °C) by following the dichroic ratio at 623 nm versus rubbing temperature (see Fig. S1, ESI†). The best observed orientation corresponds to T = 175 °C yielding a dichroic ratio of 6–7. However, at this temperature, a significant amount of material is removed from the substrate during rubbing. Therefore, a smoother rubbing, at lower T, was combined with solvent–vapor annealing to obtain highly oriented form I films (see Fig. S1 and S2, ESI†). As seen in Fig. S1 (ESI†), a significant improvement of orientation is obtained by exposure of the films rubbed at 125 °C to a saturated vapor of CHCl3. A high dichroic ratio of 8 is obtained for the films rubbed at 125 °C after 30 min exposure to CHCl3. This film was subsequently used for the structural analysis of form I by TEM.
In Fig. 1, it is worth noting that the broad structured absorption band around 450–500 nm is polarized in the same way as the dominant peak at 623 nm. As shown by Mizuguchi et al. on single crystals of the J-aggregate polymorph of another PBI derivative (N,N′-di-bis(2-phenylethyl)perylene-3,4:9,10-bis(dicarboximide)), the maximum peak at 622 nm is polarized along the π-stacking direction of the PBI molecules.46 Therefore, in form I, PBI-C10 stacks into columns parallel to the rubbing direction R, which is confirmed by TEM and polarized FTIR (vide infra).
Oriented films of form II were prepared by thermal annealing of form I films at T = 190 °C (annealing maintains the orientation of the initial films).47 As seen from Fig. 1, the resulting films are highly birefringent and they display the characteristic H-type absorption of the form II of PBI-C10. Despite their strong optical birefringence, their UV-vis spectra display a modest dichroic ratio of 2.2 (see Fig. 1e).
Having determined means to prepare highly oriented films of almost pure form I and form II of PBI-C10, the structure of both forms was determined by combining TEM and polarized FTIR spectroscopy.
(2) Molecular packing in form I (J-aggregate, dried gel phase)
 | ||
| Fig. 2 (a) Low dose electron diffraction pattern for an oriented PBI-C10 thin film. The central part of the pattern is shown with a different intensity scale to visualize the low angle reflection at 34 ± 1 Å. (b) Schematic illustration of the main reflections in the ED pattern. (c and d) GIXD 2D maps of oriented films of PBI-C10 in form I recorded for the incident beam oriented either parallel or perpendicular to the rubbing direction R as illustrated in the top sketches, respectively. The asterisk indicates a reflection from the Si(100) substrate. (e) Schematic illustration of the stacking of PBI-C10 in form I as determined from the combination of TEM, GIXD and polarized FTIR. The rubbing direction is indicated by the arrow marked R. The schematic figure shows a projection parallel and perpendicular to the rubbing direction R of the molecular arrangement in form II domains of PBIC10. The (O,x,y,z) frame associated to the PBIC10 molecule is used to index the orientations of the vibrational modes. The PBI core is sketched as a violet rectangle, the amide linker in drawn in red and the trialkoxyphenyl group in green. | ||
 | ||
| Fig. 3 Polarized infrared spectra of oriented PBI-C10 thin films of form I in the regions of the amide 1 (a) and of the amide A (b). Polarization varies by 15° steps (from 0° (black) to 90° (red)). 0° (90°) corresponds to the electric field E parallel (perpendicular) to the rubbing direction R, respectively. The inset shows the microscopy image of the investigated area whereas the white arrow highlights the rubbing direction (R). | ||
Amide vibrations (see Table S1, ESI† for assignment) are very sensitive to H bonding.62 The amide A modes shown in the Fig. 3b give rise to a broad band centered at 3250 cm−1 and a shoulder at 3317 cm−1. These two bands are typical of strongly and weakly H-bonded amide groups, respectively. In principle, PBI-C10 molecules should be involved in identical H-bonds on both sides of the PBI core in ideal J-type aggregates of form I and this should lead to a single population of amide H bonds. However, TEM and GIXD indicate substantial structural disorder in the stacks of form I, hence a mismatch between some amides groups and a proportion of weaker H-bonds. Despite this disorder, the amide A bands are strongly polarized. The intensity is maximal for E‖R (0°) and negligible for E⊥R (90°). It indicates that the H-bonds are preferentially parallel to the rubbing direction R, i.e. in the direction of columnar stacks, in agreement with the results obtained by electron diffraction. The amide 1 modes appear in the 1700–1600 cm−1 range. The bands within this range were assigned from (i) comparisons of IR spectra of model compounds (see Fig. S3, ESI†), (ii) from modeling studies (see Section 2c) and (iii) from the study of analogues in the literature.63,64 In the studied range, 3 bands are visible at 1668, 1660 and 1635 cm−1. The band at 1660 cm−1 is assigned to the asymmetric CO stretching of the imides, the other two bands to amide 1 modes from the amides. The frequencies of H bonded amide vibrations classically downshift when H bond strength increases.65 Therefore both bands can be assigned as follows: strongly H-bonded amide 1 vibrations (SA1) appear at 1635 cm−1 and weaker H bonded amide 1 vibrations (WA1) appear at 1668 cm−1 (see Table S1, ESI†). They show opposite experimental polarization dependencies (see Fig. 3a). The SA1 band is polarized essentially parallel to R whereas the WA1 band is polarized perpendicular to R, in agreement with the amide A band behavior.66 Accordingly, the strongly H-bonded amide groups are oriented mostly parallel to the stacking direction, contrary to the weakly H-bonded amides that are oriented rather perpendicular to the molecular stacks.
In other words, H-bonding is strong within the columns of PBI-C10 and much weaker between the columns, explaining the 1D columnar growth of form I.
 | ||
| Fig. 4 (a) Relaxed geometry of a cluster made of two PBI-C10 molecules using DFT prior to IR spectra calculation. The z axis is normal to the PBI macrocycle, the x axis is parallel to the short axis of the PBI and the y axis is along the long axis of the PBI macrocycle. (b) Schematic illustration of the packing of PBI molecules in the stacks of form I. Comparison of calculated (c) and experimental (d) polarized middle infrared spectra. For DFT calculation (c): (red) corresponds to electric field E perpendicular to the PBI cycle (z axis) and (black) in the plane of the aromatic core (x, y plane). For the experimental spectra (d), 0° polarization (black) corresponds to E parallel to the stacking direction and 90° (red) to E perpendicular to the stacking. | ||
To determine the preferential orientation of the PBI cores with respect to the substrate, the intense bands at 1697 and 1658 cm−1 are most useful because they display a strong dichroism. The comparison between calculations and experimental features shows (in agreement with the results reported in the literature for other PBI derivatives.64,67) that the intense bands at 1697 and 1658 cm−1 are assigned to the symmetric stretching mode νsCO and the asymmetric stretching mode νasCO of the imide groups, respectively (see Tables S1, S2 and Fig. S3, ESI† and IR model compounds). The polarization of the imide νsCO band is mainly parallel to its long axis (y axis), whereas the polarization of the imide νasCO band is in the xz plane (see Fig. 4a and Table S1, ESI†) defined by the short axis of the perylene and the axis normal to the perylene. The polarization dependence of the imide features (see Fig. 5a) reveals a maximum intensity of νsCO (νasCO) for a polarization at 0° (90°) to the stacking direction (rubbing direction). Accordingly, these results indicate that the long axis of the PBI core (y axis) has a dominant projection along the rubbing direction i.e. the tilting angle θ between the y axis and the stacking direction is below 45°. In other words, the PBI cores are strongly tilted with respect to the stacking direction. This inference is consistent with the fact that the molecular stacks of PBI cores are of J-type in form I and is also in agreement with recent findings on other amide-substituted PBI's.64 This result, when combined with the preferential contact plane suggested from GIXD, implies that the PBI cycles are oriented mainly with their short axis in the substrate plane and the long axis tilted to the substrate normal as illustrated in Fig. 2e (dominant “standing“ orientation of PBI-C10).
 | ||
| Fig. 5 Polarized infrared spectra of oriented PBI-C10 thin films of form II. Polarization varies by 15° steps (from 0° (black) to 90° (red)). 0° corresponds to the electric field E parallel to the fiber direction (rubbing direction). | ||
A tilt of the perylene core around its long axis cannot be excluded. The two aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations at 1595 and 1581 cm−1 show the same polarization as imide νsCO i.e. ‖ R. This is expected since, according the DFT model, the dipole moment for the aromatic CC stretching have their major component along the long axis of the PBI However, it must be stressed that the latter band has also a contribution of the CC stretching mode of the benzenic unit of the trialkoxybenzene dendron, (Benz. νCC in Table S1, ESI†). Since the benzenic moieties are conjugated to the amides, part of them have their plane parallel to the H bonding and have a dichroic effect with a maximal intensity when E is parallel to R i.e. to the stacking direction.
C stretching vibrations at 1595 and 1581 cm−1 show the same polarization as imide νsCO i.e. ‖ R. This is expected since, according the DFT model, the dipole moment for the aromatic CC stretching have their major component along the long axis of the PBI However, it must be stressed that the latter band has also a contribution of the CC stretching mode of the benzenic unit of the trialkoxybenzene dendron, (Benz. νCC in Table S1, ESI†). Since the benzenic moieties are conjugated to the amides, part of them have their plane parallel to the H bonding and have a dichroic effect with a maximal intensity when E is parallel to R i.e. to the stacking direction.
(3) Form II, evidence of supramolecular helical assemblies
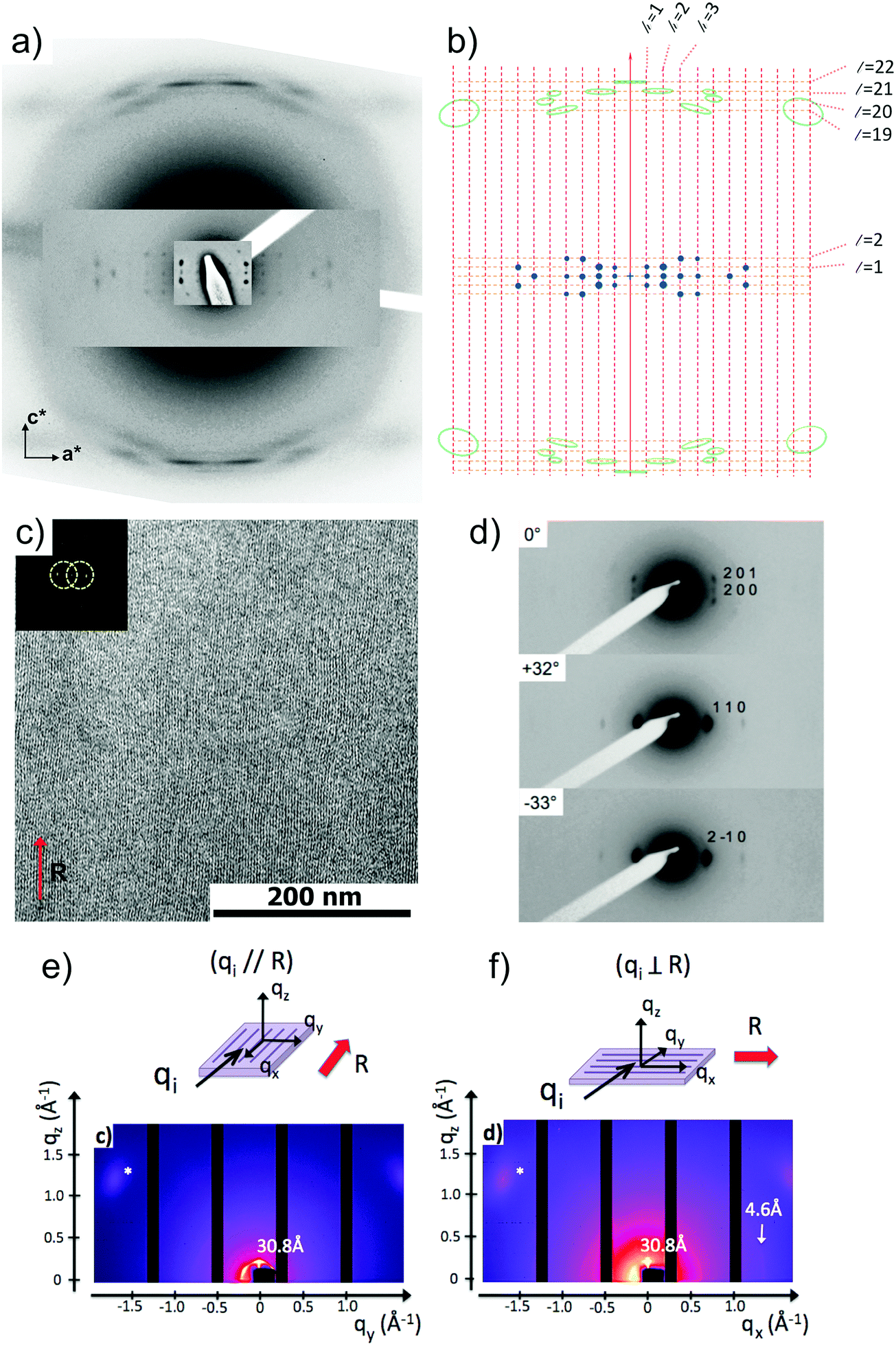 | ||
| Fig. 6 (a) ED pattern of a single contact-plane a PBI-C10 form II oriented thin film; (b) schematic representation of the ED pattern with indexed layer lines. (c) Fast Fourier filtered HR-TEM low dose image of a highly ordered PBI-C10 form II domain. The inset shows the Fourier transform of the image and the circles outline the applied filter in the FFT. (d) Evolution of the electron diffraction pattern of a PBI-C10 oriented film (form II) with the sample tilt around the rubbing direction which is vertical (c axis of helices). (e and f) GIXD 2D intensity maps of the oriented form II films recorded for the incident X-ray beam (qi) oriented parallel and perpendicular to the rubbing direction (R, red arrow). | ||
Fig. 6 shows the most characteristic experimental ED pattern (a) and its schematic interpretation (b). This pattern corresponds to a single contact plane as it displays exclusively reflections indexed as (h 0 l). All reflections belong to an orthogonal lattice with a = 26.8 Å and c = 97 Å. From this pattern, it is observed that the intense 4.6 Å reflection lies on the 21st layer line. The most intense reflections are observed on the 1st, 2nd, 20th, 21st and 22nd layer lines. The systematic absences of certain reflections is expected from the theory of Cochran, Crick and Vand on the scattering amplitudes of helices.68–70 In the present case, intense reflections are observed for all layer lines with an index l = 21n ± m where n and m are two integers. Accordingly, and despite the presence of a weak meridional 0 0 22 reflection, it is proposed that PBI-C10 form a 21/1 supramolecular helix. Contrary to a 22/1 helix, the 21/1 conformation is in line with the hexagonal symmetry of the unit cell as evidenced from rotation-tilt and GIXD experiments (see Fig. 6d). Similar helical assemblies of PBI-based molecules have been previously observed by Percec et al. for similar PBI derivatives.71,72 However, the long-range helical order persists over 97 Å, a value superior to that observed in similar systems without H-bonding.71,72 This observation suggests that H-bonding amplifies the range of inter-molecular order in PBIC10. As observed with low dose HR-TEM, (Fig. 6c) helical assemblies are ordered over very large distances up to several hundreds of nanometers.
The full unit cell parameters of form II were determined both by GIXD and rotation-tilt experiments. In the rotation-tilt experiment, a sample displaying the ED pattern shown in Fig. 6a is rotated around the c axis. As seen in Fig. 6d, a characteristic projection is observed for a tilt angle of ±32 ± 2° around c. The patterns show a strong reflection at 30.8 Å ± 1 Å with its second order on the equator. This result supports a trigonal unit cell for form II, which is further demonstrated by GIXD. Indeed, the GIXD pattern recorded when the incident beam is parallel to the rubbing direction shows a characteristic hexagonal pattern with the same 30.8 Å period (see Fig. 6e and f). When the incident X-ray beam is perpendicular to the rubbing direction, the meridional reflection along qz is still observed whereas the intensity of the reflections at ±60° has strongly decreased (although not totally disappeared). This pattern also shows the broad 4.6 Å reflection corresponding to the molecular stacking period within helices albeit less defined as in the ED patterns.
Taken together, these observations indicate that PBI-C10 molecules self-assemble into 21/1 helices that organize in a trigonal structure with a = b = 62 ± 2 Å, c = 97 ± 1 Å and γ = 120°. This unit cell allows to index all the observed reflections in the ED and GIXD patterns (see Table S2, ESI†).
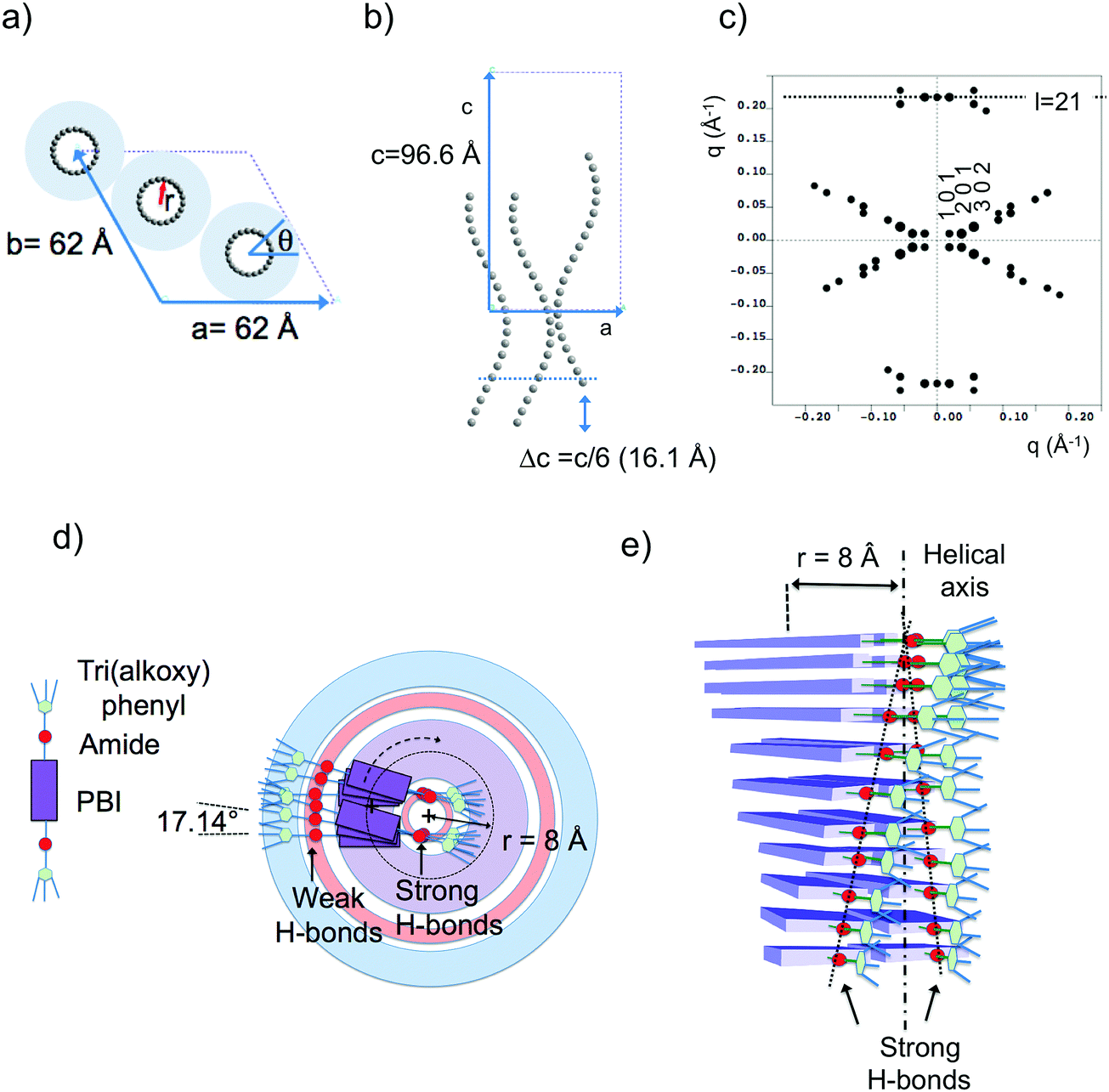 | ||
| Fig. 7 (a) Frustrated structure of form II and supramolecular organization of the PBI-C10 molecules in the helix. Projection of the PBI-C10 unit cell along the helical axis c (a) and along the b axis (b). The helices are modeled by a “bead and string” structure. The optimal radius of the helix is 8 Å (see text and ESI†). Disordered alkyl side chains of the trialkoxyphenyl dendrons are represented by a light blue corona. In this frustrated structure, one helix is azimuthally offset by θ = 180° and shifted by Δc = c/6 versus the two other helices in the unit cell. (c) Calculated ED pattern for the [0 1 0] zone. (d and e) Supramolecular organization of PBI-C10 molecules in the 211 helix of form II. The helix is off-centered so that strongly H-bonded amides are located close to the helical axis whereas weaker H-bonds between amides in the dimers are located in the outer shell of the helix. | ||
Trigonal structures bearing supramolecular helices are found in many systems e.g. thermotropic/lyotropic liquid crystals, polymers, and DNA or proteins.73–80 For instance, triplets of syndiotactic polystyrene (sPS) chains and three-fold helices of isotactic polypropylene chains are packed in a trigonal structure in the alpha form of sPS and the beta form of iPP, respectively. It is therefore tempting to assemble three 21/1 helices of PBI-C10 likewise in the trigonal unit cell with the cell parameters determined by ED and GIXD.
The present modeling supports the idea that PBI-C10 molecules organize to form supramolecular helices with a radius of 8 Å (see ESI†). Each strata of the helices consist of two PBI-C10 molecules. As a matter of fact, the distance from the PBI center to the amide group is ∼8 Å i.e. coincides with the refined helix radius. This implies that a supramolecular helix is built in such a way that one part of the amide groups involved in H-bonds are located close to the helical axis (see Fig. 7d and e) whereas the second half is located towards the exterior of the helix. This is consistent with FTIR results showing the coexistence of at least two populations of amides involved in strong or weak H-bonds in form II. Indeed, as demonstrated in ref. 40, the amide A band in Form II is characterized by two components centered at 3328 cm−1 and 3426 cm−1. The first band corresponds to amides with moderate H-bonds whereas the second band is characteristic of poorly H-bonded amides. As shown in Fig. 7, we propose that the inner (outer) amides are strongly (weakly) H-bonded. In other words, the strongly H-bonded amides constitute the spine of the helices (helical axis). This is in strong contrast to the helical assemblies reported by Frauenrath and coworkers for which the helical axis is centered on the conjugated PBI cores and symmetric H-bonds form on both sides of the central π-conjugated helical stack.62 Furthermore, we observe that the H-bonds along the helix axis are between PBI-C10 molecules of successive layers. It is proposed that the H-bonds in the outer shell of the helix involve the two side-by-side PBI-C10 molecules of a dimer in a layer.
 | ||
| Fig. 8 Low-angle electron diffraction patterns (a and e), fast Fourier transform (b and f) and corresponding low-dose HR-TEM images (c and g) and projections of the trigonal cell of the frustrated structure (d and h) for the [0 1 0] and the [1 2 0] zones, respectively. | ||
IV. Discussion
Whereas various studies have reported the growth of helical supramolecular assemblies in PBI-based systems,33,62,71,72 there has been only little attention paid to the crystalline lattices formed by the supramolecular objects. This is partly due to the fact that most previous studies have been dedicated to dilute solutions wherein individual supramolecular helices form by self-assembly.27,33,52 In this study, further insight in the original packing of a PBI-based organogelator is obtained for oriented thin films. To the best of our knowledge, this is the first evidence for a frustrated structure in the case of self-assembled conjugated semiconductors. It is also worth to stress that such a structural analysis would have been very difficult using classical fiber X-ray diffraction analysis given the usually rather large orientational distribution in drawn fibers (full width at half maximum >10–15 deg.).61 In particular, the very faint low angle and high angle details in the scattering patterns obtained by electron diffraction were only accessible from highly oriented areas (corresponding to unique contact planes) of the annealed samples prepared by high-T rubbing.This study has also demonstrated that a hydrogen bonded PBI derivative can form two distinct polymorphs whose structures imply very different H-bonding organizations. Form I is a classical rectangular columnar phase. It is the dried gel phase and it forms in solution by the buildup of strong and symmetric H-bonds between PBI-C10 molecules in the columns. The high dichroism of the amide A bands in form I indicates that the strong intra-columnar H-bonds are highly directional in the stacks. In strong contrast, in form II, the supramolecular helices are hinged on a spine made of strongly H-bonded amides and the conjugated backbones of PBI rotate around the helical axis. The outer shell of the helix involves most likely the weaker H-bonds between amides of the two PBI-C10 molecules forming the dimers. The comparison of the structures of form I and II suggests that the transformation from I to II rests on a redistribution of H bonds between nearby columns in form I. Indeed, in form I, the strong H bonds are purely intra-columnar but the columns are relatively close lying at 8.6 Å in the direction of the short axis of PBI (see Fig. 9). This close proximity implies that inter-columnar H-bonds can form quite readily upon annealing to 190 °C. These inter-columnar H bonds are at the origin of the dimers that form the layers in the helices of form II. Instead, the H-bonds close to the helical spine are a reminiscence of the original intra-columnar H-bonds in form I.
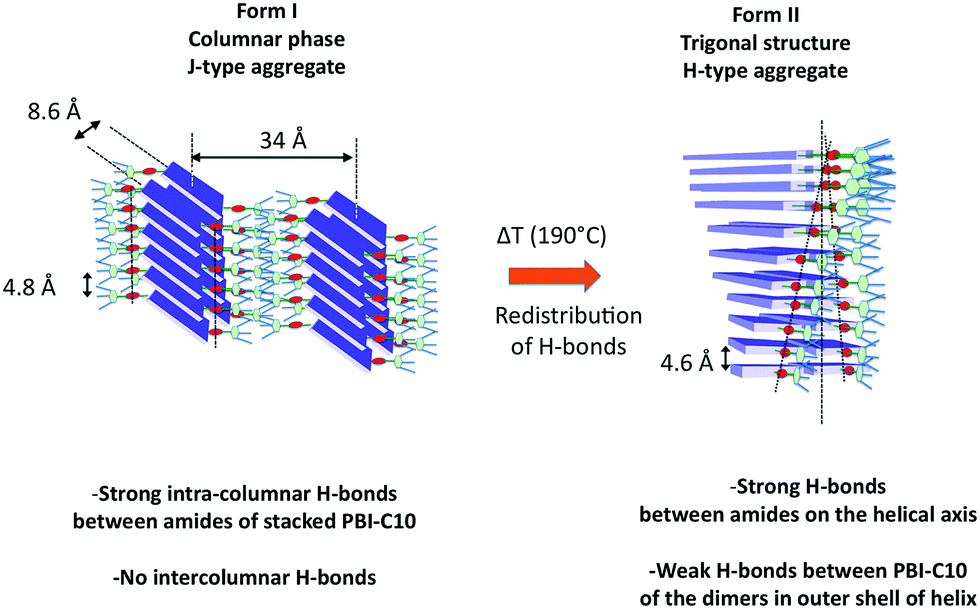 | ||
| Fig. 9 Schematic illustration of the structural reorganization between form I and form II of PBI-C10 induced by thermal annealing at 190 °C. This transformation results from a redistribution of H-bonds from purely intra-columnar in form I to a mixed mode of H-bonding in form II i.e. strong H-bonds along the spine of the helices and weak H-bonds linking the PBI-C10 molecules in the dimers. | ||
The comparison of the structures formed by the PBI-derivatives without amides by Percec et al.71,72 with PBI-C10 is interesting to evaluate the impact of H-bonding on the supramolecular organization. First, in PBI-C10, the stacking periodicity (4.6 Å) is closer to the amide⋯amide H-bonding distance in polyamides (4.8–4.9 Å) than the π-stacking distance in non hydrogen-bonded PBI systems (3.5–3.8 Å).62 This suggests that H-bonding prevails over π-stacking. As a matter of facts, none of the polymorphs shows a strong π-stacking reflection in the 3.5–3.9 Å range. Second, each PBI “ dimer“ in a given strata of the helix is rotated by 17.12° with respect to the previous strata whereas in the absence of the H-bonding amide, the rotation is by a larger angle of 36°.71 This reduced angular rotation between PBI cores might be the consequence of strong H-bonds between the amides located close to the helical axis that compete with the π-stacking tendency of the conjugated PBI cores. This result suggests that the introduction of H-bonds in molecular semiconductors is a handle to modify the extent of π-stacking between the conjugated cores.
V. Conclusion
This study has uncovered the structural features of two polymorphs of a perylene bisimide organogelator. TEM investigations show that long-range order is present in form II (H-aggregate) with the buildup of H-bonded supramolecular helices with very large helical periods of 97 Å. In strong contrast, in form I, PBI-C10 molecules arrange in columnar-like assemblies with relatively poor structural order. In form I, polarized FTIR spectroscopy revealed a high level of directionality of H-bonds, resulting in a very high corresponding dichroism of the main amide bands that are strongly polarized along the stacking direction (rubbing direction R). In form II, the lower dichroism of the amide bands is related to the helical arrangement of PBI-C10 molecules. The off-centered character of the helices of PBI-C10 relates to the asymmetry of H bonds: strong H-bonds along the helical axis and weak H-bonds in the outer shell of the helix. A detailed ED study demonstrates that the packing of 21/1 helices is according to a frustrated structure as encountered in some polyolefins. This result might be an incentive to the community to analyze the structure of other self-assembled systems that form supramolecular helices using similar frustrated structures. Preliminary results on naphthalene bisimide analogues of PBI-C10 indicate that supramolecular helices are relatively common in molecular systems where H-bonding of lateral dendrons and π-stacking of conjugated cores such as perylene bisimide compete.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Nils Blanc and Nathalie Boudet are gratefully acknowledged for their assistance for the GIXD experiments on BM02 Grenoble. Marc Schmutz and Christian Blanck are acknowledged for technical support in TEM. Bernard Lotz is acknowledged for fruitful discussions and critical reading of the manuscript. Martin Brinkmann acknowledges financial support from the European Community via the Interreg IV-A program (C25, Rhin-Solar). Philippe Mésini acknowledges financial support from ANR through project ANR14-CE08-0020 and M. D. from Région Alsace (Phd Grant). SIMS platform (Montpellier) is also thanked for providing access to IR facility.References
- F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. Schenning, J. Chem. Rev., 2005, 105, 1491–1546 CrossRef CAS PubMed.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- V. K. Praveen, C. Ranjith and N. Armaroli, Angew. Chem., 2014, 53, 365 CrossRef CAS PubMed.
- K. K. Kartha, A. Sandeep, V. K. Praveen and A. Ajayaghosh, Chem. Rec., 2015, 15, 252–265 CrossRef CAS PubMed.
- S. Ghosh, V. K. Praveen and A. Ajayaghosh, Annu. Rev. Mater. Res., 2016, 46, 235–262 CrossRef CAS.
- S. Prasantkhumar, A. Gopal and A. Ajayaghosh, J. Am. Chem. Soc., 2010, 132, 13206 CrossRef PubMed.
- S. Prasantkhumar, A. Saeki, S. Seki and A. Ajayaghosh, J. Am. Chem. Soc., 2010, 132, 8866 CrossRef PubMed.
- S.-I. Kawano, N. Fujita and S. Shinkai, Chem. – Eur. J., 2005, 11, 4735 CrossRef CAS PubMed.
- M. R. Rao and S.-S. Sun, Langmuir, 2013, 29, 15146 CrossRef CAS PubMed.
- E. R. Draper, J. J. Walsch, T. O. McDonald, M. A. Zwijnenburg, P. J. Cameron, A. J. Cowan and D. J. Adams, J. Mater. Chem. C, 2014, 2, 5570–5575 RSC.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dötz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS PubMed.
- A. S. Molinari, H. Alves, Z. Chen, A. Facchetti and A. F. Morpurgo, J. Am. Chem. Soc., 2009, 131, 2462–2463 CrossRef CAS PubMed.
- A. L. Briseno, S. C. B. Mannsfeld, C. Reese, J. M. Hancock, Y. Xiong, S. A. Jenekhe, Z. Bao and Y. Xia, Nano Lett., 2007, 7, 2847 CrossRef CAS PubMed.
- R. Steyrleuthner, M. Schubert, F. Jaiser, J. C. Blakesley, Z. Chen, A. Facchetti and D. Neher, Adv. Mater., 2010, 22, 2799–2803 CrossRef CAS PubMed.
- M. Schubert, D. Dolfen, J. Frisch, S. Roland, R. Steyrleuthner, B. Stiller, Z. Chen, U. Scherf, N. Koch, A. Facchetti and D. Neher, Adv. Energy Mater., 2012, 2, 369–380 CrossRef CAS.
- H. Yu, Z. Bao and J. H. Oh, Adv. Funct. Mater., 2013, 23, 629–639 CrossRef CAS.
- V. Kamm, G. Battafliarin, I. A. Howard, W. Pisula, A. Mavrinskiy, C. Li, K. Müllen and F. Laquai, Adv. Energy Mater., 2011, 1, 297–302 CrossRef CAS.
- L. Schmidt-Mende, A. Fechtenkötter, K. Müllen, E. Moons, R. H. Friend and J. D. MacKenzie, Science, 2001, 293, 1119 CrossRef CAS PubMed.
- F. Würthner, Chem. Commun., 2004, 1564–1579 RSC.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376 CrossRef PubMed.
- X. Zhan, A. Facchetti, S. Barlow, T. J. Marks, M. A. Ratner, M. R. Wasielewski and S. R. Marder, Adv. Mater., 2011, 23, 268–284 CrossRef CAS PubMed.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- E. Kozma and M. Catellani, Dyes Pigm., 2013, 98, 160–179 CrossRef CAS.
- F. Würthner, C. Thalacker, S. Diele and C. Tschierske, Chem. – Eur. J., 2001, 7, 2245–2253 CrossRef.
- S. Yagai, T. Seki, T. Karatsu, A. Kitamura and F. Würthner, Angew. Chem., Int. Ed., 2008, 47, 3367 CrossRef CAS PubMed.
- S. Yagai, M. Usui, T. Seki, H. Murayama, Y. Kikkawa, S. Uemura, T. Karatsu, A. Kitamura, A. Asano and S. Seki, J. Am. Chem. Soc., 2012, 134, 7983–7994 CrossRef CAS PubMed.
- J. Mizuguchi, J. Appl. Phys., 1998, 84, 4479 CrossRef CAS.
- M. Wolffs, M. Malisauskas, I. D. Cat, Z. Tomovic, J. van Herrikhuyzen, C. M. A. Leenders, E. J. Spadafora, B. Grevin, S. D. Feyter, E. W. Meijer, A. P. H. J. Schenning and P. Leclere, Chem. Commun., 2011, 47, 9333–9335 RSC.
- K. Balakrishnan, A. Datar, T. Naddo, J. Huang, R. Oitker, M. Yen, J. Zhao and L. Zang, J. Am. Chem. Soc., 2006, 128, 7390–7398 CrossRef CAS PubMed.
- B. Jancy and S. K. Asha, Chem. Mater., 2008, 20, 169–181 CrossRef CAS.
- Z. Chen, B. Fimmel and F. Würthner, Org. Biomol. Chem., 2012, 10, 5845–5855 CAS.
- L. Bu, X. Guo, B. Yu, Y. Qu, Z. Xie, D. Yan, Y. Geng and F. Wang, J. Am. Chem. Soc., 2009, 131, 13242–13243 CrossRef CAS PubMed.
- P. O. Schwartz, E. Zaborova, L. Biniek, B. Heinrich, M. Brinkmann, N. Leclerc and S. Mery, J. Am. Chem. Soc., 2014, 136, 5981–5992 CrossRef CAS PubMed.
- L. Biniek, P. O. Schwartz, E. Zabrova, B. Heinrich, N. Leclerc, S. Méry and M. Brinkmann, J. Mater. Chem. C, 2015, 3, 3342 RSC.
- N. Mizoshita, T. Tani and S. Inagaki, Adv. Mater., 2012, 24, 3350 CrossRef CAS PubMed.
- S. Ghosh, X.-Q. Li, V. Stepanenko and F. Würthner, Chem. – Eur. J., 2008, 14, 11343–11357 CrossRef CAS PubMed.
- B. Gao, D. Xia, L. Zhang, Q. Bai, L. Bai, T. Yang and X. Ba, J. Mater. Chem., 2011, 21, 15975 RSC.
- E. Hädicke and F. Graser, Acta Crystallogr., Sect. A: Found. Crystallogr., 1986, C42, 189–195 Search PubMed.
- E. Hädicke and F. Graser, Acta Crystallogr., Sect. A: Found. Crystallogr., 1986, C42, 195–198 Search PubMed.
- G. Klebe, F. Graser, E. Hädicke and J. Berndt, Acta Crystallogr., Sect. A: Found. Crystallogr., 1989, B45, 69–77 CAS.
- H.-M. Zhao, J. Pfister, V. Settels, M. Renz, M. Kaupp, V. C. Dehm, F. Würthner, R. F. Fink and B. Engels, J. Am. Chem. Soc., 2009, 131, 15660 CrossRef CAS PubMed.
- R. A. Cormier and B. A. Gregg, J. Phys. Chem. B, 1997, 101, 11004–11006 CrossRef CAS.
- S.-G. Liu, G. Sui, R. A. Cormier, R. M. Leblanc and B. A. Gregg, J. Phys. Chem. B, 2002, 106, 1307 CrossRef CAS.
- B. A. Gregg, J. Phys. Chem., 1996, 100, 852–859 CrossRef CAS.
- J. Mizuguchi, K. Hino and K. Tojo, Dyes Pigm., 2006, 70, 126–135 CrossRef CAS.
- A. Sarbu, L. Biniek, J.-M. Guenet, P. Mésini and M. Brinkmann, J. Mater. Chem. C, 2015, 3, 1235–1242 RSC.
- F. Würthner, C. Bauer, V. Stepanenko and S. Yagai, Adv. Mater., 2008, 20, 1695–1698 CrossRef.
- L. Biniek, N. Leclerc, T. Heiser and M. Brinkmann, Macromolecules, 2013, 46, 4014 CrossRef CAS.
- L. Biniek, L. S. Poujet, D. Djurado, E. Gonthier, K. Tremel, N. Kayunkid, E. Zabrova, N. Crespo-Monteiro, O. Boyron, N. Leclerc, S. Ludwigs and M. Brinkmann, Macromolecules, 2014, 47, 3871 CrossRef CAS.
- M. Brinkmann, L. Hartmann, L. Biniek, K. Tremel and N. Kayunkid, Macromol. Rapid Commun., 2014, 35, 9–26 CrossRef CAS PubMed.
- A. Hamidi-Sakr, L. Biniek, S. Fall and M. Brinkmann, Adv. Funct. Mater., 2016, 26, 408–420 CrossRef CAS.
- K. Tremel, F. S. U. Fischer, N. Kayunkid, R. D. Pietro, R. Tkachov, A. Kiriy, D. Neher, S. Ludwigs and M. Brinkmann, Adv. Energy Mater., 2014, 4, 1301659 CrossRef.
- M. Brinkmann, S. Graff, J.-C. Wittmann, C. Chaumont, F. Nuesch, A. Anver, M. Schaer and L. Zuppiroli, J. Phys. Chem. B, 2003, 107, 10531 CrossRef CAS.
- M. Brinkmann and P. Rannou, Macromolecules, 2009, 42, 1125 CrossRef CAS.
- D. Sanchez-Portal, P. Ordejon, E. Artacho and J. M. Soler, Int. J. Quantum Chem., 1997, 65, 453–461 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- R. D. King-Smith and D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 1651–1654 CrossRef CAS.
- P. Hermet, J.-L. Bantignies, A. Rahmani, J.-L. Sauvajol, M. R. Johnson and F. Serein, J. Phys. Chem. A, 2005, 109, 1684–1691 CrossRef CAS PubMed.
- M. Kohan, Nylon Plastics handbook, Hanser, Wilmington, 1995, p. 1 Search PubMed.
- R. Marty, R. Szilluweit, A. Sánchez-Ferrer, S. Bolisetty, J. Adamcik, R. Mezzenga, E. C. Spitzner, M. Feifer, S. N. Steinmann, C. Corminboeuf and H. Frauenrath, ACS Nano, 2013, 7, 8498–8508 CrossRef CAS PubMed.
- G. Creff, B. P. Pichon, C. Blanc, D. Maurin, J.-L. Sauvajol, C. Carcel, J. Moreau, P. Roy, J. R. Bartlett, M. Wong Chi Man and J.-L. Bantignies, Langmuir, 2013, 29, 5581 CrossRef CAS PubMed.
- R. Aroca, E. Johnson and A. K. Maiti, Appl. Spectrosc., 1995, 49, 466–471 CrossRef CAS.
- W. J. Doherty, A. G. Simmonds, S. B. Mendes, N. R. Armstrong and S. S. Saavedra, Appl. Spectrosc., 2005, 59, 1248–1256 CrossRef CAS PubMed.
- J.-L. Bantignies, L. Vellutini, J.-L. Sauvajol, D. Maurin, M. Wong Chi Man, P. Dieudonné and J. J. E. Moreau, J. Non-Cryst. Solids, 2004, 345–346, 605–609 CrossRef.
- The NH stretching of the weakly bonded amides should show the same dichroism as WA1, but their intensities are weaker than those corresponding the strongly bonded amides. The overall observed dichroic effect is dominated by the strongly H-bonded amides.
- F. Ambrosino and S. Califano, Spectrochim. Acta, 1965, 21, 1401–1409 CrossRef CAS.
- F. H. C. Crick and A. Rich, Nature, 1955, 176, 780 CrossRef CAS PubMed.
- W. Cochran, F. H. C. Crick and V. Vand, Acta Crystallogr., 1952, 5, 581 CrossRef CAS.
- R. E. Dickerson and I. Geis, The structure and action of proteins, Harper and Row, New York, 1969 Search PubMed.
- V. Percec, M. Peterca, T. Tadjiev, X. Zeng, G. Ungar, P. Leowanawat, E. Aqad, M. R. Imam, B. M. Rosen, U. Akbey, R. Graf, S. Sekharan, D. Sebastiani, H. W. Spiess, P. A. Heiney and S. D. Hudson, J. Am. Chem. Soc., 2011, 133, 12197–12219 CrossRef CAS PubMed.
- V. Percec, H.-J. Sun, P. Leowanawat, M. Peterca, R. Graf, H. W. Spiess, X. Zeng, G. Ungar and P. A. Heiney, J. Am. Chem. Soc., 2013, 135, 4129–4148 CrossRef CAS PubMed.
- F. Vera, J. L. Serrano and T. Sierra, Chem. Soc. Rev., 2009, 38, 781–796 RSC.
- W. Pisula, Ž. Tomović, M. D. Watson, K. Müllen, J. Kussmann, C. Ochsenfeld, T. Metzroth and J. Gauss, J. Phys. Chem. B, 2007, 111, 7481–7487 CrossRef CAS PubMed.
- M. Brinkmann, N. Charoenthai, R. Traiphol, P. Piyakulawat, J. Wlosnewski and U. Asawapirom, Macromolecules, 2009, 42, 8298 CrossRef CAS.
- B. Lotz, Macromolecules, 2012, 45, 2175–2189 CrossRef CAS.
- L. Cartier, T. Okihara and B. Lotz, Macromolecules, 1998, 31, 3303–3310 CrossRef CAS.
- B. Lotz, S. Kopp and D. L. Dorset, C. R. Acad. Sci. Paris, 1994, 319, 187 CAS.
- D. L. Dorset, M. McCourt, S. Kopp, M. Schumacher, T. Okihara and B. Lotz, Polymer, 1998, 39, 6331 CrossRef CAS.
- F. Livolant and A. Leforestier, Prog. Polym. Sci., 1996, 21, 1115 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7cp06761e |
| This journal is © the Owner Societies 2017 |