
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Neutral iodotriazole foldamers as tetradentate halogen bonding anion receptors†
Arseni
Borissov
 ,
Jason Y. C.
Lim
,
Asha
Brown
,
Kirsten E.
Christensen
,
Amber L.
Thompson
,
Martin D.
Smith
and
Paul D.
Beer
*
,
Jason Y. C.
Lim
,
Asha
Brown
,
Kirsten E.
Christensen
,
Amber L.
Thompson
,
Martin D.
Smith
and
Paul D.
Beer
*
Chemistry Research Laboratory, Department of Chemistry, University of Oxford, Mansfield Road, Oxford, OX1 3TA, UK. E-mail: paul.beer@chem.ox.ac.uk
First published on 6th February 2017
Abstract
Neutral tetradentate halogen bond donor foldamers were synthesised and exhibit enhanced anion affinities over their hydrogen bonding analogues, displaying iodide selectivity over lighter halide, carboxylate and dihydrogen phosphate anions. A foldamer with a chiral (S)-binaphthol motif was demonstrated to distinguish between enantiomers of chiral anions.
Halogen bonding (XB) is the attractive non-covalent interaction between a terminal σ-hole on an electron deficient halogen atom and a Lewis base.1 Its strength and directionality have led to several applications in materials science and, more recently, in supramolecular chemistry2,3 and organocatalysis.4 In particular, XB donors have been successfully used in anion receptors for molecular recognition and sensing applications.5 Many of these have shown enhanced binding properties over their hydrogen bonding (HB) analogues.6,7
The electron-deficient8 1,2,3-triazole motif has been exploited for anion recognition as an effective C–H hydrogen bond donor when integrated into multidentate macrocycles9 and acyclic foldamers.10–12 While the related 5-iodotriazole unit has been used as XB donor for anion binding, such XB anion hosts are rare in the literature.13 No tetradentate XB donor foldamers have been described to date; the closest examples are a tridentate halopyridinium14 and the use of triazole foldamers as XB acceptor hosts for organohalogens.15
Herein we sought to apply the potency of XB to enhance the anion affinity of the known triazole-based HB foldamer framework. Thus, XB foldamers with four 5-iodo-1,2,3-triazole XB donors were synthesized (Fig. 1) and their anion binding properties probed in comparison with HB analogues. A few variants were prepared: 1 and 2 contain triethylene glycol (TEG) chains for improved solubility, while in 3 and 4 9-anthrylmethyl termini have been introduced to provide a fluorescent response.15,16 System 4 also includes a chiral (S)-binaphthol core in order to investigate XB chiral recognition, which has only previously been observed in a bidentate receptor.17 Importantly, the XB foldamers exhibited overall stronger anion affinity than their HB analogs with the chiral XB host 4b displaying chiral discrimination with bulky amino acid anions.
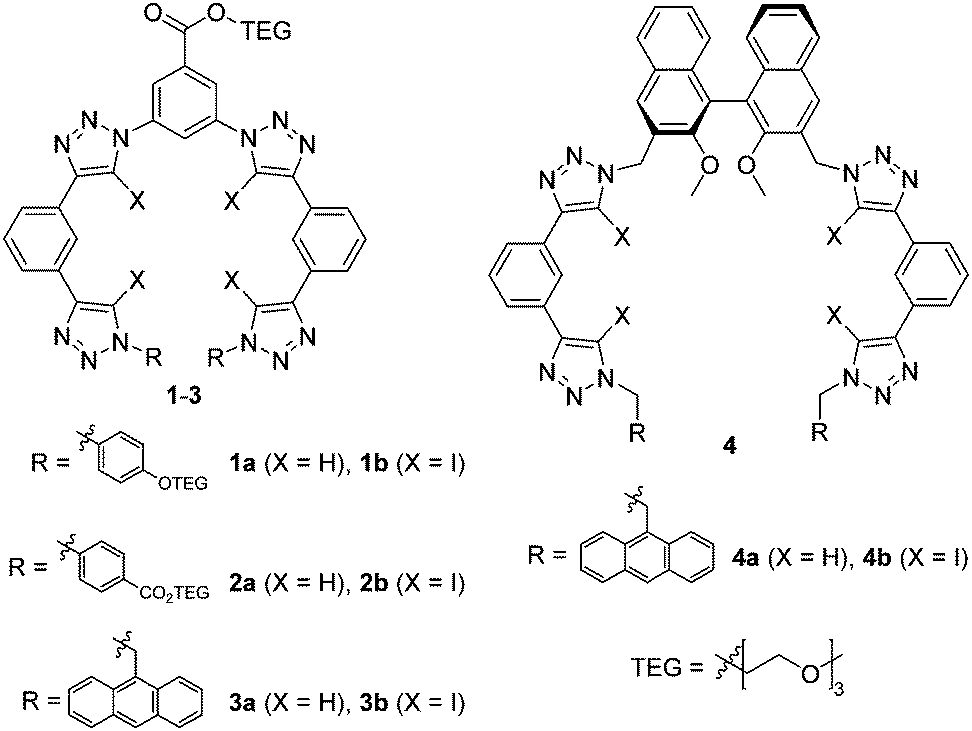 | ||
| Fig. 1 Structures of the XB and HB anion receptors 1–4. | ||
XB and HB foldamers 1–4 were synthesized via Cu(I)-catalysed azide-(iodo)alkyne cycloaddition (CuAAC) reactions (Scheme 1) using Cu(MeCN)4PF6 in the presence of tris(benzyltriazolylmethyl) amine (TBTA) ligand. For the preparation of 1a and 2a an alternative benzyltriazolylmethylamine (BTA) ligand18 was used as these compounds co-eluted with TBTA during chromatographic purification. As seen in Scheme 1, a terminal azide synthon 5–7 was reacted statistically with an excess of bis-alkyne 8a or 8b to afford an arm fragment 9–11. Two equivalents of 9–11 were then coupled under CuAAC conditions with a bis-azide core synthon 12 or 13 to give the anion receptors 1–4. The cycloadditions proceeded in moderate to high yields of 64–88% for 5H-triazole and 46–85% for 5I-triazole formation (see ESI† for full synthetic details).
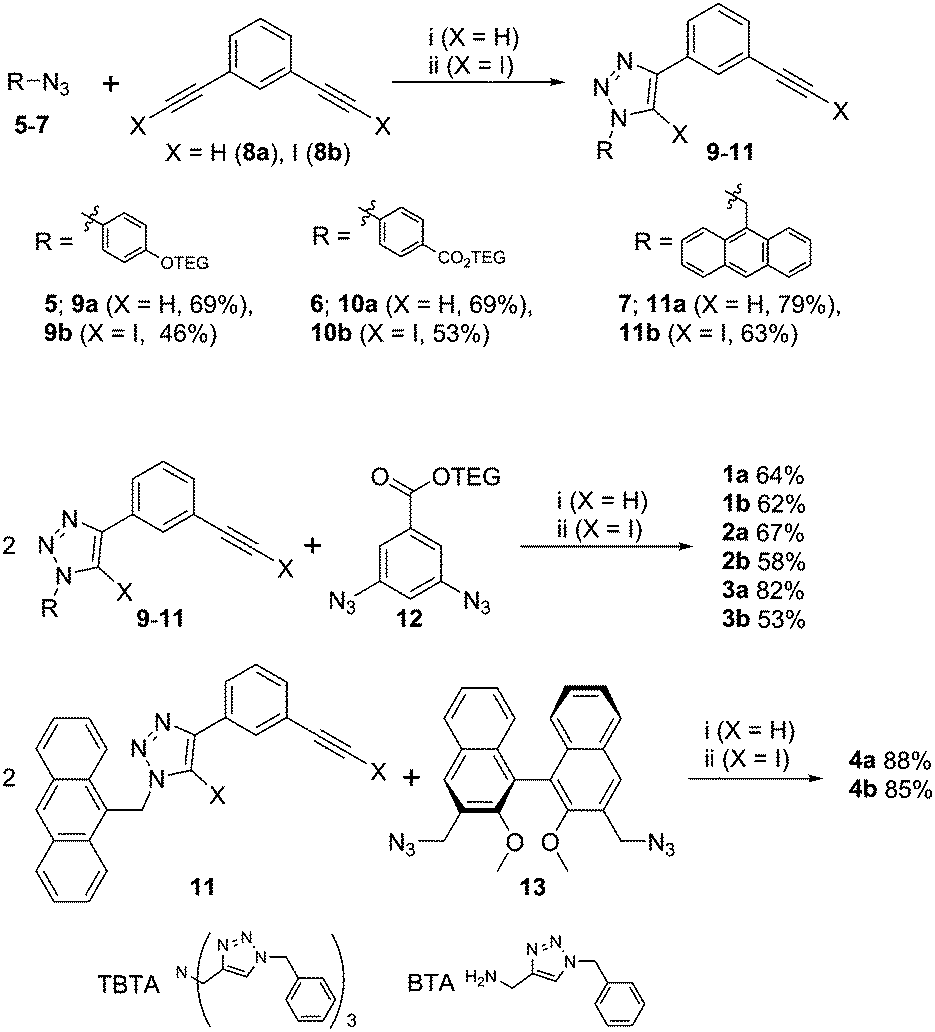 | ||
| Scheme 1 Synthesis of the anion receptors 1–4. Reaction conditions: (i) 0.05 eq. Cu(MeCN)4PF6, 0.05 eq. TBTA or BTA, 0.1 eq. DIPEA, DCM, rt; (ii) 0.1 eq. Cu(MeCN)4PF6, 0.1 eq. TBTA, THF, rt, darkness. | ||
The anion binding properties of 1–4 were studied by 1H NMR titrations by adding increasing amounts of different anions as tetrabutylammonium (TBA) salts and monitoring changes in 1H NMR spectra. For the HB receptors 1a–4a large downfield shifts (Δδ) of up to 2 ppm were observed for the triazole protons HA and HB; while smaller Δδ of 0.4–0.5 ppm occurred for the phenylene protons HC and HE (Fig. 2). This is consistent with the known binding mode of tetradentate triazole HB receptors10 wherein the triazole protons and the phenylene ortho protons all contribute to guest complexation. Binding isotherms were obtained by monitoring HA signals and 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric association constants calculated using WinEQNMR2 software.19
:1 stoichiometric association constants calculated using WinEQNMR2 software.19
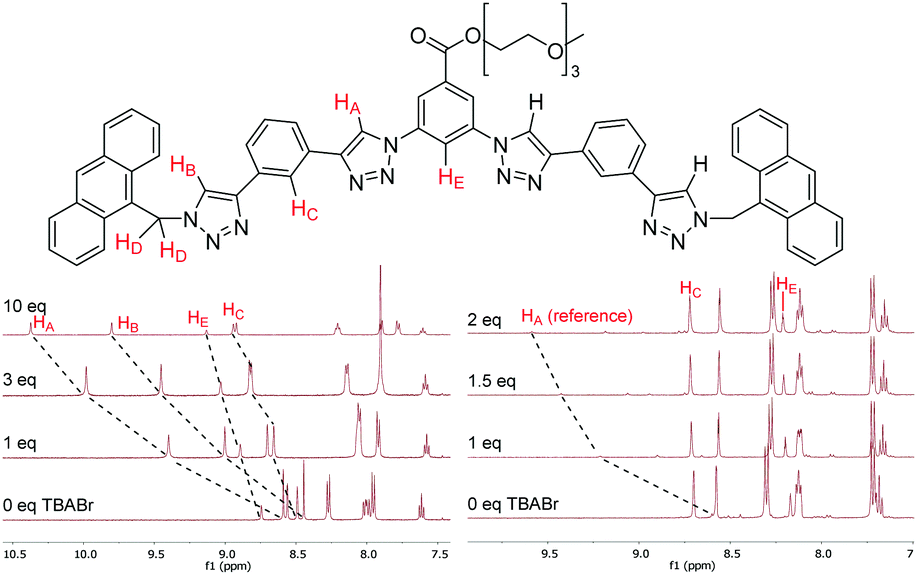 | ||
| Fig. 2 Top: Labeling of protons monitored in 1H NMR anion binding studies (consistent across 1–4). Bottom left: Partial 1H NMR spectra of 2a titrated with TBABr. Bottom right: Partial 1H NMR spectra of a competition experiment where 2b with 5 mol% of 2a as reference is titrated with TBABr (500 MHz, CDCl3, 298 K). | ||
In the case of XB foldamers 1b–4b only small downfield perturbations Δδ of 0.03–0.2 ppm were observed for the ortho phenylene protons HC and HE. This is most likely due to the large size of the receptors’ iodine atoms binding the guest anion at a significant distance from these protons (vide infra). Upfield shifts of similar magnitude were also observed for the aromatic and methylene protons in the terminal groups. Due to very small Δδ values reliable association constants could not be obtained for 1b–3b using the standard 1H NMR titration protocol. Therefore, 1b–3b were instead analysed using a host–host competition binding method similar to approaches previously employed to study alkali metal complexation with crown ethers.20 In a typical experiment a small amount (5 mol%) of HB receptor 2a was added to the solution of a XB compound 1b–3b as a reference. Changes of the reference compound HA signals upon anion addition were then monitored (Fig. 2), which enabled reliable association constant data to be determined for 1b–3b (see ESI† for full details). While 4b could not be analysed by this technique due to the lack of a suitable reference compound, it exhibited sufficient magnitude of downfield shifts in its HC protons to provide good quality data using a standard 1H NMR titration protocol.
As shown by the anion association constants for 1–4 given in Table 1, the XB foldamers 1b–4b were found to be stronger halide and oxoanion receptors than their HB analogs. This difference is especially prominent in systems 3 and 4. This is because the XB receptors rely predominantly on the iodotriazole XB donors for anion affinity, whereas their HB analogues benefit in part from secondary HB donors at the phenylene rings next to the triazoles.10,11 Deletion of these binding elements as in 3a and 4a leads to fewer convergent HB interactions and consequently a large loss of anion affinity. The XB receptors, on the other hand, are tolerant to these modifications. Thus, the anthryl-terminated 3b displayed affinity for Br− and I− that was two orders of magnitude stronger than of 3a. Likewise, XB receptor 4b exhibited notable Ka values of up to 500 M−1 in 1:1 CDCl3/acetone-d6, while 4a displayed almost no binding (too weak to be quantified).
| Cl− | Br− | I− | H2PO4− | AcO− | L-Tartrate | |
|---|---|---|---|---|---|---|
|
a 1:1 binding stoichiometry. All anions introduced as TBA salts; 1–3 studied in CDCl3; 4 in 1:1 CDCl3/acetone-d6 (500 MHz, 298 K); [receptor] = 1.5 mM. Unless stated otherwise, Ka for 1a–4a were derived from HA; for 4b from HC; for 1b–3b from HA of the reference compound 2a using the competition method. Uncertainties are given in parentheses.
b Could not be determined as HB foldamers are too strong H2PO4− binders to be used as reference compounds.
c Derived from HE.
d Too weak to be quantified.
e Derived from HC using the standard 1H NMR titration method.
|
||||||
| 1a | 232 (3) | 356 (10) | 427 (9) | 1244 (83) | 69 (1) | 331 (3) |
| 1b | 433 (44) | 600 (46) | 1202 (87) | —b | 320 (57) | 281 (27) |
| 2a | 466 (18) | 740 (29) | 960 (46) | 2751 (92) | 174 (4) | 672 (9) |
| 2b | 592 (59) | 902 (76) | 2131 (142) | —b | 504 (88) | 549 (41) |
| 3a | 20 (2) | 15 (1) | 24 (1) | 87 (11)c | —d | 64 (1) |
| 3b | 742 (65) | 1235 (79) | 2712 (187) | 493 (80)e | 753 (116) | 585 (43) |
| 4a | —d | —d | —d | 37 (3) | —d | —d |
| 4b | 335 (14) | 384 (28) | 511 (94) | 207 (4) | 165 (9) | 445 (16) |
Comparing the XB foldamers, the anion binding strength is in the order 3b > 2b > 1b > 4b. This indicates that the wider spacing of the bis-iodotriazole arms in 4b leads to less effective alignment of its XB donors with halides and carboxylates. As expected, 2b exhibited 1.5–2 times higher Ka values than 1b due to its more electron-withdrawing termini, which is also observed in the HB analogues 1a and 2a.
The XB receptors displayed the order of selectivity I− > Br− > Cl− ≈ AcO− ≈ H2PO4−, which suggests better host–guest size complementarity with larger halides. Additionally, neutral XB donors 1–4 do not display charge assistance that might favour binding of small, hard anions in cationic hosts. Interestingly, tartrate2− > AcO− selectivity was observed in HB receptors 1a–3a and XB receptor 4b but not in 1b–3b. This may indicate that with 1b–3b, the anion binding cavity is too small to simultaneously bind both anionic groups in a dicarboxylate due to the large size of the four convergent iodine atoms. Increased spacing between the bis(iodotriazole) arms in 4b allows both carboxylate groups to be bound and induces dicarboxylate selectivity. This is also seen in HB analogues which have a less crowded anion binding site.
Single crystals of 3b·NaI suitable for X-ray structural analysis were obtained by combining 3b and excess NaI in 6:4 CHCl3/acetone.‡ The structure (Fig. 3) shows the XB host encapsulating I−via four linear halogen bonds [C–I⋯I− distances: 3.484(1)–3.574(2) Å (88–90% of ΣrvdW)21 C–I⋯I− angles: 165.7(3)–178.3(3)°]. Due to the large size of the four iodine XB donor atoms the wrapping of the foldamer around the guest anion is less tight than in the analogous HB systems.22 The iodotriazoles are tilted by 29–54° relative to the neighbouring phenylene rings and the I− guest is situated 3.01 Å above the mean plane of the foldamer backbone. The Na+ countercation is complexed by the triethylene glycol chain, with an acetone solvate molecule and a triazole N3 atom from an adjacent molecule completing its coordination sphere.
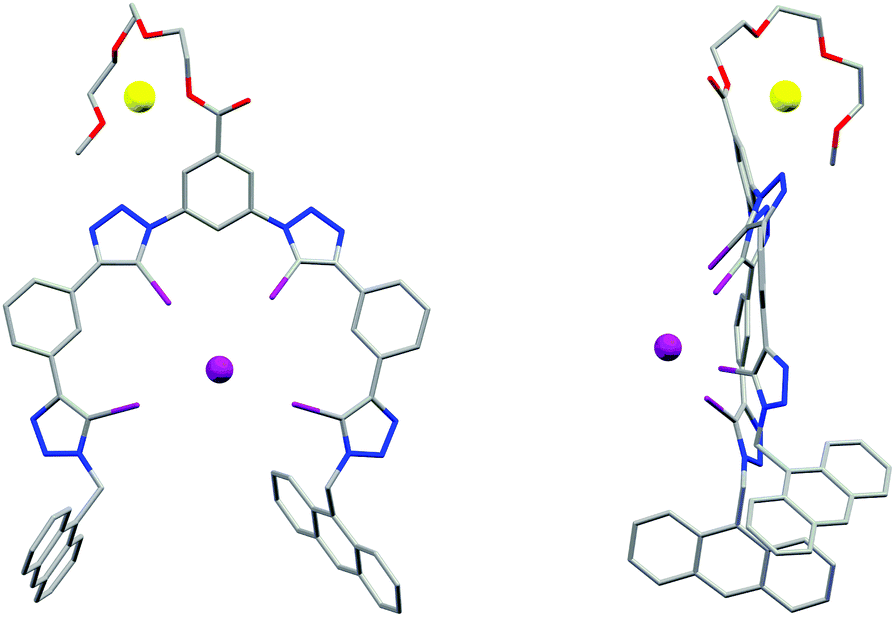 | ||
| Fig. 3 X-ray crystal structure of 3b·NaI showing the face-on (left) and side-on (right) view, visualising the folding of the receptor in a capped configuration. | ||
The XB receptors 3b and 4b displayed an intense fluorescence in 375–525 nm region arising from the emission of anthracene termini (Fig. 4). In 3b a large increase in emission intensity, without changes in wavelength, occurred upon addition of TBABr. A small increase was also observed for 4b; the most significant change occurred in the emission peak at 420 nm for both receptors. The overall increase in fluorescence intensity upon anion binding is most probably due to conformational rigidification of the receptor which suppresses non-radiative decay pathways. Thus, the larger fluorescence enhancement in 3b is likely due to 3b being less disordered in its bound state than 4b. As seen in the solid state structure of 3b·NaI, the anthracene termini are too far from each other for effective intramolecular π-stacking which accounts for the absence of excimer emission.23
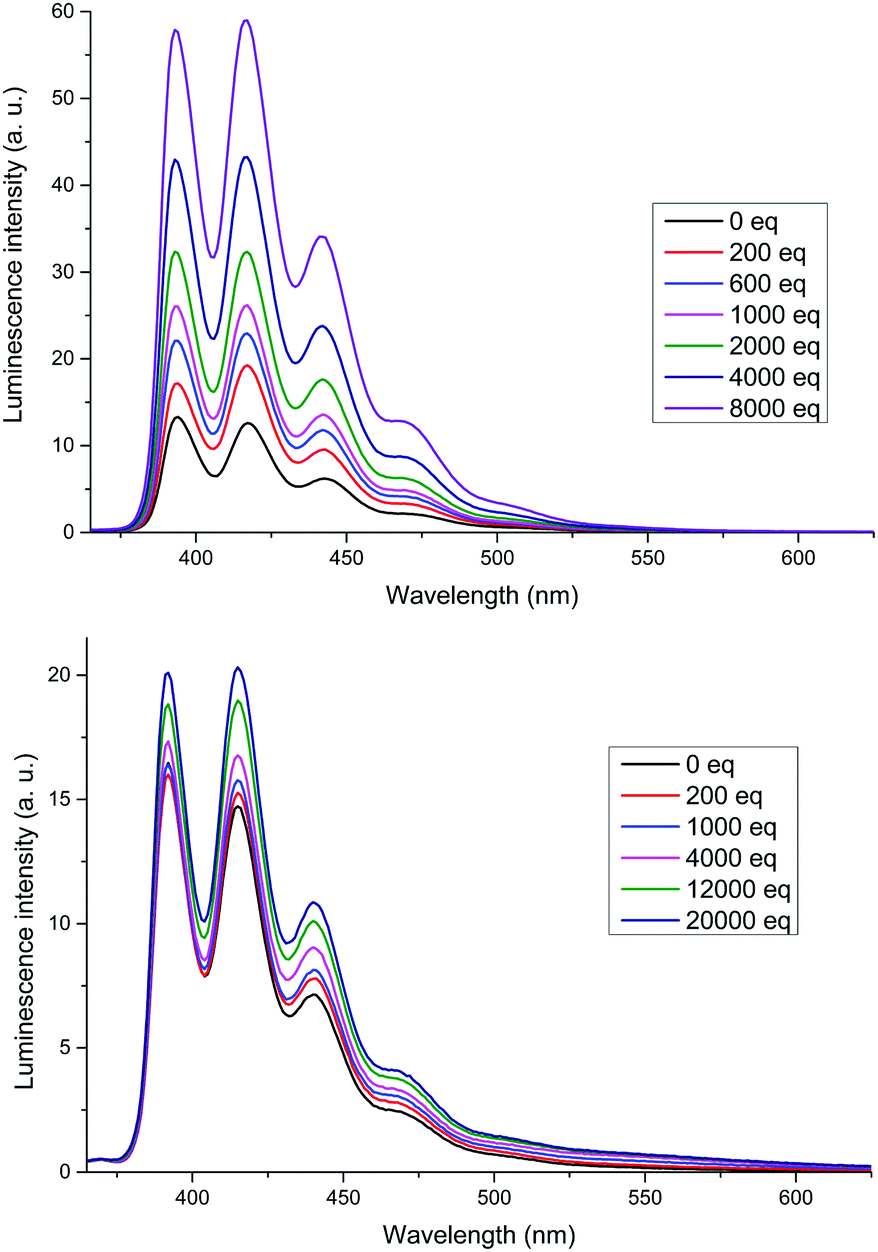 | ||
| Fig. 4 Emission spectra of 3b (top) and 4b (bottom) titrated with TBABr. Host concentration: 1 μM in CHCl3 (3b) or 1:1 CHCl3/acetone (4b), λex = 350 nm. | ||
Chiral XB 4b receptor was titrated with both enantiomers of Boc-Ala, Boc-Leu, Boc-Trp, Glu and tartrate as TBA salts. The association constant values shown in Table 2 reveal varying degrees of chiral discrimination were observed with different anions. The greatest difference in affinity was observed for tryptophan (KD/KL = 1.69), followed by leucine and alanine, which correlates with the steric bulk of the amino acid residue. In the case of the dicarboxylate guests, a modest degree of discrimination was seen for tartrate while no difference was observed for glutamate. This is once again consistent with the steric factors of guest anions.
| K a | K D/KL | |
|---|---|---|
|
a All anions were introduced as TBA salts. All titrations were undertaken in 1:1 CDCl3/acetone-d6 (500 MHz, 298 K). Uncertainties are given in parentheses.
b Determined from titration data monitoring HD.
|
||
| L-Boc-Ala | 336 (23) | 0.79 (0.17) |
| D-Boc-Ala | 265 (52) | |
| L-Boc-Leu | 287 (38) | 1.52 (0.21) |
| D-Boc-Leu | 436 (21) | |
| L-Boc-Trp | 200 (14) | 1.69 (0.12) |
| D-Boc-Trp | 337 (4) | |
| L-Tartrate | 725 (6) | 1.29 (0.04) |
| D-Tartrate | 932 (28) | |
| L-Glutamate | 1483 (220) | 0.92 (0.15) |
| D-Glutamate | 1363 (99) | |
In summary a series of novel neutral tetrakis(5-iodo-1,2,3-triazole) foldamers 1b–4b was prepared and evaluated as halogen bonding anion receptors. Importantly, compared to their HB analogues 1a–4a, the XB foldamers showed enhanced anion affinities, with a general preference for binding heavier halides over oxoanions. The advantage of XB for anion binding is especially evident in 3b and 4b where the HB analogues 3a and 4a displayed very weak affinities due to the deletion of secondary HB donors, while the XB foldamers 3b and 4b remained effective as anion receptors. The incorporation of anthracene and chiral BINOL groups into the XB foldamer structure design demonstrated fluorescent anion sensing and chiral discrimination capabilities which are currently under further investigation.
We thank the EPSRC centre for Doctoral Training in Synthesis for Biology and Medicine (EP/L015838/1), the Agency for Science, Technology and Research (A*STAR), Singapore, and the European Research Council (FP7/2007–2014, ERC Advanced Grant Agreement No. 267426) for financial support, Diamond Light Source for an award of beamtime on I19 (MT13639) and the beamline scientists for technical support.
Notes and references
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS PubMed.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed.
- S. Schindler and S. M. Huber, Halogen Bonding Ii: Impact on Materials Chemistry and Life Sciences, 2015, vol. 359, pp. 167–203 Search PubMed.
- T. M. Beale, M. G. Chudzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC.
- N. L. Kilah, M. D. Wise, C. J. Serpell, A. L. Thompson, N. G. White, K. E. Christensen and P. D. Beer, J. Am. Chem. Soc., 2010, 132, 11893–11895 CrossRef CAS PubMed.
- R. Tepper, B. Schulze, M. Jager, C. Friebe, D. H. Scharf, H. Gorls and U. S. Schubert, J. Org. Chem., 2015, 80, 3139–3150 CrossRef CAS PubMed.
- B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522–2571 RSC.
- Y. L. Li and A. H. Flood, J. Am. Chem. Soc., 2008, 130, 12111–12122 CrossRef CAS PubMed.
- H. Juwarker, J. M. Lenhardt, D. M. Pham and S. L. Craig, Angew. Chem., Int. Ed., 2008, 47, 3740–3743 CrossRef CAS PubMed.
- H. Juwarker, J. M. Lenhardt, J. C. Castillo, E. Zhao, S. Krishnamurthy, R. M. Jamiolkowski, K. H. Kim and S. L. Craig, J. Org. Chem., 2009, 74, 8924–8934 CrossRef CAS PubMed.
- J. Shang, W. Zhao, X. Li, Y. Wang and H. Jiang, Chem. Commun., 2016, 52, 4505–4508 RSC.
- A. Brown and P. D. Beer, Chem. Commun., 2016, 52, 8645–8658 RSC.
- C. J. Massena, N. B. Wageling, D. A. Decato, E. Martin Rodriguez, A. M. Rose and O. B. Berryman, Angew. Chem., 2016, 55, 12398–12402 CrossRef CAS PubMed.
- L. Y. You, S. G. Chen, X. Zhao, Y. Liu, W. X. Lan, Y. Zhang, H. J. Lu, C. Y. Cao and Z. T. Li, Angew. Chem., Int. Ed., 2012, 51, 1657–1661 CrossRef CAS PubMed.
- F. Zapata, A. Caballero, P. Molina, I. Alkorta and J. Elguero, J. Org. Chem., 2014, 79, 6959–6969 CrossRef CAS PubMed.
- J. Y. C. Lim, I. Marques, L. Ferreira, V. Felix and P. D. Beer, Chem. Commun., 2016, 52, 5527–5530 RSC.
- T. R. Chan, R. Hilgraf, K. B. Sharpless and V. V. Fokin, Org. Lett., 2004, 6, 2853–2855 CrossRef CAS PubMed.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
- J. M. Daniel, S. D. Friess, S. Rajagopalan, S. Wendt and R. Zenobi, Int. J. Mass Spectrom., 2002, 216, 1–27 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- Y. R. Hua, Y. Liu, C. H. Chen and A. H. Flood, J. Am. Chem. Soc., 2013, 135, 14401–14412 CrossRef CAS PubMed.
- K. Ghosh, A. R. Sarkar, A. Ghorai and U. Ghosh, New J. Chem., 2012, 36, 1231–1245 RSC.
- H. Nowell, S. A. Barnett, K. E. Christensen, S. J. Teat and D. R. Allan, J. Synchrotron Radiat., 2012, 19, 435–441 CrossRef CAS PubMed.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- P. Parois, R. I. Cooper and A. L. Thompson, Chem. Cent. J., 2015, 9, 30 CrossRef PubMed.
- R. I. Cooper, A. L. Thompson and D. J. Watkin, J. Appl. Crystallogr., 2010, 43, 1100–1107 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Compound data, crystal data for 3b·NaI, details of anion binding studies. CCDC 1529410. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7cc00727b |
‡ Single crystal diffraction data were collected at 100(2) K using a custom-built Crystal Logic diffractometer and synchrotron radiation (λ = 0.6889 Å) at Diamond Light Source, beamline I19.24 Unit cell parameter determination and data reduction were carried out using CrysAlisPro. The structures were solved by charge-flipping with SUPERFLIP25 and refined by full matrix least squares on F2 using CRYSTALS.26–28 Full refinement details are given in the ESI.† Single crystal data: C64H48I5N12NaO5Zn·4(C3H6O), Mr = 1954.99; triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; a = 14.3220(4) Å, b = 17.7665(5) Å, c = 17.7815(5) Å, α = 113.335(2)°, β = 100.303(2)°, γ = 93.903(2)°, V = 4038.8(2) Å3; data, restraints, parameters: 11420/955/928; Rint = 0.195, final R1 = 0.100, wR2 = 0.281 (F2; [I > 2σ(I)]); Δρmin,max = −3.28, +2.38 e Å−3. CCDC 1529410. ; a = 14.3220(4) Å, b = 17.7665(5) Å, c = 17.7815(5) Å, α = 113.335(2)°, β = 100.303(2)°, γ = 93.903(2)°, V = 4038.8(2) Å3; data, restraints, parameters: 11420/955/928; Rint = 0.195, final R1 = 0.100, wR2 = 0.281 (F2; [I > 2σ(I)]); Δρmin,max = −3.28, +2.38 e Å−3. CCDC 1529410. |
| This journal is © The Royal Society of Chemistry 2017 |