
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A combined quantum-chemical and matrix-isolation study on molecular manganese fluorides†‡
Felix
Brosi
a,
Tobias
Schlöder
a,
Alexei
Schmidt
b,
Helmut
Beckers
a and
Sebastian
Riedel
*ab
aInstitut für Chemie und Biochemie - Anorganische Chemie, Freie Universität Berlin, Fabeckstraße 34/36, D-14195 Berlin, Germany. E-mail: s.riedel@fu-berlin.de
bInstitut für Anorganische und Analytische Chemie, Albert-Ludwigs-Universität Freiburg, Albertstraße 21, D-79104 Freiburg i. Br., Germany
First published on 19th January 2016
Abstract
Molecular manganese fluorides were studied using quantum-chemical calculations at DFT and CCSD(T) levels and experimentally by matrix-isolation techniques. They were prepared by co-deposition of IR-laser ablated elemental manganese or manganese trifluoride with F2 in an excess of Ne, Ar, or N2 or with neat F2 at 5–12 K. New IR bands in the Mn–F stretching region are detected and assigned to matrix-isolated molecular MnFx (x = 1–3).
Introduction
Manganese is an essential element for all living creatures and plays a key role in the process of photosynthesis.1 Beyond that it has gained importance on a laboratory scale as a reagent for quantitative analysis2 and on an industrial scale as an alloy component.3 This is mostly due to its high bonding affinity towards oxygen that culminates in the stability of Mn2O7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 which contains manganese in its highest possible oxidation state +VII. For the stabilization of high positive oxidation states very electronegative ligands are necessary and besides oxygen also fluorine is used.5 The highest oxidation state of a manganese fluoride is +IV in solid MnF4.
4 which contains manganese in its highest possible oxidation state +VII. For the stabilization of high positive oxidation states very electronegative ligands are necessary and besides oxygen also fluorine is used.5 The highest oxidation state of a manganese fluoride is +IV in solid MnF4.
In the binary system of manganese and fluorine MnF has gained attention due to the high multiplicity of its 7Σ+ ground state. MnF has been studied extensively by theoretical methods6 as well as experimentally using mass spectrometry,7 rotational,8–10 matrix-infrared11 and electron spin resonance spectroscopy.12 Commercially available MnF2 and MnF3 have been studied in detail in both solid and gaseous forms especially with regard to Jahn–Teller-distortion12–14 using gas-phase electron diffraction,15,16 X-ray diffraction,17,18 neutron scattering,19 vibrational spectroscopy,20–22 and mass spectrometry.7,23 MnF2 and MnF3 were thermally evaporated using a Knudsen cell and subsequently deposited in solid argon and neon matrices.20,21 Another approach to study molecular MnFx species is from thermally generated manganese atoms, which were treated with elemental fluorine. MnF, MnF2 and MnF3 were formed, and an IR band obtained in solid Ar at 768.7 cm−1 was tentatively assigned to “a molecule richer in fluorine than MnF3”.11 MnF4 is a blue solid, first synthesized in 196124 and later employed in the chemical synthesis of elemental fluorine.25 It has been characterized in the solid state by X-ray diffraction,26 vibrational spectroscopy22,27 and magnetic susceptibility measurements.24,28 Since it cannot be evaporated without decomposition to MnF3 and F229 the investigation of molecular MnF4 is challenging. In an earlier study solid MnF3 was treated with TbF4 at 700 K and an IR band at 794.5 cm−1 in the MnF stretching region as well as two weaker bands at 172.9 cm−1 and 176.6 cm−1 of the Ar-matrix isolated products were claimed to be molecular MnF4.23 From the appearance of a single band in the Mn–F stretching region a tetrahedral structure was deducted.
The fact that up to now binary fluorides of manganese higher than MnF4 have not been obtained is remarkable. The heavier homologue technetium forms TcF6,30 while TcF7 is predicted to be stable,31 and ReF7 is well-known.32 FeF4 and CrF5 have been characterized,33,34 while earlier reports on the existence of CrF6 were shown to be erroneous.35 A previous report about the possible existence of manganese pentafluoride taking into account thermochemical data and sterical considerations36 stimulated us to reinvestigate the higher molecular Mn–F species using quantum-chemical calculations at DFT and CCSD(T) levels and to conduct matrix-isolation experiments in combination with IR spectroscopy.
Instead of thermally evaporating manganese, we have used the IR-laser ablation of elemental manganese to generate more reactive excited manganese atoms, which were co-condensed with fluorine diluted (0.25–3%) in neon, argon or nitrogen and neat fluorine. In addition we report about IR-laser ablation of manganese trifluoride, which has not been described previously.
Results and discussion
Calculated structures, thermochemistry and vibrational frequencies
Optimized structures for the binary manganese fluorides MnFx (x = 1–7) are shown in Fig. 1. | ||
| Fig. 1 Optimized structures of molecular manganese fluorides at the CCSD(T) (MnF–MnF5) and DFT levels (MnF6 and MnF7). Selected bond lengths (pm) and angles (°) are indicated. For a complete list see also Table S1 in the ESI.‡ | ||
The binary manganese fluorides have a high-spin ground state. The bond length of diatomic MnF calculated at the CCSD(T) level by Nhat et al. was 185 pm,6 which is fairly close to our value of 184.3 pm at the DFT level and that obtained experimentally by rotational spectroscopy (183.9 pm).10 MnF2 is computed to be linear with a CCSD(T) bond length of 181.6 pm. Gas phase electron diffraction gave a consistent value of 181.1 pm.15 MnF3 has a distorted trigonal planar structure. The quintet ground state shows two longer Mn–F bonds (175.3 pm) and one shorter Mn–F bond (173.0 pm) at the CCSD(T) level, which is in very good agreement with the results from gas phase electron diffraction (175.4 pm and 172.8 pm).16 According to our CCSD(T) calculations MnF4 has a distorted tetrahedral structure (C2v symmetry) with Mn–F bond lengths of 173.0 pm and 169.2 pm. For MnF5 a trigonal bipyramidal minimum structure (D3h symmetry) with bond lengths of 174.1 pm (Mn–Fax) and 167.4 pm (Mn–Feq) was computed at the CCSD(T) level. The d1 electron configuration of manganese in MnF6 causes a distortion from a regular octahedral structure which leads to either a structure with D4h- or D3d-symmetry. Both isomers are minima on the potential energy surface with the trigonal antiprismatic structure (bond lengths of 172.7 pm) just 0.5 kJ mol−1 in energy above the tetragonal bipyramidal structure (Mn–Fax 172.1 pm, Mn–Feq 173.4 pm) at the CCSD(T)/aug-cc-pVTZ//B3LYP/aug-cc-pVTZ level. For MnF7 also two minimum structures were computed at the DFT level. The regular D5h-symmetrical pentagonal bipyramidal isomer exhibits Mn–Fax bond lengths of 171.3 pm and Mn–Feq bond lengths of 180.4 pm. The monocapped trigonal prismatic isomer revealed four Mn–F bonds of 175.4 pm and two slightly shorter bonds of 175.3 pm, while the seventh fluorine atom is considerably weaker bound with 193.0 pm (Fig. 1). At the CCSD(T)/aug-cc-pVTZ//B3LYP/aug-cc-pVTZ level the latter isomer is favored in energy by 37.9 kJ mol−1. This is in contrast to the computed minimum structure of TcF731 and the crystal structure of ReF737 which both are (slightly) distorted pentagonal bipyramids. The reason for this disparity could be the smaller radius of the manganese atom compared to technetium and rhenium atoms.
The observability of a transient molecule in a matrix-isolation experiment requires at least a marginal thermochemical stability. This can be assessed by calculating the enthalpy of possible decomposition reactions. Table 1 lists the corresponding reaction enthalpies for the optimized manganese fluorides.
| Reaction | B3LYP | CCSD(T) | ||
|---|---|---|---|---|
| ΔE | ΔE + ΔZPE | ΔE | ΔE + ΔZPE | |
| a Values in kJ mol−1. b CCSD(T)/aug-cc-pVTZ//B3LYP/aug-cc-pVTZ. | ||||
| MnF7 → MnF5 + F2 | −394.5 | −399.9 | −357.5b | — |
| MnF7 → MnF6 + ½F2 | −271.7 | −275.6 | −195.3b | — |
| MnF7 → MnF6 + F | −194.1 | −201.1 | −119.3b | — |
| MnF6 → MnF4 + F2 | −67.9 | −76.5 | −141.5b | — |
| MnF6 → MnF5 + ½F2 | −122.8 | −124.3 | −162.2b | — |
| MnF6 → MnF5 + F | −45.2 | −49.8 | −86.6b | — |
| MnF5 → MnF3 + F2 | 217.4 | 206.5 | 131.7 | 119.9 |
| MnF5 → MnF4 + ½F2 | 54.9 | 47.7 | 20.2 | 12.9 |
| MnF5 → MnF4 + F | 132.5 | 122.2 | 96.5 | 86.4 |
| MnF4 → MnF2 + F2 | 405.0 | 397.8 | 304.1 | 295.3 |
| MnF4 → MnF3 + ½F2 | 162.5 | 158.7 | 111.4 | 107.0 |
| MnF4 → MnF3 + F | 240.1 | 233.2 | 187.7 | 180.5 |
| MnF3 → MnF2 + ½F2 | 242.6 | 239.1 | 192.6 | 188.3 |
| MnF3 → MnF2 + F | 320.2 | 313.6 | 268.9 | 261.8 |
The decomposition of MnF7 and MnF6 is strongly exothermic at all considered pathways including concerted elimination of F2, bimolecular reaction and homolytic bond fission. For MnF5 however the decomposition channels are computed to be endothermic at the CCSD(T) level. Thus, it should be possible to obtain molecular MnF5 under appropriate conditions, for example in matrix-isolation experiments. However, ΔG (298.15 K) for the gas phase reaction MnF5 → MnF4 + ½F2 is calculated to be −12.2 kJ mol−1 at the CCSD(T)/aug-cc-pVTZ level, so molecular MnF5 is not stable at ambient temperatures in the gas phase. Considering that sublimation enthalpies are usually smaller for the higher fluorides, MnF5 is most likely also not stable in the condensed phase. The value of ΔH(0 K) = 107.0 kJ mol−1 obtained for the decomposition MnF4 → MnF3 + ½F2 also suggests a low thermal stability of molecular MnF4, which is consistent with the reported vapor pressure of F2 over solid MnF4 at room temperature.29
Vibrational spectra of the manganese fluorides were calculated at the DFT (MnF–MnF7) and CCSD(T) levels (MnF–MnF5). The corresponding Mn–F stretching wavenumbers for MnF–MnF5 are listed and compared to experimentally observed IR bands in Table 2. Calculated IR wavenumbers of Mn–F stretching modes for MnF6 and MnF7 are listed in Table S2.‡
| Molecule | Mode | Calc. | Exp. | |||||
|---|---|---|---|---|---|---|---|---|
| B3LYP | CCSD(T) | Ne | Ar | N2 | ||||
| a Values in cm−1, IR intensities in km mol−1 in parentheses. b Ref. 6. c This work. d Ref. 11. e Ref. 20. f Ref. 23. g Ref. 21. | ||||||||
| MnF (C∞v, 7Σ+) | Σ+ | 607.7 | (134) | 605.9b | — | 608.5c | 589.2c | 585.8c |
| 588.6 | ||||||||
| MnF2 (D∞h, 6Σ+g) | Σ−u | 723.5 | (237) | 721.3 | (209) | 721.6c | 699.6c | |
| 722.1 | 700.1 | |||||||
| 699 | ||||||||
| 699.5 | ||||||||
| MnF3 (C2v, 5A1) | A1 | 646.1 | (9) | 656.7 | (15) | 657.1c | 643.7c | |
| 644 | ||||||||
| A1 | 720.9 | (126) | 729.9 | (134) | 726.9c | 711.2c | ||
| 728 | 712 | |||||||
| 711 | ||||||||
| 711.2 | ||||||||
| B2 | 764.2 | (256) | 779.3 | (287) | 773.4c | 758.7c | ||
| 774 | 759 | |||||||
| 758 | ||||||||
| 758.5 | ||||||||
| MnF4 (C2v, 4B2) | A1 | 675.0 | (18) | 681.5 | (13) | |||
| A1 | 749.3 | (85) | 755.5 | (83) | ||||
| B1 | 752.3 | (261) | 777.9 | (299) | 768.7 | |||
| B2 | 792.8 | (123) | 800.0 | (105) | 794.5 | |||
| MnF5 (D3h, 3A′2) | A′′2 | 742.7 | (299) | 768.5 | (331) | |||
| E′ | 816.9 | (272) | 824.4 | (235) | ||||
Matrix-isolation experiments
Molecular manganese fluorides were generated by IR-laser (λ = 1064 nm) ablation of a rotating elemental manganese target and co-condensed with fluorine diluted (0.25–3%) in neon or argon. After deposition of the sample new IR absorptions in the Mn–F stretching region were observed. Fig. 2 shows spectra obtained after deposition of the reaction products of manganese atoms with varying concentrations of F2 in Ar. The most intense band obtained with fluorine concentrations up to 1%, observed at 699.6 cm−1 in Ar and 721.6 cm−1 in Ne, respectively (ESI; Fig. S1‡), is associated with the Σ−u-mode of MnF2. Bands of MnF3 were observed at 758.7 cm−1 and 711.2 cm−1 in Ar and at 773.4 cm−1 and 726.9 cm−1 in Ne, respectively. These values agree very well with those from experiments of thermally evaporated manganese difluoride and trifluoride, respectively.20,21 While the intensity of the band due to MnF2 increased going from 0.25% to 0.5% F2 and then decreased with higher fluorine content, the intensity of the bands due to MnF3 increased steadily. Also the antisymmetric stretching band of [F3]− has been observed at 510.1 cm−1. This anion has been shown to be formed from F− and a F2 molecule during the deposition process, where the F− anion arises from a fluorine atom by electron capture in the plasma plum of an IR-laser ablated metal.38,39 A decrease in intensity with increasing fluorine concentration was observed for the band at 589.2 cm−1, which is assigned to diatomic MnF, which is in excellent agreement with a previous report.11 Its counterpart in Ne was observed at 608.5 cm−1 (ESI; Fig. S1‡). This wavenumber is consistent with an estimated value of 612.0 cm−1 obtained by rotational spectroscopy40 considering the red-shift of transition metal monofluoride bands experienced in solid noble gases.41 To the best of our knowledge this is the first report about MnF isolated in a neon matrix. It is noteworthy that IR-laser ablation of elemental manganese in the presence of fluorine did not produce any higher fluoride than MnF3. Irradiation of Ar-isolated F2 molecules with λ = 455 ± 10 nm is proved to produce reactive F-atoms, which are subject to a restricted movement within the solid Ar matrix, and thus may react with adjacent molecules such as MnF3. However, even this photolysis of the deposit failed to produce higher fluorides of manganese.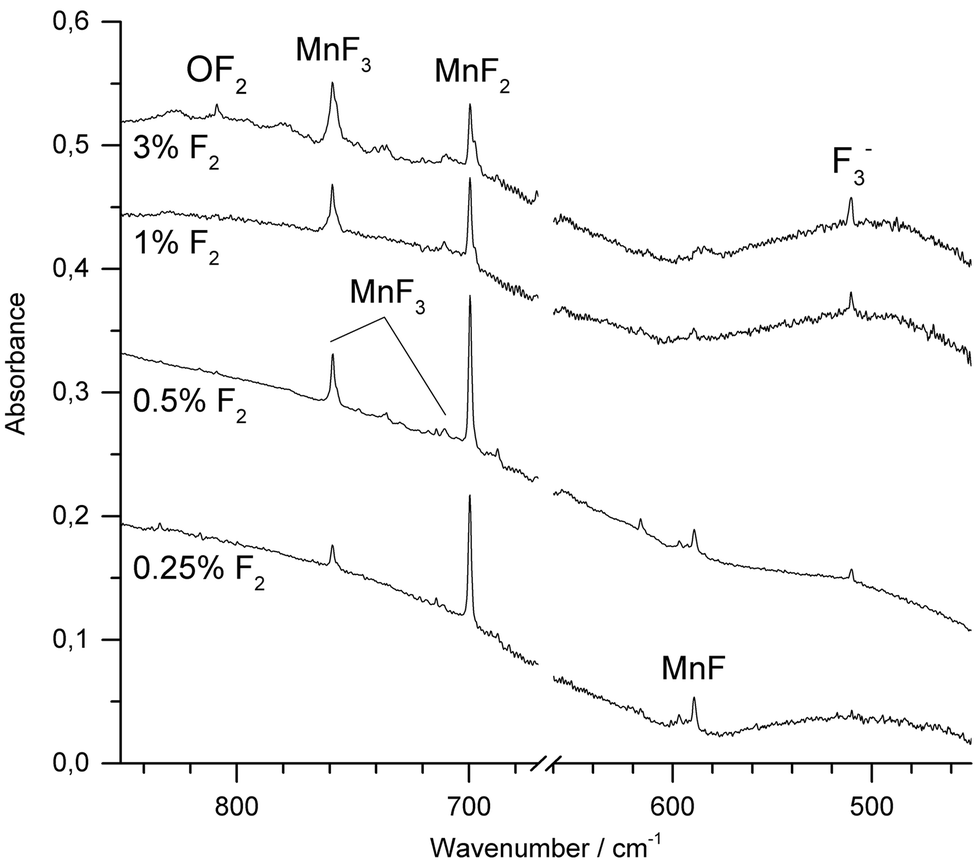 | ||
| Fig. 2 IR spectra in the 450–850 cm−1 region of the Ar matrix-isolated (12 K) reaction products of laser ablated manganese atoms in the presence of F2 (0.25–3%) after 1 h of deposition. The region of the bending mode of atmospheric CO2 (660–670 cm−1) was omitted for a better representation. | ||
When neat fluorine was used as matrix gas, several metal dependent absorptions could also be observed within Mn-F stretching range between 730 cm−1 and 780 cm−1 (ESI; Fig. S2‡). However, these absorptions were broad and relatively weak in intensity, and their assignment appeared to be difficult. Band broadening and overlapping features were also observed using N2 or 1% F2 in N2 together with IR-laser ablated manganese atoms (ESI; Fig. S3‡). This result is most likely due to the presence of several matrix sites. However, based on its behavior upon annealing and photolysis a band at 585.8 cm−1 in solid N2 could be unambiguously assigned to molecular MnF.
We have also conducted IR-laser ablation experiments using a pressed target pellet made of MnF3. Very recently we reported about the laser ablation of alkali metal fluorides,42 but, to the best of our knowledge, IR-laser ablation of solid transition metal fluorides has not yet been explored. Molecular MnF3 generated from a solid MnF3 target was first co-condensed with an excess of Ne at 5 K (Fig. 3) and produced bands at 773.4 cm−1 and 726.9 cm−1. The presence of MnF2 (Σ−u: 721.6 cm−1) indicates some decomposition of MnF3 in the plasma plum, probably by F-atom elimination.
 | ||
| Fig. 3 IR spectrum in the 700–850 cm−1 region of the Ne matrix-isolated (5 K) products of laser ablated solid MnF3 after 15 min of deposition. | ||
When Ar was used as matrix gas (Fig. 4), the band for MnF2 shifts to 699.6 cm−1 and those of MnF3 to 758.7 cm−1 and 711.2 cm−1. Additionally, the weaker third Mn–F stretching band of MnF3 was observed at 643.7 cm−1. A sharp band observed at 735.7 cm−1 could not be assigned to a mononuclear MnF2 or MnF3 species. This band was previously also observed by the thermal evaporation of solid MnF3 and tentatively assigned to a fluorine bridged dimer.11 In addition, traces of oxygen containing species such as OF2 and MnO3F43–45 were detected (ESI; Fig. S4 and S5‡). Although the target was prepared and fixed to the target holder under inert conditions, some superficial hydrolysis could probably not be prevented during the preparation of the target. The oxygen species OF2 and MnO3F might then have been formed in the plasma plum from the products of this hydrolysis.
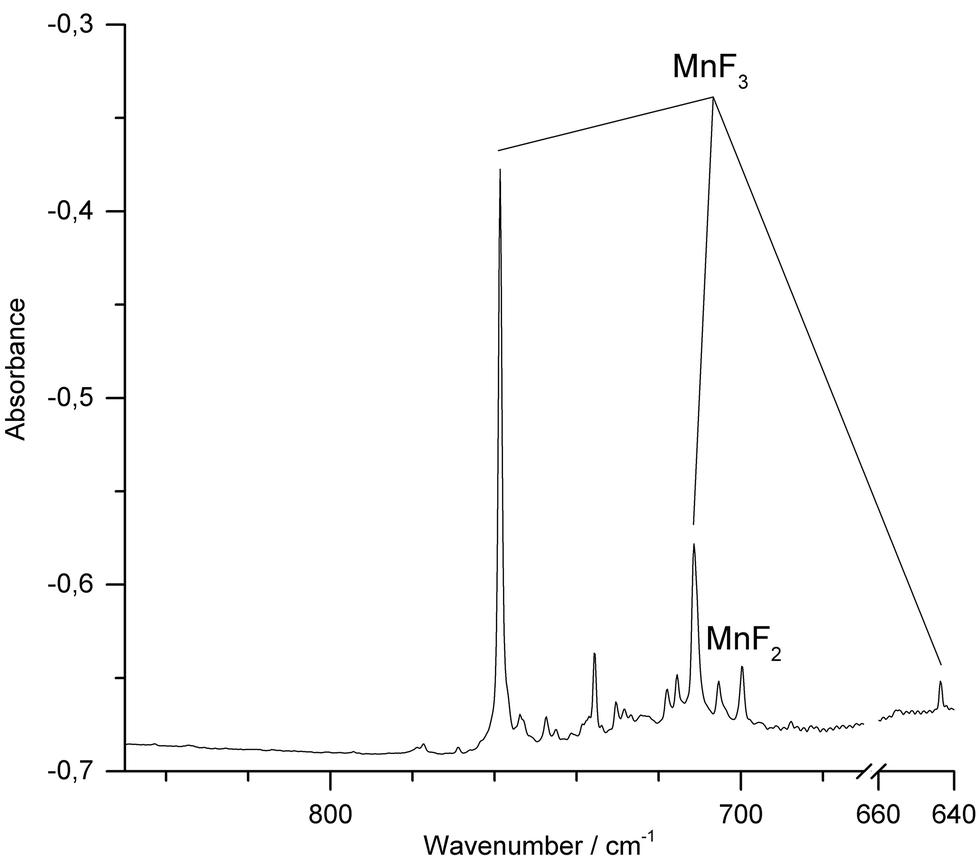 | ||
| Fig. 4 IR spectrum in the 640–850 cm−1 region of Ar matrix-isolated (12 K) products of laser ablated solid MnF3 after 15 min of deposition. The region of the bending mode of atmospheric CO2 (660–670 cm−1) was omitted for a better representation. | ||
IR-laser ablation of solid MnF3 was also carried out in the presence of fluorine (1% F2 in Ar). Apart from some line broadening, the spectrum (Fig. 5a and ESI; Fig. S6‡) is very similar to that obtained without fluorine (Fig. 4). After photolysis (λ = 455 ± 10 nm) a weak feature at 795 cm−1 appeared, which did not grow further with prolonged photolysis (Fig. 5b).
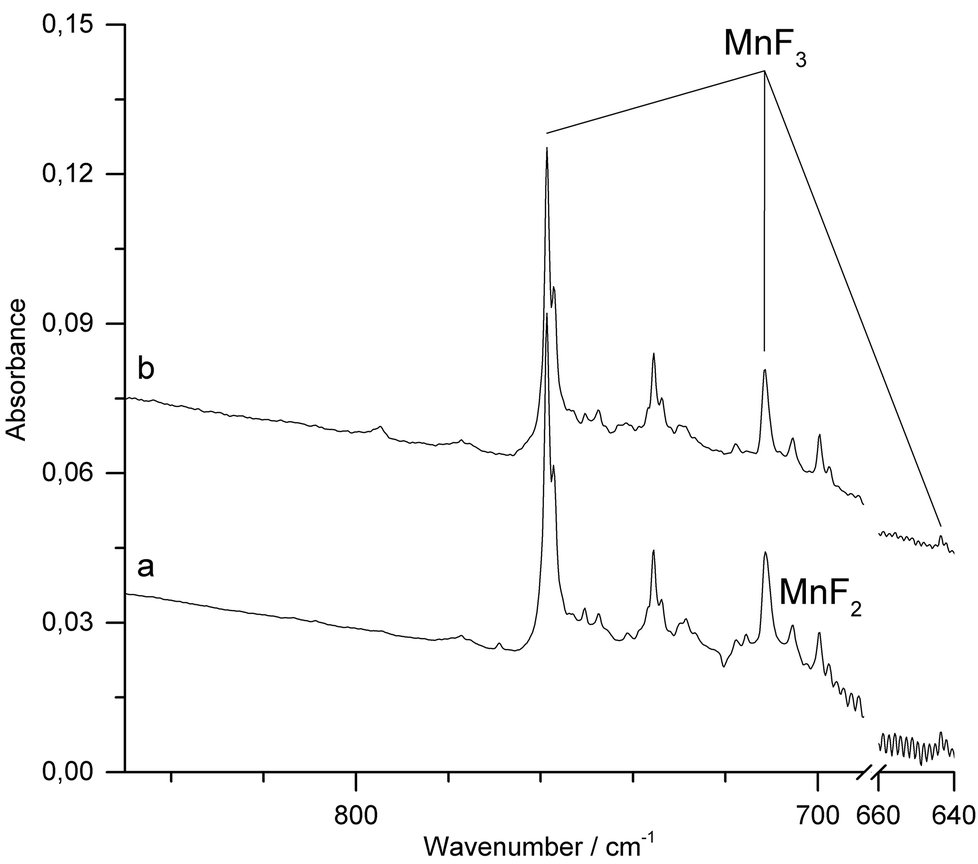 | ||
| Fig. 5 IR spectra in the 640–850 cm−1 region of Ar matrix-isolated (12 K) products of laser ablated solid MnF3 in the presence of F2 (1%). (a) After 15 min of deposition. (b) After 15 min of λ = 455 ± 10 nm irradiation. The region of the bending mode of atmospheric CO2 (660–670 cm−1) was omitted for a better representation. | ||
The carrier of this weak band is difficult to assign. A single IR band in the Mn–F stretching region at 794.5 cm−1 was previously obtained in solid Ar from the vaporization of a mixture of MnF3 and TbF4 at 700 K and assigned to molecular MnF4.23 From their observation, the authors inferred a tetrahedral structure of molecular MnF4. However, the presence of a single Mn–F stretching band is not in agreement with the calculated C2v structure of MnF4 (Fig. 1), for which at least three strong Mn–F stretching bands are predicted (Table 2). The previous assignment of molecular MnF4 to a single Mn–F stretching band at 794.5 cm−1 is therefore questionable, and the carrier of the strong IR band obtained in these experiments remains unknown.
Experimental and theoretical methods
Matrix-isolation experiments
CAUTION: Appropriate safety precautions should be taken into account when handling pure fluorine and metal fluorides. All equipment has to be adequately pretreated with fluorine prior to use. Note that sudden freezing of samples in metal cylinders to liquid nitrogen temperatures is potentially dangerous. The suitability of metal cylinders for such a cooling procedure should be proven and the cylinders should always be leak tested at liquid nitrogen temperature prior to filling with poisonous samples. The setup for matrix-isolation and IR-laser ablation experiments has been described in detail previously.35 The 1064 nm fundamental of a Nd:YAG laser was focused onto a freshly sanded rotating manganese (chemPUR, 99.99%) target or target pellets made of MnF3 (Alfa Aesar, 98%). Gaseous mixtures of various concentrations (0.25–100%) of fluorine (Solvay Fluor AG, 99.998%) in neon (Air Liquide, 99.999%), argon (SWF, 99.999%) and nitrogen (Linde, 99.999%) were prepared using a fluorine cylinder (V = 1000 cm3, thick-walled stainless steel, p298 K < 1 bar) cooled to 77 K to freeze out less volatile impurities such as traces of HF and CO2. These mixtures were than co-condensed together with the IR-laser ablated species for 45–60 min onto a CsI cold window cooled to 5 K (Ne matrices) or 12 K (Ar and F2). Pure fluorine matrices were deposited onto a protective layer of argon to prevent oxidation of the cold window material. FTIR spectra were recorded at a resolution of 0.5 cm−1 on a Bruker Vertex 70 spectrometer equipped with a KBr beam splitter using a liquid nitrogen cooled mercury cadmium telluride (LN-MCTB) detector. A common IR band observed in all spectra of such experiments using IR-laser ablated metal atoms and fluorine is due to the [F3]− anion at 510.1 cm−1 (Ar), 517.2 cm−1 (N2), or 524.6 cm−1 (Ne), respectively.46 As no isotopic splitting of the IR bands of both the monoisotopic manganese and fluorine elements can be observed, any assignment of bands of binary manganese and fluorine species mainly relies on reliably calculated spectra and meaningful variations of experimental conditions.Quantum-chemical calculations
Structures of the manganese fluorides MnFx (x = 1–7) displayed in Fig. 1 were fully optimized at the DFT level using the B3LYP functional47–49 in conjunction with the aug-cc-pVTZ basis sets for fluorine50 and manganese,51 and considering all reasonable spin multiplicities. Subsequent optimizations at the CCSD(T) level were carried out for the ground states of MnFx (x = 2–5). Harmonic frequencies were calculated for stationary points on the potential energy surface. The DFT calculations were performed with Gaussian0952 and CFOUR53 was used for the coupled-cluster calculations.Conclusion
For more than 50 years solid MnF4 has been the highest experimentally known manganese fluoride, but experimental data about its molecular structure are still missing. Quantum-chemical calculations at the CCSD(T) level predict molecular MnF4 to be stable against its decomposition according to MnF4 → MnF3 + ½F2 by ΔH(0 K) = 107 kJ mol−1. The corresponding decomposition route for the unprecedented MnF5 (MnF5 → MnF4 + ½F2) is found to be only marginally endothermic by ΔH(0 K) = 12.9 kJ mol−1. Molecular MnF6 and MnF7 are unstable with respect to their decomposition to lower manganese fluorides. The reaction of IR-laser ablated manganese atoms with elemental fluorine diluted in neon, argon and nitrogen and neat fluorine was studied. The IR analysis of the matrix-isolated products at 5–12 K revealed the formation of molecular MnF, MnF2 and MnF3. Molecular MnF3 has alternatively been produced and matrix-isolated by IR-laser ablation of a solid manganese trifluoride target. Attempts to detect IR bands of a higher manganese fluoride such as MnF4 or MnF5 by carrying out these experiments in the presence of elemental fluorine or by additional UV photolysis of the deposits failed. The presented results cast doubt on previous reports about the formation of such higher molecular manganese fluorides either from the reaction between MnF3 and TbF423 or by thermal evaporation of Mn atoms in the presence of fluorine.11
Acknowledgements
The authors thank the Fonds der Chemischen Industrie and the Deutsche Forschungsgemeinschaft (HA 5639/3-1 and GRK 1582/2 “Fluorine as a Key Element”) for financial support.References
- S. Mukhopadhyay, S. K. Mandal, S. Bhaduri and W. H. Armstrong, Chem. Rev., 2004, 104, 3981–4026 CrossRef CAS PubMed
.
- F. Margueritte, C. R. Acad. Sci., 1846, 100, 380–385 Search PubMed
- W. Zhang and C. Y. Cheng, Hydrometallurgy, 2007, 89, 137–159 CrossRef CAS
- H. Aschoff, Ann. Phys. Chem., 1860, 2(111), 217–229 CrossRef
- S. Riedel and M. Kaupp, Coord. Chem. Rev., 2009, 253, 606–624 CrossRef CAS
- P. V. Nhat, N. T. Cuong, P. K. Duy and M. T. Nguyen, Chem. Phys., 2012, 400, 185–197 CrossRef CAS
- R. A. Kent, T. C. Ehlert and J. L. Margrave, J. Am. Chem. Soc., 1964, 86, 5090–5093 CrossRef CAS
- O. Launila and B. Simard, J. Mol. Spectrosc., 1992, 154, 93–118 CrossRef CAS
- O. Launila, B. Simard and A. M. James, J. Mol. Spectrosc., 1993, 159, 161–174 CrossRef CAS
- P. M. Sheridan and L. M. Ziurys, Chem. Phys. Lett., 2003, 380, 632–646 CrossRef CAS
- S. B. Osin, D. I. Davlyatshin, V. F. Shevelkov and V. N. Mitkin, Russ. J. Phys. Chem., 1995, 69, 794–799 Search PubMed
- T. C. DeVore, R. J. Van Zee and W. Weltner Jr., J. Chem. Phys., 1978, 68, 3522–3527 CrossRef CAS
- R. J. Van Zee, C. M. Brown and W. Weltner Jr., Chem. Phys. Lett., 1979, 64, 325–328 CrossRef CAS
- P. Mondal, D. Opalka, L. V. Poluyanov and W. Domcke, Chem. Phys., 2011, 387, 56–65 CrossRef CAS
- N. Vogt, J. Mol. Struct., 2001, 570, 189–195 CrossRef CAS
- M. Hargittai, B. Réffy, M. Kolonits, C. J. Marsden and J.-L. Heully, J. Am. Chem. Soc., 1997, 119, 9042–9048 CrossRef CAS
- M. A. Hepworth and K. H. Jack, Acta Crystallogr., 1957, 10, 345–351 CrossRef CAS
- J. V. Rau, V. Rossi Albertini, N. S. Chilingarov, S. Colonna and U. Anselmi Tamburini, J. Fluorine Chem., 2001, 108, 253–256 CrossRef CAS
- Z. Yamani, Z. Tun and D. H. Ryan, Can. J. Phys., 2010, 88, 771–797 CrossRef CAS
- J. W. Hastie, R. Hauge and J. L. Margrave, J. Chem. Soc. D, 1969, 1452–1453 RSC
- V. N. Bukhmarina and Y. B. Predtechenskii, Opt. Spectrosc., 1996, 80, 684–687 Search PubMed
- Z. Mazej, J. Fluorine Chem., 2002, 114, 75–80 CrossRef CAS
- S. N. Cesaro, J. V. Rau, N. S. Chilingarov, G. Balducci and L. N. Sidorov, Inorg. Chem., 2001, 40, 179–181 CrossRef
- R. Hoppe, W. Dähne and W. Klemm, Naturwissenschaften, 1961, 11, 429 CrossRef
- K. O. Christe, Inorg. Chem., 1986, 25, 3721–3722 CrossRef CAS
- B. G. Müller and M. Serafin, Z. Naturforsch., 1987, 42b, 1102–1106 Search PubMed
- M. Adelhelm and E. Jacob, J. Fluorine Chem., 1991, 54, 21 CrossRef
- K. Lutar, A. Jesih and B. Zemva, Polyhedron, 1988, 7, 1217–1219 CrossRef CAS
- R. Hoppe, W. Dähne and W. Klemm, Justus Liebigs Ann. Chem., 1962, 658, 1–5 CrossRef CAS
- H. Selig, C. L. Chernick and J. G. Malm, J. Inorg. Nucl. Chem., 1961, 19, 377 CrossRef CAS
- S. Riedel, M. Renz and M. Kaupp, Inorg. Chem., 2007, 46, 5734–5738 CrossRef CAS PubMed
- J. G. Malm, H. Selig and S. Fried, J. Am. Chem. Soc., 1960, 82, 1510 CrossRef CAS
- T. Schlöder, T. Vent-Schmidt and S. Riedel, Angew. Chem., Int. Ed., 2012, 51, 12063–12067 (
Angew. Chem.
, 2012
, 124
, 12229–12233
) CrossRef PubMed
- A. J. Edwards, Proc. Chem. Soc., London, 1963, 205 CAS
- T. Schlöder, F. Brosi, B. J. Freyh, T. Vent-Schmidt and S. Riedel, Inorg. Chem., 2014, 53, 5820–5829 CrossRef PubMed
- M. I. Nikitin and E. G. Rakov, Russ. J. Inorg. Chem., 1998, 43, 314–318 Search PubMed
- T. Vogt, A. N. Fitch and J. K. Cockcroft, Science, 1994, 263, 1265–1267 CAS
- T. Vent-Schmidt, F. Brosi, J. Metzger, T. Schlöder, X. Wang, L. Andrews, C. Müller, H. Beckers and S. Riedel, Angew. Chem., Int. Ed., 2015, 54, 8279–8283 (
Angew. Chem.
, 2015
, 127
, 8397–8401
) CrossRef CAS PubMed
- F. Brosi, T. Vent-Schmidt, S. Kieninger, T. Schlöder, H. Beckers and S. Riedel, Chem. – Eur. J., 2015, 21, 16455–16462 CrossRef CAS PubMed
- G. D. Rochester and E. Olsson, Z. Phys., 1939, 114, 495–499 CrossRef CAS
- X. Wang, L. Andrews, F. Brosi and S. Riedel, Chem. – Eur. J., 2013, 19, 1397–1409 CrossRef CAS PubMed
- F. A. Redeker, H. Beckers and S. Riedel, RSC Adv., 2015, 5, 106568–106573 RSC
- P. J. Aymonino, H. Schulze and A. Müller, Z. Naturforsch., B: Anorg. Chem. Org. Chem. Biochem. Biophys. Biol., 1969, 24, 1508–1510 CAS
- M. J. Reisfeld, L. B. Asprey and N. A. Matwiyoff, Spectrochim. Acta, 1970, 27, 765–772 CrossRef
- E. L. Varetti and A. Müller, Z. Anorg. Allg. Chem., 1978, 442, 230–234 CrossRef CAS
- S. Riedel, T. Köchner, X. Wang and L. Andrews, Inorg. Chem., 2010, 49, 7156–7164 CrossRef CAS PubMed
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS
- C. Lee, Y. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS
- R. A. Kendall, T. H. Dunning Jr. and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS
- N. B. Balabanov and K. A. Peterson, J. Chem. Phys., 2005, 123, 064107 CrossRef
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed
- CFOUR, Coupled-Cluster techniques for Computational Chemistry, a quantum-chemical program package by J. F. Stanton, J. Gauss, M. E. Harding, P. G. Szalay with contributions from
A. A. Auer, R. J. Bartlett, U. Benedikt, C. Berger, D. E. Bernholdt, Y. J. Bomble, L. Cheng, O. Christiansen, M. Heckert, O. Heun, C. Huber, T.-C. Jagau, D. Jonsson, J. Jusélius, K. Klein, W. J. Lauderdale, D. A. Matthews, T. Metzroth, L. A. Mück, D. P. O'Neill, D. R. Price, E. Prochnow, C. Puzzarini, K. Ruud, F. Schiffmann, W. Schwalbach, C. Simmons, S. Stopkowicz, A. Tajti, J. Vázquez, F. Wang and J. D. Watts and the integral packages MOLECULE (J. Almlöf and P. R. Taylor), PROPS (P. R. Taylor), ABACUS (T. Helgaker, H. J. Aa. Jensen, P. Jørgensen, and J. Olsen), and ECP routines by A. V. Mitin and C. van Wüllen. For the current version, see http://www.cfour.de
Footnotes |
| † Dedicated to Prof. Dieter Lentz on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c5dt04827c |
| This journal is © The Royal Society of Chemistry 2016 |