
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvent effects in catalysis: rational improvements of catalysts via manipulation of solvent interactions
Paul J.
Dyson
a and
Philip G.
Jessop
b
aInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015 Lausanne, Switzerland
bDepartment of Chemistry, Queen's University, Kingston, ON K7L 3N6, Canada
First published on 17th February 2016
Abstract
In homogeneous catalysis, the main emphasis on improving catalyst performance (rate, yield and selectivity) is directed towards manipulation of the ligands. The steric and electronic effects of ligands are extremely well understood and the rational design of new ligands that leads to improved catalysts is feasible. Similarly, in heterogeneous catalysis structural changes to the catalyst are used to improve their properties. In contrast, the role that solvents play in catalytic processes is often given cursory attention. The environmental impact of solvents is often considered, as it should be, and the use of organic-immiscible solvents, e.g. water, ionic liquids, etc., is frequently considered with respect to catalyst recovery, product isolation, and recycling. Nevertheless, the direct role of solvents in reactions is often overlooked, although solvents interact directly with the catalyst, the substrates and products, and all these interactions can increase or decrease reaction rate and/or selectivity. Herein we consider the role of solvents in catalysis and illustrate the critical role of solvents viewed from a mechanistic approach. In particular, we focus largely, but not exclusively, on hydrogenation reactions and cross-coupling reactions as the main types of solvents used for these two classes of reactions tend to be very different and illustrate different functions of solvents in catalysis.
1. Introduction
Solvents are widely used at all stages of a catalytic process, including during the synthesis of the catalyst, during the catalytic reaction and often during product purification. In addition, solvents are used in the analysis of the catalyst and/or product and spectroscopy, in crystal growth that is sometimes required for a product, in product formulations and even in cleaning the reactor/instruments after use. In this review we do not intend to discuss the role of solvents in all these different areas, but instead focus on the critical role that solvents play on the catalyzed reaction with respect to the rate, yield and selectivity, an area that is often overlooked or finds only a cursory mention in research papers. Since there are many different solvents to choose from, selecting the right ones for a catalytic process can improve the outcome of the reaction considerably. From a synthetic perspective, solvents have pronounced effects on reaction equilibria; they influence the formation of different isomers and affect reaction rates and mechanisms. It is these performance aspects of solvents that we intend to discuss herein. It should be noted that performance requirements are not the only factors to consider when selecting a solvent; increasingly the choice must also be based on environmental and toxicological factors. Since these areas have been covered elsewhere,1 we do not intend to devote much time to them here. Instead, a description of key solvent parameters, mass transfer aspects, and their application in catalysis is given. This is followed by a summary of all the ways that solvents can influence catalysis and lastly case studies are given that serve to illustrate these points, ultimately showing how careful choice of a solvent or solvent mixture can significantly enhance catalytic activity.2. Solvent parameters
The solvent properties that strongly influence catalysis include dipolarity (or polarity), hydrogen-bond donating ability (proticity) and hydrogen-bond accepting ability (basicity). The performance of a catalyst is strongly influenced by these parameters and therefore finding the right solvent for a catalytic reaction, or determining how a solvent affects the reaction, is important. There are many measures of solvent properties,2 but some of them, including Reichardt's ET(30) and ETN parameters, combine two properties into one metric, which can diminish their utility when delineating solvent effects in catalysis. It is preferable to use solvent parameters that do not combine two properties. For example, dielectric constant is a common measure of solvent polarity.3 Kamlet and Taft developed three parameters, π*, β and α, that independently measure the polarity (including polarizability), basicity and proticity of a solvent,4 including ionic liquids,5 respectively. Such solvent parameters can provide useful correlations with catalytic performance.In some catalytic systems, one of these solvent properties has an over-riding influence on catalyst performance. For example, the isomerization of allylic alcohol to propanal (Fig. 1), using the photo-generated, electronically unsaturated 16 valence electron catalyst Fe(CO)4, strongly depends on the solvent basicity, β, because solvents that are able to coordinate to the unsaturated Fe(CO)4 catalytic species competitively inhibit the alkene from binding.6 In contrast, solvent polarity/polarizability (π*) and proticity (α, not shown in the figure) apparently have little impact on the reaction.
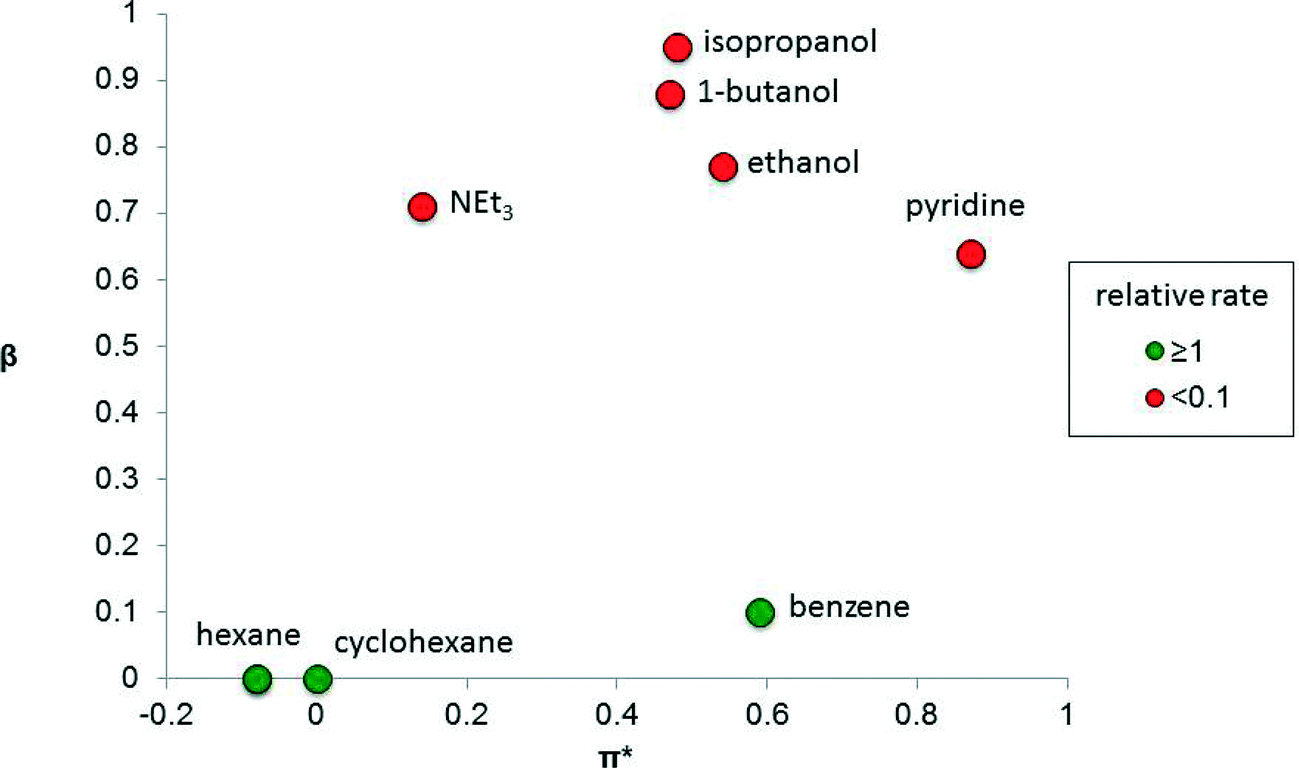 | ||
| Fig. 1 Graph showing the relative rate of isomerization of allylic alcohol to propanal catalyzed by the photo-generated Fe(CO)4 species as a function of the basicity (β) and polarity (π*) of the solvent.4 The colors indicate relative reaction rates, relative to the rate in cyclohexane. | ||
In other catalytic processes more than one solvent property may significantly influence reaction performance. For example, the enantioselectivity of the cyclopropanation shown in Fig. 2 depends on both solvent polarity and basicity.7 Highest enantioselectivities are obtained in non-basic, non-polar solvents and the lowest enantioselectivities are observed in solvents that are either basic or polar, but not both. The transition state for the selectivity-controlling step is believed to involve the approach of the styrene substrate to one of the Rh centers. Polar solvents lead to the formation of an early transition state in which the styrene substrate is quite far from the chiral environment of the catalyst, reducing the impact of the chiral ligand on selectivity.8 Lewis basic solvents could have a similar effect as they affect the electron density at the catalytic Rh centre by binding to the other Rh center.
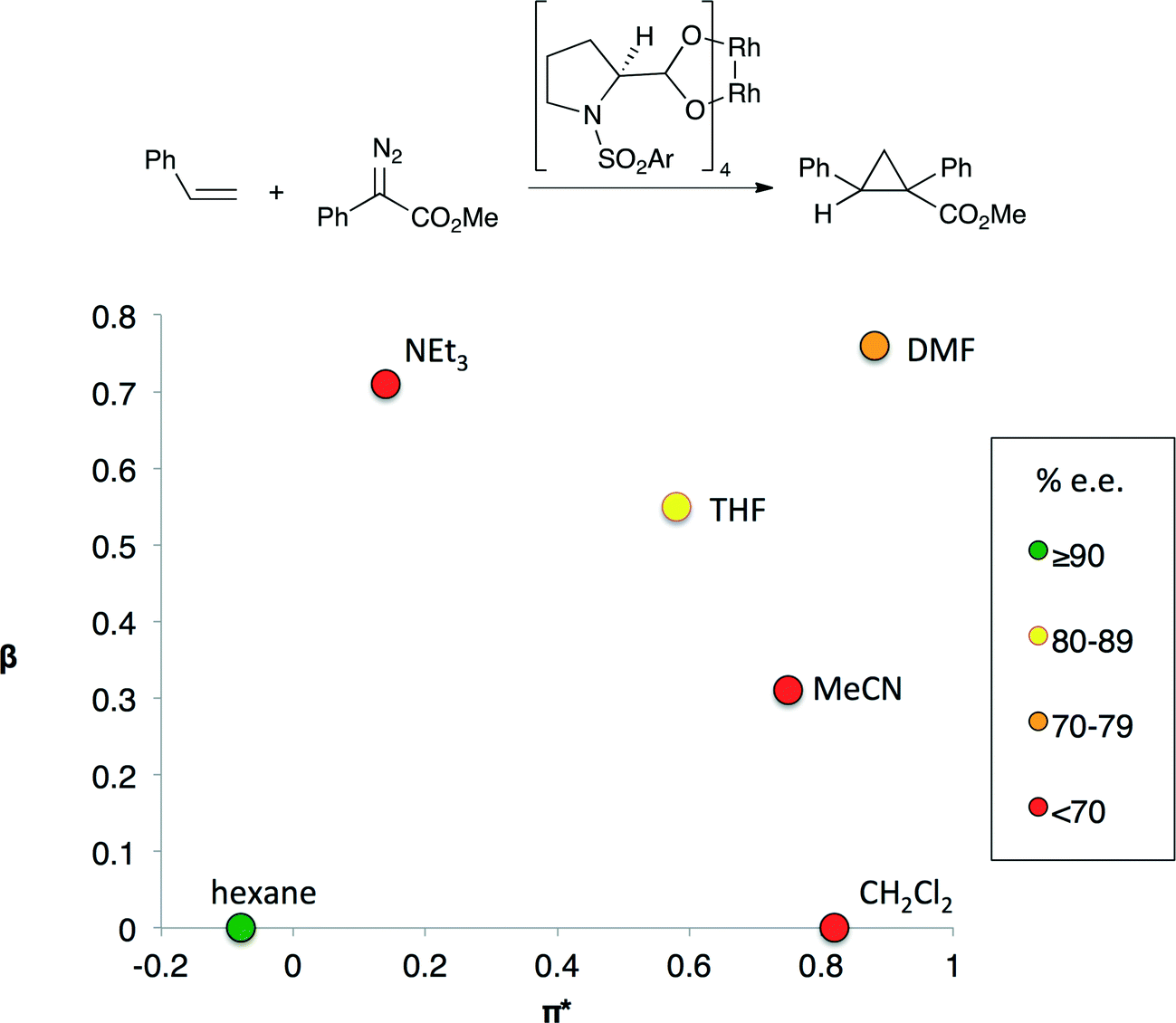 | ||
| Fig. 2 An enantioselective cyclopropanation reaction (top) and enantioselectivity (bottom) of cyclopropanation as a function of the basicity (β) and polarity (π*) of the solvent.5 The colors indicate the enantiomeric excess obtained if the reaction is performed in the indicated solvent. Only aprotic solvents were evaluated. | ||
The effect of one property, such as polarity, on catalyst performance can be determined independently of the other solvent properties using any of the four methods listed below:
1. Evaluating catalytic performance in a series of solvents that differ only in one property, e.g. cyclohexane, tetrachloroethylene, trichloroethylene and 1,2-dichloroethane which have different π values, but identical α and β parameters.
2. Evaluating catalytic performance in a series of binary solvent mixtures such as toluene/chlorobenzene mixes ranging in composition from pure toluene to pure chlorobenzene. Toluene and chlorobenzene differ only in polarity. DMF and 1,2-dichloroethane differ only in basicity (β), while 1,2-propanediol and triethylphosphate differ only in proticity (α), and can therefore be used to evaluate the effect of each parameter individually on catalytic reactions.9
3. Using supercritical fluoroform to evaluate the effect of polarity on catalysis. The polarity of fluoroform changes as a function of pressure, but its hydrogen-bond accepting ability is negligible at all pressures. The dependence of the enantioselectivity on polarity for the cyclopropanation shown in Fig. 2 was confirmed by performing the reaction multiple times in CHF3, in which the CHF3 was applied at a different polarity in each experiment (Fig. 3).
4. Mathematically fitting observed rate or selectivity data in many solvents to a linear free energy relationship that incorporates terms for each of the solvent properties of interest.5 Typically the equation takes the form shown in eqn (1) (where X is an observable such as a rate or a selectivity), but in complicated cases, where a synergistic or destructive interplay of two parameters is involved, additional (quadratic) terms are required. For example, in the case of the cyclopropanation shown in Fig. 3, eqn (2) can be derived from the published data, which has an additional term to account for the greater e.e. observed in solvents that have both polarity and basicity rather than one without the other.
| X = c + aα + bβ + dπ* | (1) |
| %ee = 91 − 49β − 25π* + 63βπ* | (2) |
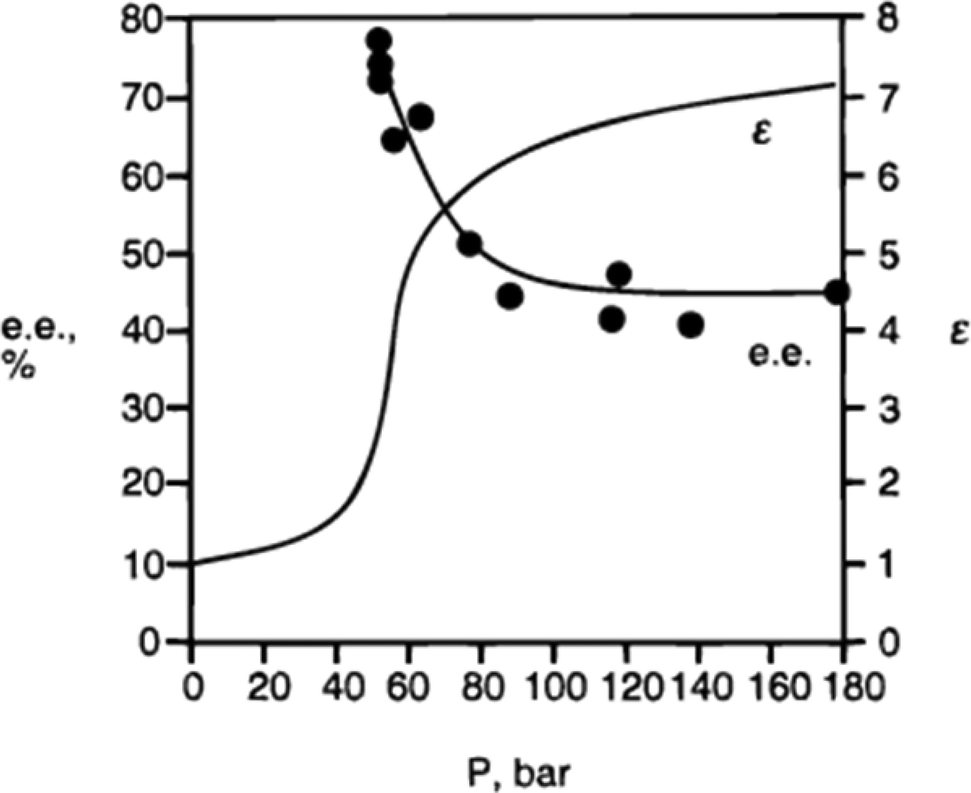 | ||
| Fig. 3 The relationship between the enantioselectivity of cyclopropanation in supercritical fluoroform and the dielectric constant of the fluoroform.5 At lower polarity (lower dielectric constant), the enantioselectivity is higher. | ||
Protic solvents are commonly used in hydrogenation reactions because they promote rapid reaction, whereas for other reactions such as C–C cross coupling reactions, protic solvents tend to be avoided because of undesirable reactivity. For example, the hydrogenation of CO2 to formic acid by Ru(X)(Y)(PMe3)4 (X, Y = H, OAc, Cl) in liquid NEt3 is slow unless a small amount of water or an alcohol is present.10 The water/alcohol is believed to facilitate the catalyst by direct coordination and the subsequent formation of H-bond interactions with CO2 (Fig. 4). As the amount of water/alcohol is increased there is relatively little additional impact on the reaction. The acidity of the alcohol also influences the reaction with more acidic alcohols leading to higher reaction rates, presumably due to more efficient H-bonding with the relatively inert CO2 substrate.11 Moreover, DFT calculations indicate that a coordinated water ligand facilitates the insertion of CO2 into the Ru–H bond, a critical step in the catalytic cycle.12
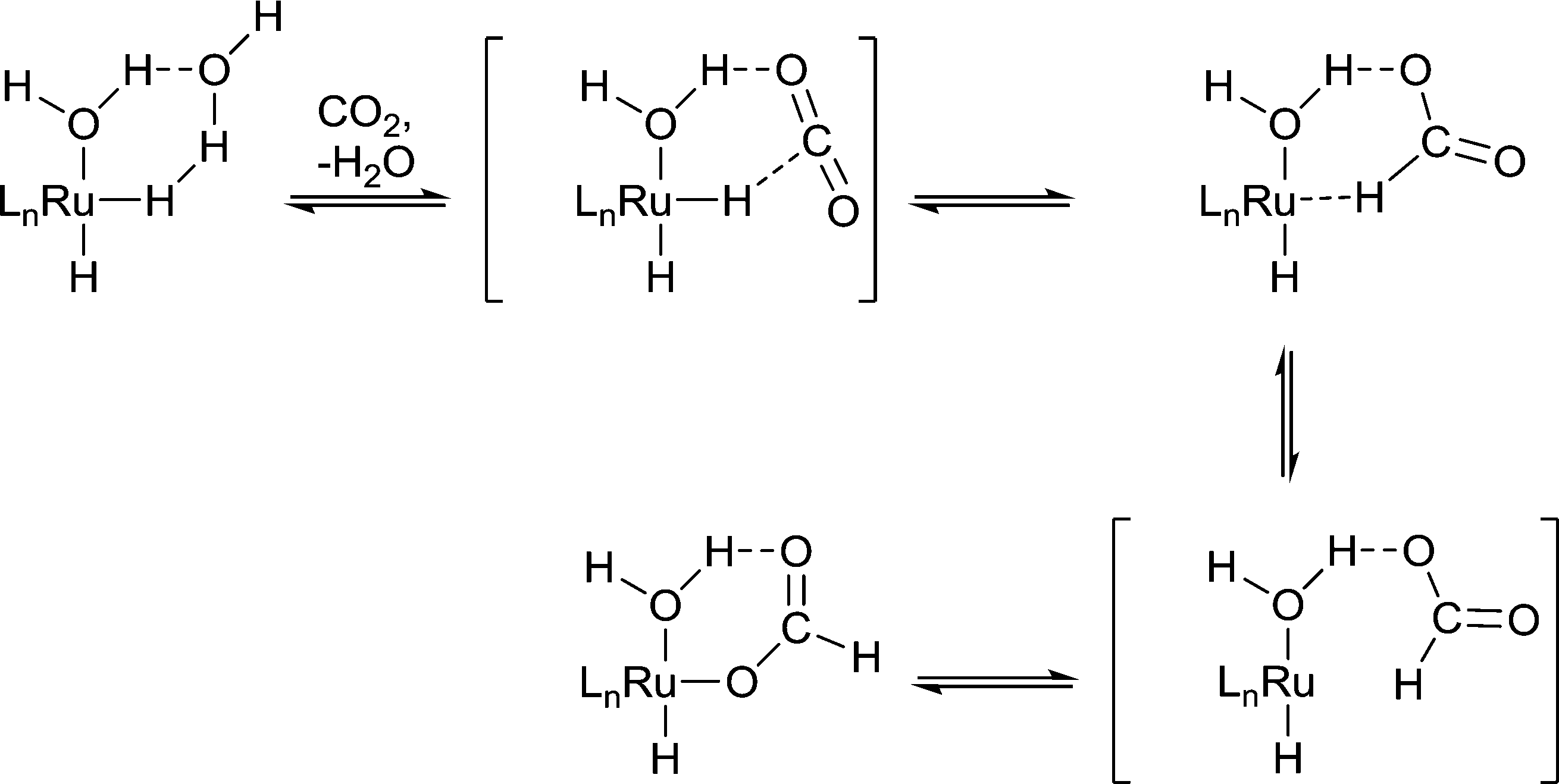 | ||
| Fig. 4 The proposed role of a water ligand during the insertion of CO2 into a Ru–H bond during the catalytic hydrogenation of CO2 by the catalytic intermediate Ru(H)2(PMe3)3(OH2). For clarity RuLn is used to represent the Ru(PMe3)3 unit. Hydrogen bonding interactions between the coordinated water and CO2 help to lock the weakly coordinating CO2 substrate at the active catalytic site. | ||
3. Solvent influences on catalysis
The various properties of solvents can influence catalyzed reactions in many different ways, which are categorized in this section. Pertinent examples are given that illustrate these solvent effects.3.1. Solvent effects on solubility
1) A solvent in which the solubility of reagents (solid, liquid or gas) is high could lead to an increased rate of reaction compared to a solvent in which reagent solubility is low. This statement may seem obvious, but it may not always be true. For example, for gaseous reagents, the thermodynamic ability of the solvent to dissolve the gas is not necessarily important. If mass transfer is fast, which is often the case for gas dissolution in non-viscous solvents that are well stirred, the extent to which a solvent can dissolve the gas (Table 1) has little impact on the rate of reaction, because rates are proportional to the activity of the gas, not the concentration, and the activity is set by the gas phase partial pressure rather than by the ability of the solvent to dissolve the gas. In contrast, if mass transfer is slow, as would be expected if stirring is inadequate or a solvent is particularly viscous, then the rates will be a function of the gas solubility and the rate of diffusion of gas from the gaseous to liquid phases. Mass transfer effects will be discussed in section 3.2.| Solvent | H2 (ref. 13) | CO (ref. 14) | O2 (ref. 15) | CO2 (ref. 16) |
|---|---|---|---|---|
| a Converted from mole fraction in the original references using C = x/(V(1 − x)) where C is the molar concentration of gas in the liquid phase, x is the mole fraction of gas in the liquid phase, and V is the molar volume of the solvent. b scCO2 = supercritical CO2, at 35 °C. The concentration of H2, CO or O2 is independent of CO2 pressure, but 15 MPa is assumed when calculating the concentration of CO2. c At 20 °C and interpolated linearly to 0.101 MPa from the solubility determined at 10.1 MPa.17 d At 22 °C and interpolated linearly to 0.101 MPa from the solubility determined at 10.1 MPa.18 e Extrapolated to 25 °C and 0.101 MPa from the literature data.19 | ||||
| scCO2b | 117 | 117 | 117 | 18 500 |
| Perfluoroheptane | 6.2 | 17.2 | 24.8 | 94 |
| Hexane | 5.0 | 13.1 | 15.1 | 92 |
| Acetone | 4.1 | 10.5 | 11.4 | 259 |
| Ethyl acetate | 3.5 | 10.4 | na | 235 |
| Methanol | 4.0 | 9.2 | 10.2 | 160 |
| Ethanol | 3.5 | 8.4 | 10.0 | 125 |
| Toluene | 3.0 | 7.5 | 8.7 | 96 |
| DMSO | 1.1 | na | 1.9 | 129 |
| [bmim][BF4] | 0.86c | 2.0d | na | 98e |
| Water | 0.78 | 0.98 | 1.3 | 34 |
2) A solvent that is unable to dissolve a kinetic product would cause it to precipitate from the reaction or lead to the formation of a separate phase and therefore not react further, allowing the kinetic product to be isolated. An elegant example of a reaction in which precipitation changes the selectivity of the reaction in favor of the kinetic product is the hydrogenation of cyclohexa-1,3-diene using the cationic catalyst [Rh(η4-nbd)(PPh3)2]+ (nbd = norbornadiene).20 In acetone (propan-2-one) the product obtained is the fully hydrogenated cyclohexane ring whereas the same catalyst in the ionic liquid [bmim][SbF6] (bmim = the 1-butyl-3-methyl-imidazolium cation) leads to partial hydrogenation with cyclohexene obtained with a selectivity of 98%. Although the nature of the catalysts in acetone and the ionic liquid are likely to differ, since the norbornadiene is hydrogenated and usually replaced by weakly coordinating solvent molecules (see Fig. 5) and it is not clear how the ionic liquid interacts with the catalyst intermediate, the reason for the high selectivity to cyclohexene is mostly likely due to the low solubility of cyclohexene in the ionic liquid, so that the cyclohexene kinetic product is eliminated from the solvent and, as it forms, creates a second phase. The more polar cyclohexa-1,3-diene substrate is considerably more soluble in the ionic liquid solvent than cyclohexene whereas in acetone the system always remains as a single phase.
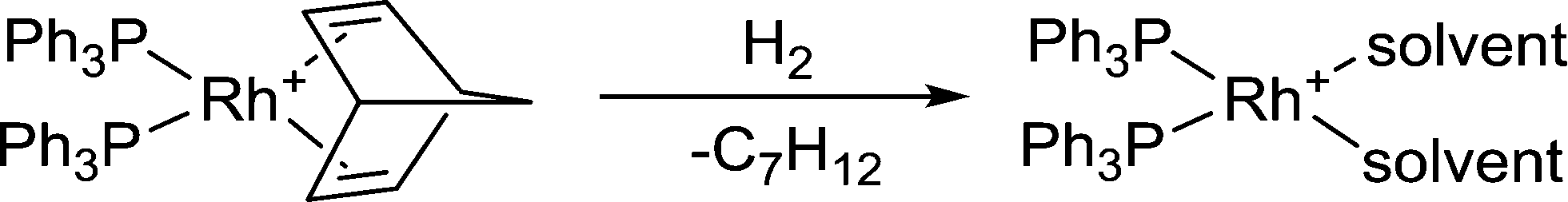 | ||
| Fig. 5 Hydrogenation of the norbornadiene ligand in the [Rh(nbd)(PPh3)2]+ pre-catalyst to afford the active catalyst with two coordinated solvent ligands. | ||
3) A solvent in which a product or by-product that autoinhibits the catalyst is insoluble would prevent autoinhibition. Alternatively, a highly solvated product/by-product may also be less reactive due to strong solvent-solute interactions. For example, reactions that eliminate halide ions can be quenched if the catalyst is poisoned (deactivated by the halide – since halides are good ligands and can preferentially coordinate to the catalyst relative to the substrate). This concept is described in detail below in section 5 on C–C coupling reactions that frequently employ aryl halide substrates.
3.2. Solvent effects on mass transfer
4) A solvent in which mass transfer is slow could reduce the rate or modify the selectivity of a reaction. Solvent viscosity and mass transfer properties can affect the rate and selectivity of catalyzed reactions due to their effect on the rate of mass transfer of substrates including gases. For example, the enantioselectivity of hydrogenation of methyl-(Z)-α-acetamidocinnamate by [Rh(dipamp)]+ (dipamp = the chiral diphosphine (R,R)-1,2-bis[(2-methoxyphenyl)(phenylphosphino)]ethane, Fig. 6) is strongly dependent on the concentration of H2 in solution, which is a function of the rate of transfer of H2 from the gas phase into the liquid phase. At high H2 concentrations (which can be achieved by using high H2 pressure with high stir rates, i.e. fast mass transfer), reaction with H2 (indicated in the figure) is fast and the enantioselectivity is determined by the ratio of rate constants k1min/k1maj. At low H2 concentrations (which can be achieved by using high H2 pressure with low stir rates, i.e. slow mass transfer), reaction with H2 is slow and the enantioselectivity is determined by the ratio k2minK1min/k2majK1maj. Thus the rate of diffusion of H2 into the solvent controls the enantioselectivity.21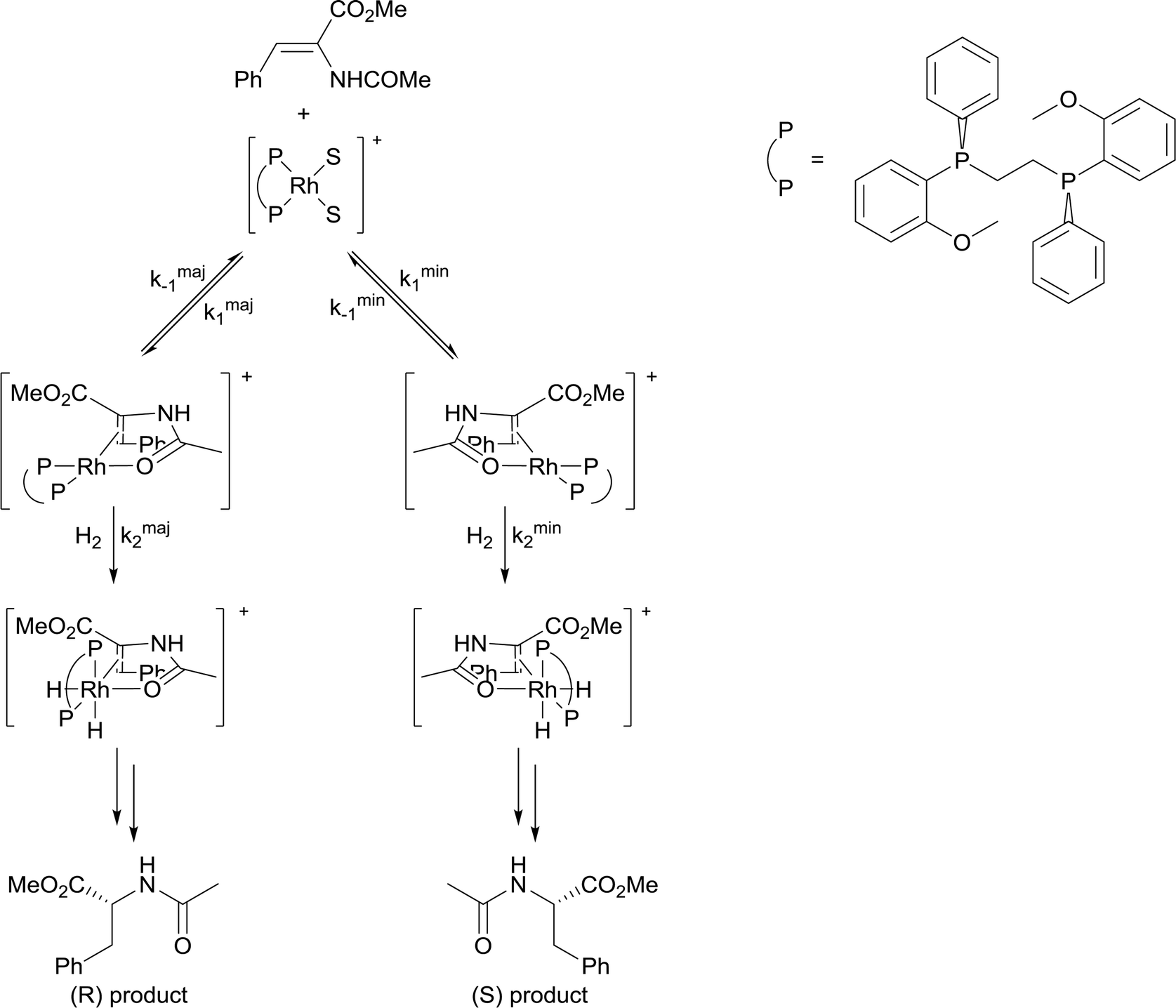 | ||
| Fig. 6 Key mechanistic steps in the asymmetric hydrogenation of methyl-(Z)-α-acetamidocinnamate by [Rh(dipamp)]+ (dipamp = the chiral diphosphine (R,R)-1,2-bis[(2-methoxyphenyl)(phenylphosphino)]ethane). | ||
Solvents having naturally high viscosities tend to have low rates of mass transfer of reagent gases which affects the rate and selectivity. For example, the asymmetric hydrogenation of atropic acid by chiral ruthenium catalysts (Fig. 7) has a higher enantioselectivity when the availability of H2 in solution is high. Thus, the enantioselectivity for this reaction is high in methanol, low in the viscous ionic liquid [bmim][PF6], and intermediate in a [bmim][PF6]/methanol mixture or CO2-expanded [bmim][PF6] (the methanol or CO2 reduces the viscosity of the ionic liquid).22 If a less viscous solvent cannot be used for the reaction, and increased stirring alone is not adequate to increase mass transfer rates, then dilution with a low-viscosity solvent, or reducing the viscosity by gas-expanding with CO2 should ameliorate or eliminate the problems associated with slow mass transfer of reagents.23
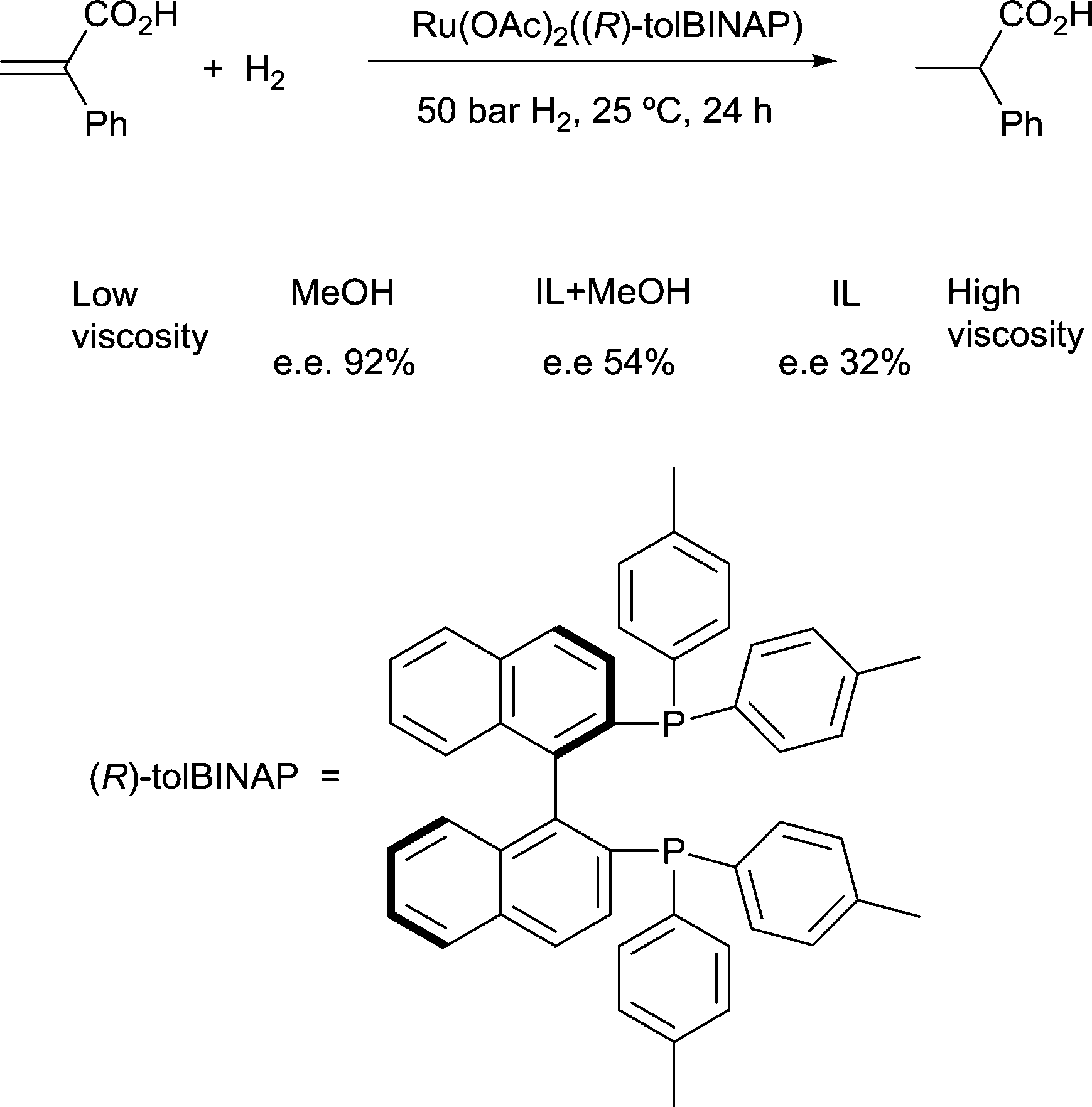 | ||
| Fig. 7 The enantioselectivity of asymmetric hydrogenation of atropic acid is dependent upon solvent viscosity. | ||
3.3. Solvent interactions with the starting materials or products that activate or deactivate the reaction or influence the selectivity
5) A solvent may interact with or react with a starting material activating it towards reaction or deactivating it. Non-covalent interactions, such as hydrogen bonds between solvents and substrates, also strongly affect the reaction, going far beyond solvation alone. Specific examples are provided in section 5. A common example rarely noted in publications, is the hydrogenation of CO2 in aqueous solutions. Carbon dioxide reacts with water upon dissolution to afford HCO3− or CO32−, depending on the pH, and these species undergo hydrogenation rather than the CO2 itself,24 although in other solvents CO2 reacts directly to give the same products.256) A solvent may react with a substrate to generate a new starting material that is more (or less) selectively transformed by catalysis, e.g. the solvent may react with a specific functional group on the starting material and temporarily protect it so that it does not interfere with the desired catalytic transformation. For example, the ring-closing hydroaminomethylation reaction (Fig. 8) gives primarily the lactam product in dioxane, because the cyclization is faster than the hydrogenolysis. However, in scCO2, the CO2 inhibits the cyclization step by reacting with the N–H group and forming, temporarily, a carbamic acid protecting group, allowing the hydrogenolysis step to proceed and affording the pyrrolidine as the major product.26
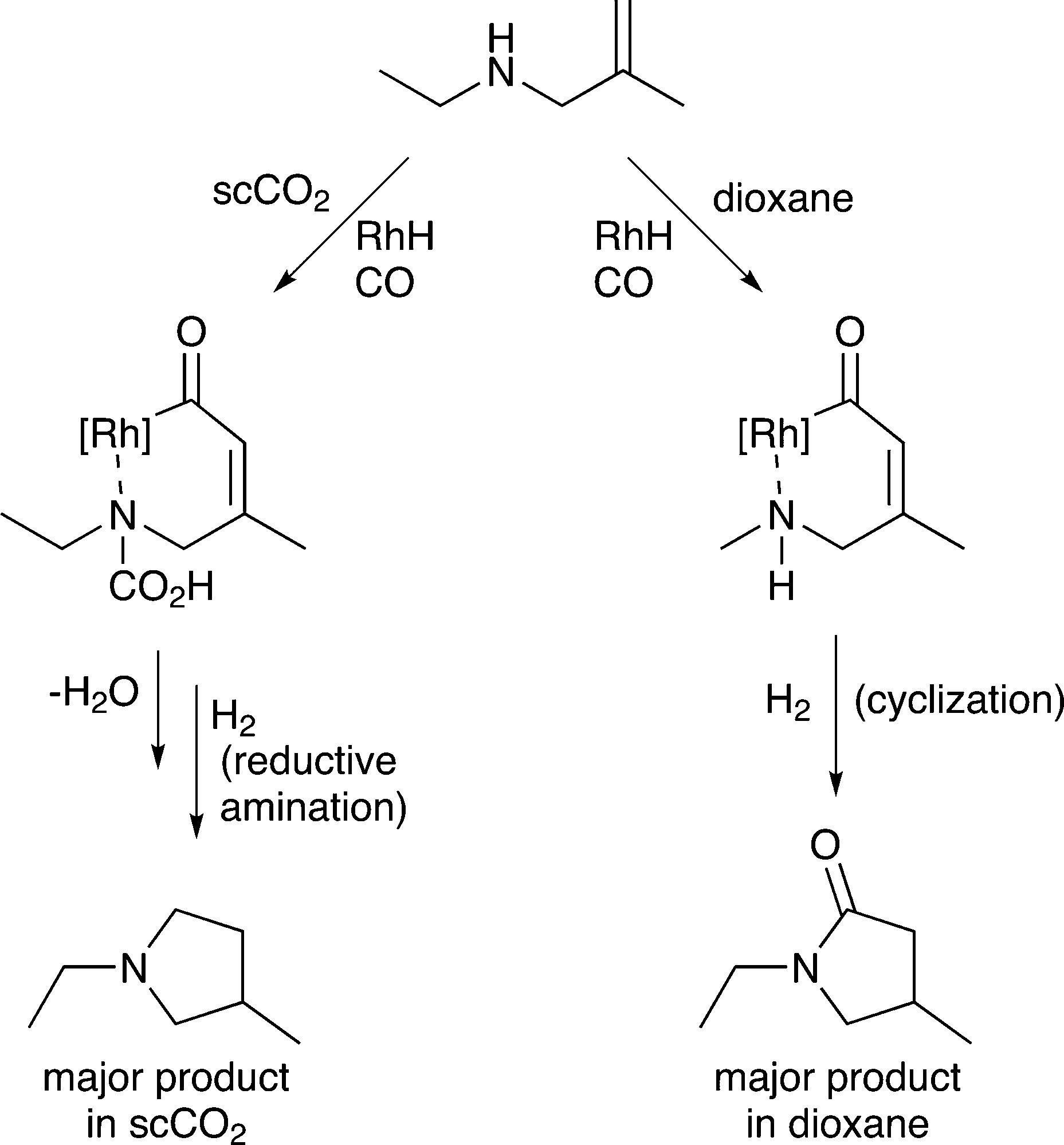 | ||
| Fig. 8 Example of a reaction in which the solvent reacts (reversibly) with the substrate to change the nature of the product. The cyclization that takes place in dioxane is inhibited in scCO2 because of the CO2 solvent reacts with the secondary amine group to form a carbamic acid protecting group leading to preferential hydrogenolysis. | ||
7) A solvent may react with a kinetic product and thereby protect it against further reaction. The hydrogenation of nitriles to primary amines using heterogeneous catalysts is difficult to control because the primary amine reacts further to give a secondary amine and ammonia by disproportionation. To avoid this problem, the hydrogenation can be performed in scCO2; the CO2 protects the primary amine kinetic product (by temporarily converting it into a carbamic acid) and prevents further reaction from taking place.27
8) A solvent that reacts with an autoinhibiting product or by-product may prevent autoinhibition. In the hydrogenation of nitriles or imines to amines using homogeneous catalysts, the primary amine product is a good ligand that can coordinate to the catalyst and reduce the activity of the catalyst or completely deactivate it. CO2-expanded THF was used to prevent such a deactivation mechanism as the CO2 reacts with the amine product.28
9) A solvent may also serve as a substrate in a reaction. For example, methanolysis and ethanolysis, more frequently termed transesterification reactions, tend to be performed in methanol and ethanol, respectively, with the solvent also serving as the substrate. Indeed, with the current surplus of bioethanol, ethanolysis reactions are of particular current interest, and are being developed to upgrade chemicals derived from renewable feedstocks such as lignin that contain a large number of C–O bonds that can undergo ethanolysis.29
3.4. Solvent interactions with the catalyst
10) A solvent may interact with or coordinate to the catalyst and thereby activate or deactivate the catalyst, influence the selectivity of the catalyst and influence catalyst stability. A classic example of such an effect involves the use of chiral solvents. Chiral solvents that impart enantioselectivity in transformations are interesting alternatives to chiral catalysts. Such solvents must have sufficiently strong interactions with key catalytic intermediates to transmit reasonable enantioselectivities. Ionic liquids containing either a chiral anion or a cation modified with a chiral group can lead to high enantioselectivites as the solvent also serves as a ligand, coordinating to the active catalyst. For example, the hydrogenation of methyl 2-acetamidoacrylate (Fig. 9) employs a chiral ionic liquid cation that appears to undergo deprotonation and then coordinates to the metal center. Addition of NEt3 enhances the enantioselectivity by promoting deprotonation of the ionic liquid to generate the ligand whereas addition of acid (preventing deprotonation) or a diphosphine (that preferentially coordinates to the catalyst) result in a racemic mixture of the product.30 In a second example, an ionic liquid with a chiral anion was used as a solvent in the dihydroxylation of 1-hexene (Fig. 9) leading to a very high enantioselectivity.31 It cannot be excluded that the chiral anion coordinates to the osmium catalyst forming a chiral catalyst that is responsible for the chiral induction.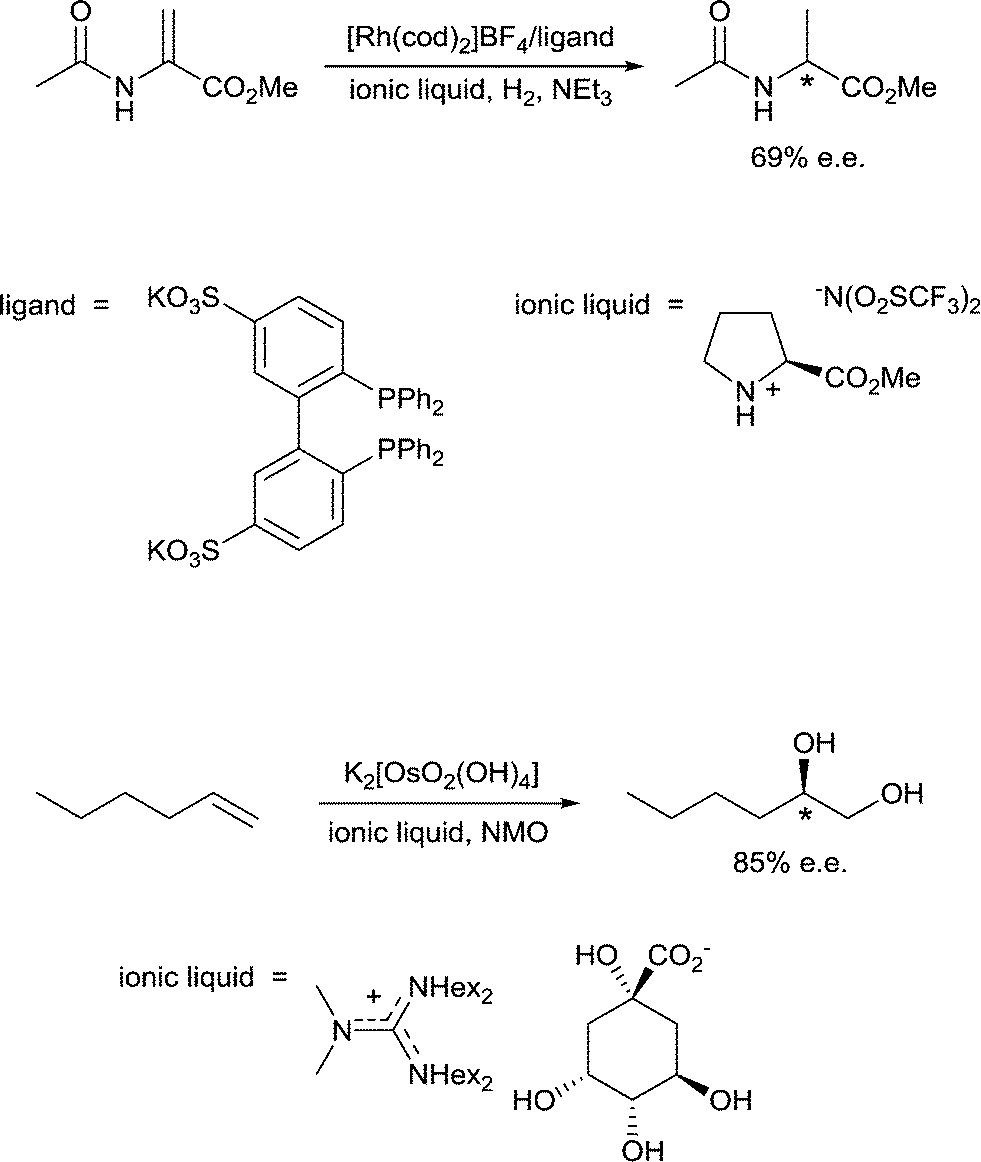 | ||
| Fig. 9 Examples of homogeneously catalyzed enantioselective reactions in chiral ionic liquids (“hex” = n-hexyl; “cod” = cycloocta-1,5-diene). Enantioselectivity (e.e.) is induced by the in situ formation of chiral catalysts by coordination of the chiral ion to the catalyst. | ||
11) A solvent can modulate the extent of ion pairing between a charged catalyst and the associated counter-ion which in turn can influence catalytic activity. Ion-pairing with cationic catalysts and their associated counter-anions, or potentially anionic catalysts and their counter-cations, can be extensive in solvents in which the ions are poorly solvated.32 Typically in low polarity solvents, the cation and anion form a strong ion-pair that can inhibit catalytic activity if the counter-ion blocks the active site of the catalyst, i.e. covers the vacant coordination site where the substrate would usually bind. The formation of aggregates is also possible.33 Conversely, in polar solvents the ions tend to be highly solvated and the cation and anion are usually completely separated from each other, allowing catalysis to proceed without the hindrance of the counter-ion. A schematic illustration of ion-pairing as a function of solvent polarity is shown in Fig. 10. Indeed, good correlations between catalytic activity and the extent and nature of ion-pairing have been reported,34 although the emphasis is usually placed on the nature of the counter-ion rather than the role of the solvent.
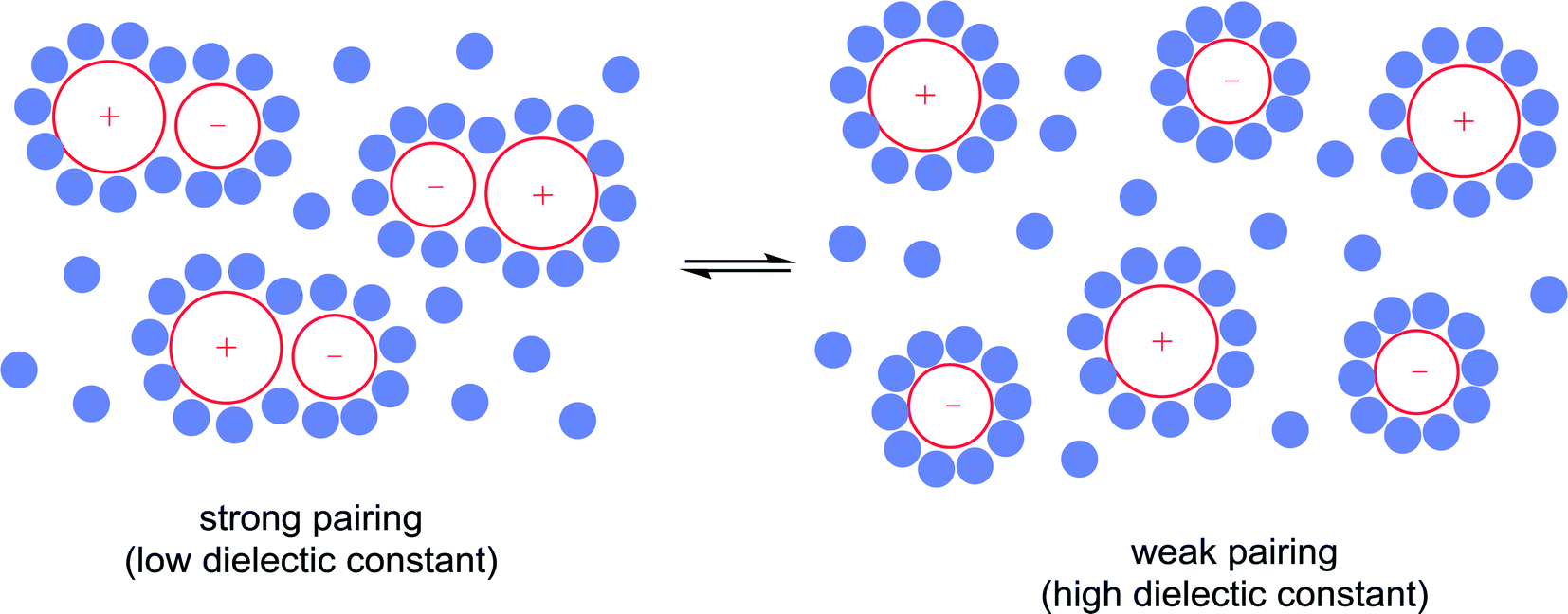 | ||
| Fig. 10 Ion-pairing in solvents as a function of the solvent polarity. Strong ion-pairing (when one of the ions is the catalyst) tends to inhibit catalytic activity whereas the presence of free ions usually leads to higher catalytic activities. | ||
Strong ion-pairing should not always be viewed as a problem with, for example, close association between chiral anions and non-chiral cationic catalysts leading to high enantioselectivities.35 In these types of reactions it is essential to use a solvent that ensures sufficient ion-pairing, to allow chiral induction, while retaining sufficient activity. It has also been found that neutral catalysts can exist in isolated or aggregated states depending upon the nature of the solvent, e.g. observed for transfer hydrogenation catalysts that interact with each other via bifurcated hydrogen bonds.36
12) A solvent can serve as a proton shuttle in a catalytic reaction. In this manner, water or alcohols can facilitate reactions without directly coordinating to the catalyst. For example, in the hydrogenation of CO2 catalyzed by an iridium complex, the energy of the transition state for the heterolytic cleavage of H2 (the step which generates the Ir–H bond into which the CO2 later inserts), decreases considerably if a water molecule is present to serve as a proton shuttle.37
13) A solvent can also be catalytically active in its own right. For example, Brønsted acids and bases are widely used as catalysts, often as alternatives to heterogeneous catalysts such as zeolites and alumina.38 Ionic liquids functionalized with Brønsted acidic or basic groups are increasingly being used as catalysts due to their low volatility.39
3.5. Transition state stabilization
14) A solvent that stabilizes the highest energy transition state in a catalytic process should accelerate the rate of reaction. This is the basis of the Hughes–Ingold rules on solvent effects.40 If the charge density of the transition state in the rate determining step is greater than the charge density of the starting materials in that step, then use of a more polar solvent should increase the reaction rate. If the charge density of the transition state is lower than that of the starting materials, then use of a more polar solvent will decrease the reaction rate. For example, the pyridine-catalyzed decomposition of t-butylperoxyformate has a greater charge separation in the transition state than in the starting material and therefore the reaction proceeds more rapidly if the polarity of the solvent increases (Fig. 11).41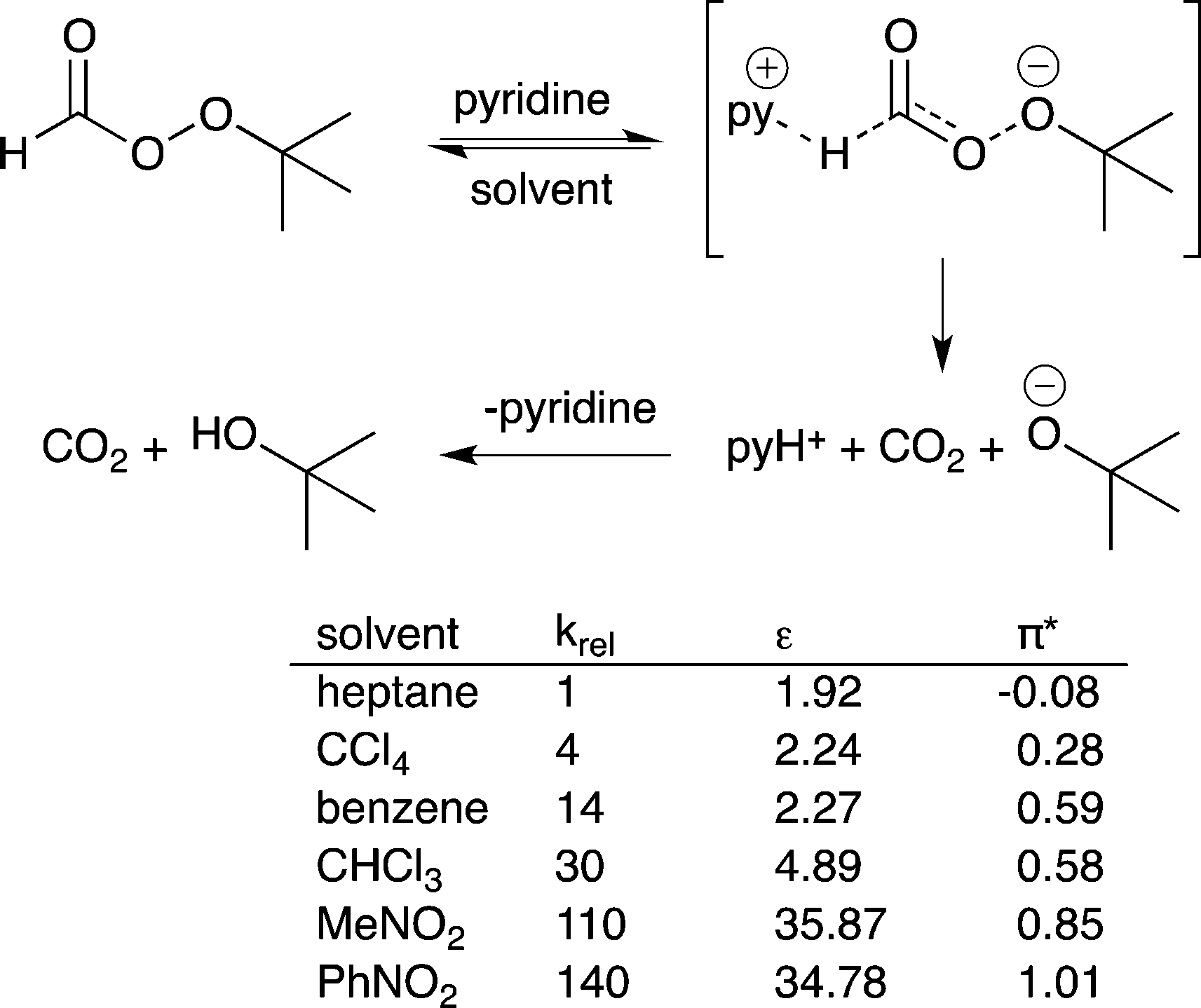 | ||
| Fig. 11 The decomposition of t-butyl peroxyformate catalyzed by pyridine. The reaction rate increases as the polarity of the solvent increases because polar solvents stabilize the transition state.37 | ||
15) A solvent that stabilizes one of two competing transition states that control the selectivity should enhance the selectivity of the product obtained via the stabilized transition state. For example, in Diels–Alder reactions higher reaction rates and selectivities are often obtained in polar solvents compared to non-polar solvents, which has been attributed to enhanced hydrogen bonding between the solvent and the transition state, as well as to enforced hydrophobic interactions when conducted in water, which facilitates alignment of the substrates.42
16) A solvent could stabilize an early transition state rather than a late transition state and thereby affect both rate and selectivity. A classic example of this phenomenon is the asymmetric cyclopropanation shown in Fig. 2 (section 2). The more polar the solvent the more an early transition state is favored, in which the incoming alkene is further from the chiral complex, and therefore results in a lower enantioselectivity.
From the list above it is evident that solvents influence catalytic reactions in many different ways. Some of these features are relatively easy to identify via experimentation, some others require in situ spectroscopic studies, and certain effects, such as solvent influence on transition states, can only be calculated. It is relatively easy to ascertain if a solvent acts as a substrate or even is the active catalyst in a reaction. Direct interactions between catalysts, substrates and products are also relatively easy to gauge using spectroscopic techniques although interactions with the active catalyst intermediates often requires exhaustive in situ spectroscopic studies. Such interactions will be described in detail below for two different types of reaction: hydrogenations and C–C cross coupling reactions.
4. Hydrogenation reactions
Pioneering studies on homogeneous hydrogenation catalysis demonstrated the important role of solvent-stabilized catalytic intermediates comprising coordinated solvent molecules. A prominent example involves the enantioselective synthesis of L-Dopa, an intermediate in dopamine biosynthesis, which is used in the treatment of Parkinson's disease. The hydrogenation of the prochiral substrate (Fig. 12) is achieved with a catalyst intermediate generated in situ from [Rh(cod)(R,R-DIPAMP)]+ (R,R-DIPAMP is a chiral diphosphine ligand and cod = cycloocta-1,4-diene), via the hydrogenation of the cod ligand to cyclooctane, analogous to the solvated intermediate described above for [Rh(nbd)(PPh3)2]+.43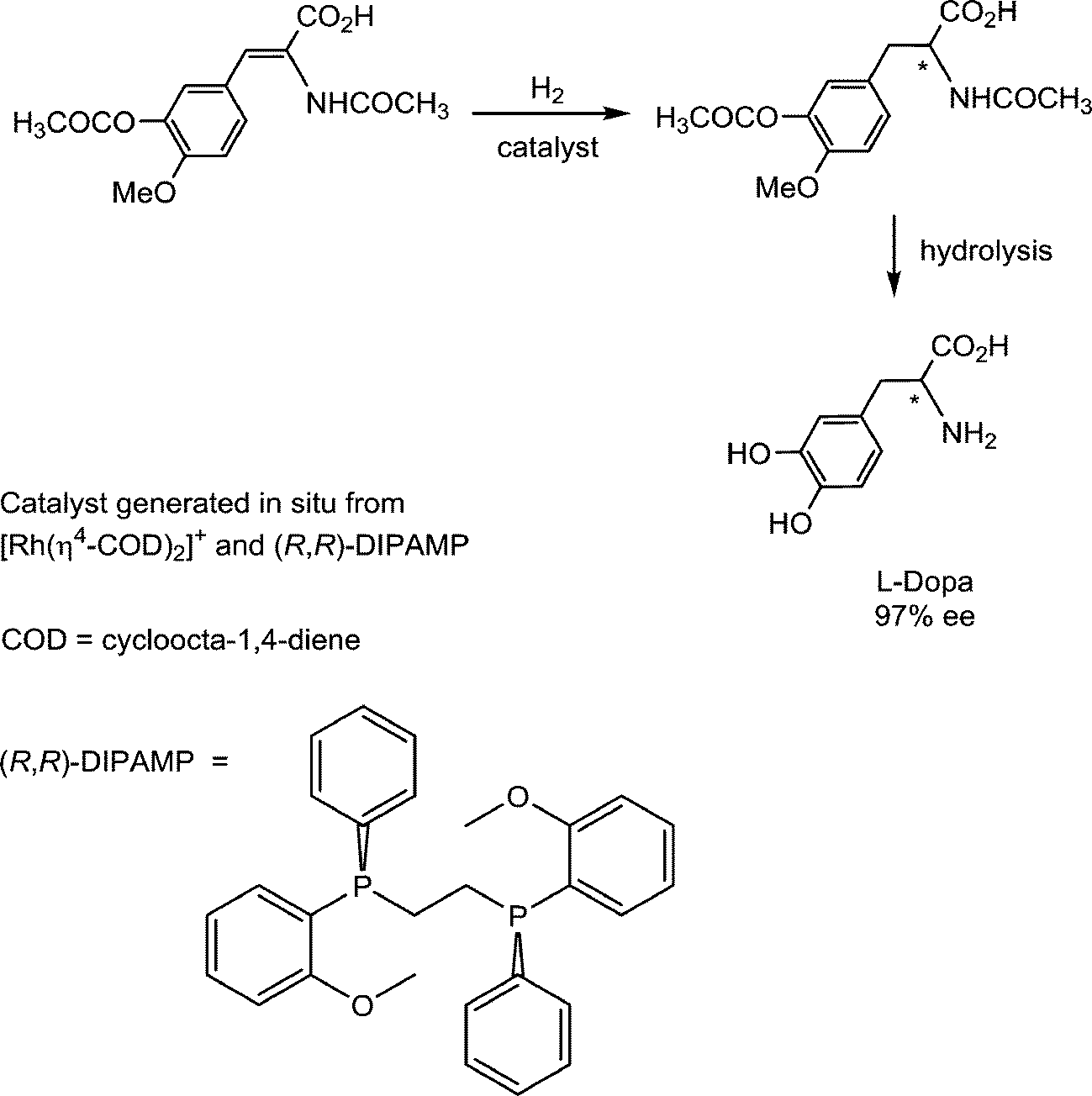 | ||
| Fig. 12 Synthesis of L-Dopa via the hydrogenation of a prochiral substrate. | ||
Since a solvated intermediate species is implicit in the catalytic cycle, the nature of the solvent is critical. Of the various solvents reported for this hydrogenation, strong donor solvents are absent, as they will coordinate too strongly to the catalyst and impede further reaction with the substrates.44 The catalyst appears to be most active in EtOH and MeOH, followed by THF, and then the non-coordinating solvents dichloromethane and toluene. This trend implies that weak coordination helps to stabilize the highly reactive catalytic intermediate, otherwise the highest activity would be expected to be obtained in dichloromethane and toluene. However, the Rh+-solvent bond must be sufficiently labile to allow facile dissociation of the solvent ligands to prevent this step from becoming rate limiting. Within the catalytic cycle, the final reductive elimination of the product tends to be rate determining. If this step is slow then β-elimination becomes more favored, which allows equilibration of the face of the substrate following migratory insertion to reform the alkyl ligand. In other words, hydrogenation of the thermodynamically preferred face rather than the kinetically preferred face takes place leading to lower enantioselectivites.
Wilkinson's catalyst is another highly important catalyst with a wide range of industrial applications45 and the implicit role of solvents has been delineated. A generic catalytic cycle for the hydrogenation of unsaturated compounds using Wilkinson's catalyst is shown in Fig. 13. In the first step the stable 16 valence electron compound, RhCl(PPh3)2, is converted to a highly reactive 14 valence electron active catalyst species, RhCl(PPh3)2, via the dissociation of a phosphine ligand. The productive part of the catalytic cycle continues with the oxidative addition of hydrogen, which was shown to be considerably faster than that determined for other species present in the reaction, e.g. the starting compound RhCl(PPh3)3 and various dimers.46 The formation of a solvated species, RhCl(PPh3)2(solvent), has tentatively been proposed, undergoing oxidative addition of hydrogen with concomitant dissociation of the solvent ligand. Indeed, if the reaction is conducted in strong donor solvents, which tends not to be the case in practice,47 such an intermediate species is not unreasonable. NMR spectroscopy in C6D6 using p-H2 revealed a number of previously unobserved species, but none involving coordination of the solvent, as expected for this solvent.48 However, a kinetic analysis using electrospray ionization mass spectrometry revealed that the slowest step in the productive part of the catalytic cycle corresponds to the association of the unsaturated substrate.49 The slow ligand association step indirectly points to a solvent molecule needing to be displaced prior to the ligand association, which is consistent with the effect of donor solvents reducing the rate of reaction. Should the solvent be an ionic liquid then the cation and/or anion could interact with the catalyst, and depending on which ions interact with the catalyst intermediate will further influence the reaction.
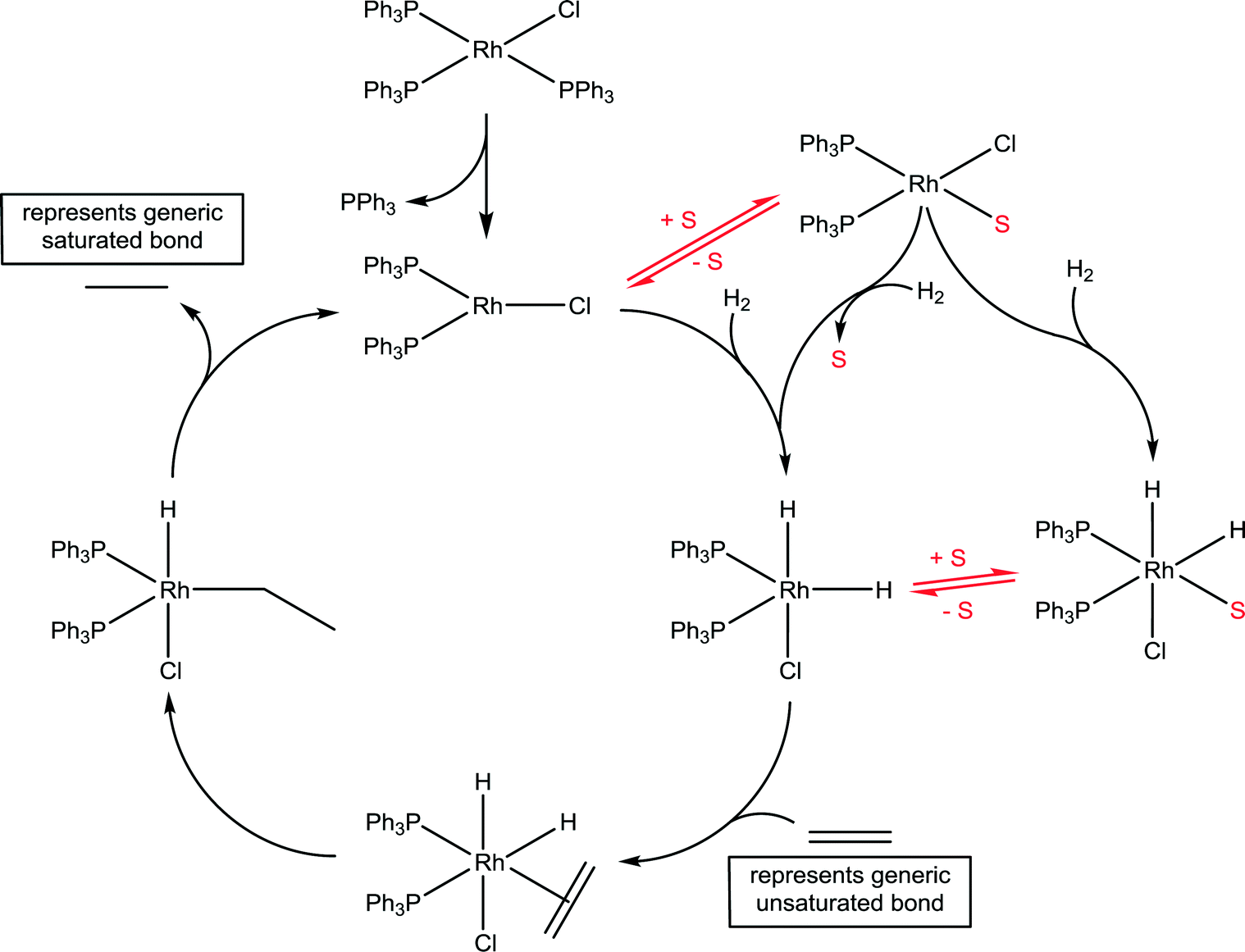 | ||
| Fig. 13 Catalytic cycle for the hydrogenation of unsaturated C–C bonds (typically alkenes and alkynes) using Wilkinson's catalyst, RhCl(PPh3)3. The widely accepted intermediates in the catalytic cycle are shown in black in the main cycle and the solvated species that have been proposed are shown to the right of the main cycle. Solvent is denoted by S. The 16 valence electron solvated species RhCl(PPh3)2(solvent) and the 18 valence electron solvated intermediate RhCl(PPh3)2(H)2(solvent) are present in donor solvents. | ||
It should be noted that the active catalyst species/mechanism is not only influenced by the solvent, but in principle by the nature of the substrate and all reaction parameters. It has been shown, for example, that at high temperatures under the reducing hydrogen environment rhodium(0) nanoparticles can form and are excellent hydrogenation catalysts, especially for aromatic and heteroaromatic substrates.50
A remarkable solvent effect was observed with Wilkinson's catalyst in the hydrogenation of alkenes containing aromatic nitro-groups (Fig. 14) and other sensitive groups such as aryl iodide that cannot be rationalized by coordination of the solvent to the catalyst. In order to prevent the reduction of the nitro group or dehalogenation from taking place very mild reactions conditions are required. In benzene, under 1 atm of H2 and at room temperature no reaction was observed, whereas in MeOH the alkene bond was reduced to afford the saturated product in 80% yield and in THF the yield increases to 91%.51 However, the highest yields were obtained in MeOH-THF and tBuOH-THF (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixtures (93 and 95%, respectively). The study also shows the importance of using mixtures of solvents rather than pure solvents, even though the latter overwhelmingly dominate the literature.
:1) mixtures (93 and 95%, respectively). The study also shows the importance of using mixtures of solvents rather than pure solvents, even though the latter overwhelmingly dominate the literature.
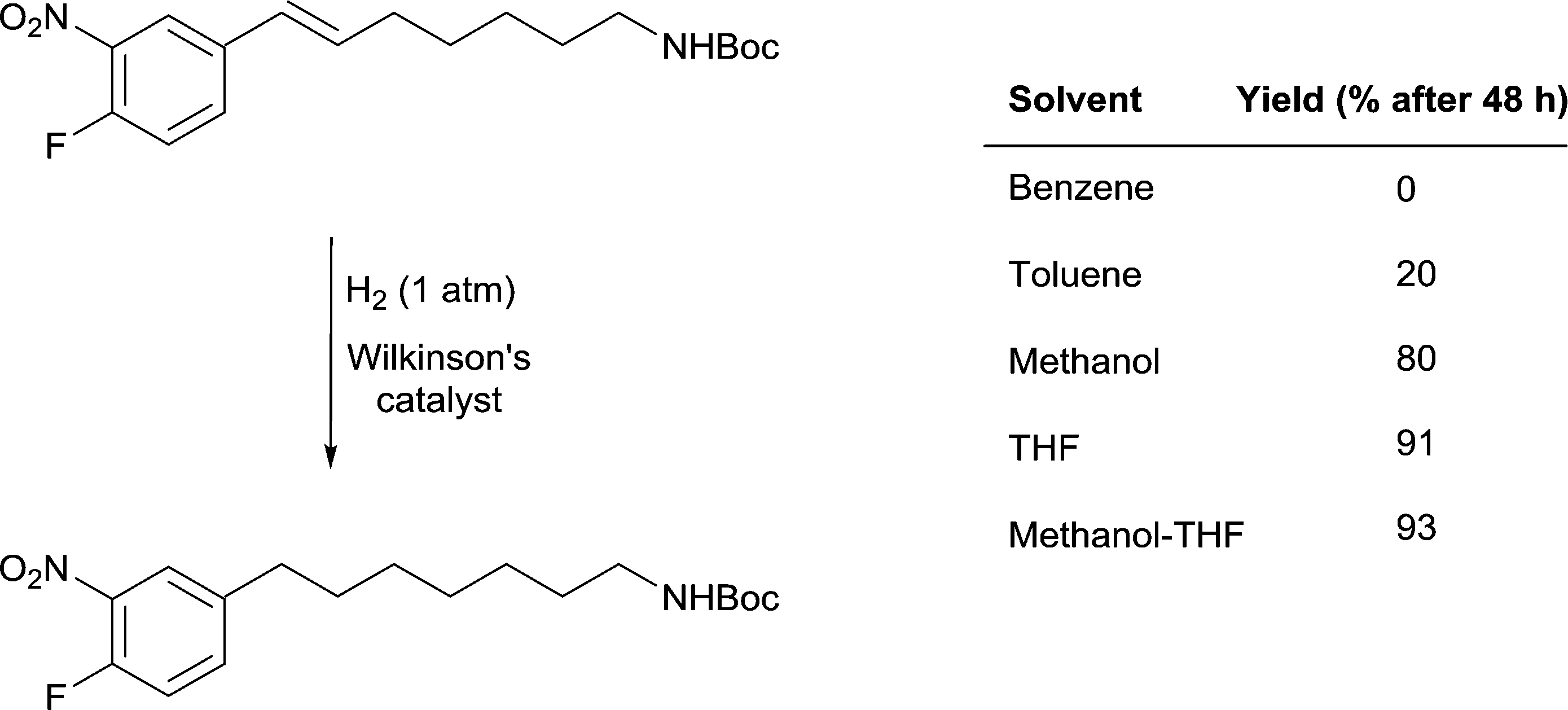 | ||
Fig. 14 Selective hydrogenation of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond in the presence of a reducible nitro-functional group using Wilkinson's catalyst. C double bond in the presence of a reducible nitro-functional group using Wilkinson's catalyst. | ||
It is unlikely that the rate of this reaction is correlated to the solubility of hydrogen gas in the different solvents evaluated (see above). However, the rate of hydrogenation does appear to be correlated with the β values of the solvents. Benzene, THF and methanol have approximately the same π* values and there is no clear trend with respect to the α parameters of the solvents. The parameter that differs between the solvents that facilitate catalytic activity under mild conditions and the solvents where the yield of the reduction is low or does not take place is β, i.e. the basicity or hydrogen-bond accepting ability of the solvent. How this parameter enhances the reaction remains a matter of speculation, but one possibility is that the alkene protons hydrogen-bond with the solvent thereby activating the unsaturated bond. These solvents would not be able to hydrogen-bond to (and activate) the nitro-group in the same way, hence endowing the system with high selectivity towards reduction of the CC double bond.
With both of the rhodium catalysts described above, the active catalyst species is generated in situ, in the case of [Rh(cod)(R,R-DIPAMP)]+via hydrogenation and elimination of the cod ligand, and with RhCl(PPh3)3via dissociation of a phosphine. The electronically unsaturated species generated are stabilized by the solvent, but if too stable, then activity is reduced. Hence, a balance between stability and activity must be found.
If catalyst activation involves dissociation of an anionic halide ligand then the nature of the solvent has a profound influence. For example, ruthenium(II) complexes [RuCl(diphosphine)(arene)]+ (the diphosphine ligand = diphenylphosphinomethane, diphenylphosphinoethane, diphenylphosphinopropane and the highly water-soluble analogue 1,2-bis(di-4-sulfonatophenylphosphino)benzene, arene = p-cymene, benzene or [2.2]paracyclophane), are efficient catalyst precursors for the hydrogenation of unsaturated bonds in an aqueous solution.52 In other solvents, where the enthalpy of chloride solvation is low, reaction rates are much lower and in some cases catalysis is completely suppressed as dissociation of the chloride anion from the ruthenium center is suppressed.53 In other words, if the halide ion is poorly solvated, it is more nucleophilic, and will coordinate to the catalyst preventing generation of the active catalyst. Solvents have also been shown to strongly influence the outcome, e.g. CC versus CO selectivity, of hydrogenation reactions catalyzed by heterogeneous systems.54
5. C–C coupling reactions
Palladium catalysts are extensively used in organic chemistry to generate new C–C bonds.55 Of the various types of C–C coupling reactions, the Suzuki reaction, which involves the cross-coupling between an aryl halide and an aryl boronic acid to form a biaryl product (Fig. 15), has been especially well studied,56 as it is a highly versatile reaction extensively used for a variety of purposes including the industrial scale production of fine chemicals.57 Despite considerable efforts to develop highly efficient catalysts for this reaction, certain coupling reactions remain challenging. In particular, the coupling of aryl chlorides (due to the high bond dissociation energy of C–Cl bonds relative to C–Br and C–I bonds) and the coupling of sterically hindered substrates. Nevertheless, enormous progress has been made improving the efficiency of Suzuki catalysts and extremely high turnover frequencies and turnover number can be achieved and the nature of the solvent plays an important role with respect to catalyst stability.58 The active catalyst in the Suzuki reaction is based on a palladium(0) species (Fig. 15 shows a generic mechanism for the reaction). | ||
| Fig. 15 Generic mechanism for the Suzuki reaction. Note that the ligands coordinated to the palladium center are not included and the palladium(0) catalyst can be generated from many different precursors. In ‘ligand-free’ reactions the solvent acts as the ligand. | ||
Irrespective of the palladium pre-catalyst used in the Suzuki reaction, (i.e. a palladium(0) complex such as Pd(PPh3)4 or Pd2(dba)3 (dba = dibenzylideneacetone), palladium(II) precursors such as Pd(PPh3)2Cl2, PdCl2 with added (phosphine) ligands, palladacycles, or palladium nanoparticles), a 14 valence electron palladium(0) complex with two trans-oriented ligands is generated that corresponds to the active starting catalyst in the catalytic cycle (Fig. 15). Such a species would be highly reactive, as it has only 14 valence electrons, is coordinatively unsaturated and is in a low oxidation state which is unstable in the absence of strongly electron withdrawing ligands. For these reasons it is not surprising that extremely high activities can be achieved. Moreover, studies on the oxidative addition of substrates to model platinum complexes show strong solvent effects on the reaction rate.59
The identification of the actual intermediates present in a catalyst cycle is extremely challenging due to their intrinsic instability, which tends to preclude their isolation and full characterization. Interestingly, recent evidence even points to a catalyst based on palladium(0) with just one coordinated phosphine ligand. A compelling mass spectrometric-based study identified mono- and bis-ligated palladium species in the gas phase and reacted them with aryl halides, i.e. simulating the initial oxidative addition step in the proposed catalytic cycle.60 The 12 valence electron palladium species with one phosphine ligand was orders of magnitude more reactive towards aryl halides compared to the complex with two coordinated phosphine ligands. Irrespective of the nature of the palladium(0) species, i.e. whether it has one or two ligands or exists in an equilibrium between the two forms, these intermediates are highly unstable and, not surprisingly, Suzuki reactions tend to be performed in strong donor solvents that can coordinate to, and stabilize, the unsaturated palladium centers present in the intermediates. Stabilization can also be obtained using very bulky, electron rich ligands often with substituent groups that can weakly coordinate to the palladium center,61 although even with these ligands strong donor solvents are invariably used and mixtures of two solvents is quite common.
With strong donor solvents comes a trade-off between activity, that is suppressed, and stability, that is enhanced. If the catalyst rapidly decomposes, which is not unexpected for such coordinately unsaturated intermediates in the absence of a donor solvent, then low activities are obtained. To circumvent this problem, high catalyst loadings are often used, whereas careful choice of the solvent may be an equally effective option.
The term ‘ligand-free catalysis’ is widely used in publications describing C–C cross coupling reactions, and is used to indicate that a metal salt was used as the pre-catalyst without the addition of a co-ligand, e.g. a phosphine, carbene or another class of ligand. However, the term is rather misleading as it implies that ligands do not bind to the metal catalyst. It is likely that in the vast majority of systems described as being ligand-free, the solvent also serves as a ligand. For example, ‘ligand-free’ (Suzuki and Negishi) couplings using Pd(OAc)2 as the pre-catalyst are efficiently catalyzed in solvents such as DMF and N-methyl-2-pyrrolidone.62 Aryl chlorides can be coupled with phenylboronic acid in high yield using Pd(OAc)2 in PEG 400, a polyethylene glycol with a molecular weight of 400 that is a viscous liquid, and also an excellent multidentate ligand.63 In certain ‘ligand-free’ reactions, the catalyst is stabilized exclusively by the substrates/product which coordinate to the catalytic metal center, such as in the polymerization of butadiene by a nickel(II) catalyst.64
Despite the fact that some catalysts enable the reaction of aryl chlorides and sterically hindered substrates, even at very low catalyst loadings and ambient temperature,65 many catalysts suffer from stability problems or require the application of very strong bases. However, the conversion of activated aryl chlorides can be enhanced by simply switching the solvent from ethanol–water to dimethylacetamide (DMA)-water, with yields of the cross-coupling products increasing from ca.10% to >90%.66 A basic solvent such as DMA can potentially coordinate to the metal catalyst and simultaneously act as a base. In this context, the Suzuki reaction has been extensively studied in ionic liquids,67 where the presence of water (or alcohols and occasionally other solvents) is essential to provide activity. For example, in [bbim][BF4] (bbim = the 1-butyl-3-butylimidazolium cation) no reaction was observed for iodobenzene whereas >90% yield can be obtained in [bbim][BF4]/methanol mixtures under equivalent conditions.68 Attempts using other neat ionic liquids also met with failure.69 Studies showed that water facilitates the Suzuki reaction by solvating the halide by-product which is otherwise poorly solvated and steadily shuts down the reaction.70 In other words, if the halide by-product is poorly solvated then it is highly nucleophilic and can react with the catalyst reducing activity (chloride is a known poison for many catalysts),71 whereas strongly solvated halide is a poor nucleophile and consequently less reactive. Based on this principle, an ionic liquid with a hydroxyl functionalized cation, able to efficiently solvate halides, was found to be an excellent solvent for the coupling of aryl chlorides and sterically hindered substrates using PdCl2 as the pre-catalyst combined with a weak base (Na2CO3). The hydroxyl group also provides other benefits including facilitating reduction of the palladium(II) salt to palladium(0), stabilizing both the palladium nanoparticle catalyst reservoirs and the active palladium(II) catalytic species and activating the aryl chloride substrate (weakening the C–Cl bond) via hydrogen bonding interactions.72 For comparison, reactions in [bmim][Tf2N] or n-propanol gave a yield of 20 and 6% in the coupling of bromobenzene with phenyl boronic acid whereas in the hydroxyl functionalized ionic liquid a yield of >90% was obtained.
The use of solvents that activate substrates leading to enhanced catalytic activities was mentioned above for both hydrogenation reactions and Suzuki cross-coupling reactions. Another area in which substrate activation could have a profound impact is in the transformations of lignocellulosic biomass into commodity chemicals, an area of growing interest.73 Here, the nature of the solvent is critical with organic solvents posing problems related to stability and recyclability,74 and biphasic aqueous-based systems often not giving adequate yields.75 Ionic liquids show considerable promise, with initial interest emanating from the observation that certain chloride-based ionic liquids have an exceptionally high capacity for dissolving complex carbohydrates, attributable to the extensive hydrogen bonding interactions that disrupt the structure of carbohydrates.76 Once dissolved, the chemical transformations of complex carbohydrates such as cellulose become far more facile. Indeed, combining a CrCl2 pre-catalyst with chloride-based ionic liquids allows carbohydrates to be converted to 5-hydroxymethylfurfural (HMF),77 a useful feedstock chemical that can be transformed into many other derivatives.78 It has been shown that introducing specific hydrogen-bond donor groups that can interact with the substrate further enhances the performance of the catalyst. Such groups also influence the intermolecular interactions between ions, favoring solvent-substrate interactions and hence substrate activation.79
6. Concluding remarks
A thorough appreciation of solvent parameters and the ways in which these parameters can influence a catalyzed reaction allows reactions to be improved by changing or modifying the solvent rather than varying, for example, the ligands attached to a homogeneous catalyst. Examples have been given that demonstrate the profound effect solvents may impart in catalyzed reactions as well as demonstrating the multiple ways in which solvents can impact on a reaction. Simple models do not necessarily provide a molecular basis for observed solvent effects, but do provide an understanding that allows solvents to be selected that provide superior reaction rates. In this review, the implicit role of the solvent with respect to its interactions with catalytic intermediates, transition states, substrates and products has been described.Although solvents play a critical role in catalysis, currently relatively little attention is paid to the role of the solvent compared to the role of a ligand, except in the context of using more sustainable solvents for industrial reactions, where there is considerable interest.1 However, in addition to manipulating the ligands around a metal center to improve catalytic activity we argue that more effort should be devoted to finding the best solvent (or solvent mixture) for a catalytic process. As there is growing interest in catalysts that exploit non-covalent interactions, the choice of solvent becomes increasingly important.
References
- P. G. Jessop, Green Chem., 2011, 13, 1391–1398 RSC; C. Capello, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC; F. M. Kerton and R. Marriott, Alternative Solvents for Green Chemistry, RSC Publishing, 2nd edn, 2013 Search PubMed.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS; Y. Marcus, Chem. Soc. Rev., 1993, 22, 409–416 RSC; C. Reichardt and T. Welton, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, Weinheim, 4th edn, 2011 Search PubMed.
- C. Daguenet, P. J. Dyson, I. Krossing, A. Oleinikova, J. Slattery, C. Wakai and H. Weingärtner, J. Phys. Chem. B, 2006, 110, 12682–12688 CrossRef CAS PubMed.
- M. J. Kamlet, J. L. Abboud, M. H. Abraham and R. W. Taft, J. Organomet. Chem., 1983, 48, 2877–2887 CAS.
- M. A. Ab Rani, A. Brant, L. Crowhurst, A. Dolan, M. Lui, N. H. Hassan, J. P. Hallett, P. A. Hunt, H. Niedermeyer, J. M. Perez-Arlandis, M. Schrems, T. Welton and R. Wildinga, Phys. Chem. Chem. Phys., 2011, 13, 16831–16840 RSC.
- H. M. Chase, T. J. McDonough, K. R. Overly and C. M. Laperle, J. Phys. Org. Chem., 2013, 26, 322–326 CrossRef CAS.
- C. Wynne, M. M. Olmstead and P. G. Jessop, J. Am. Chem. Soc., 2000, 122, 7638–7647 CrossRef.
- H. M. L. Davies, P. R. Bruzinski, D. H. Lake, N. Kong and M. J. Fall, J. Am. Chem. Soc., 1996, 118, 6897–6907 CrossRef CAS.
- M. J. Kamlet, J. L. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877–2887 CrossRef CAS; P. G. Jessop, D. A. Jessop, D. Fu and L. Phan, Green Chem., 2012, 14, 1245–1259 RSC.
- C. A. Thomas, R. J. Bonilla, Y. Huang and P. G. Jessop, Can. J. Chem., 2001, 79, 719–724 CAS.
- P. Munshi, A. D. Main, J. Linehan, C. C. Tai and P. G. Jessop, J. Am. Chem. Soc., 2002, 124, 7963–7971 CrossRef CAS PubMed.
- P. G. Jessop, Y. Hsiao, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 344–355 CrossRef CAS; Y.-Y. Ohnishi, Y. Nakao, H. Sato and S. Sakaki, Organometallics, 2006, 25, 3352–3363 CrossRef.
- C. L. Young, Hydrogen and Deuterium. Solubility Data Series, Pergamon Press, Oxford, 1981, vol. 5/6 Search PubMed.
- R. W. Cargill, Carbon Monoxide. Solubility Data Series, Pergamon Press, Oxford, 1990, vol. 43 Search PubMed.
- R. Battino, Oxygen and Ozone. Solubility Data Series, Pergamon Press, Oxford, 1981, vol. 7 Search PubMed.
- P. G. T. Fogg, Carbon Dioxide in non-aqueous solvents at pressures less than 200 kPa, Pergamon Press, Oxford, 1992, vol. 50 Search PubMed; P. Scharlin, Carbon dioxide in water and aqueous electrolyte solutions. Solubility Data Series, Oxford University Press, Oxford, 1996, vol. 62 Search PubMed.
- P. J. Dyson, G. B. Laurenczy, C. A. Ohlin, J. Vallance and T. Welton, Chem. Commun., 2003, 2418–2419 RSC.
- C. A. Ohlin, P. J. Dyson and G. B. Laurenczy, Chem. Commun., 2004, 1070–1071 RSC.
- P. Husson-Borg, V. Majer and M. F. C. Gomes, J. Chem. Eng. Data, 2003, 48, 480–485 CrossRef CAS.
- Y. Chauvin, L. Mussmann and H. Olivier, Angew. Chem., Int. Ed. Engl., 1996, 34, 2698–2700 CrossRef.
- Y. Sun, R. N. Landau, J. Wang, C. LeBlond and D. G. Blackmond, J. Am. Chem. Soc., 1996, 118, 1348–1353 CrossRef CAS.
- P. G. Jessop, R. Stanley, R. A. Brown, C. A. Eckert, C. L. Liotta, T. T. Ngo and P. Pollet, Green Chem., 2003, 5, 123–128 RSC.
- P. G. Jessop and B. Subramaniam, Chem. Rev., 2007, 107, 2666–2694 CrossRef CAS PubMed.
- S. Moret, P. J. Dyson and G. Laurenczy, Dalton Trans., 2013, 42, 4353–4356 RSC.
- S. Moret, P. J. Dyson and G. Laurenczy, Nat. Commun., 2014, 5, 4017 CAS.
- K. Wittmann, W. Wisniewski, R. Mynott, W. Leitner, C. L. Kranemann, T. Rische, P. Eilbracht, S. Kluwer, J. M. Ernsting and C. L. Elsevier, Chem. – Eur. J., 2001, 7, 4584–4589 CrossRef CAS.
- M. Chatterjee, H. Kawanami, M. Sato, T. Ishizaka, T. Yokoyama and T. Suzuki, Green Chem., 2010, 12, 87–93 RSC.
- X. Xie, C. L. Liotta and C. A. Eckert, Ind. Eng. Chem. Res., 2004, 43, 7907–7911 CrossRef CAS.
- R. Ma, W. Hao, X. Ma, Y. Tian and Y. Li, Angew. Chem., Int. Ed., 2014, 53, 7310–7315 CrossRef CAS PubMed; O. S. Stamenković, A. V. Veličković and V. B. Veljković, Fuel, 2011, 90, 3141–3155 CrossRef.
- M. Schmitkamp, D. Chen, W. Leitner, J. Klankermayer and G. Francio, Chem. Commun., 2007, 4012–4014 RSC.
- L. C. Branco, P. M. P. Gois, N. M. T. Lourenço, V. B. Kurteva and C. A. M. Afonso, Chem. Commun., 2006, 2371–2372 RSC.
- A. Macchioni, Chem. Rev., 2005, 105, 2039–2073 CrossRef CAS PubMed; P. S. Pregosin, Pure Appl. Chem., 2009, 81, 615–633 CrossRef.
- L. Rocchigiani, C. Zuccaccia, D. Zuccaccia and A. Macchioni, Chem. - Eur. J., 2008, 14, 6589–6592 CrossRef CAS PubMed; L. Rocchigiani, G. Bellachioma, G. Ciancaleoni, A. Macchioni, D. Zuccaccia and C. Zuccaccia, Organometallics, 2011, 30, 100–114 CrossRef.
- A. Moreno, P. S. Pregosin, G. Laurenczy, A. D. Phillips and P. J. Dyson, Organometallics, 2009, 28, 6432–6441 CrossRef CAS.
- K. Brak and E. N. Jacobsen, Angew. Chem., Int. Ed., 2013, 52, 534–561 CrossRef CAS PubMed; R. J. Phipps, G. L. Hamilton and F. D. Toste, Nat. Chem., 2012, 4, 603–614 CrossRef PubMed.
- G. Ciancaleoni, I. Di Maio, D. Zuccaccia and A. Macchioni, Organometallics, 2007, 26, 489–496 CrossRef CAS; G. Ciancaleoni, C. Zuccaccia, D. Zuccaccia, E. Clot and A. Macchioni, Organometallics, 2009, 28, 960–996 CrossRef.
- W.-H. Wang, J. T. Muckerman, E. Fujita and Y. Himeda, ACS Catal., 2013, 3, 856–860 CrossRef CAS.
- K. Tanabe, Solid acids and bases: their catalytic properties, Elsevier, 2012 Search PubMed.
- Q. Zhang, S. Zhang and Y. Deng, Green Chem., 2011, 13, 2619–2637 RSC.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, 3rd edn, Wiley, 2004 Search PubMed.
- R. Pincock, J. Am. Chem. Soc., 1964, 86, 1820–1826 CrossRef CAS; C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, Weinheim, 3rd edn, 2003 Search PubMed.
- R. Breslow, Acc. Chem. Res., 1991, 24, 159–164 CrossRef CAS.
- W. S. Knowles, Acc. Chem. Res., 1983, 16, 106–112 CrossRef CAS.
- Y. Chi, W. Tang and X. Zhang, in Modern Rhodium-Catalyzed Organic Reactions, ed. P. Andrew Evans, WILEY-VCH, Weinheim, 2005 Search PubMed.
- B. R. James, Homogeneous Hydrogenation, John Wiley & Sons, New York, 1973 Search PubMed.
- J. Halpern and C. S. Wong, J. Chem. Soc., Chem. Commun., 1973, 629–630 RSC.
- R. E. Harmon, J. L. Parsons, D. W. Cooke, S. K. Gupta and J. Schoolenberg, J. Organomet. Chem., 1969, 34, 3684–3685 CAS.
- S. B. Duckett, C. L. Newell and R. Eisenberg, J. Am. Chem. Soc., 1994, 116, 10548–10556 CrossRef CAS.
- J. Luo, A. G. Oliver and J. S. McIndoe, Dalton Trans., 2013, 42, 11312–11318 RSC.
- P. J. Dyson, Dalton Trans., 2003, 2964–2974 RSC.
- A. Jourdant, E. Gonzalez-Zamora and J. Zhu, J. Organomet. Chem., 2002, 67, 3163–3164 CAS.
- C. Daguenet, R. Scopelliti and P. J. Dyson, Organometallics, 2004, 23, 4849–4857 CrossRef CAS.
- C. Daguenet and P. J. Dyson, Organometallics, 2004, 23, 6080–6083 CrossRef CAS.
- I. McManus, H. Daly, J. M. Thompson, E. Connor, C. Hardacre, S. K. Wilkinson, N. Sedaie Bonab, J. ten Dam, M. J. H. Simmons, E. H. Stitt, C. D'Agostino, J. McGregor, L. F. Gladden and J. J. Delgado, J. Catal., 2015, 330, 344–353 CrossRef CAS.
- J.-L. Malleron, J.-C. Fiaud and J.-Y. Legros, Handbook of Palladium-Catalyzed Organic Reactions, Academic Press, 1997 Search PubMed.
- N. T. S. Phan, M. van der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS.
- C. Torborg and M. Beller, Adv. Synth. Catal., 2009, 351, 3027–3043 CrossRef CAS.
- K. A. Crawford, A. H. Cowley and S. Humphrey, Catal. Sci. Technol., 2014, 4, 1456–1464 CAS.
- M. Crespo, M. Martinez, S. M. Nabavizadeh and M. Rashidi, Coord. Chem. Rev., 2014, 279, 115–140 CrossRef CAS.
- K. Vikse, T. Naka, J. S. McIndoe, M. Besora and F. Maseras, ChemCatChem, 2013, 5, 3604–3609 CrossRef CAS.
- R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461–1473 CrossRef CAS PubMed.
- A. Alimardanov, L. Schmieder-van de Vondervoort, A. H. M. de Vries and J. G. de Vries, Adv. Synth. Catal., 2004, 346, 1812–1817 CrossRef CAS.
- W. Han, C. Liu and Z. Jin, Adv. Synth. Catal., 2008, 350, 501–508 CrossRef CAS.
- A. R. O'Connor, P. S. White and M. Brookhart, J. Am. Chem. Soc., 2007, 129, 4142–4143 CrossRef PubMed.
- M. Miura, Angew. Chem., Int. Ed., 2004, 43, 2201–2203 CrossRef CAS PubMed; N. Kudo, M. Perseghini and G. C. Fu, Angew. Chem., Int. Ed., 2006, 45, 1282–1284 CrossRef PubMed; J. L. Bolliger, O. Blacque and C. M. Frech, Angew. Chem., Int. Ed., 2007, 46, 6514–6517 CrossRef PubMed; K. Billingsley and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 3358–3366 CrossRef PubMed.
- C. R. LeBlond, A. T. Andrews, Y. Sun and J. R. Sowa, Jr., Org. Lett., 2001, 3, 1555–1557 CrossRef CAS.
- C. J. Mathews, P. J. Smith and T. Welton, Chem. Commun., 2000, 1249–1250 RSC; W. Miao and T. H. Chan, Org. Lett., 2003, 5, 5003–5005 CrossRef CAS PubMed; R. Wang, B. Twamley and J. M. Shreeve, J. Org. Chem., 2006, 71, 426–429 CrossRef PubMed; D. Zhao, Z. Fei, T. J. Geldbach, R. Scopelliti and P. J. Dyson, J. Am. Chem., Soc., 2004, 126, 15876–15882 CrossRef PubMed.
- R. Rajagopal, D. V. Jarikote and K. V. Srinivasan, Chem. Commun., 2002, 616–617 RSC.
- A. Corma, H. García and A. Leyva, Tetrahedron, 2004, 60, 8553–8560 CrossRef CAS; A. Corma, H. García and A. Leyva, Tetrahedron, 2005, 61, 9848–9854 CrossRef.
- N. Yan, X. Yang, Z. Fei, Y. Li, Y. Kou and P. J. Dyson, Organometallics, 2009, 28, 937–939 CrossRef CAS.
- V. Gallo, P. Mastrorilli, C. F. Nobile, G. Romanazzi and G. P. Suranna, J. Chem. Soc., Dalton Trans., 2002, 4339–4342 RSC; M. A. Klingshirn, G. A. Broker, J. D. Holbrey, K. H. Shaughnessy and R. D. Rogers, Chem. Commun., 2002, 1394–1395 RSC.
- X. Yuan, N. Yan, S. A. Katsyuba, E. E. Zvereva, Y. Kou and P. J. Dyson, Phys. Chem. Chem. Phys., 2012, 14, 6026–6033 RSC; S. A. Katsyuba, E. E. Zvereva, N. Yan, X. Yuan, Y. Kou and P. J. Dyson, ChemPhysChem, 2012, 13, 1781–1790 CrossRef CAS PubMed.
- N. Yan and P. J. Dyson, Current Opinions in Chem. Eng., 2013, 2, 178–183 CrossRef CAS; P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC; J. C. Serrano-Ruiz, R. Luque and A. Sepulveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC; S. Liu, L. P. Abrahamson and G. M. Scott, Biomass Bioenergy, 2012, 39, 1–4 CrossRef; D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- V. Choudhary, S. H. Mushrif, C. Ho, A. Anderko, V. Nikolakis, N. S. Marinkovic, A. I. Frenkel, S. I. Sandler and D. G. Vlachos, J. Am. Chem. Soc., 2013, 135, 3997–4006 CrossRef CAS PubMed.
- C. M. Cai, T. Zhang, R. Kumar and C. E. Wyman, Green Chem., 2013, 15, 3140–3145 RSC.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed; D. A. Fort, R. C. Remsing, R. P. Swatloski, P. Moyna, G. Moyna and R. D. Rogers, Green Chem., 2007, 9, 63–69 RSC; H. Wang, G. Gurau and R. D. Rogers, Chem. Soc. Rev., 2012, 41, 1519–1537 RSC; J. S. Moulthrop, R. P. Swatloski, G. Moyna and R. D. Rogers, Chem. Commun., 2005, 1557–1559 RSC; T. Stahlberg, W. Fu, J. M. Woodley and A. Riisager, ChemSusChem, 2011, 4, 451–458 CrossRef PubMed.
- M. E. Zakrzewska, E. Bogel-Łukasik and R. Bogel-Łukasik, Chem. Rev., 2010, 111, 397–417 CrossRef PubMed.
- R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113, 1499–1597 CrossRef CAS PubMed.
- S. Siankevich, Z. Fei, R. Scopelliti, G. Laurenczy, S. Katsyuba, N. Yan and P. J. Dyson, ChemSusChem, 2014, 7, 1647–1654 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |