
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A thermostable transketolase evolved for aliphatic aldehyde acceptors†
Dong
Yi
a,
Thangavelu
Saravanan
a,
Titu
Devamani
a,
Franck
Charmantray
b,
Laurence
Hecquet
b and
Wolf-Dieter
Fessner
*a
aInstitut für Organische Chemie und Biochemie, Technische Universität Darmstadt, Alarich-Weiss-Str. 4, 64287 Darmstadt, Germany. E-mail: fessner@tu-darmstadt.de; Fax: +49 6151 166636; Tel: +49 6151 166666
bClermont Université, Université Blaise Pascal, Institut de Chimie de Clermont-Ferrand, CNRS UMR 6296, ICCF, BP10448, 63177 Aubière, France
First published on 18th November 2014
Abstract
Directed evolution of the thermostable transketolase from Geobacillus stearothermophilus based on a pH-based colorimetric screening of smart libraries yielded several mutants with up to 16-fold higher activity for aliphatic aldehydes and high enantioselectivity (>95% ee) in the asymmetric carboligation step.
Enzyme-catalyzed asymmetric carboligation1 is an important tool for the development of sustainable bioprocess technologies. Transketolase (EC 2.2.1.1, TK) is a ThDP-dependent enzyme that performs the transfer of a two-carbon ketol unit among various phosphorylated sugars in the pentose phosphate pathway.2 Recently, we have identified the TK from Geobacillus stearothermophilus (TKgst) as a novel thermostable catalyst with high potential for practical applications and future biocatalyst development.3In vitro, TK has been shown to also utilize non-phosphorylated sugars and (2R)-hydroxyaldehyde acceptors with generation of a (3S)-configurated stereocenter. This makes TK a potent biocatalyst for the asymmetric synthesis of chiral threo-diols related to ketoses.4 However, specific activity towards acceptors lacking a 2-hydroxylation is about 40-fold lower than for the corresponding hydroxylated derivatives.5 From a systematic evaluation of the TK from E. coli (TKeco) by single-site saturation mutagenesis of active-site residues involved in substrate binding, several improved variants were found showing improved acceptance of non-hydroxylated propanal.6 Here we report complementary studies for directed evolution in vitro of the thermostable TKgst to increase its activity with generic aldehyde acceptors and thus broaden the product spectrum. The TKgst protein scaffold was chosen over a TK from a mesophilic organism such as E. coli because thermostability has been demonstrated to correlate well with mutational robustness, as well as with stability to organic cosolvent.7
TKgst has high protein sequence similarity to TKeco and TKs from S. cerevisiae and B. anthracis (TKban), for which protein crystal structures have been determined with bound substrates.8 Among the highly conserved active-site environment, nine amino acid residues (His462, Arg521, Asp470, His28, Gly264, His263, Leu191, Leu382 and Ser385) form a channel for substrate entrance to the catalytic core at the ThDP cofactor (Fig. 1). These residues bind the acceptor substrate and position its electrophilic carbonyl group towards a nucleophilic attack. Therefore, these residues are putative candidates to alter the TK substrate specificity. Particularly, Asp470 appears to control the enantioselective binding of 2-hydroxyaldehydes by specific hydrogen bonding to the OH group.9 The replacement of Asp470 by amino acid residues with other larger side chains were expected to narrow the acceptor access tunnel and improve the binding of aliphatic aldehyde acceptors such as propanal (Fig. 1). Leu382, which is located at a position almost opposite to Asp470, provides a hydrophobic contribution to substrate binding. Because modification of this position may complement consequences from Asp470 mutagenesis, these two sites were selected for library building by site-specific saturation mutagenesis (SSM) to sample cooperative effects in a CAST-like strategy.10 Furthermore, variation at the Leu382 position may also provide an opportunity for reversing TK's enantioselectivity with 2-hydroxyaldehydes for forming products with non-natural erythro-configuration.
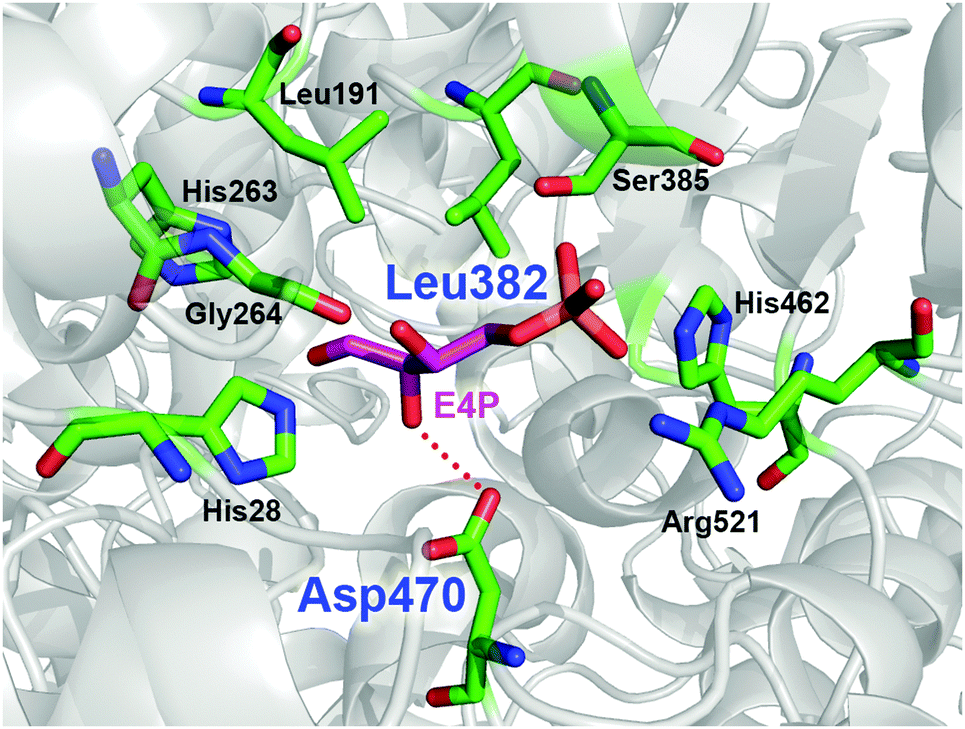 | ||
| Fig. 1 Model of the acceptor binding pocket based on the X-ray crystal structure of TK from B. anthracis (PDB entry 3M49)8k using substrate conformation of D-erythrose 4-phosphate (E4P) as determined in yeast TK (PDB entry 1NGS).8c The dotted red line indicates the hydrogen bond for hydroxyaldehyde discrimination by Asp470. Residue numbering is for TKgst.3 Graphic was created with PyMOL.11 | ||
A combined SSM library was constructed for Asp470/Leu382 replacement using NNS codon degeneracy, which requires at least 3066 clones for screening to achieve a statistical 95% coverage.12 As a control for cooperative effects, two individual single-site libraries for each Asp470 and Leu382 sites were built separately. Finally, 3456 and 2 × 96 colonies (36 plates) were randomly picked for the double site library and each of the single site libraries, respectively.
Out of the many assay methods for measuring TK activity,5,13 our recently developed colorimetric pH based assay method was chosen for library screening because of its simplicity, speed, universality, sensitivity, and low cost.5 The assay is based on conversion of hydroxypyruvate as donor with irreversible release of carbon dioxide, which in low-buffer medium causes a pH shift to alkaline and results in a color change with phenol red as indicator. This assay principle is independent of acceptor substrate structure and can be applied in microtiter plate format for the rapid multi-substrate screening of TK and reliable determination of kinetic constants. Using this high-throughput pH-assay method, the libraries were screened for activity with propanal (Scheme 1), and about 15% (192) positive clones were picked for rescreening. From this second round of screening with propanal the top 34 hits with unique sequences were finally collected for characterization. The specific activity of all enzyme variants with propanal (collected in Fig. 2 and Table S1, ESI†) was determined using samples purified by simple one-step heat-shock treatment5 taking advantage of the TKgst thermostability. Indeed, differential scanning fluorimetry14 confirmed that all of the positive variants indeed had a Tm comparable to wild-type TKgst (71 °C; Table S1, ESI†).
 | ||
| Scheme 1 Carboligation reactions studied for directed evolution of TKgst towards increased activity and stereoselectivity with generic aldehydes. | ||
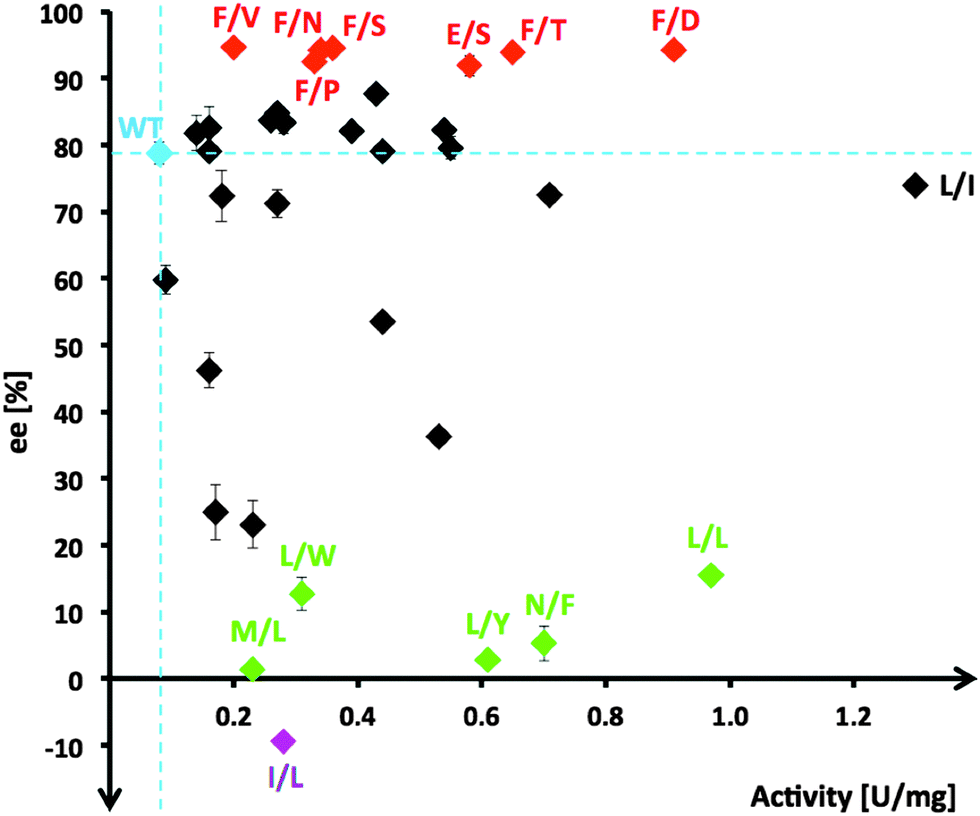 | ||
| Fig. 2 Property spread for TKgst variants from Leu382/Asp470 double-site saturation mutagenesis that were identified for activity with propanal higher than wild-type (blue). Variants showing highest (>90% ee), lowest (<15% ee) and inverted enantioselectivity are marked in red, green and purple, respectively. Variants are labeled with one-letter code for clarity. | ||
From the multiple SSM strategy, many different single-site (29%) and combined double-site variants (71%) at Asp470 (13 different residues, 11% wild-type) and Leu382 (11 different residues; >25% wild-type) positions were found that can improve catalytic rates. The replacement of Asp470 by Ile or Leu contributes with Leu382 and others to a non-polar lining of the substrate channel, which benefits the binding of the aliphatic aldehydes and leads up to 16-fold higher activity towards propanal relative to wild-type (Fig. 3). Although a considerable plasticity of the corresponding Asp469 residue in TKeco had already been noted upon single SSM,6c the much broader range observed for highly active single-site and combined variants of TKgst is remarkable.
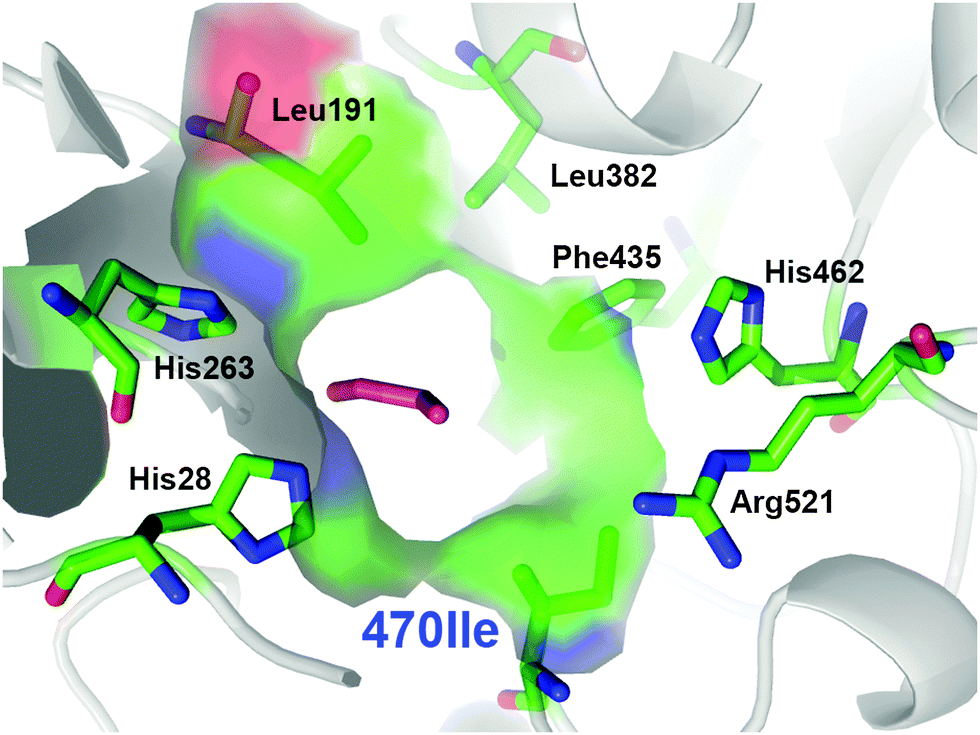 | ||
| Fig. 3 Entrance to the active site for the most active TKgst (Asp470Ile) variant with bound propanal (magenta), illustrating the compact hydrophobic substrate channel. Graphic was created with PyMOL.11 | ||
In order to evaluate the influence of small steric and/or electronic increments from the substrate structure on the activity of positive variants, the set of unique active hits was further measured for activity with the extended C4-aldehyde (butanal) and its bio-isosteric ether equivalent (methoxyethanal) as alternative acceptors (Scheme 1), together with glycolaldehyde as one of the best known substrates for wild-type TKgst (Table S1, ESI†). Remarkably, the single-site variant Asp470Ile, for which no corresponding hit had been found with TKeco,6 shows the highest activity towards propanal, butanal and methoxyethanal with 16-, 13- and 8-fold improvement against wild-type. It was assumed that structurally similar aldehydes are processed by TK variants with similar activity because of the likely similar substrate binding interactions. Indeed, this becomes evident from the highly correlated kinetic data sets determined independently for butanal and methoxyethanal (Table S1, Fig. S4 and S5, ESI†). The analysis also documents the anticipated higher rates for the latter owing to its higher electrophilic aldehyde group. Surprisingly, several of the top variants retained or even improved their activity towards glycolaldehyde (Table S1, ESI†). Particularly in case of bulky and/or hydrophobic replacements of Asp470 this indicates that the catalytic efficiency of TKgst is not impaired even if the 2-hydroxy group has to reorient to a different binding position.
Stereoselectivity in the asymmetric carboligation was subsequently analyzed for all unique variants using the ketol transfer reaction from hydroxypyruvate to propanal in deep-well format. Product formed was extracted and analyzed by chiral GC after peracetylation. Results (Table S1, ESI†) are summarized schematically in Fig. 2. The (3S)-1,3-dihydroxypentanone product was further synthesized on a preparative scale (64% yield) using the variant TKgst (Leu382Phe) to confirm product identity and enantiomeric purity (94%). As an extra control, selected high-rate variants with high, medium and poor stereoselectivity (variants Phe/Asp, Leu/Ile, Leu/Leu at 382/470, respectively) were also tested for their enantioselectivity with butanal and methoxyethanal, which confirmed their relative selectivities. The diacetates of the butanal and methoxyethanal products were insufficiently resolved by chiral GC, but trifluoroacetylation showed the TKgst (Leu382Phe) variant to yield both products with surprisingly high 99% ee. However, when re-determined by the trifluoroacetylation method the ee value for the propanal product was also found to be 99%. This indicates that some racemization occurs upon peracetylation, and that presumably all ee data determined by peracetylation here and in previous studies are generally too low.6
Stereoselectivity for most kinetically improved variants remains strongly biased towards creation of the (3S) product configuration with more than half (60%) of all top variants having enantioselectivities similar to (70–85% ee; 14 variants) or higher than wild-type. Out of the seven top candidates (21%) with enantioselectivity >90% ee (Fig. 2, red variants), six include a Leu382Phe replacement, and their second position determines the level of rate acceleration with increasing polarity of the side-chain functionality (Val/Pro → Asn → Ser → Thr → Asp). In order to understand the contribution of the Leu382Phe exchange to an enantioselectivity improvement, a 3D structure model of the mutant TKgst was simulated using the ThDP liganded structure of TKban (PDB entry 3M49). According to the modeling results, the phenyl ring of the mutated 382Phe side-chain might have π–π stacking interaction with Phe435, which jointly form a non-polar tunnel entrance and fix the propanal substrate for the ketol unit transfer. In combination with the polar interaction of the residue at position 470, attack at the carbonyl group will occur with high preference on one face only and result in improved enantioselectivity.
About 40% of variants (14 out of 34) show enantioselectivity similar to wild-type, the majority of which are double-site variants (71%). Single-site variations in this group are limited to replacement of Asp470 by Glu, Ile, Ser or Thr with rate accelerations of 4 to 14-fold. Interestingly, for TKeco a change of the corresponding Asp469 to Ala, Glu, Thr or Tyr gave 4.3- to 8-fold activity improvement towards propanal (64% and 90% ee for Thr and Glu variants, and reversed 53% ee for Tyr variant).6
Only 26% of unique hits showed enantioselectivity below 50% ee. Among those, a subset of five candidates had very poor (3S) stereoselectivity <15% ee (Fig. 2, green variants) and one variant (I/L) was identified to have inverted stereopreference for the (3R)-configuration (9.4% ee). A common motif of these variants is the presence of a bulky hydrophobic residue replacing D470 (Leu, Phe, Trp, Tyr), which is an excellent starting point for next-generation libraries to completely switch enantioselectivity towards the (3R) configuration. However, these results indicate that the modification of residues Leu382 and Asp470 alone is insufficient to reorient the binding position of propanal in the active pocket for a nucleophilic attack at the aldehyde carbonyl group to occur along an inverted trajectory.
In summary, this study underscores the strength of “smart library” concepts for the directed evolution of improved enzyme function with low screening effort.12 In combination with “smart screening” technology using our pH-based colorimetric assay principle new TKgst variants with significant improvement of activity and enantioselectivity for non-hydroxylated acceptor substrates have been discovered. These results open up a panacea of new opportunities for further studies into the engineering of thermostable TK variants for various applications in asymmetric carboligation.
This work was funded by the Deutsche Forschungsgemeinschaft (grant Fe244/9-1 to W.D.F.) and the Agence Nationale de la Recherche (grant ANR-09-BLAN-0424-CSD3 to L.H.) within the framework of Programme Blanc International, as well as by ESF project COST CM1303. D.Y. gratefully acknowledges support through a PhD scholarship administered by the DAAD. We thank D. Heyl for assistance in developing suitable methods for enantioselectivity assessment.
Notes and references
- (a) W.-D. Fessner, in Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Groeger and O. May, Wiley-VCH, Weinheim, 3rd edn, 2011, pp. 857–917 Search PubMed; (b) P. Clapes and X. Garrabou, Adv. Synth. Catal., 2011, 353, 2263–2283 CrossRef CAS; (c) M. Brovetto, D. Gamenara, P. Saenz Mendez and G. A. Seoane, Chem. Rev., 2011, 111, 4346–4403 CrossRef CAS PubMed; (d) H. C. Hailes, D. Rother, M. Mueller, R. Westphal, J. M. Ward, J. Pleiss, C. Vogel and M. Pohl, FEBS J., 2013, 280, 6374–6394 CrossRef CAS PubMed.
- G. Schenk, R. G. Duggleby and P. F. Nixon, Int. J. Biochem. Cell Biol., 1998, 30, 1297–1318 CrossRef CAS.
- J. Abdoul-Zabar, I. Sorel, V. Helaine, F. Charmantray, T. Devamani, D. Yi, V. de Berardinis, D. Louis, P. Marliere, W.-D. Fessner and L. Hecquet, Adv. Synth. Catal., 2013, 355, 116–128 CrossRef CAS.
- (a) Y. Kobori, D. C. Myles and G. M. Whitesides, J. Org. Chem., 1992, 57, 5899–5907 CrossRef CAS; (b) L. Hecquet, J. Bolte and C. Demuynck, Tetrahedron, 1994, 50, 8677–8684 CrossRef CAS; (c) K. G. Morris, M. E. B. Smith, N. J. Turner, M. D. Lilly, R. K. Mitra and J. M. Woodley, Tetrahedron: Asymmetry, 1996, 7, 2185–2188 CrossRef CAS; (d) F. T. Zimmermann, A. Schneider, U. Schörken, G. A. Sprenger and W.-D. Fessner, Tetrahedron: Asymmetry, 1999, 10, 1643–1646 CrossRef CAS; (e) G. A. Sprenger and M. Pohl, J. Mol. Catal. B: Enzym., 1999, 6, 145–159 CrossRef CAS; (f) F. Charmantray, V. Helaine, B. Legeret and L. Hecquet, J. Mol. Catal. B: Enzym., 2009, 57, 6–9 CrossRef CAS PubMed; (g) J. L. Galman, D. Steadman, S. Bacon, P. Morris, M. E. B. Smith, J. M. Ward, P. A. Dalby and H. C. Hailes, Chem. Commun., 2010, 46, 7608–7610 RSC; (h) M. Mueller, G. A. Sprenger and M. Pohl, Curr. Opin. Chem. Biol., 2013, 17, 261–270 CrossRef CAS PubMed.
- D. Yi, T. Devamani, J. Abdoul-Zabar, F. Charmantray, V. Helaine, L. Hecquet and W.-D. Fessner, ChemBioChem, 2012, 13, 2290–2300 CrossRef CAS PubMed.
- (a) E. G. Hibbert, T. Senussi, S. J. Costelloe, W. Lei, M. E. B. Smith, J. M. Ward, H. C. Hailes and P. A. Dalby, J. Biotechnol., 2007, 131, 425–432 CrossRef CAS PubMed; (b) E. G. Hibbert, T. Senussi, M. E. B. Smith, S. J. Costelloe, J. M. Ward, H. C. Hailes and P. A. Dalby, J. Biotechnol., 2008, 134, 240–245 CrossRef CAS PubMed; (c) M. E. B. Smith, E. G. Hibbert, A. B. Jones, P. A. Dalby and H. C. Hailes, Adv. Synth. Catal., 2008, 350, 2631–2638 CrossRef CAS; (d) A. Cazares, J. L. Galman, L. G. Crago, M. E. B. Smith, J. Strafford, L. Rios-Solis, G. J. Lye, P. A. Dalby and H. C. Hailes, Org. Biomol. Chem., 2010, 8, 1301–1309 RSC.
- J. D. Bloom and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9995–10000 CrossRef CAS PubMed.
- (a) M. Nikkola, Y. Lindqvist and G. Schneider, J. Mol. Biol., 1994, 238, 387–404 CrossRef CAS PubMed; (b) S. Koenig, A. Schellenberger, H. Neef and G. Schneider, J. Biol. Chem., 1994, 269, 10879–10882 CAS; (c) U. Nilsson, L. Meshalkina, Y. Lindqvist and G. Schneider, J. Biol. Chem., 1997, 272, 1864–1869 CrossRef CAS PubMed; (d) C. Wikner, U. Nilsson, L. Meshalkina, C. Udekwu, Y. Lindqvist and G. Schneider, Biochemistry, 1997, 36, 15643–15649 CrossRef CAS PubMed; (e) E. Fiedler, S. Thorell, T. Sandalova, R. Golbik, S. Konig and G. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 591–595 CrossRef CAS PubMed; (f) P. Asztalos, C. Parthier, R. Golbik, M. Kleinschmidt, G. Huebner, M. S. Weiss, R. Friedemann, G. Wille and K. Tittmann, Biochemistry, 2007, 46, 12037–12052 CrossRef CAS PubMed; (g) L. Mitschke, C. Parthier, K. Schroeder-Tittmann, J. Coy, S. Luedtke and K. Tittmann, J. Biol. Chem., 2010, 285, 31559–31570 CrossRef CAS PubMed; (h) E. Fullam, F. Pojer, T. Bergfors, T. A. Jones and T. Cole Stewart, Open Biol., 2012, 2, 110026 CrossRef PubMed; (i) S. Luedtke, P. Neumann, K. M. Erixon, F. Leeper, R. Kluger, R. Ficner and K. Tittmann, Nat. Chem., 2013, 5, 762–767 CrossRef CAS PubMed; (j) N. Maltseva, Y. Kim, K. Kwon, A. Joachimiak, W. F. Anderson, RCSB protein data bank (PDB entries 3HYL and 3M49, submitted 06/2009 and 04/2010).
- (a) U. Nilsson, L. Hecquet, T. Gefflaut, C. Guerard and G. Schneider, FEBS Lett., 1998, 424, 49–52 CrossRef CAS; (b) For a corresponding study with TKeco see U. Schoerken, PhD dissertation, University of Duesseldorf, Germany, 1997.
- M. T. Reetz, M. Bocola, J. D. Carballeira, D. Zha and A. Vogel, Angew. Chem., Int. Ed., 2005, 44, 4192–4196 CrossRef CAS PubMed.
- W. L. DeLano, The PyMOL Molecular Graphics System, Version 1.3, Schrödinger LLC., 2002, http://www.pymol.org Search PubMed.
- S. Lutz and U. T. Bornscheuer, Protein Engineering Handbook, Wiley-VCH, Weinheim, 2009 Search PubMed.
- L. Hecquet, W.-D. Fessner, V. Helaine and F. Charmantray, in Cascade Biocatalysis, ed. S. Riva and W.-D. Fessner, Wiley-VCH, Weinheim, 2014, pp. 315–337 Search PubMed.
- M. W. Pantoliano, E. C. Petrella, J. D. Kwasnoski, V. S. Lobanov, J. Myslik, E. Graf, T. Carver, E. Asel, B. A. Springer, P. Lane and F. R. Salemme, J. Biomol. Screening, 2001, 6, 429–440 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, procedures for library creation, screening and ee determination, protein modeling, product characterization and a full list of variants with sequence information, kinetic data and enantioselectivity. See DOI: 10.1039/c4cc08436e |
| This journal is © The Royal Society of Chemistry 2015 |