DOI:
10.1039/C3RA47021K
(Paper)
RSC Adv., 2014,
4, 11939-11947
Impact of alloying and lattice strain on ORR activity of Pt and Pd based ternary alloys with Fe and Co for proton exchange membrane fuel cell applications†
Received
26th November 2013
, Accepted 6th February 2014
First published on 7th February 2014
Abstract
A novel structure–catalytic activity correlation of platinum and palladium alloys is reported in this study. Metals with similar covalent radius (Pt and Pd) were alloyed with metals of lesser covalent radius (Fe and Co). The ternary alloys namely, PtFeCo and PdFeCo were synthesized by a solvothermal method. The structural properties such as lattice parameters and compressive strain of these electrocatalysts were evaluated by XRD and SAED analysis. Particle size and pattern of dispersion of these catalysts in Vulcan XC-72 carbon were characterized by TEM. The composition of the alloys was identified by ICP-OES and EDX techniques. Electrochemical properties such as ECSA, oxygen reduction reaction activity and kinetic parameters were assessed by CV and RDE studies. Structural and electroactivity of both alloys were compared with their respective bulk metal catalysts (20 wt% Pt/C, 20 wt% Pd/C). The lattice strain was calculated from the decrease in lattice parameters of alloys compared with those of pure metals. It is observed that PdFeCo/C shows more compressive strain than PtFeCo/C which is reflected in the greater improvement in ORR activity than that of PtFeCo/C catalyst. ORR studies revealed that both alloys and their pure metals follow a four electron transfer mechanism. The kinetic parameters such as mass and specific activities are calculated from the Tafel slope of ORR. Pt/C, PtFeCo/C and PdFeCo/C electrocatalysts have Tafel slope values (∼120 mV dec−1) indicative of one electron transfer as the rate determining step. The mass and specific activities of PdFeCo/C show higher increments than those of PtFeCo/C compared to their respective metals. The results of ORR studies reflect that lattice strain is an important factor to improve the catalytic activity of the electrocatalyst. Single cell polarizations were carried out at 60, 70 and 80 °C with PtFeCo/C and PdFeCo/C as cathode electrocatalysts. Both ternary alloy electrocatalysts show higher performance than pure metal catalysts. At 80 °C, PtFeCo/C and PdFeCo/C register a power density of 713 mW cm−2 and 437 mW cm−2 respectively.
1. Introduction
The proton exchange membrane fuel cell (PEMFC) is believed to be promising for mobile and transportation applications in the near future. One of the major problems facing the PEMFC system is the development of a cathode catalyst with low over potential.1,2 Currently platinum is used as the catalyst on both anode and cathode in PEMFCs. Use of expensive metals like Pt limits the prospects of commercialization of PEMFC technology. Even with platinum as cathode catalyst, the oxygen reduction reaction (ORR) is affected by an over potential of 400 mV.3 Hence various efforts are made to reduce the amount of Pt as well as to improve the ORR activity. Pt based bimetallic alloys have been synthesized with non precious transition metals such as Fe, Co, Ni, V, Cu and Cr.4–8 Recently, some trimetallic alloys such as PtVFe, PtNiFe and PtNiCo have been reported to improve the ORR activity.2,9,10 In order to control the cost of electrocatalyst, palladium has also been used as a cathode catalyst as it follows the same mechanism for oxygen reduction reaction as that of platinum.11,12 But when compared to Pt, the intrinsic catalytic activity of Pd in ORR is low. Also the long term stability is not good enough at high potentials. In order to overcome oxide formation and electro dissolution of Pd which affect the stability, it is alloyed with non precious metals such as Ti, Fe, Co, Cu and made as bimetallic alloys.13–16 Trimetallic alloys of PdCoAu and PdCoMo have also been reported with improved ORR activity compared to Pd alone.13,17 Enhanced ORR activity of both Pt alloys and Pd alloys are related to different factors such as structural properties, electronic properties and surface sensitive factors.18–20 Alloying Pt or Pd with non precious metals changes the M1–M2 bond distance through lattice contraction, which may decrease the OH adsorption energy on Pt alloy and Pd alloys, thereby increasing the ORR activities than those of the pure metals.15,21,22 Correlation of the strain properties of nanoscale particles with electrocatalytic activity of catalysts have been reported.23 Due to compressive strain effect, inter atomic distance is decreased which is considered to be an important factor in understanding the increment of ORR activity in Pt and Pd based alloys.24,25 In the theoretical study by density functional theory (DFT), the d-band center of the alloyed Pt surface was found to shift to lower position with respect to Fermi level due to the compressive strain and electronic effects produced by the alloying transition metals.26 The down shifting of d-band center favors the ORR activity by reducing the adsorption energy (AE) of oxygen and OH intermediates bonding to Pt surface.25,27
Hence, we aim to study the contribution of lattice strain on ORR activity of fuel cell catalyst. Based on our optimization study for the best composition of Pt and Pd ternary alloys for ORR activity, we report here the alloys with the ratio of Pt/Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe:Co as 7:2:1. Platinum and palladium are alloyed with small sized transition metals like iron and cobalt, and the resulting strain properties of the alloys are correlated with the enhancement in electrocatalytic activity for ORR. Carbon supported PtFeCo and PdFeCo electrocatalysts have been synthesized by solvothermal method using 1,2 propanediol and polyvinyl pyrrolidone (PVP) as solvent and surfactant respectively. The alloys have been characterized by XRD, TEM with EDX, TEM SAED and ICP-OES, while the electrocatalytic activity was analyzed by cyclic voltammetry and hydrodynamic linear sweep voltammetry. Evaluation of single cell performance of carbon supported PtFeCo and PdFeCo cathode has been carried out at different temperatures of 60, 70 and 80 °C by supplying humidified H2/O2 gases.
:Fe:Co as 7:2:1. Platinum and palladium are alloyed with small sized transition metals like iron and cobalt, and the resulting strain properties of the alloys are correlated with the enhancement in electrocatalytic activity for ORR. Carbon supported PtFeCo and PdFeCo electrocatalysts have been synthesized by solvothermal method using 1,2 propanediol and polyvinyl pyrrolidone (PVP) as solvent and surfactant respectively. The alloys have been characterized by XRD, TEM with EDX, TEM SAED and ICP-OES, while the electrocatalytic activity was analyzed by cyclic voltammetry and hydrodynamic linear sweep voltammetry. Evaluation of single cell performance of carbon supported PtFeCo and PdFeCo cathode has been carried out at different temperatures of 60, 70 and 80 °C by supplying humidified H2/O2 gases.
2. Experimental
2.1. Materials
Hexachloro platinic acid (Sigma Aldrich), palladium acetylacetonate (Alfa Aesar), anhydrous FeCl3 (Merck), cobalt acetylacetonate (Alfa Aesar), 1,2-propylene glycol, polyvinyl pyrrolidone (PVP) (MW 58000), Vulcan XC-72 carbon, ethanol, acetone, H2SO4 (Merck), Milli-Q (Millipore) water were used in the present study.
2.2. Synthesis of PtFeCo and PdFeCo alloy catalyst
Carbon supported PtFeCo and PdFeCo trimetallic alloys were synthesized by solvothermal method in the atomic ratio of 7:2:1. Hexachloro platinic acid, palladium acetylacetonate, anhydrous ferric chloride and cobalt acetylacetonate were used as precursors of Pt, Pd, Fe and Co respectively. For the synthesis of carbon supported PtFeCo catalyst, the corresponding metal precursors were dissolved in 1,2-propylene glycol and mixed well in inert (nitrogen) atmosphere. To the resultant solution, 120 mg of PVP (surfactant) was added and stirred into a homogeneous solution in N2 atmosphere. PVP was added to precursor solutions with the metal and surfactant ratio of 1:2. The homogenized solution was transferred into a PTFE-lined autoclave reactor and closed in inert atmosphere. It was heated at 200 °C in a muffle furnace for 12 hours after which the reactor was cooled to room temperature. The yellow solution was transferred into a beaker to which 100 ml of ethanol was added and mixed well. This was followed by the addition of Vulcan XC-72 carbon (20 wt% metal loading) with constant stirring. PtFeCo/C suspension was sonicated for 5 minutes and then stirred well for 3 hours by a magnetic stirrer. The PtFeCo/C was extracted by ultracentrifugation. The product was repeatedly washed with ethanol and Millipore water to remove any trace of surfactant. The product was finally rinsed with acetone and dried in vacuum oven at 80 °C for 12 hours. Similarly, PdFeCo/C was also synthesized. Pd/C was synthesised by the above procedure without non precious metals.
2.3. Characterization techniques
The catalyst samples were studied by X-ray powder diffraction technique (Rigaku Miniflex II Instrument) in the scattered angle range of 20 to 80° and in scan step of 1.5° min−1 using CuKα radiation (λ= 1.5406 Å). XRD analysis was carried out with 40 mg of catalyst spread uniformly over quartz plate. The structural and morphological characterization was conducted by high resolution transmission electron microscopy (FEI-TECNAI) at an operating voltage of 300 kV. Selected area electron diffraction (SAED) patterns were recorded in the same HRTEM instrument. Sample for TEM was prepared by dispersing 2 mg of the sample in ethanol and placing a drop of the solution on carbon coated Cu grid and drying well in air. The particle size distribution of PtFeCo/C and PdFeCo/C catalysts was calculated from the TEM image for more than 100 particles chosen at random. Energy dispersive X-ray analysis was carried out using EDAX coupled with HRTEM instrument. Bulk composition and metal loading of the catalyst were identified by Inductive coupled plasma-optical emission spectrometry (Perkin Elmer Optima 5300 Dv). The trimetalic alloy catalyst was dissolved in concentrated aqua regia and diluted to around 100 ppm. Electrochemical experiments were performed using an electrochemical workstation (CHI 608B, CH Instruments, USA), in a conventional three electrode system of saturated Ag/AgCl as reference electrode, Pt mesh as counter electrode and 0.19625 cm2 area of glassy carbon (GC) disc as working electrode. GC electrode was mirror polished with 0.05 micron alumina powder and cleaned well with Millipore water. 15 μl of catalyst in ethanol (1 μl = 1 μg catalyst) was coated on GC by drop cast method, dried slowly at room temperature and then coated with Nafion film on the uniform surface of catalyst layer (3 μl of 0.05 wt% Nafion diluted from 5 wt% Nafion, Dupont). The total metal loading on GC was 3 μg (in the case of single metal, Pt or Pd = 3 μg; in alloys, Pt or Pd + (Fe + Co) = 3 μg). Cyclic voltammetry studies were carried out in highly pure N2 saturated 0.5 M H2SO4 solution in the potential window of −0.175 V to 1.000 V vs. Ag/AgCl at a scan rate of 50 mV s−1. Oxygen reduction studies were conducted by rotating disc electrode technique (RDE) in highly pure O2 saturated 0.5 M H2SO4 solution. Prepared electrodes were polarized negatively with potential from 1.0 to 0.0 V at a scan rate of 10 mV s−1. Experiments were carried out at room temperature (30 ± 2 °C) and all the polarization curves are plotted vs. SHE.
2.4. Membrane electrode assembly (MEA) fabrication and fuel cell evaluation
A set of carbon papers (TGPH-120, Japan) were coated with 10 wt% of Teflon to provide hydrophobicity. The gas diffusion layer (GDL) was prepared by coating the slurry of Vulcan XC-72 carbon and 30 wt% of PTFE in isopropyl alcohol (IPA) by microspray method. The GDL was sintered at 250 °C for 1 hour and then at 350 °C for 30 minutes. The GDL loading was fixed as 1 mg cm−2 for all the fuel cell experiments. For cathodic electrode, the catalyst ink was prepared by sonicating the catalyst (PtFeCo/C or PdFeCo/C) and 5 wt% Nafion (Dupont) in 2 ml of IPA for half an hour to attain homogeneous dispersion of the slurry, which was spray-coated on 5 cm2 area of Nafion-212NR membrane (Dupont, 50 mm) to achieve a crack-free surface. Loading of Pt or Pd as cathode catalyst was 0.5 mg cm−2 and loading of Nafion on dried cathode and anode catalyst layers was 25 wt%. Commercial Pt/C (Sainergy Tech, USA) as anode catalyst layer with Pt loading of 0.25 mg cm−2 was coated on the anode layer of the membrane. GDL and electrodes were fixed in single cell test fixture (Fuel cell Technologies, inc. USA) by sandwiching and applying 10 lbf ft pressure with torque wrench. 5 cm2 active area of the MEA in the PEM fuel cell test fixture was connected and evaluated in fuel cell test station (K-PAS Pvt. Ltd). H2 and O2 gases were fed to the anode and cathode sides through humidifiers maintained at the cell temperature. Flow rate of H2 and O2 gases were 300 ml min−1 and the gases were fed through serpentine flow channel without back pressure. Before polarization, single cell was activated between OCP and higher current density. After stabilization, the cell was polarized at temperatures of 60, 70 and 80 °C. For comparison, commercial Pt/C and synthesized Pd/C were used as cathode catalysts with same loading as that of alloys. The Pt or Pd loading for anode was 0.25 mg cm−2 and for cathode was 0.50 mg cm−2.
3. Results and discussion
3.1. Structural characterizations
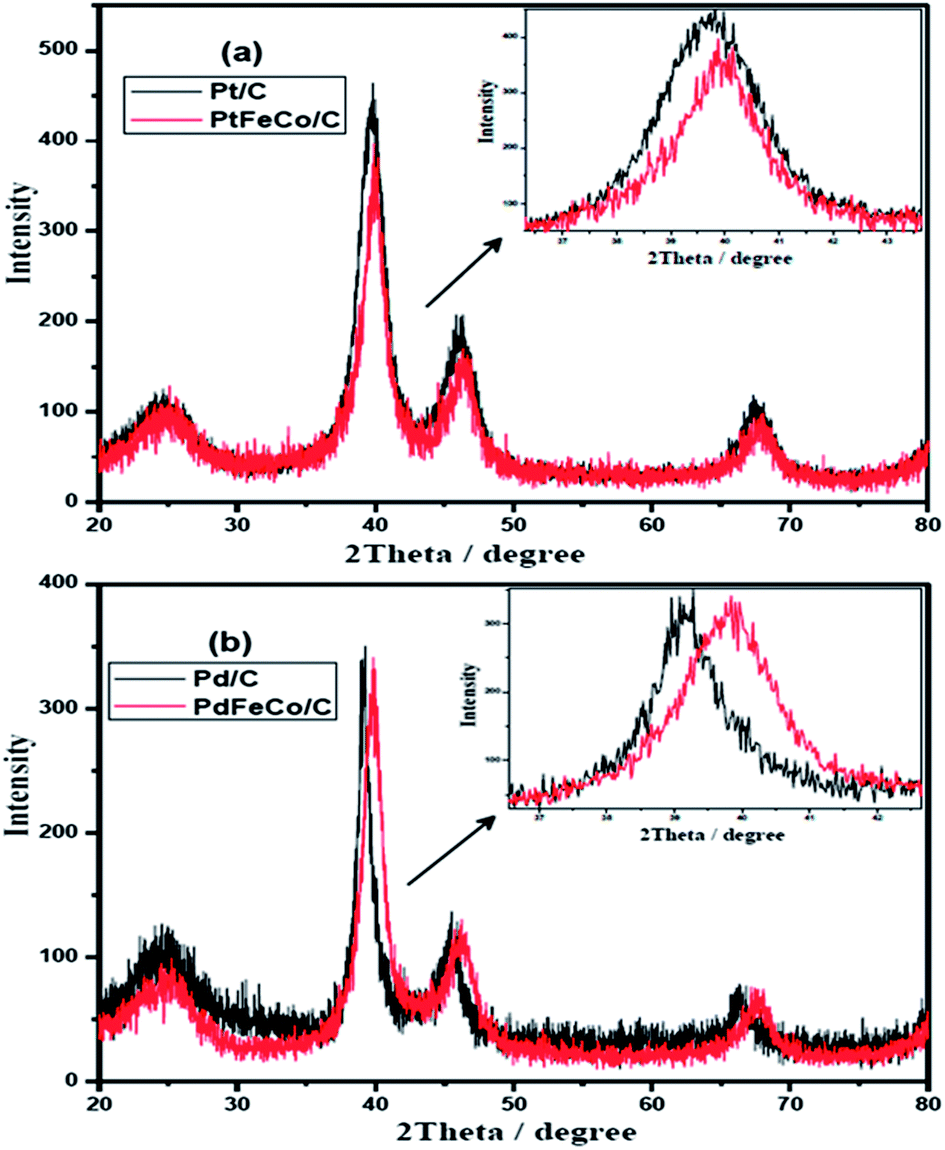
Fig. 1a and b show the XRD patterns of PtFeCo/C, PdFeCo/C electrocatalysts with their references Pt/C and Pd/C respectively. All the XRD patterns show characteristic highly crystalline face centered cubic (FCC) structure with peaks corresponding to (111), (200) and (220) planes with respect to 2θ values of 39.67, 46.17, 67.42 for Pt/C and 39.11, 45.43, 66.12 for Pd/C nano catalysts. These values correspond to the JCPDS card numbers 04-0802 of Pt and 87-0637 of Pd respectively. The diffraction peak at about 2θ = 25° corresponds to the characteristic hexagonal structure of the conducting Vulcan XC-72 carbon. There are no peaks corresponding to the metal oxides. PtFeCo/C catalyst shows relatively same diffraction patterns as that of Pt/C but with a slight shift to higher 2θ values due to formation of alloy. The inset in Fig. 1a represents the expanded (111) peaks showing a clear shift in peak position. It confirms the alloy formation due to incorporation of smaller atomic radius Fe and Co metals into FCC structure of Pt. Similar effect was observed in PdFeCo/C also (Fig. 1b). The diffraction peak (111) of PtFeCo/C shifted to slightly high 2θ value by 0.36°, while the XRD peak of PdFeCo/C shifted by a higher value of 0.78° than their corresponding pure metals namely Pt/C and Pd/C respectively. The higher angle shifts reflect changes in the lattice parameter and the same was calculated for (111) peak. These changes of the lattice parameters are denoted as strain. The strain in the alloy is calculated27 using the eqn (1).| | |
s(Pt) = {(aalloy − aPt)/aPt} × 100
| (1) |
where aalloy is the lattice parameter of PtFeCo/C and aPt is the lattice parameter of bulk Pt. Lattice parameters and compressive strain values are listed in Table 1. Though Pt (136 ± 5 pm) and Pd (139 ± 5 pm) have similar metallic radii, the effect of alloying with smaller sized Fe, Co metals produced higher compressive strain in PdFeCo/C (1.89%) than in PtFeCo/C (0.88%). The average crystallite size was calculated from the corresponding XRD patterns using Scherrer equation. Pt/C and PtFeCo/C have crystallite size of 3.0 nm and 3.5 nm respectively while Pd/C and PdFeCo/C have a crystallite size of 6.1 nm and 5.0 nm respectively. The change in crystallite size on alloying is due to the contraction caused by the smaller sized metals (Fe and Co). Solvothermal synthesis provides smaller crystallites of Pd and Pd alloys than other methods reported.28,29
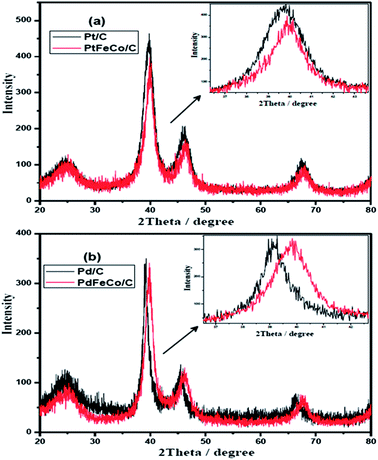 |
| | Fig. 1 XRD patterns of (a) Pt/C vs. PtFeCo/C and (b) Pd/C vs. PdFeCo/C electrocatalysts. | |
Table 1 Composition (ICP-OES and EDX), lattice constant, compressive strain (%), crystallite size (nm) and particle size (nm) of ternary alloys
| Electro catalyst |
Composition |
Lattice constant (XRD) (A°) |
Strain (XRD) (%) |
Lattice constant (SAED) (A°) |
Strain (SAED) (%) |
Crystallite size (nm) |
Particle size (nm) (TEM) |
| (ICP-OES) |
(EDX) |
| PtFeCo/C |
Pt72:Fe19:Co9 |
Pt75:Fe19:Co6 |
3.8972 |
0.88 |
3.9054 |
1.21 |
3.5 |
4.0 |
| PdFeCo/C |
Pd69:Fe20:Co11 |
Pd71:Fe20:Co9 |
3.9106 |
1.89 |
3.8878 |
2.95 |
5.0 |
7.5 |
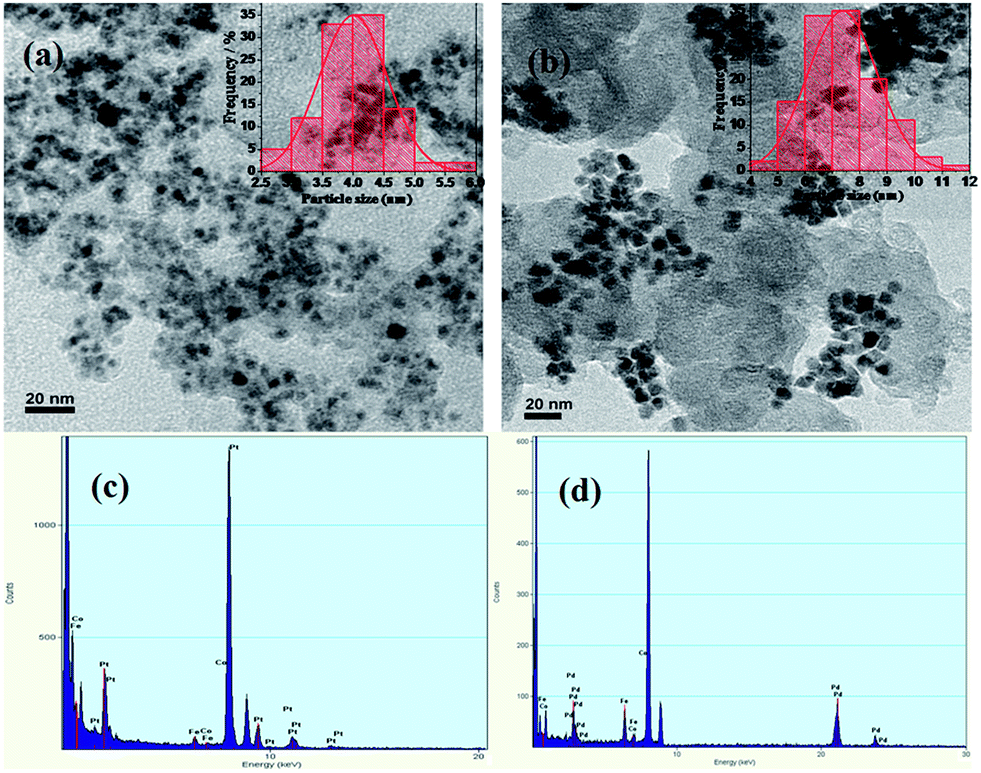
Particle size distribution and bulk composition were characterized by HR-TEM coupled with EDX which are shown in Fig. 2. The HR-TEM images were taken at 20 nm scales. TEM image of PtFeCo/C (Fig. 2a) shows that very fine particles are dispersed on carbon support. A histogram was made for a representative sample of 100 particles and the profile (inset) shows that the distribution is Gaussian and the most probable size of the particles was about 4.0 nm. PdFeCo/C image (Fig. 2b) shows the particle size of 7.5 nm (inset). SAED patterns (ESI, Fig. S1†) showed difference in lattice parameters of Pt and Pd on alloying with small sized metals, Fe and Co. From the diffraction images in Fig. 2 1/nm scale, the lattice parameter and the interplanar spacing of (111) plane was calculated from the diameter of intense (111) plane ring. The effective compressive strain was calculated from the lattice parameters. PtFeCO/C showed a compressive strain of 1.21% while PdFeCo/C registered a compressive strain of 2.95%. The strain increment trend in the cases of PtFeCo/C and PdFeCo/C were in tune with that from XRD studies. EDX spectrum reveals (Fig. 2c) the composition of Pt:Fe:Co as nearly 7:2:1 (75:19:6) atomic ratio, while Fig. 2d shows the composition of Pd:Fe:Co to be in the 7:2:1 (71:20:9) atomic ratio. This also confirms that solvothermal method is an efficient method to synthesize nano sized particles with controlled composition. Particle size obtained from TEM results match well with XRD results. As inferred from ICP-OES, the atomic composition of PtFeCo/C and PdFeCo/C in aqua regia solution are 72:19:9 and 69:20:11 respectively which matched well with those by EDX analysis.
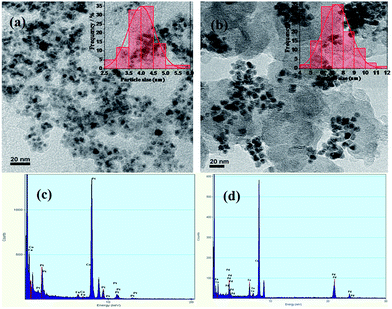 |
| | Fig. 2 HR-TEM image (a and b) with EDX spectra (c and d) of PtFeCo/C and PdFeCo/C electrocatalysts. | |
3.2. Electrochemical characterizations
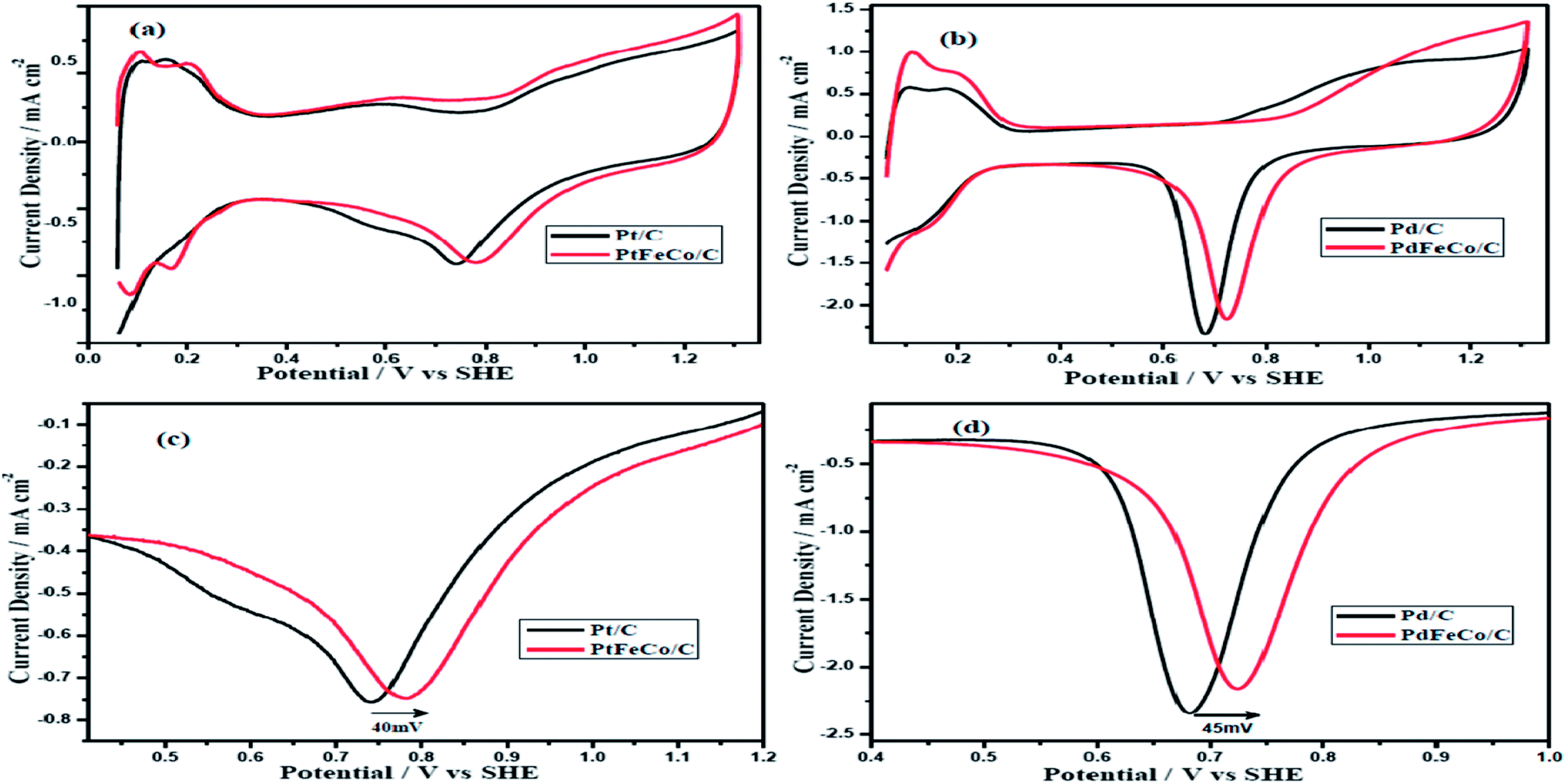
The electrochemical properties of the synthesized electrocatalysts, PtFeCo and PdFeCo trimetallic alloys were evaluated in 0.5 M H2SO4 solution by cyclic voltammetry (CV). Stabilized CV profiles were obtained after 25 continuous potential cycles in deaerated 0.5 M H2SO4 solution at a scan rate of 50 mV s−1. Fig. 3(a) and (b) show that CV plots have three distinct potential regions which are hydrogen adsorption/desorption region, double-layer region and surface oxide formation/reduction region. The electrochemical surface area (ECSA) of Pt-based electrocatalysts were calculated from the hydrogen adsorption/desorption region of the CV curves after correcting for the double layer charging current. The charge density to oxidize a monolayer of hydrogen on the Pt surface is 210 μC cm−2. The ECSA of Pd-based electrocatalyst was calculated by integrating the charge corresponding to reduction of surface Pd oxide using the charge density of 420 μC cm−2 for reduction of Pd oxide monolayer.30,31 The Integrated charge for hydrogen desorption on Pt and PtFeCo/C electrocatalysts were 239 μC, 247 μC respectively. Pt loading was 3.0 μg in Pt/C, 2.7 μg in PtFeCo/C. The oxide reduction charge of Pd/C and PdFeCo/C were 477 μC and 491 μC respectively. Pd loading of GC electrode was 3.0 μg in Pd/C and 2.4 μg in PdFeCo/C. The calculated ECSA of PtFeCo/C and PdFeCo/C were 44 m2 g−1 Pt and 49 m2 g−1 Pd respectively which were higher than that of Pt/C (38 m2 g−1 Pt) and Pd/C (38 m2 g−1 Pd). It reflects that alloying non-precious metals (Fe and Co) with precious metals (Pt and Pd) increased the ECSA. Increment in ECSA of PdFeCo/C was higher than that of PtFeCo/C.
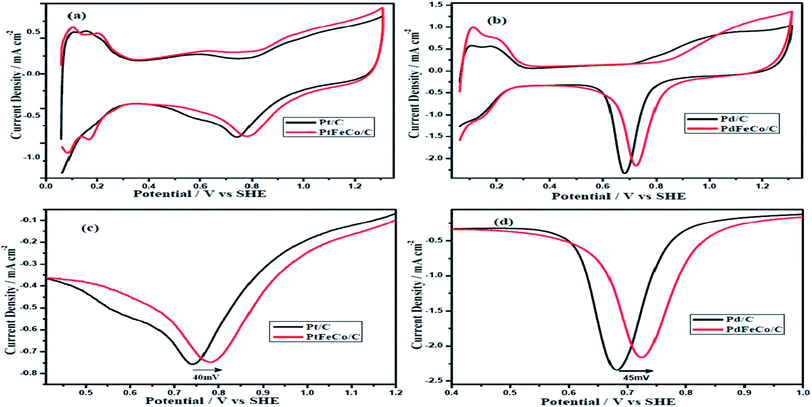 |
| | Fig. 3 Cyclic voltammograms of (a) PtFeCo/C and (b) PdFeCo/C catalysts in N2 saturated 0.5 M H2SO4 at a scan rate of 50 mV s−1. Enlarged oxide reduction peaks of (c) Pt and (d) Pd. | |
Fig. 3(c) and (d) show the enlarged CV curves of oxide reduction peaks of Pt and Pd alloys respectively. The curves reveal that Pt and Pd alloys have higher positive reduction potentials than Pt/C and Pd/C. The PtFeCo/C electrocatalyst has oxygen reduction potential at 0.782 V vs. SHE which is 40 mV higher than that of Pt/C (0.742 V). PdFeCo/C electrocatalyst has an oxygen reduction potential at 0.725 V which is 45 mV shifted in positive direction than Pd/C (0.680 V). On comparing PtFeCo/C and PdFeCo/C electrocatalysts, PdFeCo/C is having lower oxygen reduction potential. Comparison of the increments of oxygen reduction potential due to alloying shows higher for PdFeCo/C than for PtFeCo/C. These results strongly support that increasing strain could increase the ORR activity of the electrocatalysts by decreasing the adsorption energy of oxygenated species like OH.10
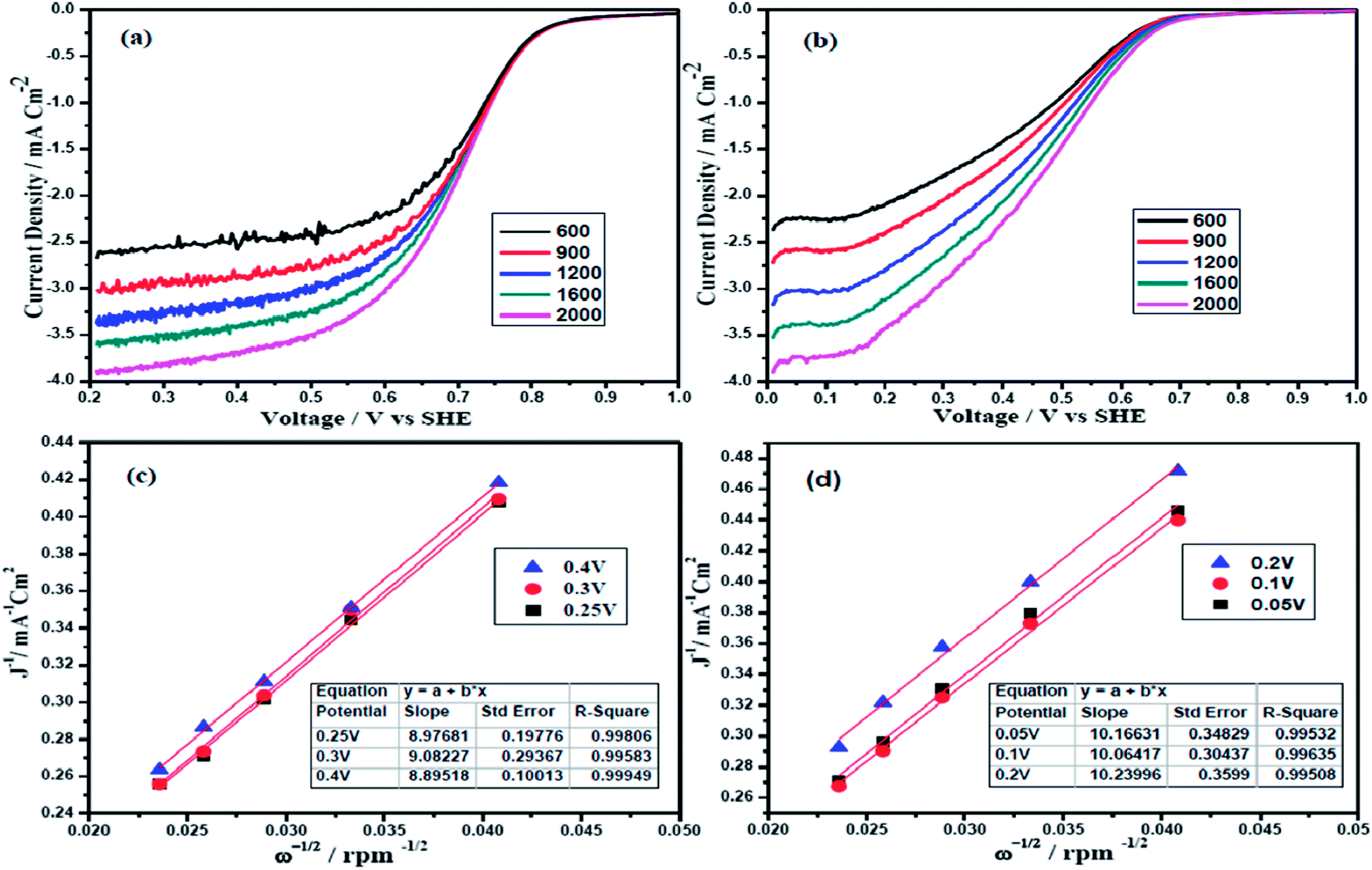
The electrocatalytic activity of the two alloys were also examined by the rotating disc electrode (RDE) technique. Fig. 4(a) and (b) show the polarization curves of Pt and Pd alloy electrocatalysts at various rotation rates from 600 to 2000 rpm in O2 saturated 0.5 M H2SO4 with a scan rate of 10 mV s−1. Potential sweep from 1.0 V to 0.0 V vs. SHE reflects well defined potential zones such as kinetic region where current density is independent of the electrode rotation rate, mixed kinetic and diffusion-controlled regions and diffusion-controlled region for both PtFeCo/C and PdFeCo/C electrocatalysts.32,33 The observed current density is normalized to the geometric area of the electrode in the respective curves. Overall current density obtained in ORR depends on the kinetic current density (jk), diffusion-limited current density (jd) and the diffusional current density in Nafion film (jf). The jf is not considered as significant while coating very thin Nafion film on catalyst (3 μl of 0.05 wt% Nafion) and hence ignored.34 According to the Koutecky–Levich eqn (2), the inverse of current density could be given as the sum of the inverses of the above current components. It could also be given in terms of jk, B and ω expressed in revolution per minute (rpm).
| | |
1/j = 1/jk + 1/jd + 1/jf = 1/jk + 1/Bω1/2
| (2) |
B is evaluated by
eqn (2a).
| | |
B = 0.2nFCO2(DO2)2/3ν−1/6
| (2a) |
where 0.2 is a constant.
n is the number of electrons transferred per mole of oxygen,
F is Faraday constant (96
485 C mol
−1),
CO2 is the bulk concentration of the oxygen (1.1 × 10
−6 mol cm
−3),
DO2 is the diffusion coefficient of oxygen in sulfuric acid (1.4 × 10
−5 cm
2 s
−1) and
ν is kinematic viscosity of sulfuric acid (1.0 × 10
−2 cm
−2 s
−1).
35,36 PtFeCo/C (
Fig. 4c) and PdFeCo/C (
Fig. 4d) electrocatalysts show the linear relationship of
j−1 vs. ω−1/2 and the parallelism of the
j−1 vs. ω−1/2 straight lines obtained at different potentials indicate the first order kinetics with respect to molecular oxygen. The slope of the straight line of
K–
L plots could be used to calculate the number of electrons involved in ORR. PtFeCo/C and PdFeCo/C show B values of 11.1 × 10
−2 mA cm
−2 rpm
−1/2 and 9.9 × 10
−2 mA cm
−2 rpm
−1/2 respectively and their corresponding
n values are 4.2 for PtFeCo/C and 3.7 for PdFeCo/C. The theoretical
B value calculated for 4-electron transfer process is 10.6 × 10
−2 mA cm
−2 rpm
−1/2.
35
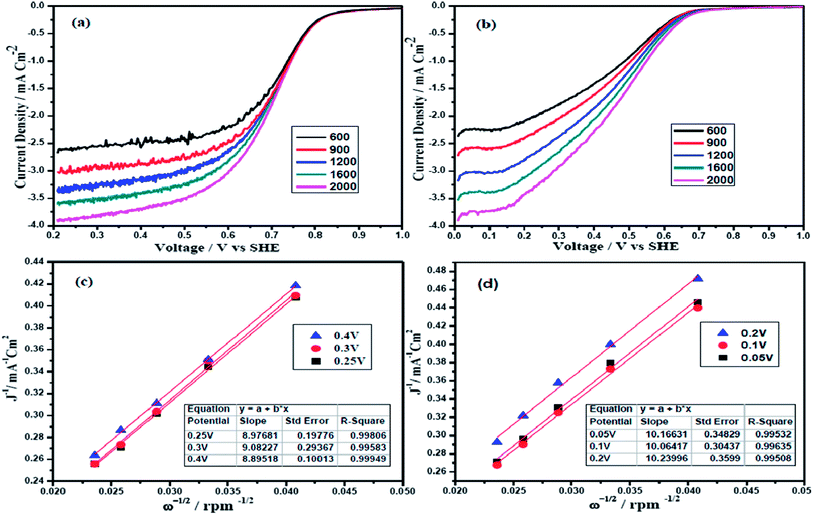 |
| | Fig. 4 Polarization curves of (a) PtFeCo/C and (b) PdFeCo/C electrocatalysts in O2 saturated 0.5 M H2SO4 at a scan rate of 10 mV s−1, K–L plots of (c) PtFeCo/C and (d) PdFeCo/C electrocatalysts. | |
The experimental B and ‘n’ values of both catalysts were very close to the theoretical values. This observation confirms that both PtFeCo/C and PdFeCo/C follow direct pathway for reduction of oxygen to water as shown below.
| | |
O2 + 4H+ + 4e− → 2H2O
| (3) |
Kinetic current density was calculated by the following eqn (4) derived from eqn (2),
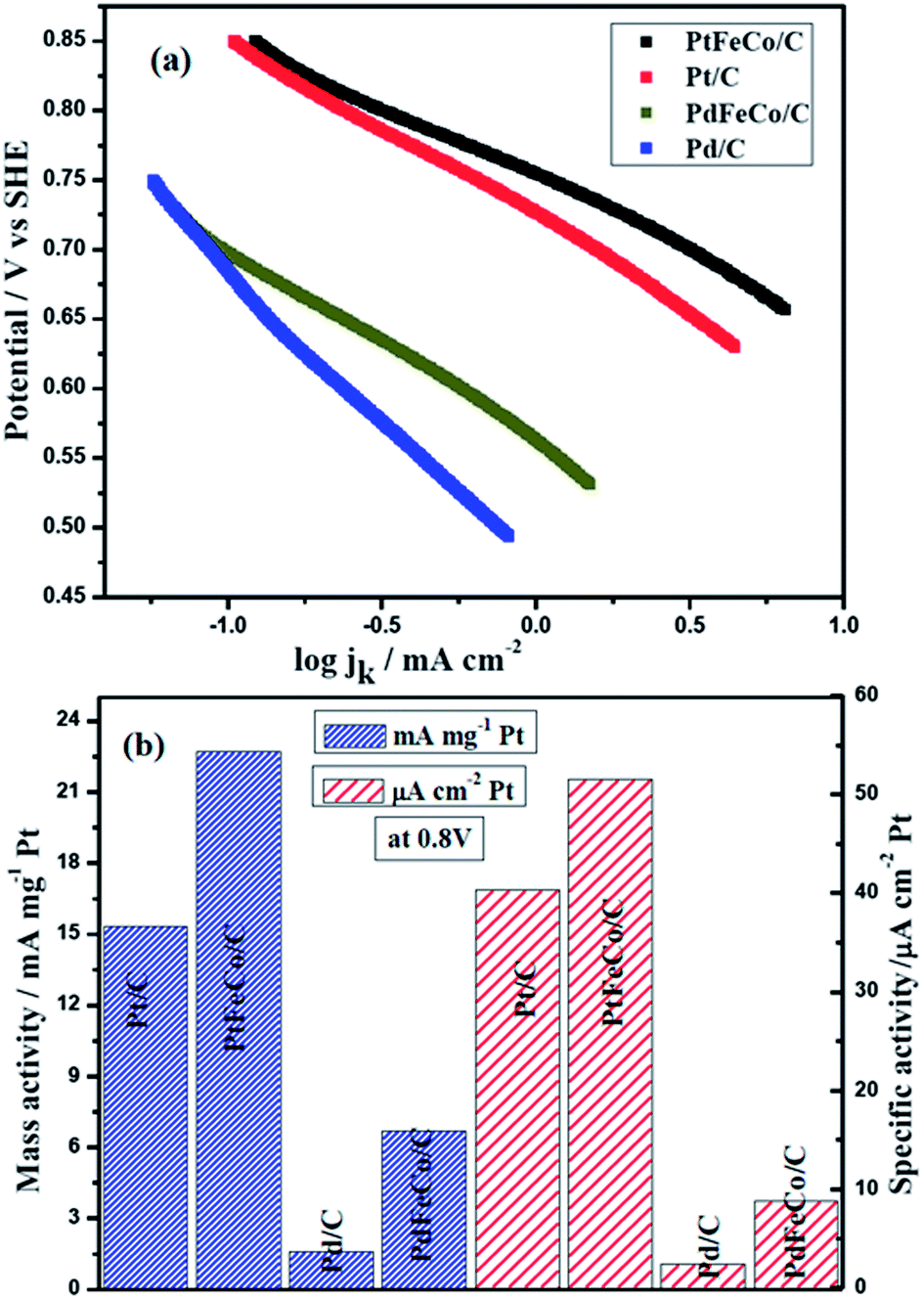
Mass transfer corrected Tafel plot was constructed (Fig. 5) from ORR data at 1600 rpm using Tafel equation η = a + blogj, where η is over potential, a = 2.303RT/αFlogjo, b = 2.303RT/αF, α is the transfer coefficient and jo is the exchange current density.37,38 From Fig. 5a, the Tafel slopes of Pt/C, PtFeCo/C and PdFeCo/C electrocatalysts were calculated as −129 mV dec−1, −109 mV dec−1and −137 mV dec−1 respectively. Pd/C shows Tafel slope value as −209 mv dec−1.39 All Tafel slopes showed good fit (R2 ≥ 0.996) in mixed kinetic and diffusion controlled region. Zhi-Pan Liu et al.,40 reported that the proton-coupled O–O bond breaking (H+ + e− + O2 (ad) → O + OH) is the major mechanism for ORR on Pt electrocatalyst. The redox nature of this mechanism leads to the strong potential dependence of the reaction and consequently a Tafel slope of −120 mV dec−1 at the low potentials. The Tafel slope of −120 mV dec−1 indicates that at low over potentials, the rate-determining step is one electron transfer step.41 Tafel slope values of −129 mV dec−1 for Pt/C and −109 mV dec−1 for PtFeCo/C are near to the −120 mV dec−1 which confirm that PtFeCo/C catalyst follows the same process of ORR as Pt/C. Since PtFeCo/C has a smaller Tafel slope value than Pt/C,42 it exhibits better oxygen reduction behavior than Pt/C. Pd has a slope value of −209 mV dec−1 which is higher than −120 mV dec−1. It indicates that the surface of Pd is not clean. This is due to strong adsorption of hydroxide anions and other oxygenated species on pure Pd surface and it implies changes in rate determining step.41 Influence of Fe and Co metals on PdFeCo/C electrocatalyst surface reduced the Tafel slope value in high current density region from −209 mV dec−1 (Pd/C) to −137 mV dec−1. The shape and slope of the Tafel plot of PdFeCo/C are very close to those of Pt/C electrocatalyst, which confirms that PdFeCo/C electrocatalyst also follows one electron transfer as rate determining step and four electron transfer pathway in ORR. The Tafel slope reflects that though Pd alloy has lesser ORR catalytic activity than Pt and its alloy, their kinetics are similar. It could be observed that alloying Fe and Co to Pd produced more effect on slope value in high current density region than that of Pt alloy with Fe and Co. This result also reflects that higher compressive strain of PdFeCo/C than that of PtFeCo/C causes higher change in ORR kinetics. Fig. 5b shows mass activity and specific activity calculated using kinetic current divided by mass of Pt or Pd and mass activity divided by real surface area of the catalyst at 0.80 V respectively. From this plot it could be inferred that mass activity of PtFeCo/C increased by 48% over that of Pt/C. But in PdFeCo/C, it is 4.1 times higher than Pd/C mass activity. The specific activity of PtFeCo/C increased by 28% over that of Pt/C, while that of PdFeCo/C was 3.6 times higher than that of Pd/C. The mass activity of Pt alloy (Pt72Fe19Co9/C) reported in this study is 220 mA mg−1 at 0.7 V which is much higher than that reported for Pt78Fe15Co7/C synthesised by electrodeposition method.43 Kinetic parameters elucidated from Tafel slope such as kinetic current, mass and specific activities are listed in Table 2. Mass and specific activity histogram (Fig. 5b) show that PdFeCo/C produced greater changes in ORR activity than PtFeCo/C while the mass activities and specific activities of Pt and Pd alloy electrocatalysts were higher than that of pure Pt and Pd electrocatalysts respectively. Based on the mass and specific activity, the overall ORR performance of the electrocatalysts may be summarized in the following order: PtFeCo/C > Pt/C > PdFeCo/C > Pd/C.
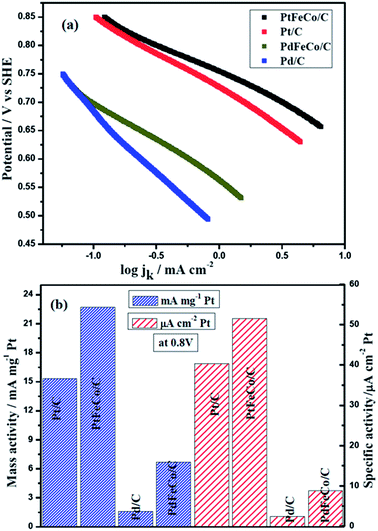 |
| | Fig. 5 (a) Tafel plots of PtFeCo/C, Pt/C, PdFeCo/C and Pd/C electrocatalyst in O2 saturated 0.5 M H2SO4 at 1600 rpm. (b) Mass activity and specific activity of Pt/C, PtFeCo/C, Pd/C and PdFeCo/C electrocatalysts in terms of kinetic current (ik) at 0.80 V. | |
Table 2 K–L slope, B constant, number of electrons (n), Tafel slope, electron transfer coefficient (α), half wave potential (E1/2), mass activity (MA), specific activity (SA) and fuel cell power density
| Electro catalyst |
K–L slope (mA−1 cm2 rpm1/2) |
B × 10−2 (mA cm−2 rpm−1/2) |
n |
Tafel slope (mV dec−1) |
α |
E1/2 (V) |
MA (0.8 V) (mA mgPt−1) |
SA (0.8 V) (μA cmPt−2) |
Power density at 80 °C (mW cm−2) |
| Pt/C |
9.17 |
10.9 |
4.1 |
−129 |
0.46 |
0.646 |
15.3 |
40.3 |
666 |
| PtFeCo/C |
8.98 |
11.1 |
4.2 |
−109 |
0.55 |
0.692 |
22.7 |
51.5 |
713 |
| Pd/C |
10.50 |
9.5 |
3.4 |
−209 |
0.22 |
0.339 |
1.6 |
2.4 |
413 |
| PdFeCo/C |
10.15 |
9.9 |
3.7 |
−137 |
0.44 |
0.441 |
6.7 |
8.8 |
437 |
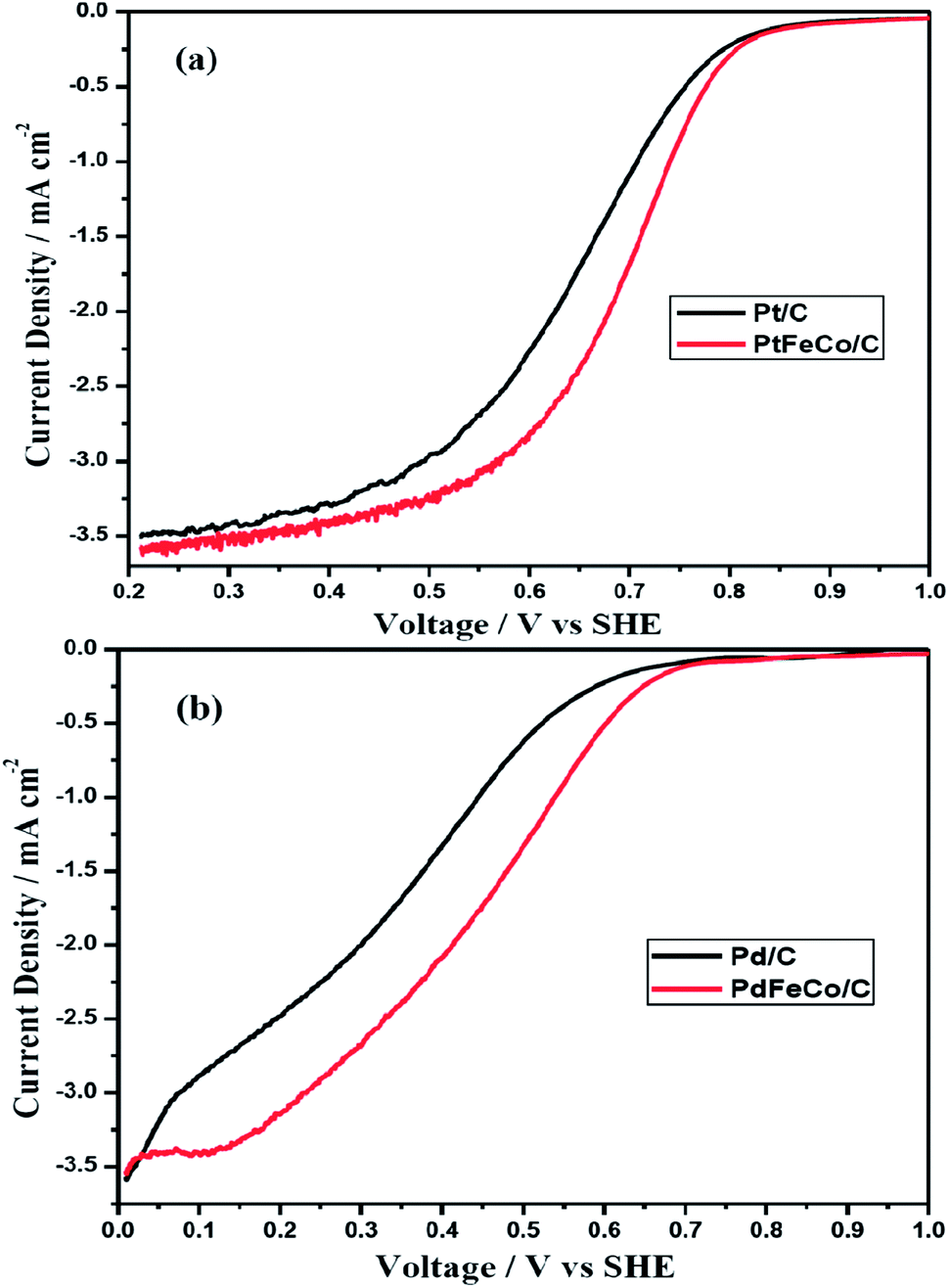
Half wave potential (E1/2) of PtFeCo/C (Fig. 6a) is 46 mV higher than that of Pt/C while the half wave potential of PdFeCo/C (Fig. 6b) is 102 mV higher than that of Pd/C electrocatalyst. This result is supported by the results obtained from CV of PtFeCo/C and PdFeCo/C in which the oxygen reduction potential shifts are higher in the case of PdFeCo/C. The half wave potentials are listed in Table 2. Summarizing the results of Tafel slope, mass and specific activities and half wave potential, it is confirmed that increased compressive strain between active metal atoms has enhanced the ORR activity. High compressive strain of PdFeCo/C results in higher increment in ORR activity than PtFeCo/C. The analysis of electrocatalysts by RDE technique implies that based on E1/2 value catalytic activity for ORR is in the order, PtFeCo/C > Pt/C > PdFeCo/C > Pd/C.
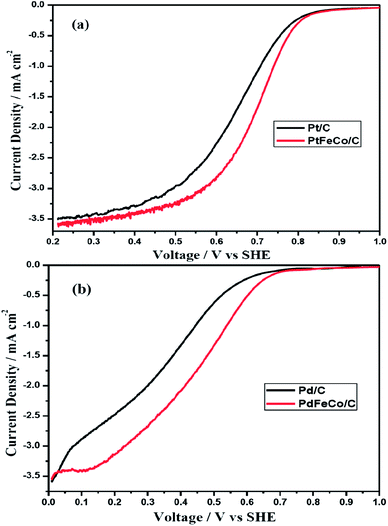 |
| | Fig. 6 Polarization curves of (a) Pt/C and PtFeCo/C, and (b) Pd/C and PdFeCo/C electrocatalysts at 1600 rpm in O2 saturated 0.5 M H2SO4. | |
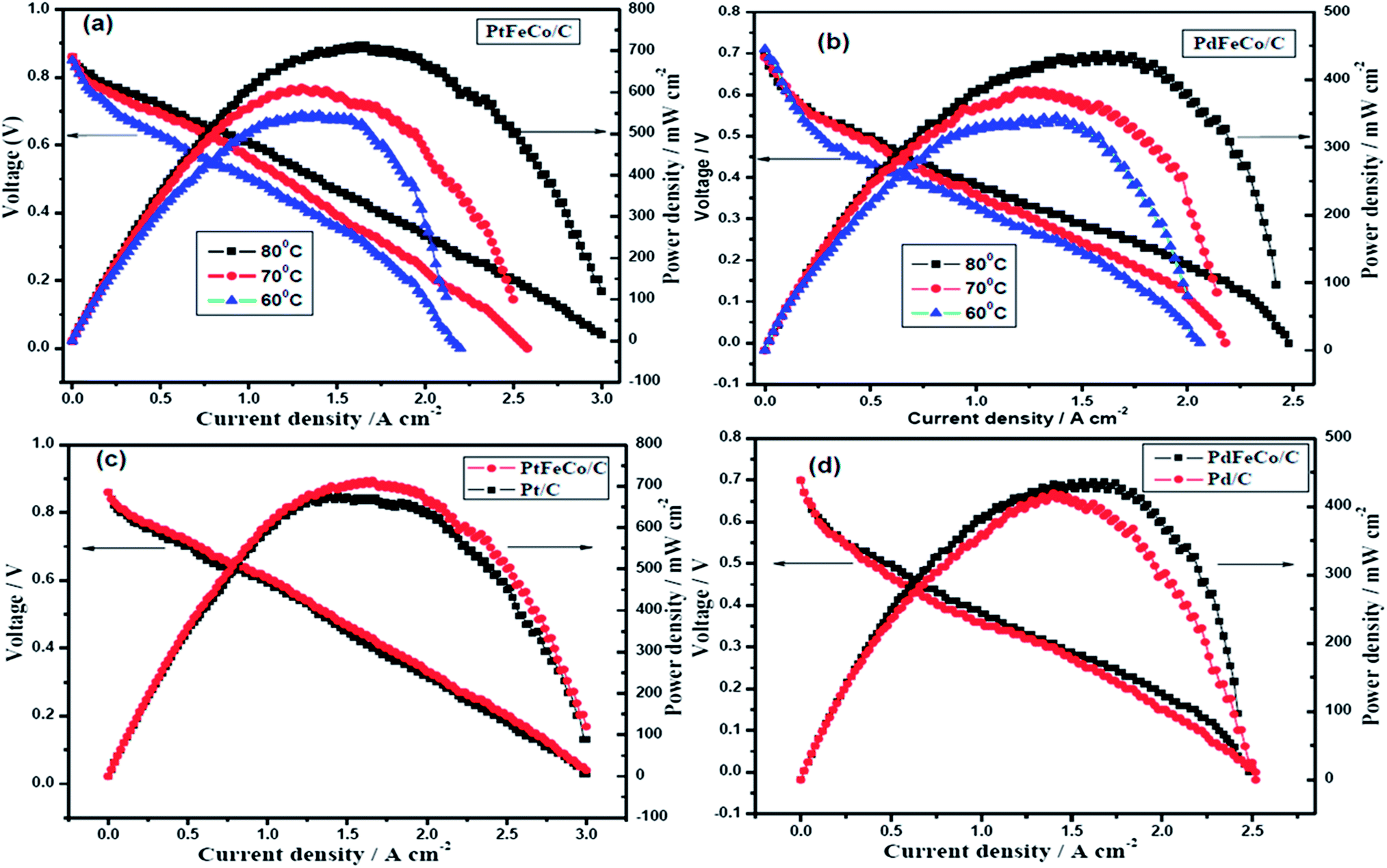
For PEM fuel cell evaluation, the MEA was fabricated using PtFeCo/C, PdFeCo/C, Pd/C electrocatalysts synthesized, and commercial Pt/C electrocatalyst for ORR at the cathode. Commercial Pt/C was used as anode catalyst for all the MEAs. Cathode and anode catalysts were homogenously coated on either side of the Nafion membrane by spray coating, followed by sandwich pressing of the GDLs and catalyst layers to form the MEA. H2/O2 gases were fed to the cell through the humidifier to keep the Nafion membrane well hydrated. Before polarization, the single cell was activated between OCP and higher current density.44 Fig. 7a shows the polarization curves of PtFeCo/C catalyst at different temperatures. The maximum power densities of 713 mW cm−2 at 80 °C, 606 mW cm−2 at 70 °C and 538 mW cm−2 at 60 °C clearly show that increasing cell temperature enhances the catalytic performance of the catalyst. PdFeCo/C (Fig. 7b) delivers maximum power density of 437 mW cm−2 at 80 °C, 380 mW cm−2 at 70 °C and 338 mW cm−2 at 60 °C. The maximum power density of PdFeCo/C (338 mW cm−2) at 60 °C is higher than those previously reported for Pd/C.29,34,42,45
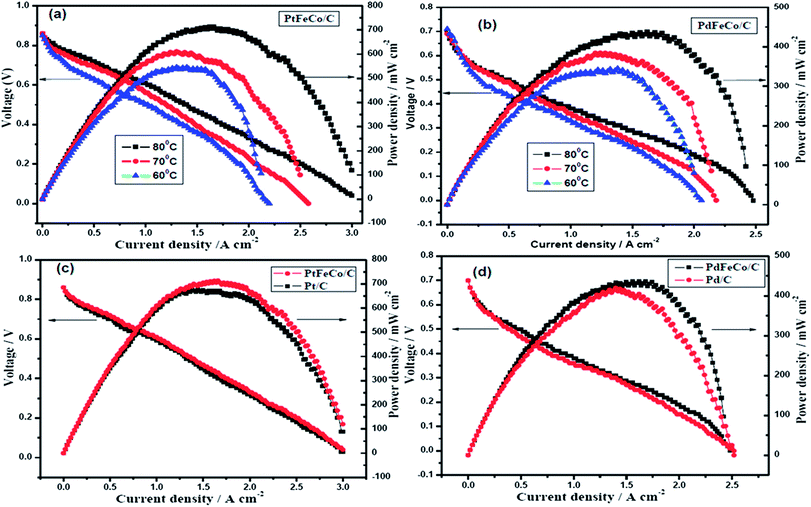 |
| | Fig. 7 Polarization curves for (a) PtFeCo/C (b) PdFeCo/C electrocatalysts as cathode with commercial Pt/C catalyst as anode, operated with humidified H2/O2 gases at different temperatures (60 °C, 70 °C, and 80 °C) without any back pressure. | |
Comparison of the polarization curves of Pt and Pd based alloys with their respective standards (Pt/C and Pd/C) at 80 °C are shown in Fig. 7(c) and (d). PtFeCo/C and PdFeCo/C deliver maximum power densities at 0.43 V and 0.27 V respectively. Pt/C cathode shows power density of 666 mW cm−2 at 80 °C which is 47 mW cm−2 lower than for PtFeCo/C. Pd/C has power density of 413 mW cm−2 which is 24 mW cm−2 lower than for PdFeCo/C. The enhancement in fuel cell performance for alloys is slightly lesser than those expected from their mass and specific activity increases. As many parameters such as humidity, gas supply to the reaction centres and water managements impact the fuel cell performance, optimization of all those conditions would reflect the real capacity of the alloys synthesized in service condition and the same is in progress. The single cell performance results fully supported the RDE results in ordering the electrocatalysts in their catalytic activity as PtFeCo/C > Pt/C > PdFeCo/C > Pd/C.
4. Conclusion
Carbon supported PtFeCo, PdFeCo and Pd electrocatalysts were synthesized by solvothermal method. XRD study of these Pt and Pd based alloys and pure metals indicated the single phase face centered cubic structure. TEM revealed the particle sizes of 4 nm (PtFeCo/C) and 7.5 nm (PdFeCo/C). Alloying changed the lattice constants of PtFeCo/C and PdFeCo/C from those of pure metals Pt and Pd respectively. PdFeCo/C showed more compressive strain value than PtFeCo/C. Cyclic voltammogram of PdFeCo/C reflected higher increment in oxide reduction potential than PtFeCo/C. Results of ORR inferred that both alloys follow four electron transfer mechanisms and one electron transfer rate determining step. Mass activity of PtFeCo/C increased by 48% over that of Pt/C. But PdFeCo/C showed 4.1 times higher value than that of Pd/C mass activity. The specific activity of PtFeCo/C increased by 28% over Pt/C. But PdFeCo/C showed 3.6 times higher activity than that of Pd/C. PdFeCo/C shows enhanced increment in half wave potential (E1/2) than PtFeCo/C. Single cell polarization was carried out for PtFeCo/C and PdFeCo/C electrocatalysts at cathode for oxygen reduction reaction at 60, 70, and 80 °C. They showed improved performance than pure metal catalysts (Pt and Pd). At 80 °C PtFeCo/C has a peak power density of 713 mW cm−2 and PdFeCo/C has peak power density of 437 mW cm−2. Results of structural and ORR characterizations imply that enhancement of ORR activity is directly related to the lattice strain. It could be highlighted that Pt metal has higher density than Pd, and hence shows less compressive strain and less shrinking on alloying as revealed by the lattice constant of PtFeCo/C.
References
- A. Serov and C. Kwak, Appl. Catal., B, 2009, 90, 313 CrossRef CAS PubMed.
- F. Bin, L. Jin, N. Peter, L. Rameshwori, M. Derrick, W. Bridgid, H. Xiang and Z. Chuan-Jian, Electrochem. Commun., 2009, 11, 1139 CrossRef PubMed.
- G. Hoogers, Fuel Cell Technology Handbook, CRC Press, Boca Raton, FL, 2003 Search PubMed.
- T. Toda, H. Igarashi, H. Uchida and M. Watanabe, J. Electrochem. Soc., 1999, 146, 3750 CrossRef CAS PubMed.
- E. Antolini, R. Passos and A. Ticianelli, Electrochim. Acta, 2002, 48, 263 CrossRef CAS.
- P. Mani, R. Srivastava and P. Strasser, J. Power Sources, 2011, 196, 666 CrossRef CAS PubMed.
- M. Oezaslan, F. Hasche and P. Strasser, J. Electrochem. Soc., 2012, 159, B444 CrossRef CAS PubMed.
- H. Yang, N. Alonso-Vante, M. Leger and C. Lamy, J. Phys. Chem. B, 2004, 108, 1938 CrossRef CAS.
- F. Bin, W. Bridgid, Y. Jun, L. Rameshwori, L. Jin, H. Xiang, L. Jordan and Z. Chuan-Jian, Int. J. Hydrogen Energy, 2012, 37, 4627 CrossRef PubMed.
- W. Bridgid, L. Rameshwori, L. Jin, N. Peter, M. Derrick and Z. Chuan-Jian, J. Phys. Chem. C, 2010, 114, 17580 Search PubMed.
- M. Vracar, D. B. Sepa and A. Damjanovic, J. Electrochem. Soc., 1986, 133, 1835 CrossRef PubMed.
- C. Xu, Y. Liu, Q. Hao and H. Duan, J. Mater. Chem. A, 2013, 1, 13542 CAS.
- J. L. Fernandez, V. Raghuveer, A. Manthiram and A. J. Bard, J. Am. Chem. Soc., 2005, 127, 13100 CrossRef CAS PubMed.
- W. E. Mustain, K. Kepler and J. Prakash, Electrochem. Commun., 2006, 8, 406 CrossRef CAS PubMed.
- M. H. Shao, K. Sasaki and R. R. Adzic, J. Am. Chem. Soc., 2006, 128, 3526 CrossRef CAS PubMed.
- X. Wang, N. Kariuki, J. T. Vaughey, J. Goodpaster, R. Kumar and D. J. Myers, J. Electrochem. Soc., 2008, 155, B602 CrossRef CAS PubMed.
- V. Raghuveer, A. Manthiram and A. J. Bard, J. Phys. Chem. B, 2005, 109, 22909 CrossRef CAS PubMed.
- N. M. Markovic, T. J. Schmidt, V. Stamenkovic and P. N. Ross, Fuel Cells, 2001, 1, 105 CrossRef CAS.
- Y. Bing, H. Liu, L. Zhang, D. Ghosh and J. Zhang, Chem. Soc. Rev., 2010, 39, 2184 RSC.
- Y. C. Wei, C. W. Liu, Y. W. Chang, C. M. Lai, P. Y. Lim, L. D. Tsai and K. W. Wang, Int. J. Hydrogen Energy, 2010, 35, 1864 CrossRef CAS PubMed.
- Y. Chen, F. Yang, Y. Dai, W. Wang and S. Chen, J. Phys. Chem. C, 2008, 112, 1645 CAS.
- Y. Xing, Y. Cai, M. B. Vukmirovic, W. P. Zhou, H. Karan, J. X. Wang and R. R. Adzic, J. Phys. Chem. Lett., 2010, 1, 3238 CrossRef CAS.
- J. X. Wang, H. Inada, L. Wu, Y. Zhu, Y. Choi, P. Liu, W. P. Zhou and R. R. Adzic, J. Am. Chem. Soc., 2009, 131, 17298 CrossRef CAS PubMed.
- Y. Suo, L. Zhuang and J. Lu, Angew. Chem., Int. Ed., 2007, 46, 2862 CrossRef CAS PubMed.
- Y. Xu, A. V. Ruban and M. Mavrikakis, J. Am. Chem. Soc., 2004, 126, 4717 CrossRef CAS PubMed.
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. M. Markovic, J. Rossmeisl, J. Greeley and J. K. Norskov, Angew. Chem., 2006, 118, 2963 CrossRef.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. Yu, Z. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nature, 2010, 2, 454 CAS.
- K. G. Nishanth, P. Sridhar and S. Pitchumani, Int. J. Hydrogen Energy, 2013, 38, 612 CrossRef CAS PubMed.
- Y. Liang, H. Zhang, Z. Tian, X. Zhu, X. Wang and B. Yi, J. Phys. Chem. B, 2006, 110, 7828 CrossRef CAS PubMed.
- C. Xu, Y. Liu, Q. Hao and H. Duan, J. Mater. Chem. A, 2013, 1, 13542 CAS.
- W. Pan, X. Zhang, H. Ma and J. Zhang, J. Phys. Chem. C, 2008, 112, 2456 CAS.
- J. Zhao, A. Sarkar and A. Manthiram, Electrochim. Acta, 2010, 55, 1756 CrossRef CAS PubMed.
- C. V. Rao and B. Viswanathan, J. Phys. Chem. C, 2009, 113, 18907 Search PubMed.
- D. C. Martinez-Casillas, G. Vazquez-Huert, J. F. Perez-Robles and O. Solorza-Feri, J. Power Sources, 2011, 196, 4468 CrossRef CAS PubMed.
- J. J. Salvador-Pascual, S. Citalan-Cigarroa and O. Solorza-Feria, J. Power Sources, 2007, 172, 229 CrossRef CAS PubMed.
- J. Zeng, S. Liao, J. Y. Lee and Z. Liang, Int. J. Hydrogen Energy, 2010, 35, 942 CrossRef CAS PubMed.
- T. Cochell and A. Manthiram, Langmuir, 2012, 28, 1579 CrossRef CAS PubMed.
- L. Wang, L. Zhang and J. Zhang, Electrochim. Acta, 2011, 56, 5488 CrossRef CAS PubMed.
- H. Erikson, A. Sarapuu, N. Alexeyeva, K. Tammeveski, J. Solla-Gullon and J. M. Feliu, Electrochim. Acta, 2012, 59, 329 CrossRef CAS PubMed.
- G. F. Wei, Y. H. Fang and Z. P. Liu, J. Phys. Chem. C, 2012, 116, 12696 CAS.
- K. Zhang, Q. Yue, G. Chen, Y. Zhai, L. Wang, H. Wang, J. Zhao, J. Liu, J. Jia and H. Li, J. Phys. Chem. C, 2011, 115, 379 CAS.
- D. C. Martinez-Casillas, H. A. Calderon, V. Collins-Martinez and O. Solorza-Feria, Int. J. Hydrogen Energy, 2012, 38, 12674 CrossRef PubMed.
- S. J. Hwang, S. J. Yoo, S. Jang, T. H. Lim, S. A. Hong and S. K. Kim, J. Phys. Chem. C, 2011, 115, 2483 CAS.
- B. P. Vinayan, R. Imran Jafri, R. Nagar, N. Rajalakshmi, N. Sethupathi and S. Ramaprabhu, Int. J. Hydrogen Energy, 2012, 37, 412 CrossRef CAS PubMed.
- J. Moreira, P. del Angel, A. L. Ocampo, P. J. Sebastioan, J. A. Montoya and R. H. Castellanos, Int. J. Hydrogen Energy, 2004, 29, 915 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SAED pattern images of Pt/C, PtFeCo/C, Pd/C and PdFeCo/C. See DOI: 10.1039/c3ra47021k |
|
| This journal is © The Royal Society of Chemistry 2014 |