Phosphorus-doped graphene nanosheets as efficient metal-free oxygen reduction electrocatalysts†
Rong
Li
ab,
Zidong
Wei
*a,
Xinglong
Gou
*b and
Wei
Xu
c
aState Key Laboratory of Power Transmission Equipment & System Security and New Technology, College of Chemistry and Chemical Engineering, Chongqing University, Chongqing 400044, China. E-mail: zdwei@cqu.edu.cn
bChemical Synthesis and Pollution Control Key Laboratory of Sichuan Province, College of Chemistry and Chemical Engineering, China West Normal University, Nanchong 637000, China. E-mail: gouxlr@126.com
cNingbo Institute of Material Technology & Engineering, Chinese Academy of Science, Ningbo 315201, China
First published on 12th April 2013
Abstract
Metal-free phosphorus-doped graphene nanosheets (P-TRG) with large surface area (496.67 m2 g−1) and relatively high P-doping level (1.16 at.%) were successfully prepared by thermal annealing a homogenous mixture of graphene oxide and 1-butyl-3-methlyimidazolium hexafluorophosphate under argon atmosphere. It was found that the P atoms were substitutionally incorporated into the carbon framework and were partially oxidized, which created new active sites for the oxygen reduction reaction (ORR). Accordingly, the ORR catalytic performance of the P-doped graphene was demonstrated to be better than or at least comparable to that of the benchmark Pt/C catalyst.
1. Introduction
The electrocatalyst for the cathodic oxygen reduction reaction (ORR) is a crucial component of a fuel cell, which determines the energy conversion efficiency, operational cost and durability of the cell.1 Currently, the most effective ORR electrocatalysts are platinum-based precious metals, but they suffer from multiple drawbacks such as slow electron transfer kinetics, high price, poor durability, severe methanol crossover effect and CO deactivation. Accordingly, numerous efforts have been devoted to the search for various alternative ORR electrocatalysts such as non-precious metals, oxides, chalcogenides, complexes, biomolecules, polymers and metal-free carbon nanomaterials.2–4 Among these candidates, heteroatom-doped nanocarbons (mesoporous carbon, nanotubes, graphene and xerogels) show the distinct advantages of low cost, long-term stability, and excellent tolerance to the crossover effect.5,6 However, their catalytic activity is still far from satisfactory for practical applications.As the mother of all carbon allotropes, graphene, a two-dimensional monolayer structure of sp2-hybridized carbon, has recently become an attractive candidate for catalysis and energy applications thanks to its extraordinary conductivity, high surface area and excellent mechanical properites.7 Theoretical calculations and experimental observations reveal that doping graphene with foreign atoms and molecules can tailor its electronic properties and chemical reactivity, including ORR activity.8 Most recently, substitutional doping of graphene with monatomic or dual heteroatoms, such as nitrogen,9 boron,10 sulfur,11 and iodine,12 has been intensively investigated for metal-free ORR electrocatalysts with enhanced electrochemical performance. In particular, the electrocatalytic activity and durability of the N-doped graphene synthesized by chemical vapor deposition of methane in the presence of ammonia or by heat treatment of thermally reduced graphene oxide using ammonia were found to be comparable to or better than that of the commercial Pt/C.13 These exciting results encouraged researchers to explore phosphorus doped carbon materials as potential ORR electrocatalysts because of the similarity of nitrogen and phosphorus in structure and chemical properites.14–17 Liu et al. synthesized P-doped graphite layers by pyrolysis of toluene and triphenylphosphine (TPP) and demonstrated that the P-doped graphite could be used as a metal-free ORR electrocatalyst in alkaline medium.14 Nevertheless, the reported electrocatalytic activity was worse than that of Pt/C because of the low P-doping level (0.26 at.%) and the very low surface area of the material (<4 m2 g−1). In contrast, P-doped nanocarbons (including multiwalled carbon nanotubes,15 nanospheres16 and ordered mesoporous carbon17) with high electroactive surface area exhibited much higher catalytic activity for O2 reduction. However, to the best of our knowledge, the preparation of metal free P-doped graphene nanosheets for application as ORR catalysts has never been reported.
In this work, metal free P-doped graphene nanosheets were facilely synthesized by annealing a homogeneous mixture of graphene oxide and an ionic liquid 1-butyl-3-methlyimidazolium hexafluorophosphate (BmimPF6), which was employed as a mild phosphorus source for in situ doping the thermally reduced graphene oxide (TRG). This novel ionic liquid assisted one-step route is very simple but scalable for simultaneous phosphorus doping and reduction of graphene oxide. The resultant P-doped graphene nanosheets (designated as P-TRG) combine the merits of large surface area (496.67 m2 g−1) and relatively high P-doping level (1.16 at.%), and are first demonstrated to be an effective ORR catalyst with enhanced catalytic activity, long-term stability, high selectivity for the four-electron reduction of dioxygen, and excellent tolerance to methanol and CO poisoning in alkaline media, outperforming or at least comparable to the commercial Pt/C catalyst.
2. Experimental section
2.1 Synthesis of graphene oxide (GO)
GO was prepared from colloidal graphite powders according to a modified Hummers method as reported elsewhere.18 In a typical procedure, 10 g of graphite and 5 g of NaNO3 were added under stirring into 230 mL of concentrated H2SO4, which was placed in a flask and cooled in an ice-water bath. 30 g of KMnO4 was then slowly added into the flask with vigorous stirring. Subsequently, the ice-water bath was replaced by a warm-water bath with a temperature of 35 °C, and the reaction mixture was stirred vigorously for 15 h to form a thick paste. Whereafter, the water bath was removed, and 460 mL of distilled water was added, causing a rapid increase in temperature near to 100 °C. After stirring for another hour, 20 mL of H2O2 (30%) and 700 mL of distilled water were sequentially added, resulting in a yellow dispersion. A yellow product (GO) was collected by centrifugation, followed by repeatedly washing with diluted HCl (1 M) until sulfates were no longer detected in the filtrate. Finally, GO was subjected to dialysis for one week to remove residual salts and acids, followed by drying under vacuum at 45 °C for 4 days.2.2 Synthesis of P-doped graphene nanosheets (P-TRG)
P-TRG was synthesized by thermally annealing a hybrid of GO and 1-butyl-3-methlyimidazolium hexafluorophosphate (BmimPF6). In a typical procedure, 70 mg BmimPF6 was dissolved in 10.0 mL alcohol–water solution with a volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, followed by adding 70 mg of GO powder. The mixture was first dispersed for 20 min in a general ultrasonic cleaning bath, and was then dried at 45 °C in vacuum for two days. The resulting GO/BmimPF6 composite was heated in a furnace under argon flow to simultaneously reduce the GO and dope it with phosphorus. The temperature was ramped at 10 °C min−1 to 550 °C and maintained for two hours. Afterwards, it was further ramped to 1000 °C at the same rate and was kept for another hour. After cooling down, the black P-TRG powder was obtained. Graphene nanosheets without P-dopant (TRG) were also prepared for comparison through the same procedure in the absence of BmimPF6.
:1, followed by adding 70 mg of GO powder. The mixture was first dispersed for 20 min in a general ultrasonic cleaning bath, and was then dried at 45 °C in vacuum for two days. The resulting GO/BmimPF6 composite was heated in a furnace under argon flow to simultaneously reduce the GO and dope it with phosphorus. The temperature was ramped at 10 °C min−1 to 550 °C and maintained for two hours. Afterwards, it was further ramped to 1000 °C at the same rate and was kept for another hour. After cooling down, the black P-TRG powder was obtained. Graphene nanosheets without P-dopant (TRG) were also prepared for comparison through the same procedure in the absence of BmimPF6.
2.3 Structural characterizations
The structure and morphology of the samples were characterized by X-ray diffraction (XRD, Rigaku Ultima IV X-ray diffractometer with Cu-Kα radiation), scanning electron microscopy (SEM, Hitachi S-4800) coupled with energy-dispersive X-ray spectroscopy (EDS, Oxford instruments X-Max), and transmission electron microscopy (TEM, FEI Tecnai F20 microscope operated at 200 kV). The elemental components were analyzed by X-ray photoelectron spectroscopy (XPS, Shimazu-Kratos AXIS UTLTRA DLD using monochromated Al-Kα X-ray source). The binding energy was referenced to the C1s line at 284.6 eV for calibration. Raman spectra were recorded by a Renishaw inVia-reflex instrument with a 532 nm excitation laser. The specific area and pore size distribution of the sample were measured by a Micromeritics ASAP 2020M with N2 as absorbate at 77 K. Before measurement, the samples were degassed in a vacuum at 200 °C for 3 h. The specific surface area was calculated using the Brunauer–Emmet–Teller (BET) method from the nitrogen adsorption data. The pore size distribution plots were derived from the adsorption branch of the isotherms based on the Barrett–Joyner–Halenda (BJH) model.2.4 Electrochemical measurements
The electrocatalytic performance of the catalysts was evaluated by cyclic voltammograms (CV), linear sweep voltammograms (LSV) and chronoamperometry in a three-electrode electrochemical cell which was connected to a computer-controlled potentiostat (CHI 760D, CH Instrument, Shanghai) coupled with a rotating disk electrode (RDE) system (ATA-1B, Jiangfen electroanalytical Instrument Co. Ltd., China). A platinum wire and a saturated calomel electrode (SCE) were used as the counter electrode and the reference electrode, respectively. A bare glassy carbon electrode (GCE, 3 mm in diameter) or modified GCE with the studied material was used as the working electrode. Prior to use, the GCE was hand-polished with chamois leather containing 0.05 μm alumina slurry to obtain a mirror-like surface, washed with ethanol and distilled water by sonication for 5 min and allowed to dry. The modified GCE was prepared by casting a 5 μL aliquot of the catalyst ink, which was obtained by ultrasonically dispersing 1 mg of the catalyst including P-TRG, TRG, or commercial Pt/C (20 wt% Pt on Vulcan XC-72R) into Nafion solution (0.5 mL, 0.05 wt% in alcohol), onto the fresh surface of the pretreated GCE electrode and dried naturally to form a uniform thin film. The loading amount of each catalyst was kept to be 141.5 μg cm−2. The experiments were carried out at room temperature in 0.1 M KOH solution, which was purged with high purity argon or oxygen with a flow rate of 20 mL min−1 for at least 30 min prior to each measurement. All potentials were measured and reported vs. the SCE reference electrode.3. Results and discussion
3.1 Composition and structural characteristics of the catalyst
GO sheets derived from the oxidization and exfoliation of graphite are usually employed to produce graphene and its derivatives on a large scale by chemical reduction or thermal annealing reduction.19 For example, N-doped graphene nanosheets were prepared by annealing GO in the presence of N-containing precursors such as NH3, polyaniline, and polypyrrole.20 Herein, BmimPF6 was chosen as the P-containing precursor for the synthesis of P-doped graphene, because BmimPF6 is one of the most common ionic liquids with nonvolatility and high thermal stability. Furthermore, GO powders can be easily dispersed with high concentration in the solution of BmimPF6–ethanol–water. After removal of the water and ethanol, a homogenous composite of GO and BmimPF6 was obtained. During the subsequent thermal annealing process, PF6− is expected to react with oxygen-containing groups on the plan and edge of the GO sheets to join into the carbon framework,21 resulting in simultaneous phosphorus doping and reduction of graphene oxide as revealed by the XRD, TEM and EDS analyses in Fig. 1. In comparison to the XRD pattern of the TRG sample, the diffraction peaks of the P-TRG (Fig. 1a) are broader and weaker, indicating that doping of graphene with P atoms can induce more defect sites or disorders in the graphitic structure due to the big difference of atomic radius between phosphorus and carbon, and thus results in a decrement in crystallinity for the P-TRG.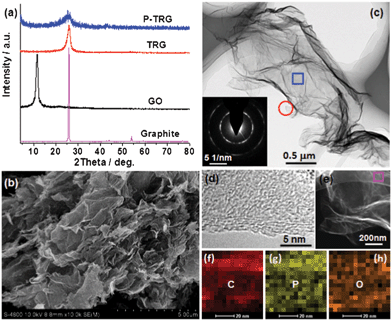 | ||
| Fig. 1 (a) XRD patterns of as-prepared P-TRG, TRG, and GO as well as the original graphite powder. (b) Typical SEM and (c) TEM images of the P-TRG sample. The inset in (c) shows the SAED pattern of the rectangle area. (d) HRTEM image of the circled area in panel (c). (e) STEM image and (f–h) the corresponding elemental mappings of the rectangle area in (e). | ||
The typical SEM image presented in Fig. 1b shows that the P-TRG sample is composed of numerous ultrathin sheets, which are randomly stacked together forming a porous network. The sheet-like morphology is further verified by TEM observation as shown in Fig. 1c. It can be seen that the transparent graphene sheets are like silk veil waves with distinctly wrinkled and folded features. These corrugations and wrinkles, most probably, originated from the intercalated P defects in the P-TRG and the structural distortion caused by the increase of bond length and decrease of bond angles when P atoms were incorporated into graphene sheets by substitution. The surface wrinkling and folding also generated lots of open edge sites, which were demonstrated to be favorable for sensing and electrocatalytic applications.22 The electron diffraction of the selected rectangle area shows a ringlike pattern with dispersed bright spots, indicating that the P-doped graphene sheets became partially misorientated because of structure distortion caused by the incorporation of phosphorus atoms into the graphitic planes. The HRTEM image (Fig. 1d) taken at the edge shows that the P-doped graphene nanosheets typically contain 3–4 layers of graphite planes. The interplanar spacing was measured to be 0.4–0.5 nm, slightly larger than the d-spacing of the (002) plane of bulk graphite due to the increase of bond length when phosphorus atoms were substitutionally incorporated into the carbon matrices. The STEM (Fig. 1e) and the corresponding elemental mapping images (Fig. 1f–h) reveal that the P-TRG sample is only composed of C, P, and O elements, and the distribution of P on the graphitic plane is relatively uniform. This fact strongly suggests that the phosphorus atoms have been doped into the graphene sheets. N and F were not detected in the P-TRG sample although both of them are constituents of the ionic liquid. Probably, BmimPF6 decomposed under the given reaction conditions and released a small amount of gaseous species such as hydrocarbons and ammoniumfluoride, which were carried out by the flowing argon.21
The successful doping of phosphorus into the graphitic matrix of the graphene sheets was verified by the XPS measurements as shown in Fig. 2. The full XPS survey spectrum (Fig. 2a) of the P-TRG sample shows a predominant peak at 284.7 eV corresponding to C1s, a peak at 532.8 eV to O1s, and a peak at 132.8 eV to P2p.14 This result confirms that the P-doped graphene is only composed of P, C and O elements, and the corresponding atomic percentage was determined to be 1.16%, 91.38% and 7.46%, respectively. The P-doping level is much higher than that (0.26 at.%) of the P-doped graphite14 and is very close to the value (1.36 at.%) of the P-doped OMC.17 The P-doping level was found to be easily tuned by adjusting the ratio of GO and BmimPF6. However, additional P-doping was proposed to make the destruction of the sp2-carbon network intensified, resulting in a decrease of the electrical conductivity of the sample.23 The high resolution spectra can provide more information about the bonding configurations and chemical environments for the P-doped graphene. As shown in Fig. 2b, the C1s peak can be deconvoluted into three different components located at about 284.6, 285.5 and 287.2 eV, which can be attributed to C–C, C–P, and C–O bonding, respectively.23 The presence of the C–P peak confirms that P atoms have been successfully intercalated into the carbon lattice of graphene; while the major component peak at 284.6 eV indicates that the graphite carbon is in the majority. The O1s peak can also be deconvoluted into three component peaks. The peak at 531 eV may result from physically absorbed oxygen molecules.24 The major peak at 532.8 eV can be assigned to the P–O bonding,14 indicating that the O1s peak arises mainly from the chemical adsorption of oxygen. The peak at 534.1 eV can be attributed to the C–O bonding,14 and its minor peak area suggests that most of the oxygen-containing groups on the carbon framework have been removed by thermal annealing reduction. The high-resolution P2p spectrum (Fig. 2d) reveals that phosphorus was doped into graphene in two main types of chemical bonding:17 P–C and P–O bonding at about 132.1 eV and 133.5 eV, respectively. The presence of both P–C and P–O stretch models in the FTIR spectrum (Fig. S1 in the ESI†) confirmed the XPS analysis. In addition, the peak area ratio of the P–C and P–O phase is close to 1:1. The presence of the P–C covalent bond, once again, confirms the successful doping of phosphorus atoms into the carbon lattice of graphene sheets. Meanwhile, the existence of the P–O phase implies that the doped P atoms have been partially oxidized by the active oxygen released from GO during the thermal reduction process. Similar phenomena were observed in the case of B-doped carbon nanotubes25 and P,N-doped carbon,26 and this kind of structure was suggested to be advantageous for the ORR application,26 which will be discussed later.
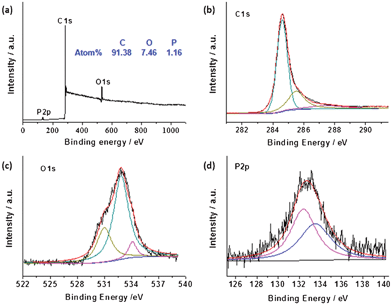 | ||
| Fig. 2 XPS survey (a) and high resolution C1s (b), O1s (c), and P2p (d) spectra of as-prepared P-TRG sample. | ||
The presence of defect sites induced by P-doping was further verified by Raman spectroscopy results as shown in Fig. 3a. Similar to the spectra of all sp2-carbons, two conspicuous peaks appear near 1340 and 1580 cm−1, corresponding to the D and G band, respectively.26 The D band of the P-TRG red-shifted slightly to 1338 cm−1 in comparison to the value of 1346 cm−1 for the TRG because of the defect sites and structural distortion caused by substitutional doping of phosphorus atoms into the carbon lattice.14 Likewise, the G band of the P-TRG also down-shifted to 1578 cm−1, compared to that of the TRG without P-dopants at 1583 cm−1. This is an important characteristic of n-type substitutional doping of graphene,27 as in the case of S-doped graphene.11 Furthermore, the ratio of ID/IG increased from 0.98 for TRG to 1.10 for P-TRG. This fact suggests that the extent of carbon disorder in P-TRG is higher than that in TRG, and thus the degree of crystallinity of P-TRG is lower than TRG, which is consistent with the XRD patterns.
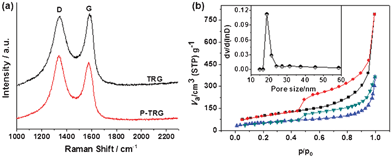 | ||
| Fig. 3 (a) Raman spectra of P-TRG and TRG. (b) Nitrogen adsorption/desorption isotherms of as-prepared P-TRG (up) and TRG (bottom). The inset shows the pore size distribution of P-TRG. | ||
P-doping has also an important effect on the surface area and pore structure of the product. The BET surface area, pore size and pore volume are determined from nitrogen adsorption/desorption isotherms as shown in Fig. 3b and Fig. S2, ESI†, the corresponding data are summarized in Table 1. As a comparison, the TRG without P-dopants is also included. Similar isotherms of type IV with a hysteresis loop were observed for both P-TRG and TRG, indicating that they possess mesoporous structure. However, the average pore size, total pore volume and BET surface of the P-doped graphene nanosheets increased remarkably compared with those of the TRG without P-dopants. These differences may be caused by the variation of bond length/angle and structural distortion due to the transformation from C–C bonds to P–C bonds after P atoms were doped into the graphene sheet. Especially, the BET surface of the P-TRG is about 125 times higher than that of the P-doped graphite layers. Generally, catalytic materials with high electroactive surface area exhibit high performance. Therefore, improved ORR catalytic activity and durability is expected for the as-prepared P-TRG.
| Average pore size (nm) | Total pore volume (cm3 g−1) | BET surface area (m2 g−1) | |
|---|---|---|---|
| TRG | 10.3 | 0.56 | 195.72 |
| P-TRG | 18.8 | 1.27 | 496.67 |
3.2 Electrocatalytic performance for ORR
To investigate the electrocatalytic activity of the P-TRG for the ORR, cyclic voltammetry (CV) experiments were performed in Ar or O2-saturated 0.1 M KOH solution with a flow rate of 20 mL min−1. The TRG as well as the bare glassy carbon electrode (GCE) were measured for comparison. In the O2-free solution, featureless CV curves were observed (data not shown) at each electrode. In contrast, the CV curves (Fig. 4a) obtained in the O2-saturated KOH solution display a well-defined ORR peak for all the electrodes tested. The ORR peak potential positively shifted from −0.439 V for GCE, to −0.307 V for TRG, and −0.215 V for the P-TRG electrode. Therefore, the P-TRG exhibited the highest catalytic activity towards the ORR. This result clearly shows that P-doping can greatly improve the ORR activity of the graphene-based materials. Meanwhile, the CV area of the P-TRG electrode is much larger than that of the TRG electrode, indicating that the P-TRG has much larger electroactive area than the TRG without P-dopants, because the CV area is closely related to the capacitance of the studied material, which is proportional to its surface area.23 This result is in good agreement with the BET data listed in Table 1. Furthermore, the peak current density at the P-TRG electrode reached 1.375 mA cm−2, which was about 6 times larger than that at the TRG electrode. Both the positively shifted peak potential and much enhanced peak current density unambiguously suggest that doping graphene with phosphorus can greatly improve its catalytic activity towards the ORR as in the case of doping graphene or carbon nanotubes with nitrogen.6,13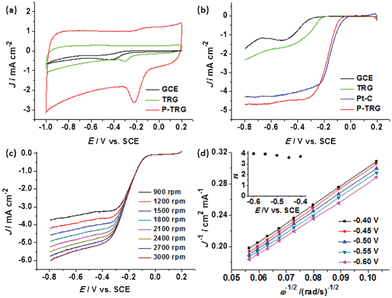 | ||
| Fig. 4 (a) CV curves of the ORR at various electrodes at a scan rate of 100 mV s−1 in O2-saturated 0.1 M KOH solution with an O2 flow rate of 20 mL min−1. (b) LSV curves of the ORR at various electrodes in O2-saturated 0.1 M KOH solution at a scan rate of 5 mV s−1 and rotation rate of 1600 rpm. (c) LSV curves of the P-TRG electrode at a scan rate of 5 mV s−1 and at different rotating rates in O2-saturated 0.1 M KOH solution. (d) Koutecky–Levich plots of J−1vs. ω−1/2 at different electrode potentials. | ||
Linear sweep voltammograms (LSV) can provide further insight into the role of the P-doping in the ORR catalysis and the ORR mechanism. Fig. 4b compares the LSV curves at different electrodes including the GCE, TRG, P-TRG and the commercial Pt/C catalyst modified GCE. The ORR onset potential of the P-TRG electrode is −0.0261 V, which is much higher than that of the GCE (−0.215 V) as well as that of the TRG electrode (−0.162 V), and is very close to that of the Pt/C electrode (−0.0026 V). It is worth noting that the onset potential difference between the P-TRG and the Pt/C electrode is only 23.5 mV, indicating the superior catalytic activity of the P-doped graphene towards the ORR. In addition, the current plateau of the LSV curves reveals that the electrochemical oxygen reduction at the GCE and TRG electrodes follows an inefficient two-step ORR process with superoxide (HO2−) as an intermediate,28 while the ORR occurs at the P-TRG electrode through a one-step pathway for the reduction of O2 to OH− with little to negligible formation of HO2− just like that at the Pt/C electrode. As a result, doping graphene with P can facilitate the adsorption of dioxygen and improve the ORR kinetics, which will be discussed later. Meanwhile, the ORR current density at the P-TRG electrode is always much larger than those at the GCE and TRG electrode, and is even larger than that at the Pt/C electrode when the potential is below −0.23 V, thereby suggesting the diffusion and transportation of the reactants, electrolyte and the reduced products have been much enhanced on the P-TRG catalyst, because it possesses high surface area, abundant mesopores and large pore volume as discussed above. Both the positive shift of onset potential and the enhanced reduction current for the ORR at the P-TRG electrode demonstrate that the ORR catalytic performance of the P-TRG is much better than that of the undoped graphene, and even is superior to or at least comparable to that of the benchmark Pt/C catalyst.
To gain more information on the ORR kinetics of the P-TRG catalyst, the rotating disk electrode (RDE) voltammetry was conducted in O2-saturated 0.1 M KOH solution with an O2 flow rate of 20 mL min−1 at a scanning rate of 5 mV s−1 and different rotation rates, and the results are presented in Fig. 4c. It is clear that the diffusion current density enhances with the increase of rotation rate from 900 to 3000 rpm due to the enhanced diffusion of electrolytes. The corresponding Koutechy–Levich curves for different potentials are plotted in Fig. 4d. The parallel and straight lines imply first-order reaction kinetics with respect to the concentration of dissolved oxygen. The transferred electron number (n) per oxygen molecule involved in the ORR process is calculated from the Koutechy–Levich equation described as below.17
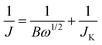
| B = 0.62nFC0D02/3v−1/6 |
485 C mol−1), C0 is the bulk concentration (1.2 × 10−3 mol L−1) of O2, D0 is the diffusion coefficient (1.9 × 10−5 cm2 s−1) of O2 in the KOH solution, v is the kinetic viscosity (0.01 cm2 s−1) of the electrolyte solution.17 The dependence of n on the potentials is presented in the inset of Fig. 4d. As can be readily seen, the n value is higher than 3.6 in the potential range of −0.4–−0.6 V, and exceeds 3.9 at more negative potentials. This fact confirms that the oxygen reduction on the P-doped graphene prefers the one-step four-electron pathway to directly form H2O/OH−, which is highly desirable for fuel cells to gain maximum energy capacity.
Resistance to fuel crossover and carbon monoxide (CO) poisoning are very important for the practical application of the newly developed catalyst to fuel cells, and are also the stern challenges facing the Pt/C electrocatalyst. Accordingly, chronoamperometric measurements were performed at the polarizing potential of −0.26 V and a rotation rate of 1600 rpm in O2-saturated KOH solution with an O2 flow rate of 20 mL min−1 to investigate possible methanol crossover and CO poisoning effects on the P-TRG catalyst as well as the Pt/C electrocatalyst for comparison. The corresponding results are presented in Fig. 5a and b, respectively. When methanol was injected into the O2-saturated KOH electrolyte, the ORR current at the P-TRG electrode remained unaltered, whereas the corresponding current at the Pt/C electrode showed a sharp shift from a cathodic current to a reversed anodic current as a result of electrochemical oxidization of methanol rather than the reduction of oxygen. These results unambiguously indicate that the P-TRG catalyst exhibits high immunity to methanol, and thus has much higher fuel selectivity towards the ORR than the Pt/C catalysts. When additional CO with the same flow of O2 was introduced into the electrolyte during the CO poisoning tests, the ORR current at the Pt/C electrode was greatly weakened by about 82.5% due to the CO poisoning. In contrast, only a slight attenuation of the ORR current was observed at the P-TRG electrode as a result of the decreased solubility of O2 in the electrolyte induced by the decreased partial pressure of O2, indicating that the P-TRG catalyst is insensitive to CO in nature.
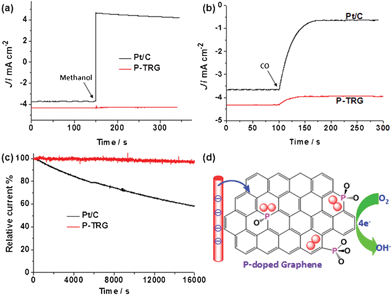 | ||
| Fig. 5 (a) Methanol crossover testes by injecting methanol into the electrolyte at 150 s to make the final concentration of methanol 3.0 M. (b) CO poisoning tests by introducing additional CO with the same flow of O2 into the electrolyte from 100 s. (c) Comparison of the stability of the P-TRG and Pt/C electrode at −0.26 V in O2-saturated 0.1 M KOH solution at a rotation rate of 1600 rpm. (d) Schematic illustration of the possible ORR process catalyzed by the P-doped graphene. | ||
Another major concern about the current fuel-cell technology is the durability. Fig. 5c compares the durability of the P-TRG and Pt/C electrodes at a constant potential of −0.26 V and a rotation rate of 1600 rpm in O2-saturated 0.1 M KOH solution. During continuous chronoamperometric measurement the current at the P-TRG electrode almost kept stable, while it decreased dramatically at the Pt/C electrode. After testing for 16000 s under the same conditions, the current loss for the P-TRG and Pt/C electrodes was 3.57% and 41.85%, respectively. This result means that the durability of the P-TRG is much superior to that of the Pt/C catalyst. It is well known that the metal-based catalysts including Pt/C frequently suffer from surface oxidation and particle dissolution/aggregation over time during fuel cell operation, resulting in gradual degradation of activity and durability.29 However, for the P-doped graphene, the doped P atoms are covalently bonded within the carbon framework, thereby the catalytic activity will not fade even in long-time operation.
The enhanced electrocatalytic performance of the doped carbon materials is usually attributed to the charge redistribution induced by the difference of electronegativity between the doped atom and carbon, which can create charged sites (C+, B+, P+, S+) favorable for O2 adsorption to facilitate the ORR process.30 For example, the active sites in the P-doped carbon should be the positively charged P atoms because the electronegativity of P is less than that of carbon. This conclusion has been made on the basis of the pesudo-planar PC3 moiety containing pure P–C bonding, which is similar to the NC3 and BC3 moieties in N and B doped carbon. Note that B, C and N lie at the same period with similar covalent radius, and tend to form sp2-hybrid orbitals to form planar structures. However, P lies at the third period, and favors the sp3-orbital configuration in molecules because its covalent radius (107 ± 3 pm) is much larger than that of C (73 pm).26 In our case, the partially oxidized P was covalently bonded with C, and existed as tetrahedral forms such as C3PO, C2PO2, and CPO3 as schematically shown in Fig. 5d. This conjecture is supported by the aforesaid XPS analysis, especially the finely split peaks in the high resolution P2p spectrum (Fig. S3, ESI†). Atomic charge will redistribute on the P-doped graphene due to the partial oxidization of P. The O atom with the highest electronegativity will first make the P atom polarized, and then withdraw electrons from the carbon atoms with the polarized P atom as a bridge, creating a net positive charge on the carbon atoms adjacent to the P atom. As a result, the positively charged carbon atoms become the active sites favorable adsorption of O2, weakening the O–O bonding, and attracting electrons from the anode to facilitate the reduction of O2 to OH− through an efficient four-electron pathway just like N-doped carbon. Further work is underway in our group to elucidate the exact ORR mechanism.
4. Conclusions
Metal-free P-doped graphene nanosheets were successfully synthesized by thermal annealing a homogenous mixture of GO and BmimPF6. This ionic liquid assisted one-step route is economical, simple but scalable for mass production of P-doped graphene. On the basis of structural characterizations and electrochemical measurements, we can draw the following conclusions.(1) The doped P atom was covalently bonded to carbon atoms, and was partially oxidized by the active oxygen released from GO during thermal reduction. (2) Significant structural variations were induced by doping P atoms into the graphene sheets. Many corrugations and open edge sites were generated, the pore size and pore volume were enlarged, and the BET surface area was greatly increased to 496.67 m2 g−1 in comparison to the value of 195.72 m2 g−1 for the pristine graphene without P-dopant. All these structural changes are favorable for the ORR applications. (3) The P-doped graphene with a P-doping level of 1.16 at% was demonstrated to be better than or at least comparable to the commercial benchmark Pt/C catalyst for the ORR applications in alkaline media. (4) The active sites for O2 adsorption and reduction in the P-doped graphene were found to be different from those reported in P-doped graphite. It seemed that the partial oxidization of P made the P-doped graphene act as N-doped carbon in the ORR process. Certainly, much more work is needed to identify the active sites and elucidate the ORR mechanism.
Acknowledgements
This work was financially supported by China National 973 Program (2012CB215500 and 2012CB720300), the National Natural Science Foundation of China (21176327, 51071131, 20936008), and the Program for New Century Excellent Talents in University (NCET-10-0890).Notes and references
- (a) M. K. Debe, Nature, 2012, 486, 43 CrossRef CAS; (b) F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172 RSC; (c) X. Zhao, M. Yin, L. Ma, L. Liang, C. Liu, J. Liao, T. Lu and W. Xing, Energy Environ. Sci., 2011, 4, 2736 RSC; (d) H. A. Gasteiger and N. M. Marković, Science, 2009, 324, 48 CrossRef CAS.
- (a) Z. Chen, D. Higgins, A. Yu, L. Zhang and J. Zhang, Energy Environ. Sci., 2011, 4, 3167 RSC; (b) A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238 RSC; (c) J. A. Cracknell, K. A. Vincent and F. A. Armstrong, Chem. Rev., 2008, 108, 2439 CrossRef CAS.
- (a) G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443 CrossRef CAS; (b) J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. S. Horn, Nat. Chem., 2011, 3, 546 CrossRef CAS.
- (a) Y. Li, W. Zhou, H. Wang, L. Xie, Y. Liang, F. Wei, J. Idrobo, S. J. Pennycook and H. J. Dai, Nat. Nanotechnol., 2012, 7, 394 CrossRef CAS; (b) M. Gao, J. Jiang and S. H. Yu, Small, 2012, 8, 12 CrossRef; (c) Y. Zheng, Y. Jiao, M. Jaroniec, Y. Jin and S. Z. Qiao, Small, 2012, 8, 3550 CrossRef CAS.
- (a) C. Huang, C. Li and G. Q. Shi, Energy Environ. Sci., 2012, 5, 8848 RSC; (b) M. Zhang and L. M. Dai, Nano Energy, 2012, 1, 514 CrossRef CAS; (c) R. Liu, D. Wu, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 2565 CrossRef CAS.
- (a) K. Gong, F. du, Z. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760 CrossRef CAS; (b) H. Jin, H. Zhang, H. Zhong and J. Zhang, Energy Environ. Sci., 2011, 4, 3389 RSC; (c) X. Wang, J. S. Lee, Q. Zhu, J. Liu, Y. Wang and S. Dai, Chem. Mater., 2010, 22, 2178 CrossRef CAS; (d) C. H. Choi, S. H. Park and S. I. Woo, Green Chem., 2011, 13, 406 RSC.
- (a) A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS; (b) Y. Sun, Q. Wu and G. Shi, Energy Environ. Sci., 2011, 4, 1113 RSC; (c) H. Wang, T. Maiyalagan and X. Wang, ACS Catal., 2012, 2, 781 CrossRef CAS; (d) L. S. Panchakarla, K. S. Subrahmanyam, S. K. Saha, A. Govindaraj, H. R. Krishnamurthy, U. V. Waghmare and C. N. R. Rao, Adv. Mater., 2009, 21, 4726 CAS.
- (a) H. Liu, Y. Liu and D. Zhu, J. Mater. Chem., 2011, 21, 3335 RSC; (b) S. Wang, L. Zhang, Z. Xia, A. Roy, D. W. Chang, J. Baed and L. Dai, Angew. Chem., Int. Ed., 2012, 51, 4209 CrossRef CAS; (c) Z. Sheng, L. Shao, J. Chen, W. Bao, F. Wang and X. Xia, ACS Nano, 2011, 5, 4350 CrossRef CAS.
- (a) Y. Zhao, C. Hu, Y. Hu, H. Cheng, G. Shi and L. Qu, Angew. Chem., Int. Ed., 2012, 51, 11371 CrossRef CAS; (b) K. R. Lee, K. U. Lee, J. W. Lee, B. T. Ahn and S. I. Woo, Electrochem. Commun., 2010, 12, 1052 CrossRef CAS; (c) Y. Li, Y. Zhao, H. Cheng, Y. Hu, G. Shi, L. Dai and L. Qu, J. Am. Chem. Soc., 2012, 134, 15 CrossRef CAS.
- (a) Z. Sheng, H. Gao, W. Bao, F. Wang and X. Xia, J. Mater. Chem., 2012, 22, 390 RSC; (b) Y. Zheng, Y. Jiao, L. Ge, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2013, 52, 3110 CrossRef CAS.
- (a) Z. Yang, Z. Yao, G. Li, G. Fang, H. Nie, Z. Liu, X. Zhou, X. Chen and S. Huang, ACS Nano, 2012, 6, 205 CrossRef CAS; (b) S. Yang, L. Zhi, K. Tang, X. Feng, J. Maier and K. Müllen, Adv. Funct. Mater., 2012, 22, 3634 CrossRef CAS; (c) J. Liang, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2012, 51, 11496 CrossRef CAS.
- Z. Yao, H. Nie, Z. Yang, X. Zhou, Z. Liu and S. Huang, Chem. Commun., 2012, 48, 1027 RSC.
- (a) L. Qu, Y. Liu, J. Baek and L. Dai, ACS Nano, 2010, 4, 1321 CrossRef CAS; (b) D. Geng, Y. Chen, Y. Chen, Y. Li, R. Li, X. Sun, S. Ye and S. Knights, Energy Environ. Sci., 2011, 4, 760 RSC.
- Z. Liu, F. Peng, H. Wang, H. Yu, W. Zheng and J. Yang, Angew. Chem., Int. Ed., 2011, 50, 3257 CrossRef.
- Z. Liu, F. Peng, H. Wang, H. Yu, J. Tan and L. Zhu, Catal. Commun., 2011, 16, 35 CrossRef CAS.
- Z. Liu, F. Peng, H. Wang, H. Yu, W. Zheng and X. Wei, J. Nat. Gas Chem., 2012, 21, 257 CrossRef CAS.
- D. Yang, D. Bhattacharjya, S. Inamdar, J. Park and J. Yu, J. Am. Chem. Soc., 2012, 134, 16127 CrossRef CAS.
- Z. Tang, S. Shen, J. Zhuang and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 4603 CrossRef CAS.
- S. Pei and H. M. Cheng, Carbon, 2012, 50, 3210 CrossRef CAS.
- L. Lai, J. R. Potts, D. Zhan, L. Wang, C. K. Poh, C. Tang, H. Gong, Z. Shen, J. Lin and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 7936 CAS.
- Y. Zhang, T. Mori, J. Ye and M. Antonietti, J. Am. Chem. Soc., 2010, 132, 6294 CrossRef CAS.
- (a) N. G. Shang, P. Papakonstantinou, M. McMullan, M. Chu, A. Stamboulis, A. Potenza, S. S. Dhesi and H. Marchetto, Adv. Funct. Mater., 2008, 18, 3506 CrossRef CAS; (b) R. Silva, J. Al-Sharab and T. asefa, Angew. Chem., Int. Ed., 2012, 51, 7171 CrossRef CAS.
- C. H. Choi, S. H. Park and S. I. Woo, J. Mater. Chem., 2012, 22, 12107 RSC.
- A. M. Puziy, O. I. Poddubnaya, R. P. Socha, J. Gurgul and M. Wisniewski, Carbon, 2008, 46, 2113 CrossRef CAS.
- L. Yang, S. Jiang, Y. Zhao, L. Zhu, S. Chen, X. Wang, Q. Wu, J. Ma, Y. Ma and Z. Hu, Angew. Chem., Int. Ed., 2011, 50, 7132 CrossRef CAS.
- C. H. Choi, S. H. Park and S. I. Woo, ACS Nano, 2012, 6, 7084 CrossRef CAS.
- S. Some, J. Kim, K. Lee, A. Kulkarni, Y. Yoon, S. Lee, T. Kim and H. Lee, Adv. Mater., 2012, 24, 5481 CrossRef CAS.
- Y. Zhang, K. Fugane, T. Mori, L. Niu and J. Ye, J. Mater. Chem., 2012, 22, 6575 RSC.
- I. E. L. Stephens, A. S. Bondarenko, U. Grønbjerg, J. Rossmeisl and I. Chorkendorff, Energy Environ. Sci., 2012, 5, 6744 CAS.
- D. Yu, Y. Xue and L. M. Dai, J. Phys. Chem. Lett., 2012, 3, 2863 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra41079j |
| This journal is © The Royal Society of Chemistry 2013 |