
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent developments in the synthesis and applications of terpyridine-based metal complexes: a systematic review
Syeda Fariha Kainat
a,
Mohammed B. Hawsawi b,
Ehsan Ullah Mughal
*a,
Nafeesa Naeem
a,
Abdulaziz M. Almohyawib,
Hatem M. Altassb,
Essam M. Husseinbc,
Amina Sadiq
d,
Ziad Moussae,
Alaa S. Abd-El-Azizf and
Saleh A. Ahmed
*b
b,
Ehsan Ullah Mughal
*a,
Nafeesa Naeem
a,
Abdulaziz M. Almohyawib,
Hatem M. Altassb,
Essam M. Husseinbc,
Amina Sadiq
d,
Ziad Moussae,
Alaa S. Abd-El-Azizf and
Saleh A. Ahmed
*b
aDepartment of Chemistry, University of Gujrat, Gujrat-50700, Pakistan. E-mail: ehsan.ullah@uog.edu.pk
bDepartment of Chemistry, Faculty of Science, Umm Al-Qura University, 21955 Makkah, Saudi Arabia. E-mail: saahmed@uqu.edu.sa; saleh_63@hotmail.com
cDepartment of Chemistry, Faculty of Science, Assiut University, 71516 Assiut, Egypt
dDepartment of Chemistry, Govt. College Women University, Sialkot-51300, Pakistan
eDepartment of Chemistry, College of Science, United Arab Emirates University, P.O. Box 15551, Al Ain, United Arab Emirates
fQingdao Innovation and Development Centre, Harbin Engineering University, Qingdao 266400, China
First published on 8th July 2024
Abstract
Terpyridine-based metal complexes have emerged as versatile and indispensable building blocks in the realm of modern chemistry, offering a plethora of applications spanning from materials science to catalysis and beyond. This comprehensive review article delves into the multifaceted world of terpyridine complexes, presenting an overview of their synthesis, structural diversity, and coordination chemistry principles. Focusing on their diverse functionalities, we explore their pivotal roles in catalysis, supramolecular chemistry, luminescent materials, and nanoscience. Furthermore, we highlight the burgeoning applications of terpyridine complexes in sustainable energy technologies, biomimetic systems, and medicinal chemistry, underscoring their remarkable adaptability to address pressing challenges in these fields. By elucidating the pivotal role of terpyridine complexes as versatile building blocks, this review provides valuable insights into their current state-of-the-art applications and future potential, thus inspiring continued innovation and exploration in this exciting area of research.
 Syeda Fariha Kainat | Syeda Fariha Kainat earned her BS in 2021 and MPhil in 2023 from the University of Gujrat, Pakistan. She has published two research articles in high-impact journals. Her research focuses on the synthesis and photocatalytic applications of terpyridine-based metal complexes. |
 Ehsan Ullah Mughal | Ehsan Ullah Mughal obtained his PhD degree in Organic Chemistry from Bielefeld University, Germany in 2013. Currently, he is working as a Tenured Associate Professor in the Department of Chemistry, University of Gujrat, Pakistan. He was a post-doc fellow at the Max-Planck Institute for Polymer Research, Mainz, Germany from 2013 to 2014. His current research interests include the design and synthesis of bioactive heterocycles, synthetic flavonoids and transition metal-based terpyridine complexes and their uses in the fabrication of DSSCs and as efficient photocatalysts. His research in the field has resulted in more than 90 published articles (research papers and review papers) in reputable high impact journals. His publications have made a great impact in the field by receiving more than 1825 citations and he has a h-index of 26. |
 Nafeesa Naeem | Nafeesa Naeem received her BS and MPhil degrees from the University of Gujrat, Pakistan, in 2018 and 2020, respectively. She is currently pursuing a PhD in Chemistry under the supervision of Dr Ehsan Ullah Mughal at the University of Gujrat, Pakistan. She has published over 35 publications (research papers and review papers) in reputable high impact international journals, receiving more than 880 citations and she has a h-index of 18. Her current research focuses on the synthesis, biological evaluation, photophysical studies, and photocatalytic applications of various heterocyclic scaffolds. |
 Amina Sadiq | Amina Sadiq is an Associate Professor of Organic Chemistry at the Government College Women University in Sialkot, Pakistan. She earned her PhD in 2013 from Bielefeld University, Germany, under the supervision of Prof. Dr Norbert Sewald, and later conducted postdoctoral research with the same group. She has published more than 65 publications in high ranked journals. Her research focuses on the design and synthesis of bioactive heterocycles, synthetic flavonoids, and chalcone-based metal complexes, along with evaluating their biological activities. |
 Saleh A. Ahmed | Prof. Dr Saleh A. Ahmed obtained his PhD in photochemistry (photochromism) under the supervision of Prof. Heinz Dürr at Saarland University, Germany. He has more than 20 years of experience as a postdoctoral fellow, senior researcher and visiting professor in France, Japan, Germany, Italy and the USA. In 2010 he was promoted to full professor of organic photochemistry. He has published more than 260 publications in high ranked journals and more than 10 US-patents. His research interests include the synthesis and photophysical properties of novel organic compounds, and development of synthetic methodologies for the synthesis of compounds with unique theoretical and biological applications. |
1. Introduction
The 2,2′:6′,2′-terpyridine (TPY) ligand 1 (Fig. 1) belongs to the class of tridentate ligands characterized by three coordination sites due to the presence of N-heteroaromatic rings within its molecular structure.1 TPY is categorized as an NNN-type pincer ligand or chelator which exhibits a strong affinity for binding with metal cations, some very strongly and others rather weakly, resulting in the formation of square planar [Pt(II)] and distorted square pyramid [Cu(II)] compounds. Moreover, TPY Ru(II) complexes are usually octahedral. Upon binding to a metal cation, TPY adopts distinct coordination modes: monodentate TPY-metal complexes, bi- and tri-dentate TPY-metal complexes.2 Prior to its coordination with a metal cation, a pyridine ring exhibits a trans–trans geometry, which minimizes the repulsion between lone pair electrons. However, upon coordination with a metal cation, TPY undergoes a structural phenomenon, adopting a cis–cis geometry. This particular arrangement ensures that the three pyridine rings present in the TPY structure align perfectly in a coplanar or twisted conformation.3 In a coplanar conformation, the π orbitals of the adjacent pyridine rings overlap effectively, leading to enhanced electronic communication and conjugation along the molecular backbone. It is worth noting that this extraordinary feature classifies TPY as a non-innocent ligand, which has the inherent capability to stabilize low-valency metals, including but not limited to Ni, Fe, Co, and Mn, among others.4 | ||
| Fig. 1 Structural representation of TPY ligand 1. | ||
The complexation of the TPY ligand with various metal ions leads to its remarkable applications in diverse fields, including therapeutic use, material sciences, and supramolecular chemistry.5–7 In the realm of therapeutic applications, TPY-based complexes are notable for their ability to intercalate with DNA and serve as potential anticancer agents.8,9 In the field of material sciences, TPY complexes find utility in photovoltaics and as sensitizers in solar cell technologies. Their unique photophysical properties make them valuable components for enhancing the efficiency of energy conversion processes.10,11 Additionally, in supramolecular chemistry, TPY and its scaffold derivatives exhibit outstanding coordination chemistry. They are known for forming strong bonds with various types of metal cations, leading to the creation of diverse supramolecular assemblies.12 These assemblies possess enchanting photophysical and redox characteristics, making them valuable tools for designing advanced materials and exploring new frontiers in molecular engineering.13
TPY derivatives, when employed in bis-TPY metal complexes, adopt an octahedral geometry wherein either the same or different TPY ligands are attached perpendicularly.14 A noteworthy point of comparison lies in the exceptional chelation ability and stability exhibited by bis-TPY metal complexes 3, especially when contrasted with tri/bipyridine complexes (Fe, Ru, Pt & Cu).15 It is worth mentioning that bis-TPY metal complexes, as exemplified by Fig. 2, are typically achiral. The primary motivation for investigating TPY ligands centers around the use of mono-TPY ligands 2 (Fig. 2) to construct supramolecular structures that mitigate the challenges associated with achieving desired stereoselectivity. Like many other nitrogen donor ligands, the stability of TPY metal complexes is intricately linked to the metal ion.12,16 Beyond their various applications, TPY metal complexes find utility in pharmaceutical and agrochemical fields. Moreover, they serve as ditopic terpyridyl units in electrochemical cells, as illustrated in Fig. 2.5,8,17 One of the outstanding qualities of TPY complexes lies in their ability to form a wide array of organometallic compounds, making them valuable catalysts for diverse organic transformations. They find application in both homogeneous and heterogeneous catalytic processes, further underscoring their versatility and importance in the realm of modern chemistry.2,18,19
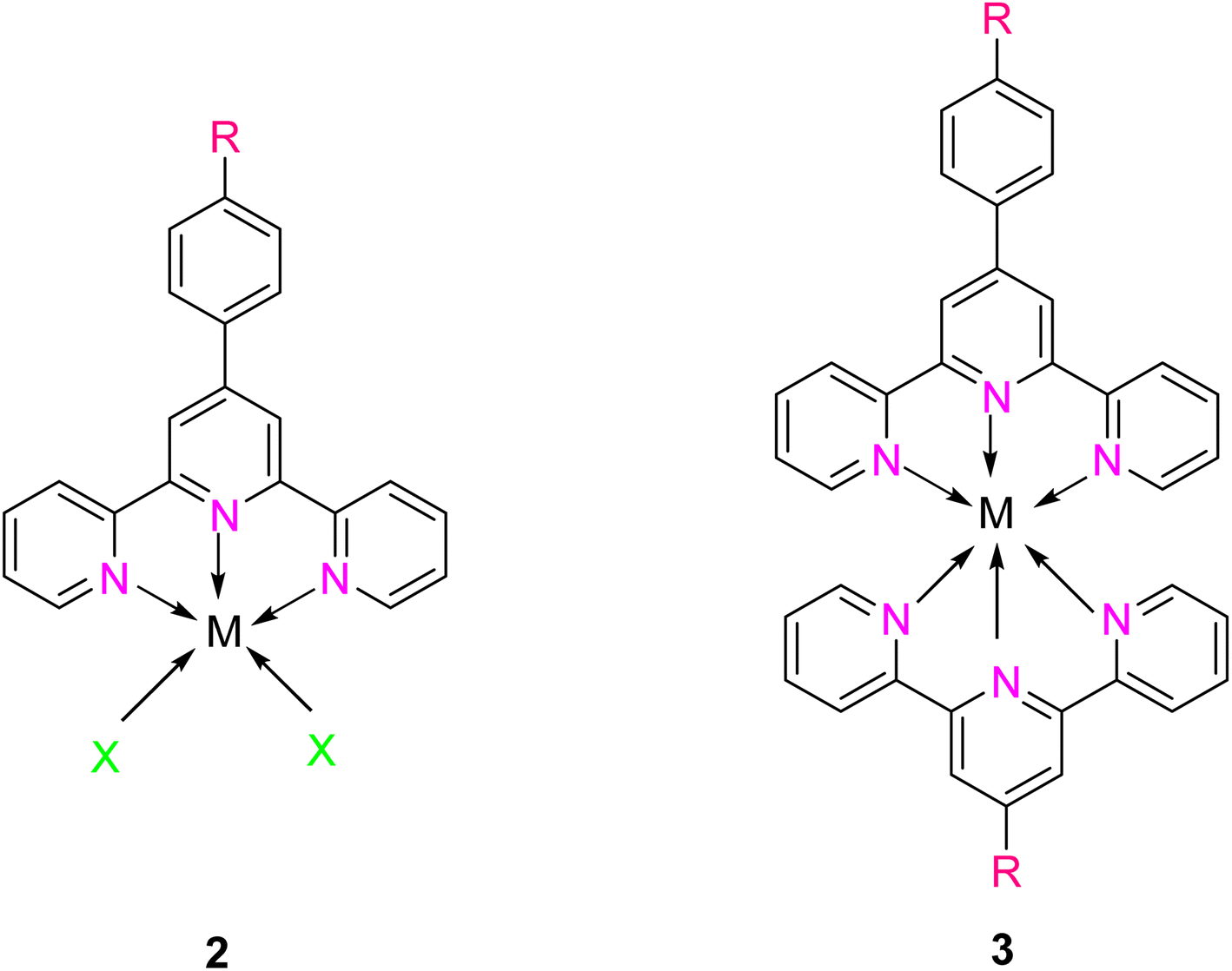 | ||
| Fig. 2 Mono- and bis-TPY metal complexes. | ||
Moreover, TPY-based metal complexes, particularly those involving iridium, ruthenium, osmium, and platinum, exhibit notable electrochemical and photophysical properties, making them crucial components in assemblies designed for charge separation.20 Researchers have explored their role as photosensitizers, electron relays, and catalysts in processes such as hydrogen evolution and singlet oxygen generation. Additionally, modifications to the ligands have been studied to enhance the stability and efficiency of these complexes.21 The literature highlights their applications in light harvesting, resistive memory devices, molecular electronics, and as luminescent probes for sensing cations and anions. Furthermore, the researchers also reported the use of machine learning and artificial intelligence techniques to enhance ion sensing capabilities. Overall, the reported literature underscores the versatility and potential of terpyridine metal complexes in a wide range of scientific and technological applications.
This comprehensive review article delves into various methodologies for the synthesis of TPY ligands and their corresponding metal complexes. Furthermore, it elucidates the diverse range of applications associated with both square planar and octahedral complexes. These applications encompass their utilization as catalysts, chemosensors, photosensitizers, supramolecular structures, photocatalysts, and in the field of biomedical applications.
The synthesis of TPY ligands and their metal complexes represents a critical aspect of their study. Understanding the various synthetic routes and methodologies is essential for harnessing the full potential of these compounds in different applications. This review compiles and discusses the state-of-the-art approaches for the preparation of TPY ligands and their subsequent coordination with metal ions.
The versatility of TPY complexes becomes evident when exploring their applications. Square planar and octahedral complexes have been found to serve as catalysts in various chemical reactions, promoting and facilitating these transformations with high efficiency. Their utility extends to chemosensors, where their distinctive properties enable selective detection and quantification of specific chemical species, making them invaluable tools in analytical chemistry.22,23 Their ability to form robust supramolecular structures enhances their value in creating intricate and functional molecular assemblies with a wide range of potential applications. Furthermore, TPY complexes demonstrate promise in biomedical applications, where they can be employed in drug delivery, imaging, and therapy due to their unique properties and biocompatibility (Fig. 3).24,25
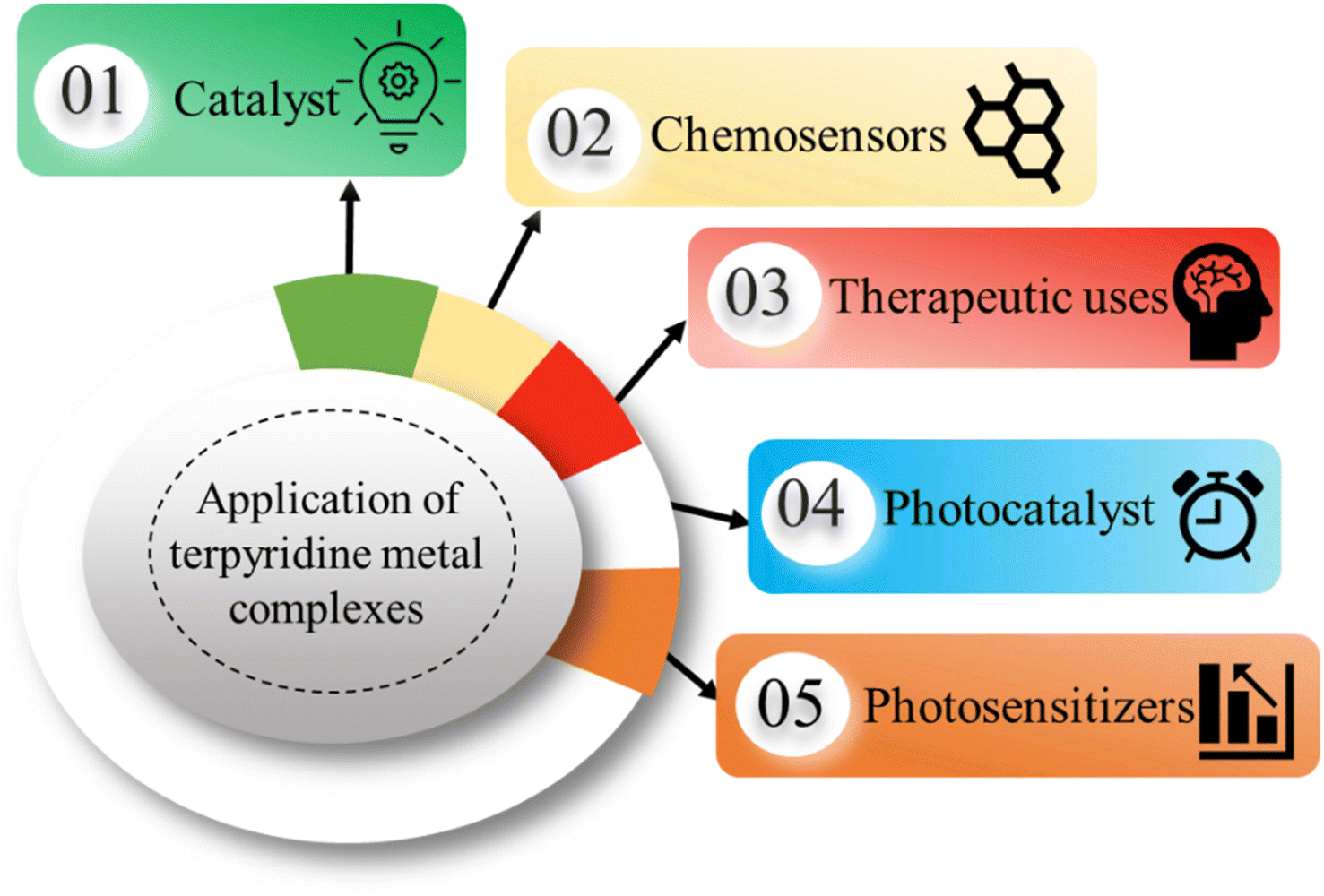 | ||
| Fig. 3 Applications of TPY metal complexes. | ||
2. Syntheses of TPY
Various methodologies are employed for the synthesis of TPY (2,2′:6′,2′-terpyridine) ligands, including coupling methodologies, ring assembly methods, one-pot synthesis, the KrӧhnkeKrӧhnke method, and Stille coupling (as illustrated in Fig. 4). Among these, the ring closure method stands out as a particularly effective approach. The ring closure method stands out as particularly effective for terpyridine ligand synthesis due to its regioselectivity, step-efficiency, high yield, functional group compatibility, reduced side reactions, and facile optimization, collectively contributing to its widespread use in the preparation of these versatile ligands. Coupling methodologies, facilitated by palladium (Pd) catalysts, have also demonstrated their utility, albeit with certain distinctions when compared to the ring assembly methodology.4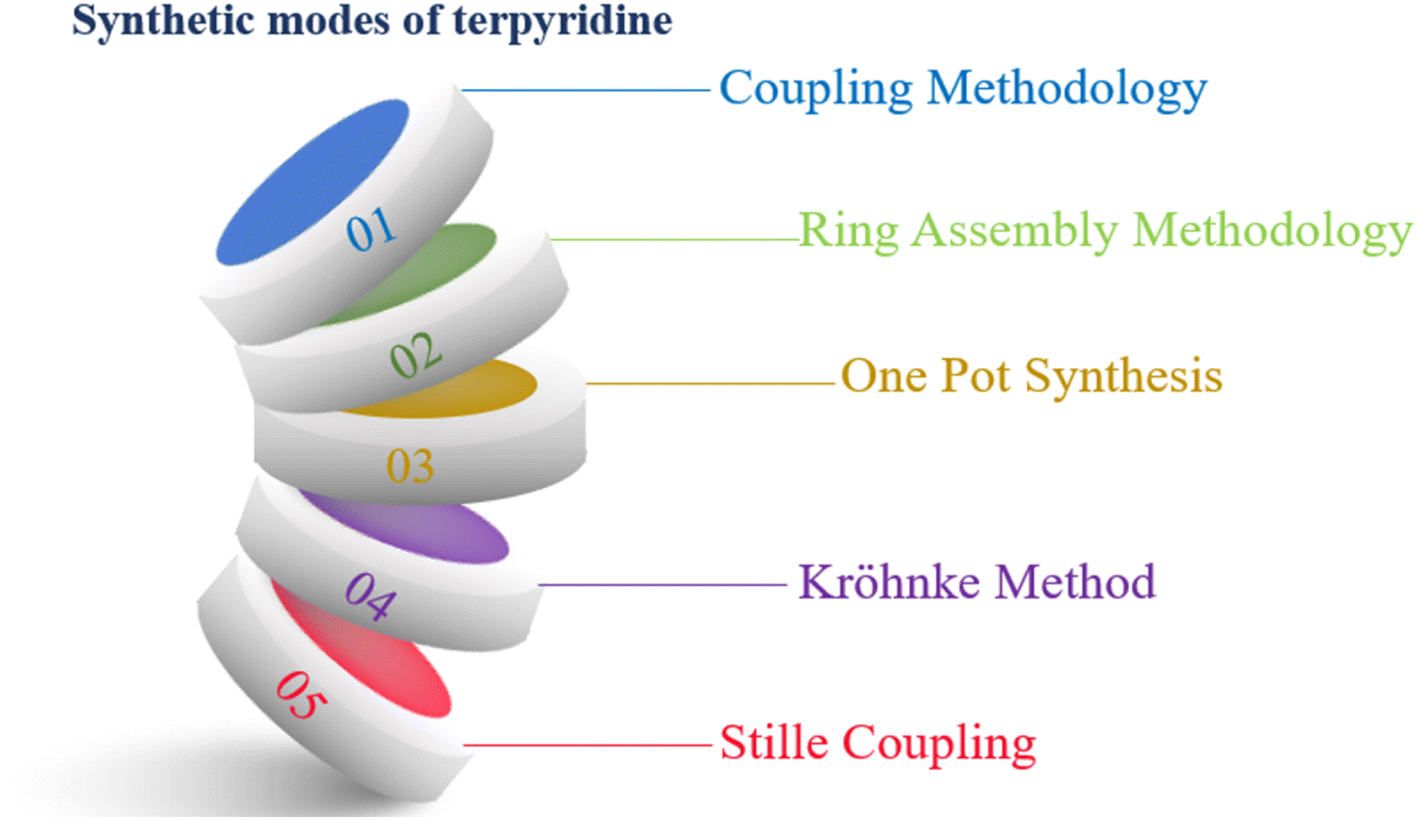 | ||
| Fig. 4 Synthetic modes of TPY. | ||
Historically, in 1932, Morgan reported a catalytic synthesis of TPY using iron chloride (FeCl3). This early work laid the foundation for subsequent advancements in TPY synthesis.26
In general, there are two main strategies for the synthesis of TPY ligands. The first strategy involves the coupling of two pyridine units to form the central pyridine ring. The second strategy entails the synthesis of either the central or two terminal pyridine rings independently, followed by their subsequent assembly to construct the complete TPY ligand structure. These synthetic approaches offer researchers diverse options for producing TPY ligands, each with its advantages and considerations. The choice of methodology depends on the specific requirements of the research and the desired properties of the TPY ligand for its intended application.27
2.1. Coupling methodology
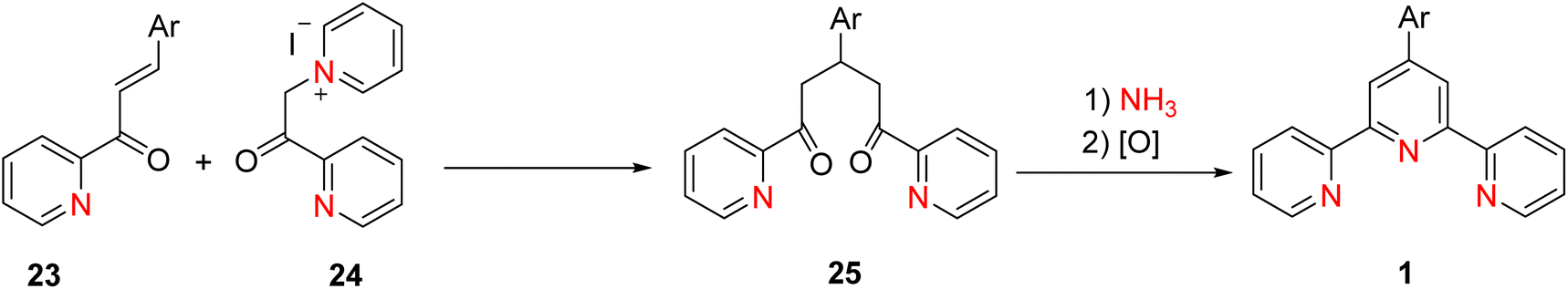
Coupling methodologies have played a pivotal role in the synthesis of terpyridine (TPY) ligands and their derivatives. Various coupling methodologies have been explored for the synthesis of oligopyridines, although these methods often proved challenging due to the complex processes required for the separation of different products from reaction mixtures. These coupling reactions primarily involve two distinct processes: oxidative dehydrogenation of pyridine or the formation of halopyridines through interaction with metal cations, sometimes accompanied by low oxidation state metal complexes.28In a significant milestone dating back to 1932, Morgan and Burstall successfully synthesized TPY (2,2′:6′,2′-terpyridine) for the first time.26 They accomplished this chemical reaction by employing the oxidative coupling of pyridine in the presence of iron chloride (FeCl3) under high-temperature and high-pressure conditions within an autoclave. This method resulted in the formation of TPY from a mixture of approximately twenty different compounds. While alternative approaches involving the dehydrogenation of various metal chlorides were explored, these methods yielded limited success in comparison to the FeCl3-coupling approach. Furthermore, researchers have investigated organometallic coupling strategies. One of the initial methods involved the reaction of 2,2′-bipyridine 4 with 2-pyridyllithium 5 in the presence of ether at a temperature of −40 °C. This reaction yielded TPY 1, as illustrated in Scheme 1, with a yield of approximately 39%. It is noteworthy that this reaction also produced dihydropyridine as a byproduct.29
 | ||
| Scheme 1 Synthesis of TPY by reaction of 2,2′-bipyridine with 2-pyridyllithium. | ||
Wakabayashi et al. (1990) achieved a notable yield improvement of 65% by employing a modified coupling reaction. This method involved the reaction between 2-pyridyllithium 5 and 6-ethylsulphinyl-2,2′-bipyridine 7, with the sulfoxide precursor obtained through the oxidation of 6-ethylthio-2,2′-bipyridine 6 using magnesium monoperoxyphthalate (Scheme 2).30,31
 | ||
| Scheme 2 Synthesis of TPY by reaction of 2-pyridyllithium and 6-ethylsulphinyl-2,2′-bipyridine. | ||
2.2. Ring assembly methodology
The ring assembly methodology represents a significant avenue for the synthesis of terpyridine (TPY) ligands, which have gained prominence for their versatility and wide-ranging applications. The ring assembly methodology stands as a coherent and fundamental procedure for the synthesis of terpyridine (TPY) ligands. This method focuses on the crucial step of constructing the pyridine ring within the ligand's structure. Specifically, it involves the condensation of ketone 8 with an aldehyde 9, resulting in the formation of the pivotal 1,5-diketone 10, as illustrated in Scheme 3. The success of this ring closure process can be influenced by the choice of suitable reagents and conditions. Notable examples include the use of ammonium acetate, hydroxylamine, acetamide, and formamide, which can serve as effective promoters or catalysts in facilitating the formation of the TPY core structure.31 | ||
| Scheme 3 Synthesis of substituted TPY by condensation of aldehyde and ketones. | ||
In the synthesis of TPY, the substituents R1 and R5 are typically represented by the 2-pyridyl group, providing the foundational structure for TPY. The remaining substituents, R2, R3, and R4, can be selected based on the desired properties and functionalities of the resulting TPY ligand.
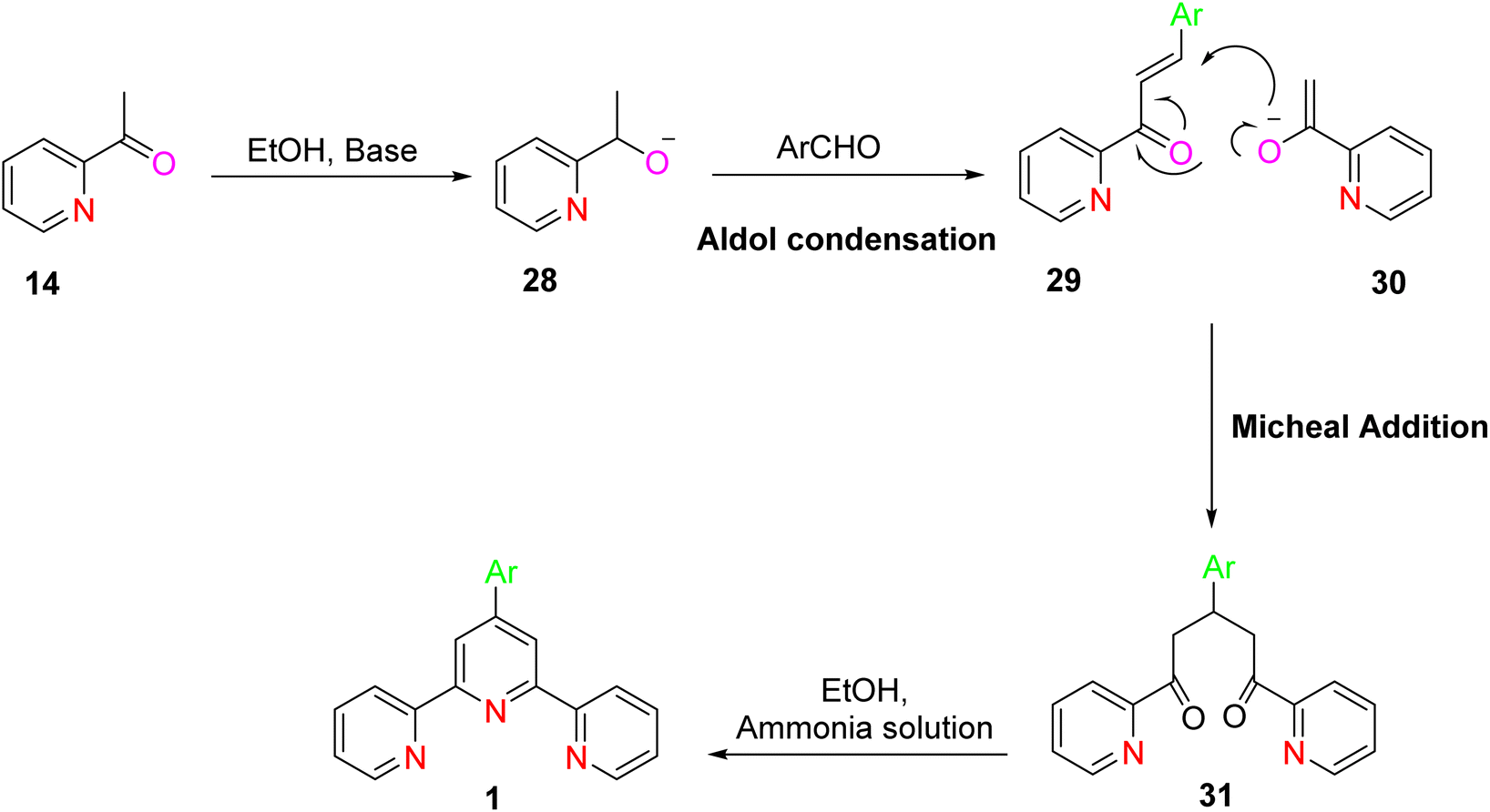
A refined and improved method for TPY synthesis involves a series of chemical transformations, primarily centered around the aldol condensation of 2-acetylpyridine. The process begins with the aldol condensation of 2-acetylpyridine. This reaction can be catalyzed by a base, acid, or even in the presence of alcohol, leading to the formation of an enone compound. Subsequently, a Michael addition reaction occurs, generating an enolate from the enone intermediate. This enolate then participates in a critical step, leading to the formation of a 1,5-diketone compound. To further advance the synthesis, potassium t-butoxide is employed as a base, converting 2-acetylpyridine 14 into potassium enolates. These enolates play a pivotal role in the subsequent ring-closure step. The key step in TPY synthesis involves the ring closure of the potassium enolates of 2-acetylpyridine. This transformation is achieved in the presence of ammonium acetate, ultimately yielding a dihydropyridine 18. The final stage of the synthesis involves the oxidation of the dihydropyridine compound, leading to the desired terpyridine (TPY) ligand, as depicted in Scheme 4.32
 | ||
| Scheme 4 Synthesis of TPY by aldol condensation. | ||
Potts et al. (1994) successfully synthesized a high-yield and versatile ligand known as 4′-methylthio-terpyridine (4′-methylthio-TPY). The synthesis commenced with the reaction of the potassium enolate of 2-acetylpyridine 14 in the presence of carbon disulfide (CS2) and methyl iodide (MeI). This reaction led to the formation of α-oxo ketene dithioacetal, specifically 3,3′-bis(methylthio)-1-(2-pyridyl)propen-1-one 20. Subsequently, the potassium enolate of 2-acetylpyridine was utilized in another reaction, resulting in the production of an enedione compound. The crucial step in the synthesis involved the ring closure of this enedione compound using ammonium acetate in glacial acetic acid under reflux conditions. Through this series of chemical transformations, 4′-methylthio-terpyridine 21 was successfully synthesized (Scheme 5).33–36
 | ||
| Scheme 5 Synthesis of 4′-methylthio-terpyridine 21 from 2-acetylpyridine 14. | ||
Jameson et al. (1991) elucidated a highly efficient two-step synthetic method for the preparation of the TPY 1. Notably, this synthetic procedure can be scaled up to produce multigram quantities of the desired compound. The first step entails the production of an enaminone compound. This is achieved by reacting N,N-dimethylformamide dimethyl acetal 22 with 2-acetylpyridine 14. The resulting enaminone serves as a pivotal intermediate in the synthesis. Subsequently, the enaminone undergoes a condensation reaction facilitated by the potassium enolate of 2-acetylpyridine. This reaction results in the formation of an enone compound. Importantly, during this transformation, dimethylamine is lost from the reaction mixture. The final critical step involves the ring closure of the enone compound. This ring closure process is initiated by the addition of ammonium acetate, and it proceeds without isolating the intermediate 1,5-dione compound (Scheme 6).32
 | ||
| Scheme 6 Synthesis of 2,2′:6′,2′′-TPY by reaction in the presence of dimethylformamide. | ||
Various approaches are employed for the synthesis of both simple terpyridine (TPY) and substituted terpyridines. These synthetic strategies, as outlined in Schemes 7–9, rely on the ring assembly method to construct the TPY framework. Here, we provide an overview of key reactions involved in the synthesis of these terpyridines via the ring assembly approach.17
 | ||
| Scheme 7 Synthesis of substituted-TPY from (E)-3-phenyl-1-(pyridin-2-yl)prop-2-en-1-one. | ||
 | ||
| Scheme 8 Synthesis of substituted-TPY from 2-acetylpyridine in the presence of Al2O3. | ||
 | ||
| Scheme 9 Base-mediated preparation of substituted-TPY from 2-acetylpyridine. | ||
The chalcone derivative is treated with an amine base to form an enaminone intermediate. This step involves deprotonation of the α-carbon adjacent to the carbonyl group, leading to the formation of the enaminone. The enaminone is reacted with an aldehyde in the presence of a base. This condensation reaction results in the formation of a compound that contains the TPY core structure and the desired substituent. This ring closure process is initiated by the addition of ammonium acetate or a related reagent. It proceeds with the formation of the substituted terpyridine (TPY) structure (Scheme 7).37
The synthesis of substituted terpyridine (TPY) from 2-acetylpyridine in the presence of Al2O3 (aluminum oxide) involves a series of chemical reactions that lead to the introduction of substituents at specific positions within the TPY framework. 2-Acetylpyridine is reacted with an amine base in the presence of Al2O3 as a catalyst. This reaction results in the formation of an enaminone intermediate. The amine base deprotonates the α-carbon adjacent to the carbonyl group, leading to the formation of the enaminone. This ring closure process is initiated by the addition of ammonium acetate or a related reagent. It proceeds with the formation of the substituted terpyridine (TPY) structure (Schemes 8 and 9).37
2.3. One pot synthesis
An efficient one-pot synthesis method has been developed for the synthesis of terpyridine (TPY) ligands. This innovative approach involves the reaction of 2-acetylpyridine 14 and ammonium acetate in the presence of glycol under microwave irradiation (MWI) conditions, and notably, it does not require the use of any catalyst (Scheme 10).38 | ||
| Scheme 10 One-pot reaction of 2-acetylpyridine and aromatic aldehydes. | ||
2.4. Krӧhnke method
The synthesis of TPY ligands via the Krӧhnke reaction is a well-established process. This reaction, first introduced by Krӧhnke in 1976, involves a sequence of chemical steps that culminate in the preparation of TPY ligands for subsequent complexation with transition metals. The Krӧhnke reaction begins with the condensation of 2-acetylpyridine 14 and various substituted benzaldehydes 32 through a straightforward oxidation reaction. This reaction is conducted in the presence of NaOH pellets and ammonium hydroxide (NH4OH) in a methanol (MeOH) solvent medium. The reaction mixture is then refluxed for a period ranging from 2 to 6 hours using a reflux assembly (Scheme 11). The progress and completion of the reaction are monitored by employing thin-layer chromatography (TLC). Once the reaction has reached its completion, the mixture is subjected to filtration to separate the precipitate. The separated precipitate is thoroughly washed to remove any residual base. This washing process is initiated with an excess amount of distilled water and continues until the washing becomes neutral. The precipitate is then subjected to a final wash with an ice-cold ethanol solution. The washed precipitate is allowed to dry in an open atmosphere.13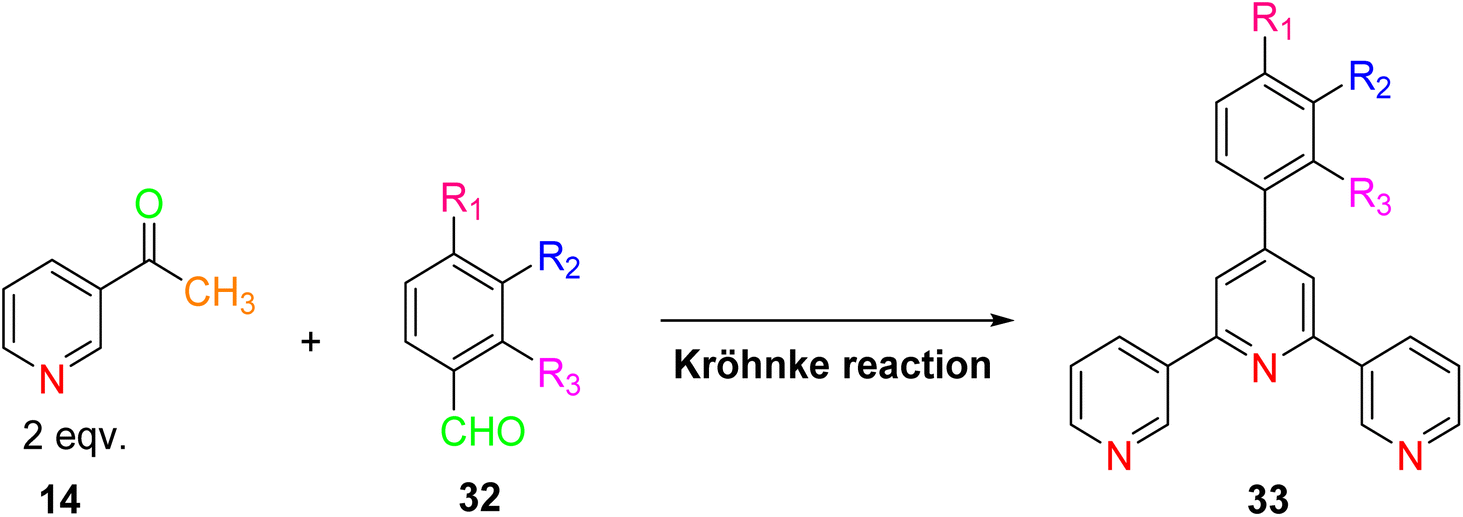 | ||
| Scheme 11 Synthesis of substituted-TPY from 2-acetylpyridine and substituted benzaldehyde. | ||
Following the synthesis of ligand 33, complexation with metal ions is achieved through a carefully controlled process. The process begins by slowly adding a methanol solution containing the targeted metal salt to dichloromethane, which contains the synthesized TPY ligand 33. This addition is performed drop by drop while maintaining constant stirring. As the reaction progresses, a noticeable color change occurs. The reaction mixture is continuously stirred at room temperature for approximately 2 hours. Ammonium hexafluorophosphate (NH4PF6) is added to the reaction mixture to induce precipitation of the TPY metal complexes 34. The precipitated complexes are washed with ice-cold methanol followed by diethyl ether to remove impurities. To obtain pure complexes, crystallization is performed using either acetonitrile or methanol solutions (Scheme 12). These complexes exhibit an octahedral geometry where metal ions are surrounded by two ligands.13
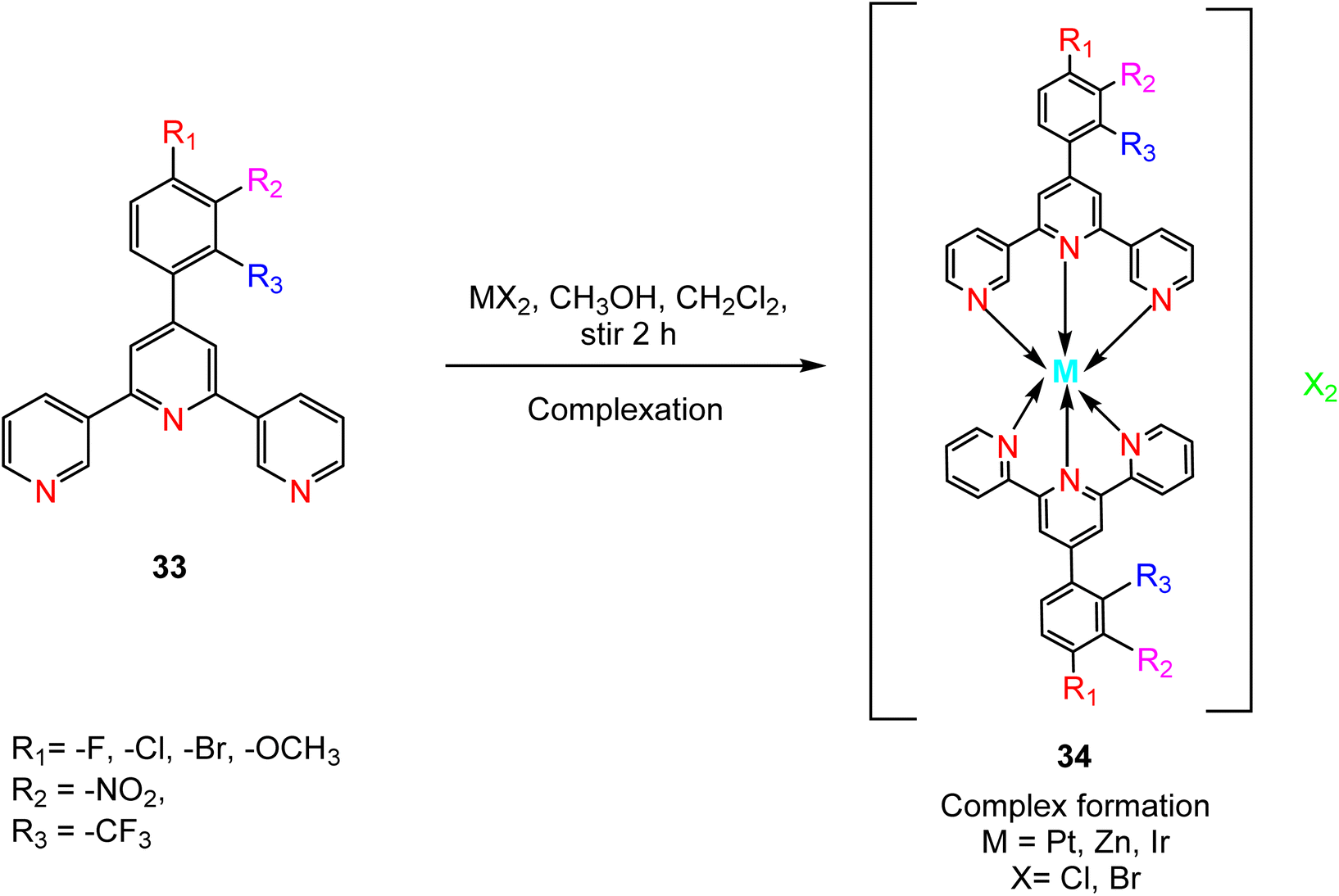 | ||
| Scheme 12 Complexation of substituted-TPY with different metals. | ||
2.5. Stille coupling
The synthesis of terpyridine (TPY) through conventional condensation processes offers simplicity but has limitations, particularly in attaching different substituents to both sides of the two pyridine rings. To overcome this challenge, a novel procedure involving palladium-catalyzed cross-coupling reactions has been investigated. Several researchers have explored conditions for Suzuki, Negishi, and Stille coupling reactions, which are known for forming carbon–carbon (C–C) bonds. However, these cross-coupling reactions have certain drawbacks, including low reactivity and challenges associated with the binding between TPY and palladium catalysts.28Among these coupling reactions, Stille cross-coupling stands out for its higher yield and tolerance toward various functional groups. This reaction involves the use of aryl tin compounds through a transmetalation process. Specifically, it can be accomplished by employing a 2,6-dihalopyridine 35 in combination with 2-stannylpyridines 36 or 2,6-distannylpyridine 40 with 2-bromopyridine 41, as depicted in Scheme 13.39
 | ||
| Scheme 13 TPY synthesis by Stille-coupling. | ||
This synthetic process allows for the introduction of a wide range of functional groups at the 2, 4, and 6 positions of the pyridine rings, leading to the production of various symmetrical and unsymmetrical TPY derivatives.40
3. Applications of terpyridine-based metal complexes
3.1. Catalysis
The TPY ligand is a tridentate pincer ligand known for its ability to stabilize various transition metal cations. This stabilization arises from the presence of three nitrogen-based coordination sites and a rotatable sigma bond within TPY. The TPY moiety comprises three electron-deficient pyridine rings, rendering it a versatile σ donor and π acceptor ligand. This property gives rise to its classification as a “non-innocent” ligand, primarily due to the phenomenon of electron delocalization from the metal atom to the lower-lying LUMO (Lowest Unoccupied Molecular Orbital) of the pyridine rings during redox chemistry. The nucleophilic characteristics of TPY as a σ donor ligand are attributed to the appearance of electron density on its constituent atoms and the steric effects associated with the pyridine rings. Because of its propensity to stabilize metal cations in lower oxidation states, TPY ligands serve as efficient catalysts in promoting the formation of carbon–carbon (C–C) bonds in cross-coupling reactions (Fig. 5).41 | ||
| Fig. 5 Application of TPY metal complexes as a catalyst. | ||
Che et al. (2010) reported the successful epoxidation and aziridination of alkene 44 using iron bis-terpyridine (TPY) metal complexes as catalysts. This transformation was achieved by employing a hybrid material, Fe(II) bis-TPY, which was immobilized onto SBA-15 (43). SBA-15 is a mesoporous material known for its large surface area and high porosity. The characterization results revealed that the average loading of ferrous ions on the catalyst was approximately 1.9 weight percent, as determined by atomic adsorption spectroscopy. This loading level was crucial for the catalytic activity of the iron bis-TPY complexes immobilized on the SBA-15 support. This catalyst exhibited excellent utility in a wide range of C–C coupling reactions, including the catalytic oxidation of various compounds, such as amines and arenes. Notably, in the epoxidation of alkenes 45, this heterogeneous catalyst delivered high yields, with the specific outcome depending on the choice of substrate and reaction conditions (as illustrated in Scheme 14). Furthermore, an essential feature of this catalyst system was its recyclability. After reloading the SBA-15-TPY support, it could be effectively reused in various reactions, demonstrating its sustainability and cost-effectiveness as a catalyst platform.43
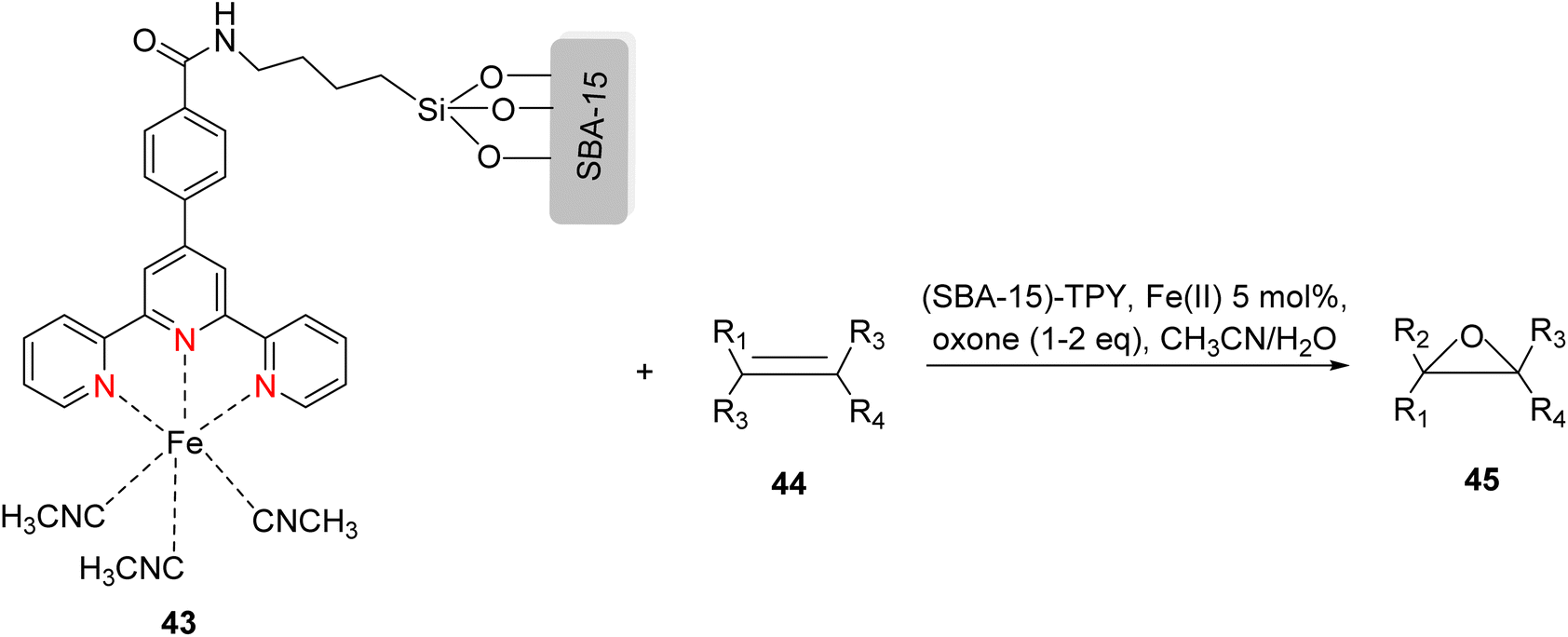 | ||
| Scheme 14 Fe(II)-TPY immobilized on SBA-15: a versatile heterogeneous catalyst for alkene epoxidation. | ||
Kamata et al. (2012) elucidated the catalytic hydrosilylation of alkenes 47 using various iron-based terpyridine (TPY) complexes with distinct substituents (designated as 49). The research findings, as illustrated in the provided Scheme 15, revealed noteworthy insights into the impact of substituent groups on the reactivity of these complexes. Specifically, when chlorine (Cl) and bromine (Br) were employed as substituents, no discernible reaction occurred under the experimental conditions. However, a significant enhancement in reactivity was observed when bulky substituents were introduced onto the iron-based TPY complexes at the 6 and 6′ positions. This modification led to significantly higher yields in the catalytic hydrosilylation reaction. The catalytic process involved the use of sodium triethylborohydride (NaHBEt3) as a reducing agent and resulted in the formation of 1-silylalkane 48, as depicted in Scheme 15. The utilization of iron salts as catalyst precursors proved to be more effective than chloride salts, resulting in improved reaction outcomes.44 Different R and X groups are used in catalysts for catalytic hydrogenation of alkenes along with their TON (s−1) (Table 1).
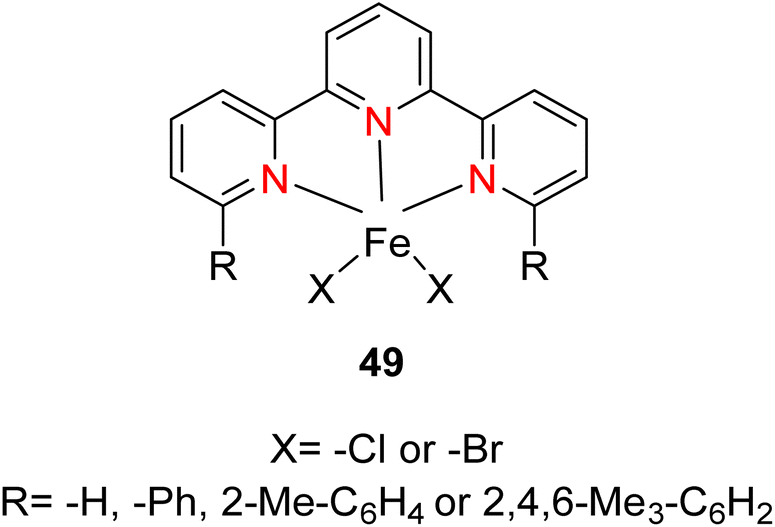
 | ||
| Scheme 15 Fe-catalyzed hydrosilylation of alkene. | ||
| Derivative number | X | R | TON (s−1) |
|---|---|---|---|
| 49a | Cl | H | 0 |
| 49b | Br | H | 0 |
| 49c | Cl | Ph | 4 |
| 49d | Br | Ph | 19 |
| 49e | Br | 2-Me-C6H4 | 4 |
| 49f | Cl | 2,4,6-Me3-C6H2 | 15 |
| 49g | Br | 2,4,6-Me3-C6H2 | 70 |
In 2018, Lin et al. introduced a novel class of catalytic materials known as TPY-based Metal–Organic Layers (MOLs), which exhibited superior performance compared to traditional Metal–Organic Frameworks (MOFs). MOFs have been widely used as catalysts; however, their catalytic efficiency is often hindered by the limited diffusion rates of reactants and products within their three-dimensional frameworks. To address this diffusion limitation, several strategies have been explored, including the construction of MOFs with larger pores and channels by modifying or diluting spectator ligands. Unfortunately, these techniques have encountered challenges such as framework distortion and difficulties in interpretation. To overcome these limitations, TPY-based Metal–Organic Layers (MOLs) were developed. These MOLs represent a reduction in dimensionality from MOFs to a scale of a few nanometers, resulting in two-dimensional structures that offer enhanced accessibility to substrates during catalytic reactions. TPY-based MOLs possess several advantageous properties, including molecular tunability, ordered structures, and heterogeneous characteristics. One specific TPY-based MOL highlighted in the study was FeBr2-TPY MOL, which was employed in the catalytic intramolecular C(sp3)–H amination of alkyl azides (substrates 50 and 52) for the synthesis of pyrrolidines and piperidines. MOLs are regarded as single-site solid catalysts that do not suffer from diffusional constraints. FeBr2-TPY MOL was synthesized through the metalation of FeBr2(THF)2. Remarkably, when FeBr2-TPY MOL reacted with NaHBEt3, it generated FeBr2(THF)2 along with the release of 1 equivalent of H2. In the catalytic process, 2% mol FeBr2-TPY MOL was utilized, along with NaHBEt3 as an activation agent and two equivalents of di-tert-butyl dicarbonate, conducted at 90 °C. This reaction led to the production of Boc-protected α-phenyl pyrrolidine 51, as illustrated in Scheme 16.45 Different catalysts used for these purposes are provided in (Table 2), along with their yield and TON (s−1).
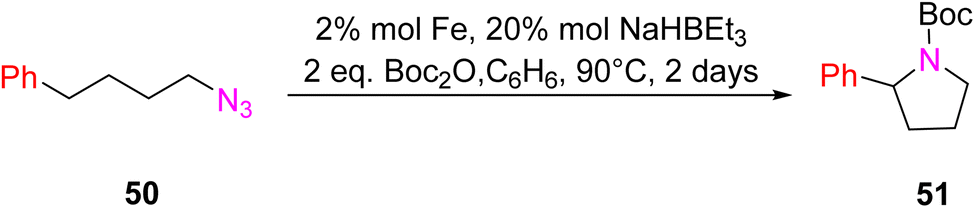 | ||
| Scheme 16 Iron-catalyzed intramolecular C–H amination. | ||
| Entry | Catalyst | Yield (%) | TON (s−1) |
|---|---|---|---|
| 50a | FeBr2-TPY MOL | 89 | 44.5 |
| 50b | FeBr2-TPY MOF | 10 | 5 |
| 50c | Homogeneous Fe(TPY)Br2 | 4 | 1.5 |
In 2020, Ye et al. presented a study focusing on the synthesis of iron TPY complexes designed for the degradation of pollutants, particularly the degradation of phenol in water. The iron(II) TPY complexes were found to generate a significantly larger quantity of high-valent iron-oxo centers and hydroxide radicals compared to their iron(II) TPY counterparts, resulting in higher catalytic activity. Notably, the research revealed that FeIII(Tpy)Cl3 exhibited superior catalytic performance in the degradation of phenol when compared to FeII(Tpy)Cl2 (52–62), as illustrated in Scheme 17. The catalytic activity of these complexes was attributed to the valence state of the metal center, where the presence of Fe(III) led to enhanced catalytic activity. These complexes were capable of converting phenols into less acidic water, with a pH ranging from approximately 4–5.87 during the degradation process. Future research endeavors are expected to involve the immobilization of this catalyst onto various semiconductors to harness its potential as a heterogeneous catalyst. The advantages of homogeneous catalysts such as these include their practicality, cost-effectiveness, ready availability, recyclability, and the ability to be easily activated by absorbing light energy.46–48
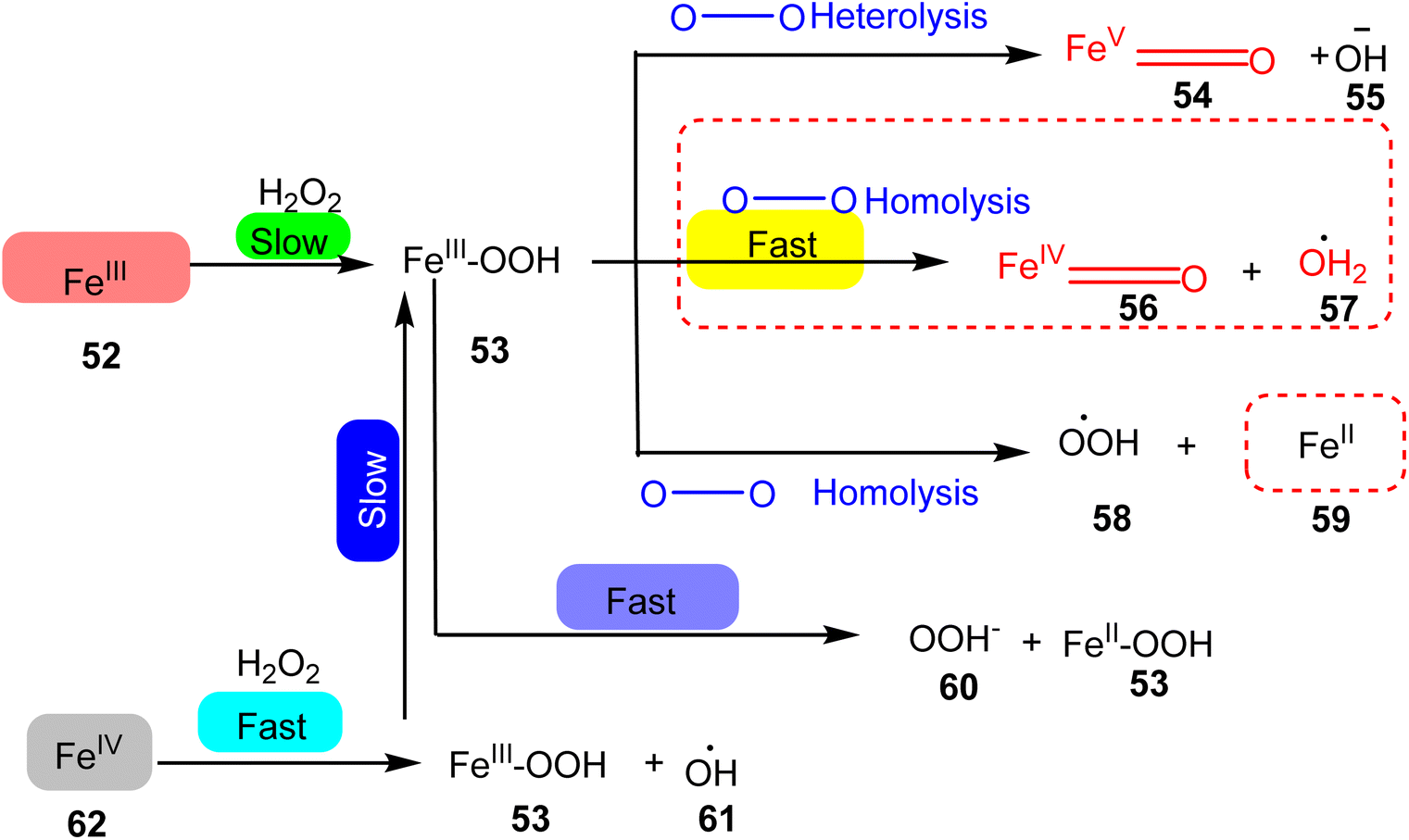 | ||
| Scheme 17 Pathways of the generation of reactive oxygen intermediates for degradation of phenol to water. | ||
In 2022, Wei et al. conducted a study focusing on the Friedel–Crafts alkylation of β-nitroalkenes and indoles in an ambient environment, utilizing iron-based TPY (terpyridine) metal complexes as catalysts. Before this research, homogeneous catalysts were commonly employed for Friedel–Crafts alkylation of β-nitroalkenes to enhance the nucleophilic properties of indoles. However, these homogeneous catalysts posed several drawbacks, including negative environmental effects, economic inefficiency, and non-recyclability. To address these limitations, the researchers developed heterogeneous catalysts that are cost-effective and environmentally friendly. These catalysts include both Brønsted and Lewis acid catalysts, which exhibit good reactivity in Friedel–Crafts alkylation reactions. Such catalysts encompass materials like MOFs (Metal–Organic Frameworks), metal-supported salts, zeolites, and mesoporous solid acid catalysts. Iron, due to its relatively nontoxic and environmentally friendly nature, is considered a vital metallic catalyst when coordinated with various ligands. The synthesis of compound 64 (as depicted in Scheme 18) involved the reaction of 4′-(3,5-dicarboxyphenyl)-2,2′:6′,2′′-TPY (H2dtp, 9.93 mg, 0.025 mmol) 63 with FeCl2·4H2O. The resulting mixture was dissolved in a solution of acetonitrile and water, stirred for 10 minutes using a magnetic stirrer, and then transferred to an autoclave under a nitrogen atmosphere. It was then heated at 150 °C for 48 hours, resulting in the formation of black lumpy crystals, referred to as compound 64. Similarly, compound 65 (as shown in Scheme 18) was synthesized using a similar approach, but in this case, reductive iron powder was employed. The reaction was carried out in deionized water and HF (hydrofluoric acid), stirred for 10 minutes with a magnetic stirrer, and then transferred to an autoclave. The mixture was heated for 48 hours in an oven at 130 °C, followed by cooling at room temperature, resulting in the formation of orange crystals, designated as compound 65.49
 | ||
| Scheme 18 Synthetic strategies for synthesis of catalyst. | ||
Before initiating catalytic reactions, both catalysts undergo an activation process under vacuum conditions to eliminate water molecules. This activation step is crucial as it provides the catalysts with active sites necessary for substrate interactions. The catalytic activity of both compounds, specifically compound 64 and compound 65, was assessed through Friedel–Crafts alkylation reactions involving indole (Scheme 19) 66 and β-nitrostyrene 67. These reactions were conducted in the presence of the respective catalysts and various solvents, yielding different levels of product yield. The experimental findings revealed that compound 65 exhibited superior catalytic activity, achieving a notably high yield of 93% in the reaction. The resulting product was characterized and studied using 1H-NMR spectroscopy.50 Different reaction conditions for Friedel–Crafts alkylation of indole 68 and β-nitrostyrene are shown in (Table 3).
 | ||
| Scheme 19 Friedel–Crafts alkylation of indole and β-nitrostyrene under different conditions. | ||
| Entry | Temperature | Cata. mol% | Cata. | Solvent | Yielda |
|---|---|---|---|---|---|
| a Isolated yield based on β-nitrostyrene.b Recycled 2 used as catalyst. | |||||
| i | 25 °C | 5 | 86 | CHCl3 | 40 |
| ii | 25 °C | 5 | 87 | CHCl3 | 93 |
| iii | 25 °C | 5 | 87 | DCM | 96 |
| iv | 25 °C | 5 | 87 | Propane | 91 |
| v | 25 °C | 5 | 87 | C2H5OH | 39 |
| vi | 25 °C | 5 | 87 | THF | — |
| vii | 25 °C | 5 | 87 | Toluene | — |
| viii | 25 °C | 5 | 87 | CH3CN | — |
| ix | 25 °C | 5 | — | CHCl3 | — |
| x | 25 °C | 5 | FeCl3 | CHCl3 | 61 |
| xi | 25 °C | 5 | Fe2(SO4)3 | CHCl3 | 87 |
| xii | 25 °C | 5 | FeCl2 | CHCl3 | 58 |
| xiii | 25 °C | 5 | FeSO4 | CHCl3 | 56 |
| xiv | 25 °C | 5 | FeCl2 + H2dtp | CHCl3 | 62 |
| xv | 25 °C | 5 | 87 | DCM | 98 |
| xvi | 25 °C | 1 | 87 | DCM | 86 |
| xvii | 25 °C | 10 | 87 | DCM | 98 |
| xviii | 25 °C | 5 | 87 (1st)b | DCM | 80 |
| xix | 25 °C | 5 | 87 (2nd)b | DCM | 79 |
| xx | 25 °C | 5 | 87 (3rd)b | DCM | 81 |
Through the optimization of reaction conditions, a range of diverse products were synthesized, and their structures were subsequently analyzed using 1H-NMR spectroscopy. Notably, the catalytic activity of compound 66, as outlined in Scheme 20, exhibited variations in response to different substituents on the indole ring. The introduction of electron-donating groups into the indole ring resulted in an increased propensity for nucleophilic attack. This enhanced nucleophilicity was attributed to the activation of the indole ring by the electron-donating substituents. However, this beneficial effect on nucleophilic reactivity was accompanied by a marginal decrease in the overall yield of the reaction. Conversely, when electron-withdrawing groups were introduced into the indole ring, a significant decrease in yield was observed. This substantial decrease in yield was primarily due to the strong electron-withdrawing nature of this substituent.50
 | ||
| Scheme 20 Friedel–Crafts alkylation of indole and β-nitrostyrene catalyzed by compound 66. | ||
Wei et al. (2022) proposed a comprehensive mechanistic pathway (as illustrated in Scheme 21) for the reaction involving indole and β-nitrostyrene 67. The mechanistic steps outlined the activation of the catalyst, substrate interactions, and the formation of key intermediates. The first step of the mechanism involved the activation of the catalyst, which included the removal of water (H2O) molecules. This activation process exposed coordinated unsaturated sites (CUS) on the catalyst, creating active sites for substrate binding. Simultaneously, the removal of the carboxylic group 69 played a pivotal role. This suspended carboxylic group acted as a hydrogen bond donor, serving as an alternative to a Brønsted acid for interaction with β-nitrostyrene. This interaction was particularly important due to the relatively weak polarity of the solvent (chloroform), which hindered proton ionization from the carboxylic group. In the subsequent steps, compound 67's nitro group interacted with the coordinated unsaturated sites of the substrate through coordinated interactions. This interaction increased the electrophilic nature of the aliphatic carbon atom in the substrate, facilitating the attack by indole 70. During this process, a water molecule was eliminated as a result of the activation step. Additionally, the carboxylic group in compound 70 temporarily lost its hydrogen bond, only to establish a new hydrogen bond with the N–H group of the incoming indole 71. This step effectively guided both substrates into the reaction framework.
 | ||
| Scheme 21 Proposed mechanism of indole with β-nitrostyrene. | ||
The discussion presented thus far highlights that the catalytic activity of compound 66 is notably enhanced due to the availability of sufficient catalytic sites. These sites facilitate the formation of intermediates through coordination with Fe(II), ultimately increasing the electrophilic character and enabling the indole attack 72. Compound 66 exhibits favorable solvent tolerance and functional group tolerance, making it versatile for various reactions. Importantly, it can be reused multiple times without a significant loss of catalytic activity. The reusability of terpyridine-based metal complexes as catalysts is an important attribute that enhances their practical utility in chemical reactions. These catalysts exhibit notable stability and can be recycled multiple times without significant loss of activity. Typically, they retain their catalytic performance through several cycles, maintaining high conversion rates and selectivity. Effective recovery methods, such as filtration or magnetic separation, facilitate their reuse, making them both economically and environmentally advantageous. This reusability not only reduces the need for frequent catalyst replacement but also contributes to more sustainable and cost-effective chemical processes. This catalyst's broad utility extends to the synthesis of diverse heterocyclic compounds using indole and β-nitrostyrene substrates under mild reaction conditions.50
In 2013, the research group led by Szymczak reported the utilization of 6,6′-dihydroxy TPY, a ligand coordinated to a ruthenium (Ru) metal center, for catalytic transfer hydrogenation of ketones 74, as illustrated in Scheme 22 with ketone substrate 73 and other substituted ketones (73a–73k). This ligand, 6,6′-dihydroxy TPY 75, possesses a bifunctional nature, allowing for facile proton transfer between the metal-coordinated substrate and other species. The structural characteristics of this catalyst were investigated through X-ray diffraction techniques, providing valuable insights into its molecular arrangement and spatial configuration. This research highlights the significance of 6,6′-dihydroxy TPY as a versatile ligand in catalytic transfer hydrogenation reactions, with the potential to drive advancements in synthetic chemistry and catalysis.52
 | ||
| Scheme 22 Catalytic transfer hydrogenation of ketones. | ||
The ruthenium complex depicted in Fig. 6 played an active role in the process of transfer hydrogenation of ketones. This reaction was facilitated using potassium tert-butoxide (KtOBu) in the presence of propanol as a co-solvent. Notably, the preceding discussion underscores the achievement of high selectivity when employing the ruthenium-based 6,6′-dihydroxy TPY complex 75. This selectivity is particularly pronounced when dealing with substituted alkenes in the presence of non-polar olefins. The mechanistic pathway underlying this catalytic process involves an outer sphere proton transfer event, wherein protons are transferred from the metal center to the ligand and subsequently to the ketone substrate (Table 4). This intricate process governs the high selectivity observed in the transformation.52
 | ||
| Fig. 6 Catalyst 75 used for transfer hydrogenation of ketone. | ||
| Compound no. | Substrate | Yield (%) | TOF (h−1) |
|---|---|---|---|
| 73a |  |
98 | 82 |
| 73b |  |
100 | 100 |
| 73c |  |
92 | 42 |
| 73d |  |
100 | 64 |
| 73e | 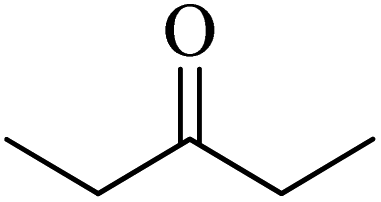 |
78 | 66 |
| 73f | 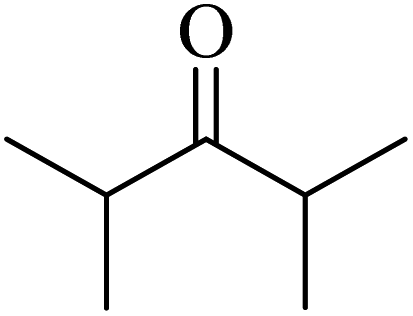 |
17 | 10 |
| 73g |  |
0 | 0 |
| 73h |  |
79 | 26 |
| 73i |  |
57 | N.D. |
| 73j |  |
66 | N.D. |
| 73k |  |
95 | N.D. |
In 2013, Cristian et al. reported the development of ruthenium-based TPY complexes designed for the direct oxidation of amines to nitriles. These ruthenium-based metal catalysts were found to be effective for the direct oxidation of both aliphatic and aromatic amines 76 into their corresponding nitriles 77. The process was characterized by its environmentally friendly and efficient nature, as it relied on the utilization of dioxygen as an oxidizing agent. The success of this oxidation reaction was attributed to the presence of the azulenyl moiety located at the 4-position of the pyridine ring, as illustrated in Scheme 23. Prior to this work, Meyer et al.53 had also reported the development of another ligand 78 capable of activating ruthenium for various reactions. Various ruthenium complexes were explored, including 2,2′:6′,2′′-terpyridine, 4′-(4-methylphenyl)-2,2′:6′,2′′-terpyridine, 4′-(4,6,8-trimethyl-azulenyl)-TPY, and 4′-azulenyl-TPY. Among these complexes, ruthenium binuclear complexes and the 4′-azulenyl-2,2′:6′,2′′-TPY complex 79 (Scheme 24) exhibited particularly high oxidation activity. It was observed that the conversion efficiency of these complexes was influenced by the length of the alkyl chain present in the reactants. Specifically, reactants with shorter alkyl chains displayed lower conversion rates compared to those with longer alkyl chains.54 Several types of substrates for direct oxidation of amines (76a–76k) to nitrile (77a–77k) are shown in Table 5.
 | ||
| Scheme 23 Direct oxidation of amines with oxygen. | ||
 | ||
| Scheme 24 Formation of dinuclear ruthenium(II) complex with 4′-azulenyl-TPY. | ||
| Entry | Substrate | Time (h) | Conversion (%) | Entry | Product | Selectivity (%) |
|---|---|---|---|---|---|---|
| 76a | n-Dodecylamine | 5 | 100 | 77a | n-Dodecane nitrile | 100 |
| 76b | n-Stearylamine | 5 | 100 | 77b | n-Stearylonitrile | 100 |
| 76c | Benzylamine | 2 | 100 | 77c | Benzonitrile | 100 |
| 76d | Ethylamine | 5 | 8 | 77d | Acetonitrile | 100 |
| 76e | n-Butylamine | 5 | 15 | 77e | n-Butanenitrile | 100 |
| 76f | iso-Butylamine | 24 | 18 | 77f | iso-Butyronitrile | 100 |
| 76g | n-Hexylamine | 5 | 58 | 77g | n-Hexanenitrile | 100 |
| 76h | n-Octylamine | 5 | 67 | 77h | n-Octanenitrile | 100 |
| 76i | 2-Methoxy benzylamine | 5 | 34 | 77i | 2-Methoxy benzonitrile | 100 |
| 76j | 4-Methyl benzylamine | 5 | 100 | 77j | 4-Methyl benzonitrile | 100 |
| 76k | 4-Chloro benzylamine | 5 | 66 | 77k | 4-Chloro benzonitrile | 100 |
In the proposed mechanism of this reaction (Scheme 25), ruthenium undergoes a crucial sequence of transformations (80–86). Initially, it replaces one of its chlorine ligands with a hydroxide (OH) ligand. This ruthenium hydroxide complex subsequently engages with the amine substrate. Through a β-hydrogen elimination reaction, an intermediate containing ruthenium and the amine is formed, leading to the production of an imine compound and a ruthenium hydride species. Following this step, the imine undergoes hydrogenation, a chemical process that converts it into a nitrile compound. Importantly, the catalyst is then reactivated through the oxidation of the formed ruthenium hydride, a reaction facilitated by the presence of dioxygen.54
 | ||
| Scheme 25 Proposed mechanism of amines oxidation with oxygen by ruthenium(II) complex with 4′-azulenyl-TPY. | ||
In 2015, Beloglazkina et al. conducted research focused on ruthenium and rhodium TPY phenanthroline complexes with catalytic activity for the oxidation of styrene 87 to styrene oxide 88. This catalytic transformation was carried out in the presence of iodobenzene under mild reaction conditions, as illustrated in Scheme 26. Electrochemical studies were conducted using cyclic voltammetry with various electrodes, including glossy carbon, platinum, and gold electrodes. These experiments aimed to monitor the extent of chemisorption occurring on the gold electrode's surface. The reported ruthenium catalysts exhibited labile bonds, facilitating their interaction with the substrate at the metal center. The presence of halides as co-ligands in the coordination sphere of ruthenium(II) catalysts notably enhanced their catalytic activity. The discussion highlights the ability of mono-TPY ruthenium complexes to readily substitute chloride ions in the coordination sphere during the oxidation of triphenylphosphine or styrene in the presence of iodobenzene under mild reaction conditions. A specific complex, [Ru(phen)(Tpy-C6H4-O-(CH2)4-SCOCH3)Cl]PF6, demonstrated high catalytic activity for the oxidation of triphenylphosphine and styrene. Furthermore, this catalyst could be conveniently recovered and reintroduced into the reaction, emphasizing its recyclability and practical utility.55
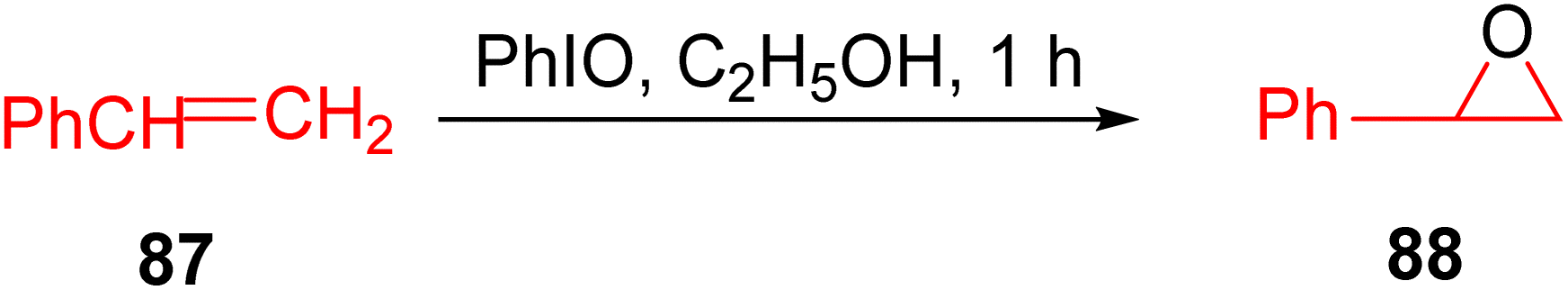 | ||
| Scheme 26 Oxidation of triphenylphosphine and styrene. | ||
Yoshida et al. (2015) conducted research focused on water oxidation, specifically the evolution of oxygen, using ruthenium-based TPY (terpyridine) complexes. These complexes were engineered to include pendant SO3-groups and were employed in the presence of Ce(IV) as an oxidant to drive this catalytic process in the secondary coordination sphere. The catalyst employed for this water oxidation process was identified as [Ru(TPY)-(bpyms)(OH2)] (with TPY denoting 2,2′:6′,2′′-terpyridine and bpyms referring to 2,2′-bipyridine-5,5′-bis(methanesulfonate)), designated as complex 90. Experimental studies revealed that water oxidation occurred more rapidly in the presence of Ce(IV) as compared to the parent ruthenium complex, [Ru(TPY)(bpy)(OH2)] 89 (where bpy = 2,2′-bipyridine), which served as a control in the study (Scheme 27a). Notably, the formation of the O–O bond crucial for oxygen evolution was promoted through the incorporation of the pendant SO3− group in the coordination sphere. Interestingly, this modification did not significantly impact the parental catalyst's behavior. This insight was derived from comprehensive investigations employing spectroscopic methods, electrochemical analyses, and X-ray crystallography. The pendant SO3− group's role in enhancing the rate of O2 evolution in the presence of Ce(IV) 91 is attributed to its capacity to repress Ce(IV)-driven water oxidation (Scheme 27b). This process operates by repressing Ce(IV) activity and facilitating the formation of a heterometallic O–O bond, as confirmed by X-ray crystallography.56
 | ||
| Scheme 27 (a) Structure of ruthenium TPY complexes (89 & 90) as a catalyst (b) scheme for the working of catalyst 91. | ||
Maity et al. (2018) conducted a study focusing on both cationic and neutral ruthenium-based TPY (terpyridine) metal complexes for transfer hydrogenation of ketones. To achieve this, they synthesized a diverse array of neutral and cationic ruthenium-based TPY metal complexes using the Krӧhnke reaction as their synthetic approach. For the cationic complexes, a comprehensive analysis was carried out employing various spectroscopic techniques, including FT-IR, NMR, and HRMS. These analyses provided detailed insights into the structural and spectroscopic properties of the cationic ruthenium complexes. In contrast, the neutral complexes were investigated using the single-crystal X-ray diffraction method, allowing for a precise determination of their molecular structures. Furthermore, to assess the impact of different aryl groups, photophysical and electrochemical studies were conducted. The results obtained from these studies revealed a notable trend: neutral ruthenium TPY complexes exhibited a more significant role in the transfer hydrogenation of ketones when compared to their cationic counterparts (as illustrated in Scheme 28). This difference in catalytic activity was attributed to the lower reduction potential exhibited by the neutral ruthenium complexes, characterized by an E1/2 = 0.30 V. This lower reduction potential facilitated the transfer hydrogenation of ketones, highlighting the importance of these neutral ruthenium TPY complexes in catalytic processes (92–99).57
 | ||
| Scheme 28 Proposed mechanism of cationic ruthenium(II) catalyst for transfer hydrogenation of ketones. | ||
Maity et al. (2018) revealed an interesting trend where electron-deficient or electron-poor ketones exhibit faster reaction rates compared to electron-rich ketones. Additionally, it was observed that ortho-substituted aromatic ketones tend to react at a slower pace in comparison to para-substituted aromatic ketones. This variation in reactivity highlights the significance of electronic properties and steric effects in influencing the kinetics of these catalytic reactions. Moreover, the neutral Ru(II) complexes demonstrated superior catalytic activity when compared to their cationic counterparts. This enhanced performance can be attributed to their lower reduction potential, approximately 0.30 V, which facilitates the reduction of ketone substrates. In addition to the ketone transfer hydrogenation, further investigations were conducted to explore the applicability of ruthenium TPY complexes in hydrosilylation reactions. These studies expand the scope of potential applications for these versatile catalysts.57
In 2022, Buhler et al. presented a study detailing the electrochemical ruthenium-catalyzed C–H activation process (100–104) in an aqueous environment, utilizing a heterogeneous catalyst. The central component of this research was a ruthenium-based molecular catalyst denoted as [Ru(TPY)(pic-PO3H2)(Cl)], where TPY represents 2,2′:6′,2′′-terpyridine, and pic-PO3H2 signifies 4-phosphonopyrid-2-yl carboxylic acid. This catalyst, labeled as complex 100, was synthesized and its characteristics were thoroughly investigated using various spectroscopic techniques. The primary objective of this catalytic system was the oxidation of benzyl alcohol 102, as illustrated in Scheme 29. To achieve this, the researchers conducted the reaction in an aqueous medium, utilizing ceric ammonium nitrate as the terminal oxidant. It's worth noting that this choice of oxidant led to the rapid deactivation of the catalyst. To facilitate the catalytic process, the catalyst was immobilized on the surface of a metal oxide. Previously, this heterogeneous catalyst had been employed for water oxidation,58 but in this study, its application was extended to the oxidation of alcohols. To gain deeper insights into the catalyst's behavior, X-ray photoelectron spectroscopy (XPS) studies were performed. These investigations revealed that the catalyst decomposed over time, ultimately limiting its overall activity. Future research in this domain will likely delve into the mechanistic aspects of the reaction, with the aim of developing more efficient catalysts for such transformations.59
 | ||
| Scheme 29 Chemical oxidation of catalyst and then oxidation of benzyl alcohol. | ||
Lewis et al. (2013) delved into the exploration of dimanganese complexes functioning as catalysts for water oxidation. Specifically, they detailed the characterization of two dimers of manganese, denoted as MnIII(μ-O)2MnVI dimer, within the context of their catalytic activity. The molecular structure of these dimers featured the ligand Py-terpy, where Py-terpy stands for 4′-(4-pyridyl)-2,2′:6′,2′′-TPY(1Py), with the pyridyl moiety serving as an electron-withdrawing group. It is noteworthy that 1Py exhibited a higher degree of stability in aqueous solutions compared to other electron-withdrawing groups such as chloride or bromide, which were found to be unstable in solution. The water oxidation processes under investigation were observed in both homogeneous and heterogeneous systems involving sponite or mica adsorbents. Intriguingly, when Ce(IV) was utilized as the oxidant in an aqueous solution, the maximum turnover number for 1Py was approximately 0.63 over a span of 4 days, indicating limited catalytic efficacy in the aqueous medium. However, in the context of the mica adsorbent system or sponite, a significant amount of molecular oxygen (O2) production was observed. In this scenario, the maximum turnover number reached 5.6 to 16 over the same 4 day period. Collectively, these investigations elucidated that 1Py effectively functioned as a heterogeneous catalyst when absorbed into sponite or mica adsorbent systems. This research sheds light on the potential of such catalysts for water oxidation processes, particularly in heterogeneous environments.62 Previously, the manipulation of selectivity in C–H oxidation reactions was primarily achieved through the utilization of non-covalent catalysts. However, recent advancements in the field, spearheaded by Lewis and his research group, have shifted the focus towards the exploration of artificial metallo-enzymes. Fig. 7a illustrates the structural features of these enzymes and underscores their fundamental roles in catalysis. In this context, Lewis and his team have particularly investigated the interaction between a mutant protein, tHisF, containing a cysteine residue, and manganese complexes. This interaction is facilitated through the thiol addition to the maleimide moiety within the complex. The conjugation of these enzymes and metallo-enzymes was meticulously studied using a combination of analytical techniques, including circular dichroism (CD), UV-visible absorption spectroscopy, and mass spectrometry. The metallo-enzyme denoted as NB-CSY [cysteine (Cys) mutants of the proteins tHisF and apo-nitrobindin (Nb)], as depicted in Fig. 7b, catalyzes C–H oxygenation reactions 105. These reactions (compounds 106–111) are conducted in the presence of oxidizing agents such as oxone or preacetic acid (Scheme 30). The scope of these transformations encompasses benzylic oxygenation, epoxidation, and the oxidation of α-C–H bonds in ether compounds. This pioneering research signifies a pivotal shift towards harnessing the catalytic potential of metallo-enzymes, offering new avenues for controlling and enhancing the selectivity of C–H oxidation processes. These endeavors hold promise for the development of more efficient and precise oxidation methodologies in organic synthesis.63
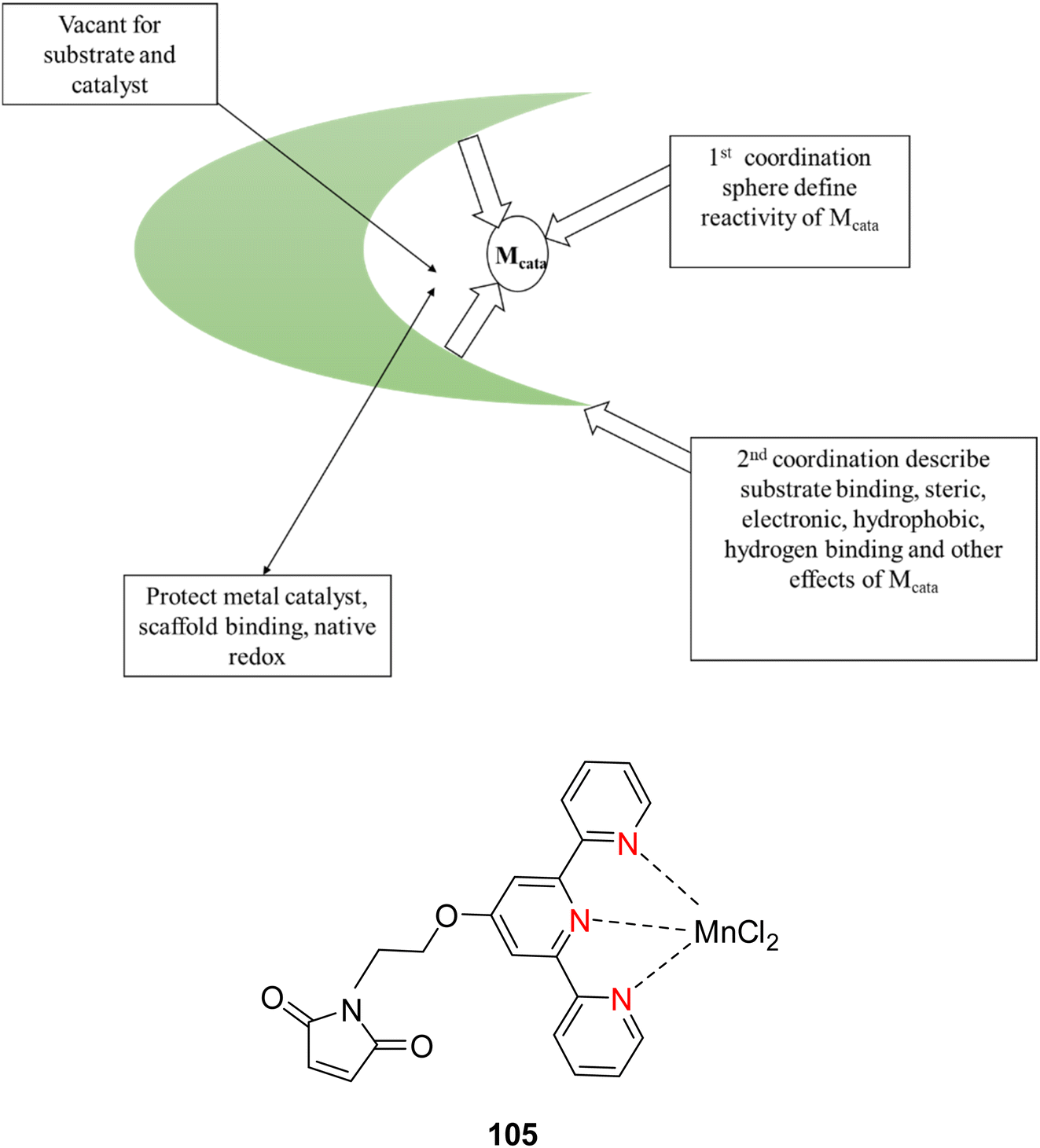 | ||
| Fig. 7 (a) Illustration of artificial metallo-enzyme along with its function. (b) Schematic representation of the Mn(II)-TPY complex 105 which was bound as a cofactor to protein substrates. | ||
 | ||
| Scheme 30 Schematic representation of various oxygenation reactions catalyzed by artificial metallo-enzyme Nb-CYS. | ||
In 2015, Liu et al. conducted a comprehensive investigation into manganese complexes 113, as illustrated in Scheme 31. These complexes were employed as catalysts in the oxidation of cyclohexene (Scheme 32) and styrene (Scheme 33) reactions, utilizing tert-butylhydroperoxide as the oxidizing agent. Notably, the study unveiled significant catalytic effects in the oxidation of olefinic compounds and the cycloaddition reaction involving styrene. Specifically, the research team synthesized five mononuclear manganese(II) complexes featuring the 4′-Ar 2,2′:6′,2′′-TPY ligand, denoted as 116, which exhibited commendable catalytic activity, particularly in the oxidation of olefins, with a special focus on styrene in the presence of tert-butylhydroperoxide (TBHP).
 | ||
| Scheme 31 Synthesis of ligands and Mn(II) complexes. | ||
 | ||
| Scheme 32 Oxidation of cyclohexene by Mn(II) complex. | ||
 | ||
| Scheme 33 Oxidation of styrene by Mn(II) complexes. | ||
In the case of the cycloaddition reaction of styrene, these complexes demonstrated remarkable performance, achieving a conversion rate of 94% and an impressive selectivity of approximately 96% when utilized as catalysts. It's worth noting that the ligands and complexes were synthesized using previously established methods. This study not only contributes to the understanding of manganese-based catalysis but also highlights the potential utility of these manganese complexes, specifically 4′-Ar 2,2′:6′,2′′-TPY ligated manganese(II) complexes, in the development of efficient and selective catalytic systems for olefin oxidation and cycloaddition reactions, with a particular emphasis on their application in the oxidation of styrene using tert-butylhydroperoxide as an oxidant.64
The results revealed that the products of cyclohexene 114 were 2-cyclohexene-1-one 115, 2-cyclohexene-1-ol 116, and cyclohexene epoxide 117.64 Effects of different Mn(II) for conversion of cyclohexene oxidation are shown in Table 6.
| Product selectivity (%) | |||||
|---|---|---|---|---|---|
| Compound no. | Conversion (%) |
|
|
|
Others |
| 113a | 90 | 5 | 38 | 51 | 6 |
| 113b | 79 | 4 | 36 | 52 | 8 |
| 113c | 88 | 4 | 34 | 55 | 7 |
| 113d | 89 | 10 | 20 | 58 | 12 |
| 113e | 90 | 7 | 31 | 55 | 8 |
Manganese complexes find application in the oxidation of aromatic olefins, with styrene 118 serving as the substrate in this context. This oxidation process yields several products, including benzaldehyde 119, styrene cyclo-oxide 120, and 1-phenylethane-1,2-ethanediol 121.64 Effect of different Mn(II) complexes for oxidation of styrene as shown in (Table 7).
| Product selectivity (%) | ||||
|---|---|---|---|---|
| Compound no. | Conversion (%) |
|
|
|
| 118a | 94 | 2 | 92 | 6 |
| 118b | 77 | 2 | 94 | 4 |
| 118c | 93 | 1 | 95 | 4 |
| 118d | 93 | 1 | 94 | 5 |
| 118e | 85 | 1 | 96 | 3 |



Zhang et al. (2016) documented the synthesis of manganese complexes 120 (Scheme 34) designed for use as catalysts in the hydroboration of alkenes, ketones, and aldehydes. These complexes were meticulously prepared by utilizing TPY metal complexes in combination with TPY ligands 119. Notably, these catalysts exhibited distinguishing efficacy in promoting hydroboration reactions under mild reaction conditions. Additionally, this particular catalyst displayed a clear chemoselectivity which favored the conversion of ketones over alkenes. Due to the paramagnetic properties inherent to Mn(II), no discernible NMR signals were detectable for these complexes.65
 | ||
| Scheme 34 Synthesis of Mn(II) complexes. | ||
The hydroboration of styrene 121, as depicted in Scheme 35, was conducted in the presence of manganese complex 113 and pinacolborane 122. Notably, complex 113 exhibited remarkable catalytic activity when employed in conjunction with diethyl ether (Et2O) as the solvent. This catalytic system yielded two distinct hydroborated products, which were subsequently identified using NMR and GC analyses. Importantly, the outcome of this reaction demonstrated excellent regioselectivity between the resulting products (123:124), with a ratio of 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5.65
:5.65
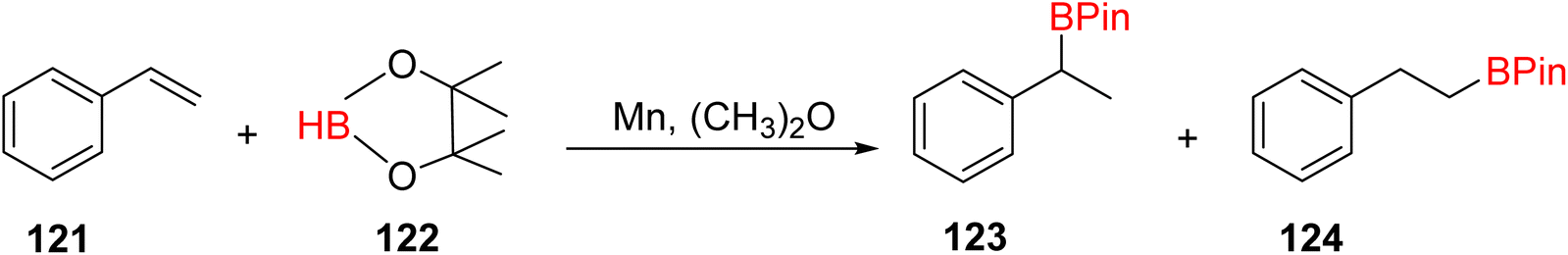 | ||
| Scheme 35 Hydroboration of styrene in the presence of manganese complexes. | ||
The hydroboration reaction of alkene 125 (depicted in Scheme 36) was conducted in the presence of pinacolborane 122 and complex 113. This reaction resulted in the formation of hydroborated products, with products 126, 127 being unequivocally identified as the predominant major product. Notably, the regioselectivity observed in this reaction was relatively lower when aromatic alkenes were used as substrates.65
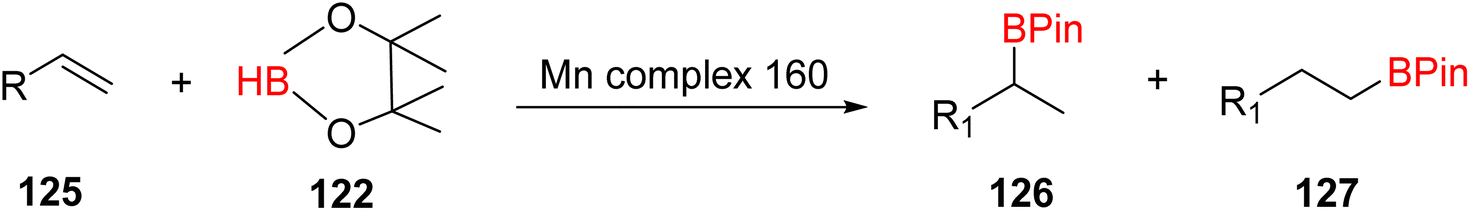 | ||
| Scheme 36 Hydroboration of alkene in the presence of manganese complexes. | ||
The hydroboration reactions involving aldehydes and ketones 128, as presented in Scheme 37, were conducted using the same methodology. These reactions were performed in the presence of pinacolborane 122, and complex 113 served as an effective catalyst for hydroborating both aldehydes and ketones. The resulting hydroborated products, designated as 129, were identified through the utilization of GC-MS analysis. Subsequently, the isolated products were primarily alcohol, with alcohol 130 emerging as the major product. Purification of these products was accomplished using column chromatography with SiO2 as the stationary phase.65 This is the first example of a metal-catalyzed hydroboration reaction for aldehyde, ketone and alkene by using manganese complexes.
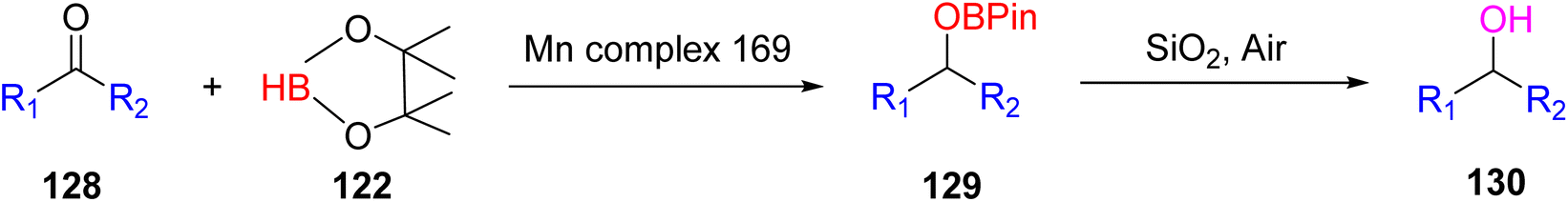 | ||
| Scheme 37 Hydroboration of aldehyde/ketone in the presence of manganese complexes. | ||
Lim et al. (2018) reported the synthesis of manganese TPY complexes designed to catalyze the dehydrogenative coupling of alcohol 131 and amines 132, resulting in the formation of aldimines 133. Notably, this reaction was conducted in the presence of atmospheric oxygen (as depicted in Scheme 38). The highest yields of aldimines were achieved when both electron-withdrawing and electron-donating groups were present on the alcohol and amine substrates. To facilitate this dehydrogenative coupling, dinuclear complexes were formed, strategically designed to regulate the oxidation state of the manganese metal.66
 | ||
| Scheme 38 Dehydrogenative coupling of alcohols with amines. | ||
Zhang et al. (2020) investigated the application of one-dimensional manganese TPY coordination polymers labeled as 136. These coordination polymers were employed as catalysts for facilitating the hydroboration and hydrosilylation reactions involving ketones 128 and aldehydes, as outlined in Scheme 39. This methodology utilized 1-D manganese coordination polymers as pre-catalysts, operating under mild reaction conditions. Furthermore, the study delved into the assessment of the regioselectivity of the hydrofunctionalization of styrene when employing 1-D manganese coordination polymers, with various ketones and aldehydes being utilized to explore this aspect.67
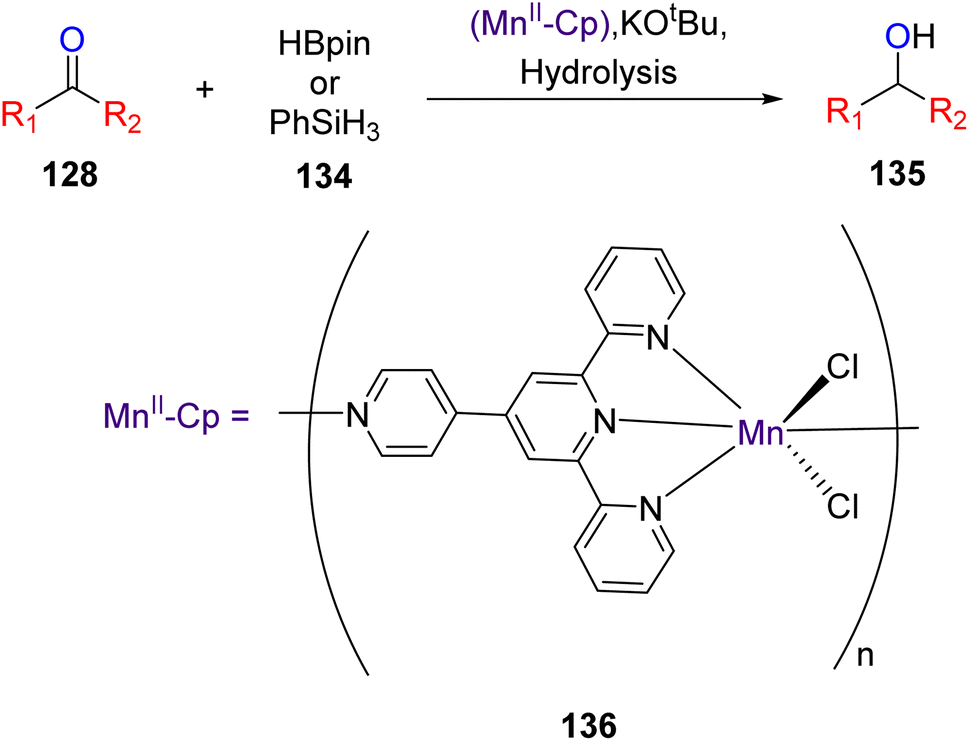 | ||
| Scheme 39 The state-of-the-art of Mn-catalyzed heterofunctionalization of carbonyl compounds. | ||
The research findings revealed that the Mn(II) pre-catalyst was subjected to characterization through the X-ray crystallography technique. This catalyst exhibited a noteworthy tolerance for various substrate types employed in hydrofunctionalization reactions. Notably, Mn–Cp represented the pioneering catalyst for hydroboration and hydrosilylation of carbonyl compounds. Subsequent efforts focused on refining the structure of the coordination polymer (Cp), and investigations extended to encompass hydrofunctionalization of alkenes. Additionally, the study involved a detailed exploration of the mechanistic underpinnings of these reactions.67
Alkyl boronates are recognized as versatile compounds characterized by their robust stability and ease of manipulation. However, they exhibit a pronounced sensitivity to the formation of C-heteroatom bonds and carbon–carbon bonds. Conventionally, the olefination of alkenes has been primarily catalyzed by expensive catalysts such as iridium (Ir) or rhodium (Rh). Nevertheless, there is a shifting focus towards the utilization of more cost-effective and readily available catalysts like cobalt and iron for this purpose.
Palmar and colleagues investigated the use of cobalt TPY complex 139 in conjunction with α-diimine ligands for the hydroboration of various alkene substrates 137, including terminal, germinal, di-substituted internal, tri-substituted, and tetra-substituted alkenes. This reaction was conducted in the presence of pinacolborane, as outlined in Scheme 40. The chelate effect was harnessed in this process to mitigate the formation of mono-anionic species, which could otherwise lead to unfavorable antiferromagnetic coupling with the Co(II) center. Both Co(I) and Co(II) species proved to be valuable in facilitating the hydroboration of sterically hindered alkenes. The α-diimine ligands played a crucial role in the reduction of highly hindered tri-substituted, tetra-substituted, and germinal-substituted alkenes. On the other hand, the TPY moiety played an essential role in the case of arenes or dienes with substituted groups that deactivate or inhibit reaction pathways due to the availability of free coordination sites.74
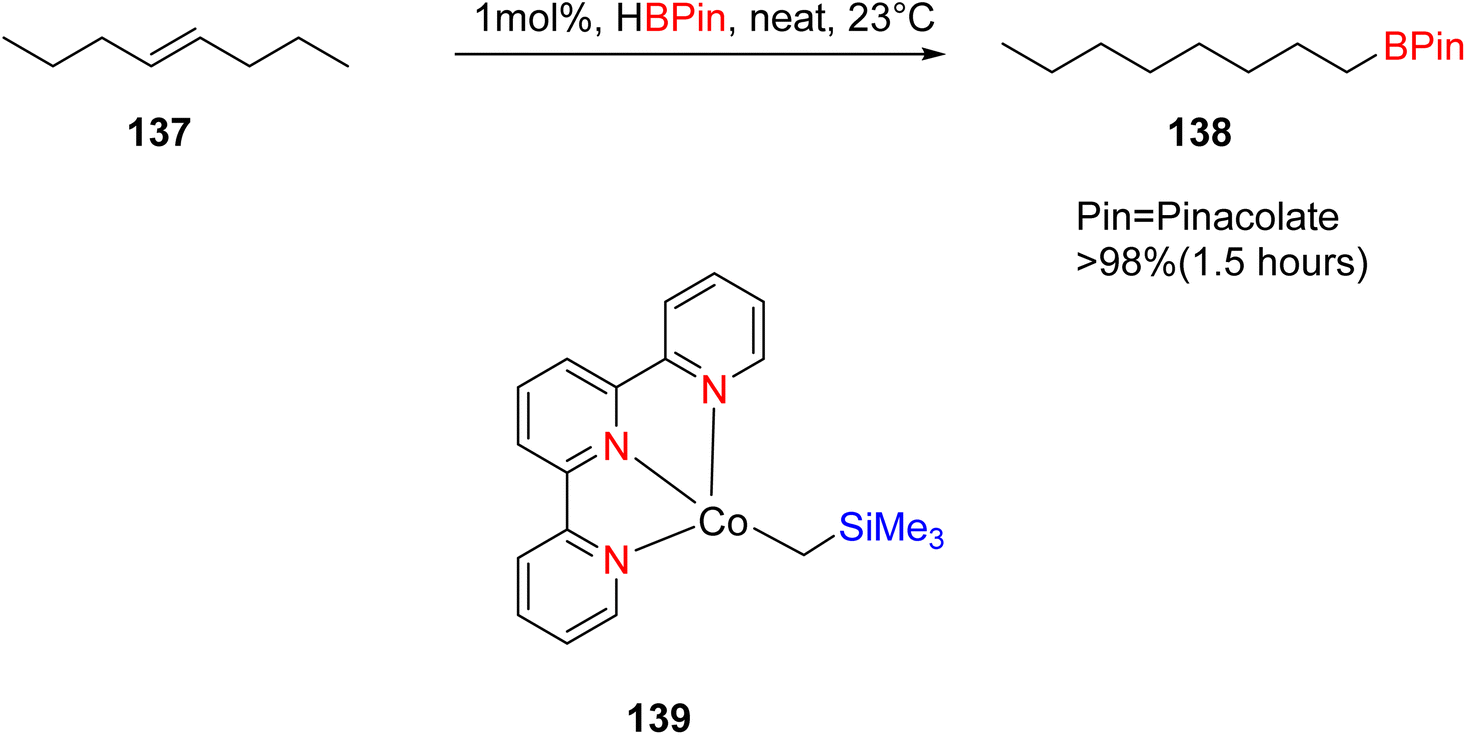 | ||
| Scheme 40 Hydroboration of alkene and complex use for this reaction. | ||
Duonog et al. (2017) introduced cobalt TPY metal complexes as catalysts for mediating the cross-coupling between aryl boronic esters and aryl halides 140, as indicated by Scheme 41. This efficient Suzuki–Miyaura cross-coupling reaction involved the use of aryl boronic esters 141 and alkyl halides in the presence of a Co(II) TPY catalyst and potassium methoxide, ultimately resulting in the synthesis of hetero biaryl compounds with high yields. This cross-coupling protocol demonstrated its versatility by successfully engaging various substrates, including π-electron-rich, π-electron-deficient heteroaryl halides, and electron-deficient aryl halides.75
 | ||
| Scheme 41 Tpy-Co catalyzed Suzuki–Miyaura cross-coupling. | ||
Lin et al. (2018) explored the use of iron and cobalt metal–organic layer (MOL) complexes for the activation of C–H bonds, with a specific focus on synthesizing pyrrolidines and piperidines, as illustrated in Scheme 42. The effectiveness of iron and cobalt MOL complexes based on TPY ligands as catalysts for C–H bond activation was established through a range of spectroscopic techniques, including UV-visible spectroscopy, X-ray absorption near-edge structure, X-ray absorption fine structure, electron paramagnetic spectroscopy, and Density Functional Theory (DFT) studies. This particular catalyst demonstrated its utility in both C(sp3)–H borylation and intermolecular amination reactions.45
 | ||
| Scheme 42 Co TPY-MOL catalyzed C–H borylation of arenes and its proposed mechanism. | ||
Bhat et al. (2018) investigated dinuclear cyclo(metallo)phosphates [M(dipp)-(Cl-terpy)]2, with two different metals Mn(1) and Ni(2), as well as the mononuclear cobalt phosphate [Co(dippH)(Cl-terpy)(MeOH)(H2O)] 204 dipp, for their catalytic potential in the oxidation of alcohols, as depicted in Scheme 43. These compounds were synthesized using 4′-chloro-2,2′:6′,2′′-TPY (Cl-terpy) and 2,6-diisopropylphenyl phosphate (dippH2), in conjunction with various metal salts including manganese, nickel, and cobalt acetate. The characterization of these compounds was performed through a combination of spectroscopic, thermogravimetric, and microanalytical techniques. The structural confirmation of the cobalt complex was achieved through X-ray diffraction, which indicated a monomeric nature. However, it was observed that the compound transformed into a dimeric form when dissolved in methanol, as confirmed by Electrospray Ionization Mass Spectrometry (ESI-MS). The oxidation of alcohols was carried out in the presence of tetra-butylhydroperoxide as the oxidant. The results demonstrated that manganese, nickel, and cobalt TPY complexes served as efficient catalysts for the conversion of alcohols into ketones.76
 | ||
| Scheme 43 Oxidation of alcohols by using cobalt TPY complexes as catalyst. | ||
Wu et al. (2018) investigated the utilization of a cobalt(II) coordination polymer as a pre-catalyst for the selective hydroboration of aldehydes 159, ketones 128, and imines 162 under specific reaction conditions as outlined in Schemes 44–46. This catalytic system employed pinacolborane 122 as the boron source. Notably, this cobalt TPY coordination polymer exhibited remarkable properties such as recyclability when paired with an air-stable activator. Furthermore, the catalyst displayed a high degree of selectivity towards various substrates bearing functional groups like carbonyls (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and imines (CN), achieving efficient hydroboration under mild reaction conditions. Importantly, this cobalt-based catalyst stands out due to its non-precious metal composition, ease of synthesis, air stability, and reusability. Additionally, it exhibited tolerance to a wide range of substituents. This research has prompted further investigations into the applicability of similar catalysts in diverse organic transformations, paving the way for potential advancements in the field of catalysis.77
O) and imines (CN), achieving efficient hydroboration under mild reaction conditions. Importantly, this cobalt-based catalyst stands out due to its non-precious metal composition, ease of synthesis, air stability, and reusability. Additionally, it exhibited tolerance to a wide range of substituents. This research has prompted further investigations into the applicability of similar catalysts in diverse organic transformations, paving the way for potential advancements in the field of catalysis.77
 | ||
| Scheme 44 CP-catalyzed hydroboration of ketone. | ||
 | ||
| Scheme 45 CP-catalyzed hydroboration of aldehyde. | ||
 | ||
| Scheme 46 CP-catalyzed hydroboration of imines. | ||
Tarriño et al. (2018) reported on the use of a cobalt catalyst for the hydrosilylation of alkenes 165 under aerobic conditions, eliminating the need for dry solvents, as illustrated in Scheme 47. Traditionally, platinum (Pt) catalysts had been employed for this purpose. However, the advent of cost-effective catalysts has enabled the achievement of anti-Markovnikov alkene hydrosilylation, a significant advancement in this field. This catalytic process was found to be applicable to both hydrosilanes and hydroalcoxysilanes, and the study involved a mechanistic investigation, shedding light on the formation of Co–H species during the reaction. This phenomenon was monitored using in situ-Raman spectroscopy, providing valuable insights into the reaction mechanism. The catalysts used for this purpose is [Co(OAc)(H2O)2(tpy)]OAc 165a and [Co(tpy)2]Cl2·6H2O 165b, cobalt(II) naphthenate 165c are most active catalyst having 22.33 TOF (min−1).78
 | ||
| Scheme 47 Cobalt pre-catalysts for the hydrosilylation of 4-vinyl cyclohexene with PhSiH3. | ||
Dai et al. (2021) introduced a novel catalytic system based on cobalt bis(2-ethylhexonate) and TPY derivatives for the hydrosilylation of olefins 125. This catalytic system demonstrated efficacy under mild reaction conditions, eliminating the need for Grignard reagents or NaHBEt3, as outlined in Scheme 48. The properties of cobalt bis(2-ethylhexonate) were notably enhanced when TPY derivatives were employed as co-catalysts. The research findings indicated that the presence of an electron-donating group on the benzene ring at the 4th position of TPY played a crucial role in catalytic performance. This innovative approach was successfully applied to the hydrosilylation of both straight-chain alkenes and aromatic alkenes. Remarkably, the reactions consistently produced high product yields even at very low reaction temperatures, primarily through the formation of β-adduct 169, without necessitating an activation step. Notably, the use of monomeric TPY derivatives was found to suppress the hydrosilylation process. Future research endeavors aim to delve into the mechanistic intricacies of this catalytic system further.79
 | ||
| Scheme 48 Hydrosilylation of alkenes along with proposed mechanism reaction in the presence of hydrosilanes. | ||
Pabst et al. (2022) reported on the utilization of a cobalt catalyst for the meta-selective borylation of fluorinated arenes (177), as illustrated in Scheme 49. Before this study, a series of screenings and stoichiometric investigations were conducted, revealing that the cobalt catalyst's efficacy could be enhanced by protecting it with a TPY moiety (specifically, 4′-(4-N,N′-dimethylaminophenyl)-5,5′′-dimethyl-TPY, denoted as 180). The TPY ligand used in this study was easily synthesized and proved to be highly effective in comparison to other TPY derivatives. Deuterium kinetic studies and stoichiometric investigations provided valuable insights into the catalytic borylation process. Analysis of the C–H activation step indicated a preference for Co–C and C–H bond activation at the meta position of the arenes, as opposed to the ortho position. It was noted that in the absence of a boron reagent, the thermodynamics favored the ortho position, making meta-selectivity challenging at room temperature for fluorinated arenes. This observation underscored the thermodynamic control of C–H bond activation. The formation of a cobalt(I) boryl intermediate was integral to the generation of the C–H intermediate in the catalytic borylation process. The results suggested that C–H activation represented the slow and kinetically controlled step in this metal-catalyzed reaction.80
 | ||
| Scheme 49 Borylation of selected fluoroarenes using 6-CH2SiMe3 as precatalyst. | ||
Suzuka et al. (2013) reported on the development of polymer-supported palladium TPY metal complexes for the facilitation of Suzuki–Miyaura cross-coupling reactions in aqueous environments. The ligands for these complexes were prepared using 4-methoxycarbonylbenzaldehyde, following a previously established procedure documented in the literature. The prepared TPY ligands were subsequently immobilized onto amphiphilic polystyrene poly(ethylene glycol) (PS-PEG) to form PS-PEG-Pd-TPY complexes, denoted as 184. These ligands featured covalent bonding rather than ionic interactions, allowing for efficient immobilization of the resin and subsequently catalyzing Suzuki–Miyaura cross-coupling reactions in an aqueous medium. The reaction involved the cross-coupling of aryl halide 181 with arylboronic acid 182 under aerobic conditions in water, resulting in the production of biaryls 183 in excellent yields, as depicted in Scheme 50. Furthermore, these complexes were employed in the synthesis of pyrimidines 188 in water, as illustrated in Scheme 51. Ongoing research efforts aim to further explore and enhance the catalytic capabilities of these Suzuki–Miyaura cross-coupling reactions.84
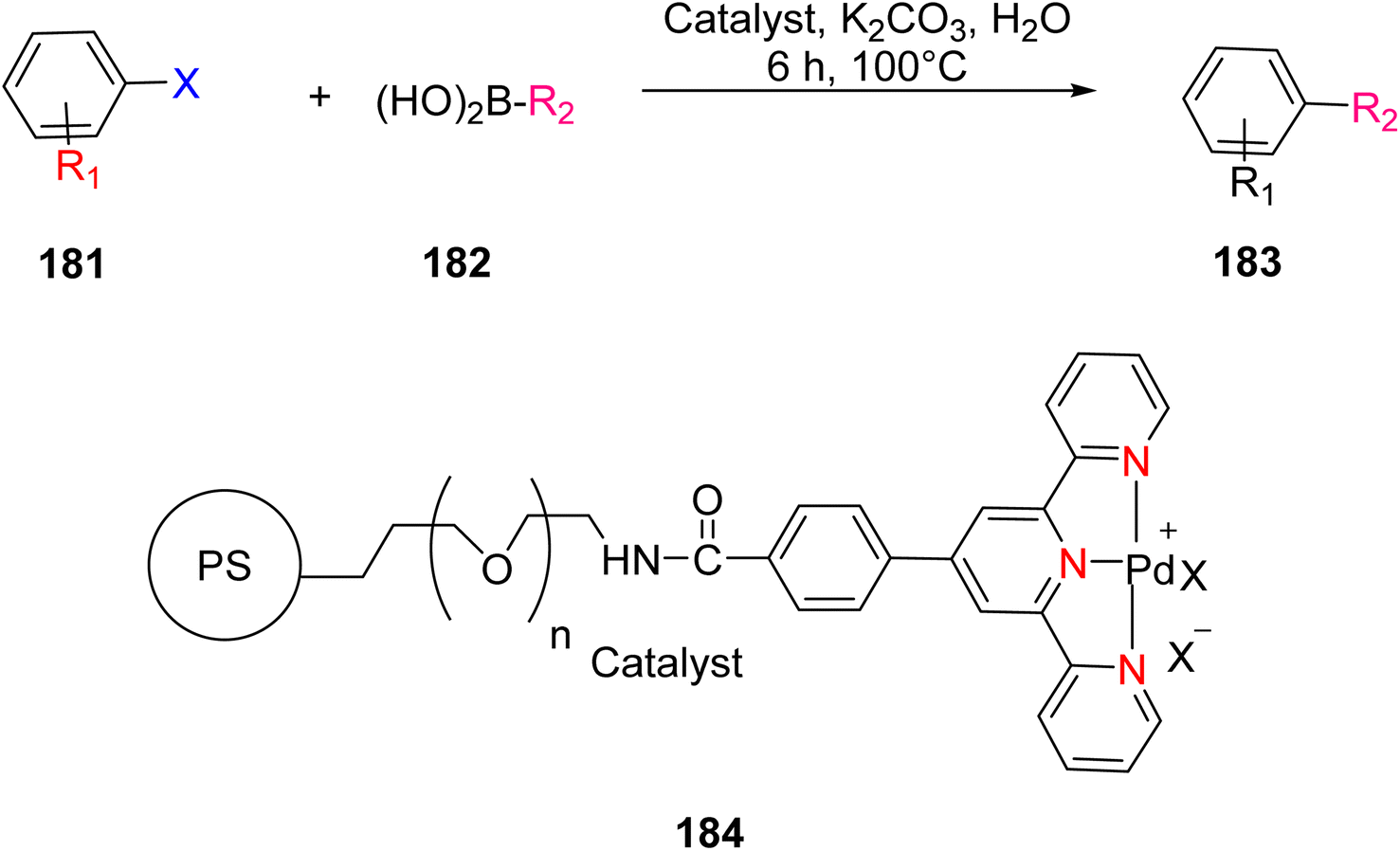 | ||
| Scheme 50 Suzuki–Miyaura cross-coupling reaction using a polymeric catalyst in water. | ||
 | ||
| Scheme 51 Synthesis of pyrimidines using a polymeric catalyst in water. | ||
Mazzotti et al. (2013) reported the use of palladium(III) TPY complex, denoted as 196, as a catalyst for the synthesis of aryl fluorides 193. This transformation involved the reaction of aryl boronic acid 194 and its derivatives, as illustrated in Scheme 53. The mechanistic analysis of this reaction pathway revealed its reliance on a single electron transfer mechanism, as depicted in Scheme 52. Remarkably, this process exhibited tolerance towards moisture and air, operating under mild conditions. The single electron transfer mechanism facilitated the formation of a palladium intermediate. Importantly, this method was operationally straightforward, and it did not yield any unwanted side products during the de-metalation step, which is a common issue encountered in the synthesis of aryl fluorides. However, it's worth noting that this method had limitations in that it was unable to fluorinate heterocyclic compounds, leading to the formation of constitutional isomers, which presented a challenge in certain cases.85
 | ||
| Scheme 52 Fluorination of aryl trifluoroborates and synthesis of catalyst. | ||
 | ||
| Scheme 53 Proposed mechanism for fluorination of aryl trifluoroborates. | ||
Amini et al. (2014) synthesized a silica-supported palladium catalyst to enable Heck and Suzuki cross-coupling reactions while ensuring its reusability. These reactions hold significant utility, particularly in the context of the Mizoroki–Heck reaction, where aryl halide 204 is coupled with olefins 205 to yield various valuable compounds (illustrated in Scheme 54A). Silica was chosen as the support material for this catalyst due to its numerous advantages, including easy accessibility, robust stability, and the ability to readily anchor organic functional groups onto its surface. This versatility made it a suitable choice for the desired applications. The Suzuki coupling reactions, specifically involving aryl iodide 207 and aryl boronic acid 208, were efficiently promoted by this catalyst (Scheme 54B). The research outcomes indicated that the catalyst denoted as 210 (as shown in Scheme 54C) could be reused up to four times with only a slight decrease in activity. Moreover, this catalyst facilitated rapid conversions within a short time frame, making it an effective and practical option for catalyzing Heck and Suzuki coupling reactions.86
 | ||
| Scheme 54 (A) Heck coupling of aryl halides with olefins (B) Suzuki coupling of aryl iodides with aryl boronic acids (C) figure indicates the catalyst used in the reaction. | ||
Suzuka et al. (2015) reported the development of a polymer-supported palladium TPY-based catalyst 188 for the hydrodechlorination of aryl chloride 211. This reaction was conducted in the presence of potassium formate 212, with the sea water serving as the reaction medium, as depicted in Scheme 55. It is noteworthy that the reduction of aryl chloride is generally considered easier compared to aryl bromide and aryl iodide. However, the observed results did not conform to this expected trend. To address this, various inhibitors were employed, and efforts were made to devise methods for their removal. The polymer support exhibited high catalytic activity and reusability in this transformation, showcasing its practical utility in transfer reductions conducted in an aqueous environment. The catalyst in this reaction was formed through the establishment of a covalent bond between the resin and the amino group. This catalytic system demonstrated excellent performance, yielding a remarkable 95% product yield. The promising results obtained in this study encouraged further investigations into the application of this catalyst in other organic transformations.87
 | ||
| Scheme 55 Hydrodechlorination of aryl chlorides using polymeric catalyst in sea water. | ||
Chen et al. (2014) reported the development of xylan-type hemicellulose polymer-supported TPY palladium complexes, 218, which served as catalysts for Suzuki–Miyaura reactions. These catalysts were thoroughly characterized in terms of their morphology and thermal stability. Nano-sized palladium catalysts were employed for the Suzuki–Miyaura reaction, involving the coupling of aryl boronic esters 216 with aryl halide 215, all under aerobic conditions. Remarkably, this catalytic system yielded a high product yield of 98%, as illustrated in Scheme 56. The catalyst played a crucial role in catalyzing and stabilizing the Suzuki–Miyaura reaction, offering significant advantages in terms of efficiency. Furthermore, this catalyst exhibited reusability, being employed up to six times and recoverable via simple filtration without any appreciable loss of its catalytic activity. This attribute has important implications for the broader utilization of polysaccharides in environmentally friendly catalytic processes, highlighting its potential significance in the field of green catalysis.88
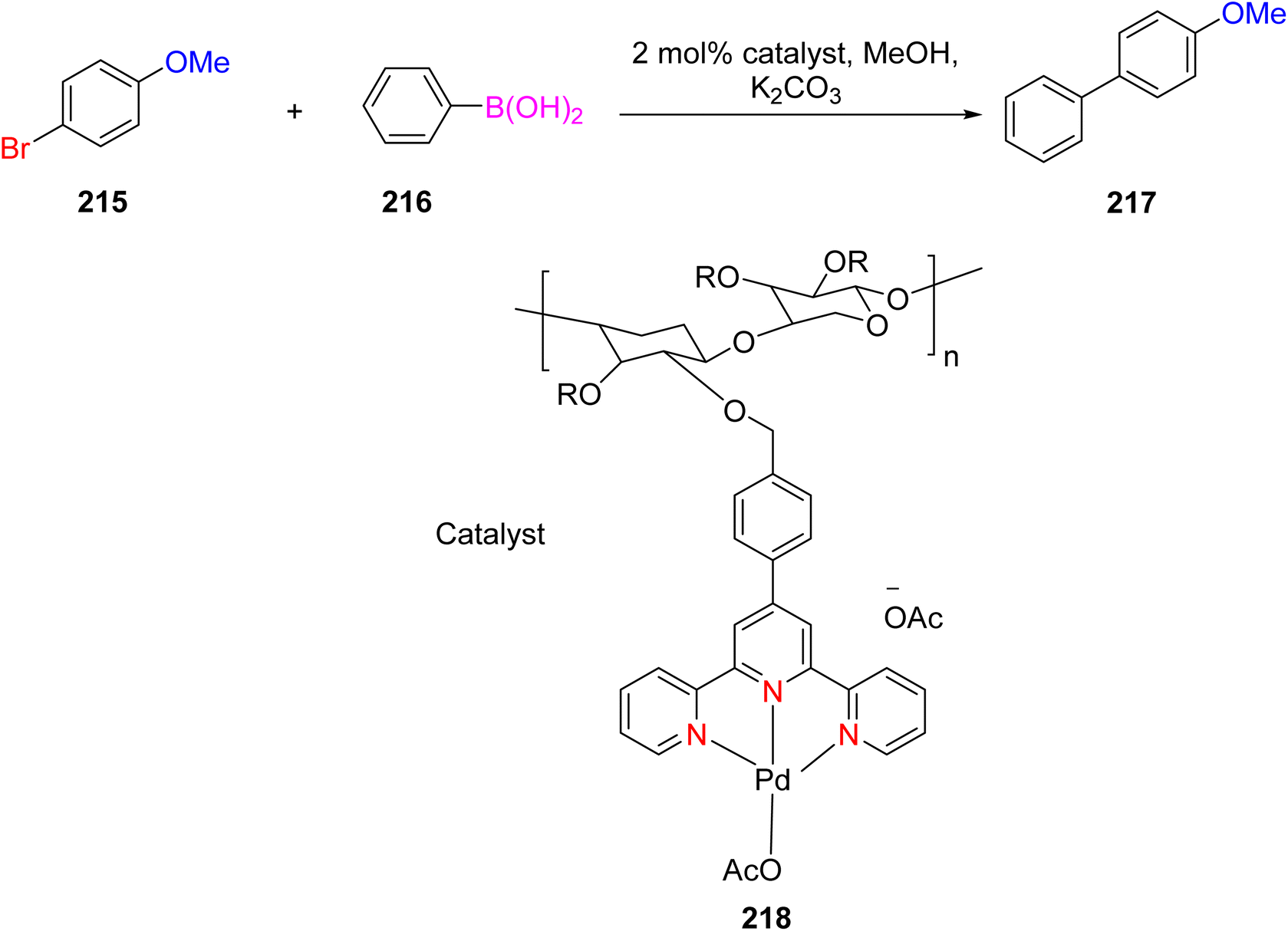 | ||
| Scheme 56 Successive trials by using recycled xylan-type hemicelluloses-supported palladium catalyst. | ||
Suzuka et al. (2015) reported the utilization of polymer-supported TPY palladium complexes, denoted as 214, for the Mizoroki–Heck reaction conducted in an aqueous environment under aerobic conditions. This catalytic system facilitated the production of various types of stilbene compounds, as illustrated in Scheme 57. The reaction involved the coupling of aryl halides 181 with styrene 118, all taking place in the presence of oxygen (aerobic conditions). The catalyst demonstrated the ability to be reused multiple times, albeit with a slight reduction in its catalytic activity upon subsequent use. This research represents a significant contribution to the field of Mizoroki–Heck reactions and their potential applications in various organic transformations.89
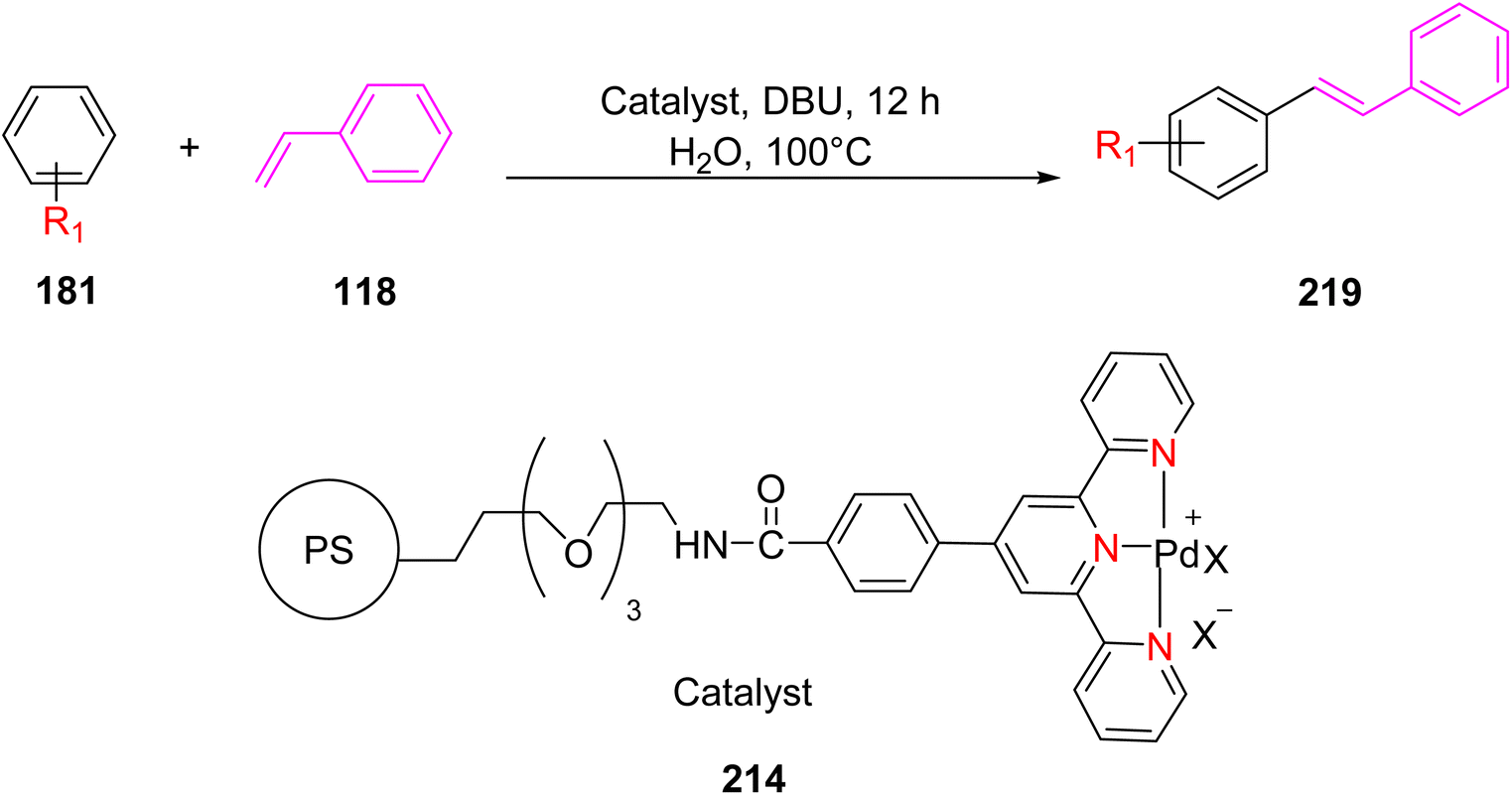 | ||
| Scheme 57 Mizoroki–Heck reaction using polymer-supported catalyst in water. | ||
Yin et al. (2019) presented palladium-containing TPY catalysts, specifically designated as 224 and 225, which were employed for the coupling of 2-iodobiphenyl 221 and iodo-benzene 222 and resulting in the production of triphenylenes 223 (as depicted in Scheme 58). Both catalysts exhibited catalytic activity in mediating the cross-coupling reaction between iodobenzene and 2-iodobiphenyl, leading to the asymmetric synthesis of triphenylenes. The observed catalytic efficiency was attributed to the square planar and unsaturated Pd2+ TPY complexes, which played a pivotal role in facilitating the reaction. Importantly, this Pd-catalyzed reaction proceeded through two distinct pathways, involving both C–C bond formation and C–H bond activation. Notably, these catalysts operated efficiently without the need for toxic ligands such as dppf and in the absence of a nitrogen atmosphere, enhancing their practical utility in environmentally friendly synthetic processes.90
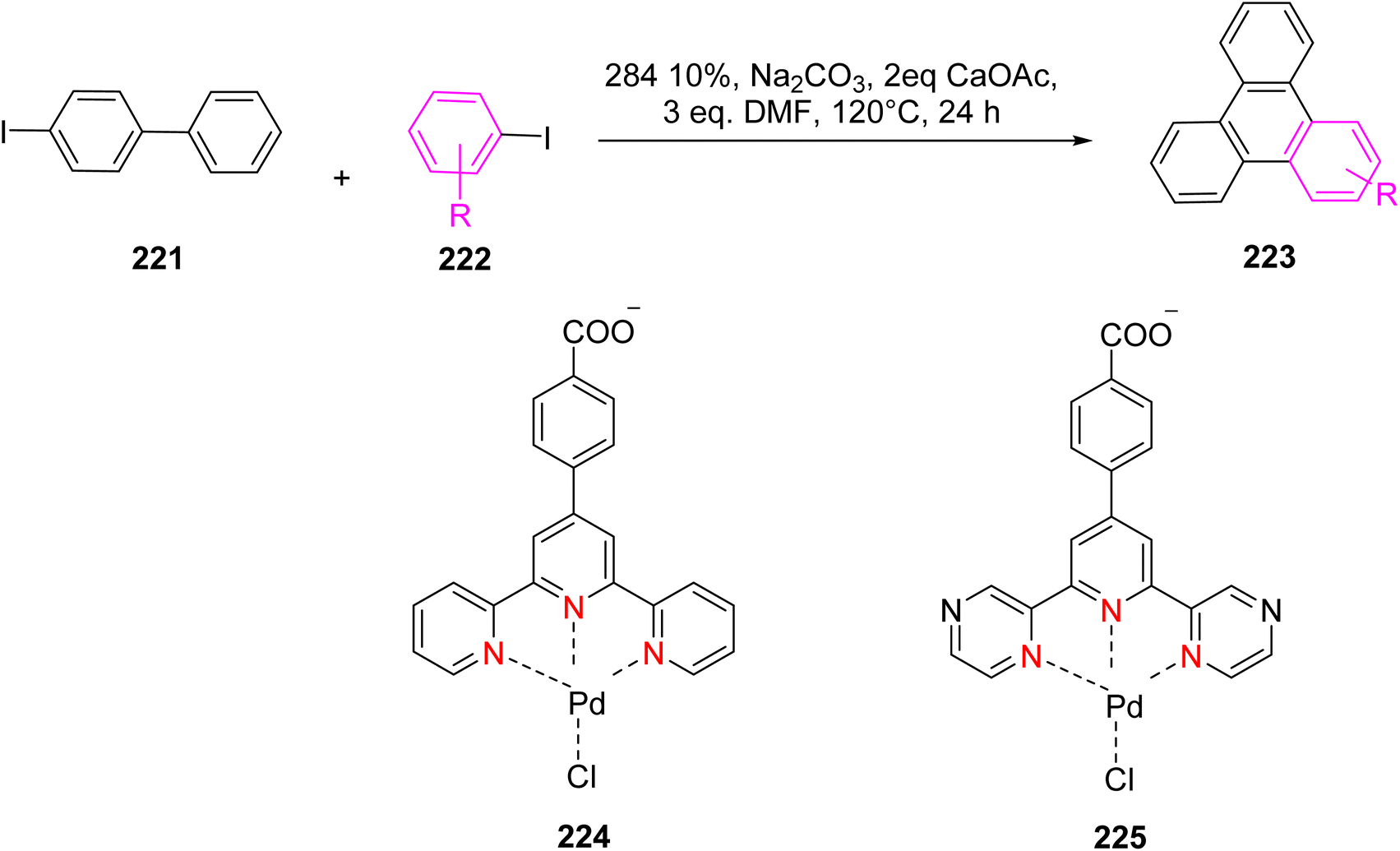 | ||
| Scheme 58 Palladium catalyst to catalyze the reaction between iodobenzene and 2-idobiphenyl for the synthesis of triphenylenes. | ||
Traghan et al. (2019) reported the synthesis of stabilized palladium nanoparticles with diameters ranging from approximately 3 to 4 nanometers, anchored on a magnetic polymer. The magnetic polymer employed for nanoparticle synthesis was 4′-(4-hydroxyphenyl)-TPY (HPTPY), denoted as 226 (Fig. 8). This ligand was prepared through the polymerization of itaconic acid, serving as the monomer, along with a cross-linking agent, trimethylolpropane triacrylate (TMPTA). These magnetic palladium nanoparticles were utilized as catalysts in cross-coupling reactions, including Suzuki–Miyaura and Mizoroki–Heck reactions. During the catalytic process, palladium ions were reduced to form Pd nanoparticles, which were stabilized in the presence of ethanol, acting as a green reducing agent. This stabilization occurred due to the interaction of the TPY ligand. These nano-sized catalysts exhibited catalytic properties, yielding products with up to 98% efficiency, and demonstrated a high turnover frequency (TOF) of 257 h−1. Moreover, the nano catalysts were easily recoverable and could be utilized even in small quantities, which is economically advantageous given the relatively high cost of palladium. This reusability aspect contributes to cost-effectiveness in various applications.91
 | ||
| Fig. 8 Structure of MP-TPY/Pd catalyst used for the above reaction. | ||
Li et al. (2022) reported on the utilization of the TPY group as an organometallic binding site for palladium (Pd) and nickel (Ni). They achieved this by grafting palladium and nickel monomers onto a silicon substrate, which was fabricated through a self-assembly process. This catalyst exhibited remarkable catalytic activity and demonstrated reusability for up to five cycles. However, the presence of active silicon palladium nanoparticles eventually led to catalyst deactivation. The catalytic mechanism for this reaction was determined to be heterogeneous, taking place on the surface of Si-TPY-Pd/Ni. This was confirmed through experiments involving hot filtrate, poison tests, and a three-phase system, as outlined in Scheme 59.92
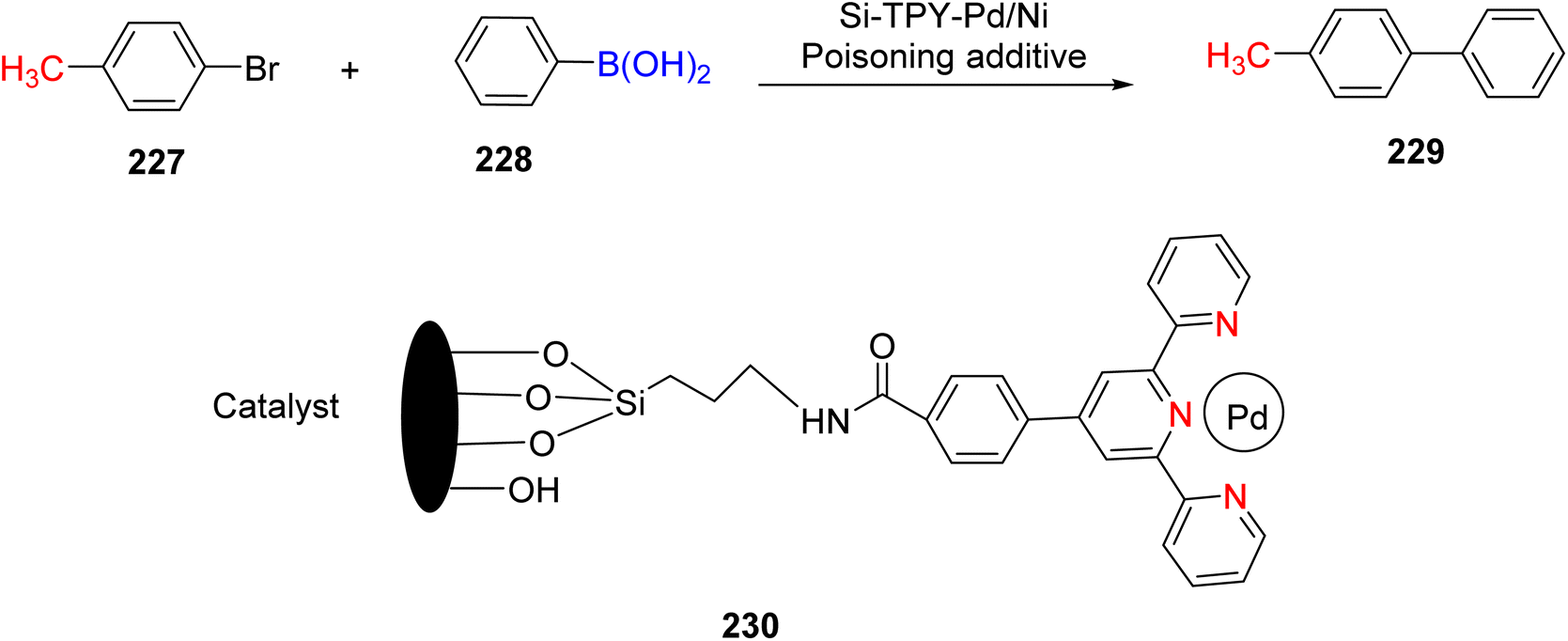 | ||
| Scheme 59 Poison experiments of Si-Tpy-Pd/Ni catalyst. | ||
 | ||
| Scheme 60 (a) Mechanism for reduction of water in the presence of ruthenium TPY complex. (b) Mechanism for reduction of carbon dioxide to carbon monoxide. | ||
Yagi et al. (2012) described dimanganese TPY complexes for water oxidation and the influence of different substituents or groups on core structure. In this phenomenon, dimanganese complexes acted as heterogeneous catalysts. [(OH2)(R-terpy)Mn(μ-O)2 Mn(R-terpy)-(OH2)]3+ 246 (R-terpy) = 4′-substituted 2,2′:6′,2′′-terpyridine, R = butoxy (BuO), propoxy (PrO), ethoxy (EtO), methoxy (MeO), methyl (Me), methylthio (MeS), chloro (Cl) groups (Fig. 9), these groups have capability of producing oxygen and characterized by using different spectroscopic technique such as UV-visible, IR spectroscopy, X-ray crystallography, magnetometric and electrochemical technique. The UV-visible spectra of derivatives are affected in the presence of 4′-substituents derivatives. In X-ray crystallographic technique, Mn center core is not distinguished in groups such as H, MeS, Me, EtO, and BuO while it is visible in groups such as MeO and PrO. The indistinguishable Mn core is due to the exchange of electrons rapidly between the center to Mn causing delocalization of Mn center. Considering its exchange internal value for the delocalized core (J = −196 to 178 cm−1) is lower than that of the localized core (J = −163 to 161 cm−1). The halfway potential for Mn pair derivatives for the delocalized core decreased with the increase in the number of electrons donating group, but this value is different for the localized core. The heterogeneous catalytic ability of water oxidation of different manganese derivatives on mica was changed by substituent groups. This is the second order rate constant (k2) for evolution oxygen, represented by the E1/2 value of an Mn(III)–Mn(IV)/Mn(IV)–Mn(IV), the value of k2 increased about 29 with an increase in E1/2 potential about 28 mV. This work might cause difficulty in the synthesis of oxygen evolution complexes but gives insight into the synthesis and mechanism for oxygen evolution.94
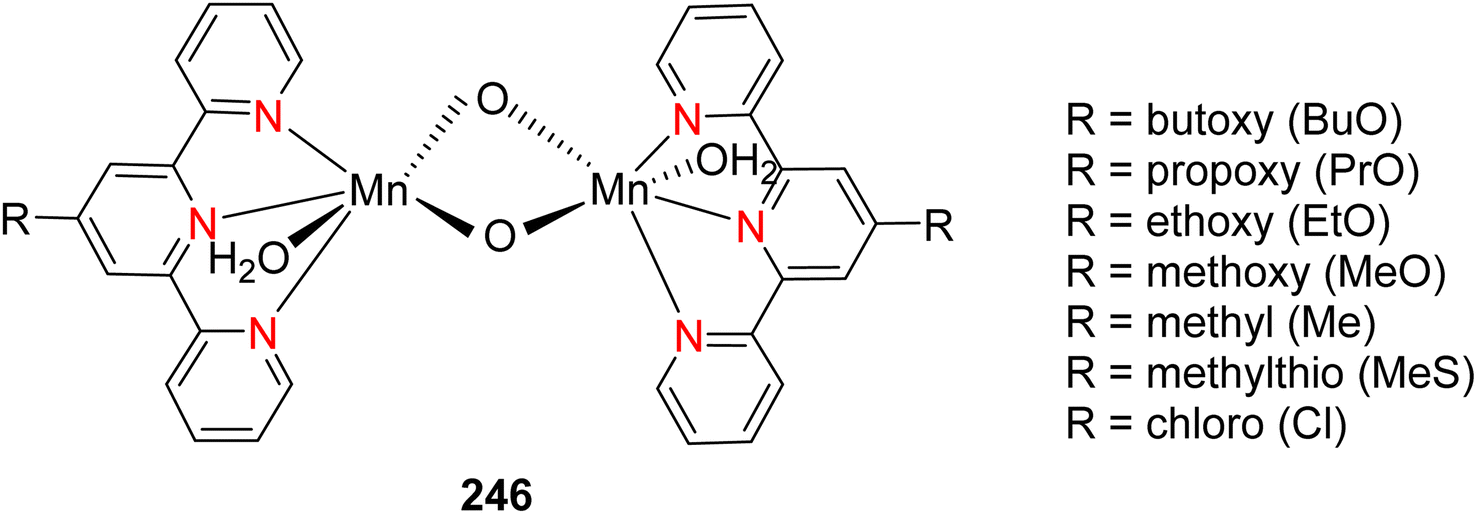 | ||
| Fig. 9 Dimanganese TPY complexes used for water oxidation. | ||
Elgrishi et al. (2014) investigated homoleptic TPY complexes featuring first-row transition metals. These complexes were employed for the electrochemical reduction of carbon dioxide to carbon monoxide, with a particular focus on nickel and cobalt TPY complexes, denoted as complex 247. The results for these two metals were distinct. Nickel complexes demonstrated a selectivity for carbon dioxide reduction over hydrogen reduction. In contrast, cobalt complexes generated a mixture of both hydrogen and carbon monoxide, and the ratio of these products could be manipulated by varying the applied potential. This phenomenon was linked to the reduction of the TPY ligand in both nickel and cobalt complexes. The resting state of nickel complexes was found to be divalent, while cobalt complexes were monovalent. Due to its divalent state, nickel was unable to form a nickel hydride intermediate necessary for hydrogen generation. Consequently, it exhibited selectivity in favor of carbon monoxide production from the reduction of carbon dioxide, rather than hydrogen production (as shown in Scheme 61). The lower oxidation state of cobalt allowed it to produce an excess amount of carbon monoxide from carbon dioxide reduction. Furthermore, the production ratio of hydrogen to carbon monoxide was modifiable through potential application. It was noted that the degradation of the polypridyl ligand limited the faradaic efficiency of cobalt complexes, ultimately impacting their intrinsic catalytic activity. This behavior was in contrast to other polypridyl catalysts.95
 | ||
| Scheme 61 Proposed mechanism for CO2 reduction to CO by Co-TPY. | ||
Elgrishi et al. (2015) described cobalt TPY complex 271 used for the reduction of carbon dioxide to carbon monoxide (Scheme 62), as an efficient catalyst for this purpose, disfavoring the production of hydrogen reported previously. First complexes were studied for the evolution of hydrogen then it was used for the reduction of carbon dioxide by electronic tuning of anchoring ligand. When these complexes were further involved in the reduction of carbon dioxide into carbon monoxide as a catalyst, this catalyst is least reactive towards the production of hydrogen and increases selectivity towards carbon dioxide reduction. This mechanism involves turning off one of the competing reactions. The activity of this reaction is evaluated by using the foot of wave analysis and catalytic Tofel plot. The rate of reaction is faster by using electron-rich ligands, faradaic studies revealed that these catalysts have higher faradaic yield and high constant for production of CO over hydrogen production. Previously it was reported that selectivity for CO is improved by disfavoring hydrogen, but this work is based on electronic modification of ligand attached to it or using foot wave analysis. Further work will be done to explore this unique relationship between steric hindrance and product selectivity (%). This work provides insight for further working on molecular CO2 catalysis which is useful in practical applications.96
 | ||
| Scheme 62 Cobalt TPY complex for CO2 reduction and proposed mechanism. | ||
Bern et al. (2016) investigated fluorinated iridium TPY complexes that functioned as photocatalysts for the generation of solar fuels and for photoredox catalysis, as depicted in Scheme 63. These complexes featured a fluorinated phenyl-pyridine ligand combined with electron-rich substituents. This combination created a push–pull effect upon the excitation of electrons, resulting in significant electrochemical and photophysical properties in the complexes. These fluorinated iridium TPY complexes were also employed as photosensitizers to produce hydrogen from water and served as redox photocatalysts for the decarboxylative fluorination of various carboxylic acids. Their performance proved efficient, often surpassing that of photosensitizers. Interestingly, as the degree of fluorination increased, there was a shift in the excited state behavior, with the complex displaying an intraligand charge transfer mechanism instead of a metal-to-ligand charge transfer character. Numerous complexes have been developed for hydrogen generation, but few have proven highly efficient for this process. Complexes labeled as 272 exhibited incomparable performance as photosensitizers in this reaction. The researchers made improvements to these complexes and envisioned future work involving the application of these compounds in situations requiring high quantum yield and photochemical stability.97
 | ||
| Scheme 63 Iridium complex C–F activation, acts as photocatalyst and solar fuel production. | ||
Kubaik et al. (2016) described the electrochemical reduction of CO2 using manganese TPY complexes (Scheme 64). The behavior of Mn was studied with TPY κ2-N, N′ and κ3-N, N′,N′′ under electrochemical reducing conditions. In carbon dioxide in the presence of Brønsted lowery acid (phenol) two-electron reduction mechanism occurs and conversion of carbon monoxide and water. According to cyclic voltammetry and infrared spectroscopy, the active catalyst is [Mn(κ3-N,N′,N′′-TPY)(CO)2]− 278. Under these conditions, the decomposition of the faradaic efficiency is reduced. Further work revealed the possibility of developing a new tridentate ligand that acted as a suitable catalyst in the presence of manganese carbonyl moiety.98
 | ||
| Scheme 64 Electrocatalytic reduction of carbon dioxide with Mn(terpyridine) carbonyl complexes. | ||
Reisner et al. (2019) described the selective reduction of carbon dioxide in cheap solar-driven photoelectrochemical devices as very challenging for the production of renewable fuel. The group discussed the synthesis of molecular catalysts, which is photocathode assembled from precious metal-free components that are active toward CO2 reduction. The catalyst is based on phosphonated cobalt TPY complex 281 which is embedded in mesoporous with a TiO2 scaffold containing light-harvesting properties. This involved reduction of carbon dioxide to carbon monoxide in the presence of water or aqueous solution. The turnover number in the reaction is ∼330 and is stable for its activity for more than a day. Furthermore, this work further explores that its selectivity towards CO is approximately 75% and states that altering the water content of photocatalysis of medium has a great effect on stability, activity and selectivity of a product of photocathode. The formation of an intermediate is suggested as an alternative carbon dioxide reduction mechanism without the loss of one TPY moiety (Scheme 65). The mechanistic pathway revealed that Co-TPY perform reduction of carbon dioxide on silicone mesoporous TiO2 is lower than that of 700 mV and improved its stability TON (s−1) up to which is about 4 times higher than that of previously reported cobalt TPY complexes. Excellent results were obtained in the presence of phosphoric acid to operate the system under these conditions. It also studies the power and precision provided by vibrational spectroscopic techniques for a better understanding of molecular catalysts based on solar devices.99
 | ||
| Scheme 65 Mechanism for photo-reduction of CO2 for immobilized CotpyP. | ||
Wang et al. (2021) described the photocatalytic reduction of CO2 in the presence of Nobel metal-free bis TPY iron complexes acting as organic photosensitizers. The bis TPY iron complexes 291 is an activated fluorescence decay compound used for selective reduction of carbon dioxide to carbon monoxide having maximum turnover which is up to 6230 and selectivity is about 99.4%. Its turnover frequency is about 127 min−1 in the presence of visible light irradiation in DMF/H2O solution and electron-donating groups are more efficient for this purpose. By using 2 h of light irradiation using 0.05 micromole of catalyst about 0.3 mmol of CO2 is produced or generated. The observed quantum yields as a result of this reaction are 9.5% at 440 nm. By using UV-visible-NIR spectroscopy, water was shown to strongly exhibit photocatalytic properties and TPY ligand rather than Fe, which was initially reduced in this process. The TADF compound 4CzIPN 292 (Fig. 10) is a more efficient photosensitizer than ruthenium TPY compounds that [Ru(bpy)3]3+. It was observed that the TADF sensitizer is more efficient for the production of carbon dioxide in the future. In this reaction, the TPY ligand is reduced and the procedure is more efficient in the presence of water.100
 | ||
| Fig. 10 Chemical structure of acts as photosensitizer for reduction of carbon dioxide. | ||
Baratta et al. (2022) described TPY di-phosphine complexes which act as efficient photocatalysts for transfer hydrogenation of carbonyl compounds. These cationic chiral and achiral diphosphine ruthenium complexes easily obtained a yield of around 85–88% and synthesis was made by using one pot synthesis method from [RuCl2(PPH3)3] 293, diphosophine and TPY in the presence of 1-butanol other derivatives are obtained by using different methods. The compounds 2, 293a and 3, 293b were characterized by using the X-ray crystallographic technique. These compounds show high photocatalytic activity for transfer hydrogenation of carbonyl compound occurred in the presence of PriOH using 1-propanol in visible light at a temperature of 30 °C (Scheme 66). The hydrogen source in this reaction was 2-propanol and turn over frequency in this reaction was about 264 h−1. The chiral compounds 2, 293a, 3 and 293b showed efficient photocatalytic activity, used for transfer hydrogenation of acetophenone. Further work is made on photocatalytic reduction and catalysts are applied for C–H activation and asymmetric transformation in the presence of irradiation of light.101 Diverse types of diphosphine ruthenium complexes as a photocatalyst, as shown in Table 8.

 | ||
| Scheme 66 Transfer hydrogenation of carbonyl compound photo-catalyzed by ruthenium. | ||
| Complex | PP |
|---|---|
| 293a | Dppp |
| 293b | (R,R)-Skewphos |
| 293c | (S,S)-Skewphos |
| 293d | (S,R)-Josiphos |
| 293e | (R) BINAP |
Cook et al. (2023) described a new TPY FeN3O catalyst (TPYthupho) 302, which involved the reduction of oxygen to water. The variable temperature and concentration spectrochemical studies using decamethylferrocene were used as a chemical reactant in acetonitrile involving catalytic reduction of O2 to water in the presence of compound Fe(TPYthupho), which acts as a catalyst in this reaction. This follows a 2 + 2 mechanism in which hydrogen peroxide is produced as an intermediate, which involves the prior reduction of water (Scheme 67). This is the first-order rate constant value for hydrogen peroxide reduction, which is larger than oxygen reduction. This hydrogen peroxide reduction is studied hydrodynamic takes place in ring disk electrode measurement. The production of the product was swept away from the cathode which swept away due to reduction. The mechanistic approach revealed that the rate of ORR was limited when binding to the Fe(II) center. The modification in the ligand was made by increasing its ORR reactivity and selectivity for further work.102
 | ||
| Scheme 67 Proposed catalytic cycle for the ORR mediated by Fe(tpytbupho)Cl2TPY metal complexes and its proposed mechanism. | ||
Mongal et al. (2023) investigated cobalt bis TPY metal complexes for photocatalytic and electrocatalytic reduction for hydrogen evolution. This process is known as artificial photosynthesis. In this work, they described cobalt bis TPY complexes acting as a photocatalyst for the production of hydrogen. This catalyst is platinized TiO2 in visible light under a sacrificial electron donor known as methanol. The cobalt bis TPY Pt/TiO2 photocatalyst exhibits remarkable activity for hydrogen evolution with high ton 5718 and quantum yield is about 5.34% compared to bare Pt/TiO2. It also shows electro-catalytically, involved in the reduction of proton in acetonitrile using trifluoroacetic acid at 0.63 V with a turnover frequency of about 18.64 s−1 with a ratio of 8:1 of acid to catalyst. The additional π-conjugated group 4′-(5-(4-diphenylamino) phenylthiophen-2-yl)-TPY 303 (Fig. 11) provides excellent surface protection with the help of steric hindrance leading to photocatalytically. The complex Pt/TiO2 provides 2859 μmol g−1 with 4 h of light irradiation. The photocatalyst shows about 5 cycles of irradiation of about 20 h. This created avenues for the generation of a new transition metal based on acting as a photo-sensitizer as well as an electro-catalyst.103
 | ||
| Fig. 11 Labelled structural representation of the cobalt complex for hydrogen evolution. | ||
Mughal et al. (2023) described different transition metal (Fe, Co, Zn) based coordinated complexes acting as efficient catalysts for photo-degradation of methylene-based pollutants in wastewater. All complexes reported had remarkable photocatalytic activity against methylene blue pollutant. Complex 304 (67.31%) and complex 305 (65.91%) show excellent photocatalytic properties. Complex 305 (Fig. 12) showed high catalytic activity of a rate constant value of about 0.011 min−1 compared to a rate constant of about 0.009 min−1 for other complex 304 (Fig. 12). This shows a pseudo-first order reaction. The reusability test revealed that 8.62% and 7.01% are lost for photocatalytic activity. These catalysts can be used about four times. This work highlights the use of TPY metal complexes for the effective removal of harmful pollutants present in water and the further use of TPY complexes for various applications.104
 | ||
| Fig. 12 Complexes acting to photo-degrade methylene blue in wastewater effluents. | ||
3.2. Chemosensors
Dendritic terpyridines are known to form polymetallic species that serve crucial roles as fluorescent sensors and in the construction of diverse supramolecular structures. Terpyridine (TPY) is recognized as a pincer ligand, capable of participating in the formation of both mono- and bis-TPY complexes. TPY comprises three nitrogen-containing rings, and to mitigate electrostatic repulsion between electrons, two pyridine groups adopt a trans geometry before coordination with metal ions. Upon chelation with metal ions, TPY undergoes a conformational change, transitioning to a cis structure, followed by further rearrangement to a planar structure. This property of TPY enhances its capability for molecular recognition and makes it well-suited for use as a sensing device.105Yin et al. (2012) researched the synthesis of siloles TPY metal complexes, with a focus on studying their optical properties and their ability to sense metal ions. Their investigations revealed that both TPY siloles exhibited a pronounced selectivity for Zn2+ ions compared to other alkali, alkaline earth, and transition metal ions when excited at a wavelength of 380 nm in tetrahydrofuran (THF). The photophysical properties of these metal ions were explored through various spectral analyses, including fluorescence and titration studies. The first compound, denoted as 306 in Fig. 13, contained two TPY groups and exhibited superior catalytic properties compared to the second compound, labeled as 307 in Fig. 13, which contained only one TPY group. This difference in catalytic performance was attributed to the formation of a metal–organic framework or coordination polymer in the former. When Zn2+ ions were introduced into THF, a significant bathochromic shift was observed, indicating an enhanced fluorescence intensity. The chemosensors showed a high fluorescence intensity when binding with metal ions, making them more sensitive than sensors that exhibited fluorescence quenching in the presence of metal ions.106
 | ||
| Fig. 13 TPY-based siloles used as chemosensor. | ||
Zhang et al. (2012) reported on the synthesis of europium complexes with TPY-capped polyethylene glycol. These complexes were prepared through the utilization of Eu(NO3)3 in conjunction with telechelic macromolecules. Various complexes were synthesized, and their properties were characterized using a range of spectroscopic techniques. Upon excitation at 300 nm, these complexes exhibited emission spectra with peaks at 593 nm and 616 nm. Notably, their fluorescent emission could be quenched in the presence of diethyl chlorophosphate, a chemical compound. The quenching process was quantified by determining the Stern–Volmer constant (Ksv), which was found to be in the range of 0.588 × 103 to 0.89 × 103 L mol−1 for quenching at the respective wavelengths of 593 nm and 616 nm. These TPY-based lanthanide complexes demonstrated effective potential for the detection of organophosphate compounds, showcasing their utility as sensors for this specific class of chemicals.107
Wang et al. (2012) described the synthesis of zinc-based TPY metal complexes for the detection of cobalt and nickel ions. The structure of complexes was determined by using X-ray crystallographic technique. The complexes revealed that complex 308 shows excellent activity and acts as fluorescence chemosensors for the detection of Ni2+ and Co2+ but not for other metal cations such as Mg2+, Pb2+, Cu2+, Fe2+, In3+, Zn2+, Cd2+ and Fe3+ and EDTA can selectively turn on and on quenched fluorescence complex 1 respond to Co2+ (on-off-on type) but does not respond to Ni2+ (on-off type sensor). They described a fluorescence-type sensor by joining terpyridine. They also show sensing activity towards Ni2+ and Co2+. The diminished property is resorted by the addition of EDTA and then it responds to Co2+ but does not towards Ni2+ metal ions. ESI spectral data shows that to complete quench of ability is due to the replacement of Zn2+. The results show that TPY coordinated compound 308 shows property towards Co2+ ions (Fig. 14).108
 | ||
| Fig. 14 Zinc-based TPY metal complex for sensing Co2+ and Ni2+ ion. | ||
Li et al. (2013) introduced a novel polyarylene ether material containing substituted TPY ligands, which functioned as a chemosensor for the detection of Ni2+ ions. The observed changes in the polyarylene ether material containing substituted TPY ligands, serving as a chemosensor for Ni2+ ions, are specifically attributable to the coordination between Ni2+ ions and TPY ligands. These changes manifest as alterations in fluorescence, color, absorption spectra, and overall material properties, providing a sensitive and selective means for Ni2+ ion detection. These chemosensors exhibited a unique response in the presence of other cations, allowing for their differentiation based on their distinct quenching behavior. The polyarylene ether, designated as PAET, served as the receptor in this system and displayed brilliant fluorescence quenching intensity when exposed to various ions. Notably, the detection of Ni2+ ions could be achieved at concentrations nearly in the sub-micromolar range. The results of this study demonstrated the effectiveness of transition metals with TPY ligands incorporated into the backbone of PAET 309. This approach provided a highly efficient, sensitive, and selective method for the analysis of Ni2+ ions, as illustrated in Fig. 15.109
 | ||
| Fig. 15 PEAT which acts chemosensor for the detection of Ni2+. | ||
Wang et al. (2013) synthesized a TPY derivative by employing 4′-4-(1,2,4-triazol-1-yl)-phenyl-2,2′′:6′,2′′-TPY in the presence of manganese nitrate. The UV-visible spectroscopy results indicated that this derivative exhibited significant calorimetric capability, particularly in the sensing of Fe2+ metal ions. Notably, the presence of Fe2+ ions could be visually observed with the naked eye, making it a promising candidate for a metal ion sensor. This research provides valuable insights for the development of sensors designed for the detection of metal ions, offering high sensitivity and selectivity in such applications.110
Jing et al. (2014) reported the synthesis of an asymmetrical diarylethene incorporating a stilbene-based TPY chromophore through a Heck reaction. This compound exhibited notable fluorescence properties that could be activated by UV-visible light and Hg2+/EDTA. Upon the introduction of Hg2+ ions, the compound's emission peak shifted by approximately 60 nm, accompanied by a significant increase in fluorescence intensity, reaching approximately four times its original level. This change in fluorescence was also associated with a noticeable alteration in color, shifting from blue to a bright green hue, indicative of the formation of a 1:2 metal/ligand complex. Diarylethene 310, illustrated in Fig. 16, served as a chemosensor capable of detecting Hg2+ ions within the solvent dichloromethane (DCM). This compound was also employed in the development of logic gates that responded to inputs from both light and chemicals, leading to fluorescence emission at 514 nm. Subsequent research efforts were directed toward refining the properties of this compound to explore its potential applications in optoelectronic devices.111
 | ||
| Fig. 16 Florescent chemosensor for detection of Hg2+ ion based on diarylethene based on stilbene linked TPY unit. | ||
Bhowmik et al. (2014) introduced a zinc-based TPY complex that demonstrated distinctive fluorescence properties, making it an efficient chemosensor for the detection of pyrophosphate in aqueous solutions at physiological pH levels. This complex exhibited a remarkable fluorescence response, with an increase in intensity of approximately 500-fold, and displayed high sensitivity, providing detectable responses at concentrations as low as 20 nm. Moreover, it proved to be highly effective in the context of fluorescence microscopy, enabling the imaging of HeLa cells. The researchers further incorporated this complex into a hydrogel matrix, which was then applied to coat paper strips for pyrophosphate detection. This development holds promise for the creation of commercially available chemosensors designed for the detection of proteins and enzymes, with potential applications in diagnostics and clinical settings.112
In the study conducted by Wu et al. in 2014, the authors investigated the supraromatic response of corannulene-based compounds to metal ions. They synthesized super aromatic TPY ligands by incorporating a corannulene unit at the 4th position of the ligand structure. Both ligands exhibited excellent properties for interacting with various metal ions, and their complexes were analyzed using UV-visible spectroscopy and fluorescence techniques. In the first compound, denoted as 311, the corannulene unit was directly linked to the 4′-phenyl TPY unit, while in the second compound, denoted as 312, an acetylene group separated the corannulene group from the 4′-phenyl TPY unit (see Fig. 17). Both synthesized complexes acted as chemosensors for metal ions. Compound 311 displayed a bathochromic shift in its absorption spectrum and a higher fluorescence quantum yield, whereas compound 312, due to the presence of the acetylene bridge, exhibited enhanced molecular conjugation, making it suitable for energy conversion applications. Both compounds were subjected to UV-visible and fluorescence measurements and were employed for the calorimetric measurement of Fe2+ ions, leading to a change in color from colorless to magenta. A comparison of the binding constants indicated that the size of the substituent played a crucial role in the conjugation of the TPY group. Additionally, these ligands served as important chemosensors for Zn2+ ions, displaying a bathochromic shift at a wavelength of approximately 60 nm, allowing for the discrimination of Zn2+ ions from other ions.113
 | ||
| Fig. 17 Super aromatic terpyridines based on corannulene responsive to metal ions. | ||
Juang et al. (2016) investigated a graphene oxide TPY conjugate and its application as a nano-chemosensor. This conjugate exhibited notable characteristics, including high sensitivity and selectivity for detecting Fe2+ and Fe3+ ions in aqueous solutions while effectively discriminating against alkali and alkaline earth metal ions. When examining the absorption spectra in the presence of Fe2+ and Fe3+ ions, the chemosensor exhibited a distinct peak at a wavelength of 568 nm, resulting in a visible change in color from light pink to a deep magenta hue. Furthermore, the graphene TPY conjugates demonstrated fluorescence emission at 473 nm, specifically quenching the fluorescence response in the presence of Fe2+, Fe3+, and Co2+ ions when combined with graphene oxide-conjugated TPY. Importantly, this quenching effect was not observed with other metal ions. The approximate detection limit for Fe2+ ions using this chemosensor in a water-based solution was estimated to be in the range of 6–10 μM.114 Yang et al. (2016) conducted a study involving TPY-based functionalized conjugated polyfluorene, synthesized through the Suzuki polymerization process. Among the compounds investigated, compound 313 displayed a notably enhanced sensing capability compared to compound 314 (Fig. 18). This heightened sensitivity was attributed to the efficient interaction between the specific sites within the triphenylamine-based TPY units and metal ions, as well as their protons. Compound 313 exhibited particularly high sensitivity, as indicated by its Stern–Volmer constants of 2.30 × 105 M−1 and 1.05 × 105 M−1. It was found to be proficient in detecting Cu2+ ions in addition to CN− ions. These findings suggest that compound 313 holds significant promise for applications in both environmental monitoring and medical contexts.115
 | ||
| Fig. 18 Chemosensors with triphenylamine-functionalized conjugated polyfluorene. | ||
Zhou et al. (2016) explored the use of coumarin-based TPY zinc complexes as a means of detecting pyrophosphate ions. The findings of their investigation demonstrated that compound 315 (Fig. 19) exhibited outstanding activity in sensing pyrophosphate ions and displayed a high degree of selectivity, particularly in the presence of other metal ions that shared a similar nucleotide environment. This selectivity was observed in aqueous media at a specified pH level. Upon interaction with pyrophosphate ions, a distinct blue shift in the absorption spectrum was observed, accompanied by a substantial increase in fluorescence intensity, reaching approximately 27-fold enhancement at a wavelength of 45 nm. Notably, this compound was effectively applied for fluorescence imaging of pyrophosphate in Hi-5 cells and C. elegans, suggesting its utility for pyrophosphate fluorescence imaging within biological contexts. These results underscore the potential utility of zinc TPY complexes for the detection of pyrophosphate ions within biological cells and their fluorescence imaging applications.116
 | ||
| Fig. 19 Chemosensor for sensing pyrophosphate. | ||
Mandal et al. (2018) investigated TPY derivatives with a particular focus on their role as turn-on fluorescent chemosensors for the detection of zinc ions. Their study highlighted the characteristic properties of ligand 316, as illustrated in Fig. 20, which displayed remarkable selectivity and sensitivity towards zinc ions, resulting in a substantial enhancement of fluorescence at a wavelength of 370 nm. Importantly, in the emission spectra of this compound, there were no observable responses to other metal ions. The detection limit for zinc cations was determined to be 9.76 μM. The complexation of Zn2+ ions 317 with this ligand occurred in a 2:1 ratio, with an association constant of approximately 1.85 × 104. These sensors were effectively employed to study the presence of Zn2+ ions in cultured MDA-MB-468 cells, showcasing their potential utility in biological applications.117
 | ||
| Fig. 20 Chemosensor which exhibits for detection of Zn2+ in solution as well as living cells. | ||
Kar et al. (2019) described cadmium-based TPY metal complexes for selective detection of pyrophosphate ions. The complexes were synthesized by using CdCl2 TPY ligand 4′-(4-N,N′-dimethylaminophenyl)-TPY 318 (Fig. 21). The synthesized complexes act as fluorescence chemosensors for the detection of pyrophosphate ions (PPi) and discriminate over the ions like F, Cl−, Br−, CO32−, SO32−, AcO−, NO2−, and H2PO4. They are also successfully employed for the detection of pyrophosphate ions in HeLa cells. These results show that it can be used as a chemosensor in a cell.118 Kang and his group described the graphene quantum dot embedded TPY known as GQDs-TPY. Graphene quantum dot TPY acts as multifunctional chemosensors for the detection of Fe2+ metal ions and is typically used for the detection of Zn2+ ions over 13 metals present in water. The TEM studies show that the diameter of GQDs-tpy is 4.0 nm. The TPY-embedded GQDs-tpy is water soluble and has improved dysfunctional chemo sensing properties as compared to other models SiO2-TPY. The GQDs-TPY is sensitive and selective for the detection of ferrous ions and its approximate detection limit is ∼1.43 × 10−7 M. It acts as a fluorescent chemosensor for the detection of Zinc ions of about 0.1–8.0 μM and a low detection limit of 5.7 × 10−8 M. It is also useful for construction of logic gates such as XOR, INHIBIT and IMPICATION logic gates for two inputs as well as OR and NOR on basis of their absorption and emission spectral properties. This is a versatile and metal coordination property of free TPY groups and is also used for not only metal sensing switching logical molecular gates and also metal functional-based hybrid materials.119
 | ||
| Fig. 21 Graphene quantum dots TPY (GQDs-terpyridine) used as selective detection of metal ion and construction of logic gates. | ||
Khavasi et al. (2019) investigated the 4′-(3,4-dimethoxy-phenyl)-TPY ligand and its interaction with copper cations as an effective sensor for tetrahedral oxometalate anions, specifically molybdate, tungstate, and vanadate. Additionally, this sensor demonstrated proficiency in detecting simple cations like halides, nitrate, nitrite, and phosphate when present in water solutions. One notable aspect of this work was the ability to detect these anions using a low molecular weight hydrogel.120
Amuthakala et al. (2020) described TPY base chemosensor for the detection of inorganic cations and fluoride ions respectively. The 2-(4-(TPY)-4′-yl-phenyl)-N-methyl-3-thiosemicarbazone (L) 319 (Fig. 22) acts as a sensor, in which TPY unit acts as cation while semicarbazone acts as an anion. This receptor molecule shows excellent properties for sensing Cu2+, Ni2+ and Mn2+ cations and F− anion, which is revealed by cyclic voltammetry and spectral technique; fluoride ions by protonation and cation sensing by using TPY moiety while anion detection is by semicarbazone moiety. Jobs plots studied revealed based on spectral data show metal–ligand interaction is through a 1:1 stoichiometric ratio. The mechanism of fluoride detection is by 1H-NMR. The fluorescence intensity shows quenching of Cu2+ ions and fluoride ions. Paper strips that are fabricated with ligand moiety show good sensitivity and selectivity towards Mn2+, Ni2+, Cu2+ cations and fluoride anions. This work reveals that TPY base semicarbazone material ligands act as efficient probes for the detection of the transition of metal cations and fluoride.121
 | ||
| Fig. 22 Receptor 2-(4-(TPY)-4′-yl-phenyl)-N-methyl-3-thiosemicarbazone for detection of inorganic cation and fluoride anions. | ||
Liu et al. (2021) reported the synthesis of a hydrophilic TPY-based copolymer composed of acrylic acid and fluorescent fragments. This copolymer was designed for the detection of various transition metal ions. Notably, the detection mechanism relied on observing color changes, indicative of metal ion chelation. The experimental results revealed distinctive color changes in the presence of specific metal ions: a violet color change in the presence of Fe2+ ions, a brown color with Co2+ ions, and a pale blue color for Cu2+ ions, all of which contrasted with the initial colorless solution. This chemo-sensor exhibited efficient absorption and luminescent properties, which were attributed to the incorporation of monomers containing the fluorene unit 320 (VOFs), as depicted in Fig. 23. The pure polymer exhibited an absorption peak at 284 nm due to π–π* transitions. Another absorption peak appeared at 335 nm with an increase in metal ion concentration in the solution. Notably, the absorption peak for Fe2+ ions was observed at 570 nm, which could be attributed to a metal–ligand charge transfer mechanism. The intensity of this peak increased proportionally with the concentration of Fe2+, allowing for quantitative detection. Furthermore, the introduction of VOFs led to intramolecular energy transfer, resulting in enhanced fluorescence and quenching responses. This property enabled the distinction between Zn2+ and cadmium ions, facilitating the detection of larger metal ions based on their concentration. These chemosensors have significant utility in aqueous environments, making them applicable in various fields, including industry, biomedicine, and environmental protection.122
 | ||
| Fig. 23 Hydrophilic terpyridine-based copolymers containing fluorescence units act as chemosensors for sensing transition metal ions. | ||
Das et al. (2021) described zinc base TPY metal complexes 321 (Fig. 24) for the detection of pyrophosphate anions in aqueous media at physiological pH value. This chemosensor has high selectivity and sensitivity over the other anions such as F−, Cl−, Br−, CO32−, SO32−, NO3, OH− HPO42−, SO42−, SO32−, S2O32−. The sensors show high fluorescence intensity in aqueous media. UV-visible and fluorescence studies were used to perform detection. The sensor showed a fluorescence intensity of about ∼50 folds on the addition of PPi anions and the sensitivity detection limit was about ∼300 nm.123
 | ||
| Fig. 24 Zinc-TPY-based chemosensor for detection of pyrophosphate. | ||
Rodrígueze et al. (2021) elucidated the properties of fluorescent zinc-based TPY complexes, exemplified by complex 322 (Fig. 25). These complexes serve as chemosensors for catecholamine amine neurotransmitters and nucleosides in aqueous media. Notably, the sensor exhibits a pronounced affinity and selectivity for L-DOPA over other common neurotransmitters such as dopamine, epinephrine, L-tyrosine, and nucleosides at a pH of 7.4. All zinc TPY complexes within the study display a robust response and significant fluorescence quenching. Optical investigations reveal that the detection limit for L-DOPA in urine and blood plasma is on the order of micromolar concentrations. Complementary analyses involving 1H-NMR, 11B-NMR, and DFT studies demonstrate that L-DOPA binds to zinc TPY complexes through the esterification between the aromatic diol moiety of L-DOPA and coordination with zinc carboxylate, facilitated by two binding sites. Spectroscopic assessments further underscore the excellent performance of complex 2-zinc, exhibiting the highest activity (logK = 6.01) for L-DOPA detection and displaying superior responsiveness among all examined complexes. The distinct structural attributes of isomer 2-zinc in its interaction with L-DOPA arise from conformational strain and the heightened acidity of the pendant phenylboronic acid group, distinguishing it from other isomers. Moreover, zinc in conjunction with eosin Y was employed as an accurate and rapidly responsive sensor for L-DOPA, a capability not shared by other anionic species. This research underscores the development of novel water-soluble fluorescent transmitters, offering effective tools for the selective detection of neurotransmitters in aqueous environments, particularly as a means of sensing transition metal ions.124
 | ||
| Fig. 25 Zinc TPY boronic acid isomer exhibits excellent activity for sensing L-DOPA among the other three reported. | ||
Zhao et al. (2022) introduced a novel approach involving zinc TPY complexes within mesoporous silica matrices. These complexes function as fluorescent chemosensors designed for the rapid detection of sulfonamide antibiotic residues in tap water and milk. The zinc groups within these complexes play a pivotal role in identifying binding sites for the recognition of sulfonamides, such as sulfathiazole. This recognition occurs through the formation of conjugates between TPY@Zn-SBA-15, represented as complex 323 (depicted in Fig. 26), and sulfathiazole (STZ). This interaction facilitates energy transfer from the organic TPY@Zn-SBA-15 component to STZ, resulting in a remarkable alteration of the fluorescence spectrum. Specifically, as the concentration of sulfathiazole increases, the emission intensity of fluorescence at 429 nm gradually diminishes. The detection capabilities of this sensor are noteworthy, with detection limits reported at approximately 0.17 μM for sulfamethizole, 0.63 μM for sulfachlorpyridazine, 0.52 μM for sulfamerazine, and 0.57 μM for sulfamethoxypyridazine. Interestingly, in the absence of introduced Zn2+ cations, these compounds exhibited high activity, although the introduction of Zn2+ cations also contributed to the determination of fluorescence signals. This research holds promise for the detection of antibiotic residues, particularly in the presence of finely tuned organic components and target-binding metal sites incorporated into mesoporous silica materials. Such innovations provide a valuable avenue for addressing the critical issue of sulfonamide contamination and residue detection in water and milk, thereby enhancing human and public health.125
 | ||
| Fig. 26 Zinc-modified TPY mesoporous silica acts as a chemosensor for the detection of sulfonamide antibiotics. | ||
3.3. Photosensitizers
Photosensitizers play a pivotal role in the realms of biology and photochemistry, yet their photophysical and molecular mechanisms have been insufficiently explored. These compounds hold particular significance in applications such as the development of dyes for dye-sensitized solar cells, light-to-energy conversion through annihilation processes, and the facilitation of charge separation to interrupt natural photosynthetic reactions.In a study conducted by Mori et al. in 2012, platinum TPY complexes were investigated when attached to a series of mesoporous silica materials, resulting in the creation of photocatalysts with distinct photoluminescent and photocatalytic properties. It was observed that these complexes exhibited strong photoluminescence, a property notably absent in the free platinum group. This enhanced luminescence intensity in isolated platinum complexes was attributed to metal–ligand charge transfer interactions. Moreover, these platinum complexes demonstrated remarkable oxidation reactivity in the presence of oxygen at room temperature. Specifically, platinum proximal complexes, exemplified by the structure represented as complex 324 (Fig. 27), exhibited high luminescence intensity attributable to metal–metal ligand charge transfer (MMLCT). This property was particularly relevant in the context of hydrogen (H2) evolution, where such complexes functioned as a single-component system. The use of a 3D structure reminiscent of MCM-48 as a host material was found to enhance phosphorescence, with implications for both reactant diffusion and the unique 1-D hexagonal structure of SBA-15 and MCM41. Moreover, MCM-48, SBA-15, and MCM41 are all types of mesoporous materials, specifically belonging to the family of Mobil Composition of Matter (MCM) and Santa Barbara Amorphous (SBA) materials. Ultimately, the observed photoluminescent and photocatalytic properties were attributed to the triplet state of the platinum complexes, along with the loading of platinum and the hexagonal structure of the host material.126
 | ||
| Fig. 27 Pt (TPY)Cl] Cl complex onto the APTMS-modified silica surface. | ||
Schubert et al. (2012) focused on heteroleptic bis(tridentate) ruthenium(II) TPY complexes derived from 1,3-bis(1,2,3-triazol-4-yl)benzene 325 through click chemistry (Fig. 28). The research involved the comprehensive optimization of the synthesis of cyclometalated ruthenium complexes. Notably, some of these complexes exhibited weak emission characteristics at room temperature, along with an unusually prolonged excited state. These complexes displayed absorption peaks at approximately 700 nm, and they possessed a high molecular extinction coefficient, roughly estimated at 2000 M−1 cm−1. This heightened absorption was attributed to metal–ligand charge transfer phenomena, leading to a color change to black in these complexes. These findings have significant implications for applications in dye-sensitized solar cells, where the unique properties of these complexes can be harnessed for their potential benefits.127
 | ||
| Fig. 28 Ruthenium cyclometalated TPY metal complexes applications for dye-sensitized solar cells. | ||
Sepehrifard et al. (2013) investigated the photophysical and electrochemical properties of novel ruthenium TPY metal complexes that were adsorbed onto P25 TiO2, and they assessed the performance of these complexes in dye-sensitized solar cells. The dyes under examination in this research possessed homoleptic characteristics, featuring tridentate chromophores. The tridentate ligands in these dyes could be either TPY or dipyrazinylpyridine, and the anchoring groups were selected from carboxylic groups or ester groups. A phenylene spacer was incorporated between the chromophore and the anchoring group. It was observed that the dipyrazinylpyridine-based dye exhibited poor performance, primarily due to its low excited state characteristics. Moreover, the phenylene spacer proved to be inefficient for dye loading. Even after efforts to promote transesterification, the ester group-based dye still exhibited suboptimal performance. Among the dyes studied, the most promising was the bis(4′-carboxy-TPY) ruthenium(II) bis(hexafluorophosphate) complex, represented as complex 326 (Fig. 29). This particular dye demonstrated an impressive solar energy conversion efficiency of approximately 1.56% under the best-measured conditions.128
 | ||
| Fig. 29 Structure of dye molecules. | ||
Mattay et al. (2014) described the zinc and ruthenium TPY metal complexes 327 (Fig. 30) as a photosensitizer for the production of oxygen. The homoleptic zinc and ruthenium complexes of bis(tridentate) 9-acridine and 10-methyl-9-acridinium-substituted TPY to check their suitability as triplet photosensitizer by photooxidation of 1,5-dihydroxynapthalene which is used as a model reaction. The generation of singlet oxygen is due to ruthenium TPY for the acridine complexes acridinium complexes are not useful in this case. The singlet oxygen generation from ZnII (1) and RuII (4) of quantum yield of about 0.82. The fact is that ZnII (1) shows poor results in the solar spectrum while RuII shows excellent results in the solar spectrum and therefore sensitizing ability is enhanced to the acridine system. The acridinum containing 2 RuII and 5 RuII as well as linker-free acridine TPY system are also ineffective sensitizers and have short-lived excited states. The metal complexes containing conjugate organic systems have long-lived triplet energy systems that tolerate great potential. The singlet oxygen production and photophysical degradation require stability consideration. Results show that it is useful as an efficient photosensitizers and photo green chemistry but not having much interest in photo dynamic therapy.129
 | ||
| Fig. 30 Ruthenium and zinc TPY complexes as photosensitizer for singlet oxygen generation. | ||
Gasser et al. (2014) introduced novel ruthenium complexes designed as photosensitizers for application in photodynamic therapy, with potential implications in dye-sensitized solar cells (DSSCs). The research focused on the synthesis and characterization of two polypridyl ruthenium complexes, with their structural properties elucidated through X-ray crystallography. These complexes were evaluated in terms of their stability and their ability to generate singlet oxygen molecules upon light absorption. Both synthesized compounds were found to be capable of producing singlet oxygen when exposed to light. The photo-toxicity of these compounds was assessed against the human cervical cancer cell line, HeLa. Compound 329 exhibited efficient photo-cytotoxicity against HeLa cells, with a detection limit in the micromolar range and no observable toxicity in the absence of light. Compound 328 displayed toxicity against HeLa cells in the nanomolar range, with a P1 80 value against HeLa cells (Fig. 31). Furthermore, these results demonstrated greater efficacy when compared to clinically approved photosensitizers, such as PS-ALA, and oxygen performers in experimental settings. Microscopy experiments revealed that the complexes accumulated predominantly in the cytoplasm, particularly within the mitochondria. These findings were validated using inductively coupled plasma mass spectrometry (ICP-MS), confirming a remarkable 67% accumulation of ruthenium in the mitochondria. Additionally, in the field of photodynamic therapy, these complexes were tested against two bacterial strains, namely S. aureus and E. coli. Photo-toxicity was observed against Gram-positive bacteria like S. aureus, while no toxicity was observed in Gram-negative bacteria. This indicated that Gram-negative bacteria were less affected by the complexes compared to their Gram-positive counterparts.130
 | ||
| Fig. 31 Polypridyl ruthenium complexes act as photosensitizers. | ||
Kassim et al. (2014) described ruthenium and tungsten metal complexes having bipyridyl and dithiolene co-ligand prepared and behavior was studied as a dye sensitizer for photochemical cells used in water splitting. The cyclic voltammetry showed multiple redox reactions, half potential for this is E1/2 = 0.652 V, E1/2 = 0.05 V and E1/2 = 0.61 V for the Ru(II)/Ru(III) reduction and for W(IV) and W(V) oxidation is and W(VI)/W(V) respectively. The bimetallic complexes were used as dye sensitizers for artificial photosynthesis. This dye is more valuable than commercially available N3 [bis(isothiocyanato)bis(2,2′-bipyridyl-4,4′-dicarboxylato)ruthenium(II)] in the presence of a homogeneous system in TiO2 by the introduction of –COOH to facilitate binding to increase the efficiency of the charge transfer process.131
Venuvanalingam et al. (2015) described rhenium(I) TPY complexes that were synthesized and studied using density functional theory (DFT) and time-dependent functional theory (TD-DFT) methods. The effect of different substituent groups on rhenium complexes and their electronic and optical property was also investigated. The rhenium complexes are better electron transport materials in organic light-emitting diode devices because of their better λelectron, the complexes having no substitution give balanced charge transfer ability with higher efficacy in organic light-emitting devices. The natural orbital theory studies show that n–π* is responsible for the ground state stabilization of complexes. Re–CO bond is a transient interaction shown by QTAIM studies. Natural transition orbital analysis showed that absorption is related to 1MLCT/1LLCT/1ILCT character, on the other hand, emission is due to 3MLCT/3LLCT/3ILCT. The complexes having higher quantum yield are smaller to ΔEs1–T1 and μs1, ET1 middling 3MLCT character. The rhenium complexes show remarkable properties for OLEDs devices.132
Kwok et al. (2016) described dinuclear alkynyl platinum base compound 330 (Fig. 32) as a sensitizer having light-harvesting properties. These dinuclear platinum complexes were studied for their application as photo sensitizers for studying light-harvesting properties. The electronic absorption properties revealed three absorption bands at wavelengths of 280 nm and 480 nm and showed a π–π* transitions among dinuclear platinum and TPY ligand and intraligand π–π* transitions occurred. The absorption band for complexes reported is a mixture of MLCT and LLCT transitions. The electrochemical properties show that complex 330 are quasi reversible reactions that occur at voltages of −1.10 and −1.60 respectively. Complex 330 also had less positive oxidation potential (+0.74 V) than complex 330 (1) (+0.95 V) and 310 (2) (+0.80 V). These complexes are used in DSSCs (dye-sensitized solar cells) and power efficiency is about 0.8%. This is the first platinum complexes used as DSSCs.133
 | ||
| Fig. 32 Platinum complexes used in dye sensitizer solar cells (DSSCs). | ||
Mongal et al. (2016) described [Ru(triOMePhtpy)(bpydca)(NCS)]PF6 (triOMePhtpy = 4′-(3,4,5-triOMe) phenyl-2,2′:6′,2′′-terpyridine; bpydca = 4,4′-dicarboxy-2,2′-bipyridine) 331 (Fig. 33), which is prepared and characterized by using IR, UV-visible, NMR and mass spectroscopy. The complex showed a metal-to-ligand charge transfer band at a wavelength of 511 nm. This Ru-based photosensitizer aims to transfer the maximum amount of light-induced charge transfer approaching dye semiconductors with different donor group substituents having phenyl TPY and NCS as facilitator ligands. The results show an increased number of metals to ligand charge transfer transition and a high extinction co-efficient. The main drawback of this work is that the transition is more toward TPY ligand which causes a reduction in photo-current conversion efficiency. To avoid this drawback further work was made using different substitution patterns on TPY moiety and performance was increased.134
 | ||
| Fig. 33 Structure of ruthenium-based dye used as sensitizer. | ||
Barbour et al. (2017) synthesized Cr(III) arylterpyridyl complexes characterized by intraligand charge transfer (ILCT) excited states. These complexes exhibited notable absorption in the visible region of the electromagnetic spectrum, owing to the presence of ILCT bands. The tunability of these ILCT bands across the UV-visible spectrum was achieved by incorporating different substituents on the aryl ring, including both electron-withdrawing and electron-donating groups. One of the synthesized complexes, Cr(4′-(4-methoxyphenyl)-2,2′:6′,2′′-terpyridine)2, denoted as complex 332 (Fig. 34), displayed a significantly higher visible region absorption. Specifically, it exhibited absorption with ε values of 11900 M−1 cm−1 at 450 nm and 5090 M−1 cm−1 at 500 nm, surpassing the parent complex Cr(tpy)2 (where tpy = 2,2′:6′,2′′-terpyridine), which had ε values of 2160 M−1 cm−1 at 450 nm and 170 M−1 cm−1 at 500 nm. Emission experiments indicated that the Cr(III) complex exhibited phosphorescence with lifetimes ranging from 140 to 600 nanoseconds upon excitation of intraligand charge transfer states. This complex displayed at least three reversible ligand-based reduction bands, with a reduction potential of −160 mV when compared to Cr(tpy)2. The excited state potential for this complex ranged from approximately +0.95 to +1.04 V versus the ferrocene/ferrocenium redox couple, which acts as a potent photo-oxidant. These complexes garnered further attention due to their cost-effectiveness and their classification within the first transition series, making them valuable subjects for continued study and exploration.135
 | ||
| Fig. 34 Chromium complex which acts as a potent photo-oxidant. | ||
Liu et al. (2014) described iridium-based TPY metal complexes 333 which possess photo-physical and photo-biological properties. The synthesized complexes were characterized using different spectroscopic techniques to study photo-physical and photo-biological properties. The results showed that these complexes have a strong bridging ligand which is visible in regions having 1π–π* transition and has red or near infrared phosphorescence showing triplet excited state absorption across NIR and visible wavelength. The iridium complexes such as Ir 334 complex (Fig. 35) having a single bond between fluorenyl group and TPY bridging ligand which extends pi-conjugation by ethynyl group of bridging ligands than that of Ir 335 shifted to red shift to the UV-visible absorption having low-level CT state and increase non-radiative decay that results in low phosphorescence. Ir 338 complex shows a bathochromic shift and has strong donating ability and transfer of negative charge from C^N^C to TPY group. All complexes show a positive absorption band in the visible region and NIR region in TA spectra. Iridium 335 and complex had stronger triplet state TA lifetime and Ir 335 and Ir 336 possessed dominate bridging ligands with localized π–π* transition. Ir 334 and Ir 337 have CT states for the other three complexes. Ir 337 complex is the strongest and most suitable agent for vitro photo-dynamic therapy towards melanoma cells when they are activated in the presence of visible light and photo-cytotoxicity and phototherapeutic agents ranging from 20 to 300. Photo-dynamic therapy effect in visible light did not produce singlet oxygen quantum yield and DNA cleaving capacity under cell-free conditions. All these Ir(III) complexes show different properties like phosphorescence brightly in compromised cells and photo-activated cellular uptake and also highlighted important properties used as photosensitizers.136
 | ||
| Fig. 35 Molecular structure of dinuclear iridium complexes used as photosensitizer. | ||
Chao et al. (2019) explored the utility of cyclometalated iridium complexes, depicted in Fig. 36, as photosensitizers in photodynamic therapy (PDT). These complexes were synthesized and comprehensively characterized using various spectroscopic techniques. The study focused on evaluating the efficiency of these complexes as PDT agents, particularly highlighting the presence of extended pi conjugation in the ligand structures and investigating the correlation between the extent of conjugation and their photodynamic performance. The research findings indicated that these complexes exhibited an affinity for accumulating in the endoplasmic reticulum (ER) of cells. Following photodynamic therapy, these complexes induced apoptosis in the cells due to ER stress. This effect was attributed to their cytotoxicity and the high singlet oxygen quantum yield stemming from the extended conjugation present in the complexes. Remarkably, these complexes demonstrated extraordinary photodynamic therapeutic efficacy against various cancer cells. For instance, Ir 339 complexes exhibited high activity against A549 cells, with an IC50 value as low as 0.65 micromolar. Importantly, after PDT, these complexes rapidly effluxed calcium ions from the endoplasmic reticulum system in A549 cells within a short timeframe of approximately 45 to 90 minutes, particularly with pre-treatment. These findings underscored the potential of the Ir 340 complex as a promising photosensitizer for application in photodynamic therapy, holding promise for further developments in cancer treatment.137
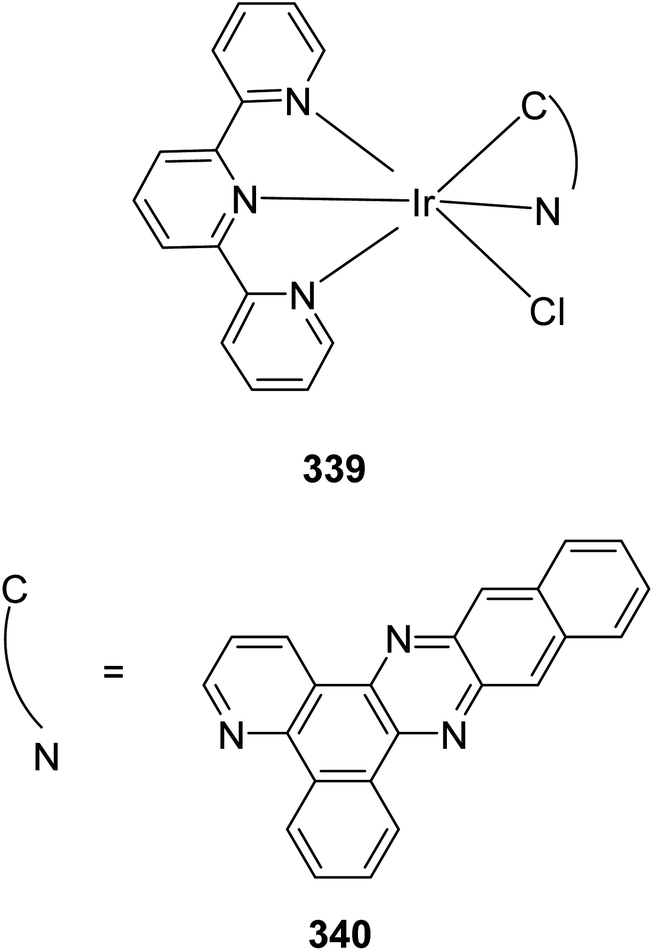 | ||
| Fig. 36 Iridium complex which acts as photosensitizer in photodynamic therapy. | ||
Wang et al. (2020) described iron TPY complexes as a photocatalytic system consisting of TADF photosensitizer used for reduction of CO2. This reaction takes place in the presence of water which enhances production of CO with about 99.3% selectivity and turn over number and frequency in this process is about 2250 and 60 min−1 respectively. This photocatalytic system consists of TPY-Fe complex 341 and TADF sensitizer 4CzIPN 342 (Fig. 37). It produces CO upon reduction of carbon dioxide upon irradiation in water or DMF. In this process, ruthenium TPY complexes exhibit very poor photophysical properties of about 20 TON (s−1).138
 | ||
| Fig. 37 Iron complex acts as a photosensitizer for the production of CO in the presence of TDAF photosensitizer. | ||
Teran et al. (2021) described rhenium TPY complexes' excited state and ground state properties. These rhenium base TPY metal complexes possessed different substituents of different electron-donating groups. Major modulations were made for electrochemical potential and almost 4-fold variations or changes were observed for metal to ligand charge transfer upon moving from CN to –OMe substituents. When moving towards electron donating group NMe2 the strong absorption band was in κ2 N complex, which shows a red shift by ca. 100 nm compared to other complexes. The CV and IR-SEC revealed that these complexes show an irreversible reaction, stepwise two-electron reduction method. The –NMe substituents have a good lifetime compared to other complexes. The complex lowest singlet and triplet state have intraligand charge transfer properties. The long triplet lifetime (ca. 380 ns obtained for NMe2 complex) indicated that strong electron donating ability shows the strongest photophysical and photochemical properties. This work further leads to the synthesis of TPY complexes with different substituent group modifications. These complexes 343 (Fig. 38) are strongly emissive and longtime having a reducing excited state with redshift absorption and would be better used as photosensitizers.139
 | ||
| Fig. 38 Rhenium complexes act as photosensitizer. | ||
4. Overall comparison of the above-mentioned TPY-based metal complexes applications
This comprehensive review explores the diverse applications of terpyridine metal complexes across various scientific domains, emphasizing their pivotal roles in catalysis, chemosensors, optoelectronic devices, photosensitizers and photocatalysis (Table 9). The catalytic efficiency of terpyridine metal complexes is assessed through a comparative analysis, revealing that cobalt-based terpyridine complexes exhibit superior catalytic performance, characterized by higher Turn Over Numbers (TON; s−1) and Turn Over Frequencies (TOF; h−1). Notably, higher TOF values indicate enhanced catalytic efficiency, while elevated TON values signify prolonged sustainability and increased substrate conversion.| Compound no. | Chemical structure | Applications | % conversion/high molar extinction coefficients (ε) | Rate constant (k) (s−1)/binding constant | Sensitivity | Selectivity (%) | Detection limit | Quantum yield (Φ) | TON (s−1)/TOF (h−1) | References |
|---|---|---|---|---|---|---|---|---|---|---|
| 43 |  |
Catalysis | 100 | — | — | — | — | — | — | 43 |
| 49 | 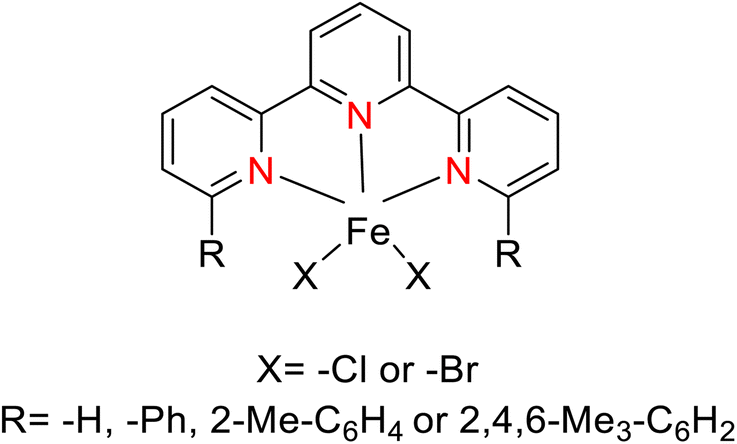 |
Catalysis | 81–97 | — | — | — | — | — | 90 | 44 |
| 65 |  |
Catalysis | 98% | — | — | — | — | — | — | 49 |
| 75 |  |
Catalysis | 100 | — | — | — | — | — | 100 TOF | 52 |
| 79 |  |
Catalysis | 8–100 | — | — | 100 | — | — | — | 54 |
| 90 | 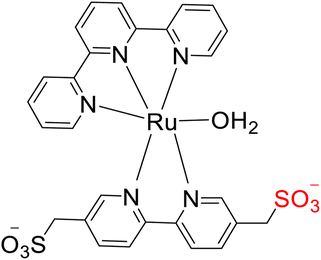 |
Catalysis | 94 | — | — | — | — | — | 47 | 56 |
| 95 |  |
Catalysis | — | — | — | — | — | — | 166 | 57 |
| 104 |  |
Catalysis | 12.1 | — | — | — | — | — | 343 | 59 |
| 113 |  |
Catalysis | 94 | — | — | — | 96 | — | — | 64 |
| 120 |  |
Catalysis | 100 | — | — | — | — | — | 65 | 65 |
| 136 |  |
Catalysis | — | — | — | — | — | — | 990/495 | 67 |
| 139 | 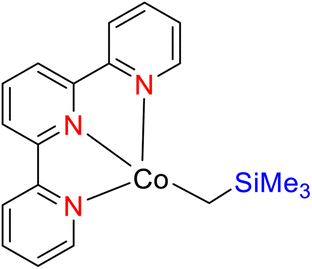 |
Catalysis | 84 | — | — | — | — | — | — | 74 |
| 144 |  |
Catalysis | 90 | — | — | — | — | — | — | 75 |
| 152 |  |
Catalysis | 70 | — | — | 4.6:1 |
— | — | — | 45 |
| 157 |  |
Catalysis | — | — | — | 86 | — | — | 107 TON | 76 |
| 171 |  |
Catalysis | 69.8 | — | — | 95.4 B-adduct | — | — | — | 79 |
| 180 | 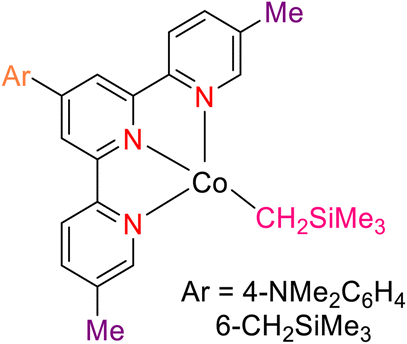 |
Catalysis | 36–49 | — | — | 92:8 meta selectivity |
— | — | — | 80 |
| 184 |  |
Catalysis | 95 | — | — | — | — | — | — | 84 |
| 184 |  |
Catalysis | 21–81 | — | — | — | — | — | — | 84 |
| 210 |  |
Catalysis | 52–98 | — | — | — | — | — | — | 86 |
| 224 | 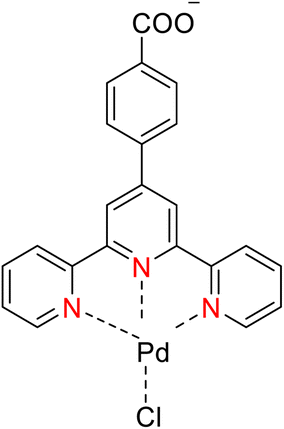 |
Catalysis | 31–71 | — | — | — | — | — | — | 90 |
| 226 | 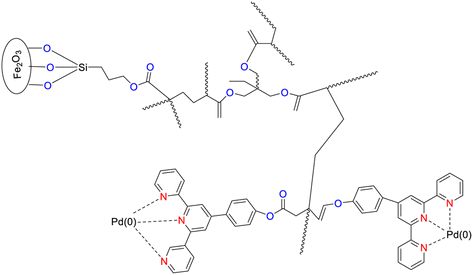 |
Catalysis | 98 | — | — | — | — | — | TOF = 257 h−1 | 91 |
| 230 |  |
Catalysis | 96 | — | — | — | — | — | TON = 595 | 92 |
| 246 |  |
Photocatalyst | — | k1 = 6.8 × 10−5 s−1 and k2 of nearly zero 2nd rate constant | — | — | — | — | — | 94 |
| 252 |  |
Photocatalyst | — | Pseudo-first order rate constant of 10.4 s−1 | — | — | — | — | TOF = 3.2 × 10−10 s−1 | 95 |
| 271 |  |
Photocatalyst | — | k = 3.2 × 10−2 s−1 rate constant | — | — | — | — | — | 96 |
| 282 | 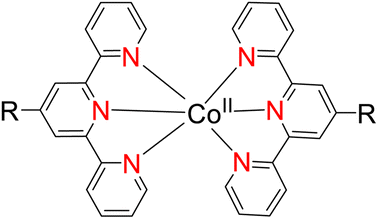 |
Photocatalyst | — | — | — | — | — | — | TON = ∼330 | 99 |
| 291, 292 | 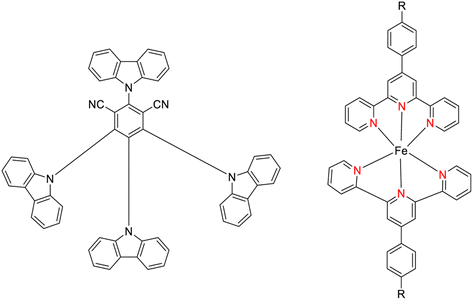 |
Photocatalyst | — | — | — | Selective reduction of CO2 to CO | — | — | TON = 6320 | 100 |
| 99.4% selectivity | TOF = 127 min−1 | |||||||||
| 293 |  |
Photocatalyst | — | — | — | — | — | — | TOF = ∼264 h−1 | 101 |
| 303 | 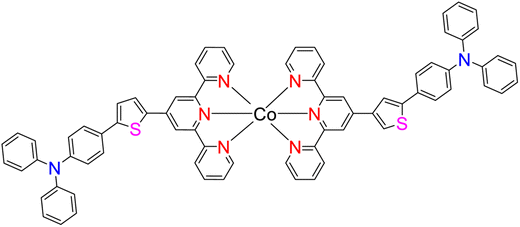 |
Photocatalyst | 45000 M−1 cm−1 molar extinction coefficient |
— | — | — | — | Apparent quantum yield = 5.34% | TON = 5718 s−1 | 103 |
| TOF = 1430 h−1 | ||||||||||
| 304, 305 |  |
Photocatalyst | Efficiency of methylene blue degradation is 67.13% and 65.91% | k = 0.0099 (304) | — | — | — | — | — | 104 |
| k = 0.0110 (305) | ||||||||||
| Rate constant follow pseudo first order | ||||||||||
| 306 | 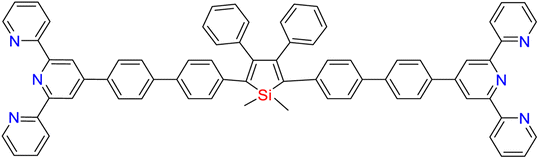 |
Chemosensor | — | — | — | (2.0 × 10−4 mol L−1) | — | 0.27% | — | 106 |
| For Zn2+ ions | ||||||||||
| 308 | 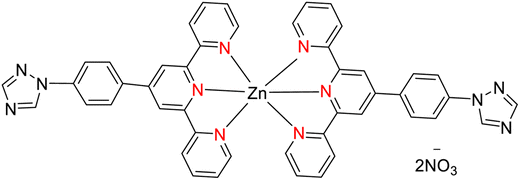 |
Chemosensor | — | — | — | Ni2+ and Co2+ selectivity | — | — | — | 108 |
| 309 |  |
Chemosensor | — | — | For Ni2+ | — | — | 7.89% | — | 109 |
| 310 |  |
Chemosensor | — | — | For Hg2+ | = | — | 0.29 | — | 111 |
| 311, 322 |  |
Chemosensor | — | — | For Fe2+ and Zn2+ | — | Fe2+ = 1.1 × 10−7 M (311) | 0.19 (311) | — | 113 |
| Fe2+ = 8.9 × 10−8 M (312) | 0.16 (312) | |||||||||
| Zn2+ = ∼1.6 × 10−8 M (311) | ||||||||||
| Zn2+ = ∼2.0 × 10−8 M (312) | ||||||||||
| 313 |  |
Chemosensor | — | — | For Cu2+ | — | 3.7 × 10−6 M−1 | 0.87 | — | 115 |
| 315 | 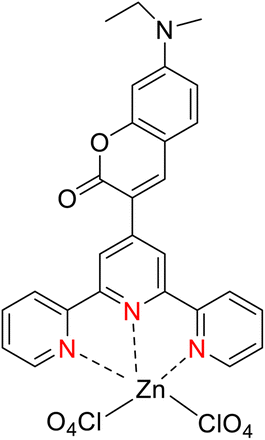 |
Chemosensor | — | — | — | Pyrophosp-hate ions | 2.18 × 10−6 M | — | — | 116 |
| 316 |  |
Chemosensor | — | — | For Zn2+ | For Zn2+ | 9.76 μM | 0.0254 | — | 117 |
| 318 |  |
Chemosensor | — | — | — | For Fe2+ | 0.14 μM | — | — | 119 |
| 319 |  |
Chemosensor | — | — | Inorganic cations (Mn2+, Fe2+, Ni2+, Cu2+, Zn2+, Pb2+ and Hg2+) | F− selectivity | Mn2+ = 2.83 μM | — | — | 121 |
| And anions (F−, Cl−, Br−, I−, HSO4− and H2PO4−) | Ni2+ = 2.67 μM | |||||||||
| Cu2+ = 1.78 μM | ||||||||||
| F− = 0.16 μM | ||||||||||
| 320 |  |
Chemosensor | — | — | — | For Zn2+ | — | — | — | 122 |
| 321 |  |
Chemosensor | — | — | — | Pyrophosp-hate ions | 300 nM | — | — | 123 |
| 322 |  |
Chemosensor | — | — | — | L-DOPA | 3.0 μmol L−1 | — | — | 124 |
| 323 | 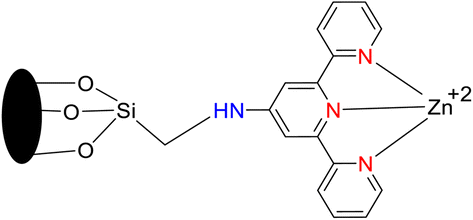 |
Chemosensor | — | — | Sulfonamide antibiotic (SA) residues | — | STZ = 0.17 sulfamethizole = 0.63, sulfachlorpyridazine = 0.52, sulfachlorpyridazine = 0.57 sulfamethoxypyridazine = 0.54 mM | — | — | 125 |
| 325 |  |
Photosensitizers | 20000 M−1 cm−1 molar extinction coefficient | — | — | — | — | — | — | 127 |
| 326 |  |
Photosensitizers | — | ca. 1 × 105 M−1 equilibrium binding constant | — | — | — | — | — | 128 |
| 327 |  |
Photosensitizers | 15200 M−1 cm−1 molar extinction coefficient |
— | — | — | — | 0.82 quantum yield for singlet oxygen generation | — | 129 |
| 328, 329 |  |
Photosensitizers | — | — | — | — | — | Quantum yield for singlet oxygen generation (ACN)indirect = 0.81 quantum yield for singlet oxygen generation (ACN)direct = 0.92 | — | 130 |
| 332 | 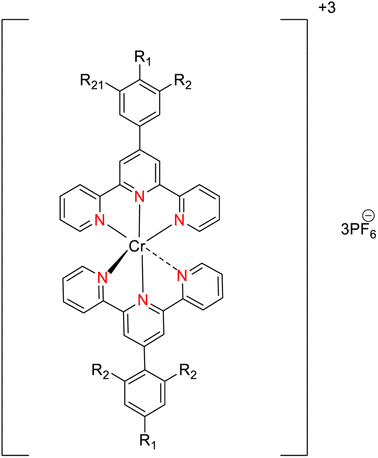 |
Photosensitizers | ε = 11900 M−1 cm−1 at 450 nm and ε = 5090 M−1 cm−1 at 500 nm molar extinction coefficient |
— | — | — | — | — | — | 135 |
| 333 | 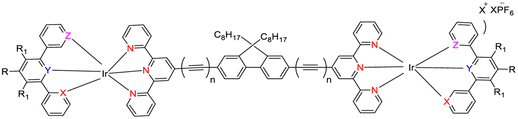 |
Photosensitizers | Molar extinction coefficients less than 100 M−1 cm−1 | — | — | — | — | 0.56 single oxygen quantum yield | — | 136 |
| 339, 340 | 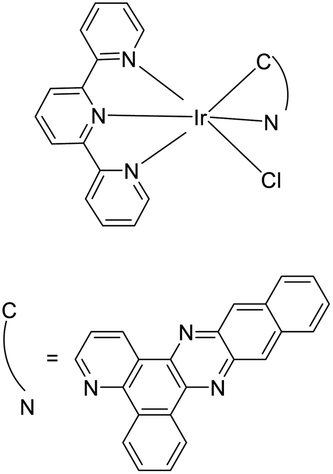 |
Photosensitizers | — | — | — | — | — | Single oxygen quantum yield | — | 137 |
| Ir341 = 0.45 | ||||||||||
| 341, 342 |  |
Photosensitizers | — | — | — | 99.3% selectivity for CO generation | — | — | TON = 2250 | 138 |
| TOF = 60 min−1 | ||||||||||
| 343 |  |
Photosensitizers | — | — | — | — | — | — | TON > 2250 | 139 |
In the realm of chemosensors, a comparative analysis highlights the efficacy of zinc-based terpyridine metal complexes, which demonstrate a lower detection limit for the identification of diverse metal cations and anions. The discussion extends to optoelectronic devices, where ruthenium terpyridine complexes emerge as efficient photosensitizers due to their superior single oxygen quantum yield (Φ) values.
Photocatalytic applications are scrutinized, revealing manganese terpyridine metal complexes as highly efficient catalysts with elevated reaction rate constants. The significance of a higher rate constant lies in the efficient generation of reactive species upon light absorption, contributing to the efficacy of photocatalytic processes such as pollutant degradation and water splitting.
The comprehensive examination of various terpyridine metal complexes presented in this review highlights the prominent role of zinc-based terpyridine metal complexes in chemosensors application. Their distinct properties, including detection limit, high sensitivity, and selectivity toward diverse metal cations and anions, underscore their significance in these fields.
Furthermore, the study reveals that zinc-based terpyridine metal complexes play a crucial role in selectively detecting various analytes in environmental and biological systems. Additional modifications in synthesis are proposed to generate novel zinc terpyridine metal complexes, expanding their applications across various scientific domains. Specifically, zinc metal–organic frameworks (MOFs) are proposed for targeted drug delivery systems and biocompatible medicine, serving as carriers for diverse drugs to be released into biological systems.
In the realm of catalysts, photocatalysts, and photosensitizers, ruthenium (Ru), cobalt (Co), zinc (Zn), manganese (Mn), and iron (Fe) terpyridine metal complexes emerge as significant contributors. Their diverse properties, including turn-over number (TON), turn-over frequency (TOF), higher rate constants, elevated single oxygen quantum yield (ΦΔ), and selectivity values, underscore their importance. Catalytically, these complexes play a role in various chemical reactions such as oxidation, C–H activation, cross-coupling reactions, and hydrogenation.28
In the field of photocatalysis, they contribute to water oxidation and water splitting, with Ru terpyridine metal complexes proving effective in water oxidation. Photosensitizers, particularly thermally activated delayed fluorescence photosensitizers with modified iron terpyridine, exhibit excellence as probes for generating cobalt with a 93.3% selectivity. Future prospects involve designing novel terpyridine metal complexes with enhanced properties related to photosensitizers, catalysts, and photocatalysts, applied in areas like green and sustainable catalysis with heightened reactivity and selectivity.
The envisaged advancements encompass enantioselective catalysis, involving asymmetric transformations, and multifunctional catalytic systems, combining multiple reactions in a single system. In response to the growing need for clean energy sources, terpyridine metal complexes play a remarkable role in the production of fuel using solar energy and exhibit captivating potential in artificial photosynthesis, particularly in the reduction of carbon dioxide to hydrogen and other valuable chemicals.
Anticipating developments in photosensitizers, the near future envisions customized photosensitizers with tunable properties such as quantum yield, long-lived excited states, and target emission wavelengths. These advancements find application in diverse processes, including energy transfer and light-driven processes. Consequently, terpyridine metal complexes stand as a captivating topic for scientific exploration, promising novel complexes with heightened tunability, versatility, and efficiency. This review underscores the versatility of terpyridine metal complexes and their potential for further advancements, paving the way for the development of innovative materials with multifaceted applications across various scientific disciplines.
5. Advantages and disadvantages of different TPY complexes in various applications
5.1. Advantages
(i) Versatility in coordination chemistry: TPY complexes exhibit a high degree of versatility in coordination chemistry due to the tridentate terpyridine ligand. This property allows for the design of a wide range of complex structures with diverse metal ions, leading to tunable properties.(ii) Photophysical properties: terpyridine ligands are known for their unique photophysical properties, such as strong absorption in the visible region and relatively long-lived excited states. This makes TPY complexes suitable for applications in sensors, imaging, and light-harvesting devices.
(iii) Catalytic activity: some TPY complexes, especially those with transition metal centers, demonstrate catalytic activity in various reactions, including oxidation and reduction processes. This catalytic behavior is valuable in applications such as homogeneous catalysis.
(iv) Modular design: the modular nature of TPY complexes allows for the easy incorporation of additional ligands or functional groups. This modularity facilitates the fine-tuning of properties, enabling the design of complexes tailored for specific applications.
(v) Biological compatibility: certain TPY complexes exhibit good biological compatibility, making them suitable for bioimaging and potential therapeutic applications. Their ability to interact with biomolecules adds an extra dimension to their utility in biological systems.
5.2. Disadvantages
(i) Low quantum yields: one of the notable challenges is the relatively low quantum yields observed in some TPY complexes, particularly in certain photophysical applications. This limitation can hinder the efficiency of devices relying on strong luminescence.(ii) Photochemical stability: TPY complexes may exhibit photochemical instability under certain conditions, leading to undesired reactions or decomposition. This can limit their application in prolonged photochemical processes or environments with intense light exposure.
(iii) Synthetic challenges: the synthesis of some TPY complexes, especially those involving multiple terpyridine units or specific metal centers, can be challenging and may have lower yields. Addressing these synthetic challenges is crucial for scalability and practical applications.
(iv) Limited redox stability: some TPY complexes may have limitations in terms of redox stability, impacting their performance in applications that involve multiple redox cycles, such as electrocatalysis or energy storage devices.
6. Exploring chemical variants in terpyridine–metal complexes: contributions, shortcomings, and future developments
6.1. Contributions of chemical variants
(i) Ligand substitution: the choice of terpyridine ligands significantly influences the properties of metal complexes. Modifications in the ligand structure, such as the substitution of hydrogen atoms with various functional groups, contribute to tunability in electronic properties, steric hindrance, and redox potentials, thus affecting the overall reactivity of the complex.(ii) Metal center variation: the type of metal center coordinated with terpyridine ligands plays a pivotal role in dictating the complex's catalytic, optical, and magnetic properties. Exploring a range of metal centers allows for tailoring complexes with specific functionalities, including enhanced catalytic activity, luminescence, and magnetic response.
(iii) Bridging ligands: introducing additional bridging ligands alongside terpyridine can lead to the formation of polynuclear complexes. The nature of these bridging ligands influences the coordination geometry, and electronic communication between metal centers, and ultimately impacts the properties of the resulting complex, opening avenues for applications in molecular magnetism and materials science.
(iv) Steric and electronic tuning: fine-tuning the steric and electronic properties of terpyridine–metal complexes through careful modification of ligands enables control over the selectivity, reactivity, and stability of the complexes. Understanding these factors is crucial for designing complexes with tailored properties for specific applications.
6.2. Shortcomings in current research
(i) Limited mechanistic understanding: despite the widespread use of terpyridine–metal complexes in catalysis and sensing, a detailed mechanistic understanding of these processes remains elusive. Addressing this gap is essential for optimizing reaction conditions, enhancing catalytic efficiency, and unlocking the full potential of these complexes in diverse applications.6.3. Developing trends in applications
(i) Bioorthogonal chemistry: an emerging trend in the application of terpyridine–metal complexes involves their use in bioorthogonal chemistry. Developing complexes that exhibit selectivity for biological targets and exploring their potential in imaging, drug delivery, and bioimaging are promising avenues for future research.(ii) Nanomaterial integration: integrating terpyridine–metal complexes into nanomaterials offers exciting possibilities for the development of multifunctional materials. This includes applications in nanocatalysis, drug delivery, and sensors, where the nanomaterial provides a platform for enhancing stability and reactivity.
(iii) Advanced functional materials: the design and synthesis of terpyridine–metal complexes for advanced functional materials, such as stimuli-responsive materials and light-emitting devices, represent a developing trend. Tailoring the electronic and optical properties of these complexes can lead to innovations in electronics, optoelectronics, and energy-related applications.
7. Synthesis and applications of terpyridine metal complexes: challenges and future research directions
The field of terpyridine metal complexes has witnessed significant growth in recent years, owing to their diverse applications in catalysis, sensing, and materials science. As we look ahead, it is essential to address the existing challenges and explore potential research directions to further advance the synthesis and applications of these intriguing complexes.7.1. Challenges in the synthesis of terpyridine metal complexes
(i) Ligand modification and design: one of the primary challenges lies in the design and modification of terpyridine ligands to enhance their coordination properties and tailor them for specific applications. Research efforts should focus on developing novel ligands with improved steric and electronic properties, allowing for fine-tuning of metal complex reactivity.(ii) Reactivity and selectivity: achieving high reactivity and selectivity in the synthesis of terpyridine metal complexes remains a challenge. Overcoming issues related to side reactions and optimizing reaction conditions to selectively form desired complexes is crucial for advancing their practical applications.
(iii) Green synthesis approaches: in line with the growing emphasis on sustainability, there is a need for the development of environmentally friendly and sustainable synthetic routes for terpyridine metal complexes. Green synthesis strategies, such as the use of renewable resources and eco-friendly solvents, should be explored to minimize the environmental impact of the synthesis processes.
7.2. Applications and future research directions
(i) Catalysis: the catalytic potential of terpyridine metal complexes in various reactions, including cross-coupling, hydrogenation, and oxidation, offers exciting possibilities. Future research should focus on understanding the underlying mechanisms and optimizing these complexes for catalytic efficiency and selectivity.(ii) Sensing and imaging: terpyridine metal complexes have demonstrated remarkable sensing capabilities for various analytes, including metal ions and small molecules. Exploring new sensing platforms and enhancing the sensitivity and selectivity of these complexes can open avenues for their application in the development of advanced sensors and imaging probes.
(iii) Materials science: the use of terpyridine metal complexes in the design of functional materials, such as luminescent materials, conductive polymers, and liquid crystals, presents a vast and unexplored landscape. Future research should delve into the development of innovative materials with tailored properties for electronic and optical applications.
(iv) Bioinorganic chemistry: understanding the interaction of terpyridine metal complexes with biological systems is an emerging area with potential applications in medicinal chemistry. Investigating their cytotoxicity, bioavailability, and targeting capabilities can pave the way for the development of novel therapeutic agents.
8. Conclusions and future perspectives
This comprehensive review highlights the recent advances in TPY complexes, spanning their synthesis and diverse applications. The review emphasizes the versatility and tunability of TPY complexes through strategic ligand design and synthesis. Researchers have developed various TPY ligands with tailored properties, enabling precise control over the complex's chemical and physical characteristics. TPY complexes exhibited rich photophysical properties, including strong absorption in the visible range and photoluminescence. These properties make them valuable in applications such as photocatalysis and photodynamic therapy. TPY complexes have found extensive utility as photocatalysts for various reactions, including the reduction of carbon dioxide and the production of solar fuels. Their ability to harness solar energy and drive chemical transformations underscores their importance in sustainable energy and environmental chemistry. The use of TPY complexes in PDT has been highlighted, particularly for cancer treatment. These complexes have shown promise as photosensitizers, inducing apoptosis in cancer cells through photodynamic processes. Their selectivity and efficient generation of reactive oxygen species make them potential candidates for future clinical applications. TPY complexes have demonstrated utility in electrochemical processes, including redox catalysis and dye-sensitized solar cells (DSSCs). Their ability to mediate electron transfer reactions and their versatility in designing efficient catalytic systems are notable advantages. The review underscores the potential for further advancements in TPY complexes, including improving their quantum yields, enhancing photochemical stability, and exploring their integration into practical devices and systems. Future research should focus on optimizing TPY complexes for specific applications and addressing challenges in their implementation. Terpyridine complexes have been subject to comprehensive scrutiny, revealing their significance as a pivotal molecular scaffold. The literature underscores the esteemed nature of these complexes. Furthermore, the exploration of terpyridine, coupled with subsequent chemical modifications, holds promise for the creation and construction of supramolecular structures or metal–organic frameworks (MOFs). The inherent potential for extensive chemical modifications further augments the versatility of this molecular entity. The ongoing investigations aim to elucidate novel applications stemming from the engineered terpyridine derivatives and their associated supramolecular architectures. The flexibility and versatility of TPY complexes make them promising candidates for addressing contemporary challenges in chemistry and materials science.Looking forward, specific research directions could include a deeper exploration of the potential bioimaging applications of TPY complexes, addressing biocompatibility concerns and developing targeted delivery systems. The integration of TPY complexes into emerging technologies, such as the design of advanced sensors for environmental monitoring or the development of efficient photocatalysts, presents promising avenues for future research. Collaborative efforts between synthetic chemists, materials scientists, and engineers are essential to bridge knowledge gaps and accelerate progress in these directions.
Moreover, a clearer delineation of practical applications for TPY complexes is crucial for guiding future research. For instance, the development of TPY-based materials with tailored electronic and optical properties holds promise in the fabrication of next-generation electronic devices and sensors. Exploring TPY complexes in the context of sustainable energy applications, such as photocatalysis for water splitting or organic photovoltaics, is an exciting frontier that merits attention. By addressing the current limitations and strategically charting research directions, the field of TPY complexes can make substantial contributions to advancing chemistry and materials science. Terpyridine complexes have been subject to comprehensive scrutiny, revealing their significance as a pivotal molecular scaffold. The literature underscores the esteemed nature of these complexes. Furthermore, the exploration of terpyridine, coupled with subsequent chemical modifications, holds promising behavior for the creation and construction of supramolecular structures or metal–organic frameworks (MOFs). The inherent potential for extensive chemical modifications further augments the versatility of this molecular entity. The ongoing investigations aim to elucidate novel applications stemming from the engineered terpyridine derivatives and their associated supramolecular architectures. Ultimately, these efforts will not only deepen our fundamental understanding of these fascinating complexes but also pave the way for innovative technologies and materials with far-reaching societal impact.
Data availability
The submitted manuscript is a review article. All the data included in the manuscript has been either cited from existing sources or written by the authors themselves.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Ziad Moussa is grateful to the United Arab Emirates University (UAEU) and to the Research Office for supporting the research developed in his laboratory and reported herein (UPAR grant code G00004605).References
- U. S. Schubert, H. Hofmeier and G. R. Newkome, Modern Terpyridine Chemistry, John Wiley & Sons, 2006 Search PubMed
.
- H. Hofmeier and U. S. Schubert, Chem. Soc. Rev., 2004, 33, 373–399 RSC
- U. S. Schubert, C. Eschbaumer, Q. An and T. Salditt, J. Inclusion Phenom. Macrocyclic Chem., 1999, 35, 35–43 CrossRef CAS
- C. Wei, Y. He, X. Shi and Z. Song, Coord. Chem. Rev., 2019, 385, 1–19 CrossRef CAS PubMed
- B. Z. Momeni, N. Davarzani, J. Janczak, N. Ma and A. S. Abd-El-Aziz, Coord. Chem. Rev., 2024, 506, 215619 CrossRef CAS
- H. He, S. Zhang, Z. Chen, H. Wang, X. Yu and X. Li, Chem. Lett., 2023, 52, 370–380 CrossRef CAS
- M. Chen, D. Sun, J. Li, Z. Wang, H. Liu, L. Pan, H. Chen and Z. Ma, New J. Chem., 2023, 47, 18785–18793 RSC
- R. Abhijnakrishna, K. Magesh, A. Ayushi and S. Velmathi, Coord. Chem. Rev., 2023, 496, 215380 CrossRef CAS
- R. Gopalakrishnan, A. I. Frolov, L. Knerr, W. J. Drury III and E. Valeur, J. Med. Chem., 2016, 59, 9599–9621 CrossRef CAS PubMed
- U. S. Schubert, A. Winter and G. R. Newkome, Terpyridine-based Materials: for Catalytic, Optoelectronic and Life Science Applications, John Wiley & Sons, 2012 Search PubMed
- G. Yin, J. Huang, D. Liu, R. Li, S. Wei, M. Si, F. Ni, Y. Zheng, Q. Yang and R. Zhou, Chin. Chem. Lett., 2023, 34, 107290 CrossRef CAS
- S. Ranjan Jena, T. Mandal and J. Choudhury, Chem. Rec., 2022, 22, e202200165 CrossRef CAS PubMed
- E. U. Mughal, M. Mirzaei, A. Sadiq, S. Fatima, A. Naseem, N. Naeem, N. Fatima, S. Kausar, A. A. Altaf, M. N. Zafar and B. A. Khan, R. Soc. Open Sci., 2020, 7, 201208 CrossRef CAS PubMed
- R. R. Panicker and A. Sivaramakrishna, Coord. Chem. Rev., 2022, 459, 214426 CrossRef CAS
- C. Kaes, A. Katz and M. W. Hosseini, Chem. Rev., 2000, 100, 3553–3590 CrossRef CAS PubMed
- A. Agosti, E. Kuna and G. Bergamini, Eur. J. Inorg. Chem., 2019, 2019, 577–584 CrossRef CAS
- R. R. Panicker and A. Sivaramakrishna, Coord. Chem. Rev., 2022, 459, 214426 CrossRef CAS
- C. Kaes, A. Katz and M. W. Hosseini, Chem. Rev., 2000, 100, 3553–3590 CrossRef CAS PubMed
- O. S. Taniya, D. S. Kopchuk, A. F. Khasanov, I. S. Kovalev, S. Santra, G. V. Zyryanov, A. Majee, V. N. Charushin and O. N. Chupakhin, Coord. Chem. Rev., 2021, 442, 213980 CrossRef CAS
- X. Yu, C. Guo, S. Lu, Z. Chen, H. Wang and X. Li, Macromol. Rapid Commun., 2022, 43, 2200004 CrossRef CAS PubMed
- S. M. Elahi, M. Raizada, P. K. Sahu and S. Konar, Chem.–Eur. J., 2021, 27, 5858–5870 CrossRef CAS PubMed
- M. A. Halcrow, Coord.
Chem. Rev., 2005, 249, 2880–2908 CrossRef CAS
- A. M. C. Thompson, Coord. Chem. Rev., 1997, 160, 1–52 CrossRef CAS
- C. Haensch, M. Chiper, C. Ulbricht, A. Winter, S. Hoeppener and U. S. Schubert, Langmuir, 2008, 24, 12981–12985 CrossRef CAS PubMed
- A. Winter and U. S. Schubert, ChemCatChem, 2020, 12, 2890–2941 CrossRef CAS
- G. T. Morgan and F. H. Burstall, J. Chem. Soc., 1932, 20–30 RSC
- A. M. W. Cargill Thompson, Coord. Chem. Rev., 1997, 160, 1–52 CrossRef
- Z. Zeng, Y. Chen, X. Zhu and L. Yu, Chin. Chem. Lett., 2023, 34, 107728 CrossRef CAS
- T. Kauffmann, J. König and A. Woltermann, Chem. Ber., 1976, 109, 3864–3868 CrossRef CAS
- J. Uenishi, T. Tanaka, S. Wakabayashi, S. Oae and H. Tsukube, Tetrahedron Lett., 1990, 31, 4625–4628 CrossRef CAS
- F. Kroehnke, Synthesis, 1976, 1976, 1–24 CrossRef
- D. L. Jameson and L. E. Guise, Tetrahedron Lett., 1991, 32, 1999–2002 CrossRef CAS
- F. H. Case and W. A. Butte, J. Org. Chem., 1961, 26, 4415–4418 CrossRef CAS
- K. T. Potts, P. Ralli, G. Theodoridis and P. Winslow, Org. Synth., 2003, 64, 189 Search PubMed
- K. T. Potts, M. J. Cipullo, P. Ralli and G. Theodoridis, J. Am. Chem. Soc., 1981, 103, 3585–3586 CrossRef CAS
- K. Hanabusa, A. Nakamura, T. Koyama and H. Shirai, Polym. Int., 1994, 35, 231–238 CrossRef CAS
- U. S. Schubert and C. Eschbaumer, Org. Lett., 1999, 1, 1027–1029 CrossRef CAS
- S. Tu, T. Li, F. Shi, Q. Wang, J. Zhang, J. Xu, X. Zhu, X. Zhang, S. Zhu and D. Shi, Synthesis, 2005, 2005, 3045–3050 CrossRef
- R.-A. Fallahpour, M. Neuburger and M. Zehnder, New J. Chem., 1999, 23, 53–61 RSC
- M. Heller and U. S. Schubert, Eur. J. Org Chem., 2003, 2003, 947–961 CrossRef
- A. Winter, G. R. Newkome and U. S. Schubert, ChemCatChem, 2011, 3, 1384–1406 CrossRef CAS
- A. Winter and U. S. Schubert, ChemCatChem, 2020, 12, 2890–2941 CrossRef CAS
- P. Liu, C.-Y. Zhou, S. Xiang and C.-M. Che, Chem. Commun., 2010, 46, 2739–2741 RSC
- K. Kamata, A. Suzuki, Y. Nakai and H. Nakazawa, Organometallics, 2012, 31, 3825–3828 CrossRef CAS
- Z. Lin, N. C. Thacker, T. Sawano, T. Drake, P. Ji, G. Lan, L. Cao, S. Liu, C. Wang and W. Lin, Chem. Sci., 2018, 9, 143–151 RSC
- Y.-X. Ye, C. Wen, J.-W. Wang, J. Pan, S. Huang, S. Liang, M. Zhou, Q. Tong, F. Zhu, J. Xu and G. Ouyang, Chem. Commun., 2020, 56, 5476–5479 RSC
- D. Yong, J. Tian, R. Yang, Q. Wu and X. Zhang, Se/ZrO2-Catalyzed Oxidation of Phenol., Chin. J. Inorg. Chem., 2024, 44(4), 1343–1347 Search PubMed
- Y. Chen, C. Chen, Y. Liu and L. Yu, Chin. Chem. Lett., 2023, 34, 108489 CrossRef CAS
- S.-L. Liu, Q.-W. Chen, Z.-W. Zhang, Q. Chen, L.-Q. Wei and N. Lin, J. Solid State Chem., 2022, 310, 123045 CrossRef CAS
- S.-L. Liu, Q.-W. Chen, Z.-W. Zhang, Q. Chen, L.-Q. Wei and N. Lin, J. Solid State Chem., 2022, 310, 123045 CrossRef CAS
- R. H. Grubbs and S. Chang, Tetrahedron, 1998, 54, 4413–4450 CrossRef CAS
- C. M. Moore and N. K. Szymczak, Chem. Commun., 2013, 49, 400–402 RSC
- B. J. Hornstein, D. M. Dattelbaum, J. R. Schoonover and T. J. Meyer, Inorg. Chem., 2007, 46, 8139–8145 CrossRef CAS PubMed
- L. Cristian, S. Nica, O. D. Pavel, C. Mihailciuc, V. Almasan, S. M. Coman, C. Hardacre and V. I. Parvulescu, Catal. Sci. Technol., 2013, 3, 2646–2653 RSC
- E. K. Beloglazkina, A. G. Majouga, E. A. Manzheliy, A. A. Moiseeva, Y. V. Lin'kova and N. V. Zyk, Polyhedron, 2015, 85, 800–808 CrossRef CAS
- M. Yoshida, M. Kondo, S. Torii, K. Sakai and S. Masaoka, Angew. Chem., Int. Ed., 2015, 54, 7981–7984 CrossRef CAS PubMed
- A. Maity, A. Sil and S. K. Patra, Eur. J. Inorg. Chem., 2018, 2018, 4063–4073 CrossRef CAS
- A. Taketoshi, T.-a. Koizumi and T. Kanbara, Tetrahedron Lett., 2010, 51, 6457–6459 CrossRef CAS
- J. Bühler, J. Zurflüh, S. Siol, O. Blacque, L. Sévery and S. D. Tilley, Catal. Sci. Technol., 2022, 12, 1512–1519 RSC
- M. Garbe, K. Junge and M. Beller, Eur. J. Org Chem., 2017, 2017, 4344–4362 CrossRef CAS
- S. Das, C. D. Incarvito, R. H. Crabtree and G. W. Brudvig, Science, 2006, 312, 1941–1943 CrossRef CAS PubMed
- H. Yamazaki, T. Ueno, K. Aiso, M. Hirahara, T. Aoki, T. Nagata, S. Igarashi and M. Yagi, Polyhedron, 2013, 52, 455–460 CrossRef CAS
- J. C. Lewis, ACS Catal., 2013, 3, 2954–2975 CrossRef CAS
- B. Liu, W. Luo, H. Li, X. Qi and Q. Hu, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2015, 45, 1097–1101 CrossRef CAS
- G. Zhang, H. Zeng, J. Wu, Z. Yin, S. Zheng and J. C. Fettinger, Angew. Chem., 2016, 128, 14581–14584 CrossRef
- H. Lim, P. Chohan, D. Moustafa, C. Sweet, B. Calalpa and P. Kaur, ChemistrySelect, 2018, 3, 9443–9447 CrossRef CAS
- G. Zhang, H. Zeng, S. Li, J. Johnson, Z. Mo, M. C. Neary and S. Zheng, Dalton Trans., 2020, 49, 2610–2615 RSC
- M. S. Kharasch and E. K. Fields, J. Am. Chem. Soc., 1941, 63, 2316–2320 CrossRef CAS
- G. Cahiez and A. Moyeux, Chem. Rev., 2010, 110, 1435–1462 CrossRef CAS PubMed
- R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed
- H. Lebel, J.-F. Marcoux, C. Molinaro and A. B. Charette, Chem. Rev., 2003, 103, 977–1050 CrossRef CAS PubMed
- B. Heller and M. Hapke, Chem. Soc. Rev., 2007, 36, 1085–1094 RSC
- A. Debuigne and R. Poli, Prog. Polym. Sci., 2009, 34, 211–239 CrossRef CAS
- W. N. Palmer, T. Diao, I. Pappas and P. J. Chirik, ACS Catal., 2015, 5, 622–626 CrossRef CAS
- H. A. Duong, W. Wu and Y.-Y. Teo, Organometallics, 2017, 36, 4363–4366 CrossRef CAS
- G. A. Bhat, A. Rajendran and R. Murugavel, Eur. J. Inorg. Chem., 2018, 2018, 795–804 CrossRef CAS
- J. Wu, H. Zeng, J. Cheng, S. Zheng, J. A. Golen, D. R. Manke and G. Zhang, J. Org. Chem., 2018, 83, 9442–9448 CrossRef CAS PubMed
- S. Gutiérrez-Tarriño, P. Concepción and P. Oña-Burgos, Eur. J. Inorg. Chem., 2018, 2018, 4867–4874 CrossRef
- Z. Dai, Z. Yu, Y. Bai, J. Li and J. Peng, Appl. Organomet. Chem., 2021, 35, e6027 CrossRef CAS
- T. P. Pabst and P. J. Chirik, J. Am. Chem. Soc., 2022, 144, 6465–6474 CrossRef CAS PubMed
- P. R. Boruah, A. A. Ali, B. Saikia and D. Sarma, Green Chem., 2015, 17, 1442–1445 RSC
- B. Suchand and G. Satyanarayana, Palladium-catalyzed acylations: one-pot synthesis of indenones, J. Org. Chem., 2017, 82(1), 372–381 CrossRef CAS PubMed
- G. Chelucci, V. Caria and A. Saba, J. Mol. Catal. A: Chem., 1998, 130, 51–55 CrossRef CAS
- M. R. S. o. Japan, Transactions of the Materials Research Society of Japan, Materials Research Society of Japan, 2007 Search PubMed
- A. R. Mazzotti, M. G. Campbell, P. Tang, J. M. Murphy and T. Ritter, J. Am. Chem. Soc., 2013, 135, 14012–14015 CrossRef CAS PubMed
- M. M. Amini, A. Feiz, M. Dabiri and A. Bazgir, Appl. Organomet. Chem., 2014, 28, 86–90 CrossRef CAS
- T. Suzuka, H. Sueyoshi, S. Maehara and H. Ogasawara, Molecules, 2015, 20, 9906–9914 CrossRef CAS PubMed
- W. Chen, L. Zhong, X. Peng, J. Lin and R. Sun, Cellulose, 2014, 21, 125–137 CrossRef CAS
- T. Suzuka, M. Adachi, Y. Nakamoto and K. Ogihara, Trans. Mater. Res. Soc. Jpn., 2015, 40, 77–80 CrossRef CAS
- S. Yin, X. Wang, J. Jiang, H. Xiao and X. Li, J. Mol. Struct., 2019, 1198, 126925 CrossRef CAS
- H. Targhan, A. Hassanpour and K. Bahrami, Appl. Organomet. Chem., 2019, 33, e5121 CrossRef
- T. Li, S. Bi, H. Li and W. Wang, Catal. Today, 2022, 2, 042 Search PubMed
- Z. Chen, J. J. Concepcion, M. K. Brennaman, P. Kang, M. R. Norris, P. G. Hoertz and T. J. Meyer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15606–15611 CrossRef CAS PubMed
- H. Yamazaki, S. Igarashi, T. Nagata and M. Yagi, Inorg. Chem., 2012, 51, 1530–1539 CrossRef CAS PubMed
- N. Elgrishi, M. B. Chambers, V. Artero and M. Fontecave, Phys. Chem. Chem. Phys., 2014, 16, 13635–13644 RSC
- N. Elgrishi, M. B. Chambers and M. Fontecave, Chem. Sci., 2015, 6, 2522–2531 RSC
- J. A. Porras, I. N. Mills, W. J. Transue and S. Bernhard, J. Am. Chem. Soc., 2016, 138, 9460–9472 CrossRef CAS PubMed
- C. W. Machan and C. P. Kubiak, Dalton Trans., 2016, 45, 17179–17186 RSC
- J. J. Leung, J. Warnan, K. H. Ly, N. Heidary, D. H. Nam, M. F. Kuehnel and E. Reisner, Nat. Catal., 2019, 2, 354–365 CrossRef CAS
- Y. Wang, T. Liu, L. Chen and D. Chao, Inorg. Chem., 2021, 60, 5590–5597 CrossRef CAS PubMed
- M. Ballico, D. Alessi, C. Jandl, D. Lovison and W. Baratta, Chem.–Eur. J., 2022, 28, e202201722 CrossRef PubMed
- E. N. Cook, S. L. Hooe, D. A. Dickie and C. W. Machan, Inorg. Chem., 2023, 62, 4745 CrossRef CAS PubMed
- B. N. Mongal, S. Sk, A. Tiwari, Y. Soujanya, C. Malapaka and U. Pal, Effective Dual Role of Cobalt Bis(Terpyridine) Complex for Electrocatalytic and Photocatalytic Hydrogen Evolution, SSRN, 2022 DOI:10.2139/ssrn.4117461
- E. U. Mughal, A. Javaid, M. Imran, M. A. Abourehab, E. B. Elkaeed, N. Naeem, A. Y. A. Alzahrani, A. Sadiq and S. F. Kainat, Inorg. Chim. Acta, 2023, 546, 121329 CrossRef CAS
- T. Wang, N. Zhang, W. Bai and Y. Bao, Polym. Chem., 2020, 11, 3095–3114 RSC
- S. Yin, J. Zhang, H. Feng, Z. Zhao, L. Xu, H. Qiu and B. Tang, Dyes Pigm., 2012, 95, 174–179 CrossRef CAS
- H. Zhang, X. Hua, X. Tuo and X. Wang, J. Rare Earths, 2012, 30, 705–708 CrossRef CAS
- P. Wang, Z. Li, G.-C. Lv, H.-P. Zhou, C. Hou, W.-Y. Sun and Y.-P. Tian, Inorg. Chem. Commun., 2012, 18, 87–91 CrossRef CAS
- H. Li, S.-J. Zhang, C.-L. Gong, Y.-F. Li, Y. Liang, Z.-G. Qi and S. Chen, Analyst, 2013, 138, 7090–7093 RSC
- P. Wang, T.-a. Okamura, H.-P. Zhou, W.-Y. Sun and Y.-P. Tian, Chin. Chem. Lett., 2013, 24, 20–22 CrossRef CAS
- S. Jing, C. Zheng, S. Pu, C. Fan and G. Liu, Dyes Pigm., 2014, 107, 38–44 CrossRef CAS
- S. Bhowmik, B. N. Ghosh, V. Marjomäki and K. Rissanen, J. Am. Chem. Soc., 2014, 136, 5543–5546 CrossRef CAS PubMed
- D. Wu, T. Shao, J. Men, X. Chen and G. Gao, Dalton Trans., 2014, 43, 1753–1761 RSC
- B. Eftekhari-Sis and S. Mirdoraghi, Nanochem. Res., 2016, 1, 214–221 CAS
- R.-S. Juang, H.-W. Wen, M.-T. Chen and P.-C. Yang, Sens. Actuators, B, 2016, 231, 399–411 CrossRef CAS
- R. R. Zhao, Q. L. Xu, Y. Yang, J. Cao, Y. Zhou, R. Xu and J. F. Zhang, Tetrahedron Lett., 2016, 57, 5022–5025 CrossRef CAS
- T. Mandal, A. Hossain, A. Dhara, A. Al Masum, S. Konar, S. K. Manna, S. K. Seth, S. Pathak and S. Mukhopadhyay, Photochem. Photobiol. Sci., 2018, 17, 1068–1074 CrossRef CAS PubMed
- A. K. Purohit, B. N. Ghosh and P. K. Kar, Spectrochim. Acta, Part A, 2018, 188, 547–550 CrossRef CAS PubMed
- S.-Y. Kang, H. Yin, K.-Q. Zhang, X. Chen and K.-Z. Wang, Mater. Sci. Eng., C, 2019, 99, 657–668 CrossRef CAS PubMed
- H. R. Khavasi and E. Jelokhani, J. Mater. Chem. A, 2019, 7, 6638–6643 RSC
- S. Amuthakala, D. S. A. Selvan and A. K. Rahiman, J. Iran. Chem. Soc., 2020, 17, 1237–1248 CrossRef CAS
- P. Liu, Z. Chi, G. Shi, H. Dong, C. Ma and X. Chen, Eur. Polym. J., 2021, 159, 110716 CrossRef CAS
- D. Das, S. Sutradhar, A. Singh and B. N. Ghosh, Z. fur Anorg. Allg. Chem., 2021, 647, 1234–1238 CrossRef CAS
- I. J. Bazany-Rodríguez, M. K. Salomón-Flores, A. O. Viviano-Posadas, M. A. García-Eleno, J. Barroso-Flores, D. Martínez-Otero and A. Dorazco-González, Dalton Trans., 2021, 50, 4255–4269 RSC
- Q. Zhao and J. Wang, Mater. Adv., 2022, 3, 7569–7578 RSC
- K. Mori, K. Watanabe, Y. Terai, Y. Fujiwara and H. Yamashita, Chem.–Eur. J., 2012, 18, 11371–11378 CrossRef CAS PubMed
- B. Schulze, D. Escudero, C. Friebe, R. Siebert, H. Görls, S. Sinn, M. Thomas, S. Mai, J. Popp and B. Dietzek, Chem.–Eur. J., 2012, 18, 4010–4025 CrossRef CAS PubMed
- A. Sepehrifard, S. Chen, A. Stublla, P. G. Potvin and S. Morin, Electrochim. Acta, 2013, 87, 236–244 CrossRef CAS
- J. Eberhard, K. Peuntinger, S. Rath, B. Neumann, H.-G. Stammler, D. M. Guldi and J. Mattay, Photochem. Photobiol. Sci., 2014, 13, 380–396 CrossRef CAS PubMed
- A. Frei, R. Rubbiani, S. Tubafard, O. Blacque, P. Anstaett, A. Felgenträger, T. Maisch, L. Spiccia and G. Gasser, J. Med. Chem., 2014, 57, 7280–7292 CrossRef CAS PubMed
- K. Arifin, W. R. W. Daud and M. B. Kassim, Sains Malays., 2014, 43, 95–101 CAS
- G. Velmurugan and P. Venuvanalingam, Dalton Trans., 2015, 44, 8529–8542 RSC
- E. C.-H. Kwok, M.-Y. Chan, K. M.-C. Wong and V. W.-W. Yam, Polyhedron, 2016, 120, 54–59 CrossRef CAS
- B. N. Mongal, S. Bhattacharya, S. Sengupta, T. K. Mandal, J. Datta and S. Naskar, Sol. Energy, 2016, 134, 107–118 CrossRef CAS
- J. C. Barbour, A. J. Kim, E. deVries, S. E. Shaner and B. M. Lovaasen, Inorg. Chem., 2017, 56, 8212–8222 CrossRef CAS PubMed
- B. Liu, S. Monro, L. Lystrom, C. G. Cameron, K. Colon, H. Yin, S. Kilina, S. A. McFarland and W. Sun, Inorg. Chem., 2018, 57, 9859–9872 CrossRef CAS PubMed
- B. Yuan, J. Liu, R. Guan, C. Jin, L. Ji and H. Chao, Dalton Trans., 2019, 48, 6408–6415 RSC
- Y. Wang, X.-W. Gao, J. Li and D. Chao, Chem. Commun., 2020, 56, 12170–12173 RSC
- R. Fernandez-Teran and L. Sévery, Inorg. Chem., 2021, 60, 1334–1343 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2024 |