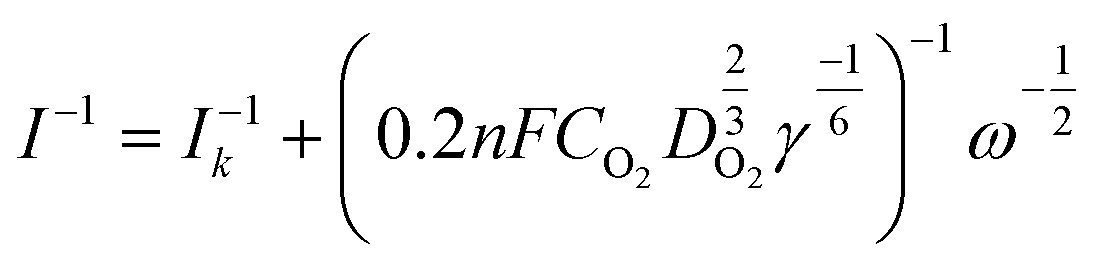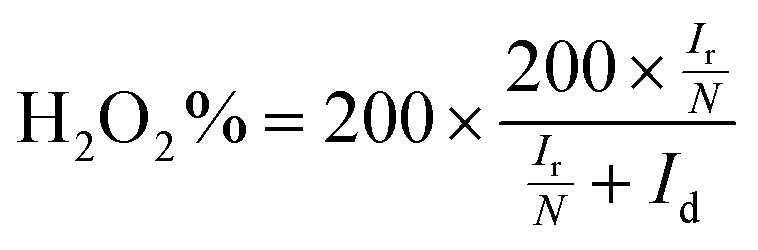DOI:
10.1039/D2NR06096E
(Paper)
Nanoscale, 2023,
15, 5448-5457
Atomically dispersed Mn atoms coordinated with N and O within an N-doped porous carbon framework for boosted oxygen reduction catalysis†
Received
1st November 2022
, Accepted 2nd February 2023
First published on 2nd February 2023
Abstract
Developing efficient and robust catalysts to replace Pt group metals for the oxygen reduction reaction (ORR) is conducive to achieving highly efficient energy conversion. Here, we develop a general ion exchange strategy to construct highly efficient ORR catalysts consisting of various atomically dispersed metal atoms anchored on N-doped porous carbon (M–SAs/NC) to investigate the atomic structure–catalytic activity relationship. The structure characterization results demonstrated that the achieved atomic structure varied due to the presence of different metal centers. Mn–SAs/NC consists of MnN3O1 centers, in which the Mn single atoms are stabilized by 3 N and 1 O. In contrast, the center metals in Fe-/Co-/Cu single-atom catalysts are coordinated by merely N atoms. Mn–SAs/NC delivers superior performance for the ORR with a half-wave potential (E1/2) of 0.91 V vs. RHE in 0.1 M KOH solution, outperforming that of the commercial Pt/C catalyst and the control Fe-/Co-/Cu single-atom catalysts. Furthermore, Mn–SAs/NC also shows excellent methanol tolerance and stability up to 5000 cycles. Density functional theory (DFT) calculations reveal that Mn single atom catalysts with MnN3O1 centers contributed to the superior ORR performance with lower energy barriers and optimized adsorption capacity of intermediates. These findings provide insights into the design and development of specific coordinated structures of atomically dispersed catalysts to facilitate the practical applications of energy conversion.
 Jinqiang Zhang | Jinqiang Zhang received his Ph.D. degree from the University of Technology Sydney in 2018. He has worked as a Postdoctoral Research Associate at the Centre for Clean Energy Technology at the University of Technology Sydney from 2018 to 2021. He is currently a Postdoc Research Fellow at the University of Toronto. His work mainly focuses on the design and synthesis of functional organic molecules and single-atom catalysts in energy conversion and storage systems such as hydrogen evolution reactions, oxygen evolution reactions, oxygen reduction reactions, CO2 capture and reduction, lithium-ion batteries, lithium–sulfur batteries, and lithium–air batteries. |
Introduction
Given their high theoretical energy density, low-cost, and zero-emission of greenhouse gases, proton exchange membrane fuel cells (PEMFCs) and zinc–air batteries (ZABs) are promising energy storage devices for next-generation portable electronics.1–3 However, their inferior energy conversion efficiency and low-lifetime are the main bottlenecks that impede their widespread applications. These shortcomings predominantly derive from the intrinsically sluggish kinetics of the oxygen reduction reaction (ORR) with a four-electron transfer process and their poor stability in corrosive electrolytes.4–6 Although platinum (Pt)-based catalysts have been identified as the best-performing ORR catalysts, their extensive commercial application is still limited by their high cost and severe scarcity.7–9 Therefore, the development of highly efficient noble metal-free ORR catalysts is significantly important for Zn–air batteries and PEMFCs.
Among single atom-based catalysts (SACs), atomically dispersed metal–Nx (M–Nx) moieties anchored on a carbon substrate electrocatalyst (M–Nx–C) have attracted extensive interest especially in electrocatalysis due to their unique physicochemical properties, such as large surface areas, high conductivity, and unsaturated coordination environments.10–12 Most of the coordination micro-environments of metal centers are porphyrin-like (M–N4) structures similar to those of Fe, Co, or Cu based biomimetic electrocatalysts.1,13,14 N atoms can not only effectively anchor and stabilize metal atoms on carbon substrates, but also modulate the electronic structures of the adjacent metal and carbon atoms to optimize the adsorption/desorption capacities of reaction intermediates for the enhancement of catalytic activities.1,14 The catalytic activity of center metals in a common M–N4 moiety with a specific coordination structure can be further boosted due to the altered local geometry of the active sites and the chemical nature of the adjacent atoms, which have been demonstrated by previously reported theoretical calculations and experiments.15 Typically, doping heteroatoms (oxygen, sulfur, phosphorus, etc.) for the replacement of one or several N atoms of the common M–N4 center structures can achieve various coordinated atomistic structures, which would be beneficial for optimizing the electronic structure of the metal center atoms, thus leading to improved electrocatalytic activity.16 For example, Wang et al. fabricated a highly efficient atomically dispersed Fe-based ORR catalyst with a unique FeN3S1 moiety by a NaCl-template pyrolysis method. DFT calculations revealed that the N and S coordinated Fe atomic sites (FeN3S1) could regulate the charge redistribution, which could weaken the binding energies of oxygen-related intermediates, boosting the fast reaction kinetics and ORR activity in 1.0 M KOH solution.17 Moreover, the incorporation of P into the atomically asymmetric Co–N3P1 structure could manipulate the electronic structure of Co sites to enhance the ORR activity. Furthermore, the tailored electronic structure can endow the asymmetric Co–N3P1 moiety with reduced reaction barriers compared to the common Co–N4 structure, enabling high ORR performance in an alkaline electrolyte.18 Thus, introducing heteroatoms can efficiently tune the coordinated micro-environment of single metal active centers, and the electrocatalytic ORR activity can be significantly enhanced. However, rational design of stable and highly efficient nanomaterials with boosted ORR electrochemical activity in a wide pH range still remains a challenge, which severely hinders the practical applications of atomically dispersed metal-based electrocatalysts.6,19,20
Herein, a series of outstanding ORR electrocatalysts consisting of atomically dispersed metal sites (Mn, Fe, Co, Cu) anchored on porous N-doped carbon (M–SAs/NC) were successfully synthesized through a simple imidazole metal organic framework (ZIF)-assisted strategy. Benefitting from the hierarchical structure, high electrical conductivity, and strong electronic interactions between the N-doped carbon framework and the atomically dispersed metal sites, the obtained SACs exhibited high ORR catalytic activities. In particular, Mn–SAs/NC with MnN3O1 centers exhibited the best catalytic activity with a high Jk of 37.85 mV cm−2 at 0.85 V vs. RHE, a half-wave (E1/2) of 0.91 V vs. RHE in 0.1 M KOH solution, and high durability (up to 95.4% current retention ratio of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s). It also shows reasonable ORR activity under acidic conditions with an E1/2 of 0.76 V vs. RHE. Furthermore, theoretical calculations confirm that the local coordination environment of 3 N and 1 O induced the boosted intrinsic catalytic activity of Mn–SAs/NC, achieving the optimized electronic structure for the reduced reaction barriers of oxygen-containing intermediates toward the ORR. This work may provide a facile new strategy for the rational design and construction of high-performance electrocatalysts with specific coordination environments for efficient electrochemical conversion reactions.
000 s). It also shows reasonable ORR activity under acidic conditions with an E1/2 of 0.76 V vs. RHE. Furthermore, theoretical calculations confirm that the local coordination environment of 3 N and 1 O induced the boosted intrinsic catalytic activity of Mn–SAs/NC, achieving the optimized electronic structure for the reduced reaction barriers of oxygen-containing intermediates toward the ORR. This work may provide a facile new strategy for the rational design and construction of high-performance electrocatalysts with specific coordination environments for efficient electrochemical conversion reactions.
Experimental section
Materials
Zinc nitrate hexahydrate (Zn(NO3)2·6H2O), nickel nitrate hexahydrate (Ni(NO3)2·6H2O), 2-methyl imidazole (2-ML), manganese chloride (MnCl2), and potassium bicarbonate (KOH) were obtained from Shanghai Chemical Reagents. Ferric chloride (FeCl3), cobalt chloride (CoCl2), and cupric chloride (CuCl2) were purchased from Alfa Aesar Co., Ltd. Nafion® solution (5 wt%, D520) was acquired from Sigma-Aldrich. All of the chemicals used in this experiment were analytical grade and used without further purification. Deionized water obtained from the Milli-DI® system was used in the whole experiments.
Synthesis of ZIF-8
In a typical procedure, 0.558 g of Zn(NO3)2·6H2O was dissolved in 15 mL methanol, which was subsequently added to 15 mL methanol containing 0.616 g of 2-MI under ultrasound for 10 min at room temperature. After stirring for another 12 h, the as-obtained ZIF-8 powder was obtained by centrifugation and washed with methanol several times, and then dried in a vacuum box at 65 °C overnight.
Synthesis of Mn–SAs/NC
Firstly, the ZIF-8 powder (100 mg) was dispersed in n-hexane (10 mL) under ultrasound for 5 min at room temperature. 50 μL of MnCl2·4H2O aqueous solution (20 mg mL−1) was then slowly injected into the above solution mixture under ultrasound for 10 min at room temperature. Next, the solution mixture was vigorously stirred for another 3 h in order to make the metal salt be absorbed completely. Then, the M@ZIF-8 sample was obtained by centrifugation and dried at 65 °C for 6 h in a vacuum box. Eventually, the M@ZIF-8 powder was heated to 900 °C in a tube furnace at a heating rate of 5 °C min−1 and maintained for 2 h under a N2 flow to yield Mn–SAs/NC catalysts, denoted as Mn–SAs/NC. For the synthesis of N-doped porous carbon (NC), the ZIF-8 powder was directly heated followed by the above procedure. For the synthesis of other control samples, various M (M = Fe, Co, and Cu)–SAs/NC were prepared via similar procedures to that for Mn–SAs/NC, except for using FeCl3, CoCl2, and CuCl2 as the metal precursors with the same mole content of metal ions, and named Fe–SAs/NC, Co–SAs/NC, and Cu–SAs/NC, respectively.
Characterization
Powder XRD patterns were recorded on a Rigaku D/Max-2500 (Rigaku Co., Japan) diffractometer equipped with Cu Kα radiation (λ = 1.54056 Å). The Raman spectra were recorded using a Renishaw inVia Raman microscope (λ = 532 nm). The morphology and structure of samples were characterized by scanning electron microscopy (SEM) on a S4800 (JEOL, Japan) and transmission electron microscopy (TEM) on a JEM-2100F (JEOL, Japan) at 200 kV equipped with an EDS detector. The nitrogen adsorption–desorption isotherms and pore structures were collected on a Quadrasorb SI-MP system (Quantachrome, USA) at 77 K. The specific surface area was calculated by the Brunauer–Emmett–Teller method. The XPS spectra were recorded on an ESCALab220i-XL electron spectrometer (VG Scientific, hν = 1486.92 eV) using Al Kα radiation.
Electrochemical measurements
All electrochemical measurements were performed on a PINE electrochemical station system equipped with high-speed rotators. A rotating disk electrode (RDE) and a rotating ring disk electrode (RRDE) (4 mm in diameter) coated with catalyst powders are used as the working electrodes. An Ag/AgCl electrode (saturated-KCl solution) and a graphite rod are used as the reference and counter electrodes, respectively. All potentials are converted into reversible hydrogen electrode (RHE) potentials using the formula: E(RHE) = E(Ag/AgC) + 0.059 pH + 0.197 V. Preparation of catalyst inks: 2 mg of catalysts was put into a solution mixture containing ethanol (500 μL) and Nafion solution (5 wt%, 5 μL). After sonication for another 30 min, the dispersed 20 μL of ink were dropped on the surfaces of the RDE and RRDE, and dried in air with a mass loading of 400 μg cm−2. For comparison, the commercial Pt/C catalyst (20 wt%) purchased from Titan was measured with a mass loading of 200 μg cm−2. All cyclic voltammetric curves were obtained between 0 V and 1.2 V vs. a reversible hydrogen electrode (RHE) at 5 mV s−1 in O2-saturated 0.5 M H2SO4 and 0.1 M KOH electrolytes, respectively. The LSV curves for the ORR were achieved at a scan rate of 5 mV s−1 and 1600 rpm after several CV cycles at 50 mV s−1 in the potential range of 0.2 to 1.2 V (vs. RHE) to activate the catalyst. Durability tests were carried out by continuously cycling the potential from 0.6 V to 1.0 V (vs. RHE) at 100 mV s−1 for 5000 cycles in 0.1 M KOH, and 3000 cycles in 0.5 M H2SO4 solution, respectively. Before the ORR test, the alkaline and acidic electrolytes were purged by oxygen injection for 30 min.
The electron transfer number (n) was determined by RRDE measurements. The electron transfer number (n) and peroxide yield (% H2O2) on the catalyst were calculated using the following equations:
Koutecky–Levich:
| | 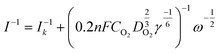 | (1) |
where
I is the measured current,
Ik is the kinetic-limiting current,
n represents the electron transfer number,
F represents the Faraday constant (96
500 C mol
−1),
CO2 represents the concentration of oxygen in 0.1 M KOH,
DO2 represents the diffusion coefficient of O
2 in 0.1 M KOH solution,
γ represents the kinematic viscosity of the 0.1 M KOH solution, and
ω is the electrode rotation rate.
| | 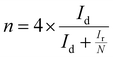 | (2) |
| | 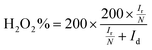 | (3) |
where
Ir and
Id are the ring and disk currents, respectively, and
N is the ring collection efficiency. The
N value is the H
2O
2 collection coefficient at the ring (
N = 0.37).
Density functional theory
All DFT calculations were carried out using the Vienna ab initio simulation package (VASP). The Perdew–Burke–Ernzerhof (PBE) functional was used to treat the exchange–correlation interactions. The plane wave basis set with a kinetic energy cutoff of 400 eV and an energy convergence criterion of 10−5 eV was used for structure relaxation. All surface calculations used a (3 × 3 × 2) Monkhorst–Pack k-point sampling. H2O and H2 were calculated in boxes of 15 Å × 15 Å × 15 Å, with the gamma point only. The free energy diagrams for the ORR were calculated with reference to the computational hydrogen electrode. The free energy of each species can be obtained using the equation:
where Eelec is the electronic energy and T was set as 298.15 K.
Results and discussion
Morphology and the structure
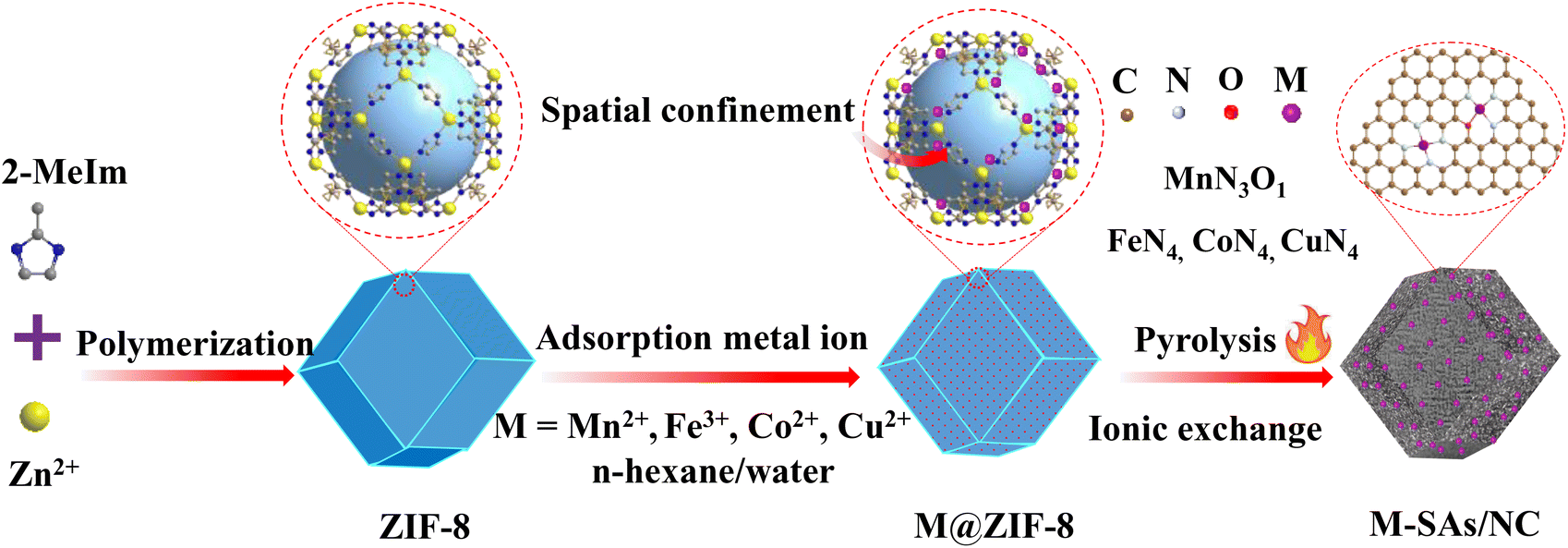
The fabrication process of atomically dispersed metal sites in an N-doped carbon framework is illustrated in Scheme 1. The precursor ZIF-8 sample was prepared by a polymerization process with Zn ions and 2-methylimidazole as the precursors, which shows a uniform rhombododecahedral shape with an average size of 200 nm (Fig. S1a and 1b†). The X-ray powder diffraction (XRD) spectrum (Fig. S1c†) of the ZIF-8 powder shows a typical crystal pattern with dominant peaks at a 2θ of 12.70, 14.80, 16.40 and 18.00°, corresponding to the (211), (220), (310), and (222) planes, respectively.21–23 The derived dodecahedral ZIF-8 powder possesses a unique cavity diameter of 11.6 Å and a pore diameter of 3.4 Å, which are perfect molecular-scale cages to trap the metal precursors (M = Fe, Co, Cu, and Mn) (molecular diameter, ca. 2.16 Å) (denoted as M@ZIF-8).24 Moreover, the presence of n-hexane and water molecules with different polarizations further facilitated the trapping of these metal ions into the pore channels of ZIFs. After the pyrolysis process, the Zn nodes (boiling point at 907 °C) can be evaporated and the nearby metal ions are easily stabilized by N or other light atoms, forming atomically dispersed metal sites in the hierarchical N-doped porous carbon network (M–SAs/NC).
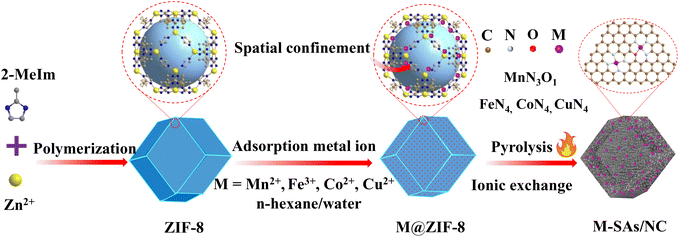 |
| | Scheme 1 Schematic illustration of the synthesis process of metal–Nx species embedded in the porous N-doped porous carbon (M–SAs/NC) composite. Brown, purple, red, and white balls represent C, metal atoms, O, and N, respectively. | |
The SEM images of NC derived from ZIF-8 show a well-maintained octahedral network after heat treatment, suggesting its robust structure (Fig. S2†). Furthermore, from the TEM images shown in Fig. 1a and b, after the incorporation of metal species, Mn–SAs/NC still retains the original morphology with a size of about 200 nm. The high-magnification TEM images of Mn–SAs/NC (Fig. S3a† and Fig. 1b) eliminate the possible presence of metal nanoparticles and the corresponding FFT image only shows the typical rings for the carbon substrate, suggesting the atomically dispersed metal species. The high-resolution transmission electron microscopy (HR-TEM) image (Fig. S3a†) also reveals the curved graphene layers and graphitic carbon layers stacked with a short-range disorder, beneficial for rapid electron transfer during the catalytic reaction process. The sub-Ångström-resolution HAADF-STEM image of Mn–SAs/NC in Fig. 1c shows the uniform brightness dots dispersed in the carbon substrate without metal crystals, ascribing to the atomically dispersed Mn atoms. Furthermore, the energy-dispersive X-ray spectroscopy (EDS) measurement (Fig. 1d and e) reveals that C, N, O, and Mn elements are uniformly dispersed in the whole carbon structure. The N2 sorption isotherms of Mn–SAs/NC show a typical type IV isotherm with a hysteresis loop, indicating a mesoporous structure (Fig. 1f). The BET surface area and the corresponding pore volume are calculated to be 2330.1 m2 g−1 and 1.51 cm3 g−1, respectively. Such a favorable hierarchical structure with sufficient micro/meso-pores and macro-pores is desirable for catalytic applications.
 |
| | Fig. 1 (a and b) TEM images at different magnifications of Mn–SAs/NC. (c) Aberration-corrected HAADF-STEM image of Mn–SAs/NC. (d and e) HAADF-STEM image and the corresponding elemental mappings of C, Mn, N, and O elements in Mn–SAs/NC. (f) N2 adsorption–desorption isotherms and pore size distribution based on the BJH method (the inset). | |
The above results demonstrate the excellent platform of ZIF-8 and the double solvents for the preparation of the atomically dispersed Mn catalyst. Other transition metal, e.g. Fe, Co, and Cu, based catalysts have also been successfully synthesized by a similar synthetic procedure with ZIF-8 as the host to trap and confine metal elements in double solvents to achieve the atomically dispersed properties. The TEM images show that Fe–SAs/NC and Cu–SAs/NC possess similar pleated structures due to the Ostwald process at high temperature as displayed in Fig. S3b and 3c.†
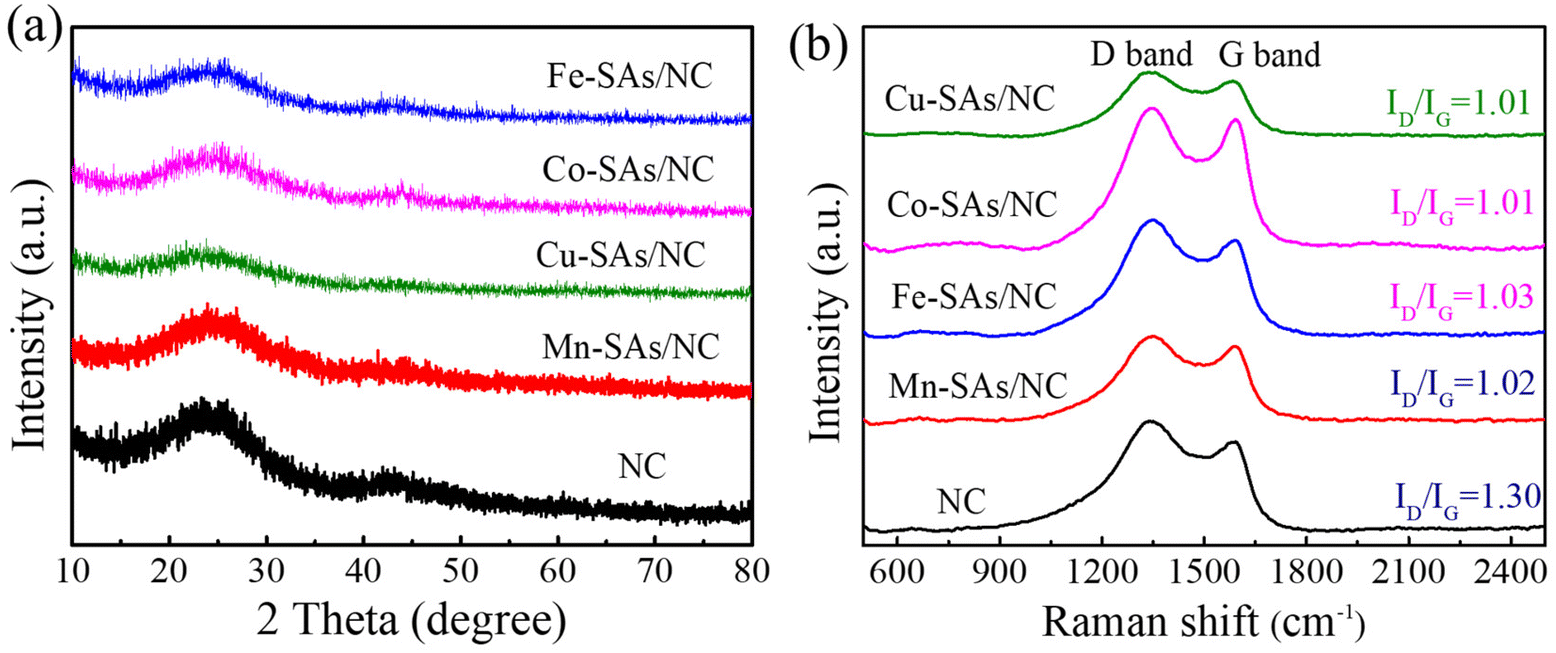
As shown in Fig. 2a, the XRD patterns of the achieved samples (NC, M–SAs/NC) revealed two distinct broad diffraction peaks at around 26° and 44°, which are assigned to the (002) and (100) crystal planes of graphitic carbon, respectively.25 No characteristic peaks related to metal oxide, metal carbide/nitride, and other metal compounds have been detected for the M–SAs/NC samples, indicating that the metal atoms (Mn, Fe, Co, Cu) are highly dispersed in the porous carbon substrate. Raman characterization was performed to characterize the vibrational properties of the M–SAs–NC samples. As shown in Fig. 2b, there are two distinct Raman peaks located at about 1350 and 1585 cm−1, which are ascribed to the D band (defects or disordered sp3-carbon) and the G band (sp2-graphitic carbon), respectively. The intensity ratio of the D to G bands (ID/IG) is always explored to estimate the graphitization degree of carbon materials. The ID/IG values of M–SAs/NC (1.02 for Mn–SAs/NC, 1.03 for Fe–SAs/NC, 1.01 for Co–SAs/NC, and 1.01 for Cu–SAs/NC) are much lower compared to that of NC (1.30), demonstrating that the incorporation of metal species highly regulates the crystalline structure and enhances the corresponding conductivity of the carbon substrate, which is beneficial for the electrocatalysis process.26 Additionally, no other peaks can be observed in the range from 500 to 2500 cm−1, suggesting that no metal compounds exist in the M–SAs/NC catalysts, further demonstrating the atomically dispersed properties of the metal species.
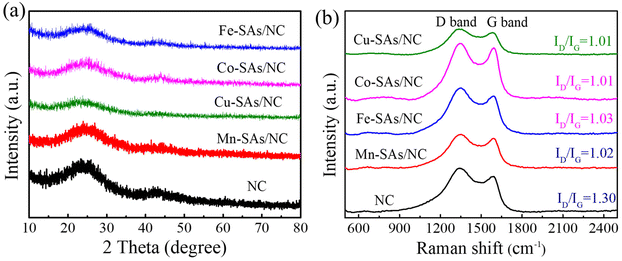 |
| | Fig. 2 (a) Powder XRD patterns and (b) Raman spectra of the as-prepared NC, Mn–SAs/NC, Fe–SAs/NC, Co–SAs/NC, and Cu–SAs/NC samples. | |
Composition analysis
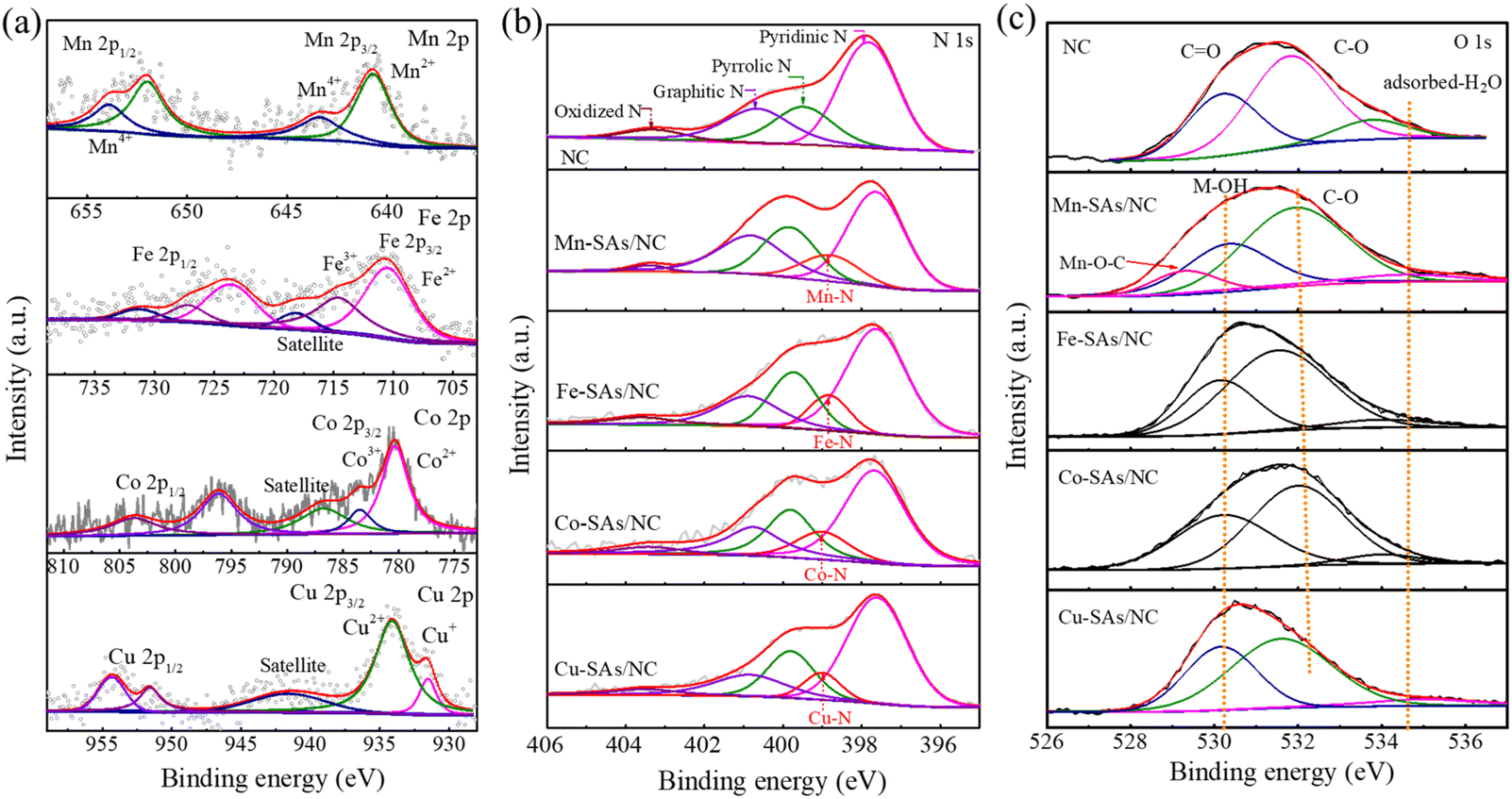
X-ray photoelectron spectroscopy (XPS) was used to deeply characterize the chemical structure of the obtained catalysts.27 As shown in Fig. S4,† the survey spectra reveal that Mn–SAs/NC contains C, N, O, and Mn elements with a Mn atomic ratio of 0.67% (Table S1†). Fig. 3a shows the high-resolution XPS spectra of Mn 2p and the results show that the two characteristic peaks at 641.1 and 652.8 eV are ascribed to Mn2+ and the peaks at 643.4 and 653.93 eV are attributed to Mn4+.27 The formation of Mn2+ and Mn4+ species in the Mn–SAs/NC sample may be originated from the different coordination effects with the surrounding light atoms after the carbonization of organic ligands. In particular, high-valence Mn4+ may contribute to the electronic interactions between O, N and Mn, in which the highly electronegative O coordinated atoms tend to attract more electrons, making the center Mn atom exist in a relatively higher valence state.28,29 The high-resolution XPS spectra of Fe, Co and Cu 2p shown in Fig. 3a indicate that the metal species possess the oxidation states instead of the corresponding metallic states, suggesting the atomically dispersed properties of these metal elements on the carbon support. As displayed in Fig. 3b, the high-resolution N 1s spectra of Mn–SAs/NC can be deconvoluted into five peaks, which are ascribed to pyridinic–N (398.4 eV), metal–N (399.3 eV), pyrrolic–N (400.1 eV), graphitic–N (400.9 eV), and oxidized–N (404.0 eV), respectively, demonstrating the successful N doping in the graphitic carbon matrix.1,30 The presence of metal–N indicates that metal Mn atoms covalently coordinate with the surrounding N atoms.31 The N 1s spectra of Fe–SAs/NC, Co–SAs/NC and Cu–SAs/NC show similar results with the formation of M–N bonds. The possible atomic model diagram of M–SAs/NC is displayed in Fig. S5.† The C 1s spectra of the prepared catalysts revealed the existence of C–C, C–N, C–O bonds (Fig. S6†). The high-resolution O 1s spectrum shown in Fig. 3c reveals the existence of C–O species at 530.2 eV, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species at 531.8 eV, and adsorbed-H2O at 533.8 eV in NC. However, the high-resolution O 1s spectra of M–SAs/NC display three different characteristic peaks with binding energies at around 530.4 eV (hydroxyl oxygen, M–OH), 533.6 eV (C–O), and 534.5 eV (surface adsorbed-H2O).32 After anchoring metal atoms, the functional group CO reduced along with the increase of M–OH. The possible reason is the CO species formed on the defects and edges of carbon layers that can easily get released without protection at high temperature.32,33 In particular, there existed a specific Mn–O binding energy situated at 529.3 eV for Mn–SAs/NC, which can be ascribed to the formation of Mn–O–C species, suggesting that the Mn atoms may be coordinated with both the N and O atoms on the carbon matrix, which is different with Fe, Co and Cu having only the M–N bonds.34–36 In order to understand the atomic structure of the as-prepared Mn–SAs/NC, the XANES and extended X-ray absorption fine structure (EXAFS) analyses were performed, as shown in Fig. S7.† As displayed in the Mn K-edge X-ray absorption near-edge structure (XANES) spectra (Fig. S7a†), the absorption edge position of Mn–SAs/NC is very closer to Mn(II) phthalocyanine (MnPc), indicating the main +2 valence state of Mn in Mn–SAs/NC. The Fourier-transformed (FT) EXAFS R-space plots in EXAFS characterization as illustrated in Fig. S7b† show a main peak situated at ≈1.46 Å, a position that coincides with the first Mn–N shell in the MnPc reference. Due to the similar bond lengths of Mn–N and Mn–O, the N and O atoms coordinated with Mn configuration cannot be obviously distinguished.33–35 Moreover, no obvious scattering path for Mn–Mn coordination is observed, indicating the atomic dispersion of Mn.34 Through XPS, we can confirm the coexistence of C–O and C–N in Mn–SAs/NC, which indicates the possibility of O and N coordination with the Mn atoms in the sample. EXAFS fitting was performed to study the structure parameters and obtain a quantitative chemical structure of Mn atoms. As shown in Fig. S8,† the first coordination shell of Mn–SAs/NC could be fitted by a mixture of Mn–N and Mn–O coordination paths, with coordination numbers of 3.3 and 0.9, respectively. This implies that the central Mn sites in the Mn–SAs/NC possessed a MnN3O1 configuration (inset image in Fig. S8†).
O species at 531.8 eV, and adsorbed-H2O at 533.8 eV in NC. However, the high-resolution O 1s spectra of M–SAs/NC display three different characteristic peaks with binding energies at around 530.4 eV (hydroxyl oxygen, M–OH), 533.6 eV (C–O), and 534.5 eV (surface adsorbed-H2O).32 After anchoring metal atoms, the functional group CO reduced along with the increase of M–OH. The possible reason is the CO species formed on the defects and edges of carbon layers that can easily get released without protection at high temperature.32,33 In particular, there existed a specific Mn–O binding energy situated at 529.3 eV for Mn–SAs/NC, which can be ascribed to the formation of Mn–O–C species, suggesting that the Mn atoms may be coordinated with both the N and O atoms on the carbon matrix, which is different with Fe, Co and Cu having only the M–N bonds.34–36 In order to understand the atomic structure of the as-prepared Mn–SAs/NC, the XANES and extended X-ray absorption fine structure (EXAFS) analyses were performed, as shown in Fig. S7.† As displayed in the Mn K-edge X-ray absorption near-edge structure (XANES) spectra (Fig. S7a†), the absorption edge position of Mn–SAs/NC is very closer to Mn(II) phthalocyanine (MnPc), indicating the main +2 valence state of Mn in Mn–SAs/NC. The Fourier-transformed (FT) EXAFS R-space plots in EXAFS characterization as illustrated in Fig. S7b† show a main peak situated at ≈1.46 Å, a position that coincides with the first Mn–N shell in the MnPc reference. Due to the similar bond lengths of Mn–N and Mn–O, the N and O atoms coordinated with Mn configuration cannot be obviously distinguished.33–35 Moreover, no obvious scattering path for Mn–Mn coordination is observed, indicating the atomic dispersion of Mn.34 Through XPS, we can confirm the coexistence of C–O and C–N in Mn–SAs/NC, which indicates the possibility of O and N coordination with the Mn atoms in the sample. EXAFS fitting was performed to study the structure parameters and obtain a quantitative chemical structure of Mn atoms. As shown in Fig. S8,† the first coordination shell of Mn–SAs/NC could be fitted by a mixture of Mn–N and Mn–O coordination paths, with coordination numbers of 3.3 and 0.9, respectively. This implies that the central Mn sites in the Mn–SAs/NC possessed a MnN3O1 configuration (inset image in Fig. S8†).
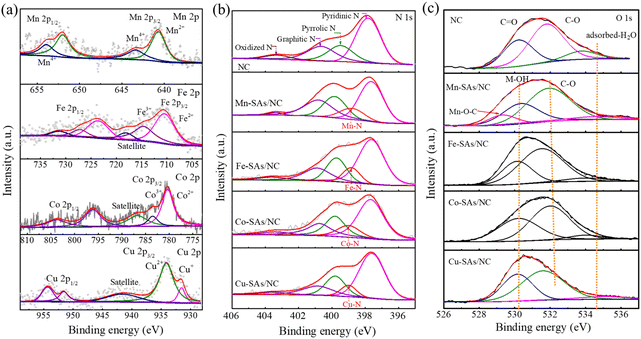 |
| | Fig. 3 (a) High-resolution XPS (a) M 2p, (b) N 1s, and (c) O 1s spectra of the as-synthesized samples. | |
Electrochemical catalytic performance
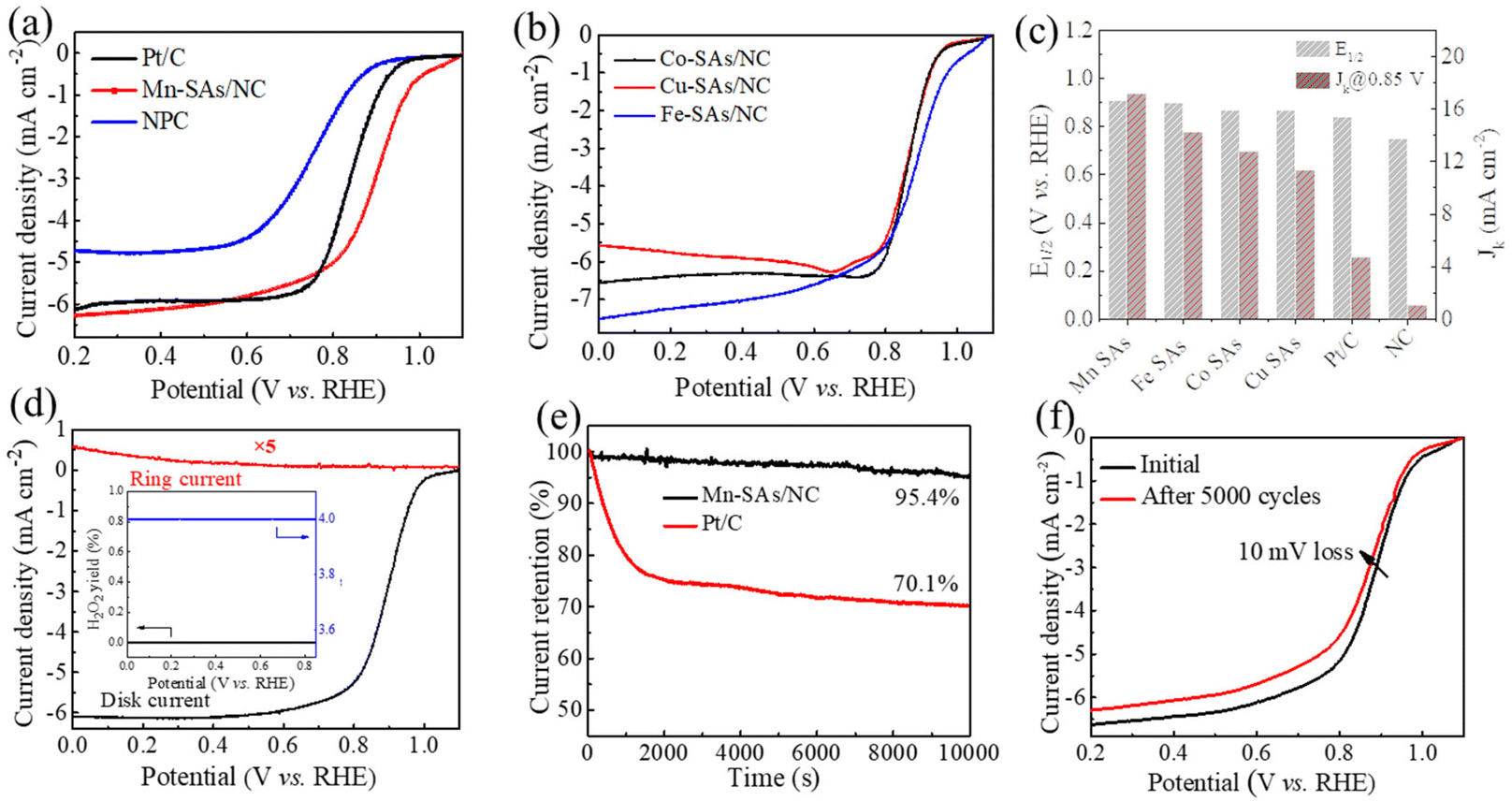
To evaluate the electrochemical ORR activity of the as-synthesized catalysts, the CV and polarization curves were obtained using rotating disk electrode (RDE) techniques in 0.1 M KOH electrolyte. As displayed in Fig. S9,† the CV curves of M–SAs/NC all exhibited sharp oxygen reduction peaks in O2-saturated alkaline solution, demonstrating their ORR catalytic capability. Moreover, Mn–SAs/NC exhibits the most positive peak at 0.93 V vs. RHE, compared to those of NC (∼0.8 V vs. RHE), Fe–SAs/NC (0.8–0.9 V vs. RHE), Co–SAs/NC (0.8–0.9 V vs. RHE), and Cu–SAs/NC (0.8–0.9 V vs. RHE). The more positive peak of Mn–SAs/NC (@0.93 V vs. RHE) further demonstrates its superior ORR capability. The LSV curves presented in Fig. 4a and b suggest that M–SAs/NC all deliver a higher ORR catalytic activity compared to that of pure NC (an Eonset of 0.89 V vs. RHE and an E1/2 of 0.75 V vs. RHE), indicating that the atomically dispersed species play a significant role in the improvement of ORR activity. In particular, Mn–SAs/NC shows the highest ORR catalytic activity and the ORR activity of M–SAs/NC follows the trend of Mn–SAs/NC (E1/2 = 0.91 V vs. RHE, Jd = 6.2 mA cm−2) > Fe–SAs/NC (E1/2 = 0.90 V vs. RHE, Jd = 7.0 mA cm−2) > Co–SAs/NC (E1/2 = 0.87 V vs. RHE, Jd = 6.3 mA cm−2) > Cu–SAs/NC (E1/2 = 0.87 V vs. RHE, Jd = 5.9 mA cm−2) (Fig. 4c). Moreover, the catalytic capability of Mn–SAs/NC is even higher than that of the commercial Pt/C catalyst (E1/2 = 0.84 V vs. RHE, Jd = 6.0 mA cm−2) and superior to those of many reported non-precious metal ORR electrocatalysts, as listed in Table S2.†37 As seen in Fig. 4c, the calculated kinetic current density (Jk) of Mn–SAs/NC is 17.13 mA cm−2 at 0.85 V vs. RHE, which is higher than those of Pt/C (4.72 mA cm−2), NC (1.07 mA cm−2), and other atomically dispersed metal catalysts Fe–SAs/NC (14.2 mA cm−2), Co–SAs/NC (12.75 mA cm−2) and Cu–SAs/NC (11.3 mA cm−2).38,39 The superior ORR catalytic activity of Mn–SAs/NC may be attributed to the specific coordination environment with both N and O atoms, the higher valence state of center metals and efficient mass transport. According to the LSV curves of Mn–SAs/NC at various rotation rates (Fig. S10a†) and eqn (1), the corresponding K–L plots at various potentials are shown in Fig. S10b.† The good linearity between the potential and the rotating rate demonstrates the four electron-transfer process and first-order reaction kinetics on atomically dispersed Mn sites in Mn–SAs/NC. The LSV curves obtained by the rotating ring disk electrode (RRDE) measurements are further used to evaluate the catalytic selectivity of Mn–SAs/NC. Based on eqn (2) and (3), Mn–SAs/NC possesses an electron transfer number (n) of ca. 4.0 for the ORR with an H2O2 yield of below 1.0%, implying the excellent ORR selectivity from O2 to H2O with the four-electron pathways (Fig. 4d). The long-term stability and methanol tolerance of catalysts are also important parameters to evaluate the practical application for the ORR. As shown in Fig. S11,† the current density–time curve of Mn–SAs/NC shows a slight decrease when 20 mL methanol was added to the electrolyte, whereas serious decay has been observed for Pt/C, revealing the high methanol tolerance of Mn–SAs/NC. The chronoamperometric (i–t) curves shown in Fig. 4e respond to the durability of Mn–SAs/NC and 20% Pt/C in an O2-saturated electrolyte. The results show that the current density for Mn–SAs/NC can remain over 95% after 10000 s, which is much higher than that for Pt/C (70.1%). In addition, the LSV curves of Mn–SAs/NC reveal that only a little decay (ΔE1/2 = 10 mV) is observed after 5000 CV cycles, further demonstrating the excellent stability of Mn–SAs/NC for the ORR (Fig. 4f). The high durability and methanol tolerance of Mn–SAs/NC for the ORR should be mainly ascribed to the unique hierarchical porous structure, and sufficient and stable Mn active sites on the carbon matrix.29,40,41
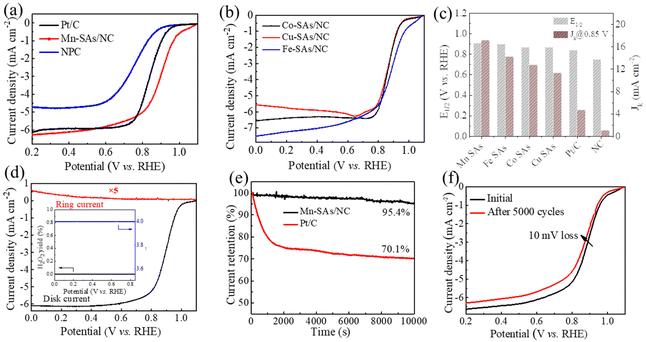 |
| | Fig. 4 ORR performance in 0.1 M KOH solutions. (a) LSV curves and (b) the corresponding Tafel plots of the as-prepared catalysts at a rotating speed of 1600 rpm. (c) The E1/2, and Jk values of the as-prepared catalysts, using commercial Pt/C forcomparison. (d) RRDE measurements of Mn–SAs/NC. (The inset shows the electron transfer number and H2O2 yield.) (e) Chronoamperometry curves of Mn–SAs/NC and Pt/C at 0.7 V at a rotating rate of 600 rpm in O2-saturated solution. (f) LSV curves of Mn–SAs/NC before and after cycling tests, respectively. | |
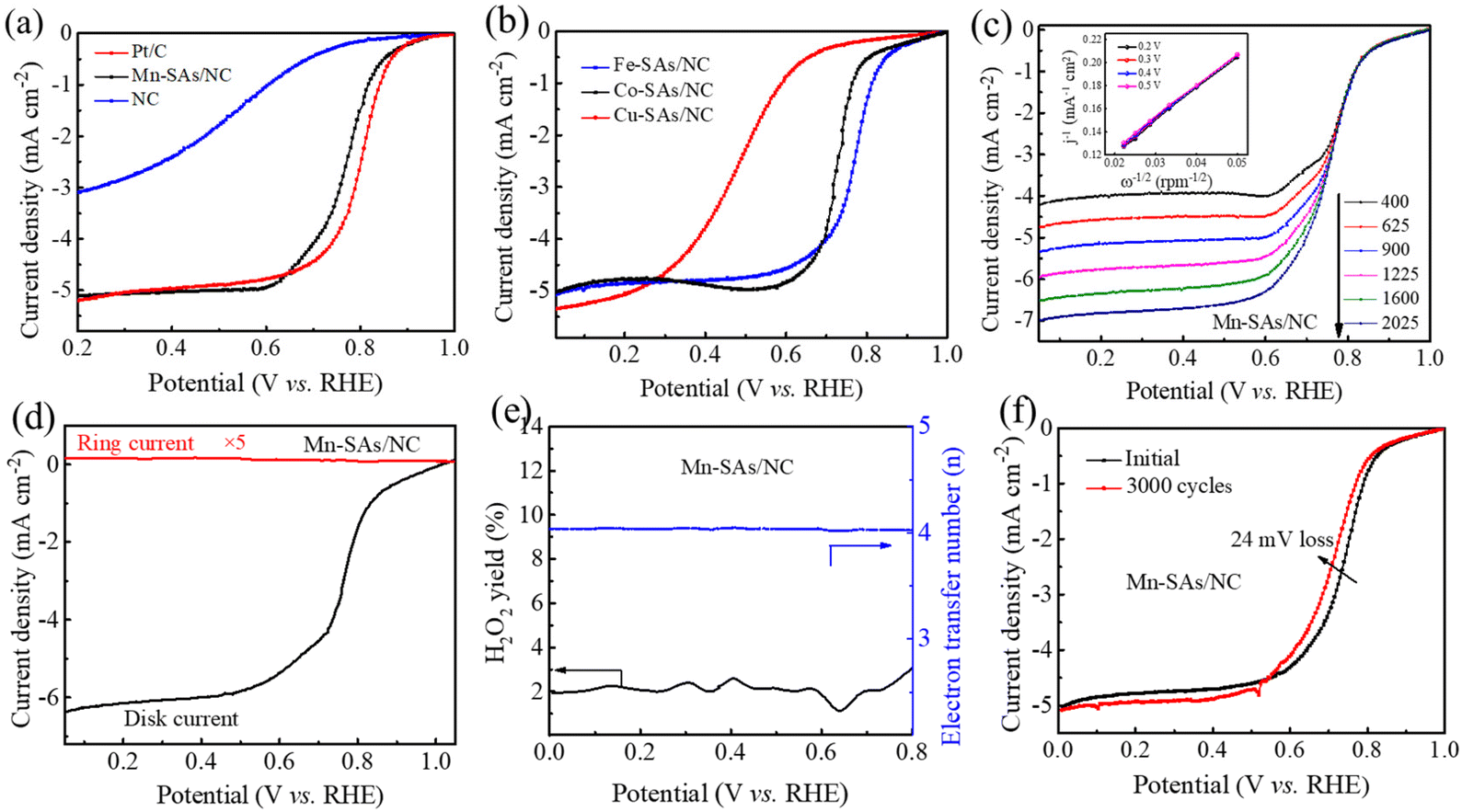
The ORR catalytic capabilities of the as-prepared catalysts have also been investigated in O2-saturated acidic electrolytes. The CV curves of the as-prepared catalysts in Fig. S12† show the cathodic reduction peak of Mn–SAs/NC at 0.76 V vs. RHE, Fe–SA/NC at 0.75 V vs. RHE, Co–SA/NC at 0.72 V vs. RHE, Cu–SAs/NC at 0.51 V vs. RHE, and NC at 0.61 V vs. RHE, indicating the superior ORR catalytic activity of Mn–SAs/NC under acidic conditions. The LSV curves shown in Fig. 5a reveal that the commercial Pt/C catalyst displays a high ORR activity with an E1/2 of (0.80 V vs. RHE). Additionally, Mn–SAs/NC possesses a more positive E1/2 (0.76 V vs. RHE) and Eonset (0.96 V vs. RHE) in 0.5 M H2SO4 solution, compared to that of NC (Eonset = 0.84 V vs. RHE, E1/2 = 0.48 V vs. RHE). In particular, these values are also superior to those of Fe–SAs/NC (Eonset = 0.95 V vs. RHE), Co–SAs/NC (Eonset = 0.95 V vs. RHE) and Cu–SAs/NC (Eonset = 0.88 V vs. RHE) as shown in Fig. 5b, and competitive to those of previously reported noble metal-free electrocatalysts (Table S2†). The LSV curves of Mn–SAs/NC obtained at different rotating rates and the corresponding K–L plots in acid solution are shown in Fig. 5c. The well-linear K–L plots derived from the LSV curves confirm the consistent first-order ORR kinetics with respect to the concentration of dissolved O2. The RRDE measurement further demonstrates that Mn–SAs/NC under acidic conditions shows a low HO2− yield of less than 2.0% and an average electron transfer number of ca. 3.95 (Fig. 5d and e). In addition, the stability test reveals that Mn–SAs/NC possesses high stability with no obvious change of Eonset and a small negative E1/2 shift of 24 mV after 3000 cycles in 0.5 M H2SO4 solution (Fig. 5f). The excellent ORR catalytic performance of Mn–SAs/NC in both acidic and alkaline solutions is closely related to the advantages of morphology, structure and the coordination environment. The hierarchical porous structure with a large surface area ensures sufficient exposed active sites for oxygen reduction. The porous carbon channels with excellent electrical conductivity can accelerate mass transport and electron transfer. Additionally, the homogeneous atomically dispersed metal active sites with specific local atomic structures provide excellent intrinsic catalytic activity to boost the ORR process. Therefore, the rationally designed porous structure and N and O co-coordinated micro-environment endow the Mn single atoms with highly efficient ORR performance.
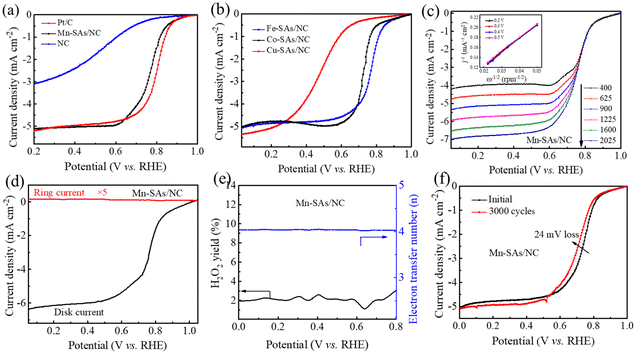 |
| | Fig. 5 ORR performance in 0.5 M H2SO4 solution. (a and b) LSV curves of various catalysts at a rotating speed of 1600 rpm. (c) LSV curves and K–L plots (the inset) of Mn–SAs/NC at different rotating rates. (d) RRDE measurements of Mn–SAs/NC and (e) the corresponding electron transfer number and H2O2 yield. (f) The LSV curves before and after the CV cycling test for Mn–SAs/NC. | |
Density functional theory calculations
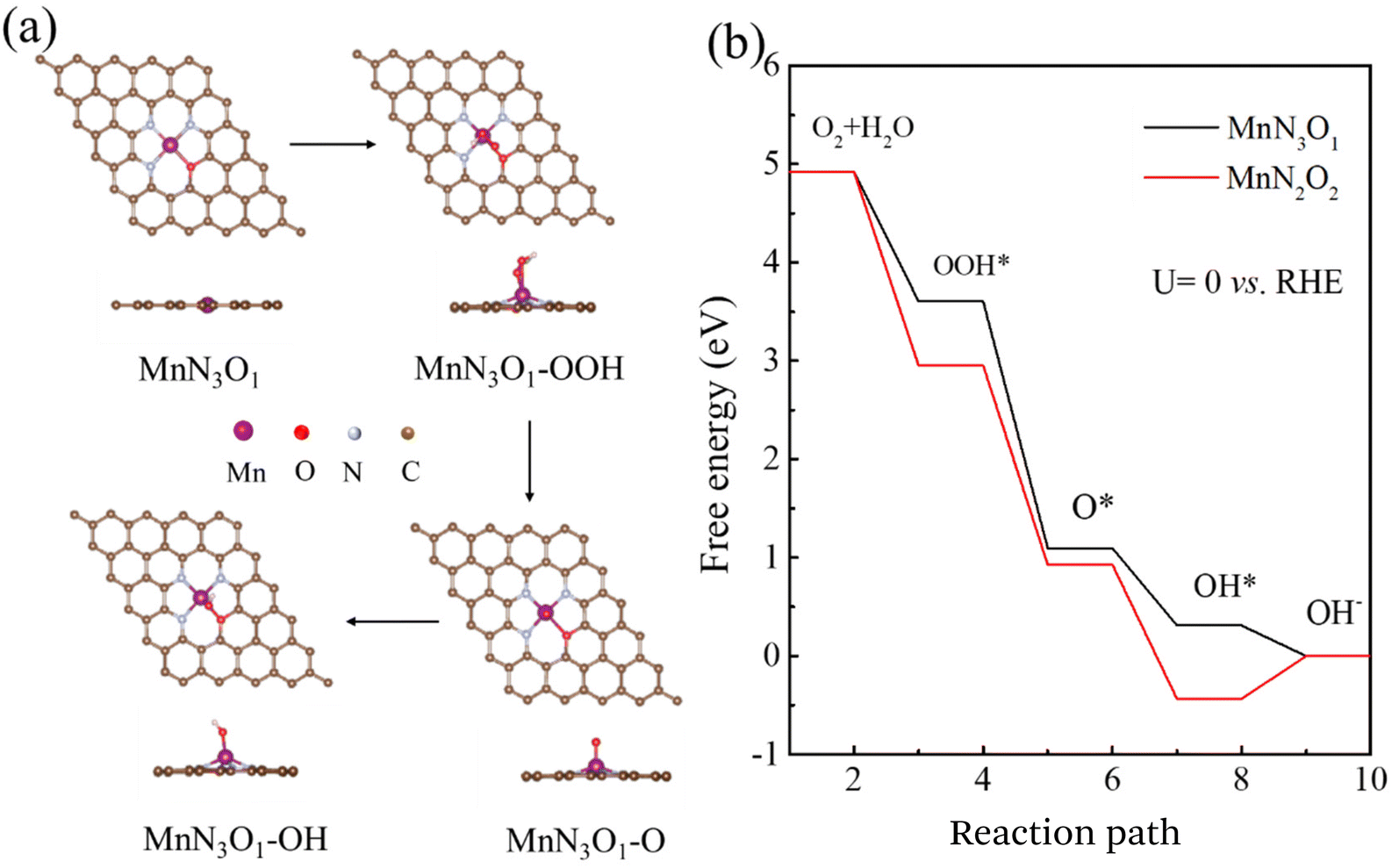
The above results demonstrate that Mn–SAs/NC is favorable for the ORR process. To further understand the correlation between the atomic structure and the catalytic mechanism for the high ORR reactivity, we further conducted DFT calculations on Mn–SAs/NC with different coordination structures. The possible models with different numbers of O and N atoms, e.g. Mn–N3O1 and Mn–N2O2, within the graphene framework have been investigated. The ORR four-electron transfer process of Mn single-atom sites is displayed in Fig. 6a and Fig. S13:† first, O2 molecules dissolved in 0.1 M KOH solution are adsorbed on Mn sites (*) to form O2*. (1) Combination with H2O to form OOH* and OH− (O2* + H2O + e− → OOH* + OH−, step I). (2) The formed OOH* can be resolved into O* and OH− (OOH* + e− → O* + OH−, step II). (3) The intermediate O* is combined with H2O to convert to OH* and OH− (O* + H2O + e− → OH* + OH−, step III). (4) OH* is further converted into OH− components and releases free radicals (*) (OH* + e− → OH− + *, step IV). The Gibbs free energy of the above-steps has been calculated to illustrate the catalytic mechanism of Mn–SAs/NC with different atomic structures. The limiting reaction barrier is the most significant parameter influencing the catalytic capability, which can be evaluated by the free energy of the rate determining step (RDS). As shown in Fig. 6b, all steps are thermodynamically downward with exothermic processes for the model Mn–N3O1, with a minimum free energy change of 0.31 eV for OH* desorption in the last step, indicating that OH* desorption is the RDS for the overall ORR. Under the same conditions, the last step of the Mn–N2O2 model is thermodynamically upward, indicating that Mn–N2O2 possesses a higher reaction barrier than Mn–N3O1. Additionally, the optimized adsorption capacities of the reaction intermediates and the free energy diagram of the ORR on the MnN4 models are shown in Fig. S14,† and the RDS for MnN4 is the third step of O* transfer to OH* by 0.27 eV. The largest reaction barrier for the MnN4 model is slightly higher than that of MnN3O1 by 0.04 eV. Therefore, the atomic structure of center metals in Mn–SAs/NC has a vital effect on the rate-determining step and the corresponding catalytic performance. In particular, the atomically dispersed Mn atoms with the 3 N and 1 O coordination environment demonstrate the lower reaction barrier and optimized interactions with the intermediates, boosting the ORR catalytic kinetics.
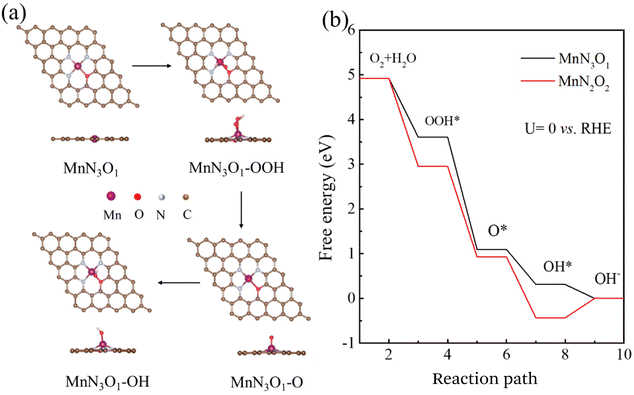 |
| | Fig. 6 Top and side views of the optimized adsorption model of O*, OH*, and OOH* on MnN3O1. Brown, purple, red, and white balls represent C, Mn, O, and N, respectively. (b) Under the condition of U = 0 V, the 4e− association pathway at the active site of MnN3O1 and MnN2O2 calculates the free energy evolution of the ORR. | |
Conclusions
In summary, we have designed a family of highly efficient ZIF-8-derived atomically dispersed metal atoms embedded in a porous carbon matrix through the combination of the conventional self-assembly of ZIF-8 and the ion-exchange strategy. ZIF-8 with sufficient organic ligands and porous channels can confine the transition metal ions by ion-exchange, which is beneficial for preventing the aggregation of metal species at high temperature. The as-prepared Mn–SAs/NC delivered superior ORR catalytic activity under both alkaline (E1/2 = 0.91 V vs. RHE) and acidic (E1/2 = 0.76 V vs. RHE) conditions, compared to those of Fe–SAs/NC, Co–SAs/NC and Cu–SAs/NC, attributing to the high valence state of Mn atoms and their specific atomic structure upon co-coordination with N and O atoms forming the MnN3O1 active sites. The DFT calculations also confirm that the MnN3O1 center ensures the lower free energy and optimized adsorption energy for intermediates, boosting the ORR process. Furthermore, Mn–SAs/NC showed excellent methanol tolerance and stability toward the ORR under both conditions with acceptable activity decay within 5000 cycles. The rational design of atomically dispersed catalysts with specific atomic structures and optimized electronic properties is promising to achieve high catalytic performance for electrochemical conversion reactions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
All authors thank the support from the “Joint International Laboratory on Environmental and Energy Frontier Materials” and the “Innovation Research Team of High-Level Local Universities in Shanghai”. This work is also supported by the National Natural Science Foundation of China (22209103).
References
- Y. Yan, S. Liang, X. Wang, M. Zhang, S.-M. Hao, X. Cui, Z. Li and Z. Lin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2110036118 CrossRef CAS.
- J. Mao, J. Iocozzia, J. Huang, K. Meng, Y. Lai and Z. Lin, Energy Environ. Sci., 2018, 11, 772–799 RSC.
- H. Yin, P. Yuan, B. Lu, H. Xia, K. Guo, G. Yang, G. Qu, D. Xue, Y. Hu, J. Cheng, S. Mu and J. Zhang, ACS Catal., 2021, 11, 12754–12762 CrossRef CAS.
- X. Wang, Z. Li, Y. Qu, T. Yuan, W. Wang, Y. Wu and Y. Li, Chem, 2019, 5, 1486–1511 CAS.
- X. Cao, J. Huo, L. Li, J. Qu, Y. Zhao, W. Chen, C. Liu, H. Liu and G. Wang, Adv. Energy Mater., 2022, 12, 2202119 CrossRef CAS.
- J. Zhang, H. Yang and B. Liu, Adv. Energy Mater., 2021, 11, 2002473 CrossRef CAS.
- X. X. Wang, J. Sokolowski, H. Liu and G. Wu, Chin. J. Catal., 2020, 41, 739–755 CrossRef CAS.
- F. Guo, M. Zhang, S. Yi, X. Li, R. Xin, M. Yang, B. Liu, H. Chen, H. Li and Y. Liu, Nano Res. Energy, 2022, 1, e9120027 CrossRef.
- Q. Qi, J. Hu, Y. Zhang, W. Li, B. Huang and C. Zhang, Adv. Energy Sustainability Res., 2021, 2, 2000067 CrossRef CAS.
- D. Liu, Q. Zeng, C. Hu, D. Chen, H. Liu, Y. Han, L. Xu, Q. Zhang and J. Yang, Nano Res. Energy, 2022, 1, e9120017 CrossRef.
- C.-X. Zhao, B.-Q. Li, J.-N. Liu and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4448–4463 CrossRef CAS PubMed.
- Y. Yan, H. Cheng, Z. Qu, R. Yu, F. Liu, Q. Ma, S. Zhao, H. Hu, Y. Cheng, C. Yang, Z. Li, X. Wang, S. Hao, Y. Chen and M. Liu, J. Mater. Chem. A, 2021, 9, 19489–19507 RSC.
- Y. He, S. Liu, C. Priest, Q. Shi and G. Wu, Chem. Soc. Rev., 2020, 49, 3484–3524 RSC.
- D. Qi, F. Lv, T. Wei, M. Jin, G. Meng, S. Zhang, Q. Liu, W. Liu, D. Ma, M. Hamdy, J. Luo and X. Liu, Nano Res. Energy, 2022, 1, e9120022 CrossRef.
- J. Huo, Z. Shen, X. Cao, L. Li, Y. Zhao, H. Liu and G. Wang, Small, 2022, 18, 2202394 CrossRef CAS PubMed.
- J. Zhang, Y. Zhao, C. Chen, Y. Huang, C. Dong, C. Chen, R. Liu, C. Wang, K. Yan, Y. Li and G. Wang, J. Am. Chem. Soc., 2019, 141, 20118–20126 CrossRef CAS PubMed.
- M. Wang, W. Yang, X. Li, Y. Xu, L. Zheng, C. Su and B. Liu, ACS Energy Lett., 2021, 6, 379–386 CrossRef CAS.
- X. Wang, X. Zhou, C. Li, H. Yao, C. Zhang, J. Zhou, R. Xu, L. Chu, H. Wang, M. Gu, H. Jiang and M. Huang, Adv. Mater., 2022, 34, 2204021 CrossRef CAS.
- L. Li, X. Cao, J. Huo, J. Qu, W. Chen, C. Liu, Y. Zhao, H. Liu and G. Wang, J. Energy Chem., 2023, 76, 195–213 CrossRef CAS.
- M. Zhao, H. Liu, H. Zhang, W. Chen, H. Sun, Z. Wang, B. Zhang, L. Song, Y. Yang, C. Ma, Y. Han and W. Huang, Energy Environ. Sci., 2021, 14, 6455–6463 RSC.
- T. Wang, Y. Wang, M. Sun, A. Hanif, H. Wu, Q. Gu, Y. S. Ok, D. C. W. Tsang, J. Li, J. Yu and J. Shang, Chem. Sci., 2020, 11, 6670–6681 RSC.
- Y. Ye, F. Cai, C. Yan, Y. Li, G. Wang and X. Bao, J. Energy Chem., 2017, 26, 1174–1180 CrossRef.
- B. Sasikumar, S. Bisht, G. Arthanareeswaran, A. F. Ismail and M. H. D. Othman, Sep. Purif. Technol., 2021, 264, 118471 CrossRef CAS.
- Z. Pu, I. S. Amiinu, R. Cheng, P. Wang, C. Zhang, S. Mu, W. Zhao, F. Su, G. Zhang, S. Liao and S. Sun, Nano-Micro Lett., 2020, 12, 21 CrossRef CAS PubMed.
- M. Liu, P. Zhang, Z. Qu, Y. Yan, C. Lai, T. Liu and S. Zhang, Nat. Commun., 2019, 10, 3917 CrossRef PubMed.
- T. F. Zhang, K.-W. Kim and K. H. Kim, J. Electrochem. Soc., 2015, 163, 54–61 CrossRef.
- Y. Fu, Q. Wei, G. Zhang, X. Wang, J. Zhang, Y. Hu, D. Wang, L. Zuin, T. Zhou, Y. Wu and S. Sun, Adv. Energy Mater., 2018, 8, 1801445 CrossRef.
- Y. Li, Y. Fang, Y. Xue, L. Hui and X. Chen, J. Mater. Chem. A, 2022, 10, 6073–6077 RSC.
- L. Yan, L. Xie, X.-L. Wu, M. Qian, J. Chen, Y. Zhong and Y. Hu, Carbon Energy, 2021, 3, 856–865 CrossRef CAS.
- R. Ge, J. Huo, J. Qu, Y. Liu, Y. Li, M. Zhu, J. Cairney, R. Zheng, J. Zhang and W. Li, Mater. Today Nano, 2022, 20, 100248 CrossRef CAS.
- X. Han, T. Zhang, W. Chen, B. Dong, G. Meng, L. Zheng, C. Yang, X. Sun, Z. Zhuang, D. Wang, A. Han and J. Liu, Adv. Energy Mater., 2021, 11, 2002753 CrossRef CAS.
- R. Ge, J. Huo, Y. Li, T. Liao, J. Zhang, M. Zhu, T. Ahamad, S. Li, H. Liu, L. Feng and W. Li, J. Alloys Compd., 2022, 904, 164084 CrossRef CAS.
- Q. Wang, Y. Yang, F. Sun, G. Chen, J. Wang, L. Peng, W. Chen, L. Shang, J. Zhao, D. Waterhouse, T. Zhang and G. Waterhouse, Adv. Energy Mater., 2021, 11, 2100219 CrossRef CAS.
- Y. Yang, K. Mao, S. Gao, H. Huang, G. Xia, Z. Lin, P. Jiang, C. Wang, H. Wang and Q. Chen, Adv. Mater., 2018, 30, 1801732 CrossRef.
- Y. Cai, J. Fu, Y. Zhou, Y. Chang, Q. Min, J. Zhu, Y. Lin and W. Zhu, Nat. Commun., 2021, 12, 586 CrossRef CAS PubMed.
- L. Jiang, J. Wang, X. Mao, X. Xu, B. Zhang, J. Yang, Y. Wang, J. Zhu and S. Hou, Carbon, 2017, 111, 207–214 CrossRef CAS.
- G. Yang, J. Zhu, P. Yuan, Y. Hu, G. Qu, B.-A. Lu, X. Xue, H. Yin, W. Cheng, J. Cheng, W. Xu, J. Li, J. Hu, S. Mu and J. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS PubMed.
- D. A. Walsh, A. Ejigu, J. Smith and P. Licence, Phys. Chem. Chem. Phys., 2013, 15, 7548–7554 RSC.
- J. Kossmann, M. L. Ortíz Sánchez-Manjavacas, H. Zschiesche, N. V. Tarakina, M. Antonietti, J. Albero and N. López-Salas, J. Mater. Chem. A, 2022, 10, 6107–6114 RSC.
- X. Liu, C. Culhane, W. Li and S. Zou, ACS Omega, 2020, 5, 24367–24378 CrossRef CAS.
- Y. Li, B. Chen, X. Duan, S. Chen, D. Liu, K. Zang, R. Si, F. Lou, X. Wang, M. Rønning, L. Song, J. Luo and D. Chen, Appl. Catal., B, 2019, 249, 306–315 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *e and
Hao
Liu
*e and
Hao
Liu