
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversible speed control of one-stimulus-double-response, temperature-sensitive asymmetric hydrogel micromotors†
Serena P.
Teora
,
Kirsten H.
van der Knaap
,
Shauni
Keller
,
Sjoerd J.
Rijpkema
and
Daniela A.
Wilson
 *
*
Department of Systems Chemistry, Institute for Molecules and Materials, Radboud University, Heyendaalseweg 135, 6525 AJ Nijmegen, The Netherlands. E-mail: d.wilson@science.ru.nl
First published on 5th August 2022
Abstract
Soft, one-stimulus-double-response, thermo-sensitive, PNIPAm-based microgels are designed for controlled autonomous motion under stimuli. At higher temperature, the motors with physically encapsulated catalase move faster, while motors in which catalase is chemically linked to PNIPAm ceased moving. The phenomenon is reversible over multiple cycles of temperature.
Artificial self-propelled micro- and nanomotors represent one of the greatest attempts in research to mimic nature. Nature presents many fascinating structures able to regulate their motion by changing the morphology of their dynamic and soft bodies in accordance with the local environment. Inspired by this, scientists are trying to design artificial, intelligent, and well-adapted motors with functionalities that resemble those of cells and microorganisms. Several geometries of micro-swimmers that convert chemical energy into autonomous motion have already been reported, including Janus particles1–6 and catalytic rods,7 asymmetric shapes8 and tubular structures.9,10 These motors can be powered with different strategies resulting in self-propelling behaviours based on bubbles formation,11 interfacial tension gradients,12 self-electrophoresis,13 or self-diffusiophoresis induced by asymmetrical chemical reactions.14,15 Despite these progresses and in contrast to biological swimmers, most artificial micromotors are still commonly formed by rigid structures.16 The lack of softness, reconfigurability and “smartness” to react to external conditions makes it challenging to gain control over the motion under certain stimuli. The use of soft materials that respond to a stimulus (e.g., light, pH and temperature) has great potential in the fabrication of motors with high functionality and adaptability in terms of movement. The effect of flexibility of the materials and how shape reconfiguration triggered by a stimulus can be used to control micromotors motion has been demonstrated by Schmdit et al. that reported microjets able to reversibly fold and unfold by applying changes in temperature to the solution in which they are immersed.17 Su et al. developed pH-responsive gelatine cartridge-case-like self-propelleded structures,18 and Dong et al. proposed a polymer-based raspberry-like micromotors which exhibits multistimuli-responsiveness.19 However, all these motors possess one-stimulus-one response behaviour based on the responsiveness of the material they are made of.
Herein, we present the fabrication of soft, biocompatible, asymmetric, thermo-responsive bubble-propelled micromotors of which the motion can be controlled by changes of temperature. These motors show one-stimulus-double response reversible behaviour depending on the strategy used for catalyst entrapment. If the catalyst is physically encapsulated in the systems, when the temperature increases, motors move faster. On contrary, if the catalyst is chemically immobilized into the system, increasing the temperature stops the motion. We took advantage of the versatility of microfluidics previously reported from our group20 implementing the microgels design with a temperature-responsive polymer, namely poly(N-isopropylacrylamide) (PNIPAm). PNIPAm has a lower critical solution temperature (LCST) in aqueous solutions at 32 °C at which it exhibits a hydrophilic-to-hydrophobic transition.21 This provides the system with additional smart behaviour that resembles functionalities of microorganisms of the biological inspiration and allows control over the motion under stimuli. To guarantee a fully biocompatible design, catalase is used as biocatalyst. The enzyme decomposes hydrogen peroxide into water and oxygen, which are responsible of the bubble-propelled mechanism of motion. The asymmetric microgels formed in the microfluidic device (Fig. 1a) consist of two-phase-separating aqueous solutions. The first aqueous solution is a mixture of poly(ethylene glycol) (PEGDA) 25 w/w% and PNIPAm 10 w/w%, the second is an aqueous solution of dextran 20 w/w%. The polymers concentrations of the first aqueous solution are fine-tuned in order to retain the properties of an aqueous-two-phase system (ATPS). To the best of our knowledge, the use of this new stimuli-responsive ATPS consisting of a PEGDA-PNIPAm composite phase and a phase separating polysaccharide has not been reported so far. The PNIPAm polymer is mixed with the PEGDA polymer in the main phase as a non-crosslinkable moiety. After collection, the PEGDA phase is photocured resulting at room temperature in a gel network in which PNIPAm is interpenetrating as a single stranded polymer. Upon polymerization, the dextran diffuses into the PEGDA-PNIPAm leaving behind a cavity. The molecular weight of the PNIPAm polymer strands in the main phase is varied between 2–80 kDa (Fig. 1b) to enlarge the effect on the decrease in water content of the microparticles when passing the LCST, upon decreasing the molecular weight. This results in an increase of molar content of the thermo-responsive polymer within the microgels, thus in a more pronounced shrinking behaviour of the particles. (Fig. 2) The size reconfiguration is demonstrated over three cycles of temperature at 20 °C and 43 °C, with volumes that reduce by −9.2%, −7.6% and −7.5% for PNIPAm 2 kDa (Fig. 2a), 10 kDa (Fig. 2b) and 80 kDa (Fig. 2c), respectively. The non-linear shrinking behaviour depending on the MW of PNIPAm is in accordance with the elastic energy stored during deformation by the hydrogel upon increasing the temperature. Above LCST of PNIPAm, the increased value of storage modulus (G′) is more pronounced in microgels with the lowest MW, while the difference between 10 and 80 kDa is less evident. (Fig. S7, ESI†) The higher stiffness of hydrogels made of PEGDA-PNIPAm 2 kDa at 43 °C can be further visualised as decrease in mesh size of the gel network (Fig. S6, ESI†). Cryo-SEM images show a change in the density of microgels due to collapse of PNIPAm within the pores of PEGDA network, thus larger pores result when higher MW are used.22 The thermo-responsiveness introduced into the system by the presence of PNIPAm is further confirmed by control experiments of size analysis over temperature cycles of PEGDA microgels (Fig. S8, ESI†). PEGDA hydrogel network is not responsive to temperature change. In fact, the measurements were performed at temperatures much lower than the LCST of poly(ethylene glycol) (PEG), which has been reported to be in the range of 100–176 °C.23 Therefore, it can be assumed that the change in volume of the PEGDA-PNIPAm microparticles can be attained to the volume phase transition of the PNIPAm chains that collapse into globules when it has passed the LCST.
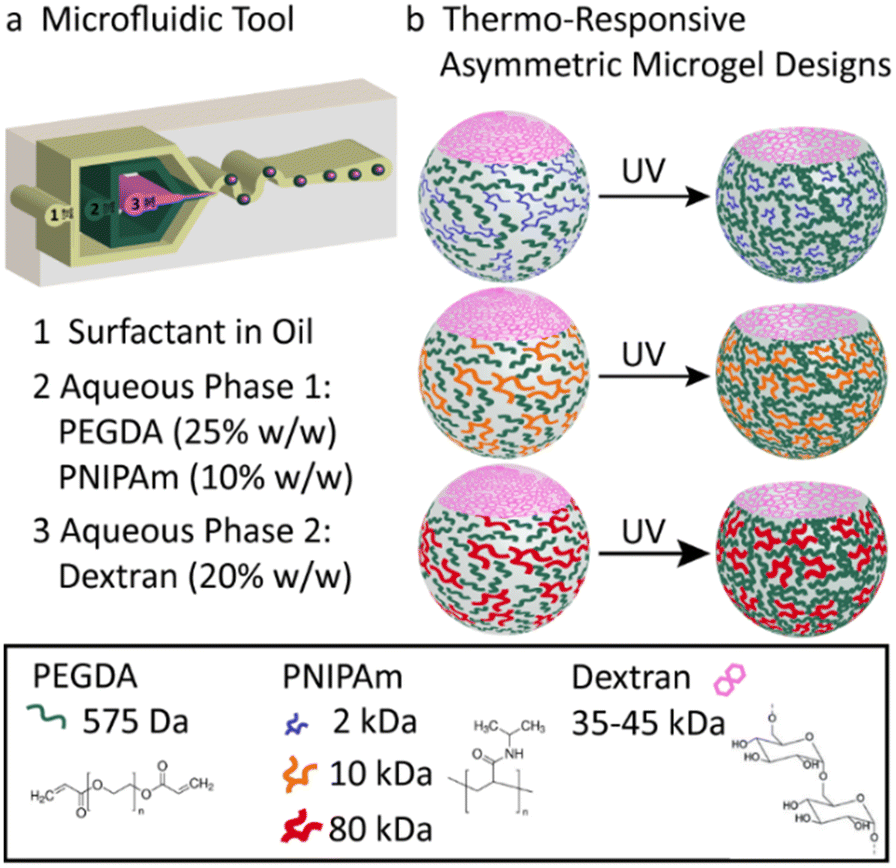 | ||
| Fig. 1 Microfluidic design of thermo-responsive, asymmetric hydrogels. (a) Microfluidic chip used for the generation of thermo-responsive ATPS droplets. The three inlets are for the injection of oil containing surfactant, temperature-responsive aqueous phase 1 and aqueous phase 2, respectively. Aqueous phase 1 consists of a mixture of PEGDA (25 w/w%) and PNIPAm (10 w/w%). Aqueous phase 2 is a 20 w/w% dextran solution. (b) Schematic representation of the two-phase droplet and asymmetric microgels made with PNIPAm at different MW (from top to bottom: 2 kDa, 10 kDa, 80 kDa). | ||
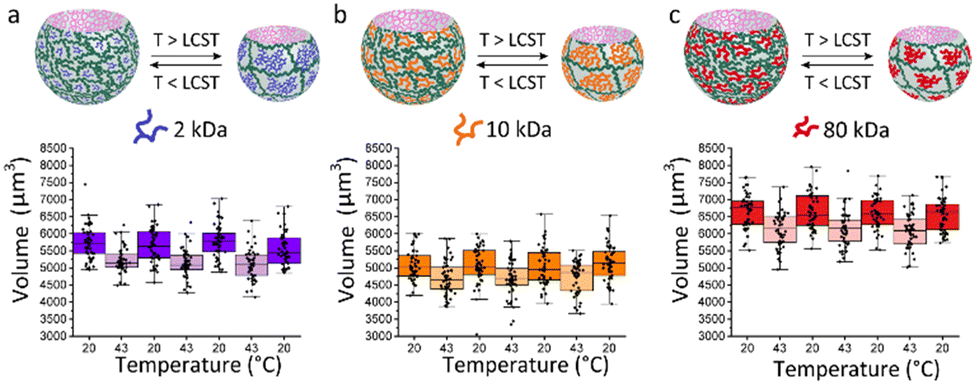 | ||
| Fig. 2 Shrinking and swelling volumes of thermo-responsive asymmetric microgels upon temperature cycles at 43 °C and 20 °C for different MW of PNIPAM. (a) PEGDA-PNIPAm 2 kDa microparticles show a shrinking in volume of 9.2%. (b) PEGDA-PNIPAm 10 kDa microparticles show a shrinking in volume of 7.6%. (c) PEGDA-PNIPAm 10 kDa microparticles show a shrinking in volume of 7.5%. | ||
Based on the design of the microparticles and the overall shrinking retaining the morphology, it is expected to observe a homogeneous distribution of the PNIPAm strands throughout the hydrogel. In order to visualize the distribution of the thermo-responsive polymer, the free chains of PNIPAm were fluorescently labelled with Cynine 3 (Cy3) dye. Confocal images confirmed the homogenous distribution within the PEGDA gel network for the three different MW of PNIPAm (Fig. 3). It was envisioned that a change in hydrophobicity within the system would influence the fluorescence properties of the Cy3. Cyanine dyes have been reported to be sensitive to their environment; decreased levels of fluorescence intensity were measured as a function of increasing solvent polarity and vice versa.24 Due to its structure, the enhanced fluorescence of Cy3 dye at room temperature decreases when we expose our microgels at 43 °C (Fig. 3). A change of surroundings of the fluorophore (Fig. S10, ESI†) causes movement of solvent out of the dye aggregates, thus the fluorophore is experiencing a different environment. Due to its conformational change,25,26 when the PNIPAm polymer undergoes a coil-to-globule transformation, most of the amide groups are buried and a significant amount of water molecules is expelled, leaving the hydrophobic chains to be exposed. As the non-sulfonated cyanine dyes (Cy3 and Cy5) are also hydrophobic, the dyes are expected to interact more with the hydrophobic moieties of the amphiphilic PNIPAm polymer, therefore also being exposed on the outside of the PNIPAm globule, away from the hydrophilic core.27 In this way the dye microenvironment is changed. Counterintuitively, the decrease in fluorescence brightness indicates that the dye is exposed to a more aqueous media and the interaction with water molecules is increased.27,28 This explains the lower fluorescence intensity and confirms the thermal collapse of transition of PNIPAm in the hydrogel microparticles at temperatures above its LCST. This phenomenon is reversible over switches of temperature at 20 °C and 43 °C (Fig. S9 and S10, ESI†).
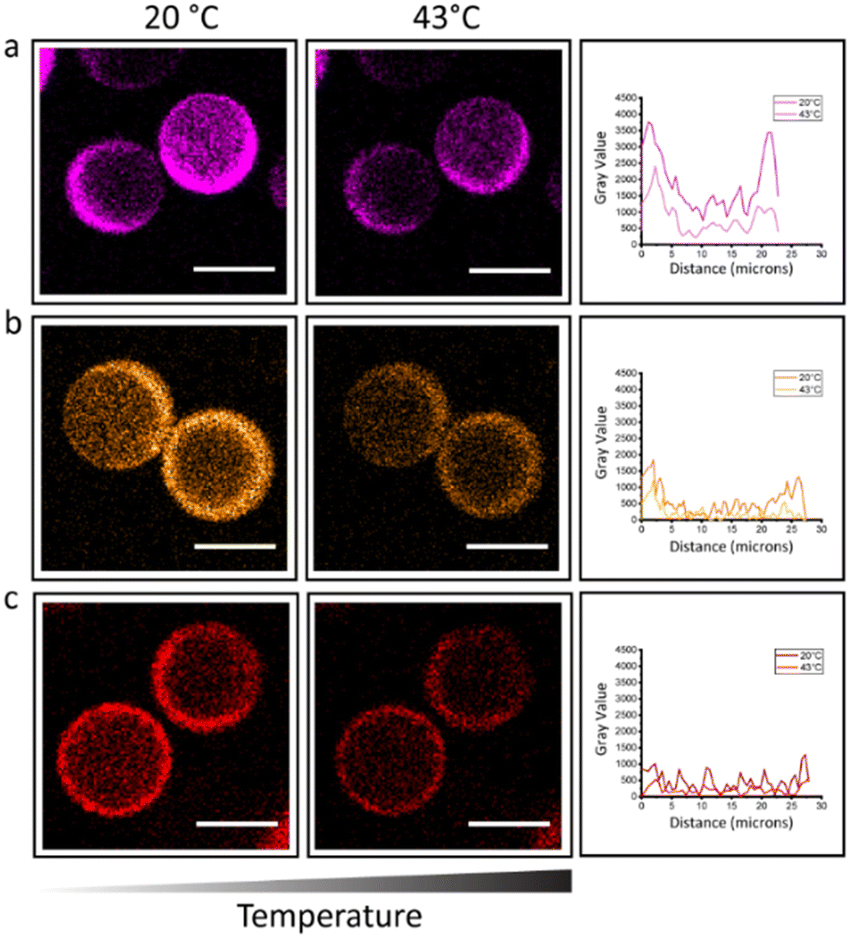 | ||
| Fig. 3 Confocal images at 20 °C and 43 °C of microgels with Cy3 labelled PNIPAm. A lower fluorescence intensity is visible at higher temperature due to changes in hydrophobicity in the microenvironment. Gray values are plotted as function of distance for microgels made of different MW PNIPAm. (a) 2 kDa, (b) 10 kDa, (c) 80 kDa. Scale bar is 20 μm. | ||
We hypothesize that shape reconfigurability at different temperatures plays an important role for controlling the self-propulsion. The presence of a stimuli-responsive polymer can introduce an additional smart behaviour to bubble-propelled micromotors. To investigate the effect of temperature on thermo-responsive motors, we used two different strategies for enzyme entrapment. With the first strategy, Catalase is physically encapsulated within the hydrogel structure of the motor. This is obtained by adding the enzyme to the PEGDA-PNIPAm phase prior to injection into the microfluidic chip. A second way is to chemically bond the catalase to the PNIPAm chains. (Fig. S1–S5, ESI†) In this case the enzyme is immobilized on the temperature-sensitive polymer, which is then mixed with the PEGDA to create a composite main aqueous phase for the generation of ATPS droplets. This was done, in both cases, with PNIPAm at three different MW. Ten of the fastest motors were tracked for 20 seconds after addition to a solution 4 wt% of hydrogen peroxide fuel over cycles of temperature. Motors in which the catalase is physically encapsulated show increased speeds at 43 °C (Fig. 4). This happens due to the shrinking of the motors at higher temperature, which causes a reduction in volume, thus a higher concentration of enzyme exposed to the fuel. At room temperature, motors made of PNIPAm 2 kDa (Fig. 4a) and 10 kDa (Fig. 4b) and 80 kDa (Fig. 4c) show speeds ∼40 μm s−1. The values of speeds become almost double at 43 °C, with fastest speed for 2 kDa PNIPAm motors (Fig. 4a). The higher molar ratio of thermo-responsive strands leads to the formation of more collapsed globules above LCST, which, in turns, means more compact and sftiff motors that swim faster. For higher PNIPAm MW, there are less strands that collapse within the PEGDA network, so the motors are less dense at higher temperature, thus they swim slower. A control experiment is performed to rule out the behaviour of physically encapsulated catalase in the PEGDA:Dextran hydrogel micromotors over cycles of temperatures (Fig. S11, ESI†). A clear difference is visible, since these motors at 43 °C show non-significant increased speeds that can be explained with a slightly more active enzyme, of which the optimum activity is reported around 40 °C.29 On contrary, motors in which the catalase is chemically linked to PNIPAm surprisingly stop moving at 43 °C (Fig. 5). At room temperature the motors have speed ∼80 μm s−1, ∼35 μm s−1 and ∼30 μm s−1 for catalase immobilized on 2 kDa (Fig. 5a), 10 kDa (Fig. 5b) and 80 kDa (Fig. 5c), respectively. It is reasoned that the molar percentage of PNIPAm is decreased for the micromotors which incorporate PNIPAm at higher MW as single stranded polymer, which decreases the bonded amount of catalyst, explaining faster motors at room temperature for lower PNIPAm MW. When passing its LCST, PNIPAm strands switch from swollen coils to collapsed globules that tangle around the catalase creating a hydrophobic microenvironment. The aqueous content of the microparticles is reduced within these hydrophobic globules and consequently the catalase does not have access to the fuel anymore. The inflow of fuel to the globules is hindered thus no fuel reaches the catalase enzyme, which is now enclosed into the PNIPAm globules. In this way, no bubbles are formed leading to complete impairment of the movement of the motors at 43 °C. This behaviour is reversible: going to temperatures below its LCST, PNIPAm strands change their conformation back to linear and flexible chains30 allowing the catalase to be in contact with the fuel, thus produce oxygen bubbles for the autonomous motion.
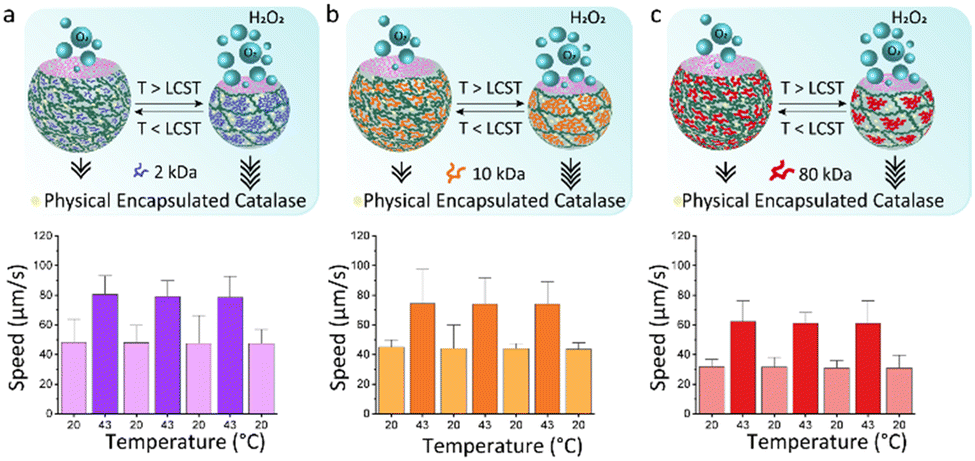 | ||
| Fig. 4 Speeds of PEGDA-PNIPAm micromotors over cycles of temperature at 20 °C and 43 °C in a 4 wt% hydrogen peroxide solution. The catalase is physically encapsulated in the gel network. PNIPAm polymer has different MW: (a) 2 kDa, (b) 10 kDa, (c) 80 kDa. Error bars indicate the range error. | ||
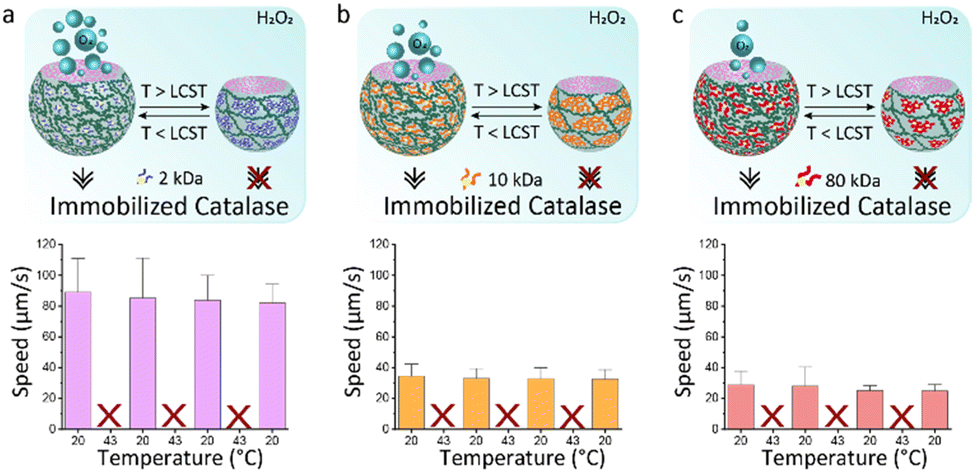 | ||
| Fig. 5 Speeds of PEGDA-PNIPAm micromotors over cycles of temperature at 20 °C and 43 °C in a 4 wt% hydrogen peroxide solution. The catalase is bonded to PNIPAm prior to mixing with PEGDA. PNIPAm polymer has different MW: (a) 2 kDa, (b) 10 kDa, (c) 80 kDa. Error bars indicate the range error. | ||
In summary, we have reported the first one-stimulus-double response bubble-propelled micromotor based on a new ATPS in which one of the two aqueous solutions is a composite phase consisting of photocrosslinkble PEGDA and the thermo-responsive polymer PNIPAm. We showed the size reconfiguration of PEGDA-PNIPAm asymmetric microgels for different MW of PNIPAm over cycles of temperature below and above its LSCT. At 43 °C, the volumes shrinking decreases for higher MW, as decreases the molar ratio of thermo-responsive strands undergoing to thermal collapse. With fluorescence experiments we further proved changes in hydrophobicity in the microenvironment due to the presence of PNIPAm at temperatures above its LCST. The PNIPAm successfully adds a break to the system that can be used to gain control over the motion. The smartness of this new thermo-responsive architecture allows for almost two times faster speeds at 43 °C if the enzyme is physically encapsulated in the microgels, with higher values for motors made of lower MW PNIPAm. In this case, the denser gel results in more compact and stiff motors that swim faster. Oppositely, when the enzyme is chemically attached to the thermo-sensitive polymer, at higher temperature the thermal collapse to globules of PNIPAm strands avoids the exposure of catalase to the fuel, preventing the bubble formation, thus the motion of micromotors. The reconfigurability and smartness to have a double response to one stimulus of our system can be used in future studies for enhanced adaptability in terms of motion and function. The shrinking behaviour can be used as breath-in effect for encapsulation of proteins and other models of drugs. Due to the self-propulsion, micromotors could be targeted to a specific location where the cargo could be released as a result of the breath-out effect induced by temperature-triggered re-swelling of the microgels.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. R. Howse, R. A. L. Jones, A. J. Ryan, T. Gough, R. Vafabakhsh and R. Golestanian, Phys. Rev. Lett., 2007, 99, 048102 CrossRef PubMed.
- B. Jurado-Sánchez, S. Sattayasamitsathit, W. Gao, L. Santos, Y. Fedorak, V. V. Singh, J. Orozco, M. Galarnyk and J. Wang, Small, 2015, 11, 499–506 CrossRef PubMed.
- W. He, J. Frueh, N. Hu, L. Liu, M. Gai and Q. He, Adv. Sci., 2016, 3, 1600206 CrossRef PubMed.
- C. Fiedler, C. Ulbricht, T. Truglas, D. Wielend, M. Bednorz, H. Groiss, O. Brüggemann, I. Teasdale and Y. Salinas, Chem. – Eur. J., 2021, 27, 3262–3267 CrossRef CAS PubMed.
- J. Zhu, H. Wang and Z. Zhang, Langmuir, 2021, 37, 4964–4970 CrossRef CAS PubMed.
- Y. Feng, X. Chang, H. Liu, Y. Hu, T. Li and L. Li, Appl. Mater. Today, 2021, 23, 101026 CrossRef.
- W. Wang, L. A. Castro, M. Hoyos and T. E. Mallouk, ACS Nano, 2012, 6, 6122–6132 CrossRef CAS PubMed.
- S. Keller, S. P. Teora, G. X. Hu, M. Nijemeisland and D. A. Wilson, Angew. Chem., Int. Ed., 2018, 57, 9814–9817 CrossRef CAS PubMed.
- W. Gao, A. Uygun and J. Wang, J. Am. Chem. Soc., 2012, 134, 897–900 CrossRef CAS PubMed.
- A. A. Solovev, Y. Mei, E. Bermúdez Ureña, G. Huang and O. G. Schmidt, Small, 2009, 5, 1688–1692 CrossRef CAS PubMed.
- R. F. Ismagilov, A. Schwartz, N. Bowden and G. M. Whitesides, Angew. Chem., Int. Ed., 2002, 41, 652–654 CrossRef CAS.
- C. Maggi, F. Saglimbeni, M. Dipalo, F. De Angelis and R. Di Leonardo, Nat. Commun., 2015, 6, 7855 CrossRef CAS PubMed.
- A. M. Brooks, M. Tasinkevych, S. Sabrina, D. Velegol, A. Sen and K. J. M. Bishop, Nat. Commun., 2019, 10, 495 CrossRef CAS PubMed.
- R. Golestanian, T. B. Liverpool and A. Ajdari, Phys. Rev. Lett., 2005, 94, 220801 CrossRef PubMed.
- F. Yang, S. Qian, Y. Zhao and R. Qiao, Langmuir, 2016, 32, 5580–5592 CrossRef CAS PubMed.
- W. Gao, S. Sattayasamitsathit, J. Orozco and J. Wang, J. Am. Chem. Soc., 2011, 133, 11862–11864 CrossRef CAS PubMed.
- V. Magdanz, G. Stoychev, L. Ionov, S. Sanchez and O. G. Schmidt, Angew. Chem., Int. Ed., 2014, 53, 2673–2677 CrossRef CAS PubMed.
- Y. Su, Y. Ge, L. Liu, L. Zhang, M. Liu, Y. Sun, H. Zhang and B. Dong, ACS Appl. Mater. Interfaces, 2016, 8, 4250–4257 CrossRef CAS PubMed.
- L. Zhang, H. Zhang, M. Liu and B. Dong, ACS Appl. Mater. Interfaces, 2016, 8, 15654–15660 CrossRef CAS.
- S. Keller, G. X. Hu, M. I. Gherghina-Tudor, S. P. Teora and D. A. Wilson, Adv. Funct. Mater., 2019, 29, 1904889 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- K. N. Plunkett, X. Zhu, J. S. Moore and D. E. Leckband, Langmuir, 2006, 22, 4259–4266 CrossRef CAS PubMed.
- S. Saeki, N. Kuwahara, M. Nakata and M. Kaneko, Polymer, 1976, 17, 685–689 CrossRef CAS.
- H. S. Muddana, T. T. Morgan, J. H. Adair and P. J. Butler, Nano Lett., 2009, 9, 1559–1566 CrossRef CAS PubMed.
- H. Yim, M. S. Kent, S. Mendez, G. P. Lopez, S. Satija and Y. Seo, Macromolecules, 2006, 39, 3420–3426 CrossRef CAS.
- G. Zhang, Macromolecules, 2004, 37, 6553–6557 CrossRef CAS.
- D. Poplinger, A. Bazylevich, M. Bokan, G. Gellerman and L. Patsenker, J. Photochem. Photobiol., A, 2021, 408, 113113 CrossRef CAS.
- I. Yapici, K. S. S. Lee, T. Berbasova, M. Nosrati, X. Jia, C. Vasileiou, W. Wang, E. M. Santos, J. H. Geiger and B. Borhan, J. Am. Chem. Soc., 2015, 137, 1073–1080 CrossRef CAS PubMed.
- R. M. Daniel, M. E. Peterson, M. J. Danson, N. C. Price, S. M. Kelly, C. R. Monk, C. S. Weinberg, M. L. Oudshoorn and C. K. Lee, Biochemical Journal, 2009, 425, 353–360 Search PubMed.
- K. Jain, R. Vedarajan, M. Watanabe, M. Ishikiriyama and N. Matsumi, Polym. Chem., 2015, 6, 6819–6825 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc02854a |
| This journal is © The Royal Society of Chemistry 2022 |