
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total synthesis, isolation, surfactant properties, and biological evaluation of ananatosides and related macrodilactone-containing rhamnolipids†
Maude
Cloutier
 a,
Marie-Joëlle
Prévost
a,
Serge
Lavoie
b,
Thomas
Feroldi
b,
Marianne
Piochon
a,
Marie-Christine
Groleau
a,
Jean
Legault
b,
Sandra
Villaume
c,
Jérôme
Crouzet
c,
Stéphan
Dorey
c,
Mayri Alejandra
Dìaz De Rienzo
ad,
Eric
Déziel
*a and
Charles
Gauthier
*a
a,
Marie-Joëlle
Prévost
a,
Serge
Lavoie
b,
Thomas
Feroldi
b,
Marianne
Piochon
a,
Marie-Christine
Groleau
a,
Jean
Legault
b,
Sandra
Villaume
c,
Jérôme
Crouzet
c,
Stéphan
Dorey
c,
Mayri Alejandra
Dìaz De Rienzo
ad,
Eric
Déziel
*a and
Charles
Gauthier
*a
aCentre Armand-Frappier Santé Biotechnologie, Institut National de la Recherche Scientifique (INRS), 531, Boulevard des Prairies, Laval (Québec), H7V 1B7, Canada. E-mail: charles.gauthier@inrs.ca
bLaboratoire d'Analyse et de Séparation des Essences Végétales (LASEVE), Département des Sciences Fondamentales, Université du Québec à Chicoutimi, 555, Boulevard de l'Université, Chicoutimi (Québec), G7H 2B1, Canada
cUniversité de Reims Champagne-Ardenne, INRAE, USC RIBP 1488, SFR Condorcet-FR CNRS 3417, 51100 Reims, France
dSchool of Pharmacy and Biomolecular Sciences, Liverpool John Moores University, L3 3AF, Liverpool, UK
First published on 4th May 2021
Abstract
Rhamnolipids are a specific class of microbial surfactants, which hold great biotechnological and therapeutic potential. However, their exploitation at the industrial level is hampered because they are mainly produced by the opportunistic pathogen Pseudomonas aeruginosa. The non-human pathogenic bacterium Pantoea ananatis is an alternative producer of rhamnolipid-like metabolites containing glucose instead of rhamnose residues. Herein, we present the isolation, structural characterization, and total synthesis of ananatoside A, a 15-membered macrodilactone-containing glucolipid, and ananatoside B, its open-chain congener, from organic extracts of P. ananatis. Ananatoside A was synthesized through three alternative pathways involving either an intramolecular glycosylation, a chemical macrolactonization or a direct enzymatic transformation from ananatoside B. A series of diasteroisomerically pure (1→2), (1→3), and (1→4)-macrolactonized rhamnolipids were also synthesized through intramolecular glycosylation and their anomeric configurations as well as ring conformations were solved using molecular modeling in tandem with NMR studies. We show that ananatoside B is a more potent surfactant than its macrolide counterpart. We present evidence that macrolactonization of rhamnolipids enhances their cytotoxic and hemolytic potential, pointing towards a mechanism involving the formation of pores into the lipidic cell membrane. Lastly, we demonstrate that ananatoside A and ananatoside B as well as synthetic macrolactonized rhamnolipids can be perceived by the plant immune system, and that this sensing is more pronounced for a macrolide featuring a rhamnose moiety in its native 1C4 conformation. Altogether our results suggest that macrolactonization of glycolipids can dramatically interfere with their surfactant properties and biological activity.
Introduction
Bacteria represent a rich reservoir of structurally diverse glycosylated metabolites.2 Among these compounds, microbial glycolipids show considerable potential for biomedical and biotechnological applications.3 Microbial glycolipids are surfactants, i.e., amphiphilic surface-active compounds, which are made by the combination of a lipidic chain covalently linked to a carbohydrate moiety. Because of their ability to form pores and destabilize biological membranes, microbial glycolipids have attracted increased attention as therapeutic agents.4 Glycolipids exhibit a wide range of pharmaceutical activities, including antibacterial,5 antifungal,6 antiviral,7 hemolytic,8 anticancer,9 and adjuvant10 activities. In addition, surfactants of microbial origin are increasingly considered for use in diverse biotechnological applications such as in the food and cosmetic industries as well as in bioremediation technologies.3Rhamnolipids are a specific class of microbial biosurfactants that have been intensively investigated in recent years.11 Structurally, rhamnolipids are α-configured mono- or di-L-rhamnose residue(s) O-linked to an (R)-β-hydroxyalkanoic acid dilipidic chain of C6 to C14 carbon length (see RhaC10C103 in Fig. 1). As compared to the commercially available petroleum-derived synthetic surfactants, the biodegradability, low critical micelle concentrations (CMCs), and high tension surface activity make rhamnolipids exquisite environmental alternatives for industrial applications.12 Rhamnolipids are also of interest because they exhibit a plethora of intriguing pharmacological activities. They inhibit the formation of microbial biofilms,13–15 which play crucial role in infections caused by many pathogenic bacteria. They exhibit antimicrobial activities against both Gram-positive and Gram-negative bacteria as well as against plant pathogenic fungi.12 Because of their ability to interact with biological membranes, rhamnolipids can trigger the death of cancer cells16 and induce hemolysis of red blood cells.17,18 Furthermore, as invasion pattern molecules (also known as elicitors),19 rhamnolipids can stimulate the plant immune system resulting in the strengthening of plant cell walls along with the production of antimicrobial compounds.20–23
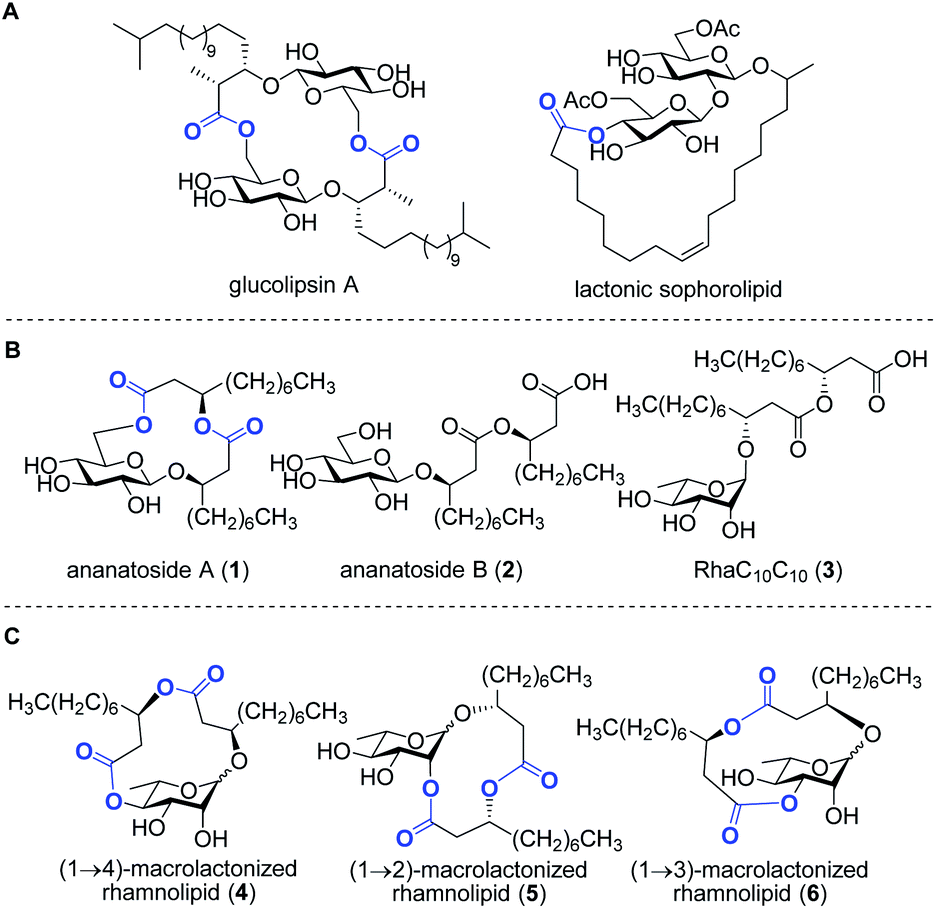 | ||
| Fig. 1 Examples of macrolactone-containing glucolipids produced by microbes (A); structures of target ananatoside A (1) and ananatoside B (2) produced by Pantoea ananatis along with the related RhaC10C10 (3) (B); structures of target macrodilactone-containing rhamnolipids 4–6 (C). Lactone functionalities are highlighted in blue. | ||
Rhamnolipids are mainly produced by pathogenic Gram-negative bacteria belonging to the Pseudomonas and Burkholderia genera,11,24 which hampers their exploitation at the industrial level since several are human pathogens. As such, there is an increased interest to identify non-pathogenic bacteria that can produce high concentrations of rhamnolipids and structurally-related biosurfactants. Our group has recently identified the bacterium Pantoea ananatis BRT175 as an alternative non-pathogenic producer of biosurfactants.25,26 The biosynthesis of rhamnolipids involves the successive function of three enzymes: RhlA, which directs the biosynthesis of the lipidic precursor, along with RhlB and RhlC, which are two rhamnosyltransferases.27,28 Although the production of biosurfactants in P. ananatis is also catalyzed by RhlA and RhlB homologues, we unexpectedly identified a gluco-rather than a rhamnolipid from P. ananatis ethyl acetate extracts.25 The isolated compound, that we called ananatoside A (1, Fig. 1), features an unprecedented 15-membered macrodilactone ring comprising a β-D-glucose residue linked to a C10C10 dilipid chain through both C1 and C6 positions.26 Biologically active glucolipids containing mono-, di-, and trilactones of different macrocyle sizes have been identified from other microorganisms29–31 including, to name a few examples, lactonic sophorolipids (Fig. 1), glucolipsins (Fig. 1), cycloviracins, fattiviracins, macroviracins, and anthrobacilins. By restricting the rotation of the substituents and stabilizing the active conformation of the glycolipid, the presence of the macrocycle is a prerequisite to the bioactivity of these constrained saccharides.30,32
The identification of ananatoside A (1) as a novel microbial macrolide prompted us to investigate P. ananatis extracts with the aim of identifying structurally similar biosurfactants. As part of our research program on the synthesis of microbial glycans,33–36 we are interested in developing synthetic routes that would allow an alternative and straightforward access to these macrodilactone-containing glycolipids, and enable the assessment of their tensioactive properties and biological activities. Within this framework, we herein present the total synthesis of ananatoside A (1), its newly identified open-chain congener ananatoside B (2), the related RhaC10C10 (3) as well as five unprecedented, anomerically pure (1→2)-, (1→3)-, and (1→4)-macrodilactone-containing rhamnolipids (4–6) (Fig. 1). We show that ananatoside A (1) can be efficiently obtained via three different pathways implying either a chemo- or enzymatic macrolactonization, or an intramolecular glycosylation as the key steps of these synthetic sequences. DFT calculations were used to decipher the ring conformations and anomeric configurations of synthetic macrolactonized rhamnolipids 4–6, which were readily obtained through intramolecular glycosylation. Their surfactant properties and biological activity, i.e., antimicrobial activity, cytotoxicity, hemolytic activity, as well as their interaction with the plant immune system, were also investigated, providing meaningful fundamental insights into the impact of the presence of the macrodilactonic ring on the physical and biological properties of this relevant class of microbial glycolipids.
Results and discussion
Isolation of ananatoside A (1) and ananatoside B (2)
The non-pathogenic bacterium P. ananatis BRT175 was found to be a producer of surface-active glycolipids based on its genetic homologies with rhamnolipid biosynthetic genes. The microbial biosurfactants were extracted with EtOAc from the supernatant of a liquid culture of BRT175, which was acidified to pH ∼3 prior the extraction. Purification of the crude extract was performed using a semi-preparative reversed-phase HPLC system equipped with a charged aerosol detector (CAD).37 The use of the CAD detector was particularly appealing over traditional UV/visible detectors as it allows the detection of molecules lacking chromophore groups such as glycolipids (see Fig. S1†). Through this optimized procedure, ananatoside A (1) was isolated as a yellow oil and its physical and analytical data (Rf, specific rotation, HRMS, and 1D and 2D NMR) were in perfect agreement with those we recently published.26 A more polar and major congener (see Fig. S1†), that we have named ananatoside B (2), was also isolated from the EtOAc extract as a white amorphous powder. This compound was analyzed by HR-ESI-TOF-MS in the positive mode, yielding pseudomolecular ion peaks at m/z 538.3587 [M + NH4]+ and m/z 543.3142 [M + Na]+, pointing towards the presence of a glycolipid featuring a hexose residue linked to two hydroxydecanoic acid chains. Comparison of the 1D and 2D NMR data of ananatoside B (2, see Fig. S2† for main COSY and HMBC correlations) with our previously reported data for ananatoside A (1) suggested the presence of an open-chain β-linked glucolipid congener without the macrolide ring (no HMBC cross-peak between H6 and C1′′). The absolute configuration of both the glucose and β-hydroxydecanoic acid chains were determined following acid hydrolysis and measurement of specific rotation in comparison with an authentic D-glucose sample and literature data for the lipid chain, as we previously described.26 Based on these chemical and spectroscopic evidences, we established the complete structure of ananatoside B (2) as shown in Fig. 1. The structural determination of ananatoside A (1) and ananatoside B (2) were further confirmed through total synthesis as described in the next section.Total synthesis of ananatoside A (1) and ananatoside B (2)
Along these lines and as depicted in our retrosynthetic analysis strategy (Fig. 2), we envisioned to build the macrolide skeleton of ananatoside A (1) via three parallel synthetic pathways (routes A–C) as to maximize the chances of efficiently reaching our target. We first hypothesized that ananatoside A (1) would be formed through the intramolecular glycosylation of derivative 7 following chemoselective cleavage of the C6-O-TBS group (route A). Notably, precursor 7, activated in the form of an STol glycoside,45 would be equipped with a (2-azidomethyl)benzoyl (AZMB)46 group at C2 that would act as a neighboring participating group enabling the formation of the 1,2-trans-glucosidic linkage. The β-selectivity would also be favored by the steric constraint exerted by the resulting macrolide.44 The propensity of the AZMB group to be orthogonally cleaved by means of, for instance, Staudinger reduction36,47 without affecting the macrolide functionality represents a further advantage of using this protecting group at this specific position. Precursor 7 would be readily synthesized by Steglich esterification48 between thioglucoside 9 and dilipid 10.
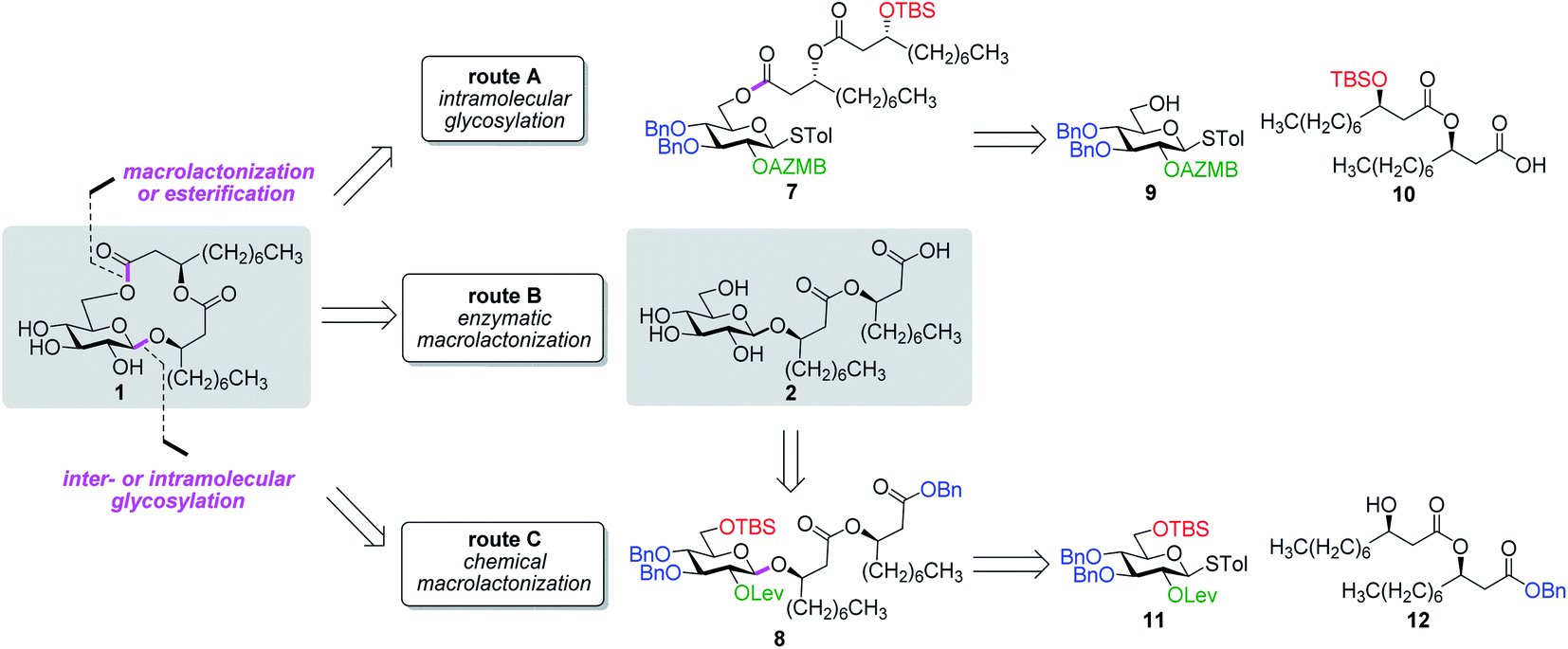 | ||
| Fig. 2 Retrosynthetic disconnection strategy for the total synthesis of ananatoside A (1) and ananatoside B (2) according to three different pathways: chemical macrolactonization, enzymatic macrolactonization, and inter- or intramolecular glycosylation. AZMB: 2-azidomethylbenzoyl; Bn: benzyl; Lev: levulinoyl; STol: thiotolyl; TBS: tert-butyldimethylsilyl. Blue: permanent protecting groups (Bn); red: temporary protecting groups (TBS); green: esters used as temporary protecting groups enabling neighboring group participation (AZMB and Lev). | ||
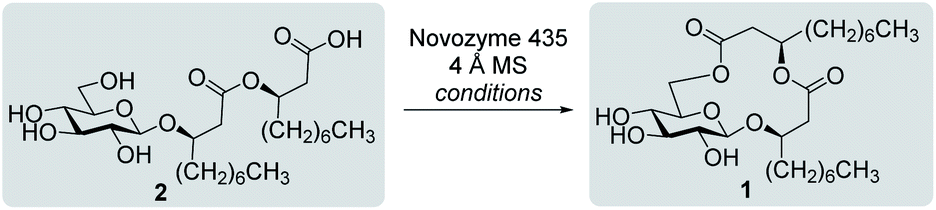
Inspired by the success story of the total synthesis of several macrolide-containing natural products29,38,49–52 and by way of comparison with the intramolecular glycosylation strategy, we also wanted to construct the 15-membered ring of ananatoside A (1) through both enzymatic and chemical macrolactonizations (Fig. 2). Regarding the chemical macrolactonization (route C), glucolipid 8 would act as an exquisite acyclic precursor for this intramolecular transformation following unmasking of the seco acid functionality. Moreover, global deprotection of this compound (8) would complete the total synthesis of ananatoside B (2). Glucolipid 8 would be prepared through the stereocontrolled glycosylation of thioglucoside 11 with benzylated dilipid 12. Here again, the use of a neighboring participating group, i.e., levulinoyl (Lev),36,47,53 that could be selectively cleaved in the presence of an ester functionality would insure a successful outcome for the final steps of the synthetic route. Finally, we hypothesized that ananatoside A (1) could be enzymatically synthesized from unprotected ananatoside B (2) using commercially available solid-supported lipase such as Novozyme 435 (route B).41 If successful, this enzymatic process would allow the direct and straightforward conversion of ananatoside B (2) into ananatoside A (1) from the isolated natural product or the synthetic compound.
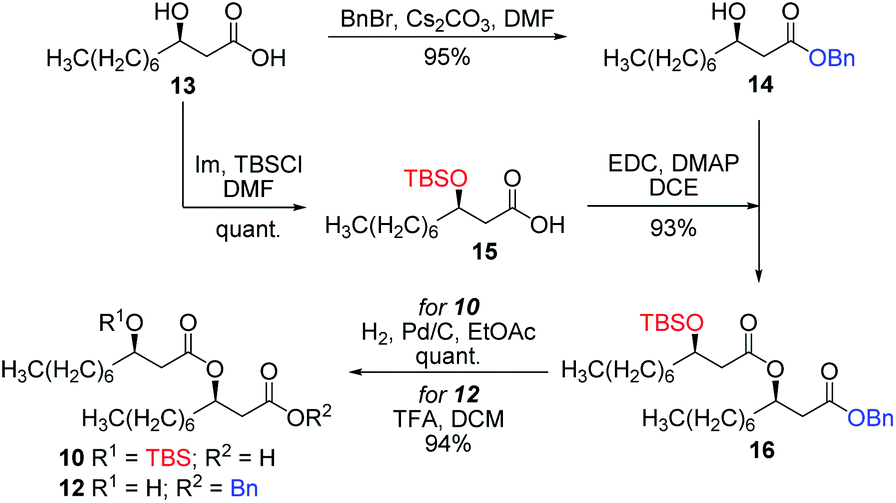 | ||
| Scheme 1 Synthesis of β-hydroxydecanoic acid derivatives. | ||
Having completed the synthesis of the dilipid derivatives, we then focused our attention on the synthesis of thioglucosides 9 and 11, which were either used for subsequent Steglich esterification or glycosylation reactions, respectively. These two compounds were prepared from diol 17,60 which was obtained in a three-step, one-pot sequence from the corresponding pertrimethylsilylated thioglucoside. As revealed in Scheme 2, regioselective silylation at the C6 position of diol 17 followed by esterification with AZMBOH and subsequent desilylation under the action of in situ generated HCl led to derivative 9 bearing a free OH at C6 in 74% yield over three steps. Alternatively, a levulinoyl group was installed at C2 by treatment of the C6-O-TBS-protected derivative with levulinic anhydride to provide fully protected thioglucoside 11 in 90% yield over two steps.
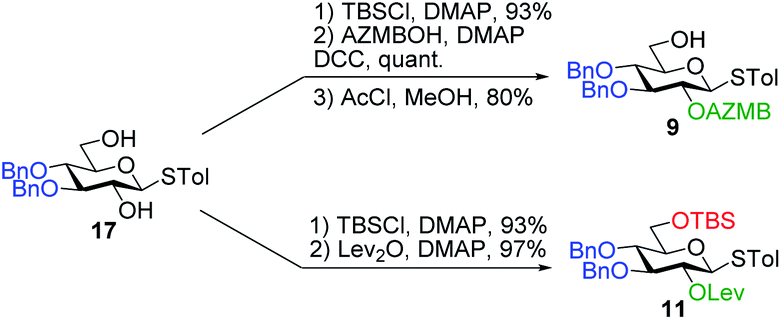 | ||
| Scheme 2 Synthesis of thioglucoside derivatives 9 and 11. | ||
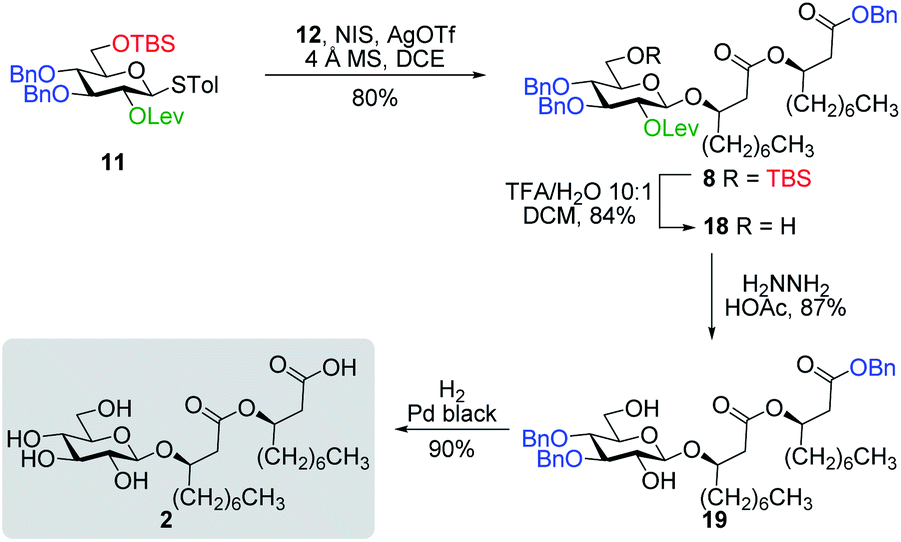 | ||
| Scheme 3 Total synthesis of ananatoside B. | ||
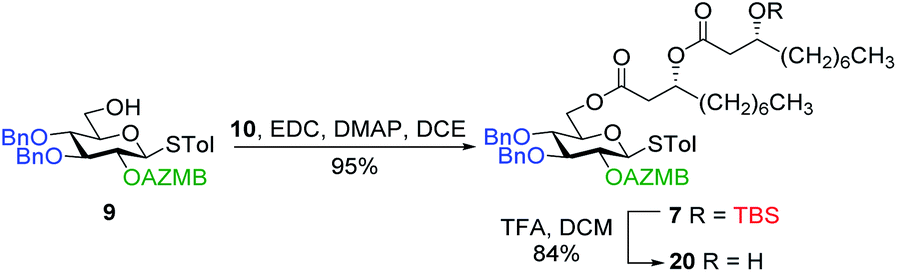 | ||
| Scheme 4 Synthesis of alcohol 20 ready for intramolecular glycosylation. | ||
As shown in Table 1, different conditions were screened for the intramolecular glycosylation of acyclic precursor 20, i.e., promoter, molar volume, and additive. Initially, the concentration was set to 0.01 M (corresponding molar volume of 100 mL mmol−1) to minimize intermolecular interactions and thus preventing the formation of di- and oligomeric byproducts. At this concentration, AgOTf and TMSOTf were first evaluated as catalysts in combination with NIS (entries 1 and 2).61 In both conditions, target macrolide 21 was isolated in good yields with full β-stereoselectivity while no traces of diolide or higher oligomers were detected. Encouraged by these results, the impact of the concentration of precursor 20 on the reaction outcome was investigated by conducting the intramolecular glycosylation at 0.001 and 0.1 M (entries 3 and 4). We were pleased to find that macrolide 21 was formed quantitatively when the concentration of the acyclic precursor was set to 0.001 M. Moreover, even at a concentration of 0.1 M, which could have favored intermolecular interactions, only monomer 21 was isolated although the yield was significantly lower (47%). The use of excess amounts of TMSOTf (entry 5) or other triflates such as Yb(OTf)3 and Zn(OTf)2 (entries 6 and 7, respectively) also resulted in the exclusive formation of target macrolide 21. Inspired by Fürstner and co-workers50 who completed the total synthesis of cycloviracin B1 through metal-templated macrolactonization, we then sought to evaluate the impact of an excess of metallic ions on the outcome of the glycosylation and to observe if oligomeric macrolides could be generated. As it is known that potassium and sodium are effective ions for such templated reactions,50 KOTf and NaOTf were employed as additives in the presence of catalytic amounts of TMSOTf (entries 8–10). Once again, the intramolecular reaction was highly favored as no other products than macrolide 21 were isolated from the reaction mixture.
|
|
||||
|---|---|---|---|---|
| Entry | Triflate (equiv.) | Molar volume (mL mmol−1) | Additive | Yielda (%) |
| a Isolated yield. | ||||
| 1 | AgOTf (0.4) | 100 | — | 70 |
| 2 | TMSOTf (0.2) | 100 | — | 86 |
| 3 | TMSOTf (0.2) | 1000 | — | Quant. |
| 4 | TMSOTf (0.2) | 10 | — | 47 |
| 5 | TMSOTf (2.1) | 100 | — | 85 |
| 6 | Yb(OTf)3 (1.6) | 100 | — | 21 |
| 7 | Zn(OTf)2 (1.6) | 100 | — | 75 |
| 8 | TMSOTf (0.2) | 100 | NaOTf | 78 |
| 9 | TMSOTf (0.2) | 100 | KOTf | 63 |
| 10 | TMSOTf (0.2) | 50 | KOTf | 61 |
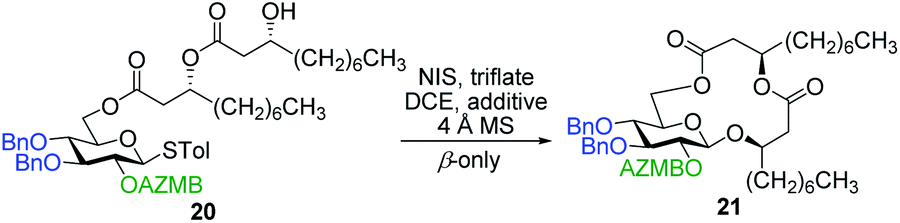

With protected macrolides 21 and 23 in hand, we were able to complete the total synthesis of ananatoside A (1) (Scheme 5). Therefore, the AZMB group of macrolide 21 and the Lev group of macrolide 23 were orthogonally removed through Staudinger reduction or treatment with hydrazine acetate, respectively. Pd-catalyzed hydrogenolysis of the resulting alcohols cleanly led to the formation of ananatoside A (1) in 56 or 90% yield over two steps from macrolide 21 or 23, respectively. Physical and analytical data (Rf, specific rotation, HRMS, and NMR) of synthetic ananatoside A (1) were in perfect agreement with the isolated natural product26 (see ESI† for details). The overall yields and number of steps for both synthetic sequences were very similar, i.e., 33 and 35% over eight steps from known diol 17 for the intramolecular glycosylation and macrolactonization pathways, respectively.
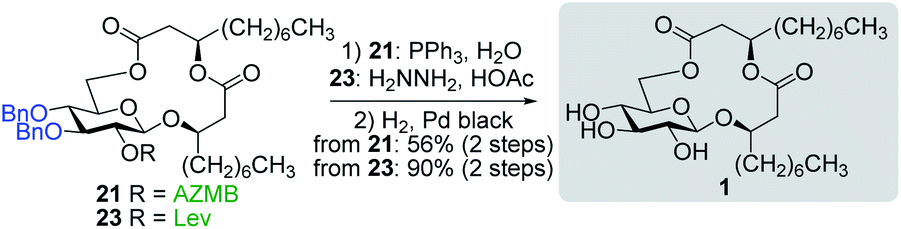 | ||
| Scheme 5 Deprotection of macrolides 21 and 23 into ananatoside A (1). | ||
Synthesis of RhaC10C10 (3) and macrolactonized rhamnolipids 4–6
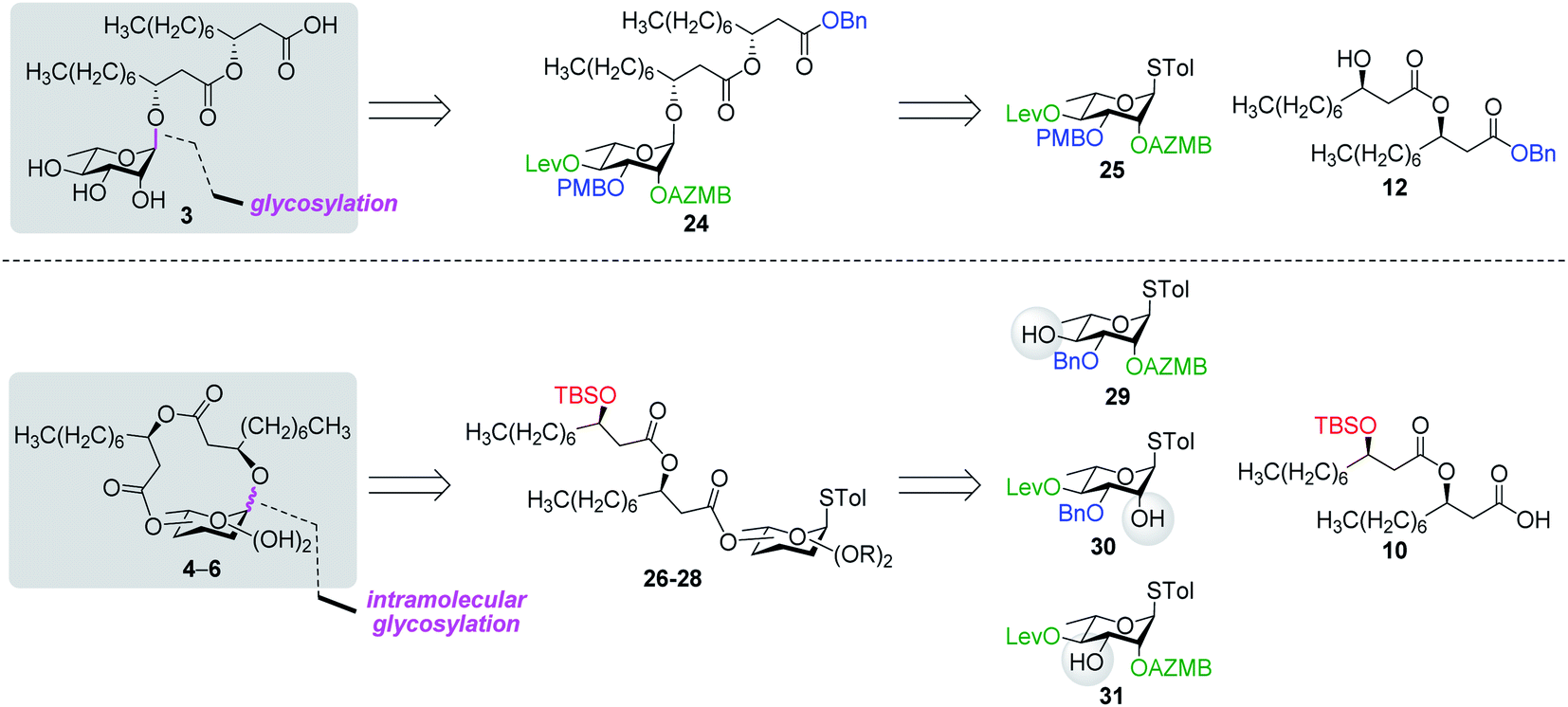 | ||
| Fig. 3 Retrosynthetic analysis of rhamnolipid 3 and macrodilactone-containing rhamnolipids 4–6. PMB: para-methoxybenzyl. Blue: permanent protecting groups (Bn and PMB); red: temporary protecting groups (TBS); green: esters used as temporary protecting groups enabling neighboring group participation when branched at C2 (AZMB and Lev). | ||
 | ||
| Scheme 6 Synthesis of thiorhamnoside building blocks 25, 29, 30, and 31. | ||
 | ||
| Scheme 7 Total synthesis of RhaC10C10 (3). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :85). Following cleavage of both the Lev and Bn groups, α- and β-(1→2)-macrolides, i.e., compounds 5α and 5β, respectively, were isolated in pure forms in a combined yield of 83%. Intramolecular glycosylation of precursor 38 also generated a mixture of anomers although this time they were easily separable by silica gel chromatography under their protected forms (39β, 24%, β-anomer; and 39α, 39%, α-anomer). Orthogonal cleavage of both the AZMB and Lev groups completed the total synthesis of target (1→3)-macrolide 6 in pure α- and β-anomeric forms. The determination of the anomeric configurations in macrolides 4, 5β, 5α, 6β, and 6α was accomplished by NMR with the help of DFT calculations as described in the next section.
:85). Following cleavage of both the Lev and Bn groups, α- and β-(1→2)-macrolides, i.e., compounds 5α and 5β, respectively, were isolated in pure forms in a combined yield of 83%. Intramolecular glycosylation of precursor 38 also generated a mixture of anomers although this time they were easily separable by silica gel chromatography under their protected forms (39β, 24%, β-anomer; and 39α, 39%, α-anomer). Orthogonal cleavage of both the AZMB and Lev groups completed the total synthesis of target (1→3)-macrolide 6 in pure α- and β-anomeric forms. The determination of the anomeric configurations in macrolides 4, 5β, 5α, 6β, and 6α was accomplished by NMR with the help of DFT calculations as described in the next section.
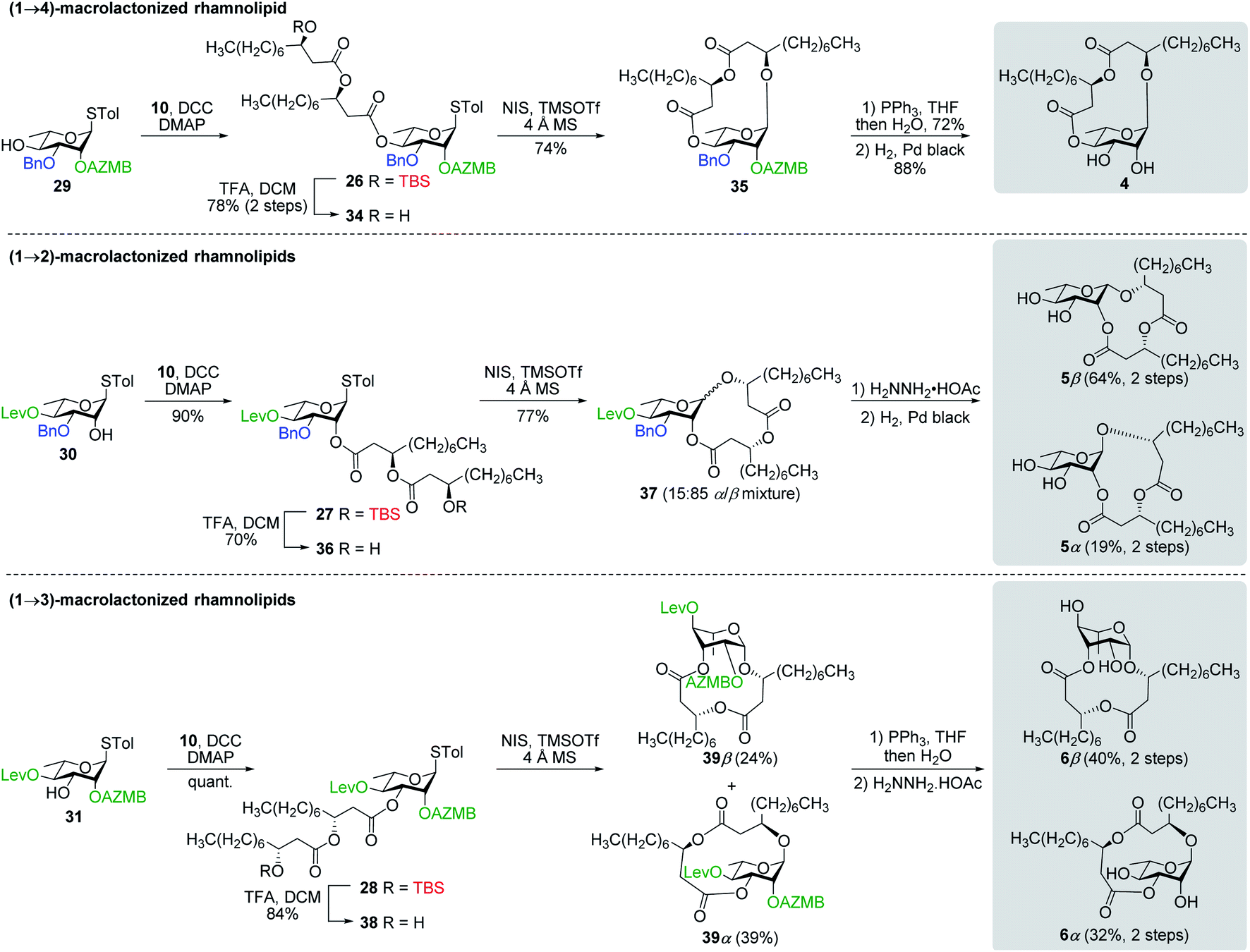 | ||
| Scheme 8 Synthesis of (1→4), (1→2), and (1→3)-macrolactonized rhamnolipids 4–6. | ||
Molecular modeling of macrolactonized rhamnolipids
To determine the anomeric configuration of a monosaccharide, the 3JH1,H2 coupling constant is commonly measured by 1H NMR.70 In the case of rhamnopyranosides, the J values for the two anomers are too low to be a suitable selection criterion. Therefore, the δC at C5 (70.0 ppm for α-Rhap or 73.2 ppm for β-Rhap)71 or the 1JC1,H1 coupling constant (167.2–172.3 Hz for α-Rhap or 152.3–159.8 Hz for β-Rhap) are preferred for rhamnopyranosides.72 These methods are valid only because rhamnopyranosides usually adopt a typical 1C4 conformation. However, in the case of macrolactonized rhamnolipids 4–6, other conformations are likely to appear due to the steric constraints imposed by the bicyclic nature of these compounds, rendering useless these empirical rules for identifying the proper anomer. To overcome this problem, in silico models of synthetic rhamnolipids were prepared with truncated alkyl chains as to minimize the number of conformers to be considered (Fig. S4†). The conformational space of these models was explored in depth (Fig. S5†) using the improved RDKit algorithm, which is based on the geometry of distances and empirical preferences for certain angles of torsion (ETKDGv2).73 The geometry of the unique conformers was optimized by molecular mechanics (MMFF94) followed by quantum molecular modeling using density functional theory (mPW1PW91) and the basis set 6-31G(d, p). The thermochemical parameters were calculated to deduce the abundance of conformers in solution using the Boltzmann equation, thus eliminating marginal conformers (<1%). The retained conformers were inspected so that no imaginary vibrational frequency could be found. Examination of the sugar conformation of the conserved structures confirmed the irregular nature of these glycosides. Indeed, three different skew boat (1S3 for 4, 2S0 for 5α, and 5S1 for 6β) along with two chair conformations (4C1 for 6α and 1C4 for 5β) were found for the main conformers of the modelized compounds (Fig. 4). The shielding tensors were calculated with the same level of theory and then converted to chemical shifts using the multiple reference method.74,75 First, the means of the absolute values of the errors (MAE) were calculated between each synthetic molecules 4–6 and the corresponding in silico anomer pairs 40–42 (Table S1†). In most cases, it was possible to assign the α- and β-anomers to a single model, but the case of compound 5β was found to be ambiguous at the B97-2/pVTZ level of theory. Indeed, when only the 1H NMR data were considered, we found that 5α and 5β would both be of α-configuration. To ensure the validity of these assignments, the pooled comparison method proposed by Lauro et al.76 was performed for the 5α/5β and 6α/6β anomeric pairs (Table S1†). In this way, the anomeric configuration of each synthetic macrolactone was unambiguously assigned as depicted in Scheme 8. | ||
| Fig. 4 3D Structure of the macrolactonized rhamnolipids models 40–42. Only the most stable structure of each conformer is depicted. Pie chart gives Boltzmann population for standard six-membered ring1 for the three levels of theory used in this study (from top to bottom: mPW1PW91/6-31G(d,p), mPW1PW91/6-311+G(d,p) and B97-2/pVTZ). | ||
Surfactant properties of natural glycolipids
Biosurfactants are molecules that can associate with each other leading to the formation of micelles. They can also interact with surfaces between phases of different polarity such as the air/water or oil/water interface.77 To gain quantitative insights into the surface-active properties of microbial glycolipids, the surface tensions at CMC (γCMC) of ananatoside A (1) and ananatoside B (2) were measured using a Fisher tensiometer by means of the du Noüy ring method. The CMC, referring to the concentration of surfactant at which micelles start to form, was determined from a plot of surface tension again each concentration. These data were compared to those obtained for synthetic RhaC10C10 (3). Due to the presence of a free carboxylic acid moiety in biosurfactants 2 and 3, the surface tensions were measured at neutral pH, which corresponds to the presence of their anionic forms in solution based on an estimated pKa of 5.5 for RhaC10C10 (3).78 As shown in Table 4, the CMC value for ananatoside B (2) was similar to the one measured for rhamnolipid 3, denoting that ananatoside B (2) can be considered a potent biosurfactant. In the case of ananatoside A (1), the measured CMC value was even lower than for RhaC10C10 (3). However, the latter value must be taken with cautious as the presence of the macrolactone functionality decreased considerably the water solubility of ananatoside A (1) as compared to its open-chain congener. Consequently, the surface tension measurements were performed quickly to avoid the precipitation of ananatoside A (1) into the aqueous medium. It is interesting to note that the surface tension values measured for biosurfactants 2 and 3 were on the same order of magnitude than those measured by Pemberton and co-workers59 for structurally similar synthetic rhamnolipids bearing C10C10 lipid chains.| Glycolipida | CMCb (μM) | γ CMC (nM m−1) | Z-Averageb,c (nm) |
|---|---|---|---|
| a Synthetic samples. b Data taken at pH 7.0. c Hydrodynamic diameter of micelles measured with a Zetasizer instrument at concentrations of 80 μM for ananatoside B (2) and RhaC10C10 (3), and 100 μM for ananatoside A (1). d Size distribution showed the formation of aggregated particles. | |||
| 1 | 43 ± 2 | 37 ± 1 | >800d |
| 2 | 63 ± 2 | 28 ± 1 | 99.0 |
| 3 | 58 ± 1 | 30 ± 1 | 92.1 |
In parallel to these experiments, we took advantage of dynamic light scattering (DLS) to evaluate the hydrodynamic diameter of biosurfactant micelles formed at concentrations above the CMC (Table 4).79 DLS considers the intensity of light scattered by spherical particles, which prevents its application for highly polydisperse and non-spherical colloidal systems. Using a Zetasizer instrument, the results showed that the Z-average, i.e., the mean diameter of the micelles, was in the same range for ananatoside B (2) and for rhamnolipid 3 (see Fig. S6†). Once again, due to its hydrophobicity, ananatoside A (1) was too polydisperse for conducting a proper distribution analysis, showing a Z-average value above 800 nm, which meant that the particles were aggregated in water at the tested concentration. Similar results were obtained for ananatoside A (1) when the DLS experiments were conducted in DMSO as the solvent. These results showed that ananatoside B (2), similarly to rhamnolipid 3, formed monodisperse colloidal particles at concentrations above its CMC.
The propensity of surfactants to stabilize emulsions shows promising for biotechnological applications such as bioremediation of contaminated soils and water.77 Therefore, the emulsifying properties of microbial biosurfactants 1–3 were evaluated against light as well as heavy hydrocarbon contaminants such as cyclohexane and kerosene, respectively (see Fig. S7†). The emulsifying activity results showed that ananatoside B (2) and RhaC10C10 (3) were equally able to generate emulsions with cyclohexane and kerosene while ananatoside A (1) was unable to form any emulsion, mainly because of its lack of affinity for water. Altogether, these results once again highlighted that ananatoside B (2), but not its macrolide counterpart 1, exhibits potent surfactant properties that could be advantageously used for biotechnological applications.
Antimicrobial activity, cytotoxicity, and hemolytic activity of natural and synthetic glycolipids
As previously mentioned in the introduction section, numerous studies have reported the antimicrobial potential of rhamnolipids against several pathogenic bacteria and fungi.12 However, as rhamnolipids were usually tested as mixtures of different congeners and not as pure synthetic compounds, there seems to be no clear consensus regarding the antimicrobial activity of one specific congener. Hence, as an initial investigation toward their biological functions in bacteria, the in vitro antimicrobial activity of ananatoside A (1), ananatoside B (2), RhaC10C10 (3), and macrolides 4–6 was evaluated against Gram-negative, i.e., Pseudomonas aeruginosa PA14, Pseudomonas aeruginosa LESB58, and Escherichia coli DH5α, and Gram-positive bacteria, i.e., Staphylococcus aureus MRSA, Staphylococcus aureus Newman, and Bacillus subtilis PY79, as well as against fungi, i.e., Candida albicans ATCC 10231 and Candida albicans LSPQ 0199. At the maximum tested concentration (37.5 μg mL−1), glycolipids 1–6 were unable to inhibit the growth of these microbes. These results suggest that the biosynthetic production of ananatoside A (1) and ananatoside B (2) by P. ananatis is not aimed for antimicrobial purposes.As the anticancer potential of microbial rhamnolipids has previously been reported,16 we next sought to evaluate the in vitro cytotoxicity of our synthetic and natural glycolipids 1–6 against cancerous cell lines, i.e., human lung carcinoma (A549) and colorectal adenocarcinoma (DLD-1). The compounds were also tested against human normal skin fibroblasts (WS1) to estimate their selectivity towards cancer cells. As revealed in Table 5, very interesting conclusions can be derived from these cytotoxicity results. First, it appears that only macrolactones, as compared to their open forms, were able to inhibit the growth of human cell lines. Second, the cytotoxic activity of the active macrolides was not specific to cancer cell lines as comparable IC50 values were measured against healthy cells (WS1), pointing toward a general mechanism involving the formation of pores in cell membrane.16 Specifically, ananatoside A (1) was cytotoxic against human cell lines (IC50 = 50−58 μM) while its open form counterpart ananatoside B (2) was inactive at the maximum tested concentration (IC50 > 200 μM). Furthermore, macrolactonization of the inactive RhaC10C10 (3, IC50 > 200 μM) generated cytotoxic macrolides 4, 5β, and 6β (IC50 = 50–112 μM). While (1→4)-macrolactonized rhamnolipid 4 was the most active congener, only the β-anomers of (1→2)- and (1→3)-macrolactonized rhamnolipid 5β and 6β, respectively, were found to inhibit the growth of human cell lines. These results showed that both the position of the macrolactone ring and the anomeric configuration of the glycosidic bonds can impact the cytotoxicity of the rhamnolipids.
| Glycolipida | Cytotoxicity | Hemolysis | ||
|---|---|---|---|---|
| (IC50 in μM)b | (HC50 in μM)c | |||
| A549 | DLD-1 | WS1 | Erythrocytes | |
| a Synthetic samples. b Half maximal inhibitory concentration measured via the resazurin assay. Etoposide was used as a positive control80 showing IC50 values of 1.2, 27, and 34 μM against A549, DLD-1, and WS1 cell lines, respectively. c Half maximal inhibitory concentration measured on sheep blood erythrocytes. Triton X-100 was used as a positive control showing an HC50 value of 52 ± 2 μM. d No inhibition or activity at the maximum tested concentration (IC50 or HC50 > 200 μM). | ||||
| 1 | 50 ± 7 | 58 ± 2 | 52 ± 4 | 12.3 ± 0.3 |
| 2 | >200d | >200d | >200d | >200d |
| 3 | >200d | >200d | >200d | 65 ± 6 |
| 4 | 56 ± 2 | 70 ± 10 | 57 ± 1 | 12.0 ± 0.7 |
| 5β | 103 ± 7 | 110 ± 2 | 105 ± 5 | 14 ± 2 |
| 5α | >200d | >200d | >200d | >200d |
| 6β | 112 ± 2 | 112 ± 3 | 110 ± 1 | 12.3 ± 0.5 |
| 6α | >200d | >200d | >200d | >200d |
Owing to their surfactant properties, glycolipids can break the membrane of red blood cells causing hemolytic activity.18 To understand if the cytotoxicity of macrolactones 1, 4, 5β, and 6β was due to their surface tension activity, we evaluated the hemolytic potential of synthetic glycolipids 1–6 against sheep erythrocytes (Table 5). As anticipated, all the cytotoxic glycolipids were shown to exhibit hemolytic activities on red blood cells with HC50 ranging from 12 to 14 μM. The non-cytotoxic rhamnolipid 3 was also found to exert a weak hemolytic activity (HC50 = 65 ± 6 μM). Altogether, these data suggest a non-specific mechanism of cytotoxicity involving the intercalation of macrolactones 1, 4, 5β, and 6β into the lipid constituents of the cell membranes.
Interaction of natural and synthetic glycolipids with the plant immune system
Bacterial rhamnolipids, such as compound 3, trigger the plant immune system, which can ultimately lead to plant protection against diseases caused by fungal and bacterial pathogens.23 As ananatoside A (1) and ananatoside B (2) share structural similarities with rhamnolipid 3 and because they have been isolated from P. ananatis cultures, an emerging plant pathogen causing important agricultural damage worldwide,81 we wanted to investigate whether these bacterial glycolipids, along with our unprecedented series of synthetic rhamnolactones, could be perceived by the plant immune system. We relied on measuring the extracellular content of reactive oxygen species (ROS) produced by tomato plants (Solanum lycopersicum) following treatment with the synthetic compounds, as an early and important marker of plant immunity.20,22,82 Tomato leaves were challenged with synthetic glycolipids 1–6 at concentrations of 100 μM and the production of ROS was monitored over a 720 min time frame (Fig. 5). As expected, rhamnolipid 3, used as a positive control in this study, triggered a long-lasting and strong production of ROS in tomato leaves.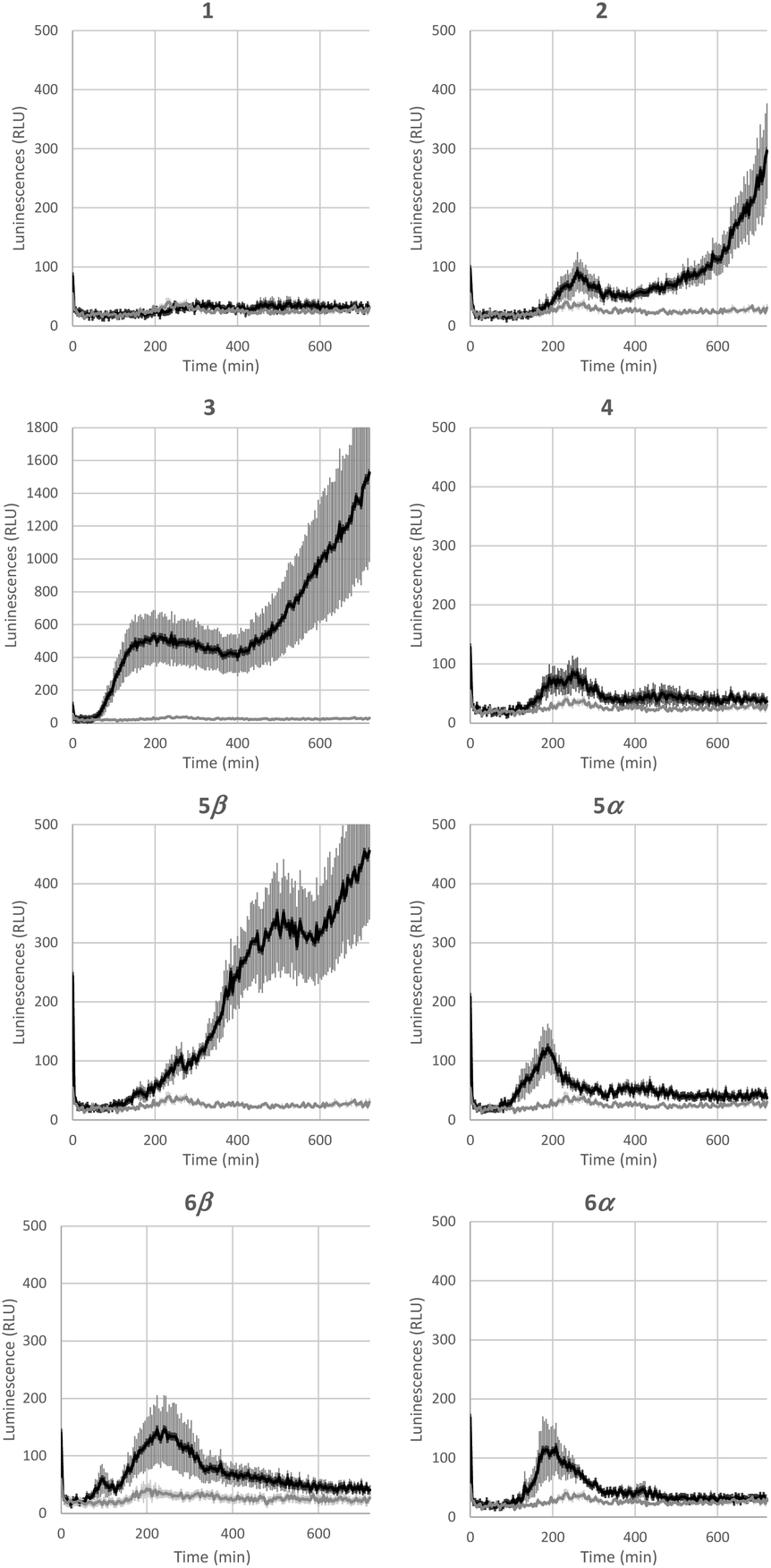 | ||
| Fig. 5 Extracellular reactive oxygen species (ROS) production following treatment of tomato leaf disks with ananatoside A (1), ananatoside B (2), RhaC10C10 (3), and related macrodilactone-containing rhamnolipids (4–6). Production of ROS was measured in tomato leaf disks following treatment at 100 μM with synthetic glycolipids (1–6). MeOH (0.5%) was used as a control. ROS production was measured using the chemiluminescence of luminol and photon counts were expressed as relative luminescence units (RLUs). Data are mean ± SEM (n = 6). Experiments were independently realized three times with similar results. | ||
Although less strong in amplitude, a similar pattern of ROS production was measured for ananatoside B (2), but not for ananatoside A (1), its macrolactonic counterpart. These responses seem to be however plant-specific because, when tested in Arabidopsis thaliana, both natural glycolipids 1 and 2 were perceived by plants (see Fig. S8†). As for rhamnolactones 4–6, all of them were sensed by tomatoes: the luminescence curves showed a small burst at 200 min followed by a return to basal levels over the next 200 min. The only exception to this general trend was compound 5β. Indeed (1→2)-macrolactonized rhamnolipid 5β induced a strong and long-lasting production of ROS following the initial burst at 200 min. This response was stereospecific as corresponding α-anomer 5α did not show a similar sensing pattern. Interestingly, our molecular modeling showed that macrolide 5β was the only one to be found in the native 1C4 conformation for the rhamnose ring (see Fig. 4), which could be part of the explanation for these intriguing results. Taken together, these results highlight that (1), glycolipids sharing the same 3-hydroxyalkanoate dilipidic chain than rhamnolipids but bearing a glucose instead of a rhamnose moiety can be perceived by plants; and (2) macrolactonization of these glycolipids alter the sensing patterns according to the plant species as well as the conformation of the rhamnopyranose ring.
Conclusions
In summary, we have isolated and structurally characterized rhamnolipid-like ananatoside A (1) and ananatoside B (2) from an organic extract of P. ananatis, a non-human pathogenic producer of biosurfactants. We have accomplished, for the first time, the total synthesis of ananatoside A (1) and ananatoside B (2), confirming the structure of these bacterial glucolipids. The macrodilactone-containing ananatoside A (1) was efficiently synthesized according to three alternative pathways: (1) via the stereoselective intramolecular glycosylation of a thioglucoside donor; (2) via the chemical macrolactonization of a seco acid precursor; and (3) via the direct enzymatic macrolactonization of its open form congener ananatoside B (2) using a solid-supported lipase. Capitalizing on the expeditious intramolecular glycosylation of orthogonally protected thiorhamnoside donors, we have accomplished the synthesis of diastereoisomerically pure (1→2), (1→3), and (1→4)-macrolactonized rhamnolipids. We have determined the anomeric configuration of these unprecedented macrolides through molecular modeling and found that the rhamnose ring adopts unusual conformations including skew boat and flipped chair conformations. Determination of the surfactant properties of bacterial glucolipids 1 and 2 in comparison with rhamnolipid 3 has revealed that ananatoside B (2), but not its macrolide counterpart 1, represents a potent biosurfactant, as shown by its efficient emulsifying activity. Furthermore, we have shown that synthetic glycolipids 1–6 do not exhibit any significant antimicrobial activity against Gram-negative and Gram-positive bacteria as well as against fungi. We have also highlighted that the presence of the macrodilactone functionality can convert non-cytotoxic glycolipids into cytotoxic ones according to the position and anomeric configuration of the macrolides. We have revealed a direct correlation between the cytotoxicity and the hemolytic activity of these macrolides pointing towards a mechanism involving the formation of pores into the lipidic cell membrane. Finally, we have demonstrated that natural glucolipids 1 and 2 as well as unnatural macrolides 4–6 can be perceived by the plant immune system, and that this sensing was long-lasting for macrolide 5β featuring a rhamnose moiety in its native 1C4 conformation. Altogether our results suggest that macrolactonization of glycolipids can dramatically interfere with their surfactant properties and biological activity. Our interdisciplinary study could serve as a foundation for the rational design of rhamnolipid-like biosurfactants with improved properties for biotechnological and/or therapeutic applications.Author contributions
M. C. and M.-J. P. synthesized the compounds. M. P., S. L., and C. G. performed the isolation and structural elucidation of ananatosides. S. L. and T. F. conducted the DFT calculations. M.-C. G. performed the antimicrobial and hemolytic assays. J. L. performed the cytotoxicity assays. S. V., J. C., and S. D. assessed the compounds against the plant immune system. M. A. D. D. R. conducted the surfactant assays. All authors analysed and discussed the results. M. C., S. L., M. P., E. D., and C. G. wrote the manuscript. E. D and C. G. secured funding for this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This article is dedicated to the memory of Pr. Éric Marsault, University of Sherbrooke, who passed away in early 2021. This work was supported by Discovery grants from the Natural Sciences and Engineering Research Council of Canada (NSERC) under award number RGPIN-2016-04950 (to C. G.) and RGPIN-2020-06771 (to E. D.). C. G. was supported by a Fonds de recherche du Québec – Santé (FRQS) Research Scholars Junior 2 Career Award. S. V., J. C., and S. D. thank the SFR Condorcet FR CNRS 3417 (project P3IA) and French MESRI for financial support. M. A. D. D. R. thank the Microbiology Society (Round 1 Research Visit Grant 2019) and Liverpool John Moores University (ECR Fellowships 2018–2019) for travel funds. M. C. thanks NSERC and Fonds de recherche du Québec – Nature et technologies (FRQNT) for M.Sc. and Ph.D. scholarships. M. J. P. thanks NSERC for an undergraduate scholarship. We thank Sylvain Milot, INRS, for valuable discussions.Notes and references
- A. Bérces, D. M. Whitfield and T. Nukada, Tetrahedron, 2001, 57, 477–491 CrossRef.
- S. I. Elshahawi, K. A. Shaaban, M. K. Kharel and J. S. Thorson, Chem. Soc. Rev., 2015, 44, 7591–7697 RSC.
- I. Mnif and D. Ghribi, J. Sci. Food Agric., 2016, 96, 4310–4320 CrossRef CAS.
- M. Inès and G. Dhouha, Carbohydr. Res., 2015, 416, 59–69 CrossRef PubMed.
- P. Saravanakumari and K. Mani, Bioresour. Technol., 2010, 101, 8851–8854 CrossRef CAS PubMed.
- R. Sha, L. Jiang, Q. Meng, G. Zhang and Z. Song, J. Basic Microbiol., 2012, 52, 458–466 CrossRef CAS PubMed.
- V. Shah, G. F. Doncel, T. Seyoum, K. M. Eaton, I. Zalenskaya, R. Hagver, A. Azim and R. Gross, Antimicrob. Agents Chemother., 2005, 49, 4093–4100 CrossRef CAS PubMed.
- S. Hewald, K. Josephs and M. Bölker, Appl. Environ. Microbiol., 2005, 71, 3033–3040 CrossRef CAS.
- J. Chen, X. Song, H. Zhang and Y. Qu, Enzyme Microb. Technol., 2006, 39, 501–506 CrossRef CAS.
- V. M. F. Lima, V. L. D. Bonato, K. M. Lima, S. A. Dos Santos, R. R. Dos Santos, E. D. C. Gonçalves, L. H. Faccioli, I. T. Brandao, J. M. Rodrigues-Junior and C. L. Silva, Infect. Immun., 2001, 69, 5305–5312 CrossRef CAS PubMed.
- A. Abdel-Mawgoud, F. Lépine and E. Déziel, Appl. Microbiol. Biotechnol., 2010, 86, 1323–1336 CrossRef CAS PubMed.
- J. Chen, Q. Wu, Y. Hua, J. Chen, H. Zhang and H. Wang, Appl. Microbiol. Biotechnol., 2017, 101, 8309–8319 CrossRef CAS PubMed.
- H. Zheng, N. Singh, G. S. Shetye, Y. Jin, D. Li and Y.-Y. Luk, Bioorg. Med. Chem., 2017, 25, 1830–1838 CrossRef CAS PubMed.
- I. Aleksic, M. Petkovic, M. Jovanovic, D. Milivojevic, B. Vasiljevic, J. Nikodinovic-Runic and L. Senerovic, Front. Microbiol., 2017, 8, 2454 CrossRef PubMed.
- A. Nickzad and E. Déziel, Lett. Appl. Microbiol., 2014, 58, 447–453 CrossRef CAS.
- L. Jiang, C. Shen, X. Long, G. Zhang and Q. Meng, Appl. Microbiol. Biotechnol., 2014, 98, 10187–10196 CrossRef CAS PubMed.
- G. G. F. Leite, J. V. Figueirôa, T. C. M. Almeida, J. L. Valões, W. F. Marques, M. D. D. C. Duarte and K. Gorlach-Lira, Biotechnol. Prog., 2016, 32, 262–270 CrossRef CAS PubMed.
- M. Manaargadoo-Catin, A. Ali-Cherif, J.-L. Pougnas and C. Perrin, Adv. Colloid Interface Sci., 2016, 228, 1–16 CrossRef CAS PubMed.
- M. Zhou and W. Wang, Front. Plant Sci., 2018, 9, 1613 CrossRef PubMed.
- W. P. Luzuriaga-Loaiza, R. Schellenberger, Y. De Gaetano, F. O. Akong, S. A. Villaume, J. Crouzet, A. Haudrechy, F. Baillieul, C. Clément, L. Lins, F. Allais, M. Ongena, S. Bouquillon, M. Deleu and S. Dorey, Sci. Rep., 2018, 8, 8534 CrossRef PubMed.
- A. Kutschera, C. Dawid, N. Gisch, C. Schmid, L. Raasch, T. Gerster, M. Schäffer, E. Smakowska-Luzan, Y. Belkhadir, A. C. Vlot, C. E. Chandler, R. Schellenberger, D. Schwudke, R. K. Ernst, S. Dorey, R. Hückelhoven, T. Hofmann and S. Ranf, Science, 2019, 364, 178–181 CAS.
- M. Robineau, S. Le Guenic, L. Sanchez, L. Chaveriat, V. Lequart, N. Joly, M. Calonne, C. Jacquard, S. Declerck, P. Martin, S. Dorey and E. A. Barka, Molecules, 2020, 25, 3108 CrossRef CAS.
- J. Crouzet, A. Arguelles-Arias, S. Dhondt-Cordelier, S. Cordelier, J. Pršic, G. Hoff, F. Mazeyrat-Gourbeyre, F. Baillieul, C. Clément, M. Ongena and S. Dorey, Front. Bioeng. Biotechnol., 2020, 8, 1014 CrossRef.
- G. Soberon-Chavez, F. Lépine and E. Déziel, Appl. Microbiol. Biotechnol., 2005, 68, 718–725 CrossRef CAS PubMed.
- D. D. N. Smith, A. Nickzad, E. Déziel and J. Stavrinides, mSphere, 2016, 1, e00075-15 CrossRef PubMed.
- C. Gauthier, S. Lavoie, M. Piochon, S. Martinez, S. Milot and E. Déziel, Carbohydr. Res., 2019, 471, 13–18 CrossRef CAS PubMed.
- U. A. Ochsner, A. Fiechter and J. Reiser, J. Biol. Chem., 1994, 269, 19787–19795 CrossRef CAS.
- R. Rahim, U. A. Ochsner, C. Olvera, M. Graninger, P. Messner, J. S. Lam and G. Soberon-Chavez, Mol. Microbiol., 2001, 40, 708–718 CrossRef CAS PubMed.
- J. Xie and N. Bogliotti, Chem. Rev., 2014, 114, 7678–7739 CrossRef CAS PubMed.
- J. Rodriguez, S. O'Neill and M. A. Walczak, Nat. Prod. Rep., 2018, 35, 220–229 RSC.
- A. M. Abdel-Mawgoud and G. Stephanopoulos, Synth. Syst. Biotechnol., 2018, 3, 3–19 CrossRef PubMed.
- R. Pereda-Miranda and M. Bah, Curr. Top. Med. Chem., 2003, 3, 111–131 CrossRef CAS PubMed.
- M. Tamigney Kenfack, Y. Blériot and C. Gauthier, J. Org. Chem., 2014, 79, 4615–4634 CrossRef CAS PubMed.
- A. Laroussarie, B. Barycza, H. Andriamboavonjy, M. Tamigney Kenfack, Y. Blériot and C. Gauthier, J. Org. Chem., 2015, 80, 10386–10396 CrossRef CAS PubMed.
- M. Tamigney Kenfack, M. Mazur, T. Nualnoi, T. L. Shaffer, A. Ngassimou, Y. Blériot, J. Marrot, R. Marchetti, K. Sintiprungrat, N. Chantratita, A. Silipo, A. Molinaro, D. P. AuCoin, M. N. Burtnick, P. J. Brett and C. Gauthier, Nat. Commun., 2017, 8, 115 CrossRef PubMed.
- M. Cloutier, E. Delar, K. Muru, S. Ndong, R. R. Hoyeck, T. Kaewarpai, N. Chantratita, M. N. Burtnick, P. J. Brett and C. Gauthier, Org. Biomol. Chem., 2019, 17, 8878–8901 RSC.
- T. Vehovec and A. Obreza, J. Chromatogr. A, 2010, 1217, 1549–1556 CrossRef CAS PubMed.
- A. Fürstner, Eur. J. Org. Chem., 2004, 943–958 CrossRef.
- K. S. Bisht, S. Bhatt and K. Muppalla, Tetrahedron Lett., 2006, 47, 8645–8649 CrossRef CAS.
- C. Gagnon, É. Godin, C. Minozzi, J. Sosoe, C. Pochet and S. K. Collins, Science, 2020, 367, 917–921 CrossRef CAS PubMed.
- C. Ortiz, M. L. Ferreira, O. Barbosa, J. C. S. dos Santos, R. C. Rodrigues, A. Berenguer-Murcia, L. E. Briand and R. Fernandez-Lafuente, Catal. Sci. Technol., 2019, 9, 2380–2420 RSC.
- R. Das and B. Mukhopadhyay, ChemistryOpen, 2016, 5, 401–433 CrossRef CAS PubMed.
- Y. Yang, X. Zhang and B. Yu, Nat. Prod. Rep., 2015, 32, 1331–1355 RSC.
- X. G. Jia and A. V. Demchenko, Beilstein J. Org. Chem., 2017, 13, 2028–2048 CrossRef CAS.
- G. Lian, X. Zhang and B. Yu, Carbohydr. Res., 2014, 403, 13–22 CrossRef PubMed.
- T. Wada, A. Ohkubo, A. Mochizuki and M. Sekine, Tetrahedron Lett., 2001, 42, 1069–1072 CrossRef CAS.
- C. Gauthier, P. Chassagne, F.-X. Theillet, C. Guerreiro, F. Thouron, F. Nato, M. Delepierre, P. J. Sansonetti, A. Phalipon and L. A. Mulard, Org. Biomol. Chem., 2014, 12, 4218–4232 RSC.
- B. Neises and W. Steglich, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef.
- A. Parenty, X. Moreau and J. M. Campagne, Chem. Rev., 2006, 106, 911–939 CrossRef CAS PubMed.
- A. Fürstner, M. Albert, J. Mlynarski, M. Matheu and E. DeClercq, J. Am. Chem. Soc., 2003, 125, 13132–13142 CrossRef PubMed.
- S.-Y. Zhu, J.-S. Huang, S.-S. Zheng, K. Zhu and J.-S. Yang, Org. Lett., 2013, 15, 4154–4157 CrossRef CAS PubMed.
- J. R. Mateus-Ruiz and A. Cordero-Vargas, J. Org. Chem., 2019, 84, 11848–11855 CrossRef CAS.
- A. Hassner, G. Strand, M. Rubinstein and A. Patchornik, J. Am. Chem. Soc., 1975, 97, 1614–1615 CrossRef CAS.
- B. Menhour, P. Mayon, K. Plé, S. Bouquillon, S. Dorey, C. Clément, M. Deleu and A. Haudrechy, Tetrahedron Lett., 2015, 56, 1159–1161 CrossRef CAS.
- A. A. Compton, B. S. Deodhar, A. Fathi and J. E. Pemberton, ACS Sustainable Chem. Eng., 2020, 8, 8918–8927 CrossRef CAS.
- J. Bauer, K. Brandenburg, U. Zähringer and J. Rademann, Chem.–Eur. J., 2006, 12, 7116–7124 CrossRef CAS PubMed.
- M. De Vleeschouwer, D. Sinnaeve, J. Van den Begin, T. Coenye, J. C. Martins and A. Madder, Chem.–Eur. J., 2014, 20, 7766–7775 CrossRef CAS PubMed.
- C. Coss, T. Carrocci, R. M. Maier, J. E. Pemberton and R. Polt, Helv. Chim. Acta, 2012, 95, 2652–2659 CrossRef CAS.
- R. P. Pacheco, R. J. Eismin, C. S. Coss, H. Wang, R. M. Maier, R. Polt and J. E. Pemberton, J. Am. Chem. Soc., 2017, 139, 5125–5132 CrossRef PubMed.
- Y.-P. Hu, S.-Y. Lin, C.-Y. Huang, M. Zulueta, J.-Y. Liu, W. Chang and S.-C. Hung, Nat. Chem., 2011, 3, 557–563 CrossRef CAS PubMed.
- P. Konradsson, U. E. Udodong and B. Fraser-Reid, Tetrahedron Lett., 1990, 31, 4313–4316 CrossRef CAS.
- A. M. Felix, E. P. Heimer, T. J. Lambros, C. Tzougraki and J. Meienhofer, J. Org. Chem., 1978, 43, 4194–4196 CrossRef CAS.
- J. S. Bajwa, Tetrahedron Lett., 1992, 33, 2299–2302 CrossRef CAS.
- E. P. Boden and G. E. Keck, J. Org. Chem., 1985, 50, 2394–2395 CrossRef CAS.
- T. Fujisawa, T. Mori, K. Fukumoto and T. Sato, Chem. Lett., 1982, 11, 1891–1894 CrossRef.
- P. Westerduin, P. E. De Haan, M. J. Dees and J. H. van Boom, Carbohydr. Res., 1988, 180, 195–205 CrossRef CAS.
- H. I. Duynstee, M. J. van Vliet, G. A. van der Marel and J. H. van Boom, Eur. J. Org. Chem., 1998, 303–307 CrossRef CAS.
- S. David and S. Hanessian, Tetrahedron, 1985, 41, 643–663 CrossRef CAS.
- I. Cumpstey, A. J. Fairbanks and A. J. Redgrave, Tetrahedron, 2004, 60, 9061–9074 CrossRef CAS.
- J. Ø. Duus, C. H. Gotfredsen and K. Bock, Chem. Rev., 2000, 100, 4589–4614 CrossRef CAS PubMed.
- L. Feng, S. N. Senchenkova, W. Wang, A. S. Shashkov, B. Liu, S. D. Shevelev, D. Liu, Y. A. Knirel and L. Wang, Gene, 2005, 355, 79–86 CrossRef CAS PubMed.
- K. Bock and C. Pedersen, J. Chem. Soc., Perkin Trans., 1974, 2, 293–297 RSC.
- S. Wang, J. Witek, G. A. Landrum and S. Riniker, J. Chem. Inf. Model., 2020, 60, 2044–2058 CrossRef CAS PubMed.
- A. M. Sarotti and S. C. Pellegrinet, J. Org. Chem., 2009, 74, 7254–7260 CrossRef CAS PubMed.
- A. M. Sarotti and S. C. Pellegrinet, J. Org. Chem., 2012, 77, 6059–6065 CrossRef CAS PubMed.
- G. Lauro, P. Das, R. Riccio, D. S. Reddy and G. Bifulco, J. Org. Chem., 2020, 85, 3297–3306 CrossRef CAS PubMed.
- A. Daverey and K. Pakshirajan, in Microbes and Microbial Technology, ed. I. Ahmad, F. Ahmad and J. Pichtel, Springer, New York, NY, 2011 Search PubMed.
- A. Lebrón-Paler, J. E. Pemberton, B. A. Becker, W. H. Otto, C. K. Larive and R. M. Maier, Anal. Chem., 2006, 78, 7649–7658 CrossRef PubMed.
- H. Zhong, L. Yang, X. Yang, G. Zeng, Z. Liu, Y. Liu and X. Yuan, RSC Adv., 2015, 5, 88578–88582 RSC.
- C. Gauthier, J. Legault, S. Lavoie, S. Rondeau, S. Tremblay and A. Pichette, J. Nat. Prod., 2009, 72, 72–81 CrossRef CAS PubMed.
- T. A. Coutinho and S. N. Venter, Mol. Plant Pathol., 2009, 10, 325–335 CrossRef CAS PubMed.
- J. Qi, J. Wang, Z. Gong and J. M. Zhou, Curr. Opin. Plant Biol., 2017, 38, 92–100 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc01146do |
| This journal is © The Royal Society of Chemistry 2021 |