
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOlefin metathesis: what have we learned about homogeneous and heterogeneous catalysts from surface organometallic chemistry?†
Christophe
Copéret‡
 *a,
Zachariah J.
Berkson‡
a,
Ka Wing
Chan‡§
a,
Jordan
de Jesus Silva‡
a,
Christopher P.
Gordon‡
a,
Margherita
Pucino‡
a and
Pavel A.
Zhizhko‡
b
*a,
Zachariah J.
Berkson‡
a,
Ka Wing
Chan‡§
a,
Jordan
de Jesus Silva‡
a,
Christopher P.
Gordon‡
a,
Margherita
Pucino‡
a and
Pavel A.
Zhizhko‡
b
aETH Zürich, Department of Chemistry and Applied Biosciences, Vladimir Prelog Weg 2, CH-8093 Zürich, Switzerland. E-mail: ccoperet@ethz.ch
bA. N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, Vavilov Str. 28, 119991 Moscow, Russia
First published on 9th February 2021
Abstract
Since its early days, olefin metathesis has been in the focus of scientific discussions and technology development. While heterogeneous olefin metathesis catalysts based on supported group 6 metal oxides have been used for decades in the petrochemical industry, detailed mechanistic studies and the development of molecular organometallic chemistry have led to the development of robust and widely used homogeneous catalysts based on well-defined alkylidenes that have found applications for the synthesis of fine and bulk chemicals and are also used in the polymer industry. The development of the chemistry of high-oxidation group 5–7 alkylidenes and the use of surface organometallic chemistry (SOMC) principles unlocked the preparation of so-called well-defined supported olefin metathesis catalysts. The high activity and stability (often superior to their molecular analogues) and molecular-level characterisation of these systems, that were first reported in 2001, opened the possibility for the first direct structure–activity relationships for supported metathesis catalysts. This review describes first the history of SOMC in the field of olefin metathesis, and then focuses on what has happened since 2007, the date of our last comprehensive reviews in this field.
1 Historical views on SOMC in metathesis: from early days until 2007
Olefin metathesis was discovered more than half a century ago1–3 and is today a key enabling technology applied in the petrochemical as well as the fine chemical and polymer industries.4,5 Some of the catalysts discovered in the sixties are still used today.5–10 For instance, the Olefin Conversion Technology (OCT) process, which converts ethene and butene to propene at ca. 400 °C, still relies on the original silica-supported tungsten oxide (WO3/SiO2).2,6,9–12 Molybdenum catalysts supported on alumina (or silica–alumina) can operate at significantly lower temperatures (50–250 °C) and have been developed to convert liquid olefins in processes such as the Shell Higher Olefin Process (SHOP).5,6,9,10,13,14 Alumina-supported rhenium oxides (Re2O7/Al2O3) and related systems can operate at room temperature without pre-activation but have only been developed in pilot plants due to their relatively fast deactivation, sensitivity to poisons, and the price and volatility of Re oxides making regeneration problematic.5,6,9,10,15,16 After years of research, today we still have very little understanding regarding the structure of the active surface sites in these catalysts and the mechanisms of their formation under reaction conditions.In parallel, numerous ill-defined homogenous catalysts based on group 6 metal chlorides (e.g. MoCl5 and WCl6) and organometallic activators (e.g. AlEt3 and SnMe4) have been developed.17–19 One may note that earlier reports demonstrated that Ti could promote metathesis polymerization.20,21 These catalysts are still used today, mostly in the polymer industry.5,6,10,22 Following the proposal of Chauvin (1971)23 that metal carbenes (alkylidenes) and metallacyclobutanes are the reaction intermediates in these transformations, a tremendous research effort of organometallic chemists has eventually led to the development of highly active and selective well-defined homogeneous olefin metathesis catalysts (Fig. 1) that have revolutionized the way complex molecules and polymers are constructed today. These catalysts can be divided in two main classes: (i) the d0 high-oxidation-state metal alkylidenes, mostly based on Mo and W, and (ii) the Ru alkylidenes.4,24,25
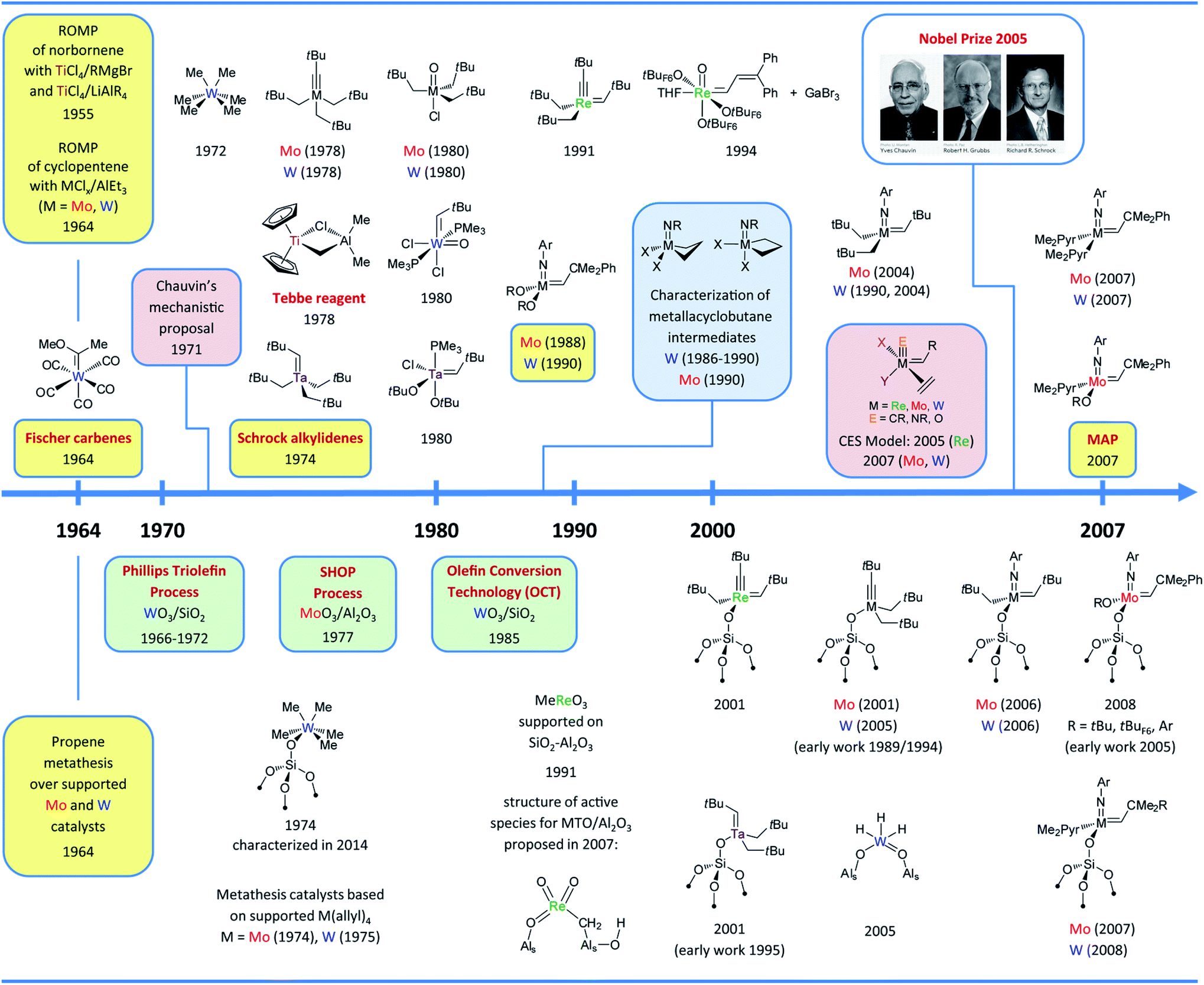 | ||
| Fig. 1 Timeline of the development of molecular (upper part) and supported (bottom part) group 6–7 olefin metathesis catalysts (until 2007–2008). Colour codes: selected key experimental findings (yellow), selected mechanistic studies (pink), observation of metallacycle intermediates (blue), important industrial processes (green). Abbreviations: Ar = 2,6-iPr2C6H3, Me2Pyr = 2,5-dimethylpyrrolyl, tBuF3 = CMe2(CF3), tBuF6 = CMe(CF3)2, tBuF9 = C(CF3)3. The complementary time (starting in 2007/2008) can be found in Fig. 9 and the whole timeline figure (1964–2020) can be found in ESI.† | ||
The development of both molecular26–29 and surface organometallic chemistry (SOMC)30–32 in the seventies also led to the discovery of highly active supported olefin metathesis catalysts generated by grafting organometallic precursors on various supports. Among them the noteworthy examples are WMe6 supported on silica (1974),33,34 whose structure, [(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)WMe5], and thermal transformations on the surface were only elucidated recently using highly dehydroxylated silica and state of the art NMR spectroscopic techniques (2014),35 and supported Mo and W allyl complexes, [M(allyl)4] (ca. 1974–1978).36–40 All of these surface compounds probably form the active alkylidene species in situ. An alternative strategy focused on generating supported alkylidenes via protonation of molecular alkylidynes ([W(CtBu)X3], X = OtBu and CH2tBu) with surface silanols (1989–1994).41–43 Later studies showed however that, while active in olefin metathesis, the corresponding surface species in these systems were alkylidynes, [(SiO)W(CtBu)X2] (2005).44 The same applies to Mo alkylidynes (2001) that turned out to be active in both alkene and alkyne metathesis.45–47 Similarly, the silica-supported Mo imido alkylidene [(SiO)Mo(
SiO)WMe5], and thermal transformations on the surface were only elucidated recently using highly dehydroxylated silica and state of the art NMR spectroscopic techniques (2014),35 and supported Mo and W allyl complexes, [M(allyl)4] (ca. 1974–1978).36–40 All of these surface compounds probably form the active alkylidene species in situ. An alternative strategy focused on generating supported alkylidenes via protonation of molecular alkylidynes ([W(CtBu)X3], X = OtBu and CH2tBu) with surface silanols (1989–1994).41–43 Later studies showed however that, while active in olefin metathesis, the corresponding surface species in these systems were alkylidynes, [(SiO)W(CtBu)X2] (2005).44 The same applies to Mo alkylidynes (2001) that turned out to be active in both alkene and alkyne metathesis.45–47 Similarly, the silica-supported Mo imido alkylidene [(SiO)Mo(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHtBu)(NH)(CH2tBu)], the supported analogue of Schrock catalysts, has been proposed as putative intermediate formed in situ upon grafting [Mo(N)(CH2tBu)3] on silica via sublimation (1996).48,49 The first reported well-defined surface-supported alkylidene characterized at the molecular level was based on Re, [(SiO)Re(CtBu)(CHtBu)(CH2tBu)] (2001),50 and obtained by grafting [Re(CtBu)(CHtBu)(CH2tBu)2] on SiO2-700 (silica containing mostly isolated silanols as a result of partial dehydroxylation at 700 °C under vacuum). Besides being fully characterized by solid-state NMR and X-ray Absorption Fine Structure (EXAFS) spectroscopy51 complemented by the synthesis of molecular models, it showed very high activity in olefin metathesis, exceeding that of the most active molecular catalysts at the time. This unexpected activity (considering that silica-supported Re oxides are inactive) led to detailed computational studies (vide infra).52,53 It is noteworthy that the corresponding silica-supported Ta alkylidene [(SiO)Ta(CHtBu)(CH2tBu)2], also characterized in 2001 and first prepared several years earlier as a mixture of mono- and bis-grafted surface species on silica dehydroxylated at 500 °C, showed low activity,54 paralleling what was known in molecular chemistry.25,55,56 These discoveries and the development of numerous well-defined molecular alkylidene precursors have opened the way to develop in the following years several generations of highly active silica-supported metathesis catalysts based on well-defined surface alkylidenes, prepared by grafting the corresponding molecular precursors: first with the isoelectronic Mo and W imido alkyl alkylidene complexes (2006), [(SiO)M(NAr)(CHtBu)(CH2tBu)] (M = Mo57 and W58) followed by the Mo and W imido amido alkylidenes (2007), [(SiO)M(NAr)(CHtBu)(NR2)] (M = Mo with NR2 = NPh2,59 pyrrolyl,59,60 or pyrazolyl,61 and M = W with NR2 = 2,5-dimethylpyrrolyl62), as well as Mo imido alkoxo alkylidene complexes (2008), [(SiO)Mo(NAr)(CHtBu)(OR)] (R = tBu, tBuF6, 2,6-iPr2C6H3).63–65 Moving away from having a pendant alkyl to amido and later alkoxide ligands was a key to the development of highly active, selective and stable silica-supported metathesis catalysts as the former invariably showed lower selectivity and fast deactivation due to the formation of hydrides (vide infra). In many of these systems the cross-metathesis initiation products were observed and quantified, demonstrating that up to 100% of the surface sites were catalytically active. The above summary briefly reviews the field of olefin metathesis from an SOMC perspective until ca. 2007–2008 and is depicted in Fig. 1.66 This summary focuses on early transition metal catalysts in relation to the corresponding supported metal oxides (Mo, W and Re); Ru-based catalysts are not discussed because SOMC had so far only very little influence on their understanding and development.67
CHtBu)(NH)(CH2tBu)], the supported analogue of Schrock catalysts, has been proposed as putative intermediate formed in situ upon grafting [Mo(N)(CH2tBu)3] on silica via sublimation (1996).48,49 The first reported well-defined surface-supported alkylidene characterized at the molecular level was based on Re, [(SiO)Re(CtBu)(CHtBu)(CH2tBu)] (2001),50 and obtained by grafting [Re(CtBu)(CHtBu)(CH2tBu)2] on SiO2-700 (silica containing mostly isolated silanols as a result of partial dehydroxylation at 700 °C under vacuum). Besides being fully characterized by solid-state NMR and X-ray Absorption Fine Structure (EXAFS) spectroscopy51 complemented by the synthesis of molecular models, it showed very high activity in olefin metathesis, exceeding that of the most active molecular catalysts at the time. This unexpected activity (considering that silica-supported Re oxides are inactive) led to detailed computational studies (vide infra).52,53 It is noteworthy that the corresponding silica-supported Ta alkylidene [(SiO)Ta(CHtBu)(CH2tBu)2], also characterized in 2001 and first prepared several years earlier as a mixture of mono- and bis-grafted surface species on silica dehydroxylated at 500 °C, showed low activity,54 paralleling what was known in molecular chemistry.25,55,56 These discoveries and the development of numerous well-defined molecular alkylidene precursors have opened the way to develop in the following years several generations of highly active silica-supported metathesis catalysts based on well-defined surface alkylidenes, prepared by grafting the corresponding molecular precursors: first with the isoelectronic Mo and W imido alkyl alkylidene complexes (2006), [(SiO)M(NAr)(CHtBu)(CH2tBu)] (M = Mo57 and W58) followed by the Mo and W imido amido alkylidenes (2007), [(SiO)M(NAr)(CHtBu)(NR2)] (M = Mo with NR2 = NPh2,59 pyrrolyl,59,60 or pyrazolyl,61 and M = W with NR2 = 2,5-dimethylpyrrolyl62), as well as Mo imido alkoxo alkylidene complexes (2008), [(SiO)Mo(NAr)(CHtBu)(OR)] (R = tBu, tBuF6, 2,6-iPr2C6H3).63–65 Moving away from having a pendant alkyl to amido and later alkoxide ligands was a key to the development of highly active, selective and stable silica-supported metathesis catalysts as the former invariably showed lower selectivity and fast deactivation due to the formation of hydrides (vide infra). In many of these systems the cross-metathesis initiation products were observed and quantified, demonstrating that up to 100% of the surface sites were catalytically active. The above summary briefly reviews the field of olefin metathesis from an SOMC perspective until ca. 2007–2008 and is depicted in Fig. 1.66 This summary focuses on early transition metal catalysts in relation to the corresponding supported metal oxides (Mo, W and Re); Ru-based catalysts are not discussed because SOMC had so far only very little influence on their understanding and development.67
On the one hand, this summary showed how surface organometallic chemistry has helped to bridge the gap between homogeneous and heterogeneous catalysts by providing supported catalysts with unprecedented activities and fully characterized active site structures. On the other hand, these well-defined catalysts did not bare much resemblance with and seem to be unrelated to the supported industrial catalysts based on supported metal oxides, even though the attempts to shed light on the classical heterogeneous systems using SOMC approach have been undertaken since the seventies.36–40
In this review, we want to discuss first what we have learned at the molecular level from these well-defined silica-supported catalysts and how this knowledge has been used to develop new generations of molecular and supported catalysts; we also want to draw clear reactivity trends and detailed structure–activity relationships based on the molecular understanding of large libraries of supported catalysts (Section 2). Second, we would like to discuss how surface organometallic chemistry is being finally used to understand the supported metal oxides that comprise industrial catalysts (Section 3). We will then conclude by reflecting on how the field might develop in the future.
2 Bridging the gap between molecular and supported catalysts
2.1 From well-defined supported metathesis catalysts to general guideline principles and consequences for metathesis catalysts
As discussed above, the first highly active and well-defined supported olefin metathesis catalyst was [(SiO)Re(CtBu)(CHtBu)(CH2tBu)]. The reactivity of this catalyst was noteworthy, not only because the molecular precursor [Re(CtBu)(CHtBu)(CH2tBu)2] is inactive but also because its activity exceeded that of all reported heterogeneous and well-defined homogeneous catalysts known at the time, e.g. Re2O7/Al2O3 and [Mo(NAr)(CHR)(OtBuF6)2].50 The remarkable metathesis activity of such well-defined silica-supported Re alkylidenes entailed the question regarding the exact role of the surface siloxy ligand in such catalysts. A DFT-computational study examined a series of [Re(CR)(CHR)(X)(Y)] complexes and evaluated the influence of various X, Y ligand pairs (X = Y = alkyl or alkoxide vs. X = alkyl and Y = siloxide) on each elementary step comprising the metathesis pathway (Scheme 1): olefin coordination, [2 + 2]-cycloaddition, cycloreversion and olefin de-coordination (the last two ones being micro-reverse of the first two). It was found that one of the key factors driving the reactivity of these d0 catalysts was the asymmetry at the metal centre (X ≠ Y).53,68 Indeed, the presence of a strong σ-donor X ligand together with a weaker Y ligand favours olefin coordination due to the distortion of the metal site in the complex prior to olefin binding and disfavours the formation of overly stabilized metallacyclobutane intermediates, leading to an overall increase in activity.
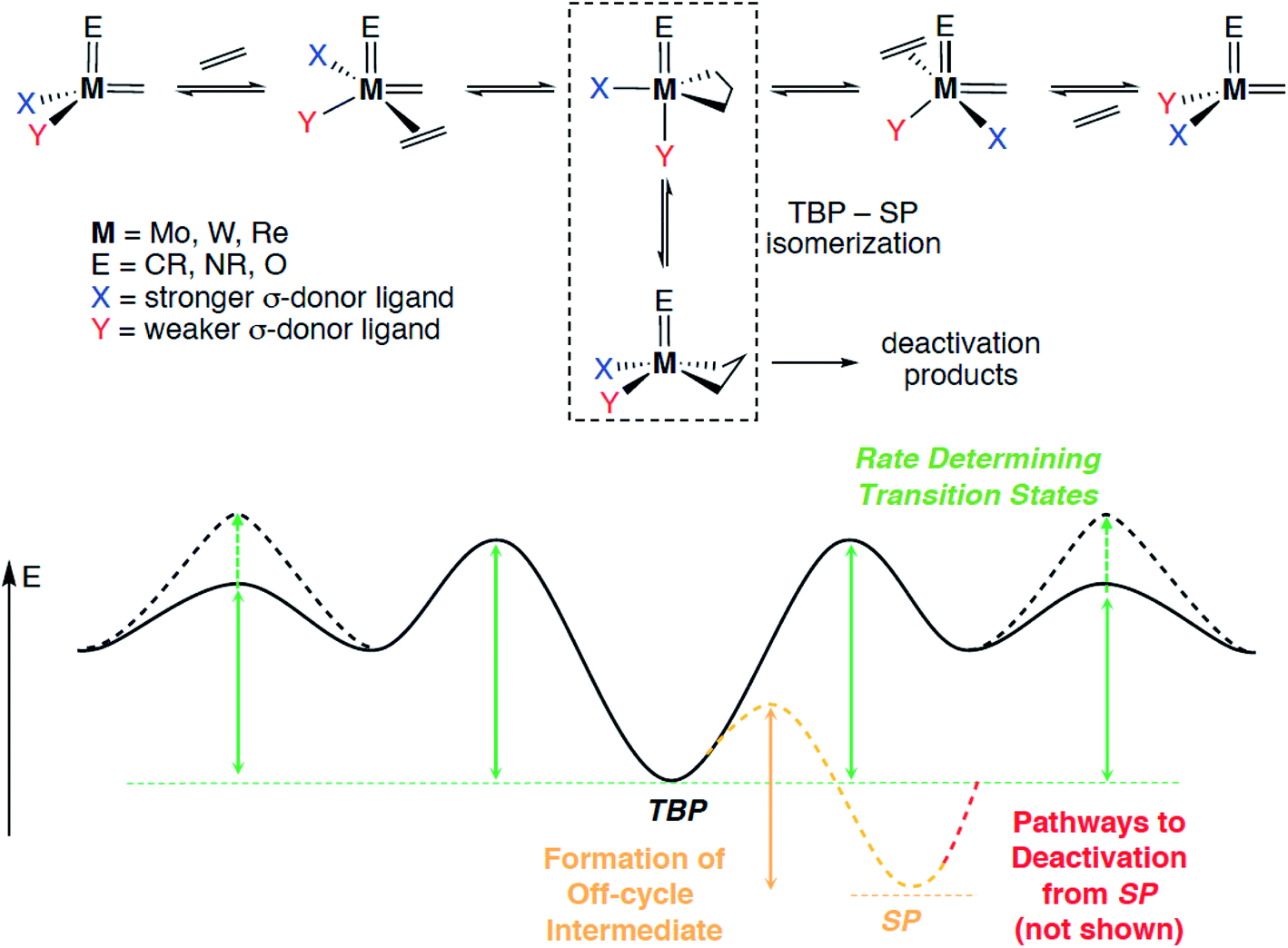 | ||
| Scheme 1 Mechanism of olefin metathesis and corresponding schematic potential energy surface, indicating the key energy barriers and reaction energies that are subject to catalyst design. | ||
More precisely, the presence of two anionic ligands of different σ-donating strength leads to a distortion from a tetrahedral structure to a (more) trigonal-pyramidal structure, in which the stronger ligand X occupies the apical position. This distortion opens a coordination site trans to X, thus favouring olefin coordination prior to [2 + 2] cycloaddition of the olefin. The [2 + 2] cycloaddition yields a metallacyclobutane intermediate with a trigonal-bipyramidal (TBP) geometry, in which the stronger σ-donor X ligand is in the equatorial plane, together with the two metal–carbon bonds of the flat metallacyclobutane. This TBP intermediate can isomerize through a turnstile process into an often more stable square-pyramidal (SP) metallacyclobutane with a puckered structure, that corresponds to an off-cycle reaction intermediate, susceptible to further catalyst deactivation (vide infra). The overall metathesis involves a retrocyclization from the TBP, that leads to an inversion of configuration at the metal centre (when X ≠ Y) for each productive metathesis cycle. One should note that in many cases the highest transition state corresponds to coordination/de-coordination and that the most stable intermediate is usually the SP off-cycle metallacyclobutane, so that the rate of metathesis is directed by the stability of the latter intermediate and the ease of the olefin coordination step. This concept, that showed the importance of dissymmetry at the metal centre, was soon after generalized to d0 Mo and W catalysts, [M(E)(CHR)(X)(Y)] with M(E) = Mo/W(imido/oxo) (Scheme 1),68,69 and shown to be a predictive guiding principle for the metathesis catalyst design not only in heterogeneous but also in homogeneous systems such as MAP,25,70–78 MAC,79,80 or NHC-stabilized cationic catalysts.81–85
Metallacyclobutane intermediates of W-based catalysts are usually more stable than those of their Mo-based congeners and are often observed; the Mo analogues are rarely reported for either homogeneous or well-defined silica-supported systems.25,86 Computational results indicate that weaker σ-donating ligands X and Y lead to more stable metallacyclobutane intermediates, and the isomerization between the two metallacyclobutane isomers (TBP and SP) via a turnstile process tends to be more facile for W as compared to Mo and for weaker σ-donor ligands.87 It is thus not surprising that most reported metallacyclobutanes of silica-supported alkylidene catalysts are based on W. Both TBP and SP W metallacyclobutanes were observed by NMR spectroscopy alongside the corresponding W alkylidene in a study of silica-grafted W imido alkylidenes.62 Destabilization of metallacyclobutanes, the most stable intermediates in the system, by introducing strongly σ-donating ligands has a major consequence on the catalyst performance because it should lower the overall energy span of the reaction. This principle has been recently exploited in designing Mo and W catalysts containing N-heterocyclic carbenes (NHC)—very strong σ-donor ligands (see Section 2.2).81–85 The effect of the ancillary X ligand on the relative stability of metallacyclobutanes derived from the reaction of such silica-supported W imido alkylidenes with ethylene has been studied in detail by solid-state NMR spectroscopy. With X = OtBu, a strong σ-donor ligand, the SP metallacyclobutane was formed exclusively. In contrast, with X = OtBuF9, a weak σ-donor ligand, the TBP/SP ratio was found to be 83/17, favouring the TBP structure. The OtBuF3 and OtBuF6 ligands fell in between these two extreme cases, showing the sensitivity of the TBP/SP ratio to the σ-donor strength of the X ligand.88 Interestingly, the only reported silica-supported molybdacyclobutane so far adopts a TBP geometry and contains a strong σ-donor NHC ligand.89 In addition to being off-cycle resting states, SP-metallacyclobutane intermediates also open catalyst deactivation pathways, in particular β-H transfer; the resulting allyl hydrides can lead to de-grafting or promote olefin isomerization.90,91 β-H transfer is particularly favoured with X = alkyl, while it is suppressed by X = OR and NR2, consistent with the higher selectivity and stability of the latter types of catalysts.91 It was also demonstrated that replacing an imido by a stronger σ-donating oxo ligand in Mo and W systems strongly disfavours β-H transfer in the SP structure,69 increasing the catalyst stability and thus explaining the generally superior performance of Mo and W oxo alkylidenes as compared to their imido analogues (see Section 2.2). These studies highlight the complexity of the olefin metathesis mechanism and the necessity to account for multiple factors that contribute to the overall catalyst efficiency, the actual performance of a given catalyst with particular structure resulting from a combination of all these features.
The reaction mechanism in Scheme 1 infers that a metallacyclobutane with TBP-structure is the key intermediate in olefin metathesis for d0 metals. Notably, Ru-based metathesis catalysts involve a similar TBP metallacyclobutane intermediate that has been observed experimentally, while the corresponding SP structure is predicted to be highly unstable in this case.92–96 It is noteworthy that all metallacyclobutanes that engage in olefin metathesis show a TBP geometry or a geometry that provides similar frontier molecular orbitals – e.g. Cp2M(C3H6) – with a flat metallacycle. In addition, all metathesis-active metallacyclobutane intermediates display similar 13C NMR chemical shift patterns, regardless of whether they are based on Ti, Ta, Mo, W, Re, or Ru. In all known cases, these metallacyclobutanes feature α- and β-carbon atoms with isotropic 13C chemical shifts of around 100 ppm and 0 ppm, respectively (Fig. 2).87,97 This stands in sharp contrast to metallacyclobutanes that do not engage in olefin metathesis, which typically show isotropic 13C chemical shifts of below 50 ppm for their α-carbons and above 30 ppm for their β-carbons. The origin of this empirical correlation was elucidated in 2017 in a detailed analysis of the chemical shift tensors of metathesis active and inactive metallacyclobutanes.98 It was pointed out that the large deshielding on the α-carbon atom (around 100 ppm) is mainly due to a remarkably large deshielding in the direction perpendicular to the metallacyclobutane plane (the direction of the most deshielded component of the 13C chemical shift tensor – δ11, see Fig. 2). Orbital analysis of the 13C chemical shift tensor shows that this specific deshielding and orientation of the tensor is linked to an electronic structure that is specific and necessary to olefin metathesis; in metathesis active metallacyclobutanes with deshielded α-carbons, there is an empty metal d-orbital that points into the plane of the metallacycle and engages in π-interactions with the α-carbon atoms. This orbital interaction introduces a π-character into the metal–carbon single bond, making the M–C bond alkylidene-like. The presence of this vacant metal d-orbital enables the retrocyclization of the metallacyclobutane to yield the alkylidene and olefin product. In other words, the presence of alkylidene character at the metallacyclobutane stage entails a low-energy pathway to alkylidene formation via retrocyclization because the electronic structures of these metathesis intermediates (i.e. metallacyclobutane and alkylidene) are quite similar.
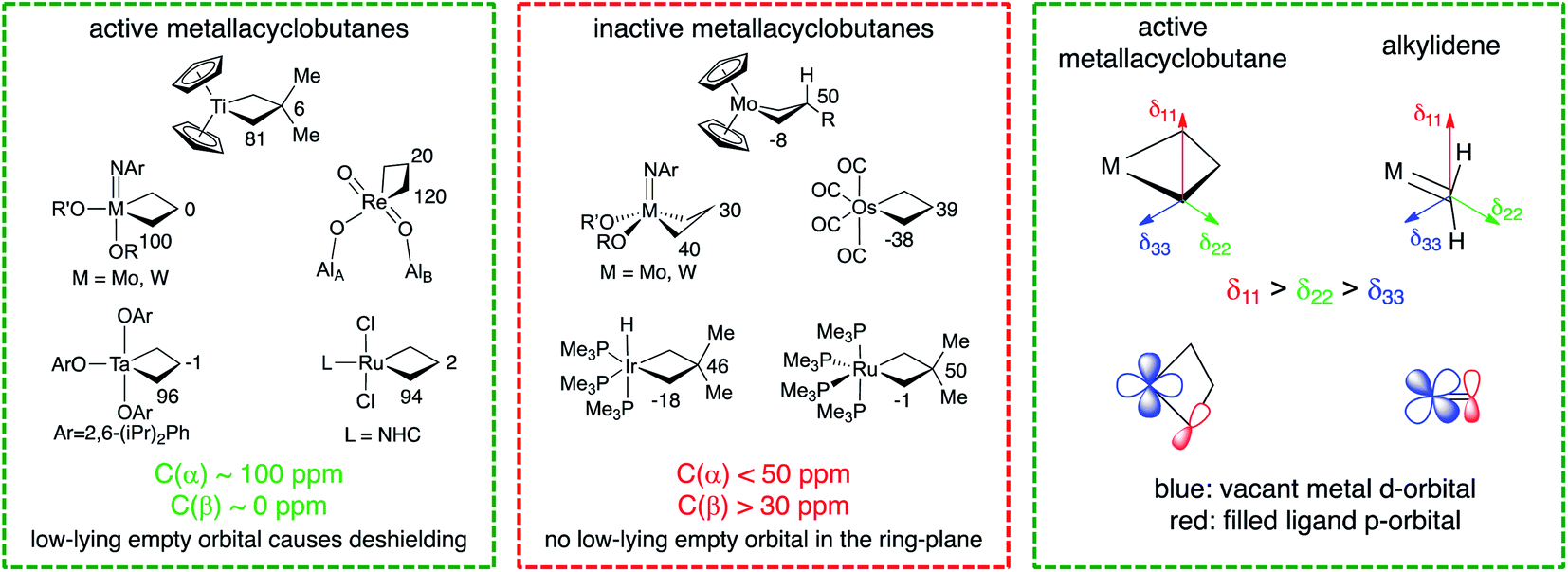 | ||
| Fig. 2 Chemical shifts of selected metallacyclobutanes and their connection to activity in olefin metathesis. | ||
The presence of a vacant metal d-orbital pointing into the metallacycle plane is hence a necessary requirement for efficient olefin metathesis catalysts. Understanding this electronic feature enables rationalization of the observation that certain metallacyclobutanes remain inactive or are not on the olefin metathesis pathway. In particular, square-pyramidal (SP) metallacyclobutanes are commonly observed with d0 catalysts based on Mo, W, or Re. In these metallacyclobutanes no empty metal d-orbital is available to engage in a π-type interaction with the α-carbon atoms. Accordingly, these carbon atoms have typically 13C shifts of 30–50 ppm and the orientation and magnitude of the 13C chemical shift tensors do not evidence residual π-bonding character in the metal–carbon bond. For these species, no low-energy pathway for cycloreversion exists, as the metal–carbon single bonds cannot easily convert into double bonds due to the absence of a suitably oriented metal d-orbital. Only upon isomerization to the TBP structure can these intermediates enter the catalytic cycle and undergo retrocyclization. Similarly, ruthenacyclobutanes with d6 electron configuration are not able to participate in olefin metathesis, in contrast to their d4 counterparts. While the d4 metallacyclobutanes feature a vacant metal d-orbital pointing into the ring, this orbital is occupied in the d6 case, impeding the occurrence of cycloreversion.98
Overall, the study of electronic and structural features of olefin metathesis catalysts reveals that (i) the electronic dissymmetry at the metal centre can be beneficial for low-energy cycloaddition/cycloreversion processes, as the geometric distortion occurring in these steps is minimized by dissymmetry-induced preorganization, and (ii) introducing strong σ-donor ligand in the coordination sphere of the metal leads to destabilization of the metallacyclobutane intermediates and thus prevents the formation of overly stable resting states. In addition, olefin metathesis catalysts need to feature a low-lying vacant metal d-orbital that points into the plane of the metallacyclobutane intermediate, usually in a TBP geometry. Only in the presence of such an orbital are efficient cycloaddition and cycloreversion possible from an electronic point of view. Olefin metathesis catalysts hence require a specific ligand field that provides precisely such electronic structure. The chemical shift of the metallacyclobutane intermediate is a key descriptor to detect the presence of such a ligand field from which the corresponding alkylidene can be derived.
2.2 Comparative performance and detailed structure–activity relationships in well-defined silica-supported alkylidenes and related systems
In particular, the SOMC approach was extensively used to investigate the catalytic performance of silica-supported 4-coordinated Schrock-type alkylidenes [M(E)(CHR)(X)(Y)] (where M = Mo or W; X, Y = anionic ligands; E = imido or oxo ligand) with the goal to understand the role of each ligand, as well as the effects of the structure of the olefin substrate. Due to the factors discussed above (high reactivity, sensitivity to poisons, interference of multiple deactivation pathways) and complexity of the olefin metathesis mechanism these systems are very sensitive to the particular reaction conditions (the use of specific substrates, purification procedures and catalytic test set-ups), which often results in apparent contradictions between different studies (e.g. former conclusions about the activity of Mo vs. W catalysts). In particular, one of the general effects that was established via comparison of vast arrays of experimental data and rationalized based on the DFT models described in Section 2.1 is a marked difference in catalytic behaviour between internal versus terminal olefins for Mo and W catalysts. The case studies discussed below focus on the self-metathesis of cis-4-nonene and 1-nonene as prototypical internal and terminal olefins, respectively, carried out under batch reactor conditions after similar purification protocols (see Fig. 3a for reaction equations). The descriptors used to characterize the performance of the catalysts are the initial turnover frequency (typically measured after 3 min of the reaction, TOF3 min) that serves as a measure of catalyst activity (when reaction occurs without initiation), and the time required to reach equilibrium or the highest achievable conversion (TON) that depends on both activity and stability. Note that for terminal olefins the conversion can theoretically reach 100% if ethylene is being removed from the reaction medium (non-equilibrium conditions), while metathesis of 4-nonene reaches equilibrium at ca. 50% conversion.
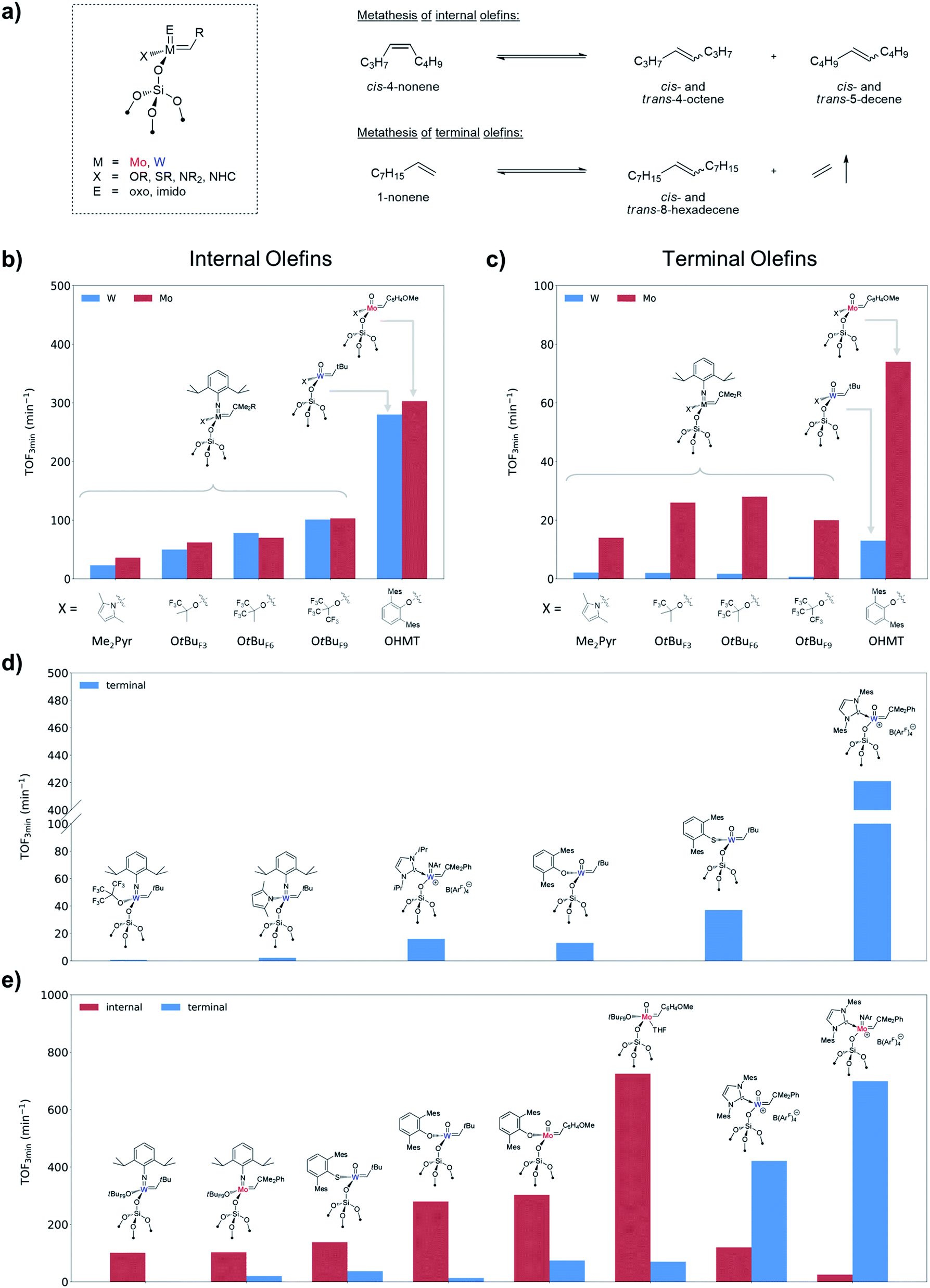 | ||
| Fig. 3 (a) Typical test substrates for the metathesis of internal (cis-4-nonene) and terminal (1-nonene) olefins. (b) Comparative activity of selected silica-supported W and Mo alkylidenes in metathesis of internal olefins. (c) Comparative activity of selected silica-supported W and Mo alkylidenes in metathesis of terminal olefins. (d) Design of the W catalysts with improved performance in metathesis of terminal olefins. (e) Overall comparative activity of selected silica-supported W and Mo alkylidenes in metathesis of internal and terminal olefins. | ||
SiO)W(NAr)(CHCMe3)(X)] (Ar = 2,6-iPr2C6H3) bearing X ligands with different σ-donating ability (X = OtBu, OtBuF3, OtBuF6, OtBuF9, OSi(OtBu)3, S-2,4,6-iPr3C6H2, Me2Pyr)88,101,102 clearly shows that the activity in these systems increases with decreasing σ-donating ability of the X ligand, similarly to what is found for some of the molecular analogues.103,104Fig. 3b highlights this trend for selected catalysts tested under strictly identical conditions: the initial activity increases in a row Me2Pyr < OtBuF3 < OtBuF6 < OtBuF9.102 This trend is in line with what is expected from the computational model described in Section 2.1 and reflects on the one hand the increasing electronic dissymmetry between the surface siloxy and the X ligand, and on the other hand the preference for the TBP over the SP metallacycle that increases as a function of the electron-withdrawing nature of the X ligand. The latter is consistent with the TBP/SP ratios of tungstacyclobutane intermediates observed in these systems by solid-state NMR (vide supra).88 Noteworthy, the same trend, very similar activities and equilibrium times are observed for the corresponding Mo imido alkylidene analogues [(SiO)Mo(NAr)(CHCMe2Ph)(X)] (Fig. 3b).102 Note however that, in contrast to W, no metallacycle intermediates in this series have been observed so far for Mo.
The study of a broader series of W imido alkylidenes [(SiO)W(NAr)(CHCMe2R)(X)], where the nature of both imido and X ligands was varied (Ar = AriPr, ArCl, ArCF3, and ArF5; X = OtBuF9, OtBuF6, OtBu, OSi(OtBu)3, Me2Pyr; R = Me or Ph; Fig. 4), revealed however a more complex picture, with an interplay between E and X ligands.105 In particular, changing electronic properties of the imido ligand away from the classically used electron-donating N-2,6-iPrC6H3 leads to very different orders of activity depending on the electronic nature of the X ligand. Multivariate linear regression analysis tools were applied to systematically investigate the catalyst performance by correlating the initial TOF with electronic and steric descriptors (such as NBO charge and Sterimol parameters) of each member of the ligand set. The resulting quantitative structure–activity relationship (Fig. 4) showed that the highest activity is achieved when X and NAr present opposite electronic character (in terms of electron-donating ability) and that simple evaluation of easily accessible electronic and steric ligand descriptors allows for rational catalyst design.
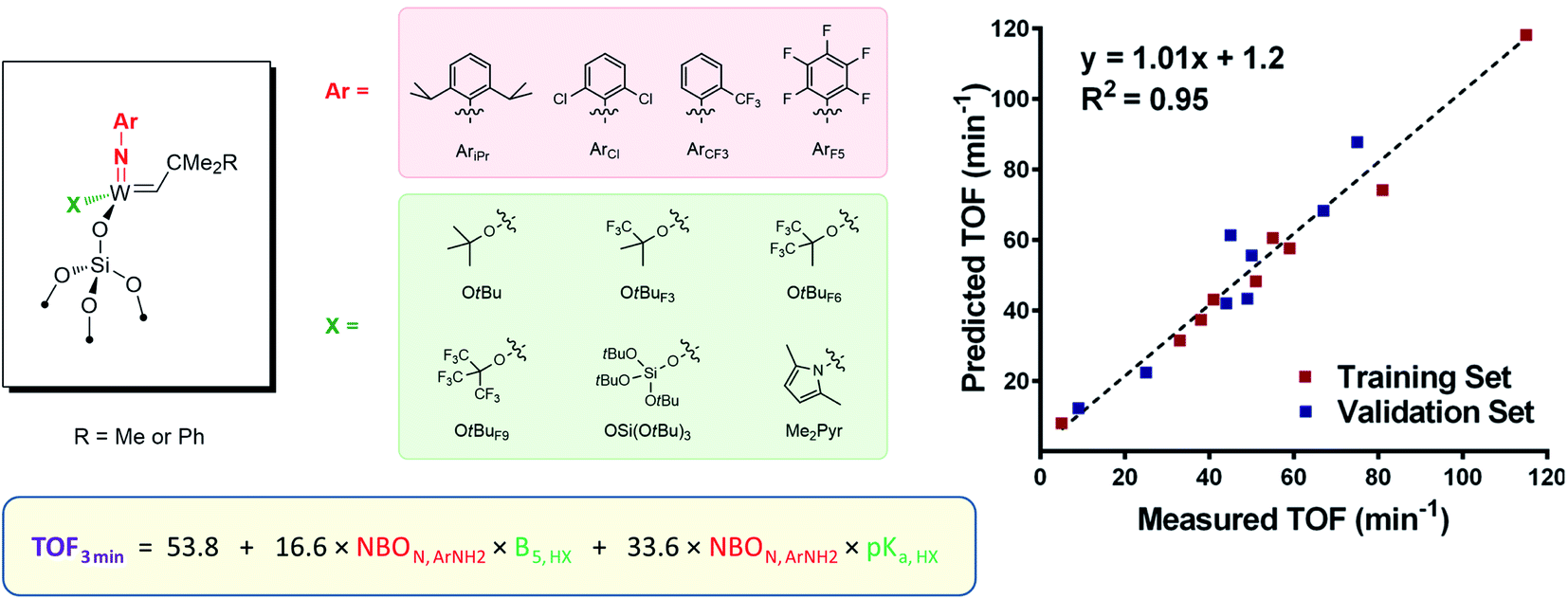 | ||
| Fig. 4 Structure–activity relationship for silica-supported tungsten imido alkylidenes varying X and NAr ligands. | ||
Although supported oxo alkylidenes have long been considered prototypes of the active sites in industrial heterogeneous metathesis catalysts based on supported Mo and W oxides, the preparation of oxo analogues of well-defined silica-supported imido alkylidenes for both W and Mo, [(SiO)M(O)(CHR)(X)] (M = W: X = OHMT (HMT = 2,6-Mes2C6H3),106 OdAdP (dAdP = 2,6-Ad2-4-MeC6H2),107 R = tBu; M = Mo: X = OHMT,108 OtBuF9,109 OTPP (TPP = 2,3,5,6-Ph4C6H),109 R = 4-MeOC6H4; Fig. 5), was achieved only recently due to the advances in the synthesis of molecular precursors.76,110–114 This discovery allowed a direct comparison between oxo and imido supporting E ligands and confirmed a significantly improved catalytic activity for the oxo in both Mo and W cases (Fig. 3b), consistent with the DFT studies on these systems.69 The improved performance of the oxo ligand can be attributed to the combination of its smaller size, which enables an easier access of the incoming di-substituted olefin to the metal centre thereby increasing the initial rate of metathesis, and its strong σ-donating ability, which concurrently improves the stability of the catalyst (due to disfavoured β-H transfer in the corresponding SP metallacycles, vide supra Section 2.1). Paralleling the observations in the imido systems, Mo and W oxo catalysts with the same X ligand display very similar activities in metathesis of internal olefins (e.g. for X = OHMT, Fig. 3b), where frameworks containing electron-withdrawing X ligands (OtBuF9) display the highest turnover frequencies.109 Notably, the activities of the corresponding molecular aryloxide precursors, [M(O)(CHR)(OAr)2], appear to be drastically lower because of the presence of two identically bulky ancillary aryloxide ligands in place of only one in the supported system. It is worth stressing at this point that a surface siloxy group should be viewed as a rather small ligand, whose buried volume115 is estimated to be roughly 20.6%, that is to be compared with 24.3% for HMTO and 36.8% for dAdPO.107 In fact it was shown that introducing a very large siloxide ligand (tBu3SiO) in the coordination sphere of molecular Schrock-type alkylidenes drastically suppresses their metathesis reactivity even towards ethylene.116 Having a smaller X ligand facilitates distortion of the metal complex to coordinate the olefin. Thus, in the case of OtBuF9 both the supported and molecular Mo alkylidenes demonstrate very high activity.109
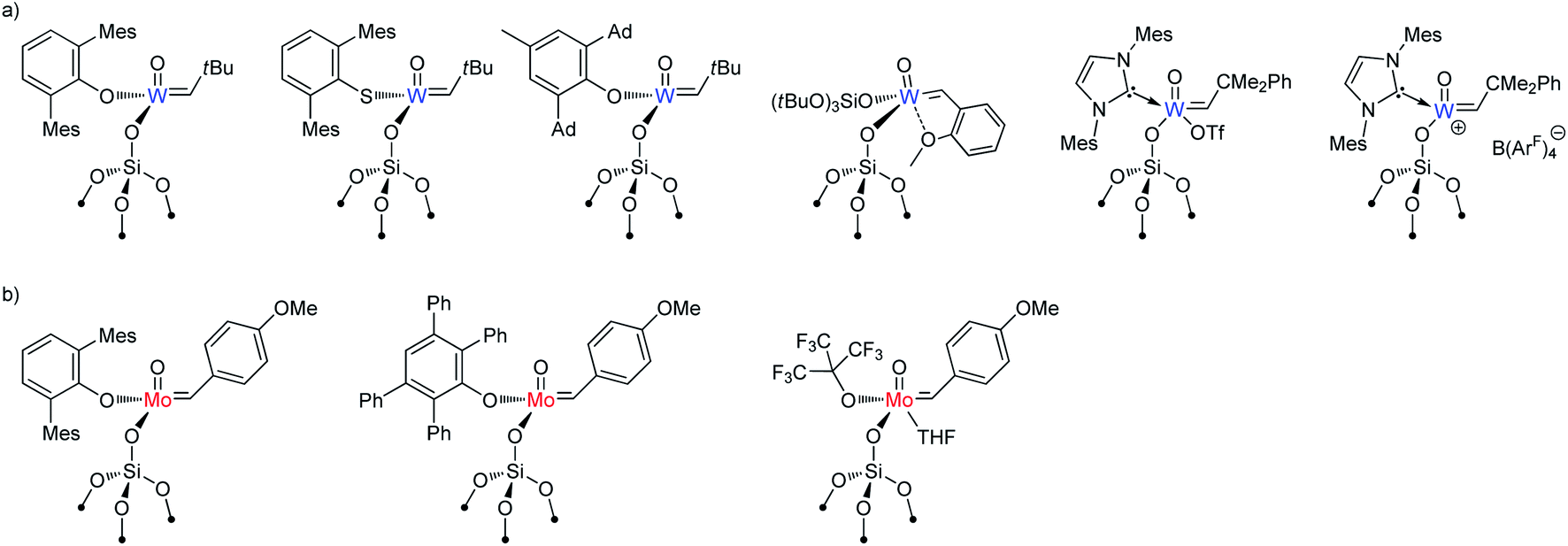 | ||
| Fig. 5 Well-defined supported W (a) and Mo (b) oxo alkylidenes reported to date. | ||
SiO)M(NAr)(CHR)(X)] with X = CH2tBu (W58vs. Mo64) and Me2Pyr (W62vs. Mo60,64)). The same trend was confirmed in the metathesis of 1-nonene in batch conditions for the pairs of analogous Mo and W imido and oxo catalysts (Fig. 3c). At first sight, these observations may seem contradictory with respect to what is found in the metathesis of internal olefins (Fig. 3b). However, one should recall that a key disparity between Mo and W is the distinct difference in the relative stabilities of their corresponding metallacyclobutane intermediates (see Section 2.1). Metallacycles are significantly more stable for W than for Mo and are rarely observed for the latter in either homogenous or supported well-defined catalysts. In fact, a series of parent (i.e. unsubstituted) tungstacyclobutanes were prepared and observed by solid-state NMR upon reaction of 13C-labelled ethylene with silica-supported alkylidenes (Scheme 2).88,105,107 These species were shown to be slow to initiate metathesis even in the case of internal olefins, whereas the initial alkylidenes are highly active.88,107 Therefore these very stable parent metallacyclobutanes (that can be significantly more stable than the substituted ones involved in metathesis of internal olefins) correspond to catalytic off-cycle resting states and can be identified as a main reason for the lower apparent activity of the W-based systems towards metathesis of terminal olefins, where ethylene is present as a co-product. The better performance of the oxo systems as compared to the imido ones (3–5 times higher initial TOF, Fig. 3c) can be explained by the strong σ-donating ability of the oxo ligand that results in the destabilization of these metallacyclobutanes. Noteworthy in this context are the supported oxo alkyl systems [(SiO)xM(O)(CH2R)3−x] (M = Mo and W; R = CMe3, SiMe3, x = 1, 2) that probably generate alkylidenes in situ via α-H abstraction117–120 and show relatively good performance in propene metathesis at 60–80 °C, the Mo analogues still being significantly more efficient than the W ones. Another difference between Mo and W that should be pointed out is the lower selectivity of Mo systems, which often show formation of olefin isomers at higher conversion. This isomerization is likely associated with the formation of Mo hydrides or related species and is presumably related to the easier reducibility of Mo.
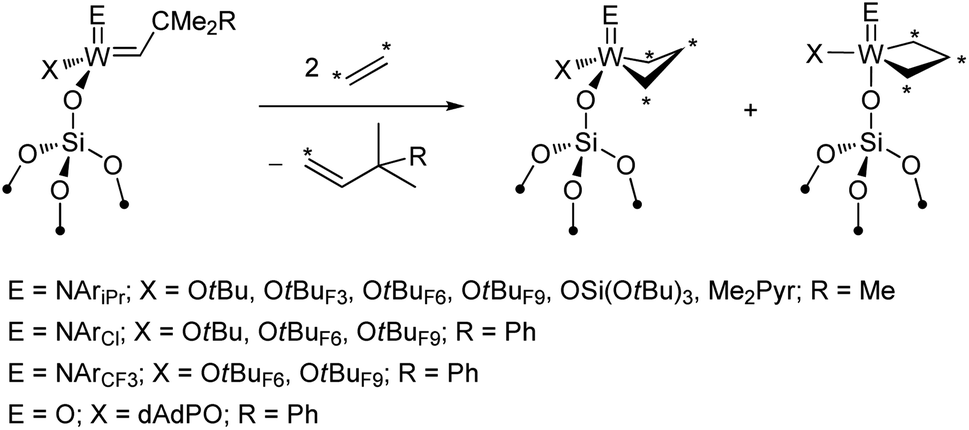 | ||
| Scheme 2 Preparation of 13C-labelled W metallacyclobutanes on silica. | ||
As discussed above, the lower activity of W-based supported metathesis catalyst towards terminal olefins originates from the formation of very stable SP metallacyclobutanes. In view of the influence of ancillary ligands on the stability of metallacyclobutane intermediates (see Section 2.1) a possible strategy to improve their catalytic performance is to destabilize such intermediates by introducing strong σ-donating ligands (Fig. 3d). As already shown in the series of W imido systems [(SiO)W(NAr)(CHtBu)(X)], the catalyst bearing the stronger σ-donating pyrrolyl X ligand displays appreciably better performances towards 1-nonene as compared to the fluorinated alkoxide analogues.102 Incorporating a very strong neutral σ-donor N-heterocyclic carbene (NHC) ligand in tetra-coordinated cationic imido alkylidene species81 leads to a significant increase in activity in the metathesis of terminal olefins (Fig. 3d).121 Similarly, for the oxo systems replacement of the aryloxide ligand in [(SiO)W(O)(CHtBu)(OHMT)] by a more σ-donating thiolate (SHMT) gives a substantially more active catalyst (Fig. 3d).122 Ultimately, the tetra-coordinated cationic NHC-bearing oxo catalysts display a tremendous boost in activity in the metathesis of terminal olefins and even surpass values registered for internal olefins. Noteworthy, the supported cationic W oxo alkylidene NHC complex [(SiO)W(O)(CHCMe2Ph)(IMes)]+[B(3,5-(CF3)2C6H3)4]− not only showed very high activity (Fig. 3d), but also appeared to be remarkably stable, reaching TON over 1 million in the metathesis of propene over six days under flow conditions.123 Subsequently, the utilization of a strong σ-donor NHC ligands was also investigated for the corresponding Mo imido systems (Fig. 3e).89 Within this ligand framework the reactivity towards metathesis of terminal olefins is increased by almost one order of magnitude, with TOFs significantly exceeding the ones obtained with internal olefins (probably as a consequence of increased steric bulk from the NHC and BArF ligands).
More recently, a combination of high-throughput experimentation, surface organometallic chemistry and statistical data analysis was employed to gain a better understanding of key parameters that drive the efficiency of silica-grafted molybdenum imido alkylidene catalysts for the homometathesis of 1-nonene.124 Evaluation of in situ prepared formulations from a library of 35 phenols and two molybdenum bis-pyrrolide precursors allowed quantitative relation of the productive turnover number and turnover frequency to structural features of the aryloxy ligands by examining their computed electronic and steric characteristics. Apart from σ-donor electronic effects, dispersive interactions were found to be the key driver for high activity in the homodimerization of 1-nonene. The catalytic activity descriptor TON1 h correlates predominantly with attractive noncovalent interactions (NCIs) when phenols bear ortho aryl substituents and, conversely, with repulsive NCIs when the phenol has no aryl ortho substituents. In line with the outcome of analysis performed on the molecular monoaryloxide pyrrolide analogues,125 noncovalent interactions are thus an important synthetic handle to consider for the design of active d0 metathesis catalysts. Furthermore, the comparison of both molecular complex and surface grafted analogue revealed a loss of product stereoselectivity for (Z)-selective molecular catalyst upon grafting (vide infra).
Another type of interaction in these heterogeneous catalysts that has being uncovered by SOMC studies is related to surface heterogeneity and its potential consequences for the catalytic behaviour.126 What has been observed for several species and studied in details for [(SiO)W(O)(CHCMe2Ph)(IMes)]+[B(3,5-(CF3)2C6H3)4]−, is a peculiar kinetic profile comprised of two consecutive regimes, which supports the presence of two types of active sites on the surface with very different initiation rates. It has been proposed that these two types of sites result from different interaction of the W centre with an oxygen atom of a siloxane bridge in its proximity (Scheme 3). This data highlights the effects that may arise from the amorphous nature of the supports like silica and the difficulties to obtain truly single-site catalysts—one of the ultimate goals of SOMC.
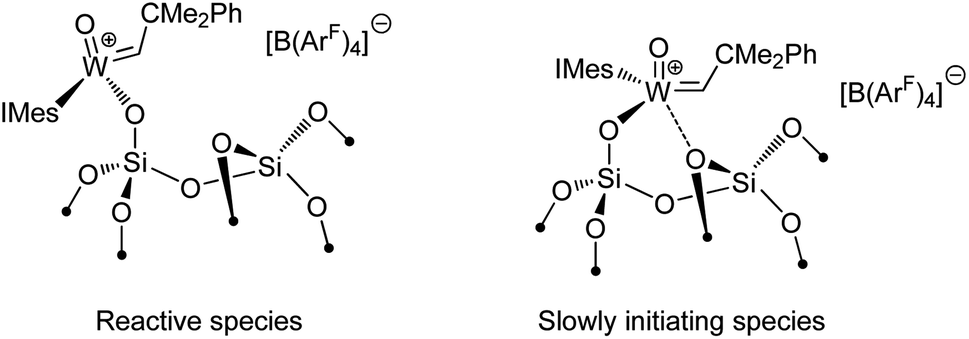 | ||
| Scheme 3 Possible explanation of the presence of different types of sites on the surface of silica-supported catalysts. | ||
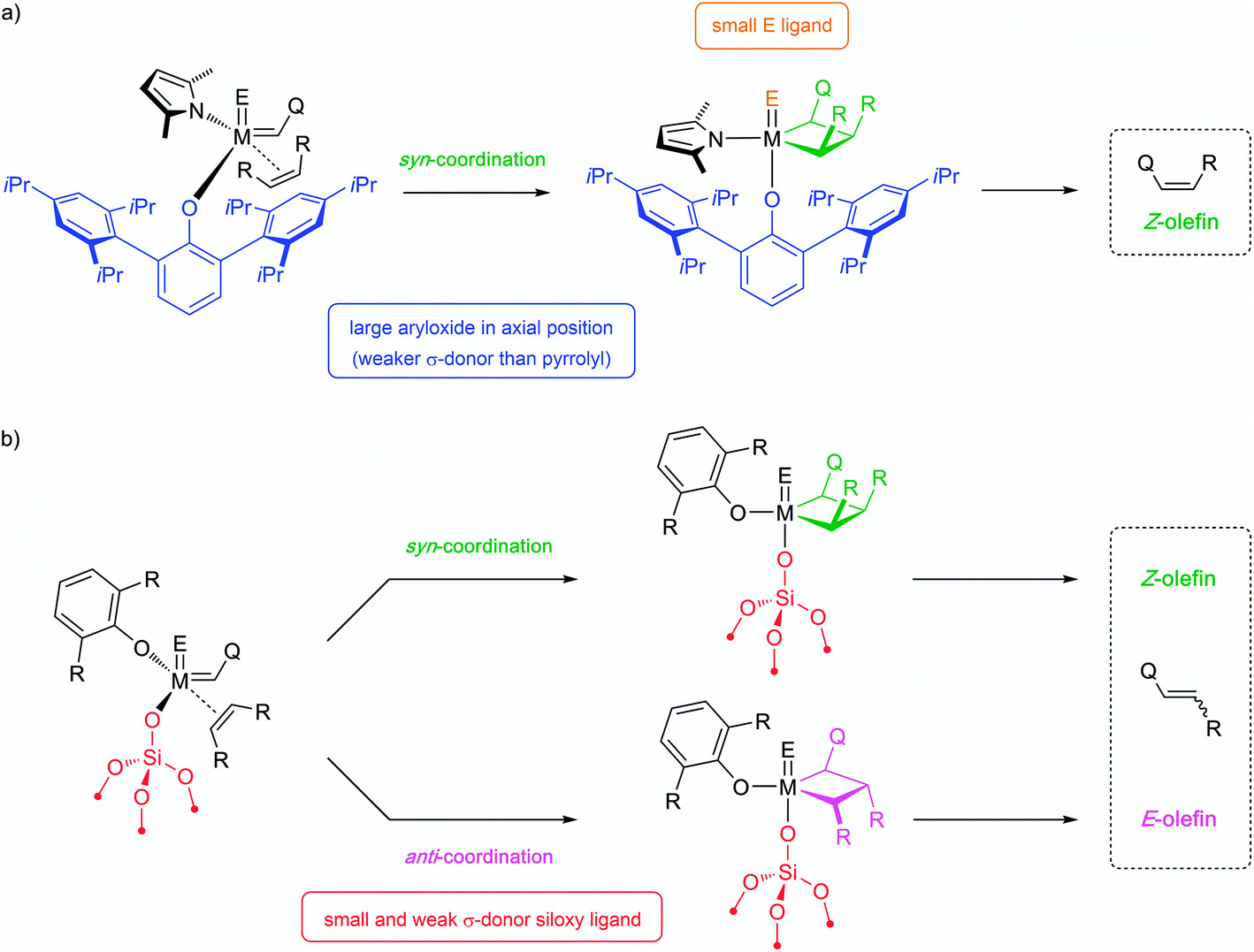 | ||
| Scheme 4 E/Z-selectivity in molecular and supported systems. | ||
Another important aspect of industrial interest is the metathesis of functionalized olefins, such as fatty esters. While among classical heterogeneous catalysts only Re-based systems are compatible with such substrates, homogeneous group 6–7 catalysts often also suffer from low activity and side reactions leading to fast deactivation, and thus until recently only Ru-based catalysts were considered as the main candidates for these reactions. However, recent advances have shown the d0 Schrock-type catalysts are particularly efficient for the metathesis of functionalized olefins, even allowing the formation of fluoro- and chloro-alkenes in a stereoselective way.77,78,80,131–133 To probe the applicability of SOMC catalysts towards this type of olefins, ethyl (or methyl) oleate was typically used as a test substrate in some of the studies cited above. While it is very early to draw any structure–activity relationships in this area, these results have generally proven that well-defined silica-supported catalysts based on Re,134 Mo,59,60,63,64,108,135 and W101,106,107,122,123,135 alkylidenes are sufficiently functional group tolerant and can easily reach TONs of 500–1000, with the best record so far being the cationic W oxo NHC complex on silica reaching TON of ca. 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 at 10 ppm catalyst loading.123
000 at 10 ppm catalyst loading.123
3 Understanding classical metathesis catalysts via SOMC—bridging the gap between molecular and industrial catalysts
As discussed above, industrial catalysts are based on group 6 (Mo or W) and Re supported metal oxides for which the nature and the formation of the active sites remain unknown, even if isolated high-valent metal oxo sites are likely the precursors of the putative active species, an oxo alkylidene. This proposal has led to the development of strategies to prepare model systems based on well-defined metal oxo sites at the surface of oxide supports. One typically distinguishes group 6 (Mo or W) from Re-based systems as the latter is active at room temperatures without the need of activation step (besides the initial calcination at high temperatures that is used for all supported metal oxide catalysts) and only works when supported on alumina-containing supports. In addition, Re-based catalysts have been shown to be compatible with olefin substrates containing ester functionality upon activation with SnMe4. These systems (group 6 vs. 7) will thus be discussed separately, though we will draw parallels between the two systems when possible.3.1 Supported group 6 metal oxides
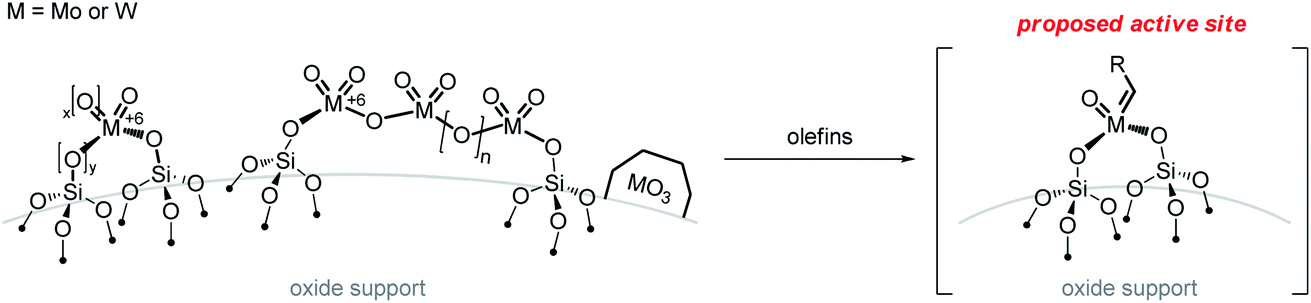 | ||
| Scheme 5 Formation of metathesis active sites from supported group 6 metal oxides. | ||
The most commonly proposed initiation mechanisms include: (i) allylic C–H activation,136–139 (ii) vinylic C–H activation,140,141 (iii) H-assisted mechanism,142 (iv) oxidative coupling/ring contraction,5 and (v) pseudo-Wittig mechanism143,144 (Scheme 6). Most of these mechanisms involve reduction of the high-valent metal oxo centre prior to the reaction with olefins to yield the putative oxo alkylidenes (Routes i–iv). The exception is the pseudo-Wittig mechanism, which allows the direct formation of the oxo alkylidenes from high-valent isolated metal oxo sites (Route v).
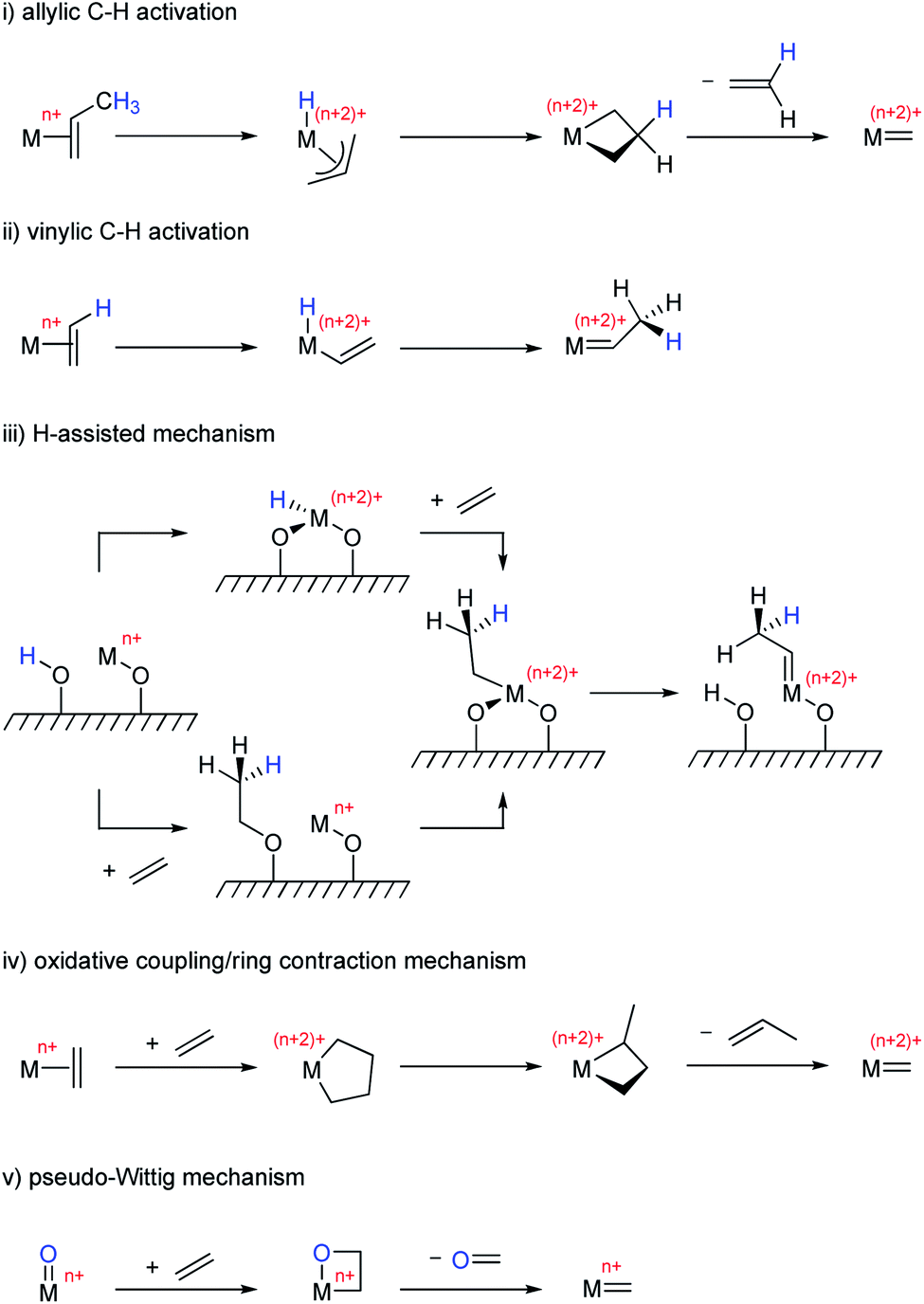 | ||
| Scheme 6 Initiation mechanisms commonly proposed for heterogeneous olefin metathesis catalysts. | ||
Reduced metal centres can be generated during pre-treatment under reducing conditions (e.g. alkenes, H2, N2, etc.) at high temperatures; this process is accompanied by the formation of various oxygenate products (e.g. acetaldehyde, formaldehyde, acetone, water and carbon dioxide). These high temperature pretreatments are typically used in activating WO3/SiO2 to increase olefin metathesis activity allowing metathesis reactions to be performed at lower temperatures.145–149 A similar pretreatment effect has also been reported for supported Mo-based catalysts.150,151 Early studies have shown that MoO3/SiO2 could be reduced by photoreduction in the presence of H2 or CO, where Mo in +4 oxidation state was proposed to be the precatalytic species that leads to active site formation.152 Further studies on the photoreduced species revealed that propylene is directly formed from ethylene; in view of the kinetic isotope effect observed, a vinylic C–H activation mechanism was proposed in the formation of alkylidenes from Mo(IV) species.153 Studies on MoO3/SBA-15 suggest the involvement of surface Brønsted acid sites, possibly associated with Mo–OH groups that can protonate propene to reduce Mo(VI) sites to Mo(IV) with the release of acetone (Scheme 7). Here, too, the reduced Mo(IV) centres were proposed to undergo a vinylic C–H activation leading to the formation of Mo alkylidenes.150 However, the understanding of the mechanisms of M(VI) (M = Mo or W) alkylidenes formation from fully oxidized precursor sites at the molecular level still remains very limited.
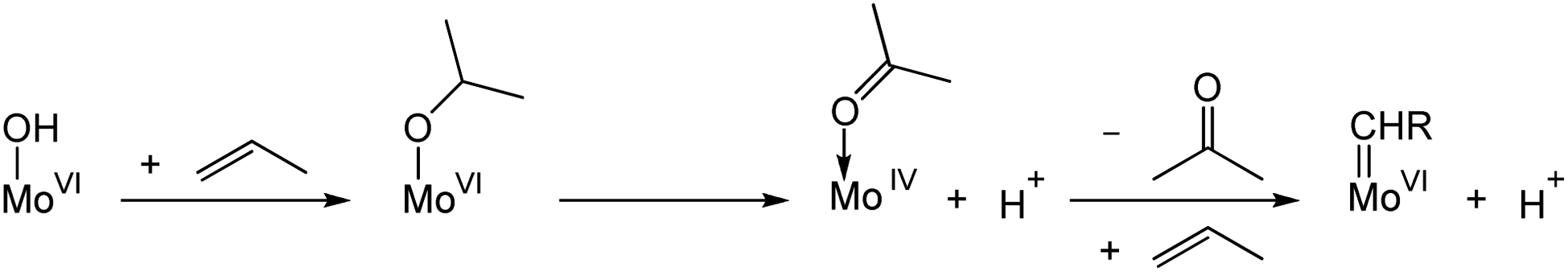 | ||
| Scheme 7 Proposed initiation mechanism for MoO3/SBA-15 in the presence of propylene. | ||
Access to well-defined supported metal oxo sites have been enabled by combining SOMC with the use of Thermolytic Molecular Precursors (TMPs). This approach involves (i) grafting molecular precursors containing thermally labile organic ligands such as (tBuO)3SiO– or tBuO– on the surfaces of supports, followed by (ii) thermolysis of the grafted materials to remove the ligands as volatile organics, leaving isolated metal oxo sites. This process can take place under either vacuum or oxidative conditions.154,155
For instance, grafting of [W(O)2(OSi(OtBu)3)2(DME)]156 on silica partially dehydroxylated at 700 °C (SiO2-700), followed by a thermal treatment at 400 °C under high vacuum (10−5 mbar) yields isolated W oxo species. Extended X-ray absorption fine structure (EXAFS) spectroscopy suggests the presence of a ca. 1:1 mixture of mono-oxo and di-oxo surface species, [(SiO)2W(O)2] and [(SiO)4W(O)] (Scheme 8).157 It is worth noting that similar surface species—[(SiO)2W(O)2] and [(SiO)4W(O)] sites—can also be prepared through the SOMC/TMP approach starting from a mono-oxo molecular precursor [W(O)(OSi(OtBu)3)4], as determined by EXAFS.158 These W oxo sites are inactive for olefin metathesis below 400 °C, similar to what is observed for industrial catalysts based on WO3/SiO2 prepared by impregnation for instance. However, upon activation with an organosilicon reductant (2,3,5,6-tetramethyl-1,4-bis(trimethylsilyl)-1,4-diaza-2,5-cyclohexadiene; Me4-BTDP),159 materials containing isolated W oxo sites become active in olefin metathesis at low temperature (70 °C). Detailed structural characterization of the activated well-defined W oxo material suggests that reduced W(IV) species formed upon activation are the precatalytic sites which initiate at low temperatures to enable olefin metathesis. Exposure of the activated materials to 13C-dilabeled ethylene followed by characterization with 1H and 13C solid state NMR indicates the formation of metallacyclopentane and metallacyclobutane species; based on 13C chemical shift the latter adopt an SP-geometry and can thus be associated with an off-cycle olefin metathesis intermediate that would need to isomerize into a TBP-geometry to become part of the productive catalytic cycle (see Section 2.1). In addition, it was proposed that the metallacyclobutanes could result from ring contraction of metallacyclopentanes based on earlier reports on Ta and Re-based molecular systems (Scheme 6, Route iv).160,161 However, ring contraction of metallacyclopentanes to metallacyclobutanes is calculated to be quite unfavourable according to computational studies (vide infra for further discussion on initiation mechanisms of W(IV) species).90
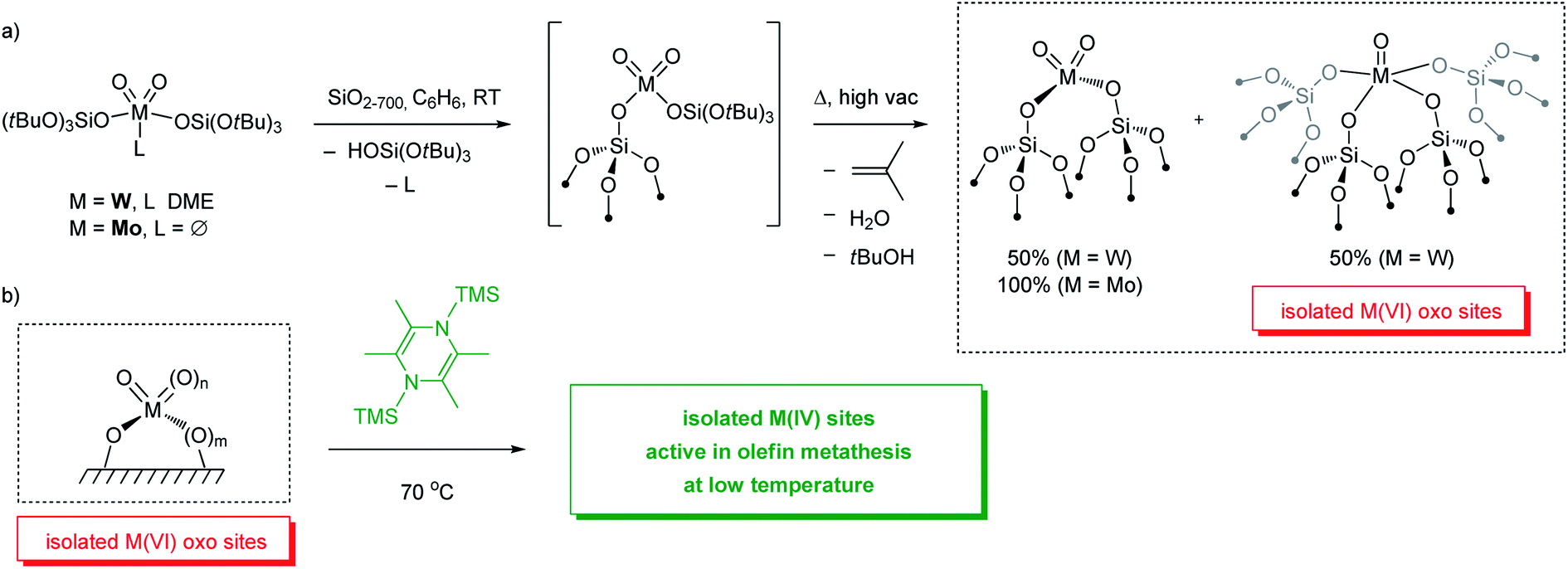 | ||
| Scheme 8 Preparation (a) and activation (b) of molecularly-defined isolated M(VI) oxo (M = W or Mo) sites on silica. | ||
Similarly, well-defined silica-supported Mo oxo sites can be generated and activated using an analogous approach (Scheme 8). Upon grafting [Mo(O)2(OSi(OtBu)3)2] on SiO2-700 followed by thermolysis and calcination under synthetic air, only di-oxo species [(SiO)2Mo(O)2] are formed on the surface according to EXAFS.162 In contrast to W, Mo undergoes partial reduction under vacuum. The Mo-based materials obtained after calcination become metathesis active at 70 °C upon activation with organosilicon reductants, similar to the well-defined W-oxo catalyst discussed above. While more detailed mechanistic investigations are still underway for the Mo-based materials, it has also been proposed that Mo(IV) species are likely the “activated” precatalysts that generate supported Mo alkylidenes in situ from olefins.
In order to further understand the formation of alkylidenes from the putative M(IV) oxo species, tailored W(IV) oxo complexes, [WO(OR)2(py)3] (R = Si(OtBu)3, tBuF6, and tBuF9), and the corresponding well-defined supported [(SiO)WO(OtBuF6)(py)3] were synthesized as molecular and supported model systems, respectively.163,164
With the molecular system, it was shown that addition of tris(pentafluorophenyl)borane (B(C6F5)3) or alternative Lewis acids was essential to switch on metathesis activity for olefins containing allylic C–H bonds, though interestingly no metathesis activity was observed for olefins without allylic C–H bonds. Detailed spectroscopic, kinetic, and computational studies suggest that B(C6F5)3 removes the pyridine ligands from [WO(OR)2(py)3] generating in situ highly active low-coordinate W(IV) species that can activate olefins and form metathesis active species. While contacting a solution of [WO(OtBuF6)2(py)3] and B(C6F5)3 in the presence of ethylene yields unsubstituted metallacyclopentanes having no pyridine ligands, no metallacyclobutanes were observed. Detailed mechanistic studies combined with DFT calculations suggest that the formation of alkylidenes from the W(IV) precatalyst involves two key steps: (i) the C–H bond activation of an allylic C–H group in the olefins and (ii) a proton transfer process facilitated by pyridine and B(C6F5)3 (Scheme 9).
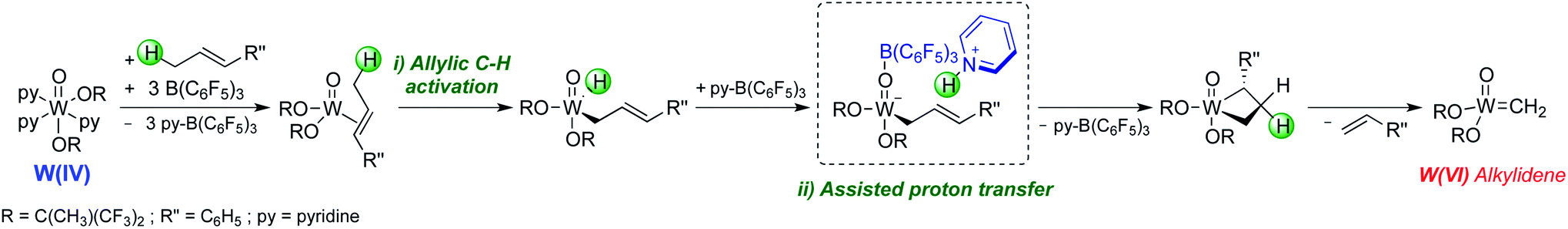 | ||
| Scheme 9 Proposed initiation mechanism for W(IV) oxo complex [WO(OtBuF6)2(py)3]. | ||
The corresponding silica-supported [(SiO)WO(OtBuF6)(py)3], that was prepared by grafting [WO(OtBuF6)2(py)3] on SiO2-700 (Fig. 6a) was characterized by spectroscopic techniques (IR, NMR, and EXAFS) and shown to initiate metathesis in the presence of Lewis acids like B(C6F5)3 for all types of olefins, including those without allylic C–H bonds, suggesting the opening of alternative and/or additional initiation mechanisms in the supported system.164
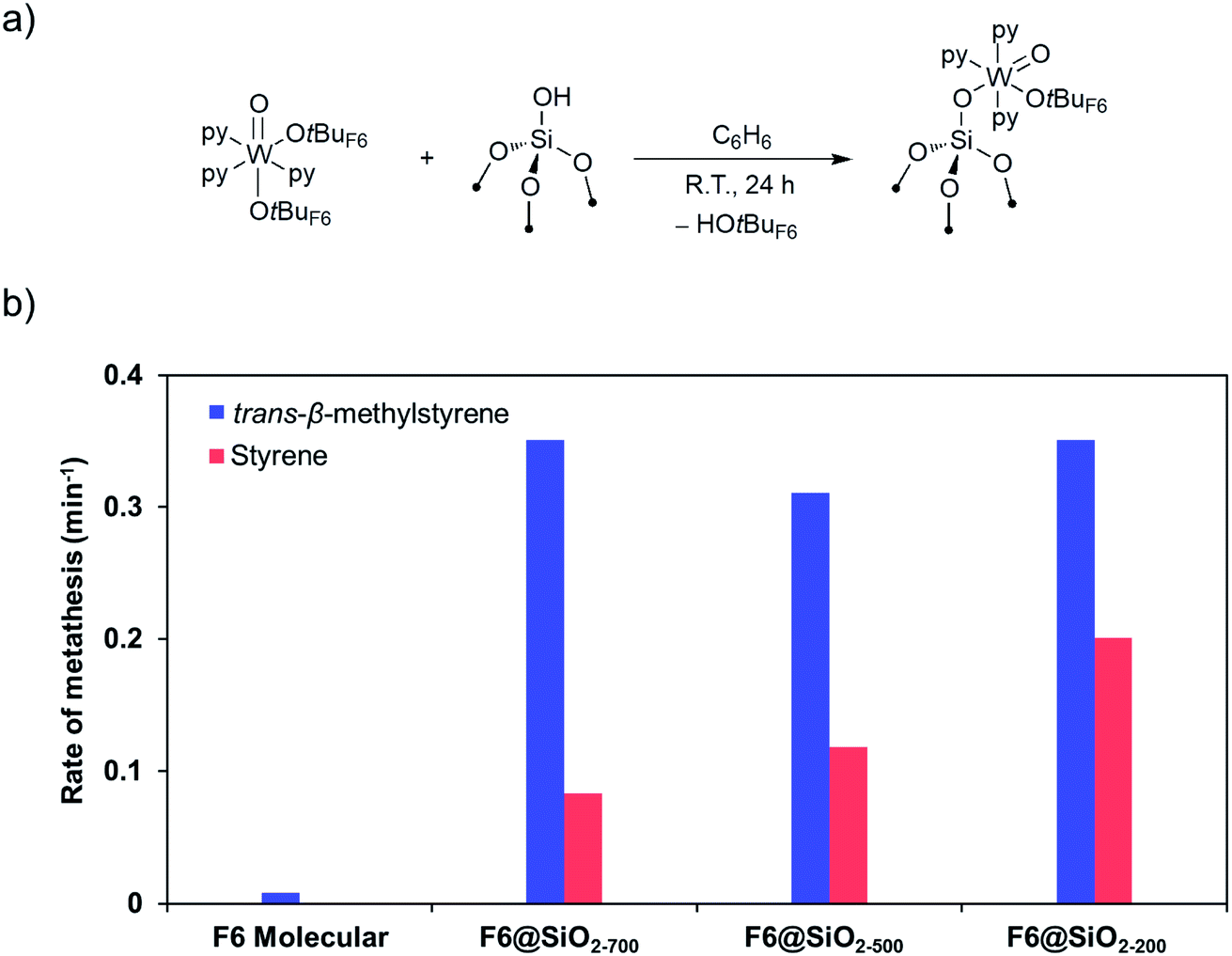 | ||
| Fig. 6 (a) Preparation of well-defined silica-supported W(IV) oxo species. (b) Rates of metathesis of trans-β-methylstyrene (blue) and styrene (red) catalysed by W(IV) oxo species, molecular and supported on silica prepared at different dehydroxylation temperatures. Reprinted with permission from ref. 164. Copyright 2019 American Chemical Society. | ||
Investigation of this system on silica supports prepared at different dehydroxylation temperatures shows that the rate of metathesis for olefins having no allylic C–H bond increases with increasing amounts of residual OH groups, while olefins having allylic C–H bonds do not show such behaviour (Fig. 6b), suggesting that surface protons can also be involved in the initiation, similar to what was suggested earlier for supported Mo metathesis catalysts.150 In fact, detailed spectroscopic studies (including observation of pyridinium species on the surface by IR and solid-state NMR using 15N-labeled pyridine) show that the residual surface silanols (SiOH) display strong Brønsted acidity, likely due to the presence of proximal W species. One can propose that for olefins having no allylic C–H groups, these acidic silanols can assist the initiation by protonating the olefins or the W centre (Scheme 10). Protonation of the olefins coordinated to W centres can directly lead to the formation of tungsten–carbon bonds, i.e. a tungsten alkyl species. Alternatively, protonation of free olefin can yield transient carbocations that can either generate surface alkoxy groups, that are observed experimentally, or react further with the W(IV) centres, forming the tungsten alkyl species. Another possibility would be protonation of the W centres to generate W–H species that can undergo insertion with an olefin to yield tungsten alkyl species. Subsequent deprotonation of the tungsten alkyl species by the silanolate ligand leads to the formation of an alkylidene. Hence, in contrast to the molecular [WO(OtBuF6)2(py)3] analogue, supported [(SiO)WO(OtBuF6)(py)3] is able to initiate metathesis of the substrates without allylic C–H bonds. These studies showed that the initiation of metathesis catalysts (whether involving substrates with allylic C–H group or not) involves key proton transfer steps and that oxide supports in heterogeneous olefin metathesis catalysts might not always be inert, even for a support like silica.
 | ||
| Scheme 10 Proposed initiation mechanisms for olefin without allylic C–H bonds. Reprinted with permission from ref. 164. Copyright 2019 American Chemical Society. | ||
3.2 Supported group 7 metal oxides
Since their discovery in the late sixties, supported Re-based oxide catalysts have attracted much attention for olefin metathesis processes because of their low temperature activity (25–80 °C).165 In addition, they catalyse the metathesis of functionalized olefins when activated by organotin reagents.166 Another noteworthy feature specific to these catalysts is that metathesis active species are not generated in the presence of ethylene alone but require higher olefins, as determined by labelling experiments.138,144 Despite these properties, the industrial viability of supported Re oxide catalysts has remained limited due to their high cost, short term stability, and sensitivity to poisons. In contrast to group 6 systems, rhenium oxides are inactive when supported on silica and require Lewis acidic supports, e.g. alumina or silica–alumina, to show activity. Monomeric Re oxo surface species are generally agreed to be the active precatalytic sites.167 Several types of surface perrhenates have been proposed as precatalytic sites, the coordination environments of which depend on the specific binding mode to surface Al sites (Fig. 7). Most recently, based on comprehensive in situ UV-vis, Raman, IR, and XANES/EXAFS spectroscopic analyses, complemented by DFT calculations, the precatalytic sites have been proposed to be dioxo (AlSO)2Re(O)2 surface species formed by reaction with acidic μ2 and μ3 Al–OH sites and interacting with tri-coordinate Al sites available on the 110 facet of alumina (vide infra).167–169
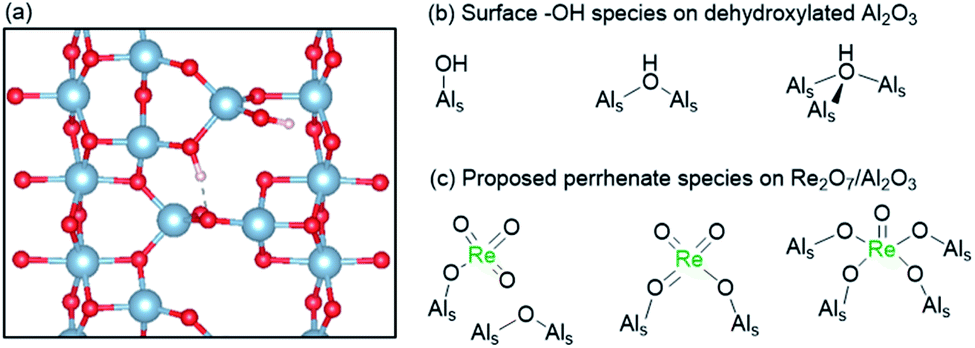 | ||
| Fig. 7 (a) Periodic model of the (110) surface of partially dehydroxylated γ-Al2O3 showing different proposed μ1 and μ2 surface Al–OH species.170 (b) Different surface Al–OH species present on dehydroxylated γ-Al2O3 and (c) proposed perrhenate species on Re2O7/Al2O3.167 | ||
Despite this improved molecular understanding of surface sites in Re2O7/Al2O3, the structure of the active sites is still unclear, as are the mechanisms by which the necessary alkylidenes (or metallacyclobutanes) for the catalytic metathesis cycle are generated from olefins. In parallel, it was shown that methyltrioxorhenium (CH3ReO3) can catalyse olefin metathesis in the presence of Lewis acid co-catalysts or when contacted with supports containing Lewis acid sites.171,172 It may be worth noting that well-defined pentacoordinated silica-supported Re oxo alkylidenes are also inactive in metathesis without the presence of Lewis acid sites.173 In addition, the similar reactivity patterns of CH3ReO3/Al2O3 and organotin-activated Re2O7/Al2O3,166,174 including metathesis activity at low temperature and compatibility with functional groups, led to further detailed investigations of the nature of the active sites in CH3ReO3/Al2O3.
First, CH3ReO3 is metathesis active only on supports with Lewis acidic sites such as γ-Al2O3 and the support needs to be activated at high temperatures (calcination and/or thermal treatment >400 °C) prior to grafting CH3ReO3. The high temperature thermal treatment leads to dehydroxylation of the alumina surfaces (removal of chemisorbed H2O) with concomitant exposure of highly Lewis acidic Al sites that seem to be essential to form the active sites. Titration studies point to the presence of only relatively small quantities of active sites (10–15%).175–177 Detailed experimental and computational studies on alumina have revealed that high-temperature dehydroxylation exposes highly reactive tricoordinate AlIII sites that react with H2 and CH4via heterolytic splitting of the H–X (X = H or CH3) on Al,O pairs, highlighting their particularly high reactivity.170,178,179 EXAFS analyses indicate that the structure of CH3ReO3 is only slightly affected by grafting, with the possible presence of an interaction with an additional oxygen. However, solid-state NMR studies using 13C labelled CH3ReO3 with complementary DFT calculations showed that the major species on CH3ReO3/Al2O3 corresponds to chemisorbed CH3ReO3 interacting with Lewis acidic Al sites via its oxo ligands, while a minor species involves the C–H bond activation of CH3ReO3 on Al,O pairs, similarly to what was observed for CH4 on dehydrated Al2O3. Using labelling experiments, it was shown that the active species (or more precisely the resting state of the active species) corresponds to a bridging μ2-methylene Al–CH2ReO3 complex (Scheme 11).175,180 This complex was proposed to generate the active alkylidene in situ on reaction with olefins, as evidenced by the metathetical exchange between the grafted CH3ReO3 and gas phase ethylene.175 Subsequent in situ solid-state 13C NMR analyses of CH3ReO3/Al2O3 in the presence of 13C2H4 revealed the formation of TBP and SP metallacyclobutanes, providing experimental evidence that metathesis catalysed by Al2O3-supported Re-based materials proceeds through a similar reaction mechanism as the molecular and supported group 6 catalysts (Scheme 11c).177 Detailed DFT calculations showed that the active site involves a very specific configuration for the chemisorption of the tetracoordinate Re centre on alumina, with one oxo ligand bound to the tricoordinate Al site (AlIII in Scheme 11a) and the methylene carbon to a tetracoordinate Al site (AlIVb in Scheme 11a).177 This geometry maximizes the alkylidene character of the adsorbed μ-methylene species as evidenced by the calculated 13C chemical shift, favouring the formation of the alkylidene and a low-energy metathesis pathway (vide supra Section 2.1). Alternative configurations of CH3ReO3 on alumina lead to either inactive or poorly active metal sites, illustrating the importance of the AlIII sites found on specific facet of the alumina surface. Further studies of CH3ReO3 on different oxide supports including silica–alumina,181 chlorinated alumina,182 and amorphous alumina183 show that CH3ReO3 is activated only by strongly Lewis acidic surface sites that are removed from surface Brønsted acid sites and that the activity and stability of the catalytic sites may be tuned by adjusting the Lewis acidity of the support. The importance of the Lewis acidity of the support was also illustrated by a study on the immobilization of CH3ReO3 within the pores of the metal–organic framework (MOF) NU-1000,184 where dehydration of the zirconia nodes to generate highly Lewis acidic sites was shown to be a crucial step for the activation of CH3ReO3 for metathesis. In this case, however, it was not possible to obtain further information about the structure of the active sites.
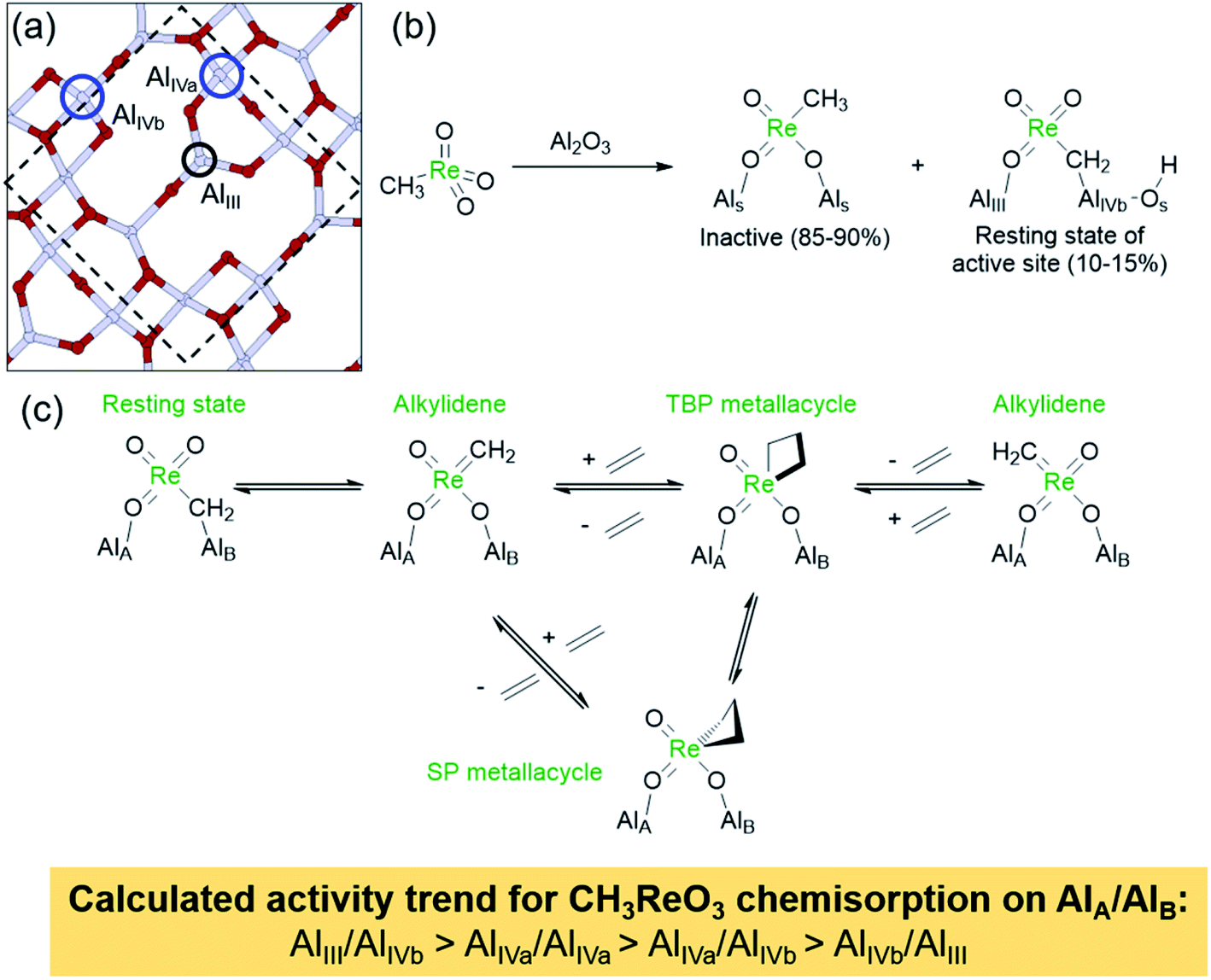 | ||
| Scheme 11 (a) Periodic model of the (110) surface of fully dehydroxylated γ-Al2O3 showing different tetra- (blue circles) and tri- (black circle) coordinate surface Al sites. (b) Species formed on deposition of CH3ReO3 onto Al2O3, including the bridging μ2-methylene Al–CH2–ReO3 complex proposed to be the resting state of the active site. (c) Proposed activation pathway of CH3ReO3/Al2O3.175,177 | ||
Analyses of the propene metathesis kinetics and selectivity have also been used to probe the structure of active sites in Re-based metathesis catalysts. Stereoselectivity at low conversions has been broadly demonstrated as a tool to characterize the active site structures in olefin metathesis catalysts.128,129,186 Indeed, the intrinsic stereoselectivity of a catalyst at low conversions (i.e. far from equilibrium) depends on the local environment of the active sites, while at high conversions the stereoselectivity tends toward thermodynamic equilibrium of (E) and (Z) isomers of the products due to isomerization via secondary metathesis.127 For instance, propene metathesis reactions catalysed by alumina-supported materials typically yield thermodynamic (E/Z) ratios of 2-butenes due in part to strong olefin adsorption on alumina,187 which favours secondary metathesis isomerization over product desorption.144 Indeed, the rates of formation of (E)- and (Z)-2-butene are governed by the relative rates of surface adsorption, metallacyclobutane formation, secondary metathesis isomerization, and desorption, as illustrated in Fig. 8a. The rate laws can be described by Langmuir–Hinshelwood kinetics (Fig. 8b), where the rates of primary propene metathesis, rmeta, and secondary metathesis isomerization, risom, depend on the surface coverages of propene (θC3) and butenes (θC4), respectively. The surface coverages in turn depend on the partial pressures Pn and the equilibrium adsorption coefficients λn. For a catalyst where olefins adsorb strongly, the rate of isomerization through secondary metathesis exceeds the rate of metathesis favouring a thermodynamic (E)/(Z) 2-butene ratio of ∼3 with a hyperbolic dependence on the propene conversion.185 This is exactly what is observed in the propene metathesis behaviour of CH3ReO3/Al2O3 (Fig. 8d, green). This behaviour also provides opportunities to tune the (E/Z) selectivity of heterogeneous olefin metathesis catalysts olefin adsorption properties, for instance by surface modification.185 In fact, generating active sites on passivated alumina with surface trimethylsiloxy group (Fig. 8c) leads to greatly enhanced (Z) selectivities by favouring the desorption of primary metathesis products. Indeed, the extrapolated selectivity at very low conversion is the same for both catalysts, indicating the presence of the same active sites. However, the presence of surface trimethylsilyloxy groups favours olefin desorption (λiPi ≪ 1) such that the secondary metathesis isomerization is disfavoured and the (E)/(Z) ratio is only linear as a function of conversion, as is observed for CH3ReO3/Me3Si–Al2O3 (Fig. 8d, blue). This suggests that developing more selective heterogeneous catalysts will require development of tuneable surfaces where the first coordination sphere of the metal centre, and corresponding olefin adsorption properties, can be controlled.
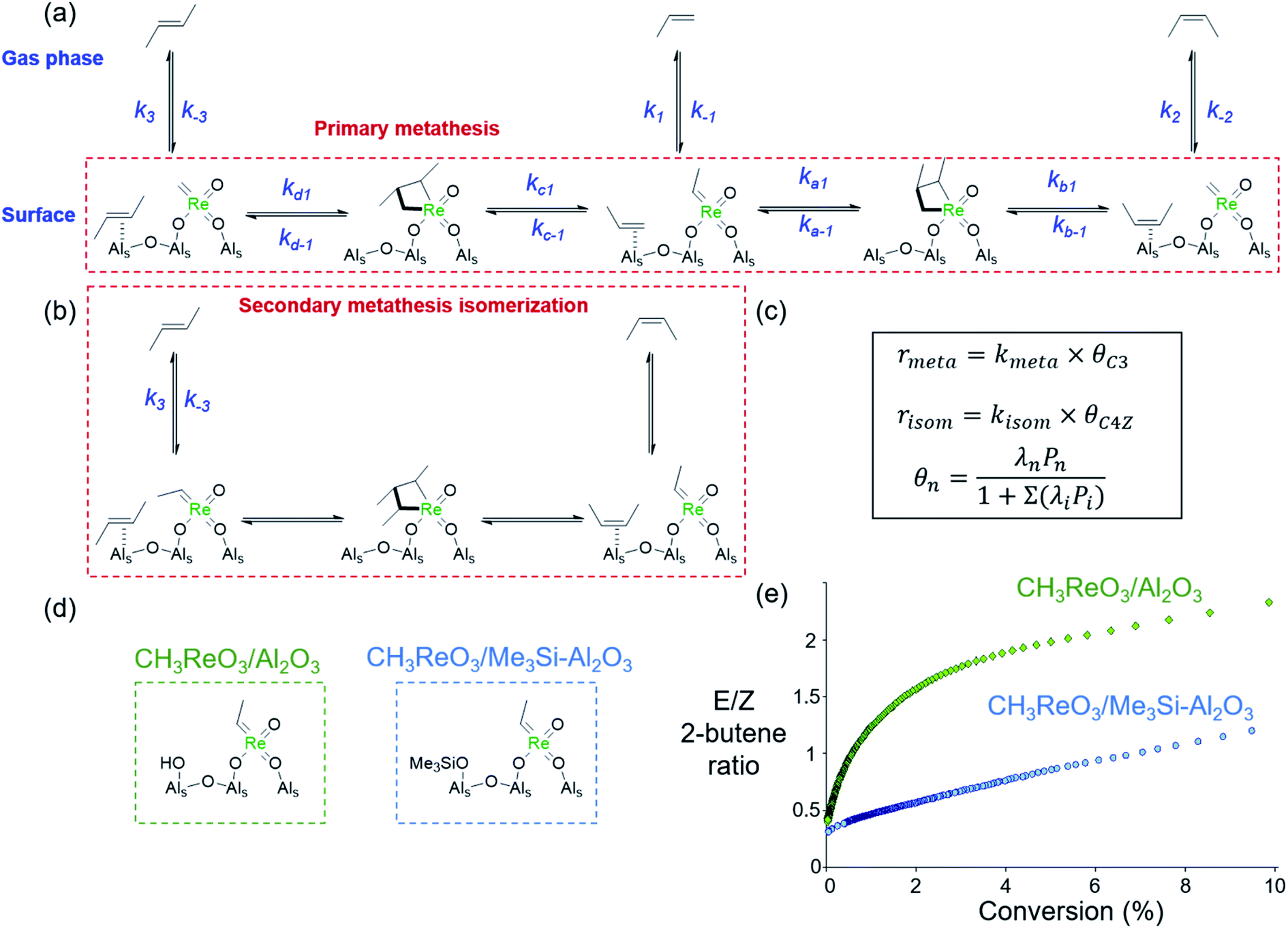 | ||
| Fig. 8 Origin of (E/Z) selectivity of CH3ReO3/Al2O3 for propene metathesis. (a) Scheme showing the adsorption, metathesis reaction, and desorption processes of propene and its 2-butene metathesis products. (b) Scheme showing the isomerization of (E)-2-butene to (Z)-2-butene by secondary metathesis. (c) Equations for the rates of primary metathesis and secondary metathesis isomerization and surface coverages of the relevant olefins derived from Langmuir–Hinshelwood theory. (d) Proposed active sites of CH3ReO3/Al2O3 and CH3ReO3 on Me3Si-treated Al2O3. (e) (E/Z) selectivity ratios for propene metathesis catalysed by CH3ReO3/Al2O3 (green) and CH3ReO3/Me3Si–Al2O3 (blue) as a function of propene conversion.185 | ||
As mentioned above, CH3ReO3/Al2O3 exhibits similar reactivity patterns to Re2O7/Al2O3 activated by organotin additives. Specifically, both types of catalysts are active for the metathesis of functionalized olefins, while Re2O7/Al2O3 on its own is not.138,171,175,188 In fact, similar 13C NMR signals are observed in the solid-state 13C MAS NMR spectra of Re2O7/Al2O3 activated by SnMe4 and CH3ReO3/Al2O3, indicating the formation of similar μ-methylene (Al–CH2ReO3) intermediate species in both materials.189 Contacting Re2O7/Al2O3 with SnMe4 was proposed to form CH3ReO3in situ, which could then generate the μ-methylene species.189
Despite the detailed studies on the related CH3ReO3/Al2O3 system, the precise activation mechanism of Re2O7/Al2O3 remains unknown. Oxygenated products, primarily aldehydes, have been observed during the activation of Re2O7/Al2O3, leading to the hypothesis that the metathesis active Re methylidene forms via a pseudo-Wittig mechanism.168 However, it is also noteworthy that this catalyst does not initiate the degenerate ethene metathesis without being contacted first with propene, which may indicate that intermediates coming from propene are important to generate the active sites. One possibility is the formation of surface propoxy species190 that can be used to reduce the metal sites, hence opening reaction pathway possibly related to what is proposed on Mo and W-based catalysts.150,164 However, further spectroscopic and computational studies are clearly required to solve this longstanding problem and to allow the further development and implementation of Re-based metathesis catalysts.
4 Conclusions, generalizations and perspectives
By 2007, surface organometallic chemistry (SOMC) had established that it was possible to generate well-defined and fully characterized supported alkylidenes by grafting the corresponding molecular alkylidene precursors via protonolysis of a variety of anionic ligands (alkyl, amido, alkoxo) and to obtain highly active heterogeneous metathesis catalysts. Since then, this approach has enabled the synthesis of very large libraries (≫100) of silica-supported Schrock-type metathesis catalysts based on Mo and W, with remarkable activity, selectivity and stability. This has been possible thanks to the concurrent development of (i) novel molecular precursors, (ii) advanced analytical techniques, in particular solid-state NMR spectroscopy, (iii) guideline principles derived from computational modelling, and (iv) high throughput experimentation. The original structure–activity relationship—established via computational modelling based on well-defined and fully characterized surface structures—has shown its robustness over the years and still constitutes a guideline to develop catalysts libraries (Fig. 9). The concept of asymmetry at the metal centre, derived from well-defined silica-supported catalysts, have been successfully transferred to their homogeneous counterparts, thanks to the detailed understanding of the specific role of each ligand on the activity, selectivity and stability of Schrock-type metathesis catalysts. This has triggered the development of novel homogeneous and consequently supported metathesis catalysts (Fig. 9), whose catalytic performances have reached unexpectedly high-level performances, that includes stability, functional group tolerance and stereoselectivities, that explain their use and development of industrial settings.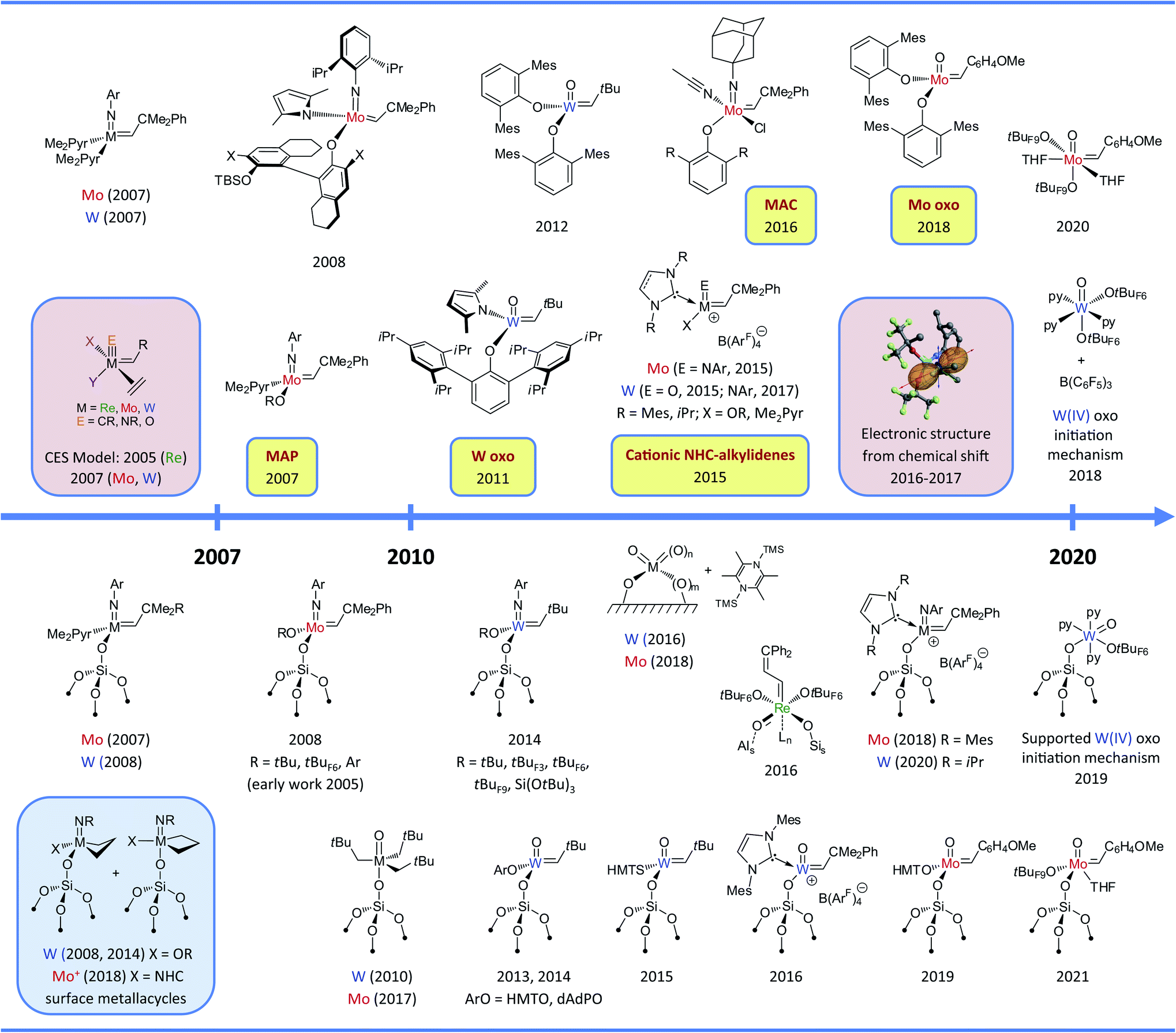 | ||
| Fig. 9 Timeline of the development of molecular (upper part) and supported (bottom part) group 6–7 olefin metathesis catalysts after 2007. Colour codes: selected key experimental findings (yellow), selected mechanistic studies (pink), observation of metallacycle intermediates (blue). Abbreviations: Ar = 2,6-iPr2C6H3, Me2Pyr = 2,5-dimethylpyrrolyl, tBuF3 = CMe2(CF3), tBuF6 = CMe(CF3)2, tBuF9 = C(CF3)3, HMT = 2,6-Mes2C6H3, dAdP = 2,6-Ad2-4Me-C6H2. The whole timeline figure (1964–2020) can be found in ESI.† | ||
SOMC has also permitted observation and characterization—for the first time in 2008—of the TBP and SP metallacyclobutane reaction intermediates in silica-supported W-based metathesis catalysts.62 The first—and only—reported supported Mo metallacyclobutane intermediate was only observed in 2018, with the highly active cationic Mo imido catalyst stabilized by NHC ligand;89 this parallels the difficulty of observing such intermediates for the corresponding homogeneous Mo-based metathesis catalysts, presumably because of their poor stability. Furthermore, trapping these reaction intermediates in a series of silica-supported W-based metathesis catalysts has also highlighted the effect of ancillary ligands on the relative stability of TBP and SP intermediates and enabled a better understanding of the nature of the surface siloxy ligands, whose σ-donating ability falls in between OtBuF3 and OtBuF6 ligands.88
Understanding the relative stability of metallacyclobutanes has been instrumental for explaining the reactivity difference between Mo- and W-based metathesis catalysts for terminal vs. internal olefins. Indeed, while both metals demonstrate very similar activities with internal olefins (cis-4-nonene), Mo catalysts greatly outperform their W analogues in the case of terminal olefins. This can be attributed to a large extent to the formation of very stable parent tungstacyclobutane intermediates in the presence of ethylene, a co-product of the metathesis of terminal olefins. This off-cycle reaction intermediate does not readily release ethylene via cycloreversion to ensure productive metathesis for W. This problem can be partially alleviated by tuning the catalysts, e.g. by introducing strong σ-donor ligands such as oxo, thiolate or NHC, or by working under conditions that favour the removal of ethylene. It is however noteworthy that in few cases the presence of ethylene can be used to accelerate the metathesis of internal olefins.191 This is particularly pronounced for the tungsten oxo cationic catalysts that contain a bulky NHC stabilizing ligand, where ethylene is thought to help interconversion of the metal centre between resting and active states.126
Moreover, detailed NMR studies have revealed the parallel between the NMR spectroscopic signatures of molecular and silica-supported TBP and SP metallacyclobutanes, pointing in particular to the observation of very deshielded α-carbon and shielded β-carbon in TBP structures. This is particularly noteworthy since Ru-based metathesis catalysts also display the same peculiar NMR signatures for the metallacyclobutane intermediates, which also adopt a TBP geometry. While our original solid-state NMR studies focused on understanding the electronic structures and dynamics of supported metal alkylidenes,192 the investigation of metallacyclobutane intermediates by solid-state NMR augmented by computational studies revealed the unique electronic structure imposed by the TBP geometry;98 this geometry entails the presence of an empty orbital that points into the metallacyclobutane ring for both d0 and d4 electronic configurations. This electronic structure is reminiscent of the originating alkylidene, explaining why such TBP metallacyclobutanes are key reaction intermediates in metathesis, while SP structures are just off-cycle resting states that are formed via isomerisation from the TBP into SP through a turnstile process and that must isomerize back into the TBP structure to undergo cycloreversion. The unique electronic nature of the TBP structure indicates that it is likely involved as a key intermediate for all metathesis catalysts including those arising from ill-defined systems such as supported metal oxides, as well as metal chlorides (e.g., WCl6) activated with alkylaluminium or tin reagents, or even well-defined penta-coordinated Re molecular systems, e.g. Re(O)(CHR)(OtBuF6)3 or silica-supported analogues, that are typically activated with Lewis acids.173,193 In fact, such TBP intermediates have also been observed in CH3ReO3 supported on alumina contacted with ethylene.177 These intermediates have yet to be observed in ill-defined catalysts based on supported metal oxides, but their specific spectroscopic signatures and the recent development in solid-state NMR194,195 give hope that they should be accessible in the near future and provide the additional information for rational improvement of these systems. Furthermore, studies directed at understanding electronic structures from solid-state NMR have also shown that olefin metathesis is formally isolobal to σ-bond metathesis196 and olefin insertion197 with d0 metal alkyl compounds, suggesting that one can transfer concepts developed from one reaction to another.
SOMC has also helped to better understand the classical supported metal oxide catalysts by studying well-defined supported group 6 high-valent metal oxo species and their conversion into active sites. Such studies have included the development of well-defined low-valent M(IV) oxo molecular and supported compounds.163,164 From these studies, one can propose with some degree of certainty that the active sites in supported metal oxides are formed in situ by first reducing the high-valent isolated metal(VI) oxo by a 2-electron reduction process into low-valent M(IV) sites. Such M(IV) sites are then further converted in situ into alkylidenes and metallacyclobutanes via processes involving the C–H bond activation of olefins and/or proton transfers that can also involve surface OH groups, consistent with earlier studies discussing the importance of Brønsted acidity in supported metal oxides. The formation of the alkylidene from M(IV) is calculated to be slightly endoergic; this could explain the small fraction of active sites and the need to constantly (re)generate them under reaction conditions. In addition, such initiation likely involves very specific environment such as the proximity of metal sites and OH groups, further explaining the low amounts of active sites. It is likely that similar processes are also involved for supported Re oxides, where the alumina support likely plays an important role in these processes. In addition, alumina has been shown to be particularly important to stabilise the rhenium oxo and alkylidene intermediates for the related CH3ReO3/Al2O3 model system. While the molecular and supported model systems have helped to clarify the initiation of supported metal oxide metathesis catalysts, identifying the structure of the active sites, the state of working catalysts, and their deactivation pathway by spectroscopic means remains a challenge. In addition, SOMC has helped to uncover a low temperature activation process, based on organic reducing agents, that enabled the metathesis of liquid olefins with supported Mo and W oxides at 70 °C in contrast to the much higher temperatures used in current processes (typically >250 °C).157,162
In the course of these studies, we have shown that supported catalysts prepared via SOMC can produce more robust catalytic systems that can reach very high turnover numbers (above 1 million in some cases). This is at least in part due to the stabilization of reaction intermediates through site isolation, mitigating bimolecular decomposition pathways that often limit the performance of homogeneous catalysts. However, in few instances, the surface is not innocent and additional interactions (e.g. coordination) between the metal centre and adjacent surface oxygen functionalities can limit dynamics and introduce heterogeneity that can inhibit the catalytic event by decreasing the number of available active sites, slowing down productive metathesis, and opening deactivation pathways. While the active sites are directly at the surface, a situation that has been shown to slightly favour Z-selectivity (because the substituents on the alkylidene and olefins points away from the surface), high stereoselectivity has so far never been achieved in contrast to the molecular analogues, where tuning the aryloxide ligands in MAP and MAC catalysts has allowed stereoselectivity to reach values >95% at high conversion.73–80,198 This illustrates that surface siloxy ligands can be considered as locally small, even though one might consider surfaces as rather bulky ligands hence precluding deactivation through dimerization (concept of site isolation). In fact, a buried volume as evaluated from a cristobalite model shows that the surface indeed behaves as a rather small ligand (20.6%).107 With alumina surfaces, the observed stereoselectivities are often close to thermodynamic values, mostly due to stronger interactions of olefin products with the surface and post-metathesis isomerization prior to desorption.185 Finally, surface interactions of substrates/products play an important role in stereoselectivity and probably in the overall activity of supported metathesis catalysts and are likely a factor limiting so far their applications beyond hydrocarbon substrates. Understanding the metal and substrate/product interactions with the surfaces is likely a key to designing the supported catalysts with improved performances towards particular classes of olefin substrates.
While this review has concentrated on olefin metathesis and demonstrated that SOMC can generate highly performant catalysts that can surpass in some cases their homogeneous analogues, one can see that this methodology can be readily transposed to related reactions, such as alkyne,46,47,199,200 imine,201,202 and oxo/imido203,204 metathesis, which involve cleavage of π-bonded systems. These concepts can also be used to develop alkane metathesis reactions that involve similar elementary steps.205 For instance alumina-supported W hydrides formed upon hydrogenolysis of organometallic W surface species are proposed to generate upon exposure to olefins alkylidene hydride species that operate as “bifunctional single active sites” and catalyse olefin metathesis as well as variety of other hydrocarbon transformations such as methane coupling and cross-metathesis of methane and higher alkanes.205–207 Since highly reactive intermediates can be made readily accessible on surfaces through site isolation, this methodology can also be extended to a broader variety of catalytic organic transformations.208,209
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank CNRS, ETH Zürich, and SNF, as well as BASF, BP, and XiMo, for the financial support of our metathesis program over the years. We are also grateful to many collaborators, in particular the late Prof. R. A. Andersen (UC Berkeley), Profs M. Buchmeiser (Univ. Stuttgart), O. Eisenstein (Univ. Montpellier), C. Raynaud (Univ. Montpellier), M. Sigman (Univ. Utah), X. Solans-Monfort (UAB), and R. R. Schrock (MIT/UC Riverside). J. D. J. S. was supported by the National Research Fund, Luxembourg (AFR Individual PhD Grant 12516655). P. Z. was supported by the Ministry of Science and Higher Education of the Russian Federation.References
- R. L. Banks and G. C. Bailey, Ind. Eng. Chem. Prod. Res. Dev., 1964, 3, 170–173 CrossRef CAS.
- R. L. Banks, Chemtech., 1986, 16, 112–117 CAS.
- H. S. Eleuterio, J. Mol. Catal., 1991, 65, 55–61 CrossRef CAS.
- Handbook of Metathesis, ed. R. H. Grubbs, A. G. Wenzel, D. J. O'Leary and E. Khosravi, Wiley-VCH, 2015 Search PubMed.
- K. J. Ivin and J. C. Mol, Olefin Metathesis and Metathesis Polymerization, Academic Press, London, 2nd edn, 1997 Search PubMed.
- J. C. Mol, J. Mol. Catal. A: Chem., 2004, 213, 39–45 CrossRef CAS.
- S. Lwin and I. E. Wachs, ACS Catal., 2014, 4, 2505–2520 CrossRef CAS.
- R. Streck, J. Mol. Catal., 1988, 46, 305–316 CrossRef.
- J. C. Mol and P. W. N. M. van Leeuwen, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schuth and J. Weitkamp, Wiley-VCH, 2 edn, 2008, ch. 14.8, pp. 3240–3256 Search PubMed.
- C. L. Dwyer, in Metal-Catalysis in Industrial Organic Processes, ed. G. P. Chiusoli and P. M. Maitlis, The Royal Society of Chemistry, 2006, ch. 6, pp. 201–217 Search PubMed.
- R. L. Banks, J. Mol. Catal., 1980, 8, 269–276 CrossRef CAS.
- P. P. Company, Hydrocarbon Process., 1967, 46, 232 Search PubMed.
- W. Keim, Angew. Chem., Int. Ed., 2013, 52, 12492–12496 CrossRef CAS.
- E. R. Freitas and C. R. Gum, Chem. Eng. Prog., 1979, 75, 73–76 CAS.
- P. Amigues, Y. Chauvin, D. Commereuc, C. C. Lai, Y. H. Liu and J. M. Pan, Hydrocarbon Process., 1990, 69, 79 CAS.
- J. Cosyns, J. Chodorge, D. Commereuc and B. Torck, Hydrocarbon Process., 1998, 77, 61 CAS.
- G. Natta, G. Dall'Asta and G. Mazzanti, Angew. Chem., Int. Ed., 1964, 3, 723–729 CrossRef.
- N. Calderon, H. Y. Chen and K. W. Scott, Tetrahedron Lett., 1967, 8, 3327–3329 CrossRef.
- N. Calderon, Acc. Chem. Res., 1972, 5, 127–132 CrossRef CAS.
- A. W. Anderson and N. G. Merckling, US Pat., 2721189, 1955 Search PubMed.
- W. L. Truett, D. R. Johnson, I. M. Robinson and B. A. Montague, J. Am. Chem. Soc., 1960, 82, 2337–2340 CrossRef CAS.
- V. Dragutan and R. Streck, Catalytic Polymerization of Cycloolefins, Elsevier, Amsterdam, 2000 Search PubMed.
- J. L. Hérisson and Y. Chauvin, Makromol. Chem., 1971, 141, 161–176 CrossRef.
- R. R. Schrock and A. H. Hoveyda, Angew. Chem., Int. Ed., 2003, 42, 4592–4633 CrossRef CAS.
- R. R. Schrock, Chem. Rev., 2009, 109, 3211–3226 CrossRef CAS.
- G. Wilkinson, Science, 1974, 185, 109 CrossRef CAS.
- R. R. Schrock and G. W. Parshall, Chem. Rev., 1976, 76, 243–268 CrossRef CAS.
- F. N. Tebbe, G. W. Parshall and G. S. Reddy, J. Am. Chem. Soc., 1978, 100, 3611–3613 CrossRef CAS.
- G. W. Parshall and S. D. Ittel, Homogeneous Catalysis: The Applications and Chemistry of Catalysis by Soluble Transition Metal Complexes, John Wiley & Sons Inc, New York, 2nd edn, 1992 Search PubMed.
- J. P. Candlin and H. Thomas, Adv. Chem. Ser., 1974, 132, 212–239 CrossRef CAS.
- D. G. H. Ballard, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 2191–2212 CrossRef CAS.
- Y. I. Yermakov, Catal. Rev.: Sci. Eng., 1976, 13, 77–120 CrossRef.
- W. Mowat, J. Smith and D. A. Whan, J. Chem. Soc., Chem. Commun., 1974, 34–35 RSC.
- J. Smith, W. Mowat, D. A. Whan and E. A. V. Ebsworth, J. Chem. Soc., Dalton Trans., 1974, 1742–1746 RSC.
- M. K. Samantaray, E. Callens, E. Abou-Hamad, A. J. Rossini, C. M. Widdifield, R. Dey, L. Emsley and J. M. Basset, J. Am. Chem. Soc., 2014, 136, 1054–1061 CrossRef CAS.
- Y. I. Ermakov, B. N. Kuznetsov, L. G. Karakchiev and S. S. Derbenev, React. Kinet. Catal. Lett., 1974, 1, 307–313 CrossRef CAS.
- A. N. Startsev, B. N. Kuznetsov and Y. I. Yermakov, React. Kinet. Catal. Lett., 1975, 3, 321–327 CrossRef CAS.
- B. N. Kuznetsov, A. N. Startsev and Y. I. Yermakov, J. Mol. Catal., 1980, 8, 135–145 CrossRef CAS.
- Y. Iwasawa, S. Ogasawara and M. Soma, Chem. Lett., 1978, 7, 1039–1042 CrossRef.
- Y. Iwasawa, H. Ichinose, S. Ogasawara and M. Soma, J. Chem. Soc., Faraday Trans. 1, 1981, 77, 1763–1777 RSC.
- K. Weiss and G. Lössel, Angew. Chem., Int. Ed., 1989, 28, 62–64 CrossRef.
- R. Buffon, M. Leconte, A. Choplin and J.-M. Basset, J. Chem. Soc., Chem. Commun., 1993, 361–362 RSC.
- R. Buffon, M. Leconte, A. Choplin and J.-M. Basset, J. Chem. Soc., Dalton Trans., 1994, 1723–1729 RSC.
- E. Le Roux, M. Taoufik, M. Chabanas, D. Alcor, A. Baudouin, C. Copéret, J. Thivolle-Cazat, J.-M. Basset, A. Lesage, S. Hediger and L. Emsley, Organometallics, 2005, 24, 4274–4279 CrossRef CAS.
- R. P. Saint-Arroman, M. Chabanas, A. Baudouin, C. Copéret, J.-M. Basset, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2001, 123, 3820–3821 CrossRef CAS.
- H. Weissman, K. N. Plunkett and J. S. Moore, Angew. Chem., Int. Ed., 2006, 45, 585–588 CrossRef CAS.
- D. P. Estes, C. P. Gordon, A. Fedorov, W.-C. Liao, H. Ehrhorn, C. Bittner, M. L. Zier, D. Bockfeld, K. W. Chan, O. Eisenstein, C. Raynaud, M. Tamm and C. Copéret, J. Am. Chem. Soc., 2017, 139, 17597–17607 CrossRef CAS.
- W. A. Herrmann, A. W. Stumpf, T. Priermeier, S. Bogdanović, V. Dufaud and J.-M. Basset, Angew. Chem., Int. Ed., 1996, 35, 2803–2805 CrossRef CAS.
- F. Blanc, M. Chabanas, C. Copéret, B. Fenet and E. Herdweck, J. Organomet. Chem., 2005, 690, 5014–5026 CrossRef CAS.
- M. Chabanas, A. Baudouin, C. Copéret and J. M. Basset, J. Am. Chem. Soc., 2001, 123, 2062–2063 CrossRef CAS.
- M. Chabanas, A. Baudouin, C. Copéret, J. M. Basset, W. Lukens, A. Lesage, S. Hediger and L. Emsley, J. Am. Chem. Soc., 2003, 125, 492–504 CrossRef CAS.
- X. Solans-Monfort, E. Clot, C. Copéret and O. Eisenstein, Organometallics, 2005, 24, 1586–1597 CrossRef CAS.
- X. Solans-Monfort, E. Clot, C. Copéret and O. Eisenstein, J. Am. Chem. Soc., 2005, 127, 14015–14025 CrossRef CAS.
- M. Chabanas, E. A. Quadrelli, B. Fenet, C. Copéret, J. Thivolle-Cazat, J.-M. Basset, A. Lesage and L. Emsley, Angew. Chem., Int. Ed., 2001, 40, 4493–4496 CrossRef CAS.
- R. R. Schrock, Acc. Chem. Res., 1979, 12, 98–104 CrossRef CAS.
- R. R. Schrock, ACS Symp. Ser., 1983, 211, 369–382 CrossRef CAS.
- F. Blanc, C. Copéret, J. Thivolle-Cazat, J. M. Basset, A. Lesage, L. Emsley, A. Sinha and R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 1216–1220 CrossRef CAS.
- B. Rhers, A. Salameh, A. Baudouin, E. A. Quadrelli, M. Taoufik, C. Copéret, F. Lefebvre, J. M. Basset, X. Solans-Monfort, O. Eisenstein, W. W. Lukens, L. P. H. Lopez, A. Sinha and R. R. Schrock, Organometallics, 2006, 25, 3554–3557 CrossRef CAS.
- F. Blanc, J. Thivolle-Cazat, J. M. Basset, C. Copéret, A. S. Hock, Z. J. Tonzetich and R. R. Schrock, J. Am. Chem. Soc., 2007, 129, 1044–1045 CrossRef CAS.
- F. Blanc, R. Berthoud, A. Salameh, J. M. Basset, C. Copéret, R. Singh and R. R. Schrock, J. Am. Chem. Soc., 2007, 129, 8434–8435 CrossRef CAS.
- D. Gajan, N. Rendon, K. M. Wampler, B. Jean-Marie, C. Copéret, A. Lesage, L. Emsley and R. R. Schrock, Dalton Trans., 2010, 39, 8547–8551 RSC.
- F. Blanc, R. Berthoud, C. Copéret, A. Lesage, L. Emsley, R. Singh, T. Kreickmann and R. R. Schrock, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12123–12127 CrossRef CAS.
- F. Blanc, N. Rendon, R. Berthoud, J.-M. Basset, C. Copéret, Z. J. Tonzetich and R. R. Schrock, Dalton Trans., 2008, 3156–3158 RSC.
- N. Rendon, R. Berthoud, F. Blanc, D. Gajan, T. Maishal, J. M. Basset, C. Copéret, A. Lesage, L. Emsley, S. C. Marinescu, R. Singh and R. R. Schrock, Chem.–Eur. J., 2009, 15, 5083–5089 CrossRef CAS.
- H. Balcar, N. Žilková, J. Sedláček and J. Zedník, J. Mol. Catal. A: Chem., 2005, 232, 53–58 CrossRef CAS.
- C. Coperet, Dalton Trans., 2007, 5498–5504 RSC.
- B. Marciniec, S. Rogalski, M. J. Potrzebowski and C. Pietraszuk, ChemCatChem, 2011, 3, 904–910 CrossRef CAS.
- A. Poater, X. Solans-Monfort, E. Clot, C. Copéret and O. Eisenstein, J. Am. Chem. Soc., 2007, 129, 8207–8216 CrossRef CAS.
- X. Solans-Monfort, C. Copéret and O. Eisenstein, Organometallics, 2012, 31, 6812–6822 CrossRef CAS.
- R. Singh, R. R. Schrock, P. Müller and A. H. Hoveyda, J. Am. Chem. Soc., 2007, 129, 12654–12655 CrossRef CAS.
- S. J. Malcolmson, S. J. Meek, E. S. Sattely, R. R. Schrock and A. H. Hoveyda, Nature, 2008, 456, 933–937 CrossRef CAS.
- A. H. Hoveyda, S. J. Malcolmson, S. J. Meek and A. R. Zhugralin, Angew. Chem., Int. Ed., 2010, 49, 34–44 CrossRef CAS.
- I. Ibrahem, M. Yu, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 3844–3845 CrossRef CAS.
- M. M. Flook, A. J. Jiang, R. R. Schrock, P. Muller and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 7962–7963 CrossRef CAS.
- A. J. Jiang, Y. Zhao, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 16630–16631 CrossRef CAS.
- D. V. Peryshkov, R. R. Schrock, M. K. Takase, P. Muller and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 20754–20757 CrossRef CAS.
- T. T. Nguyen, M. J. Koh, X. Shen, F. Romiti, R. R. Schrock and A. H. Hoveyda, Science, 2016, 352, 569–575 CrossRef CAS.
- X. Shen, T. T. Nguyen, M. J. Koh, D. M. Xu, A. W. H. Speed, R. R. Schrock and A. H. Hoveyda, Nature, 2017, 541, 380–385 CrossRef CAS.
- J. K. Lam, C. Zhu, K. V. Bukhryakov, P. Müller, A. Hoveyda and R. R. Schrock, J. Am. Chem. Soc., 2016, 138, 15774–15783 CrossRef CAS.
- M. J. Koh, T. T. Nguyen, J. K. Lam, S. Torker, J. Hyvl, R. R. Schrock and A. H. Hoveyda, Nature, 2017, 542, 80–86 CrossRef CAS.
- M. J. Benedikter, F. Ziegler, J. Groos, P. M. Hauser, R. Schowner and M. R. Buchmeiser, Coord. Chem. Rev., 2020, 415, 213315 CrossRef CAS.
- M. R. Buchmeiser, Chem.–Eur. J., 2018, 24, 14295–14301 CrossRef CAS.
- M. J. Benedikter, J. V. Musso, W. Frey, R. Schowner and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2021, 60, 1374–1382 CrossRef CAS.
- K. Herz, M. Podewitz, L. Stöhr, D. Wang, W. Frey, K. R. Liedl, S. Sen and M. R. Buchmeiser, J. Am. Chem. Soc., 2019, 141, 8264–8276 CrossRef CAS.
- R. Schowner, W. Frey and M. R. Buchmeiser, J. Am. Chem. Soc., 2015, 137, 6188–6191 CrossRef CAS.
- R. R. Schrock, in Handbook of Metathesis, ed. R. H. Grubbs, A. G. Wenzel, D. J. O'Leary and E. Khosravi, Wiley-VCH, 2015, vol. 1, ch. 1, pp. 1–32 Search PubMed.
- X. Solans-Monfort, C. Copéret and O. Eisenstein, Organometallics, 2015, 34, 1668–1680 CrossRef CAS.
- V. Mougel and C. Copéret, Chem. Sci., 2014, 5, 2475–2481 RSC.
- M. Pucino, M. Inoue, C. P. Gordon, R. Schowner, L. Stöhr, S. Sen, C. Hegedüs, E. Robé, F. Tóth, M. R. Buchmeiser and C. Copéret, Angew. Chem., Int. Ed., 2018, 57, 14566–14569 CrossRef CAS.
- A. M. Leduc, A. Salameh, D. Soulivong, M. Chabanas, J. M. Basset, C. Copéret, X. Solans-Monfort, E. Clot, O. Eisenstein, V. P. W. Bohm and M. Roper, J. Am. Chem. Soc., 2008, 130, 6288–6297 CrossRef CAS.
- X. Solans-Monfort, C. Copéret and O. Eisenstein, J. Am. Chem. Soc., 2010, 132, 7750–7757 CrossRef CAS.
- L. Cavallo, J. Am. Chem. Soc., 2002, 124, 8965–8973 CrossRef CAS.
- C. Adlhart and P. Chen, J. Am. Chem. Soc., 2004, 126, 3496–3510 CrossRef CAS.
- P. E. Romero and W. E. Piers, J. Am. Chem. Soc., 2005, 127, 5032–5033 CrossRef CAS.
- A. G. Wenzel and R. H. Grubbs, J. Am. Chem. Soc., 2006, 128, 16048–16049 CrossRef CAS.
- E. F. van der Eide, P. E. Romero and W. E. Piers, J. Am. Chem. Soc., 2008, 130, 4485–4491 CrossRef CAS.
- J. Feldman, W. M. Davis, J. K. Thomas and R. R. Schrock, Organometallics, 1990, 9, 2535–2548 CrossRef CAS.
- C. P. Gordon, K. Yamamoto, W.-C. Liao, F. Allouche, R. A. Andersen, C. Copéret, C. Raynaud and O. Eisenstein, ACS Cent. Sci., 2017, 3, 759–768 CrossRef CAS.
- R. R. Schrock and C. Copéret, Organometallics, 2017, 36, 1884–1892 CrossRef CAS.
- L. P. H. Lopez, R. R. Schrock and P. Muller, Organometallics, 2006, 25, 1978–1986 CrossRef CAS.
- F. Allouche, V. Mougel and C. Copéret, Asian J. Org. Chem., 2015, 4, 528–532 CrossRef CAS.
- P. A. Zhizhko, V. Mougel, J. De Jesus Silva and C. Copéret, Helv. Chim. Acta, 2018, e1700302 CrossRef.
- R. R. Schrock, R. T. Depue, J. Feldman, C. J. Schaverien, J. C. Dewan and A. H. Liu, J. Am. Chem. Soc., 1988, 110, 1423–1435 CrossRef CAS.
- R. R. Schrock, J. S. Murdzek, G. C. Bazan, J. Robbins, M. DiMare and M. O'Regan, J. Am. Chem. Soc., 1990, 112, 3875–3886 CrossRef CAS.
- V. Mougel, C. B. Santiago, P. A. Zhizhko, E. N. Bess, J. Varga, G. Frater, M. S. Sigman and C. Copéret, J. Am. Chem. Soc., 2015, 137, 6699–7604 CrossRef CAS.
- M. P. Conley, V. Mougel, D. V. Peryshkov, W. P. Forrest Jr, D. Gajan, A. Lesage, L. Emsley, C. Copéret and R. R. Schrock, J. Am. Chem. Soc., 2013, 135, 19068–19070 CrossRef CAS.
- M. P. Conley, W. P. Forrest, V. Mougel, C. Coperet and R. R. Schrock, Angew. Chem., Int. Ed., 2014, 53, 14221–14224 CrossRef CAS.
- M. Pucino, F. Zhai, C. P. Gordon, D. Mance, A. H. Hoveyda, R. R. Schrock and C. Copéret, Angew. Chem., Int. Ed., 2019, 58, 11816–11819 CrossRef CAS.
- J. De Jesus Silva, M. Pucino, F. Zhai, D. Mance, Z. J. Berkson, D. F. Nater, A. H. Hoveyda, C. Coperet and R. R. Schrock, Inorg. Chem., 2021 DOI:10.1021/acs.inorgchem.1020c03173.
- D. V. Peryshkov and R. R. Schrock, Organometallics, 2012, 31, 7278–7286 CrossRef CAS.
- V. Mougel and C. Copéret, ACS Catal., 2015, 5, 6436–6439 CrossRef CAS.
- K. V. Bukhryakov, R. R. Schrock, A. H. Hoveyda, C. Tsay and P. Müller, J. Am. Chem. Soc., 2018, 140, 2797–2800 CrossRef CAS.
- F. Zhai, K. V. Bukhryakov, R. R. Schrock, A. H. Hoveyda, C. Tsay and P. Müller, J. Am. Chem. Soc., 2018, 140, 13609–13613 CrossRef CAS.
- F. Zhai, R. R. Schrock, A. H. Hoveyda and P. Müller, Organometallics, 2020, 39, 2486–2492 CrossRef CAS.
- A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759–1766 CrossRef CAS.
- K. M. Wampler, R. R. Schrock and A. S. Hock, Organometallics, 2007, 26, 6674–6680 CrossRef CAS.
- E. Mazoyer, N. Merle, A. de Mallmann, J. M. Basset, E. Berrier, L. Delevoye, J. F. Paul, C. P. Nicholas, R. M. Gauvin and M. Taoufik, Chem. Commun., 2010, 46, 8944–8946 RSC.
- D. Grekov, Y. Bouhoute, K. C. Szeto, N. Merle, A. De Mallmann, F. Lefebvre, C. Lucas, I. Del Rosal, L. Maron, R. M. Gauvin, L. Delevoye and M. Taoufik, Organometallics, 2016, 35, 2188–2196 CrossRef CAS.
- Y. Bouhoute, D. Grekov, K. C. Szeto, N. Merle, A. De Mallmann, F. Lefebvre, G. Raffa, I. Del Rosal, L. Maron, R. M. Gauvin, L. Delevoye and M. Taoufik, ACS Catal., 2016, 6, 1–18 CrossRef CAS.
- N. Merle, F. Le Quéméner, Y. Bouhoute, K. C. Szeto, A. De Mallmann, S. Barman, M. K. Samantaray, L. Delevoye, R. M. Gauvin, M. Taoufik and J. M. Basset, J. Am. Chem. Soc., 2017, 139, 2144–2147 CrossRef CAS.
- J. De Jesus Silva, D. Mance, M. Pucino, M. J. Benedikter, I. Elser, M. R. Buchmeiser and C. Copéret, Helv. Chim. Acta, 2020, 103, e2000161 CrossRef CAS.
- V. Mougel, M. Pucino and C. Copéret, Organometallics, 2015, 34, 551–554 CrossRef CAS.
- M. Pucino, V. Mougel, R. Schowner, A. Fedorov, M. R. Buchmeiser and C. Copéret, Angew. Chem., Int. Ed., 2016, 55, 4300–4302 CrossRef CAS.
- J. De Jesus Silva, M. A. B. Ferreira, A. Fedorov, M. S. Sigman and C. Copéret, Chem. Sci., 2020, 11, 6717–6723 RSC.
- M. A. B. Ferreira, J. De Jesus Silva, S. Grosslight, A. Fedorov, M. S. Sigman and C. Copéret, J. Am. Chem. Soc., 2019, 141, 10788–10800 CrossRef CAS.
- M. Pucino, W.-C. Liao, K. W. Chan, E. Lam, R. Schowner, P. A. Zhizhko, M. R. Buchmeiser and C. Copéret, Helv. Chim. Acta, 2020, 103, e2000072 CrossRef CAS.
- C. Copéret, Beilstein J. Org. Chem., 2011, 7, 13–21 CrossRef.
- M. Leconte, J. L. Bilhou, W. Reimann and J. M. Basset, J. Chem. Soc., Chem. Commun., 1978, 341–342 RSC.
- M. Leconte and J. M. Basset, J. Am. Chem. Soc., 1979, 101, 7296–7302 CrossRef CAS.
- M. Renom-Carrasco, P. Mania, R. Sayah, L. Veyre, G. Occhipinti, D. Gajan, A. Lesage, V. R. Jensen and C. Thieuleux, Dalton Trans., 2019, 48, 2886–2890 RSC.
- T. T. Nguyen, M. J. Koh, T. J. Mann, R. R. Schrock and A. H. Hoveyda, Nature, 2017, 552, 347–354 CrossRef CAS.
- Y. C. Mu, T. T. Nguyen, M. J. Koh, R. R. Schrock and A. H. Hoveyda, Nat. Chem., 2019, 11, 478–487 CrossRef CAS.
- Y. C. Mu, T. T. Nguyen, F. W. van der Mei, R. R. Schrock and A. H. Hoveyda, Angew. Chem., Int. Ed., 2019, 58, 5365–5370 CrossRef CAS.
- M. Chabanas, C. Copéret and J. M. Basset, Chem.–Eur. J., 2003, 9, 971–975 CrossRef CAS.
- P. A. Zhizhko, F. Toth, C. P. Gordon, K. W. Chan, W.-C. Liao, V. Mougel and C. Copéret, Helv. Chim. Acta, 2019, 102, e1900190 CrossRef CAS.
- M. F. Farona and R. L. Tucker, J. Mol. Catal., 1980, 8, 85–90 CrossRef CAS.
- R. H. Grubbs and S. J. Swetnick, J. Mol. Catal., 1980, 8, 25–36 CrossRef CAS.
- J. R. McCoy and M. F. Farona, J. Mol. Catal., 1991, 66, 51–58 CrossRef CAS.
- M. Ephritikhine, M. L. H. Green and R. E. MacKenzie, J. Chem. Soc., Chem. Commun., 1976, 619–621 RSC.
- Y. Iwasawa and H. Hamamura, J. Chem. Soc., Chem. Commun., 1983, 130–132 RSC.
- Y. Iwasawa, H. Kubo and H. Hamamura, J. Mol. Catal., 1985, 28, 191–208 CrossRef CAS.
- D. T. Laverty, J. J. Rooney and A. Stewart, J. Catal., 1976, 45, 110–113 CrossRef CAS.
- A. K. Rappe and W. A. Goddard, J. Am. Chem. Soc., 1982, 104, 448–456 CrossRef CAS.
- A. Salameh, C. Copéret, J. M. Basset, V. P. W. Bohm and M. Roper, Adv. Synth. Catal., 2007, 349, 238–242 CrossRef CAS.
- A. G. Basrur, S. R. Patwardhan and S. N. Was, J. Catal., 1991, 127, 86–95 CrossRef CAS.
- K. Ding, A. Gulec, A. M. Johnson, T. L. Drake, W. Wu, Y. Lin, E. Weitz, L. D. Marks and P. C. Stair, ACS Catal., 2016, 6, 5740–5746 CrossRef CAS.
- J. G. Howell, Y.-P. Li and A. T. Bell, ACS Catal., 2016, 6, 7728–7738 CrossRef CAS.
- S. Lwin, Y. Li, A. I. Frenkel and I. E. Wachs, ACS Catal., 2016, 6, 3061–3071 CrossRef CAS.
- S. Lwin and I. E. Wachs, ACS Catal., 2017, 7, 573–580 CrossRef CAS.
- K. Amakawa, S. Wrabetz, J. Kröhnert, G. Tzolova-Müller, R. Schlögl and A. Trunschke, J. Am. Chem. Soc., 2012, 134, 11462–11473 CrossRef CAS.
- A. Chakrabarti and I. E. Wachs, ACS Catal., 2018, 8, 949–959 CrossRef CAS.
- B. N. Shelimov, L. V. Elev and V. B. Kazansky, J. Catal., 1986, 98, 70–81 CrossRef CAS.
- B. N. Shelimov, I. V. Elev and V. B. Kazansky, J. Mol. Catal., 1988, 46, 187–200 CrossRef.
- K. L. Fujdala and T. D. Tilley, J. Catal., 2003, 216, 265–275 CrossRef CAS.
- C. Copéret, Acc. Chem. Res., 2019, 52, 1697–1708 CrossRef.
- J. Jarupatrakorn, M. P. Coles and T. D. Tilley, Chem. Mater., 2005, 17, 1818–1828 CrossRef CAS.
- V. Mougel, K.-W. Chan, G. Siddiqi, K. Kawakita, H. Nagae, H. Tsurugi, K. Mashima, O. Safonova and C. Copéret, ACS Cent. Sci., 2016, 2, 569–576 CrossRef CAS.
- K. W. Chan, PhD thesis, ETH Zürich, 2019.
- T. Saito, H. Nishiyama, H. Tanahashi, K. Kawakita, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2014, 136, 5161–5170 CrossRef CAS.
- S. J. McLain, J. Sancho and R. R. Schrock, J. Am. Chem. Soc., 1979, 101, 5451–5453 CrossRef CAS.
- G. K. Yang and R. G. Bergman, Organometallics, 1985, 4, 129–138 CrossRef CAS.
- K. Yamamoto, K. W. Chan, V. Mougel, H. Nagae, H. Tsurugi, O. V. Safonova, K. Mashima and C. Copéret, Chem. Commun., 2018, 54, 3989–3992 RSC.
- K. W. Chan, E. Lam, V. D'Anna, F. Allouche, C. Michel, O. V. Safonova, P. Sautet and C. Copéret, J. Am. Chem. Soc., 2018, 140, 11395–11401 CrossRef CAS.
- K. W. Chan, D. Mance, O. V. Safonova and C. Copéret, J. Am. Chem. Soc., 2019, 141, 18286–18292 CrossRef CAS.
- J. C. Mol, Catal. Today, 1999, 51, 289–299 CrossRef CAS.
- E. Verkuijlen, F. Kapteijn, J. C. Mol and C. Boelhouwer, J. Chem. Soc., Chem. Commun., 1977, 198–199 RSC.
- S. Lwin, C. Keturakis, J. Handzlik, P. Sautet, Y. Li, A. I. Frenkel and I. E. Wachs, ACS Catal., 2015, 5, 1432–1444 CrossRef CAS.
- S. Lwin, Y. Li, A. I. Frenkel and I. E. Wachs, ACS Catal., 2015, 5, 6807–6814 CrossRef CAS.
- S. Lwin and I. E. Wachs, ACS Catal., 2016, 6, 272–278 CrossRef CAS.
- R. Wischert, P. Laurent, C. Copéret, F. Delbecq and P. Sautet, J. Am. Chem. Soc., 2012, 134, 14430–14449 CrossRef CAS.
- W. A. Herrmann, W. Wagner, U. N. Flessner, U. Volkhardt and H. Komber, Angew. Chem., Int. Ed., 1991, 30, 1636–1638 CrossRef.
- R. Buffon, A. Auroux, F. Lefebvre, M. Leconte, A. Choplin, J. M. Basset and W. A. Herrmann, J. Mol. Catal., 1992, 76, 287–295 CrossRef.
- M. Valla, D. Stadler, V. Mougel and C. Copéret, Angew. Chem., Int. Ed., 2016, 55, 1124–1127 CrossRef CAS.
- I. Wohrle, A. Reckziegel, P. Esser and M. Sturmann, US Pat., US20030054956A1, 2001 Search PubMed.
- A. Salameh, J. Joubert, A. Baudouin, W. Lukens, F. Delbecq, P. Sautet, J. M. Basset and C. Copéret, Angew. Chem., Int. Ed., 2007, 46, 3870–3873 CrossRef CAS.
- A. Salameh, A. Baudouin, D. Soulivong, V. Boehm, M. Roeper, J. M. Basset and C. Copéret, J. Catal., 2008, 253, 180–190 CrossRef CAS.
- M. Valla, R. Wischert, A. Comas-Vives, M. P. Conley, R. Verel, C. Copéret and P. Sautet, J. Am. Chem. Soc., 2016, 138, 6774–6785 CrossRef CAS.
- J. Joubert, A. Salameh, V. Krakoviack, F. Delbecq, P. Sautet, C. Copéret and J. M. Basset, J. Phys. Chem. B, 2006, 110, 23944–23950 CrossRef CAS.
- R. Wischert, C. Copéret, F. Delbecq and P. Sautet, Angew. Chem., Int. Ed., 2011, 50, 3202–3205 CrossRef CAS.
- R. Wischert, C. Copéret, F. Delbecq and P. Sautet, ChemCatChem, 2010, 2, 812–815 CrossRef CAS.
- A. W. Moses, C. Raab, R. C. Nelson, H. D. Leifeste, N. A. Ramsahye, S. Chattopadhyay, J. Eckert, B. F. Chmelka and S. L. Scott, J. Am. Chem. Soc., 2007, 129, 8912–8920 CrossRef CAS.
- A. Gallo, A. Fong, K. C. Szeto, J. Rieb, L. Delevoye, R. M. Gauvin, M. Taoufik, B. Peters and S. L. Scott, J. Am. Chem. Soc., 2016, 138, 12935–12947 CrossRef CAS.
- F. Zhang, K. C. Szeto, M. Taoufik, L. Delevoye, R. M. Gauvin and S. L. Scott, J. Am. Chem. Soc., 2018, 140, 13854–13868 CrossRef CAS.
- M. D. Korzyński, D. F. Consoli, S. Zhang, Y. Román-Leshkov and M. Dincă, J. Am. Chem. Soc., 2018, 140, 6956–6960 CrossRef.
- A. Salameh, A. Baudouin, J. M. Basset and C. Copéret, Angew. Chem., Int. Ed., 2008, 47, 2117–2120 CrossRef CAS.
- A. Salameh, A.-M. Leduc, M. Chabanas, J.-M. Basset and C. Copéret, Chim. Oggi, 2007, 25, 84–86 CAS.
- V. E. Grinev, V. A. Khalif, E. L. Aptekar and O. V. Krylov, Russ. Chem. Bull., 1981, 30, 1338–1341 CrossRef.
- A. M. J. Rost, H. Schneider, J. P. Zoller, W. A. Herrmann and F. E. Kuhn, J. Organomet. Chem., 2005, 690, 4712–4718 CrossRef CAS.
- M. Valla, M. P. Conley and C. Coperet, Catal. Sci. Technol., 2015, 5, 1438–1442 RSC.
- A. A. Gabrienko, S. S. Arzumanov, A. V. Toktarev and A. G. Stepanov, J. Phys. Chem. C, 2012, 116, 21430–21438 CrossRef CAS.
- A. H. Hoveyda, Z. Liu, C. Qin, T. Koengeter and Y. Mu, Angew. Chem., Int. Ed., 2020, 59, 22324–22348 CrossRef CAS.
- S. Halbert, C. Copéret, C. Raynaud and O. Eisenstein, J. Am. Chem. Soc., 2016, 138, 2261–2272 CrossRef CAS.
- B. T. Flatt, R. H. Grubbs, R. L. Blanski, J. C. Calabrese and J. Feldman, Organometallics, 1994, 13, 2728–2732 CrossRef CAS.
- T. Kobayashi, F. A. Perras, I. I. Slowing, A. D. Sadow and M. Pruski, ACS Catal., 2015, 5, 7055–7062 CrossRef CAS.
- C. Copéret, W.-C. Liao, C. P. Gordon and T.-C. Ong, J. Am. Chem. Soc., 2017, 139, 10588–10596 CrossRef.
- C. P. Gordon, D. B. Culver, M. P. Conley, O. Eisenstein, R. A. Andersen and C. Copéret, J. Am. Chem. Soc., 2019, 141, 648–656 CrossRef CAS.
- C. P. Gordon, S. Shirase, K. Yamamoto, R. A. Andersen, O. Eisenstein and C. Copéret, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E5867–E5876 CrossRef CAS.
- S. J. Meek, R. V. O'Brien, J. Llaveria, R. R. Schrock and A. H. Hoveyda, Nature, 2011, 471, 461–466 CrossRef CAS.
- D. P. Estes, C. Bittner, Ò. Àrias, M. Casey, A. Fedorov, M. Tamm and C. Copéret, Angew. Chem., Int. Ed., 2016, 55, 13960–13964 CrossRef CAS.
- M. Genelot, N. P. Cheval, M. Vitorino, E. Berrier, J.-M. Weibel, P. Pale, A. Mortreux and R. M. Gauvin, Chem. Sci., 2013, 4, 2680–2685 RSC.
- B. Hamzaoui, J. D. A. Pelletier, E. Abou-Hamad and J.-M. Basset, Chem. Commun., 2016, 52, 4617–4620 RSC.
- M. A. Aljuhani, S. Barman, E. Abou-Hamad, A. Gurinov, S. Ould-Chikh, E. Guan, A. Jedidi, L. Cavallo, B. C. Gates, J. Pelletier and J.-M. Basset, ACS Catal., 2018, 8, 9440–9446 CrossRef CAS.
- P. A. Zhizhko, A. V. Pichugov, N. S. Bushkov, F. Allouche, A. A. Zhizhin, D. N. Zarubin and N. A. Ustynyuk, Angew. Chem., Int. Ed., 2018, 57, 10879–10882 CrossRef CAS.
- P. A. Zhizhko, A. V. Pichugov, N. S. Bushkov, A. V. Rumyantsev, K. I. Utegenov, V. N. Talanova, T. V. Strelkova, D. Lebedev, D. Mance and D. N. Zarubin, Organometallics, 2020, 39, 1014–1023 CrossRef CAS.
- J.-M. Basset, C. Coperet, D. Soulivong, M. Taoufik and J. T. Cazat, Acc. Chem. Res., 2010, 43, 323–334 CrossRef CAS.
- C. Coperet, Chem. Rev., 2010, 110, 656–680 CrossRef CAS.
- M. K. Samantaray, V. D'Elia, E. Pump, L. Falivene, M. Harb, S. Ould Chikh, L. Cavallo and J.-M. Basset, Chem. Rev., 2020, 120, 734–813 CrossRef CAS.
- C. Copéret, F. Allouche, K. W. Chan, M. P. Conley, M. F. Delley, A. Fedorov, I. B. Moroz, V. Mougel, M. Pucino, K. Searles, K. Yamamoto and P. A. Zhizhko, Angew. Chem., Int. Ed., 2018, 57, 6398–6440 CrossRef.
- C. Copéret, A. Comas-Vives, M. P. Conley, D. P. Estes, A. Fedorov, V. Mougel, H. Nagae, F. Núñez-Zarur and P. A. Zhizhko, Chem. Rev., 2016, 116, 323–421 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc06880b |
| ‡ All authors contributed equally. |
| § Current address: Department of Chemistry, University of Oxford, Oxford, United Kingdom. |
| This journal is © The Royal Society of Chemistry 2021 |