
ADMET polymers: synthesis, structure elucidation, and function
Julia
Pribyl
 a,
Kenneth B.
Wagener
a and
Giovanni
Rojas
*ab
a,
Kenneth B.
Wagener
a and
Giovanni
Rojas
*ab
aThe George and Josephine Butler Polymer Research Laboratory, Department of Chemistry and Center for Macromolecular Science and Engineering, University of Florida, Gainesville, FL 32611-7200, USA
bUniversidad Icesi, Facultad de Ciencias Naturales, Departamento de Ciencias Químicas, Calle 18 No. 122-135, Cali, Colombia. E-mail: grojas@icesi.edu.co
First published on 7th September 2020
Abstract
Acyclic Diene Metathesis polycondensation produces materials with well-defined primary structures, perfection that is the consequence of symmetry imparted by monomer design. Diverse functionalities can be incorporated either in the polymer backbone or as pendant groups; chemical compatibility of the functional group with the catalyst is a requirement. Structural perfection led to structure/property investigations that would be difficult in imprecise systems. This comprehensive review summarizes the synthetic strategies for production of ADMET polymers. Further presented are the effects on secondary structure of controlling branch identity, in-chain functional groups, and frequency along the polymer chain. Results of spectroscopic, thermal and imaging techniques have been key for understanding the relationships between structure and properties.
1. Introduction
Acyclic diene metathesis polymerization (ADMET) is a step growth polycondensation reaction which releases ethylene as a byproduct. Elimination of ethylene by applying vacuum to the reaction vessel is the driving force of the reaction. Polymerization occurs either in bulk at high temperature up to 190 °C or with the use of solvents at considerably lower temperatures via metathesis using an appropriate catalyst.The first well-defined metathesis catalyst was created by Schrock in 1986 based on tungsten1 after which molybdenum-containing Schrock catalysts became prevalent.2 Both are most effective for producing high molecular weight polymers with degrees of polymerization higher than 150 for hydrocarbon polymers, while being less useful for polymers containing functional groups. Catalysts containing late transition metals, primarily ruthenium, developed by Grubbs and coworkers are best used when functional groups are present;3–6 their use has expanded metathesis chemistry considerably. The first-generation ruthenium catalyst, known as Grubbs’ first-generation, led to a variety of new ruthenium structures featuring diverse reactivity and functional group tolerance, while maintaining low rates of olefin isomerization and consequently, chain walking. Fig. 1 shows the structures of Schrock's molybdenum and Grubbs’ first-generation catalyst.
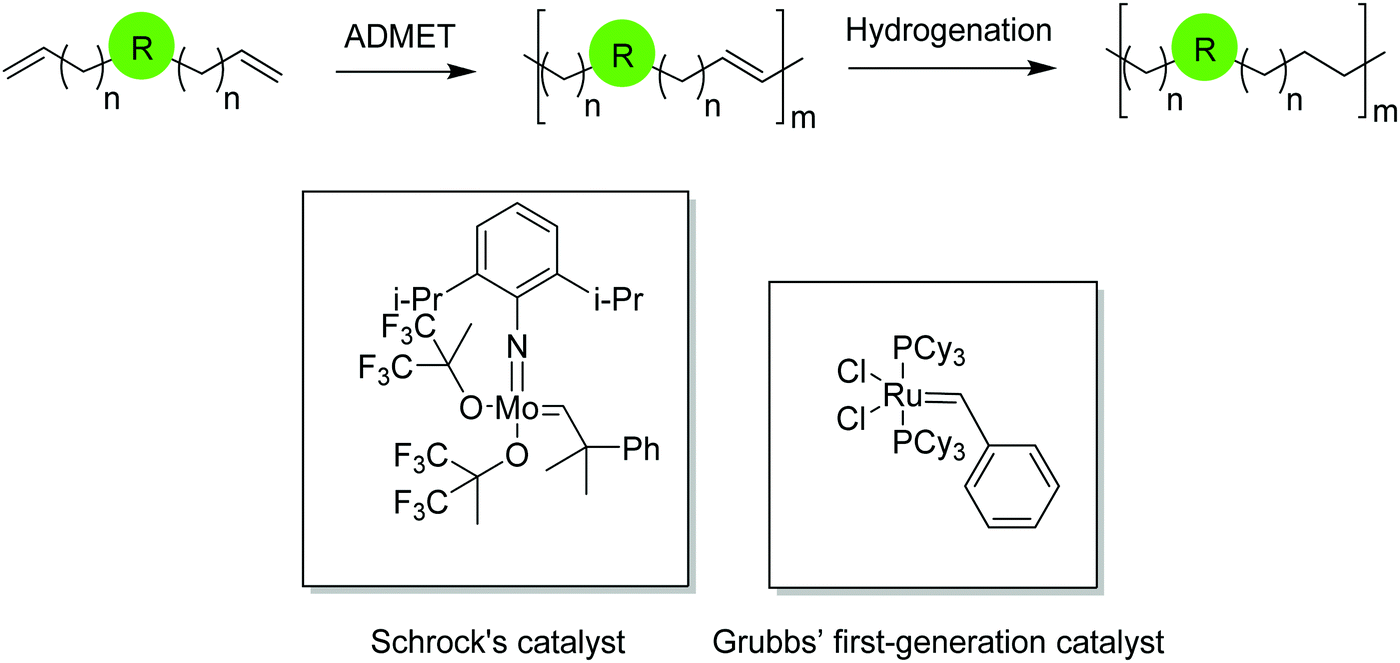 | ||
| Fig. 1 The ADMET reaction and commonly used catalysts (inset). | ||
ADMET monomers normally are α,ω-dienes that produce unsaturated ADMET polymers. The major advantage of ADMET chemistry is the formation of repeat units with symmetrically disposed functionalities (both hydrocarbon and otherwise) along the polymer backbone, a key feature that is a direct consequence of the monomer symmetry. The unsaturated polymer can be further saturated by exhaustive hydrogenation of the polymer chain. Fig. 1 shows a symmetric α,ω-diene that yields an unsaturated ADMET polymer with a generic functionality. Functionality is precisely placed along the polymer backbone by controlling symmetrically the length between terminal olefins. Depending upon the nature of the introduced functionality and the design of the α,ω-diene monomer, a diverse library of saturated and unsaturated ADMET polymers can be obtained with the functionality either incorporated in the polymer chain or pendant along the chain.
The first significant ADMET polymerization work appeared in 19914 followed by the creation of diverse polymers containing precisely placed in-chain or pendant functional groups. For example, when the R group is an alkyl chain, the resultant polymer can be seen as a model polyolefin; many other polymers containing sulfur, boron, silicon, and amino acids have been synthesized. The following sections review the synthetic methods used for the preparation of those monomers and polymers, along with discussion of secondary structure elucidation and applications of ADMET polymers as functional materials.
2. Synthesis
As is true for any step polymerization, performing an ADMET reaction is straightforward requiring highly pure monomer and quantitative conversion.5 Monomer synthesis can be accomplished in a number of ways. The following sections show different approaches for obtaining α,ω-diene monomers containing alkyl chains, halogens, carboxylic and phosphonic acids, sulfur, boron, amino acid and amphiphile branches and their use in ADMET polymerization.Polyethylene with precisely placed alkyl branches
Polyethylene (PE) is the highest volume polymer produced worldwide; its industrial production is currently an indicator of a country's development.6 While polyethylene's repeat unit usually is shown in its most simple form, in fact the primary, secondary, tertiary structures are complex. Consequently, careful studies of structure–property relationships, thermal behavior, and morphology of this polymer remain useful. Various forms of polyethylene are synthesized commercially via chain propagation chemistry using free-radical initiation, heterogeneous Ziegler–Natta catalysis, metallocene-based catalysis and most recently, late transition metal catalytic systems.7 Linear low density polyethylenes, copolymers of ethylene and α-olefins are of interest since their thermal and mechanical properties can be tuned for specific applications. The type, concentration, and distribution of the α-olefin monomer along the polymer chain determines the physical properties of the material. For example, propene, 1-butene and 1-octene result in methyl, ethyl and hexyl branches, respectively. Although polymer behavior also can be manipulated by controlling the polymerization process, LLDPE structures nicely tune material properties created by statistical incorporation of the desired comonomer.ADMET avoids the random nature of branching in polyethylene by generating precision repeat units, again, a consequence of step rather than chain polymerization methodology. The most significant early ADMET reference was reported in 1991 creating perfectly linear, nonbranched ADMET polyethylene (PE) via the polymerization of 1,9-decadiene, followed by exhaustive saturation with hydrogen.4 Since then ADMET has been used to produce functionalized unsaturated polyolefins8 as well as saturated PE models with perfect primary structures.7,9–11 The first ADMET synthetic and thermal studies yielded precision PEs with methyl, ethyl and hexyl branches;10–12 these ADMET polymers modeled commercial versions made via Ziegler–Natta or metallocene chemistry. Specifically they were precision examples duly named ethylene/1-propene (EP), ethylene/1-butane (EB), and ethylene/1-octene (EO).
ADMET monomer synthesis initially comprised reaction schemes requiring several steps.13 Since then, monomer synthesis for such alkyl branched precision polymers has been reduced to just two steps. The following sections describe the history of monomer synthesis including the preferred two step approach. The result has been the preparation of a family of alkyl α,ω-diene monomers for short chain branching (methyl, ethyl, etc.) to long chain branching (up to 21 carbons).
Monomers for modeling ethylene/1-propene copolymers
Modeling alkyl branched noncommercial LLDPE started by synthesizing an α,ω-diene possessing a methyl branch symmetrically disposed in the monomer. A series of methyl-centered monomers was obtained by varying the methylene spacers between the center carbon and the olefins in the diene (n = 2, 3, 4, 6, 8 and 9).7Fig. 2 displays the synthetic steps for obtaining such methyl α,ω-diene monomers. Ethyl acetoacetate reacted with an alkenyl bromide possessing appropriate methylene spacing to produce the disubstituted β-keto product, which was deacylated by retro-Claisen condensation to yield the ester. Reduction using lithium aluminum hydride yielded the respective alcohol, which was tosylated, followed by reduction via hydride displacement to produce the methyl symmetrical diene of interest.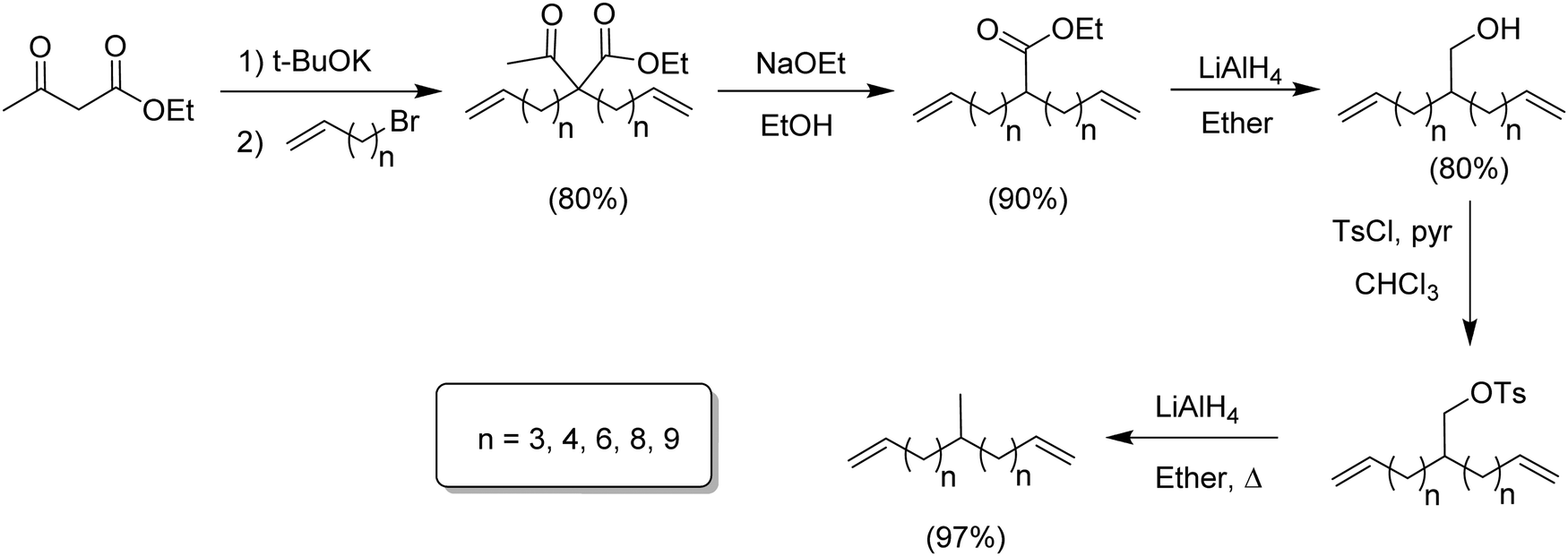 | ||
| Fig. 2 Preparation of methyl-substituted α,ω-dienes. | ||
Monomers for modeling ethylene/1-butene copolymers
The second group of alkyl monomers modeled PE bearing ethyl instead of methyl branches, again varying the number of methylene spacers along the chain. The synthetic route (Fig. 3) was comparable to that in Fig. 2, yet was modified to enhance yield.9 The synthesis started with the dialkenylation of diethyl malonate with potassium tert-butoxide, followed by decarboxylation of the resulting diacid. The monoacid was reduced to the primary alcohol and directly converted to the respective bromide using CBr4 in dichloromethane. A Grignard of the respective bromide was produced, and then a single carbon homologation was performed by the addition of dry ice yielding the respective carboxylic acid. Further reduction with lithium aluminum hydride produced the alcohol. Reaction of the alcohol with carbon tetrabromide and triphenylphosphine in dichloromethane led to the respective brominated monomer, which was then eliminated by forming the respective Grignard, followed by addition of acidic water to produce the series of ethyl-α,ω-diene monomers.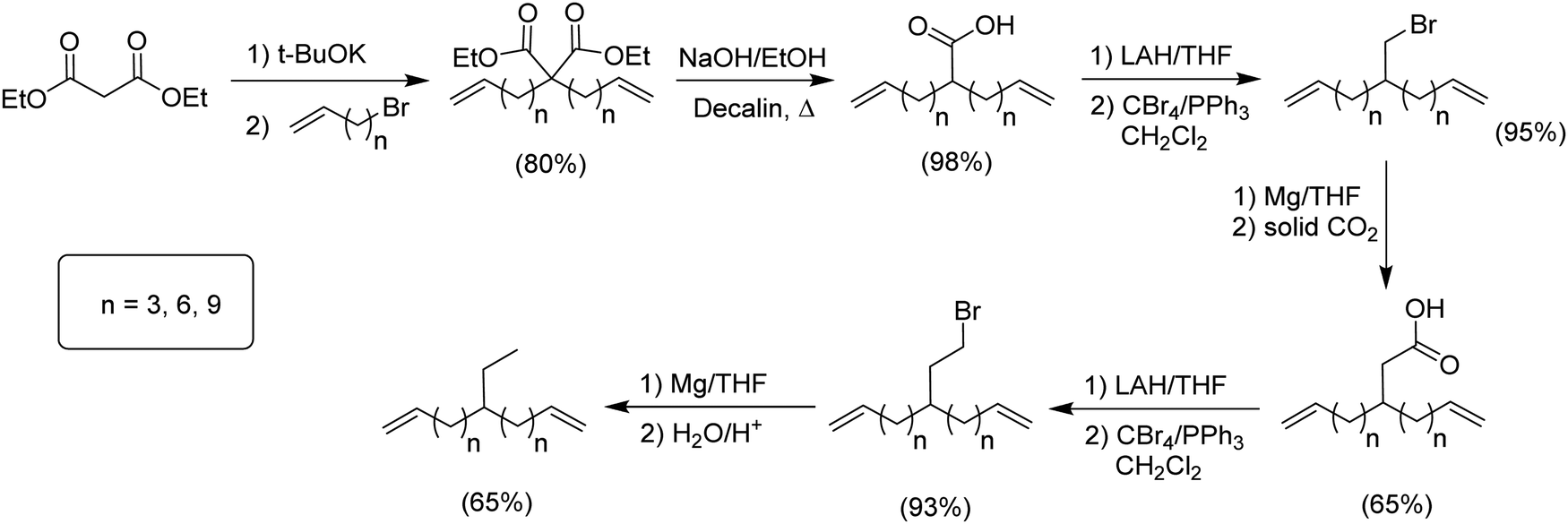 | ||
| Fig. 3 Preparation of ethyl-substituted α,ω-dienes. | ||
Monomers for modeling ethylene/1-octene copolymers
Fig. 4 defines an effort to create a universal methodology generating any alkyl branch symmetrically located in an α,ω-diene monomer, a hexyl branch used in this case to model commercial LLDPE versions.12 Starting with diethylmalonate, dialkenylation using sodium hydride and the respective bromoalkene (n = 3, 6, 9) was followed by decarboxylation of the resulting diacid. The monoacid was reduced to the primary alcohol and directly converted to the sulfonic acid ester using mesyl chloride. The major achievement of this synthetic route was the coupling carbon–carbon reaction, which as any other C–C coupling involves a metal mediated mechanism. Typically, the coupling of carbon moieties to existing structures is accomplished via a transition metal complex having a high α-olefin binding affinity using nickel or palladium, for example. However, these complexes can result in substitution competing with C–C coupling leading to undesired olefin isomerization even with metal concentrations as low as 0.5 mol%. The problem was solved using of a softer metal (copper) and low temperatures; olefin isomerization was avoided. The last coupling step involved the respective Grignard of 1-bromopentane and CuBr·S(CH3)2, yielding the respective hexyl α,ω-diene monomers in 57 to 69% yield.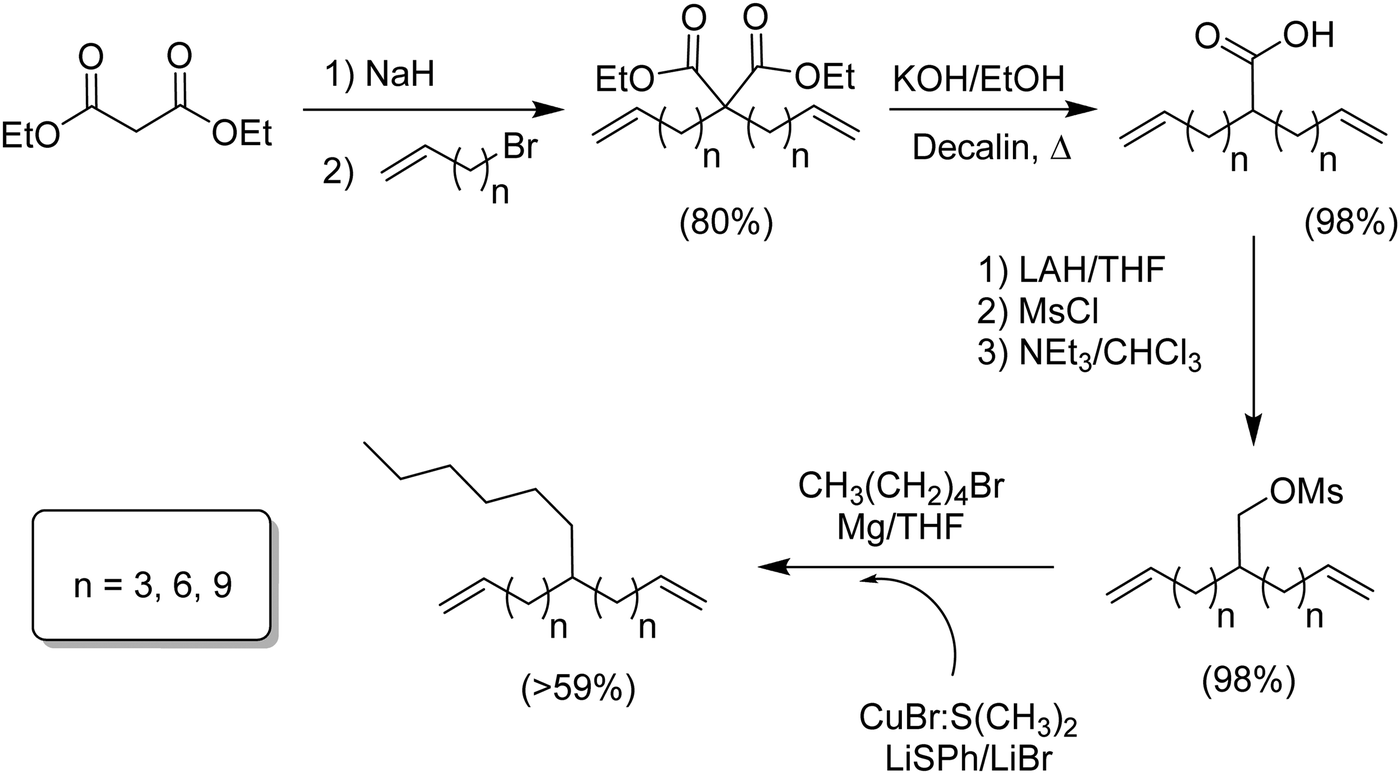 | ||
| Fig. 4 Preparation of n-hexyl-substituted α,ω-dienes. | ||
Two step method to monomers modeling ethylene/α-olefin copolymers
Although the synthetic route in Fig. 4 prepared hexyl-α,ω-dienes with any alkyl branch, yields were less than desired especially for branches longer and shorter than n-hexyl. Fig. 5 ultimately led to the most efficient monomer synthesis procedure for such monomers. Just two steps – alkylation and decyanation—resulted in nearly quantitative yields of a large number of ADMET starting monomers.14–16 The approach is based on the double alkenylation of the carbon α to the nitrile, followed by the reductive elimination of the nitrile moiety. A set of diverse alkyl-substituted monomers, from methyl to hexyl, and branched and bulkier groups such adamantyl were obtained. Alkenylation of nitriles was carried out in the presence of lithium di-isopropyl amide and alkenyl bromides creating the alkylcyano-α,ω-dienes in quantitative yields. Decyanation of nitriles was achieved with potassium metal via radical chemistry. The resulting tertiary radical after decyanation was further quenched by abstraction of hydrogen from tert-butanol, yielding the desired alkyl-α,ω-diene monomers in quantitative yields.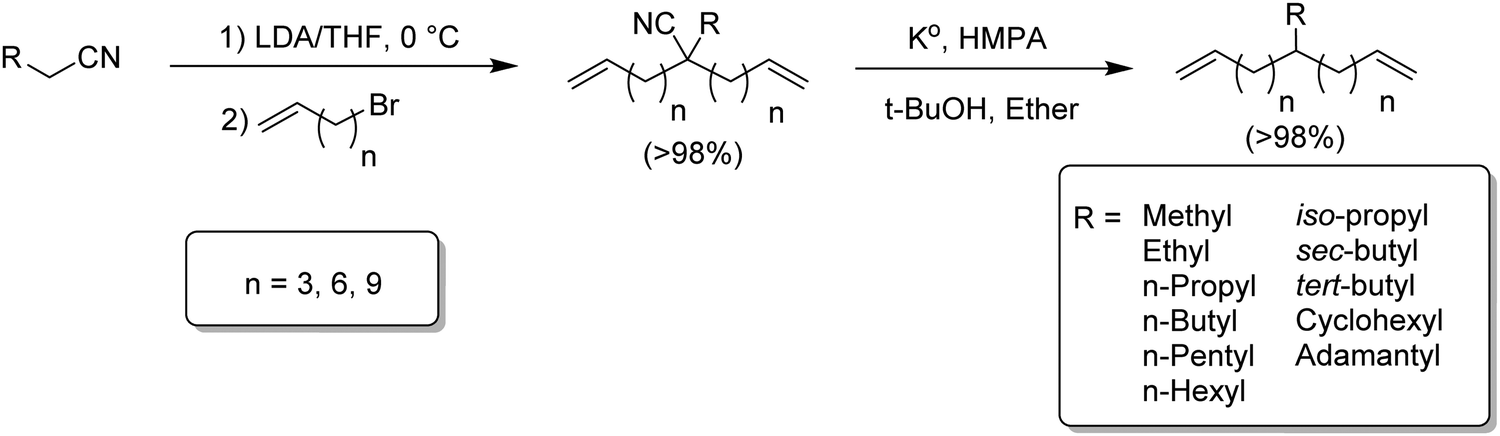 | ||
| Fig. 5 Two-step preparation of alkyl-substituted α,ω-dienes via a universal synthesis. | ||
The same alkylation/decyanation approach was employed for the synthesis of α,ω-dienes having a long chain branch (21 carbons, Fig. 6).17 which model the copolymerization of ethylene with long α-olefins. These compounds play a significant role in metallocene PE as they can help ease processability while maintaining the mechanical properties.11,18
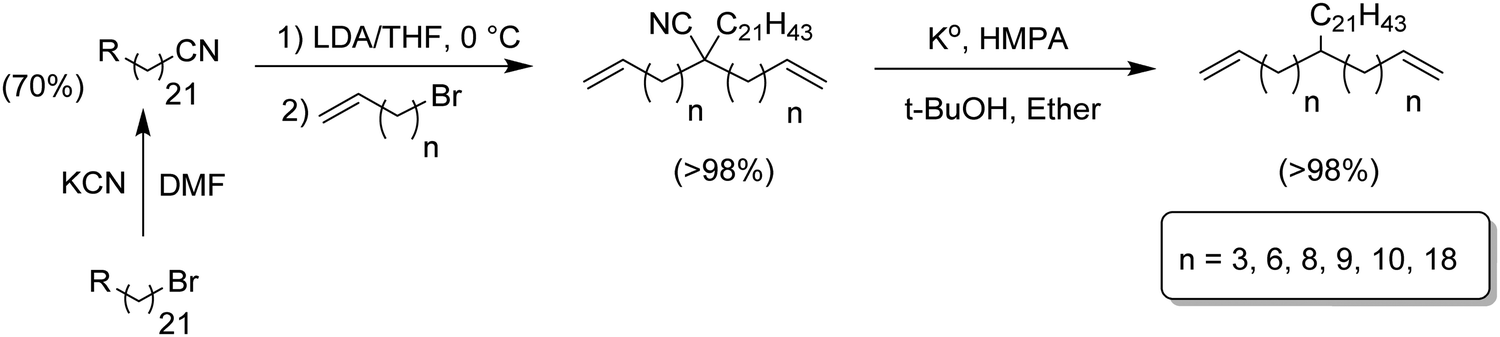 | ||
| Fig. 6 Preparation of n-henicosane-substituted α,ω-dienes. | ||
ADMET monomers and polymers with short methylene run lengths
The alkylation–decyanation universal monomer synthesis produced most precision alkyl monomers with certain exceptions. Competing cyclization chemistry can pose a special challenge when attempting to prepare ADMET polymers with short methylene run lengths between functional groups. Ring closing to form small molecules (not ADMET polymers) occurs preferentially when the formation of 5-, 6- and 7-membered rings is possible, an example of ring closing metathesis (RCM). When viewing Fig. 6, the shortest methylene spacer for ADMET chemistry is n = 3. If instead n = 1 or 2, RCM creates 5- and 7-membered rings, respectively.Overcoming this RCM phenomenon was achieved as illustrated in Fig. 7.15,19,20 Monomers contained two precisely substituted carbon centers with different length methylene run lengths present. The strategy creates an ADMET polymer that models a noncommercial LLDPE containing a methyl group on every 7th carbon (EP7, Fig. 7a), while Fig. 7b yields a methyl group on every 5th carbon (EP5). Synthesis of the requisite monomers started via monoalkenylation of diethylmalonate in the presence of sodium hydride, followed bis nucleophilic attack on 1,6-dibromohexane (Fig. 7a) or 1,4-dibromobutane (Fig. 7b) respectively. Hydrolysis using NaOH in water/ethanol yielded tetracarboxylic acids. Decarboxylation was achieved by heating the product in decalin, and further reduction with lithium aluminum hydride formed the respective diol. Reduction to the respective dialkyl-α,ω-diene was carried out by mesylation of the hydroxyl groups, followed by hydrogen incorporation with LiAlH4 in tetrahydrofuran.
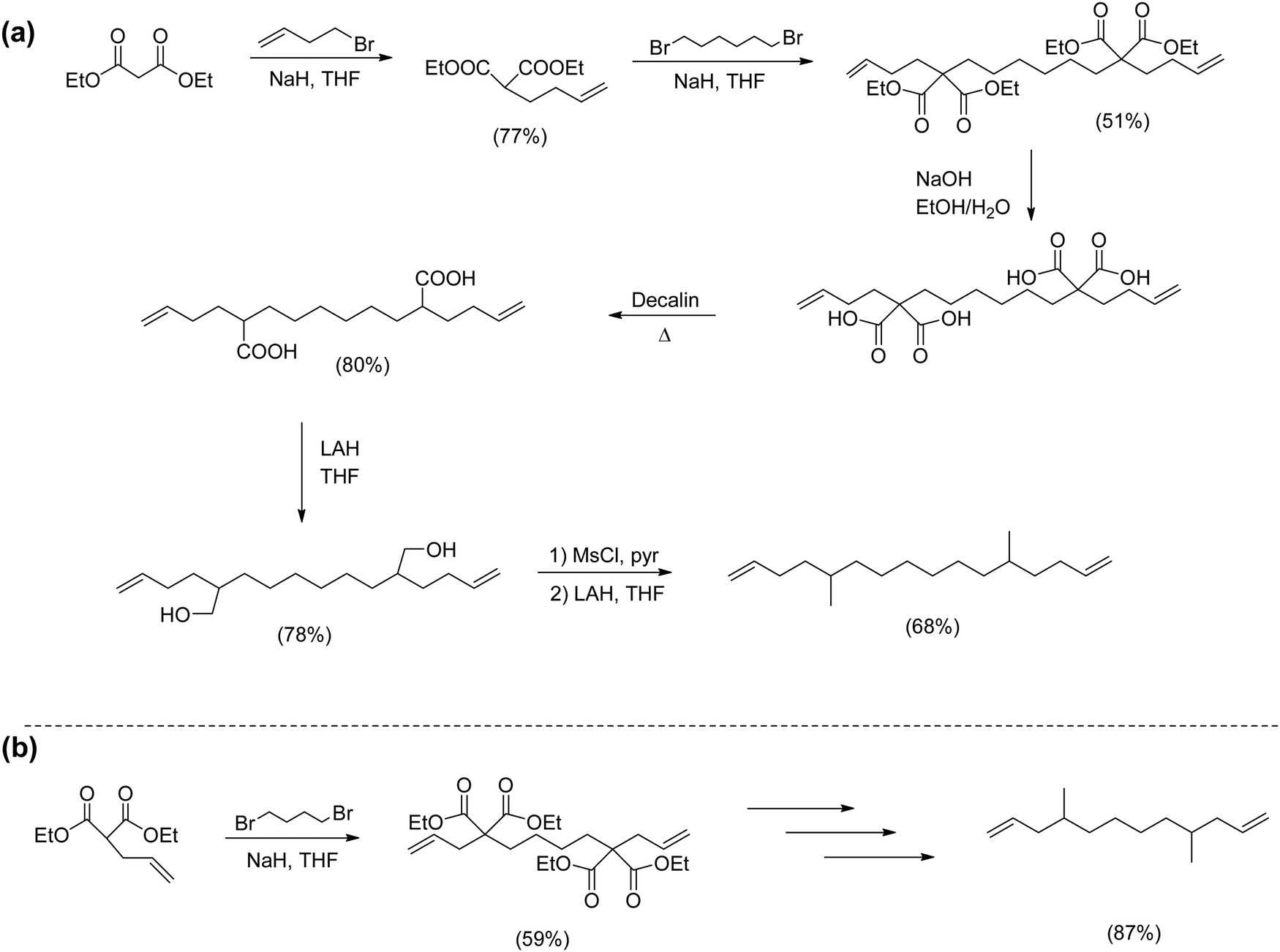 | ||
| Fig. 7 Preparation of short methylene run length methyl-substituted α,ω-dienes for modeling EP7 (a) and EP5 (b). | ||
Monomers for modeling ethylene/α-olefin copolymers with long in-chain methylene run lengths
While ADMET polymerization creating short in-chain methylene run length polymers was restricted by ring closing metathesis, synthesis of long methylene run lengths was equally interesting since long alkenyl halides are not easily commercially available. The following synthetic route represented the first systematic attempt to produce ADMET monomers and polymers modeling LLDPE with alkyl branches on every 39th carbon. Here, the synthesis of 20-bromoicos-1-ene as alkenylating agent was required.16Fig. 8 shows the synthetic steps for producing such alkyl-α,ω-diene monomers. The most important synthetic contribution involved the cross-metathesis of 11-bromoundec-1-ene, thereby doubling the chain carbon content to yield 1,20-dibromoicos-10-ene. Further hydrogenation yielded 1,20-dibromoicosane; elimination of HBr was done with potassium tert-butoxide in THF at 0 °C. Finally, the production of the respective alkyl-α,ω-diene monomers was done via quantitative alkylation–decyanation methodology, yielding a diverse set of ADMET monomers containing pendant alkyl chains from methyl to pentadecyl. Again, this chemistry is shown in Fig. 8.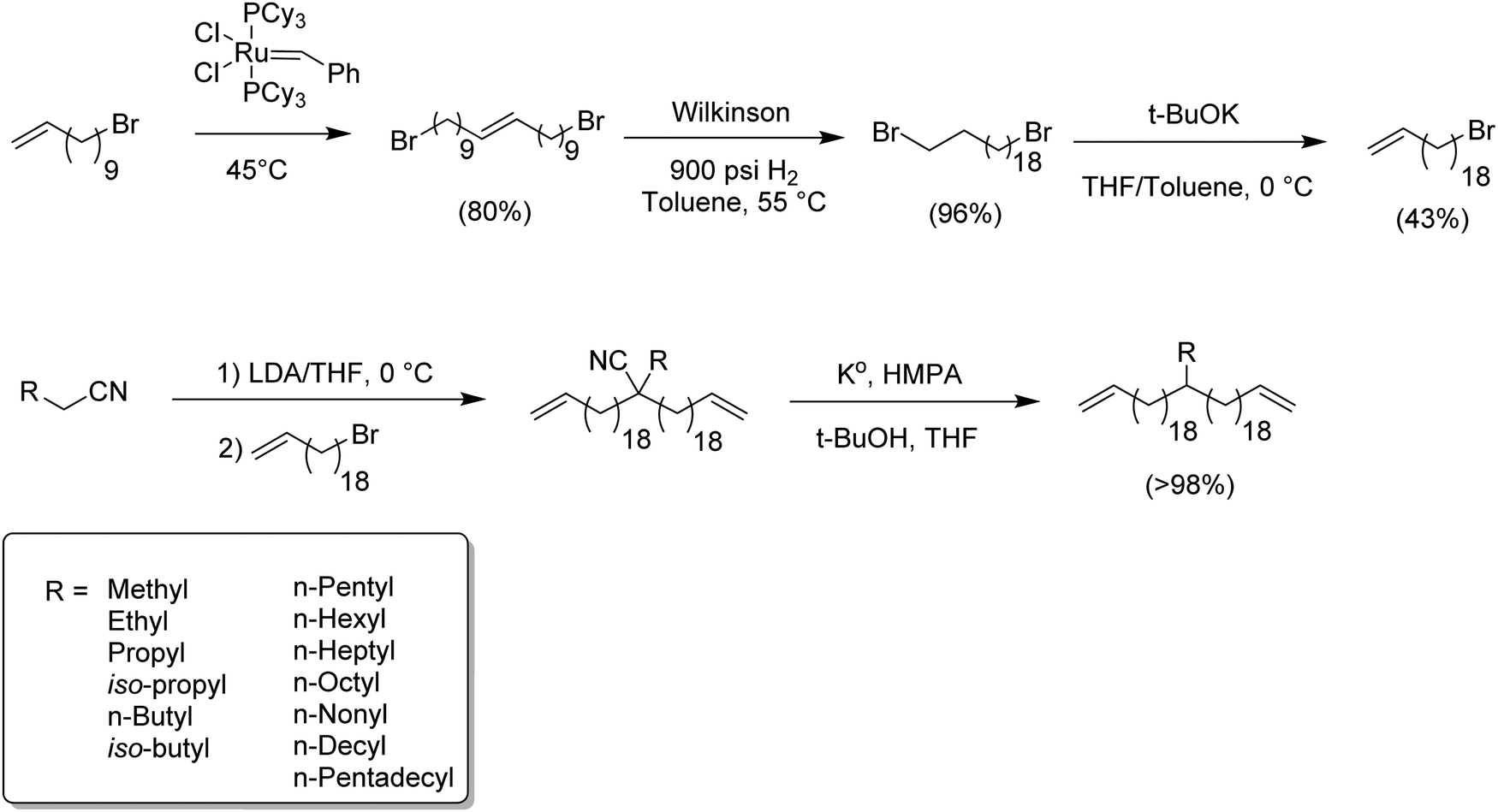 | ||
| Fig. 8 Preparation of long methylene run length alkyl-substituted α,ω-diene ADMET monomers. | ||
The second approach to synthesize long methylene run lengths was reported in 2014,21 presenting a methodology for extending the distance between the halogen and the terminal olefin alkenylating reagent. Extension of the chain was achieved for methylene units longer than 18 of them, in particular 27 and 36. Fig. 9 illustrates the synthetic approach for extending the length of the alkenyl moiety, beginning with an α-olefin-ω-ol containing p methylene units extending to p + q units using an ω-bromoalcohol containing q carbons. The appropriate α-olefin-ω-alcohol was oxidized with pyridinium chlorochromate in dichloromethane to yield the respective aldehyde. A previously prepared Grignard reagent (a protected alcohol containing q methylene units) was added to the aldehyde to produce the respective alcohol and consequently extend the main chain. Once the chain was extended the resulting product contained two alcohol functionalities, one internal and free and another terminal and protected. In general terms, the product required reduction of the free OH and final deprotection of the terminal alcohol.
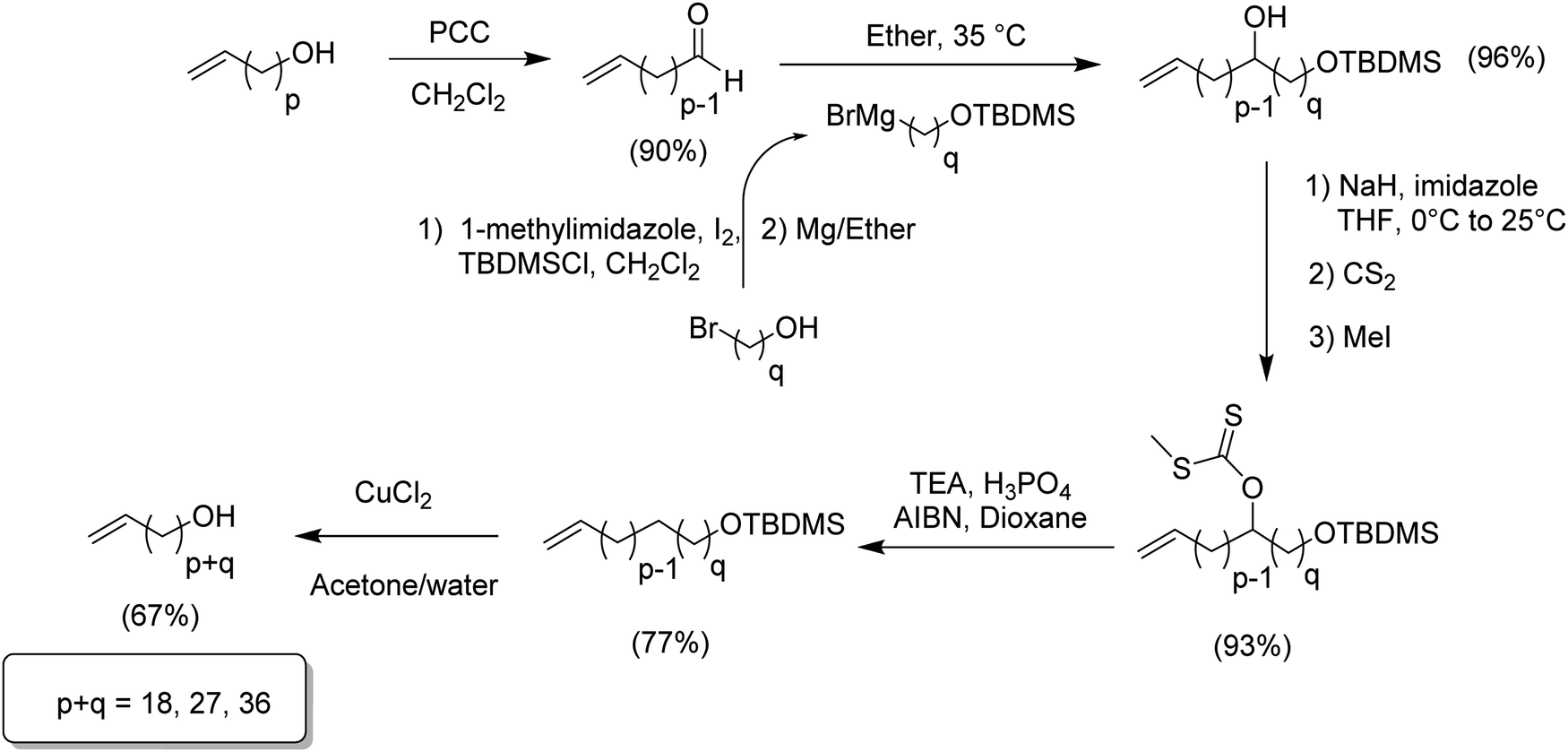 | ||
| Fig. 9 Preparation of long methylene run-length synthons for ADMET. | ||
Reduction of the internal secondary alcohol was done by forming the respective xanthate with carbon disulfide and methyl iodide, followed by a further radical termination with phosphonic acid initiated by AIBN. Finally, the resulting α-olefin-ω-protected alcohol was deprotected with copper(II) chloride in a mixture of acetone and water. The significant advantage of this methodology is that the procedure can be repeated as many times as necessary for extending the main chain as desired.
Commercial LLDPE possesses two major structural defects: incorporation of the comonomer in a random fashion and chain walking promoted by the catalyst yielding structures with branches of different lengths in the main PE chain. The ADMET approach for modeling LLDPE circumvents both problems by introducing only and exactly the branch identity selected, and by placing the desired alkyl branch exactly in a specific place along the PE chain. ADMET polyolefins have the most perfect primary structure possible.
That said, more realistic models can be prepared by systematically introducing randomness. ADMET copolymerization chemistry allows this to be so. For example, copolymerization of alkyl-α,ω-dienes with 1,9-decadiene yields a model of LLDPE that statistically distributes the alkyl branch along the PE chain. This method of randomizing ADMET model polyolefins was introduced in 2003.11Fig. 10 shows the ADMET copolymerization of 6-methylundeca-1,10-diene with 1,9-decadiene to produce unsaturated EP models, and further exhaustive hydrogenation yielded nine EP models. Different materials were obtained by simply controlling the comonomer content during polymerization: EP0 is the ADMET polymer with no alkyl branch, while homopolymerization of 1,9-decadiene, and EP-111 corresponds to the maximum possible of branches obtained by this polymerization, corresponding to homopolymerization of 6-methylundeca-1,10-diene. The suffix numbers correspond to the number of methyl branches randomly incorporated in the PE chain per thousand polymer carbons. A series of ADMET models presenting 1.5, 7.1, 13.6, 25, 43.3, 55.6, and 111 methyl branches per 1000 of carbons was reported.
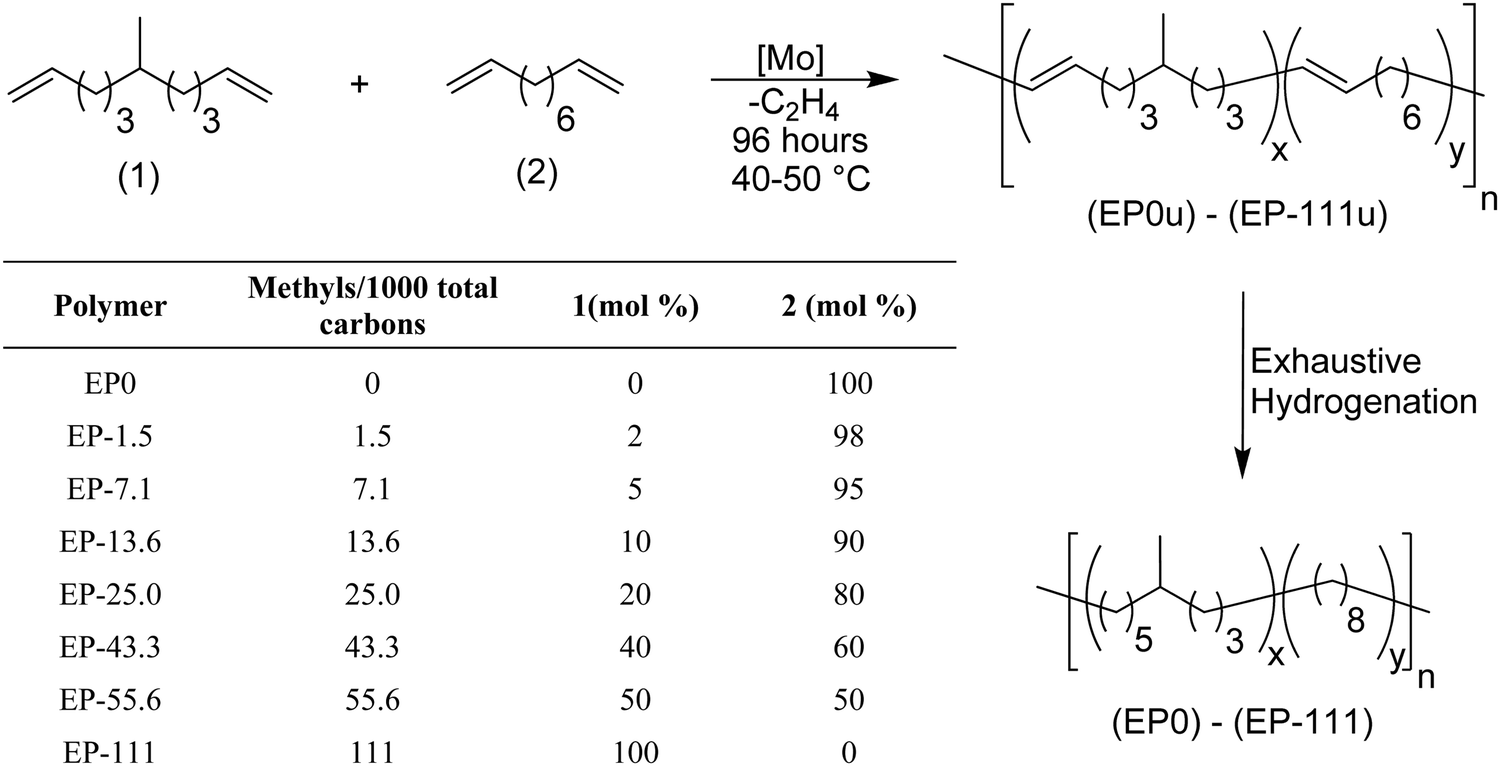 | ||
| Fig. 10 Random EP model copolymers prepared by ADMET polymerization. | ||
Using the same ADMET copolymerization approach models of ethylene/1-hexene (EH) copolymers were synthesized, i.e., polyethylene bearing a butyl branch randomly placed along the PE backbone.13,15Fig. 11 shows the copolymerization of 9-butylheptadeca-1,16-diene with 1,9-decadiene for the production of eight ADMET random EH models. By controlling the comonomer content during polymerization, model polymers were produced containing 2.5, 6, 11.5, 21.3, 37, 43.5 and 66.7 butyl branches per thousand PE carbons.
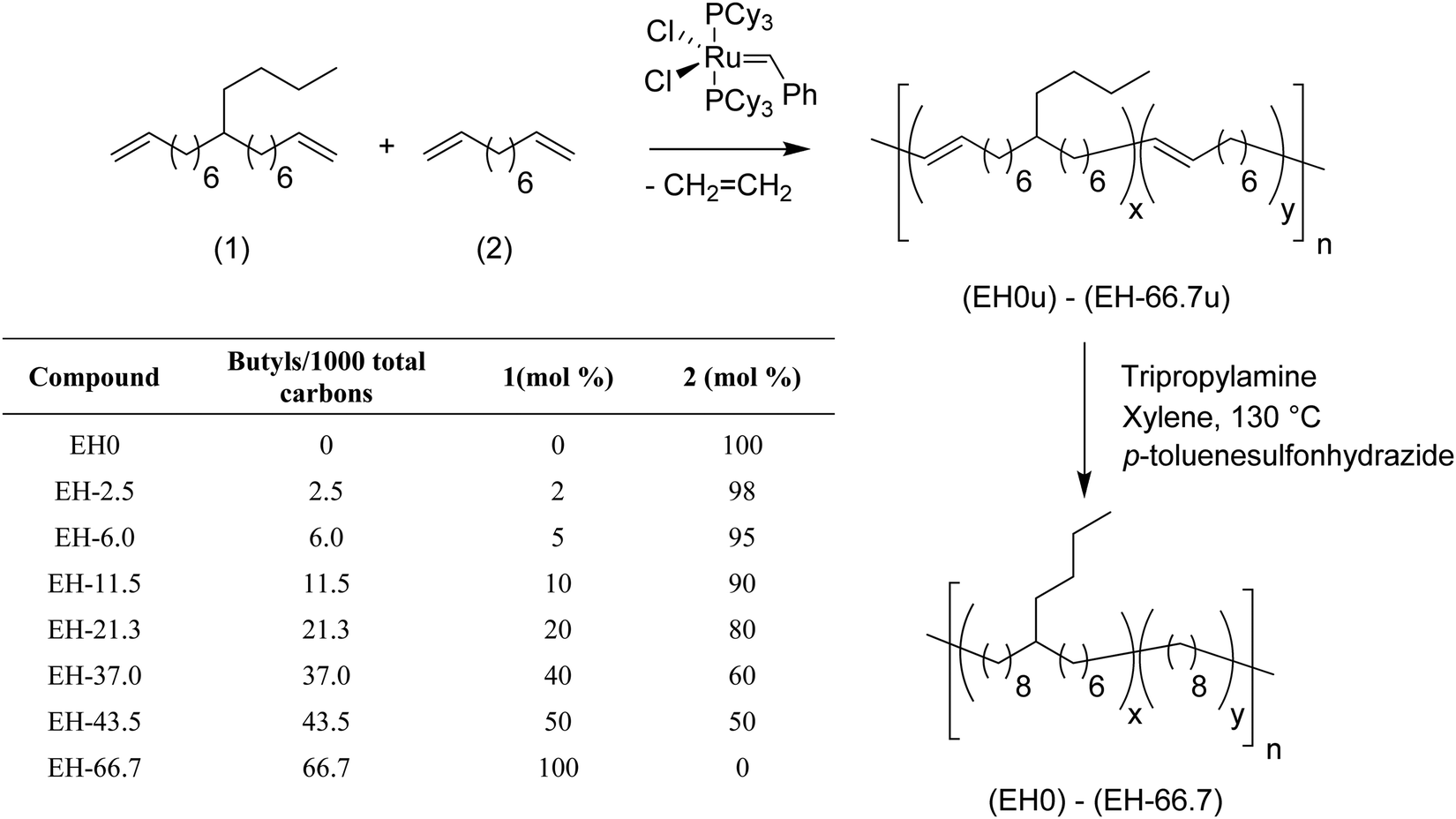 | ||
| Fig. 11 Random EH model copolymers prepared by ADMET polymerization. | ||
While these previous systematic studies of random methyl and butyl branches expanded the scope of ADMET polyolefins, it was important to prove that ADMET polymers containing longer branches could be modeled. Fig. 12 shows the same synthetic approach, but now containing a randomly placed pendant alkyl branch of 21 carbons. Thus an ADMET model of ethylene/1-tricosene (ETC) copolymer was generated in this manner.17 Control over the comonomer content during polymerization yielded materials containing 2.4, 5.8, 13.4, 22.2, 34.5 and 48 long branches (21 carbon branches) per thousand PE carbons.
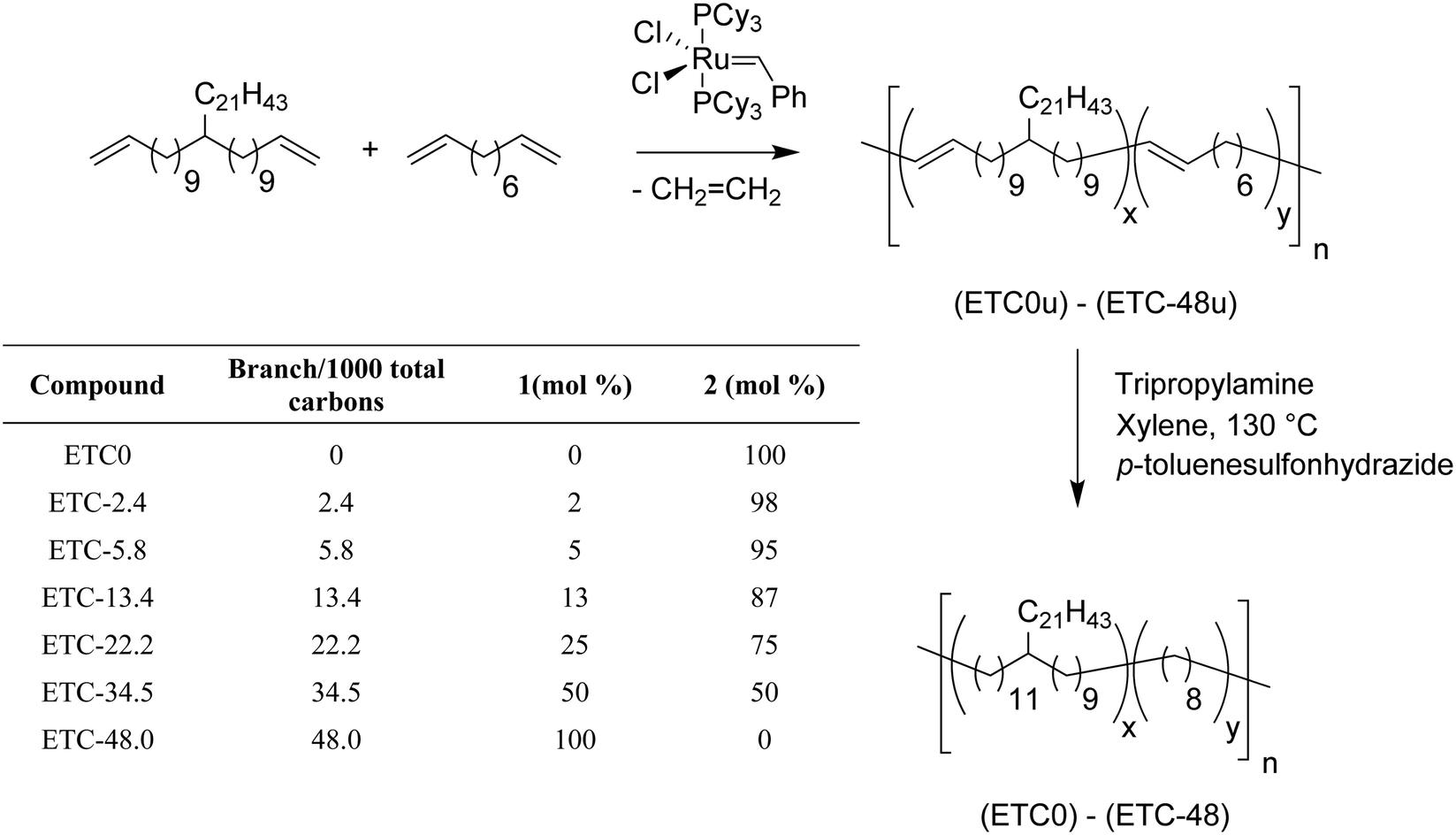 | ||
| Fig. 12 Random ETC model copolymers prepared by ADMET polymerization. | ||
ADMET polymers containing halogens
Although modeling ethylene/α-olefin copolymers was first of highest priority, modeling of other ethylene copolymers naturally followed. Ethylene/vinyl halide model copolymers were of interest due to the structural precision ADMET provides.22–24 Ethylene/vinyl chloride copolymers offer improved thermal stability relative to PVC; ethylene/vinyl fluoride polymers are of great commercial value, and ethylene/vinyl bromide copolymers are structurally important but relatively understudied. Fig. 13a shows three synthetic routes for the preparation Br, Cl and F containing α,ω-dienes, and Fig. 13b shows their respective ADMET polymerization, followed by exhaustive hydrogenation.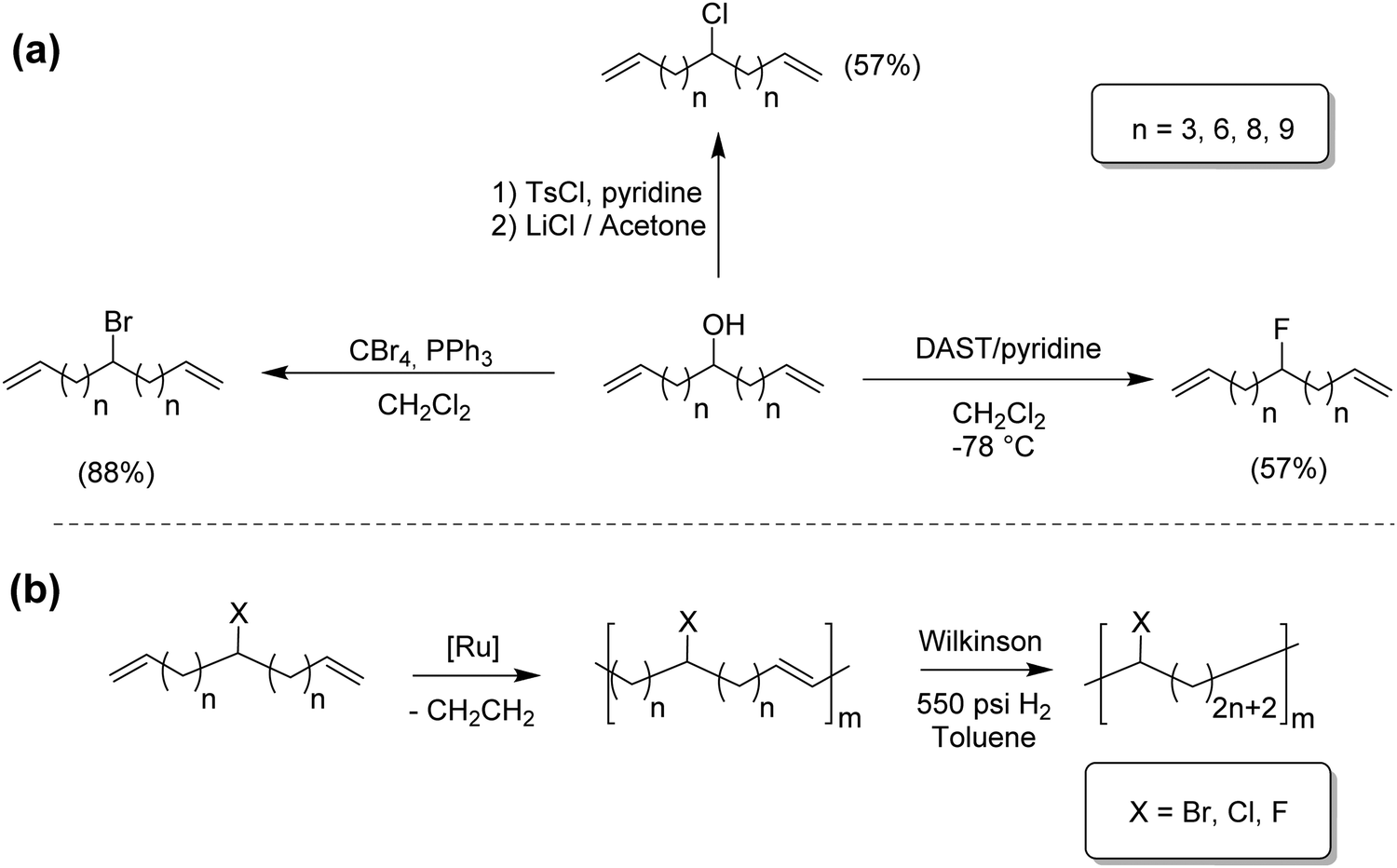 | ||
| Fig. 13 Preparation of halogen-substituted α,ω-diene monomers (a) and resulting ADMET polymers (b). | ||
All monomer syntheses started from a common material, an α,ω-diene-alcohol: the chlorinated monomer by substitution of the respective tosylated monomer with lithium chloride, the brominated monomer by addition of carbon tetrabromide and triphenylphosphine in dichloromethane, and the fluorinated monomer by adding diethylaminosulfur trifluoride in pyridine and dichloromethane at −78 °C.
ADMET polymers containing carboxylic acids
While the two step synthesis scheme worked well for alkyl-α,ω-dienes,9 study of ADMET modeling of ethylene/acrylic acid copolymers (EAA) required a different approach.25 In this case, free carboxylic acids inactivate both ruthenium and molybdenum catalysts. Circumvention was achieved by protecting the acid group; Fig. 14 outlines the approach to EAA copolymers. Ethyl vinyl ether, which alone poisons Grubbs catalysts, indeed protects the acid group quite well; ADMET polymerization was smoothly achieved employing Grubbs’ first-generation catalyst. Deprotection and olefin saturation was carried out simultaneously by hydrogenation at 550 psi in toluene using Wilkinson's catalyst yielding precision carboxylic acid containing polyolefins. Three different EAA models were obtained containing a carboxylic acid group on every 9th, 15th and 21st carbon. Conventional EAA copolymers are of industrial importance as are their neutralized versions, called ionomers, where precision versions might be used as battery electrolytes. A set of two different ionomers was obtained by neutralizing the acid polymer with sodium acetate or zinc acetate, yielding the respective sodium and zinc ionomers.26,27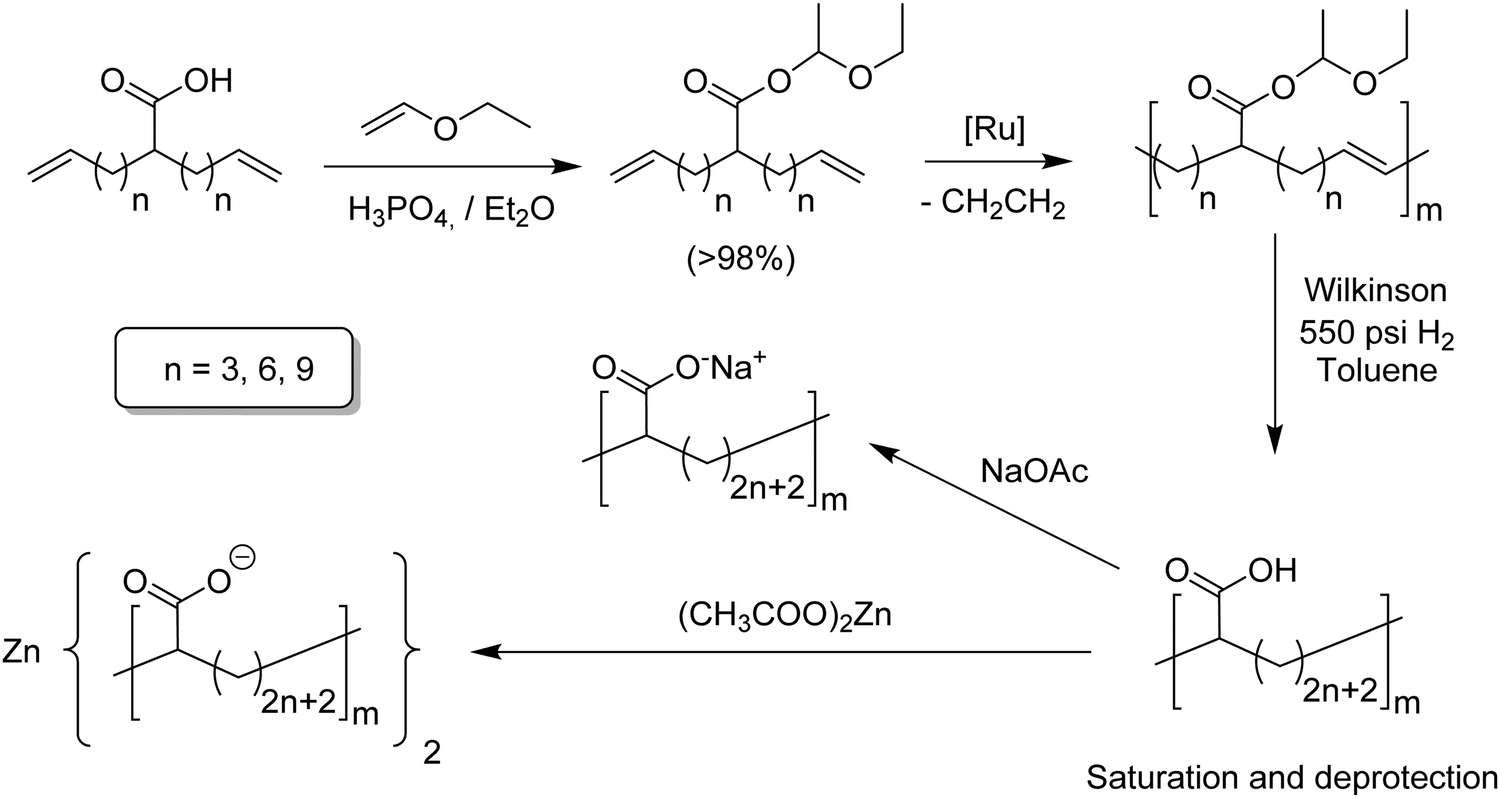 | ||
| Fig. 14 Preparation of EAA copolymers via ADMET polymerization and their respective sodium and zinc ionomers. | ||
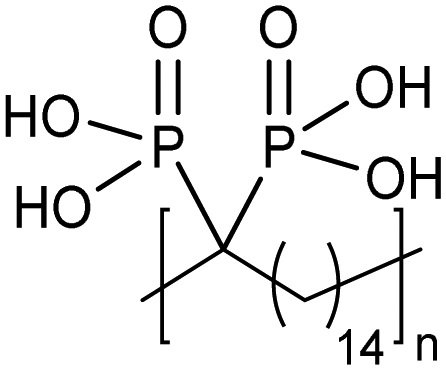
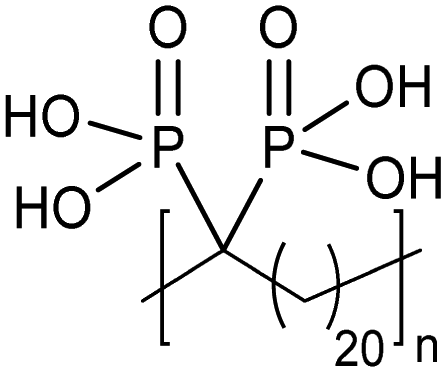
ADMET polymers containing phosphonic acids
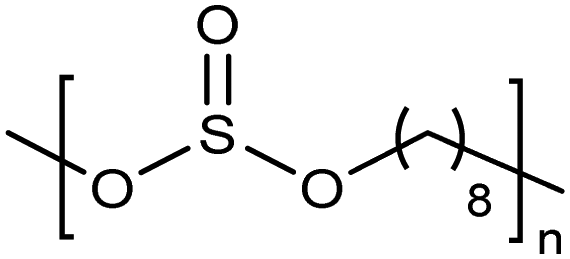
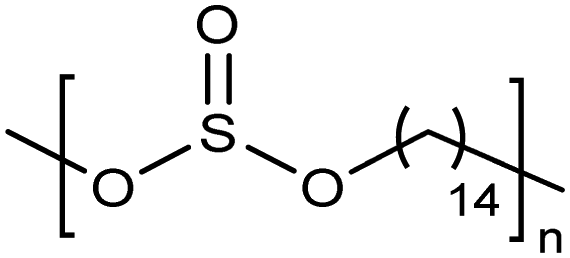
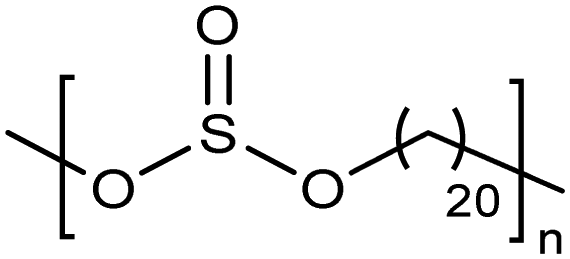
Opper et al., expanded the carboxylic acid chemistry to include phosphonic acid moieties;28,29Fig. 15 shows three approaches for synthesis of such phosphonic acid ADMET polymers. Fig. 15a illustrates the production of ADMET polymers with a phosphonic acid on every 7th, 15th and 21st carbon. Monomer synthesis started from the alkenylation of diethyl methylphosphonate with LDA, followed by ADMET polymerization with Grubbs’ first-generation catalyst, that step followed by saturation of the double bond with Wilkinson's catalyst. Deprotection of the phosphonate to yield the respective phosphonic acid polymer was performed using bromotrimethylsilane in methanol. Fig. 15b shows the production of a geminal phosphonic acid by the same approach but instead starting from tetraethyl methylenediphosphonate treated with sodium hydride. Fig. 15c depicts the production of a polymer containing a benzyl ring bearing a methyl phosphonic moiety. Synthesis starts with double etherification of the phenol groups with 3,5-dihydroxybenzyl alcohol with potassium carbonate and potassium iodide, followed by conversion of the free hydroxyl groups to phosphonate esters and further ADMET polymerization, saturation and deprotection.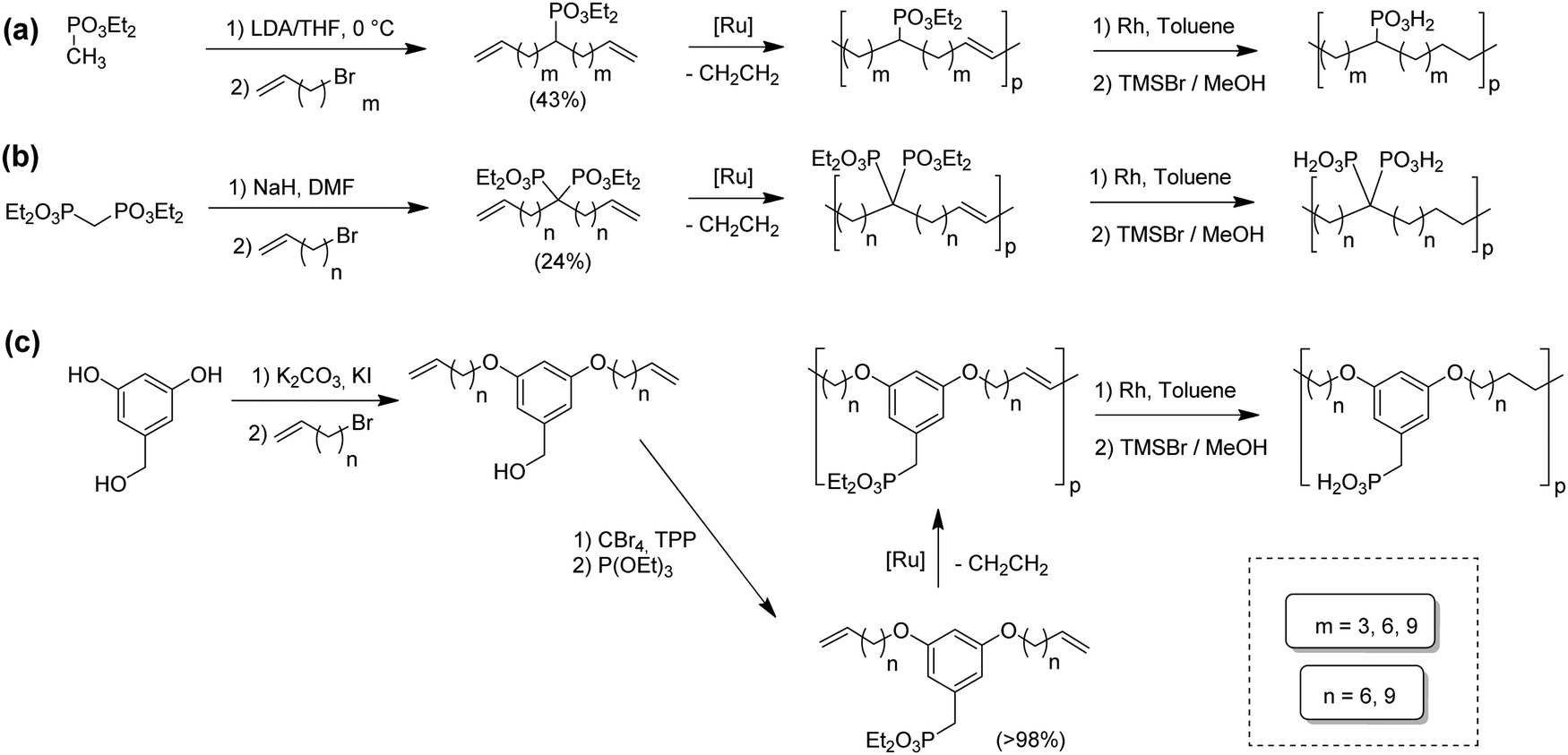 | ||
| Fig. 15 (a)–(c). Preparations of ADMET polymers bearing phosphonic acid moieties. | ||
ADMET polymers containing sulfonic acid groups
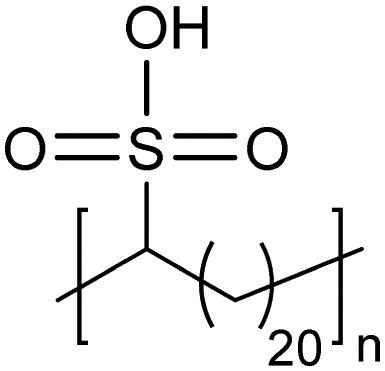
Similar to carboxylic and phosphonic acid polymers, sulfonic acid polymers such as Nafion® are excellent proton exchange membranes.30 Commercially produced sulfonic acid polymers lack definition and uniformity, inevitable features induced by defects arising from the random polymerization and, more importantly, incomplete sulfonation after polymerization.31,32 Consequently, ADMET polymerization of monomers containing sulfonates and sulfonic acids create perfect primary structures to model commercially produced materials.33,34Fig. 16 shows the synthetic pathway for the production of polyethylene containing sulfonic acid groups on every 7th and 21st carbon. Monomers were obtained by alkenylation of ethyl methanesulfonate with lithium diisopropylamide. After ADMET polymerization using Grubbs’ first-generation catalyst and further exhaustive hydrogenation, deprotection of the sulfonated polymer was attempted using sodium methoxide in methanol, then to be followed by protonation with hydrochloric acid.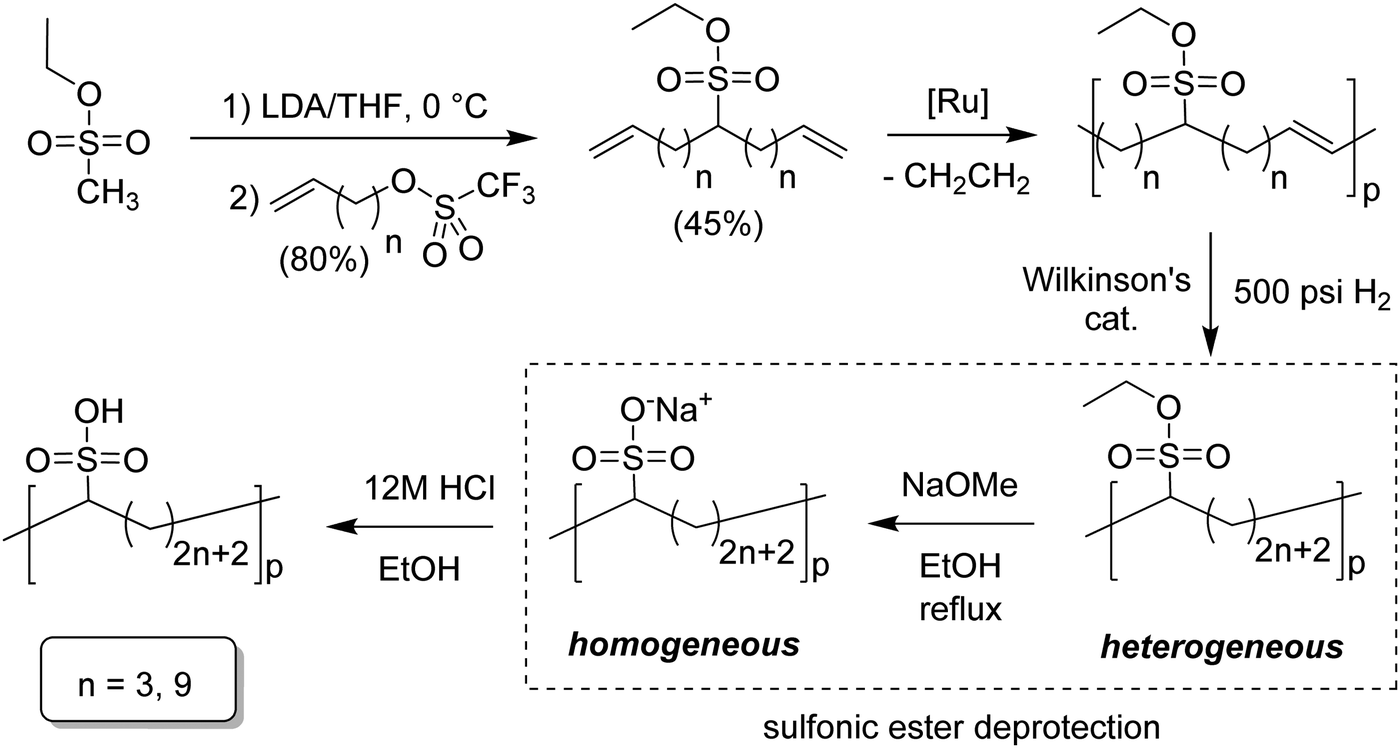 | ||
| Fig. 16 Preparation of precision sulfonic acid-functionalized polyolefins via ADMET. | ||
As suggested above, deprotections typically are done homogeneously in organic solution (e.g., tetrahydrofuran, toluene, dichloromethane, etc.). However, sulfonic ester polymers deprotected in this way produced a partially deprotected mixture of sulfonic ester and sulfonate functionality along the polymer backbone, resulting in a loss of precision. Partial deprotection occurred because while the sulfonic ester exhibits good solubility in organic solvents, as hydrolysis to the sulfonate occurred solubility decreased and the polymer began to precipitate, rendering full deprotection infeasible.
Quantitative heterogeneous-to-homogeneous deprotection: ADMET polymers containing sulfonic acid functional groups
Precisely sulfonated polyethylenes finally were synthesized via a new concept called “heterogeneous-to-homogeneous deprotection”.33 Initially, the ester-protected polymer was floated on a solution of wet dimethylsulfoxide (DMSO) (the protected polymer was insoluble), and sodium hydroxide pellets were added to the heterogeneous mixture at 80 °C. Gradually, the sulfonic esters were converted to the sulfonic acid salt. As this process progressed, the polymer became increasingly soluble, in turn further enabling the deprotection chemistry. Eventually, the deprotected polymer became completely soluble, and the reaction solution became completely homogeneous. Deprotection was quantitative.Due to the difficulty of fully removing DMSO after deprotection, a more convenient procedure involving refluxing a heterogeneous polymer/ethanol solution in the presence of sodium methoxide was devised to achieve the same result (emphasized in Fig. 16). Infrared spectroscopy of the deprotected polymer displayed no residual sulfonic ester stretches, indicating quantitative deprotection.
ADMET polymers containing sulfone groups via solution chemistry
While sulfonic acid ADMET polymers exhibit a sulfur moiety pendant on the main polymer chain, sulfone polymers present the sulfur atom in the main polymer chain. Commercial aromatic polysulfones normally exhibit excellent chemical and thermal stability for applications in the aerospace, medical, and automotive industries.35Fig. 17 shows the preparation of three ADMET aliphatic polysulfones that contain a sulfone group on every 7th, 15th and 21st position along the main chain. High melting aliphatic polysulfones were unknown prior to this work. Preparation of sulfones started with a double addition of sulfide to an alkenyl bromide, followed by oxidation of the sulfur atom to the respective sulfone with hexachlorophosphazene in water and hydrogen peroxide.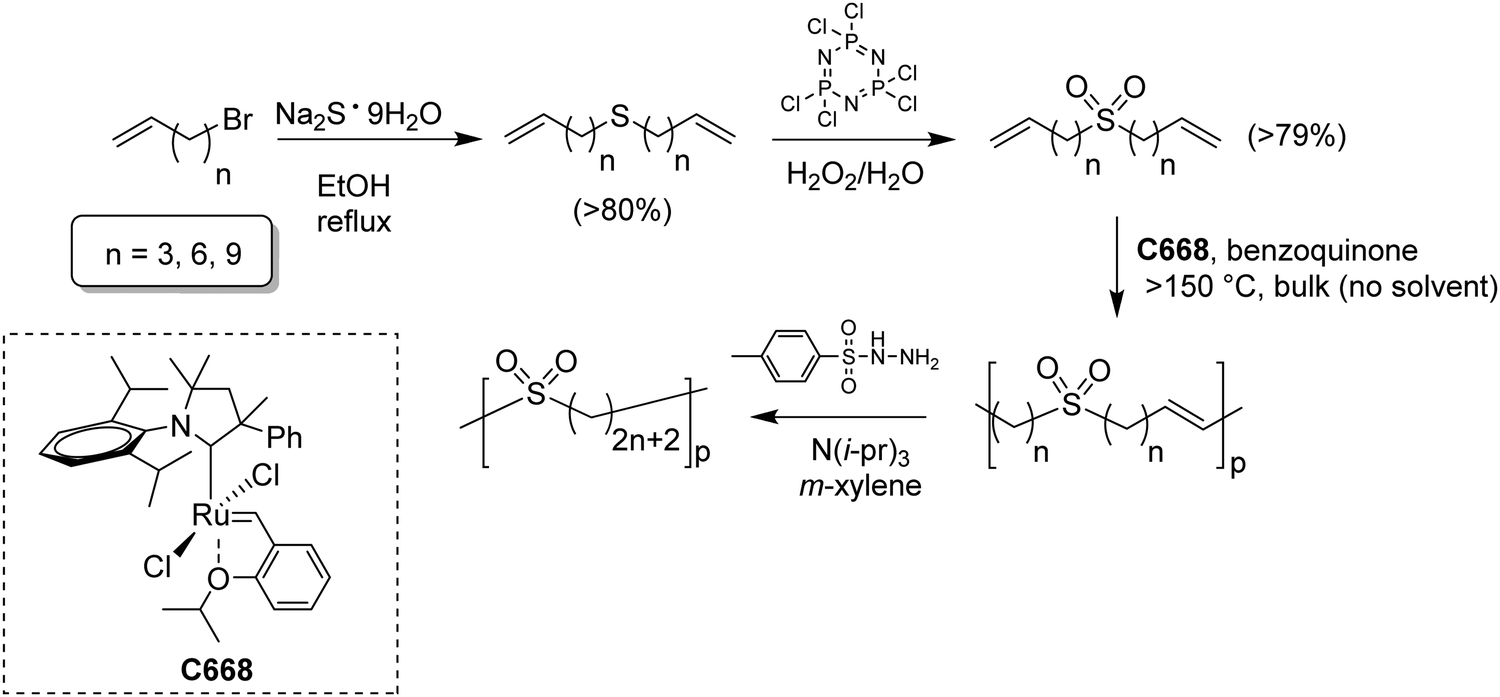 | ||
| Fig. 17 Preparation of symmetric sulfone-α,ω-dienes and bulk high temperature ADMET polymerization of high Tm precision polysulfones. | ||
The first ADMET polymerizations of sulfone-α,ω-dienes, done in solution with Grubbs’ first-generation catalyst, yielded low molecular weight polymers due to limited solubility of the precision polysulfones in an organic solvent.35 Even so, the saturated precision polysulfones exhibited high melting points (up to 175 °C), inspiring further investigation.
High temperature bulk metathesis polycondensation yielding aliphatic polysulfones
High temperature bulk (no solvent) metathesis polycondensation avoids the solubility issues mentioned above, producing high molecular weight precision polysulfones. Bulk metathesis polycondensation at high temperature had never been attempted previously. It became essential to identify a metathesis catalyst which was stable above the melting point of the resulting unsaturated precision polysulfone (>130 °C) so as to keep the reaction mixture in the melt during polymerization. A high temperature-stable catalyst (see Fig. 17 inset) from a class of cyclic alkyl amino carbene (CAAC)-ligated ruthenium catalysts reported by Bertrand and Grubbs36 was selected. Bulk high temperature (up to 190 °C) ADMET polymerization of precision polysulfones with the specified CAAC ruthenium catalyst generated precision polysulfones with Mw values up to 87 kDa.37 A small amount of benzoquinone (ca. 4 mol%) added to the polymerization mixture effectively suppressed undesired 1,2-olefin isomerization reactions, leading to precisely spaced sulfone groups. Bulk polycondensation of this nature mimics large scale, commercial processes such as those used to produce polyester.ADMET polymers containing boron
Polymers having boron moieties as pendant groups have been used as catalyst supports, glucose sensors, and drug delivery vehicles, as well as in film technology and other areas.38–41 Boron-containing molecules are Lewis acids, which have proven to increase metathesis yield while decreasing olefin isomerization. Fig. 18 shows two pathways for the synthesis of boron-containing polymers: at the top an ADMET polymer with a boronic ester pendant to the main chain, and at the bottom an in-chain phenyl boronic acid polymer.42,43 The monomer for the reaction in Fig. 18a was obtained from the respective alcohol-α,ω-diene with sodium hydride and 4-benzyl bromide to form the respective ether. The boronic ester was then attached using a Grignard reagent with 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane. Monomer for the in-chain phenyl boronic acid polymer, in Fig. 18b, was carried out by reacting an alkenyl bromide with 1-bromo-3,5-dihydroxybenzene, followed by introduction of the boronic acid via a standard lithiation procedure; triisopropyl borate was used as the boron source, yielding the free boronic acid monomer.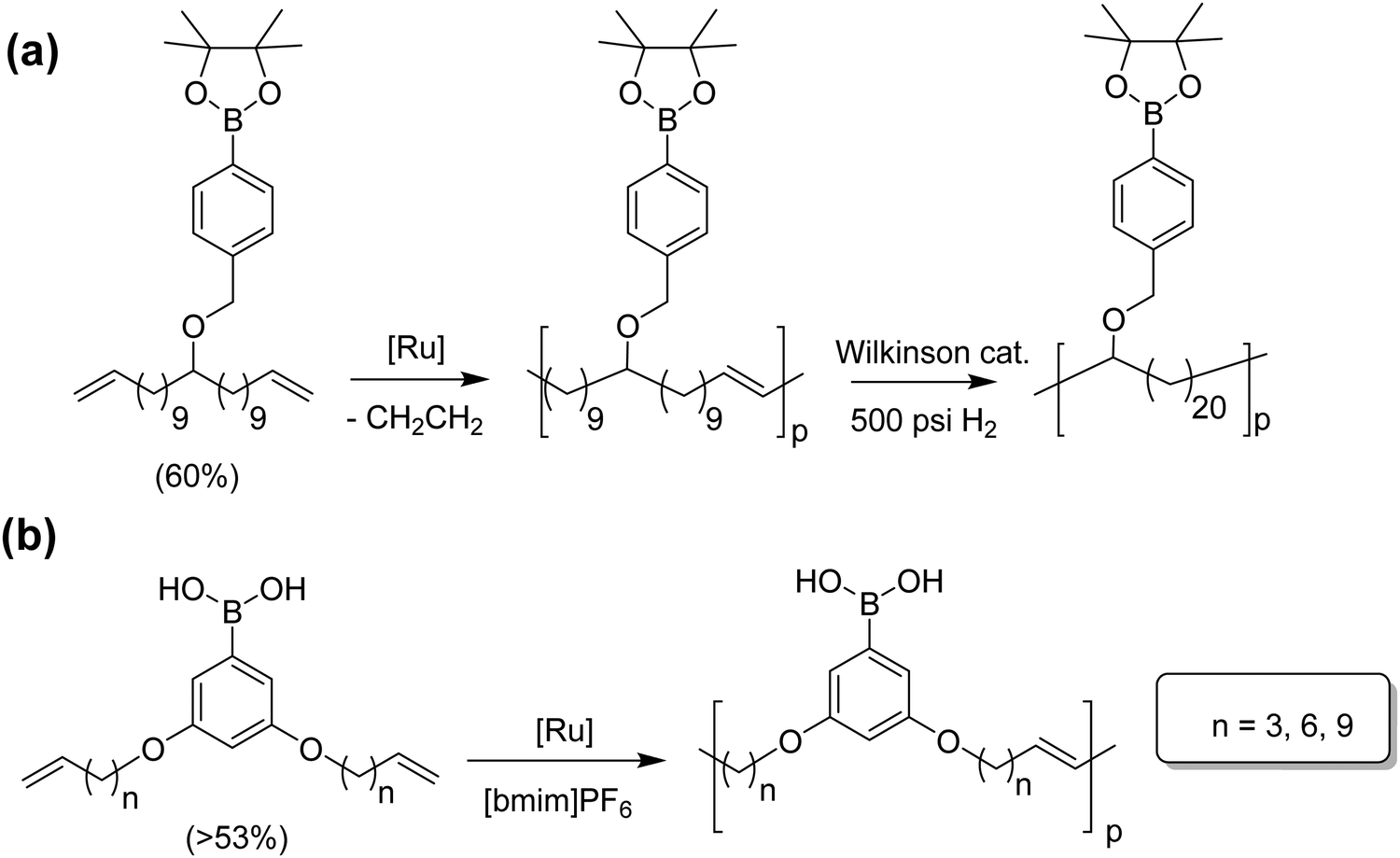 | ||
| Fig. 18 Preparation of boronic ester-containing polymer (a), and phenyl boronic acid polymer (b) via ADMET polymerization. | ||
ADMET polymers containing silicon
Silicon has a rich and known chemistry; more importantly, silicon is compatible with the catalysts used for ADMET polymerization. Silicon atoms can be incorporated in ADMET monomers and polymers as part of diverse functionalities, including carbosilanes44,45 and carbosiloxanes,46 both in-chain, pendant along the polymer chain, or even as terminal groups, as in telechelic polymers.46,47Fig. 19 shows the synthesis of an ABA block copolymer by the ADMET polymerization of 1,9-decadiene, followed by end-capping with dimethylchlorosilane. ABA block formation was carried out via double substitution of the terminal chlorine (block B) by the terminal OH from polydimethylsiloxane (PDMS, block A).47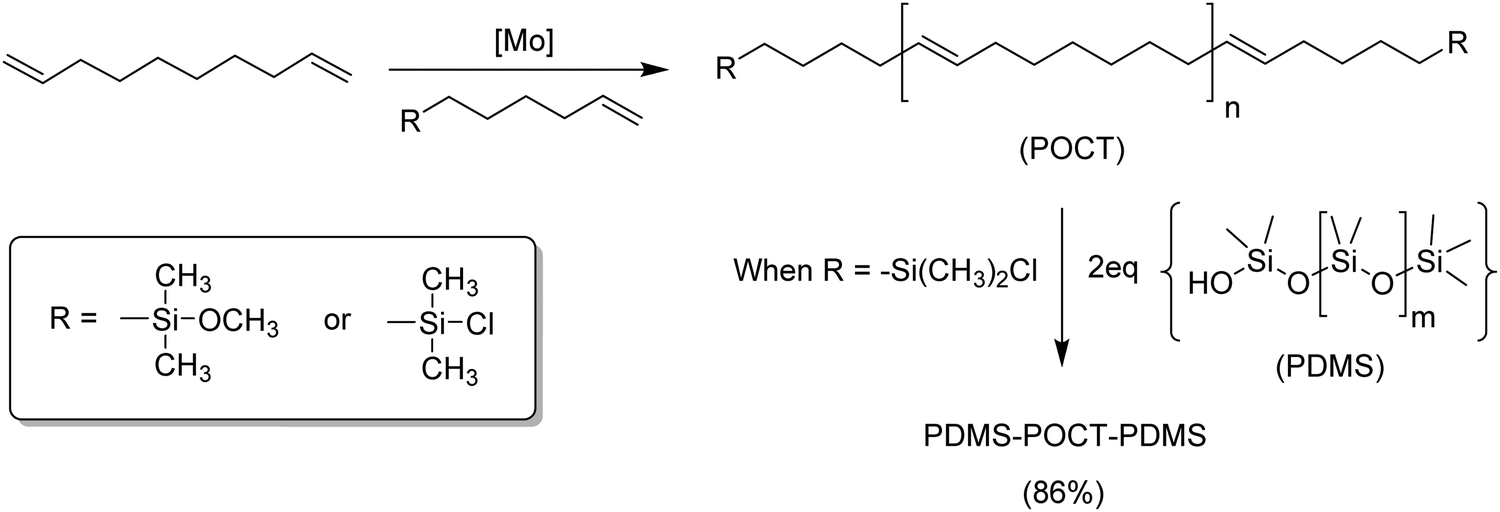 | ||
| Fig. 19 Preparation of telechelic polyoctenamer oligomers using ADMET chemistry (top) and copolymerization of the telechelic polymer with polydimethylsiloxane (bottom). | ||
ADMET polymers containing carbosilanes were obtained by addition of magnesium in ether to 5-bromopent-1-ene to prepare the respective Grignard reagent, which was added to silicon tetrachloride to form the disubstituted product, as shown in Fig. 20. Dichlorosilane-α,ω-diene monomer was polymerized using Schrock's molybdenum catalyst. After polymerization the material was transformed into the respective dimethyl, diphenyl, or dibutyl polycarbosilane by simply adding the corresponding alkyl lithium.44,45 Both Schrock's and Grubbs’ catalysts have been used for production of ADMET polycarbosiloxanes, yielding diverse materials and applications.46Fig. 21 shows an example of six carbosiloxane monomers, all synthesized using Grignard techniques and the appropriated chlorosilane, that were easily polymerized via ADMET.
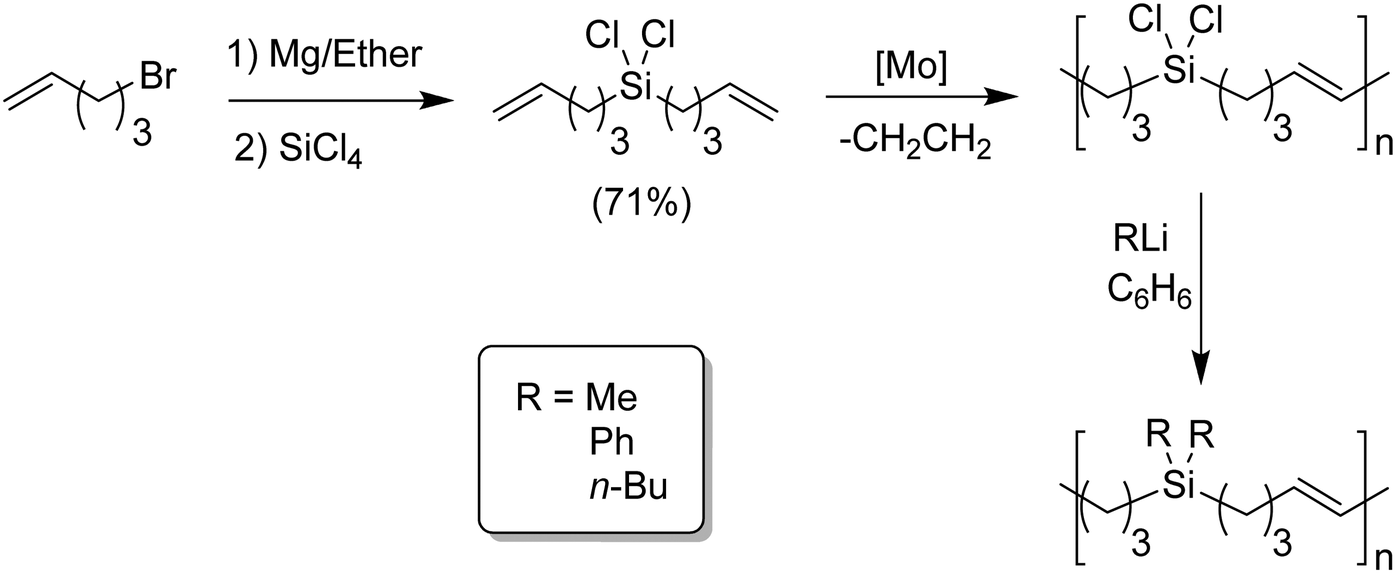 | ||
| Fig. 20 Preparation of polychlorosilanes and subsequent polysilanes via ADMET polymerization. | ||
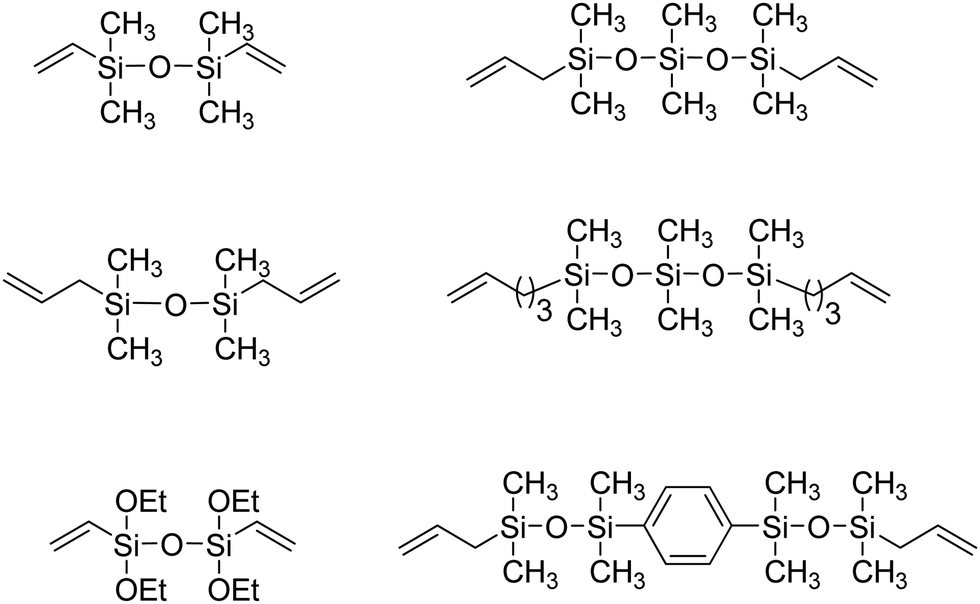 | ||
| Fig. 21 Chemical structures of carbosiloxane α,ω-diene ADMET monomers. | ||
ADMET polymers containing amino acids
Amino acids have been incorporated in ADMET polymers as pendant groups or in the main chain. The major advantage of placing an amino acid as part of the main chain is biodegradability; for example, microorganisms can hydrolyze peptide bonds, consequently decreasing the molecular weight of the polymer and shortening the main chain. On the other hand, amino acids or short peptides pendent along the PE chain imparts biocompatibility to a material that is not biocompatible by nature. The major bottleneck for the incorporation of amino acids into α,ω-diene monomers is reactivity. Amino acids generally present one carboxylic acid and one amine group available for reaction. Both termini can be used to link the amino acid to the α,ω-diene monomer, but regardless of which end is used for linking, the other functionality must be protected.48–51Fig. 22 shows the incorporation of a generic O-protected amino acid to a carboxylic acid α,ω-diene, Fig. 22a, and the incorporation of a generic N-protected amino acid to an amino-α,ω-diene, Fig. 22b. In both cases, coupling the protected amino acid was carried out by mixing the α,ω-diene in a THF solution containing 1-hydroxybenzotriazole (HOBt) and diisopropylcarbodiimide (DIC). Both protected monomers yielded a set of ADMET polymers containing a variety of amino acids precisely distributed along the polymer chain.48,49,51 While previous examples showed the possibility of coupling amino acids along the polymer backbone, incorporation of amino acids into the main chain requires a different synthetic approach that has proved to be difficult, due to reactivity of both termini groups. Instead of using the respective amino acid, a different approach to synthesize chiral amino acid-based monomers was used starting from α-amino alcohols.48 Monomers shown in Fig. 23 were synthesized according to the procedure by Koyama et al.52 The alkenyl acid was reacted with the coupling reagents l-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC-HC1) and 1,3-dicyclohexylcarbodiimide (DCC) to form the activated ester. Then the amino alcohol was added, followed by the addition of 4-(dimethylamino)pyridine (DMAP) to catalyze the reaction, yielding a set of five monomers, as shown in Fig. 23.
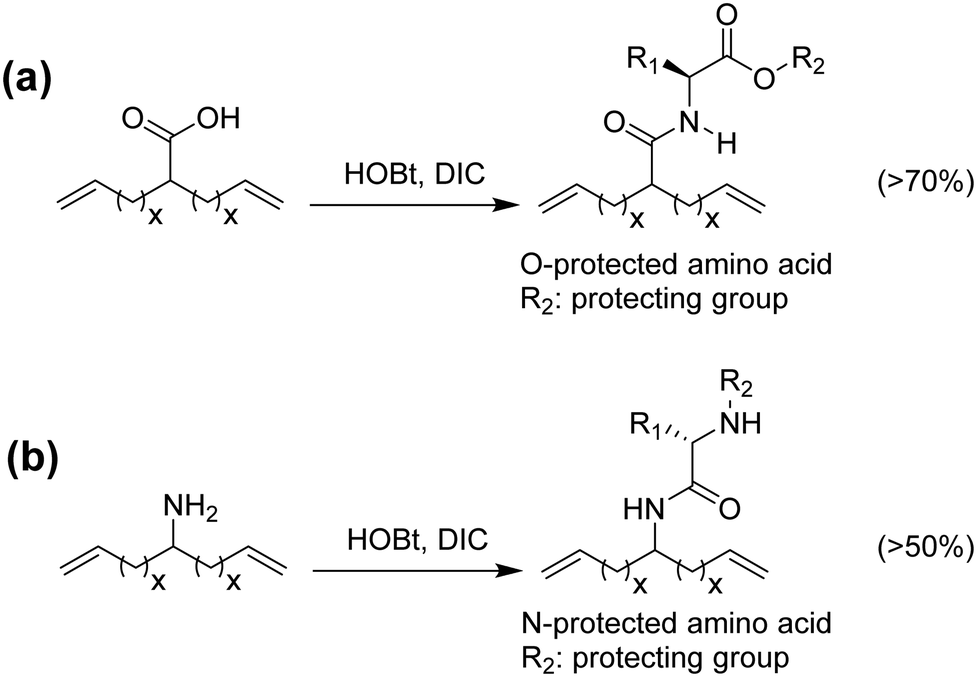 | ||
| Fig. 22 Preparation of amino acid-containing ADMET monomers via N-terminus (a) and C-terminus (b). | ||
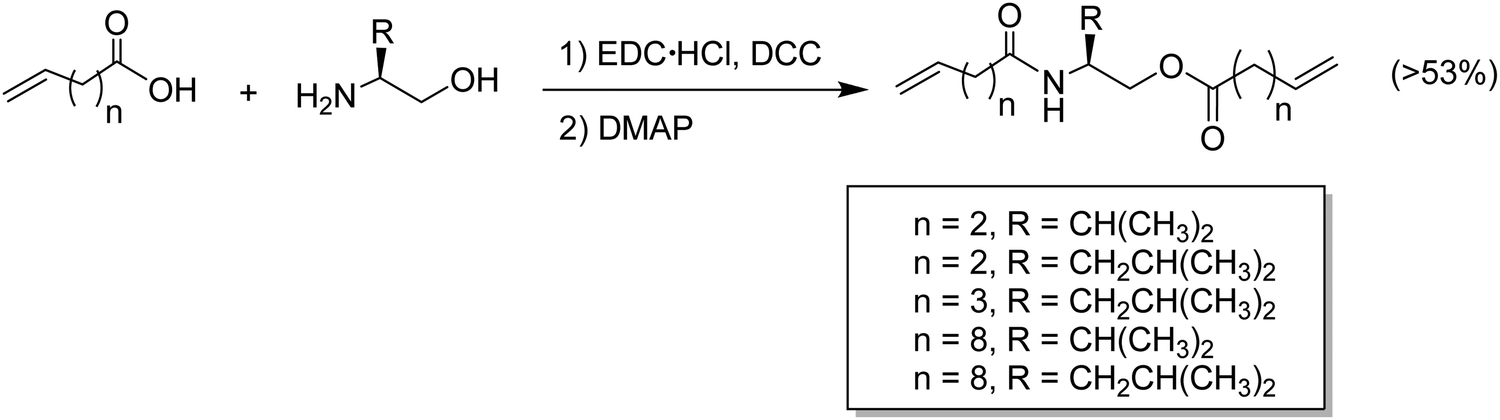 | ||
| Fig. 23 ADMET monomers bearing amino acid units in the backbone. | ||
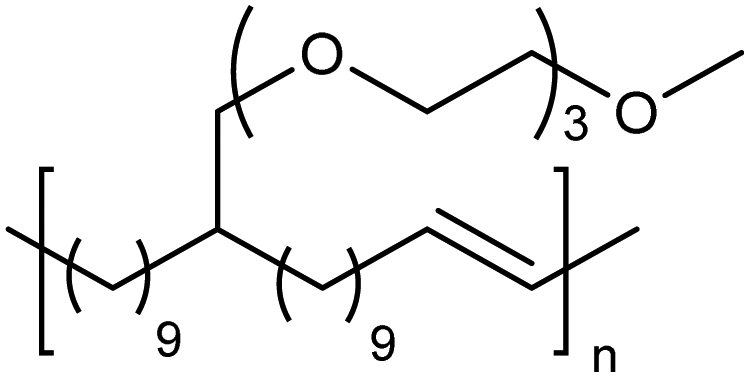
ADMET amphiphiles
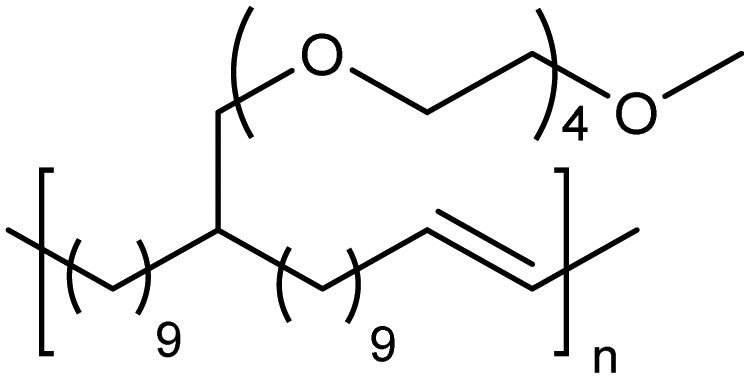
Amphiphilic copolymers have been extensively studied due to the vast array of compositions, morphologies, and properties possible. Systems featuring hydrophilic segments, for example poly(ethylene glycol) (PEG), and lipophilic segments, polyethylene, are of importance because of biocompatibility and possibility of phase segregation, and more importantly, since they can self-assemble into supramolecular structures. ADMET polycondensation is an excellent tool for modeling polymeric systems that lack structural regularity when synthesized through other means. Fig. 24 shows a set of ethylene-based copolymers bearing PEG branches. Both the length of the PEG branch and frequency of the branch along the PE backbone can be manipulated by monomer design.53–55 Monomer synthesis was carried out by coupling the appropriate PEG segment to an α,ω-diene alcohol using sodium hydride in dimethyl formamide. ADMET polymerization was performed using Grubbs’ first-generation catalyst, and further hydrogen saturation with p-toluenesulfonyl hydrazide yielded a set of five model polymers.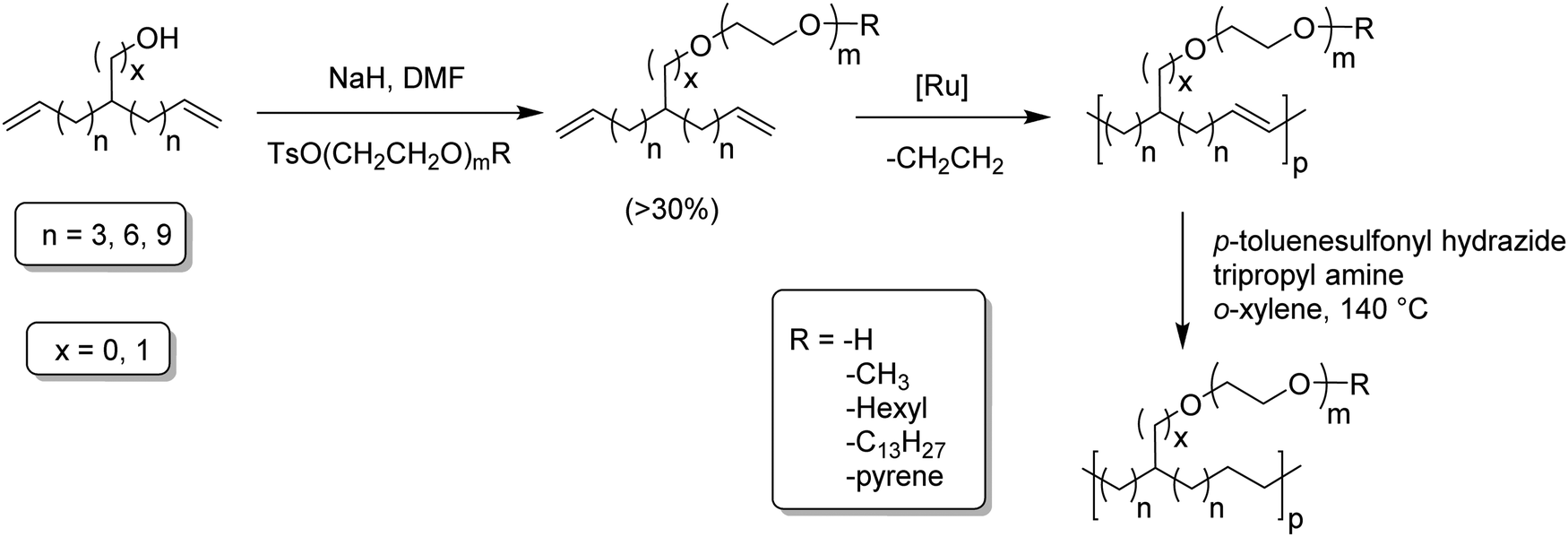 | ||
| Fig. 24 Preparation of amphiphilic ADMET polymers. | ||
3. Secondary structure determination of ADMET polymers
Precision placement of functional groups inherent in ADMET polymerization presents the opportunity to address how precision molecular structure impacts secondary structure. Understanding the parameters that promote the formation of different secondary structures will enable the design of functional materials that take advantage of useful nanoscale morphologies. Ideally, self-assembled secondary structures may be leveraged to target specific and useful nanoscale morphologies. Elucidation of nanoscale structures formed by ADMET polymers has been tremendously aided by X-ray scattering,56 800 MHz solid state nuclear magnetic resonance (NMR),57,58 differential scanning calorimetry (DSC),59,60 and other instrumental methods.61,62 The functional group size, polarity, frequency, and location relative to the linear polyethylene backbone each have pronounced effects on the observed semi-crystalline secondary structure.In polymers synthesized by ADMET polymerization, three distinct crystal motifs are possible (Fig. 25). The thickness of thin crystals (Fig. 25a) is defined by the length of a single alkyl run length with the functional groups excluded from the crystalline domain. The rest of the chain may enter the amorphous phase or bend and adjacently re-enter the crystalline domain. Extended chain (EC, Fig. 25b) crystals have longer-range ordering of polymer chains, and the functional group is incorporated into the crystalline domain. Because the crystals must accommodate the functionality, the functional groups are generally aligned at an angle to the vector normal to the polyethylene crystal direction. In adjacent re-entry (AR) crystals (Fig. 25c) the polymer chain preferentially undergoes a hairpin turn at the location of the functional group, so that the chains predominantly crystallize intramolecularly with the functionality on the edges of the crystalline polyethylene regions. Note that in the cases of EC and AR crystals (Fig. 25b and c) that amorphous regions also exist but are not shown.
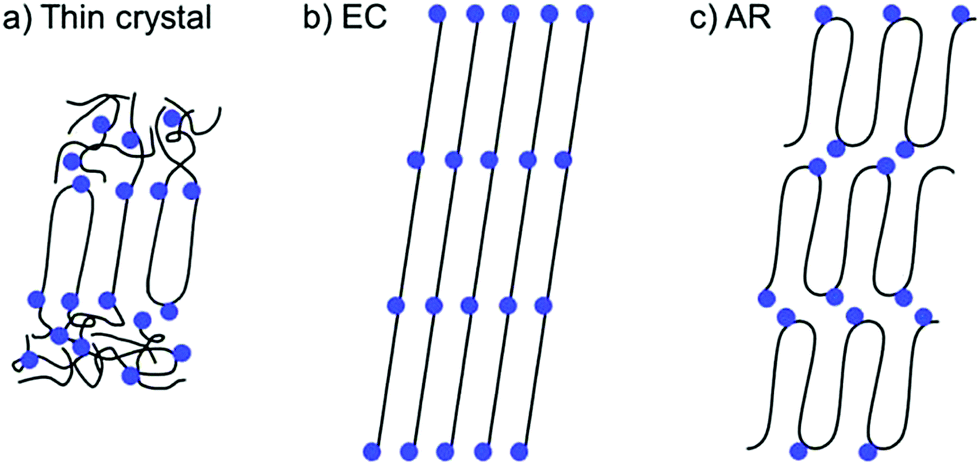 | ||
| Fig. 25 Three types of crystals formed by precisely functionalized PE (Reprinted from ref. 63.) (a) Thin crystal (b) extended chain (EC), and (c) adjacent reentry (AR). Black lines represent polyethylene segments, and blue dots represent the incorporated functional groups. (Reprinted with permission from ref. 63. Copyright©2017 American Chemical Society.) | ||
ADMET polymers prepared with alkyl groups pendant to the polyethylene backbone generally adopt the thin crystal motif if the pendant groups become sufficiently large that they cannot be reasonably incorporated into the crystal structure. The effect of branch size on crystal morphology was shown elegantly by comparing polyethylene with an ethyl or n-hexyl branch on every 21st carbon (EB21 and EO21, respectively).64 In the wide-angle X-ray diffraction (WAXD) pattern of EB21, a prominent peak located at 20.1° accompanied by a minor reflection peak at 22.5° (Fig. 26a) suggests a dominant hexagonal crystal structure which includes the ethyl branches but some triclinic character of the structure is also noted. It was reasoned that if the ethyl branches were all aligned horizontally in the crystal lattice, there would be crowding of these branches in the crystal lattice, leading to instability. Instead, it is likely that ethyl groups are staggered within the crystal structure to avoid lattice distortion. The observed effect of ethyl group staggering is tilting of the a–b plane relative to the CH2 sequences (in the c plane), leading to the observed triclinic character. Alternatively, if the pendant aliphatic group becomes so large that it cannot be accommodated within the crystal structure, it is simply excluded as shown for EO21. Fig. 26b shows the WAXD pattern of EO21. Sharp reflections at 19.6° and 23.1° differ only nominally from those recorded for monoclinic polyethylene (19.45° and 23.17°). The lack of an observed a-axis lattice expansion relative to monoclinic polyethylene is evidence that the n-hexyl branches must be excluded from the crystalline domain, resulting in thin crystals.
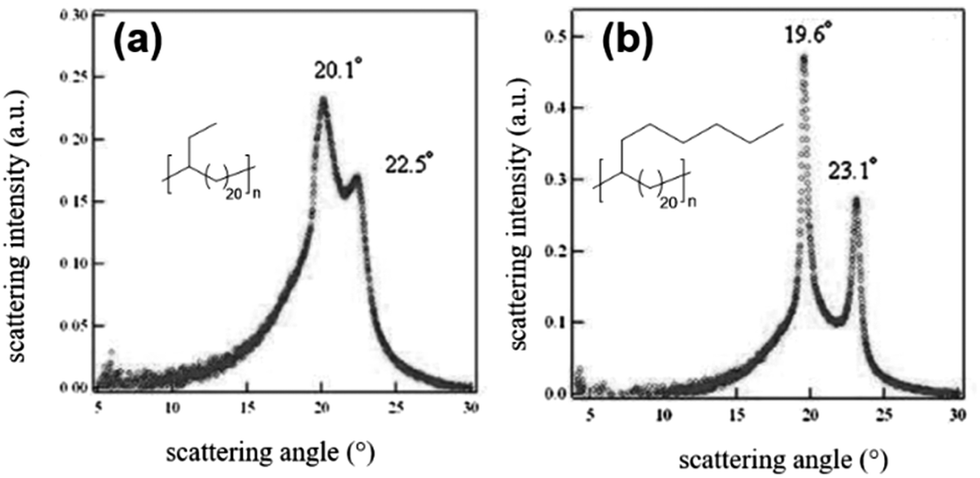 | ||
| Fig. 26 WAXD patterns of slowly cooled EB21 (a) and EO21 (b). (Adapted with permission from ref. 64. Copyright©2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.) | ||
Another important consideration is the effect of precision spacing of the ethyl and n-hexyl groups compared to randomly incorporated alkyl branches (e.g., those in commercial LLDPE). Pristine branch spacing afforded by ADMET polymerization resulted in narrow lamellar thickness distributions (LTD), which are positively correlated to mechanical properties such as tensile strength. A comprehensive study of the effect of alkyl branch size on the crystal morphology found that all branches larger than ethyl groups are excluded from the crystalline domains, resulting in a thin crystal type secondary structure.13 In addition to WAXD analysis which showed the inclusion of methyl and ethyl branches in the crystalline domain and the exclusion of all others, differential scanning calorimetry (DSC) showed that all of the polymers with propyl and larger alkyl pendant groups exhibited a melting endotherm at essentially the same temperature (Fig. 27), evidence in further support that bulky groups are excluded from the crystalline regions. Further, the heats of fusion are all essentially the same. The trend of bulky alkyl group exclusion also holds when the methylene spacer length between groups is increased.65,66
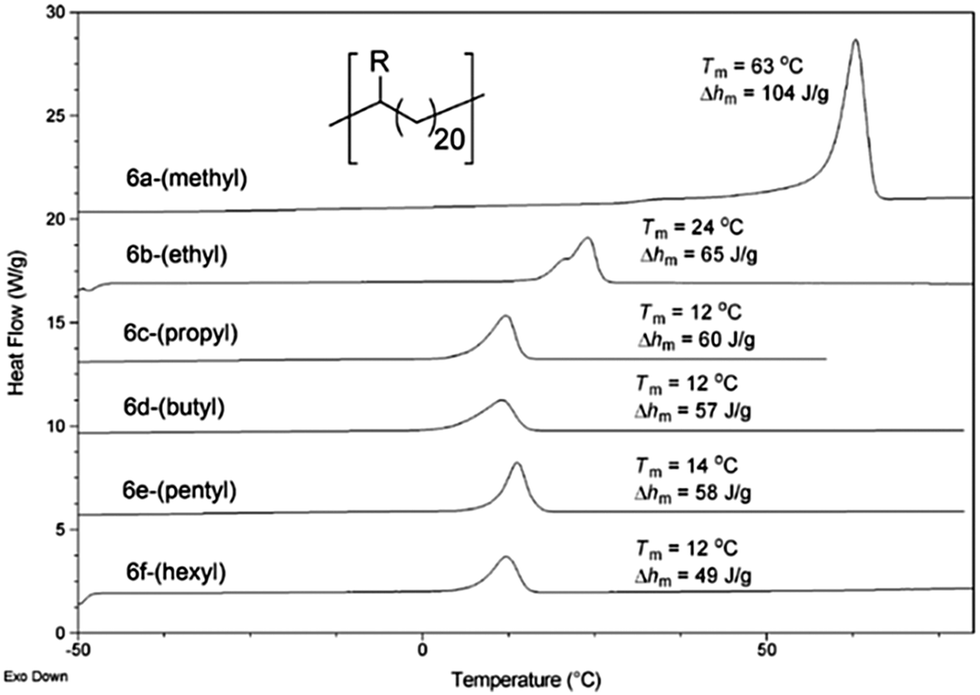 | ||
| Fig. 27 DSC curves of linear alkyl-substituted ADMET polymers (R group is specified in parentheses next to each curve). (Reprinted with permission from ref. 13. Copyright©2009 American Chemical Society.) | ||
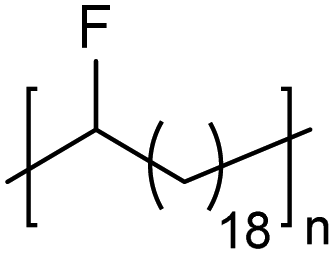
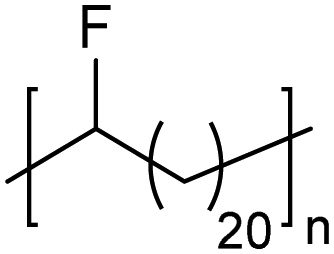
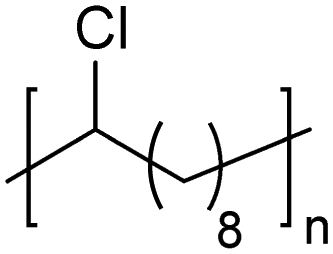
In addition to methyl- and ethyl-substituted precision polyethylenes, halogen-containing precision polymers form EC crystals. The crystal structures and crystallization thermodynamics of halogen-containing precision polymers have been investigated in detail.22,67–71 Some trends can be distilled from this work, particularly based on the halogen which is incorporated. When a halogen (F, Cl, or Br) is substituted on every 19th carbon, each of the WAXD patterns displays sharp diffraction peaks often associated with homopolymer crystallization rather than copolymer-like behavior (Fig. 28).22 A purely orthorhombic unit cell (isomorphous with that of linear polyethylene) was observed for polyethylenes with either F or O substituted for H on every 19th carbon (Fig. 28). Substitution with the larger halogens Cl and Br led to slightly broadened WAXD patterns and evidence of lattice expansion resulting in a less-symmetric triclinic unit cell.
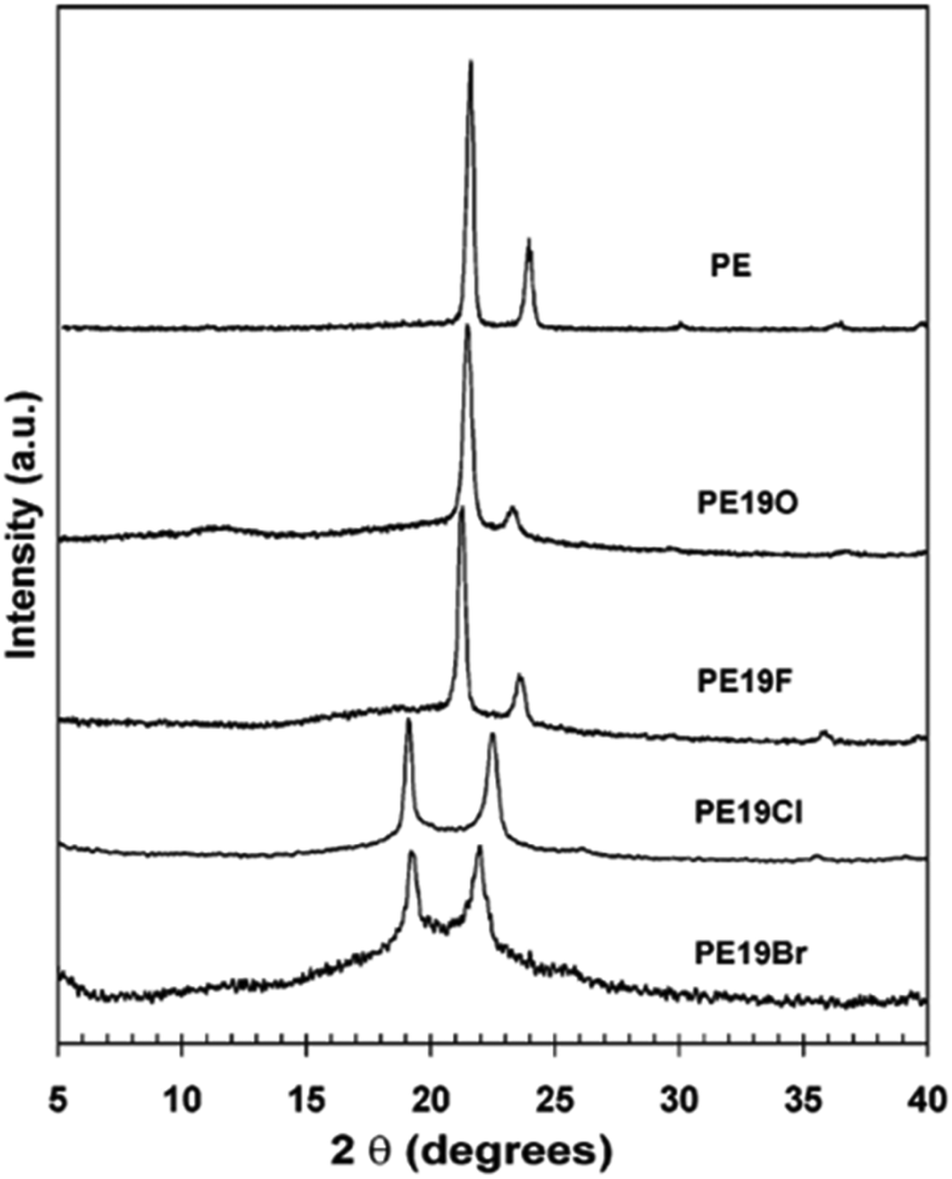 | ||
| Fig. 28 WAXD patterns of linear polyethylene (top) and precision substituted polyethylenes prepared via ADMET polymerization. Pendant groups (X = H, O, F, Cl, Br) are indicated next to each pattern. (Reprinted with permission from ref. 22. Copyright©2006 American Chemical Society.) | ||
Interestingly, pendant groups larger than Cl, Br or ethyl branches can seemingly be incorporated into the crystal structure of precise polyethylenes, forming an EC crystal structure while retaining an orthorhombic unit cell. Precision poly(ethylene-co-acrylic acid) polymers were prepared by ADMET polymerization with the carboxylic acid pendant group located on every 9th, 15th, and 21st carbon (p9AA, p15AA, and p21AA, respectively).25 Based on DSC experiments of these polymers, the methylene spacer length had a profound effect on the thermal behavior. Similar to other ADMET polymers with the functional group pendant to the polyethylene backbone, placement of functional groups closer together yields less crystalline material because a higher fraction of steric bulk prevents crystallization. In this case, both p9AA and p15AA showed completely amorphous behavior (no melting endotherm seen via DSC). However, an ordered secondary structure for p21AA was evident from the sharp melting endotherm in the DSC trace. These DSC results agreed with the X-ray scattering data with respect to semi-crystalline versus amorphous behavior.
To induce further anisotropy in the p21AA sample, it was stretched and then analyzed by X-ray scattering. The scattering pattern of the stretched sample is shown in Fig. 29a. Drawing the sample in the direction of the arrow in Fig. 29a promoted alignment of the polyethylene segments (seen as multiple reflections of the (110) plane) which were oriented at a 90° angle to the low-angle acid reflections. A proposed schematic indicating the arrangement of the carboxyl groups in the crystal structure is presented in Fig. 29b. Based mainly on the X-ray scattering pattern in Fig. 29a, it was reasoned that the combination of low acid group frequency and crystalline stabilization led to hydrogen bonding between in-plane acid neighbors. A secondary structure consisting of EC crystals which do not undergo a crystal lattice change to accommodate the carboxylic acid groups was assigned in the absence of other evidence to suggest otherwise. This result is contrary to what one might predict based on previous studies with large aliphatic and large halogen pendant groups.13,22
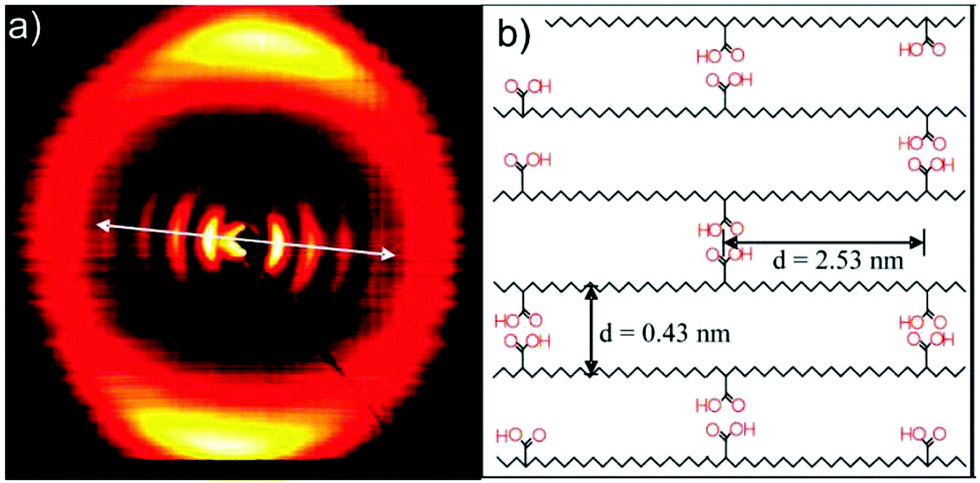 | ||
| Fig. 29 (a) X-ray scattering pattern of p21AA stretched along the arrow direction. Multiple reflections of 2.48 nm−1 correspond to a layered structure with 2.53 nm spacing oriented perpendicular to the polyethylene backbone (b) Proposed schematic of the arrangement of p21AA chains within a crystalline domain showing hydrogen bonds between acid groups in the same plane. Hydrogen bonds may form with chains above and below the plane shown. (Reprinted with permission from ref. 25. Copyright©2007 American Chemical Society). | ||
Until recently, polymers with AR-type secondary structures (Fig. 25c) were thought to be rare in the large library of materials prepared by ADMET polymerization. In 2008, careful DSC studies of ADMET polymers with precision-spaced short pendant chains of poly(ethylene glycol) (PEG) were published. Modulated DSC and selective annealing experiments on an ADMET polymer with PEG pendant groups on every 21st carbon confirmed two distinct crystalline populations.53 It was reasoned that the bimodal melting behavior corresponded to nanoscale segregation between the polyethylene backbone and PEG side-chains. For this to occur, the pendant PEG groups (which are chemically immiscible with the PE backbone) must have induced a fold in the polyethylene backbone, and the pendant groups were crystallizing amongst themselves separately. In 2017, the Winey group performed a closer examination of the p21AA nanostructure (originally reported in 2007)25 using molecular dynamics simulations, wide angle X-ray scattering (WAXS), and Raman spectroscopy. Both EC and AR structures of p21AA were simulated, and the experimental data, particularly the structure factor peak deconvoluted from the experimental WAXS, matched closely with the calculated AR results (Fig. 30).
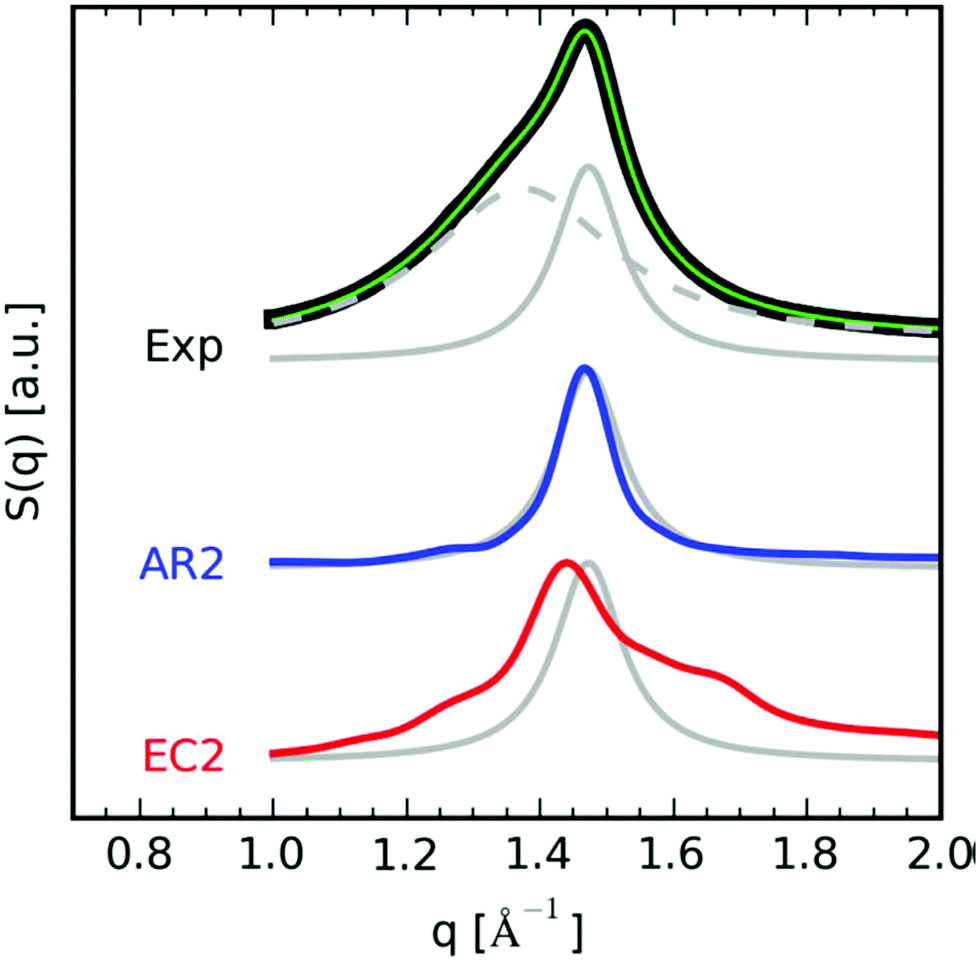 | ||
| Fig. 30 Structure factor comparison between simulations and experimentally obtained WAXS profile of p21AA. (top) Experimental WAXS profile deconvoluted into amorphous and crystalline peaks (black = experimental, green = total fit, dashed gray = amorphous halo, solid gray = crystalline peak). (middle) Calculated AR structure factor peak (blue). (bottom) Calculated EC structure factor peak (red). Both calculated structure factors were overlaid onto the experimental crystalline peak (solid gray) for comparison. (Reprinted with permission from ref. 63. Copyright©2017 American Chemical Society.) | ||
The agreement between the modeled AR data and experimental data rectified the puzzling result that the bulk-crystallized p21AA seemed to perfectly integrate the carboxylic acid groups into an orthorhombic crystal structure despite the large size of the functional group. Winey and co-workers noted that further comparison between p21AA and similar ADMET polymers support the AR structural hypothesis. For example, the layer spacings of p21AA were compared with those of the ADMET polymer with two geminal acid groups on every 21st carbon (p21gAA) as well as the polymer with a phosphonic acid on every 21st carbon (p21PA). Despite the very large van der Waals volume differences between the functional groups on these three polymers, their layer spacings are within 7% of one another. If these polymers were each arranged in an EC structure, it would stand to reason that the more space a functional group occupies, the larger the required tilt angle between the normal to the functional group layer and the chain axes for efficient packing (decreasing the layer spacing). This is not the observed trend. Also, the melting temperatures of p21AA, P21gAA, and P21PA are similar (47.7, 46.1, and 47.4 °C, respectively),60 and very similar to the melting point of PEG-grafted polyesters with 20-carbon-long alkyl segments (between 44 and 46 °C).72 In contrast, in purely aliphatic systems, increasing the functional group size in EC drastically lowers the melting point.
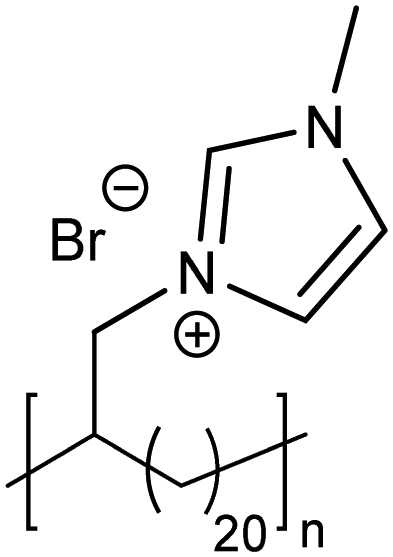
The exciting implications of the AR-type secondary structure lead to consideration of how this nanoscale arrangement might result in channels of ion-conducting functionality for efficient shuttling of protons or other ions in electrochemical devices. Use of these types of materials as ion-transport membranes is only possible if long range ordering of the polymer chains can be achieved. Yan and co-workers demonstrated long-range ordering through the carefully controlled solution growth of single crystals made from precise acid and ion-containing polymers.73 Specifically, p21AA, p21gAA and a precise polymer with imidazolium bromide on every 21st carbon (p21ImBr) were chosen to demonstrate the growth of large single crystals from dilute solutions at appropriate crystallization temperatures. The p21AA single crystals were quite uniform in size and generally square-shaped (Fig. 31a and b), while the p21ImBr polymer formed remarkably large crystals with a few different shapes (one is shown in Fig. 31c and d). In addition to ample Raman spectroscopy and scattering data which suggested an AR-type secondary structure, the P21ImBr crystal thicknesses exceeded the fully extended contour length of the polymer (based on average molecular weight) by as much as a factor of three. Since it is incredibly unlikely that the polymer chains would be stretched out end-to-end in an EC-conformation to form these large crystals, this serves as another piece of evidence in support of the AR-type secondary structure in these polymer crystals.
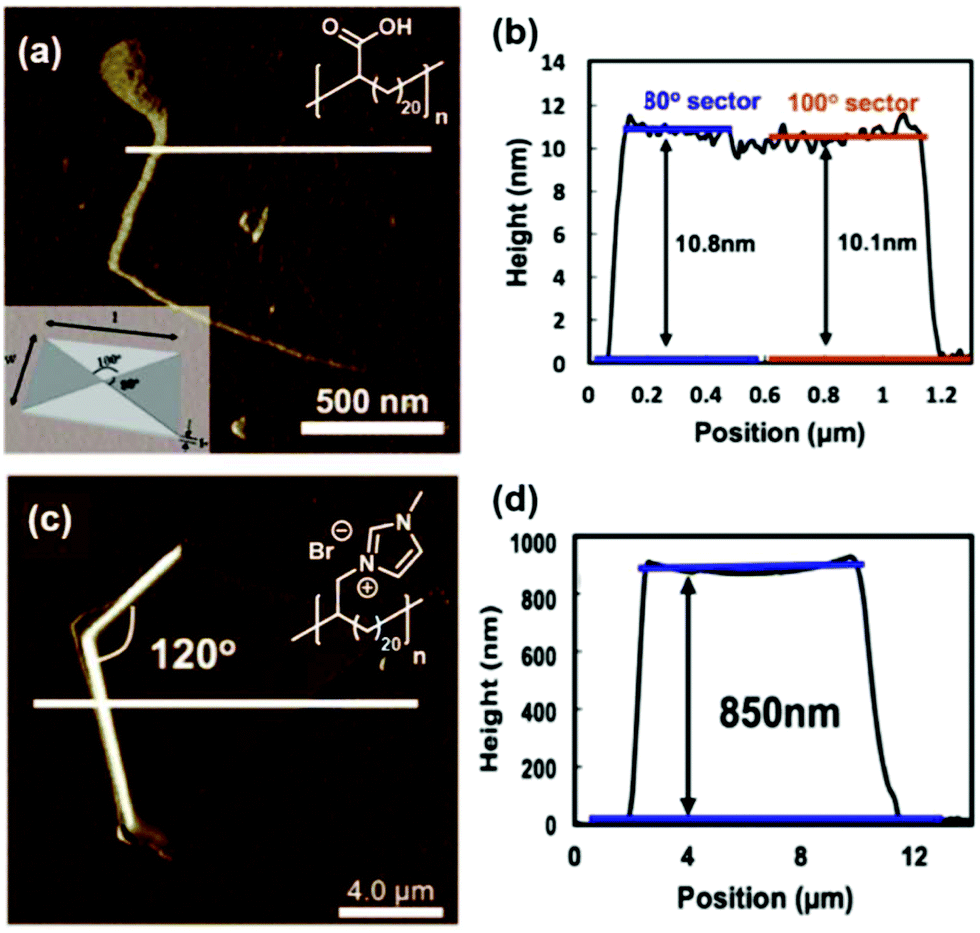 | ||
| Fig. 31 (a) AFM image of a p21AA single crystal (polymer structure and schematic of p21AA crystal showing 80° and 100° growth sectors inset). (b) Height profile corresponding to the path drawn in white in image (a). (c) AFM image of p21ImBr (polymer structure inset). (d) Height profile corresponding to the path drawn in white in image (c). (Adapted with permission from ref. 73 Copyright©2017 Elsevier Ltd.) | ||
Recently, an exciting advance showing the advantages of an AR-type secondary structure was observed for precision polyethylenes with a sulfonic acid on every 21st carbon (p21SA).34 The folded chain structure formed channels of sulfonic acid groups which are highly hygroscopic and lead to swelling of the acid-enriched regions in the presence of humidity (Fig. 32). Sulfonic acids are favored for many ion-conducting membranes mainly due to their hygroscopicity. Perhaps the most prevalent commercial sulfonic acid-containing membranes are made of Nafion®, which has been used as the benchmark material in fuel cells and other electrochemical devices for decades. However, the microstructure and the path that protons take through the material is not without controversy.74 To demonstrate whether the AR-type ordered morphology of p21SA membranes would lead to desirable proton conductivity, electrochemical impedance spectroscopy (EIS) was performed at varying degrees of humidity on both Nafion® 117 and a membrane made of p21SA. The conductivities of the two materials were very similar above 60% relative humidity, and this was the first reported instance of such proton conductivity in a polyethylene-based crystalline structure.
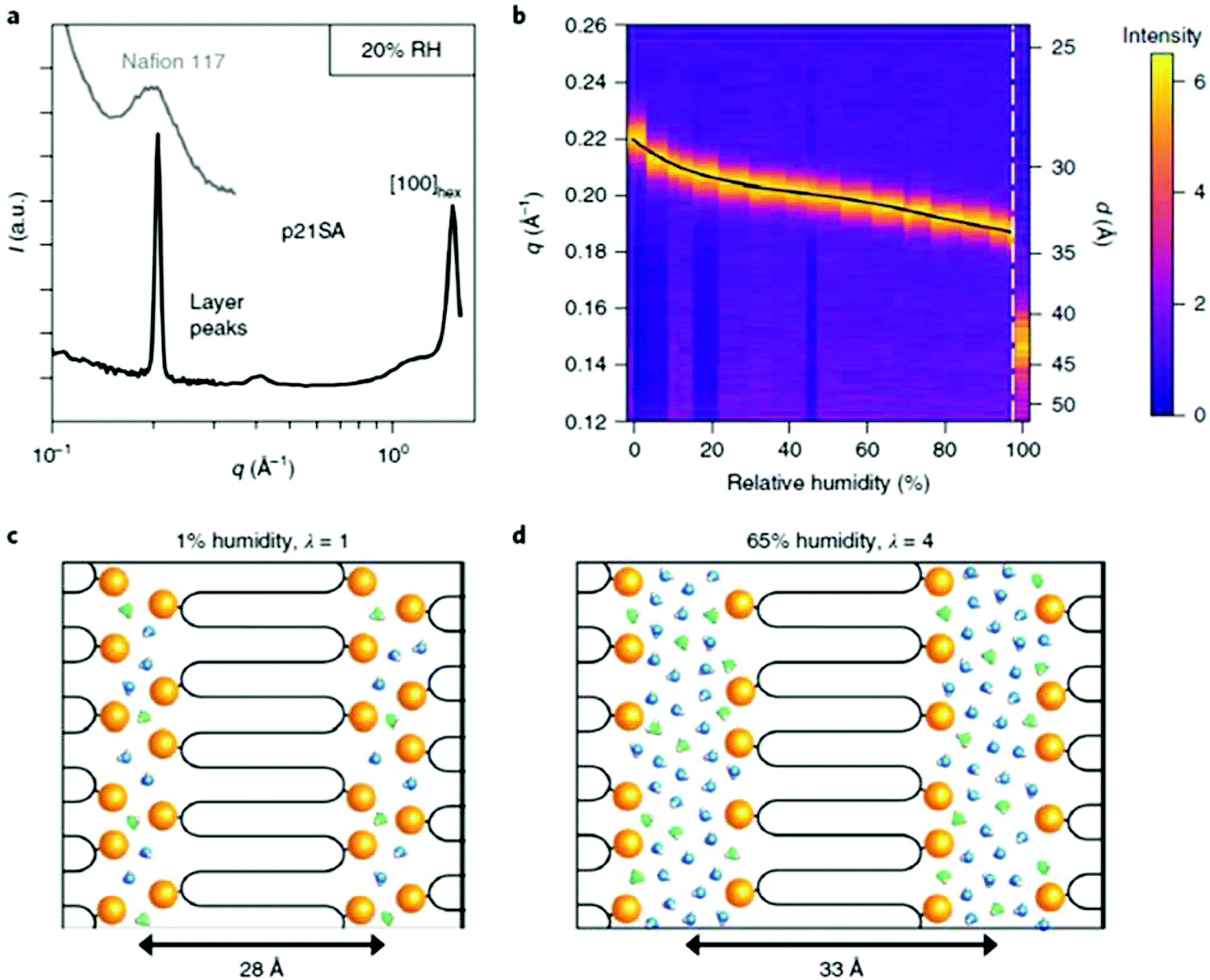 | ||
| Fig. 32 (a) Intermediate- and wide-angle X-ray scattering profiles of p21SA at 20% relative humidity (RH) at room temperature (intermediate X-ray scattering of commercial Nafion® 117 is shown in light gray for comparison). (b) Primary layer peak of p21SA as a function of RH at 40 °C and immersed in water (far right vertical strip). Each vertical strip is an I versus q curve, with the far-right vertical strip displaying the primary layer peak of p21SA submerged in water. Images (c) and (d) are schematic representations of the layered structure with increasing humidity. (Reprinted with permission from ref. 34. Copyright©2018 Springer Nature Limited.) | ||
Based on the work discussed in this section, the implications of harnessing predictable secondary structures are becoming clearer. The ability to extract the effects of chemical structure on nanoscale morphology has enabled the construction of a set of design rules for precision materials with predictable structures. These rules will serve as a powerful toolbox for the design of precision polymers which can help solve contemporary problems as well as those yet undiscovered (Table 1).
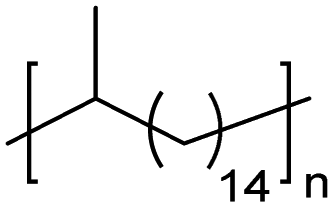
| Polymer structure | Secondary structurea | Functional group frequency (on and every # of carbons) | T m (°C) | Ref. |
|---|---|---|---|---|
| a TC = thin crystals, EC = extended chain crystals, AR = adjacent reentry. b Secondary structure originally assigned as EC, but updated to AR after additional molecular dynamics simulations, Raman spectroscopy, and WAXD experiments.63 c Based on selective annealing DSC experiments. d X-ray scattering performed at room temperature which was above the melting point of the polymer. e Determined based on DSC results. | ||||
|
EC | 15 | 397 | 75 |
|
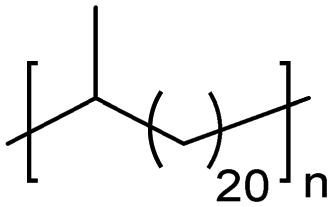
EC | 21 | 63 | 13 |
|
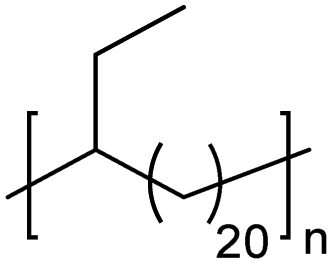
EC | 21 | 2413 22.264 | 13 and 64 |
|
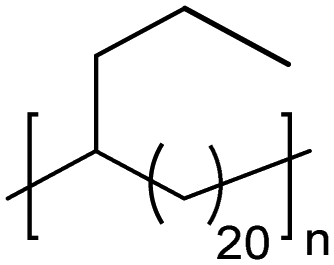
TC | 21 | 12 | 13 |
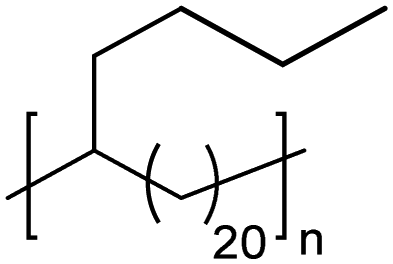
|
TC | 21 | 12 | 13 |
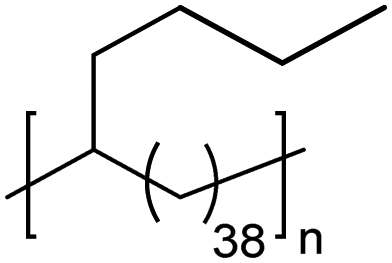
|
TC | 39 | 75.3 | 65 |
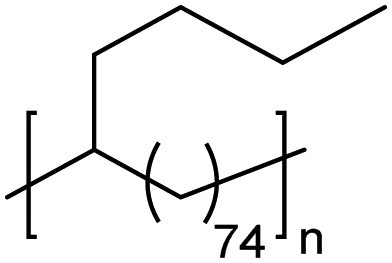
|
TC | 75 | 104 | 66 |
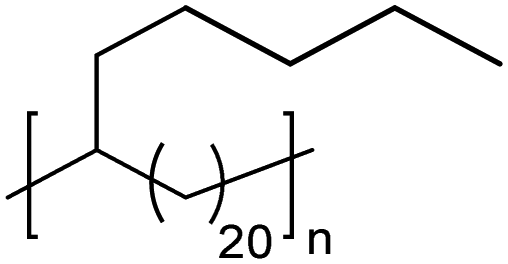
|
TC | 21 | 14 | 13 |
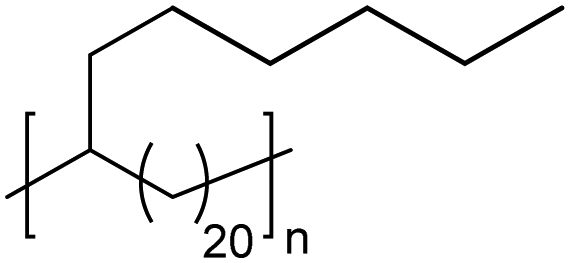
|
TC | 21 | 1213 10.364 | 13 and 64 |
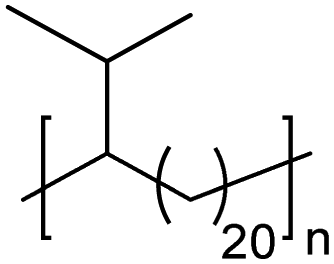
|
TC | 21 | 11 | 13 |
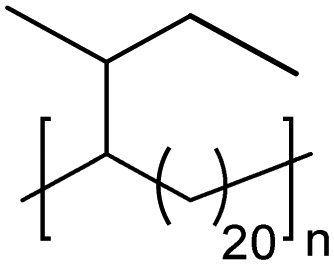
|
TC | 21 | 9 | 13 |
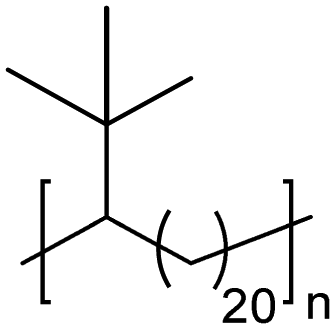
|
TC | 21 | 13 | 13 |
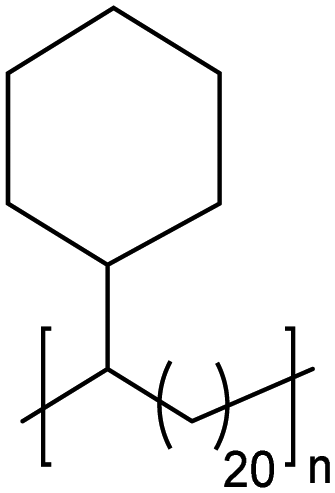
|
TC | 21 | 9 | 13 |
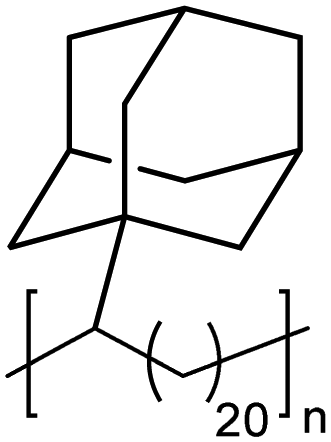
|
TC | 21 | −8, 17 | 13 |
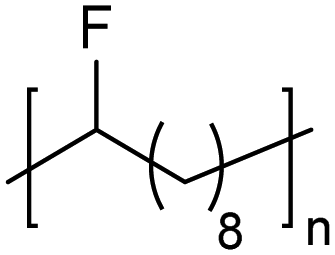
|
EC | 9 | 124.3 | 67 |
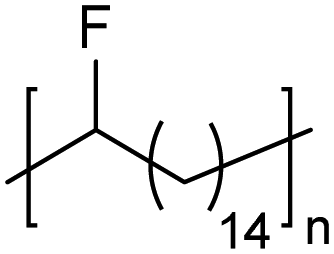
|
EC | 15 | 125.7 | 67 |
|
EC | 19 | 127.5 | 22 |
|
EC | 21 | 127.7 | 67 |
|
EC | 19 | 134.7 | 22 |
|
EC | 9 | 15 | 68 |
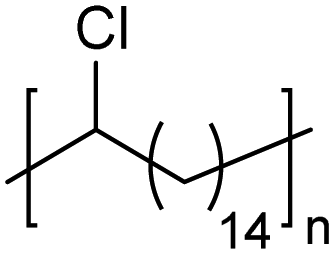
|
EC | 15 | 54 | 68 |
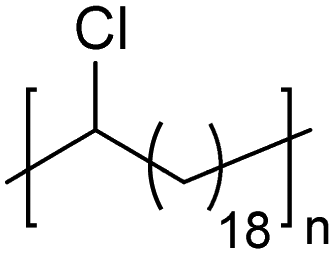
|
EC | 19 | 72.7,22 6368 | 22 and 68 |
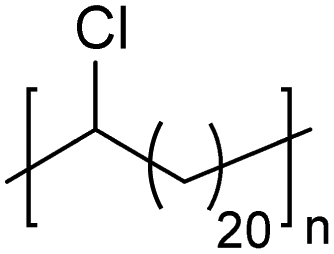
|
EC | 21 | 70 | 68 |
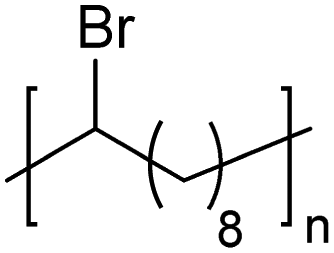
|
EC | 9 | −13 | 71 |
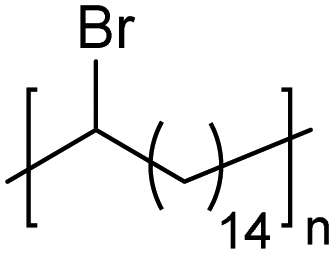
|
EC | 15 | 49 | 71 |
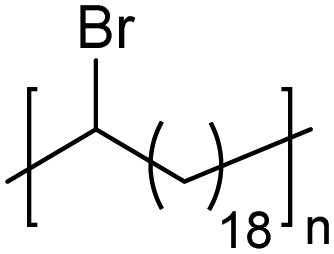
|
EC | 19 | 61.5 | 22 |
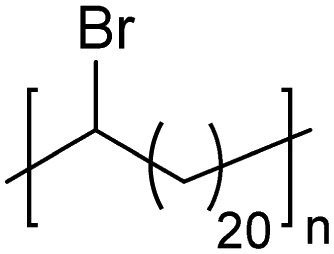
|
EC | 21 | 70 | 71 |
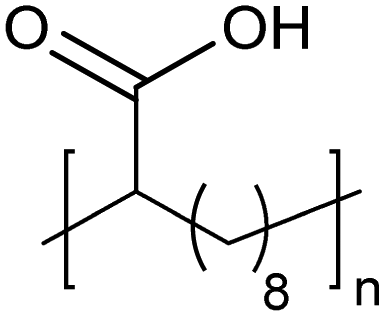
|
Amorphous | 9 | — | 25 |
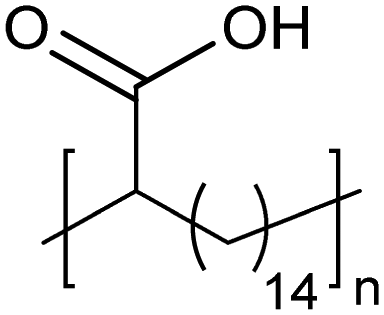
|
Amorphous | 15 | — | 25 |
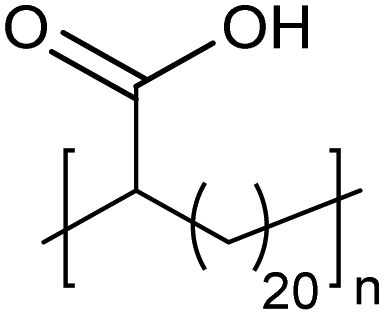
|
ARb | 21 | 45 | 25 |
|
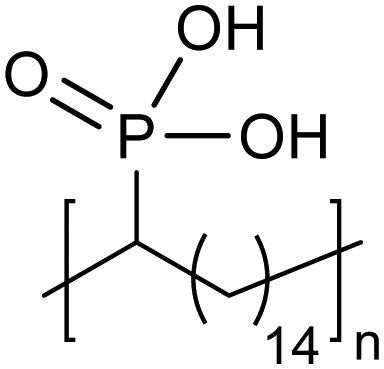
Amorphous | 15 | — | 28 |
|
AR73 | 21 | 48 | 28 |
|
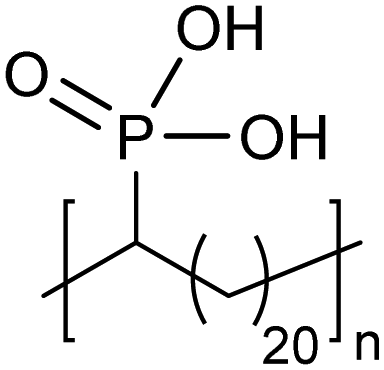
Amorphous | 15 | — | 28 |
|
AR73 | 21 | 87 | 28 |
|
AR | 21 | 90 | 73 |
|
ARc | 21 | Approx. −47 and −25 | 53 |
|
ARc | 21 | Approx. −47 and −25 | 53 |
|
Amorphous | 8 | — | 76 |
|
Amorphousd | 14 | 7 | 76 |
|
EC | 20 | 37 | 76 |
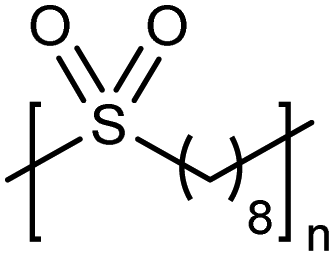
|
EC | 8 | 175 | 35 |
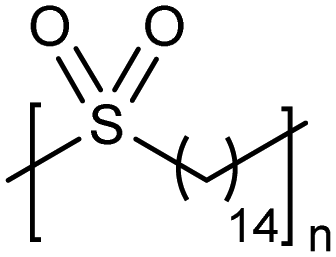
|
EC | 14 | 162 | 35 |
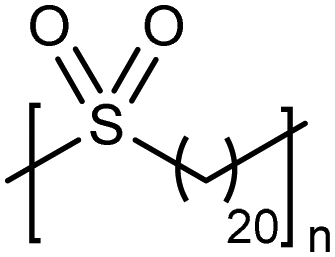
|
EC | 20 | 150 | 35 |
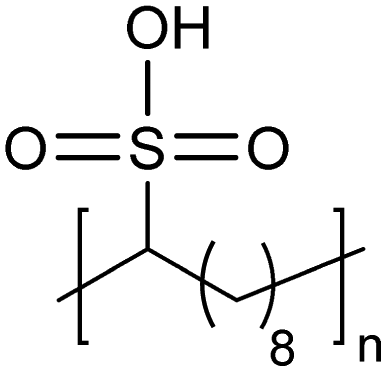
|
Amorphouse | 9 | — | 33 |
|
AR34 | 21 | 65 | 33 |
4. Functional materials from ADMET polymerization
In addition to the rich body of work focused on characterizing the secondary structures formed by ADMET polymers, much attention has been given to the use of ADMET polymerization to prepare polymers with a vast array of functional features. In this section, these functional materials and how the precision derived from ADMET polymerization chemistry contributes to their performance will be discussed.Polymers for ion-transport and small-molecule releasing ADMET polymers
Similar to the precision p21SA work discussed above,34 Lv and co-workers were inspired by the chemistry of Nafion® membranes containing sulfonic acid groups as the proton-conducting functionality accompanied by fluorinated alkyl regions for thermal and chemical durability.77 Due to the solvophobicity of fluorinated reagents, fluorination is generally performed as a post-polymerization modification. In this work, Lv et al., cleverly incorporated small fluorinated segments in the centers of symmetric α,ω-diene monomers. Since the monomers were only partially fluorinated, they remained soluble during a solution ADMET polymerization in toluene, yielding molecular weights up to Mn = 28.5 kDa. The sulfonic acid functionality was incorporated as a post-polymerization modification by treating the internal olefins with a stoichiometric amount of thioacetic acid and radical initiator, forming thioacetate-functionalized polymers. The thioacetate groups were oxidized to sulfonic acid groups by treatment with formic acid and hydrogen peroxide. The chemical structures of some of the polymers prepared with fluorination and/or sulfonation are shown in Fig. 33. It should be noted that the sulfonic acid groups were not precision-spaced throughout the polymer because the thioacetate group could add to either side of the internal olefins, and semi-crystallinity was only observed in the polymers with the lowest fraction of thioacetate/sulfonic acid incorporated. The resulting polymers did exhibit high degradation temperatures (generally >400 °C) compared to sulfonated polyethylenes.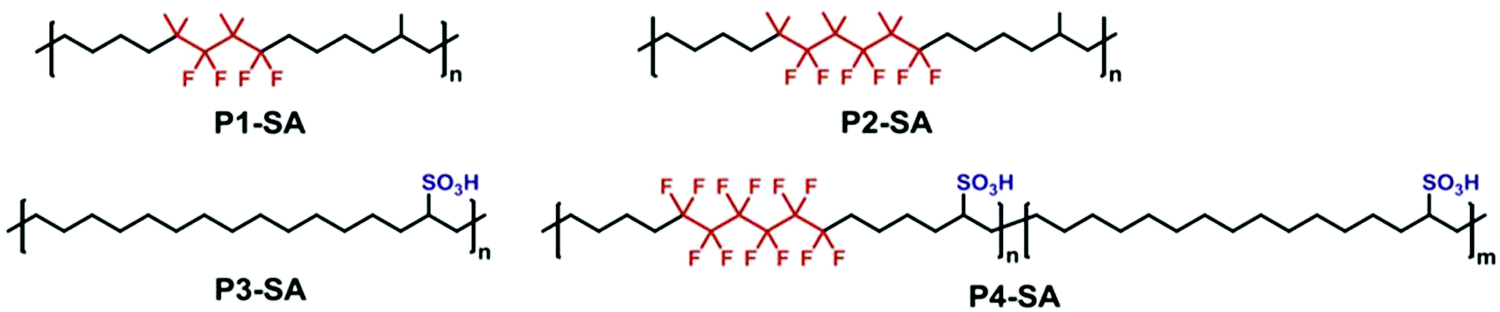 | ||
| Fig. 33 Chemical structures of partially fluorinated and/or sulfonated precision polymers as precise analogs of commercial proton-conducting membranes. (Reprinted with permission from ref. 77. Copyright©2017 Elsevier Ltd.) | ||
ADMET polymers have also found use as platforms for controlled molecule release and/or stimulus-triggered degradation. In one such study, ADMET polymerization was used to prepare polymeric prodrugs consisting of polyethylene backbones with pendant PEG or methylene spacer groups bearing a therapeutic agent (the non-steroidal anti-inflammatory drugs ibuprofen or naproxen in this case, see Fig. 34).78 Surprisingly, each of the tested variations of pendant type (hydrophilic or hydrophobic and length) seemed to generate similar drug release profiles characterized by an initial rapid release of the drug followed by a tailing-off of release. It was hypothesized that solubility of the polymer was the main determining factor in the release, where surface available drug groups were cleaved first. Perhaps the polymer chains collapse as a result of reduced solubility which contributed to the decrease in drug release after the initial burst.
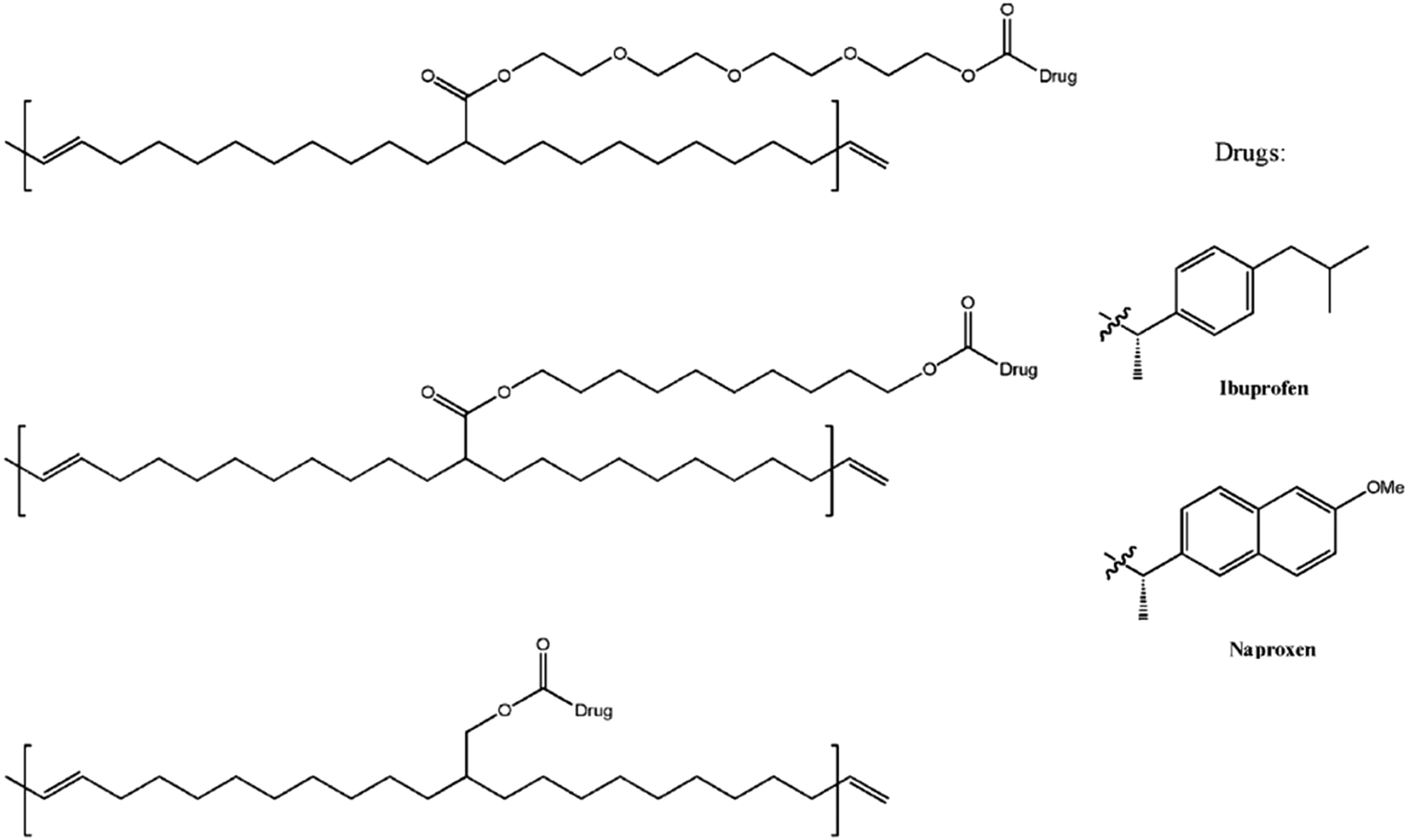 | ||
| Fig. 34 Chemical structures of prodrug precision polymers. (Adapted with permission from ref. 78. Copyright©2010 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim.) | ||
A similar area of interest consists of polymers which degrade in response to environmental stimuli into small molecules which are ideally benign and/or simple to re-use. This field is generally divided among two strategies: self-immolative or chain-shattering degradation. Self-immolative polymers are defined by an incorporated group at the chain end that, once activated, triggers a cascade reaction causing the polymer chain to “unzip” into small molecules that may or may not be the same as the monomer used to construct the chains. Chain-shattering approaches require triggering reactions to occur at each repeating unit, often requiring stoichiometric amounts of the stimulating molecule for full degradation. Miller and co-workers recently published an approach for a self-amplifying chain-shattering system which needs only small amounts of weak acid to begin degradation.79 Their synthetic approach is shown in Fig. 35a. Briefly, the synthesis of the iodo acetal monomer (1) started with the treatment of acrolein (2) with TMSBr. Then acetalization with alcohol (3) resulting in bromo acetal (4). The bromo acetal was simply converted to the iodo acetal (1) under standard Finkelstein conditions in good yield. ADMET polymerization afforded structure (5), and the internal olefins were hydroxylated via Upjohn-dihydroxylation to yield (6).
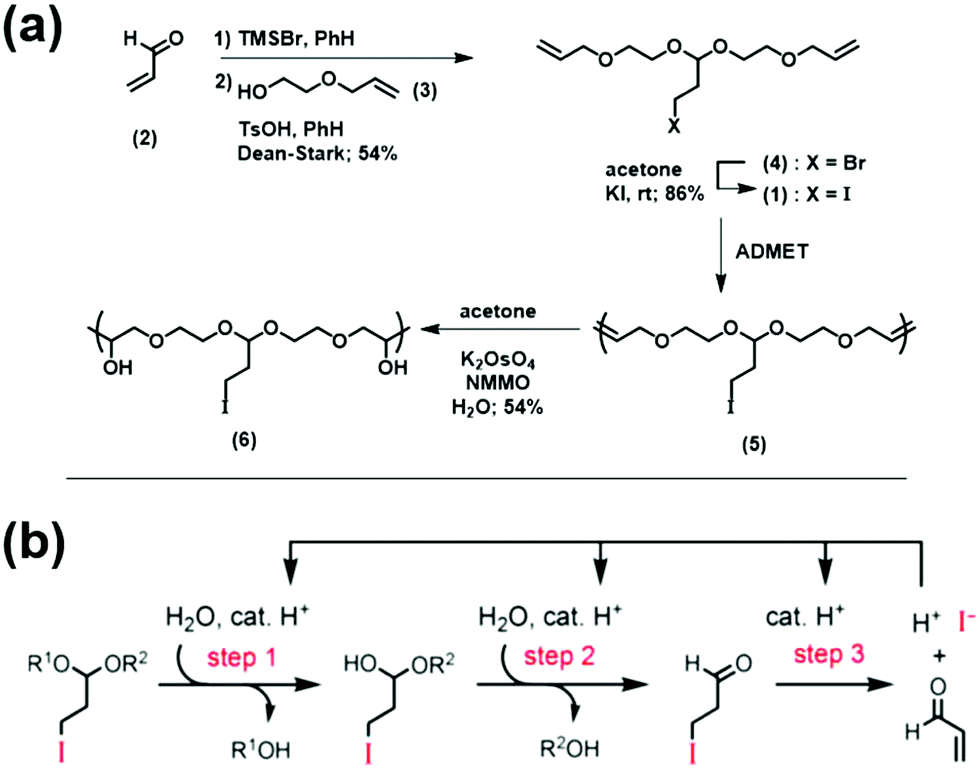 | ||
| Fig. 35 (a) Synthetic approach to form 3-iodopropyl acetal-containing polymer; (b) mechanism of 3-iodopropyl acetal hydrolysis with self-amplified cleavage after generation of HI in step 3 (this process happens within the polymer chain, depicted here as R1 and R2 for clarity). (TMSBr = bromotrimethylsilane, PhH = benzene, TsOH = p-toluene sulfonic acid, NMMO = N-methylmorpholine N-oxide). (Adapted with permission from ref. 79. Copyright©2019 American Chemical Society.) | ||
A proposed general mechanism of self-amplifying chain-shattering is shown in Fig. 35b. The reaction begins with activation of the 3-iodopropyl acetal with weak acid, eventually releasing HI which then serves as the self-amplifying degradation reagent. The chain-shattering of structure (6) was studied by monitoring the disappearance of the acetal proton via1H NMR spectroscopy in a 40% (v/v) CD3CN in D2O solution (pD0 = 5.5). A sigmoidal disappearance of the acetal proton along with a sigmoidal shift in the GPC trace to lower MW species suggested that the proposed self-amplifying mechanism was correct. A hydrogel made with a similar acetal motif also showed similar sigmoidal decomposition behavior. It is important to note that the two molecules released from the decomposition are hydroiodic acid, a very strong acid that accelerates further degradation, and (2) acrolein, a potent biocide. These aspects provide the potential for these polymers to serve as benign vehicles for controlled release of biocidal acrolein, for instance.
Conjugated polymers
ADMET polymerization is particularly useful in preparing conjugated polymers (CPs), because an olefin bond is left behind after the metathesis reaction. The result of the metathesis can be the linkage of two π-systems in a conjugated fashion when an α,ω-diene monomer is strategically designed (see Fig. 36). CPs with a vast array of conjugated functionality have been prepared via ADMET polymerization including poly(acetylenes),80 poly(fluorene vinylenes),81–84 poly(carbazole vinylenes),85 poly(phenylene vinylenes),86 poly(thienylene vinylenes),87 poly(selenylene vinylenes),88 and ferrocene-containing CPs.89 Poly(thienylene vinylene) and poly(selenylene vinylene) homopolymers are known for their low band gaps, allowing for efficient absorption of visible light across a large portion of the visible spectrum and making them attractive candidates for application to organic light-emitting diodes90 and organic photovoltaics.91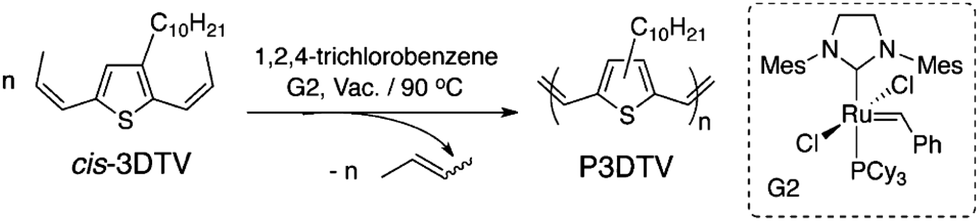 | ||
| Fig. 36 Synthesis of conjugated polymer poly(3-decylselenylene vinylene) (P3DSV) via ADMET polymerization. (Reprinted with permission from ref. 92. Copyright©2015 American Chemical Society.) | ||
Hillmyer and co-workers investigated how the optoelectronic characteristics (e.g., band gap, photoluminescence, absorption strength, charge transport) of poly(thienylene vinylenes) could be tuned through statistical ADMET copolymerization between structurally varied thienylene vinylene monomers.93 A study by Zhang and Qin demonstrated similar optoelectronic tunability by copolymerizing thienylene vinylenes with selenylene vinylenes.92 They proposed that by replacing S for Se in the CP structure, additional tunability of optoelectronic properties would be accessible due to the larger atom size, enhanced polarizability, and stronger spin−orbit coupling of Se. Further work from the same group presented a versatile synthetic route toward derivatized selenylene vinylene monomers resulting in some fundamental understanding of structure/property relationships of these unique polymers.88
A novel CP which functioned as a photoswitchable polyelectrolyte was recently reported by Joo and Bielawski.94 The α,ω-diene monomer was derived from a 4,5-dithienylimidazolium salt (see resulting ADMET polymer structure before and after UV-irradiation in Fig. 37a). The resulting polymer displayed impressive emission properties resulting from the conjugated structure. Fig. 37b and c show the absorption spectra of the monomer and polymer prior to irradiation and the emission performance of each after irradiation, respectively. These results are presented photographically in Fig. 37d. The authors noted that this photochromic polymer motif may serve as an interesting alternative to other CPs such as poly(thienylene vinylenes) which have already been explored in-depth. For example, adding donor/acceptor groups or changing the conjugation length of photochromic monomers may provide tunability in the resulting polymeric optoelectronic properties.
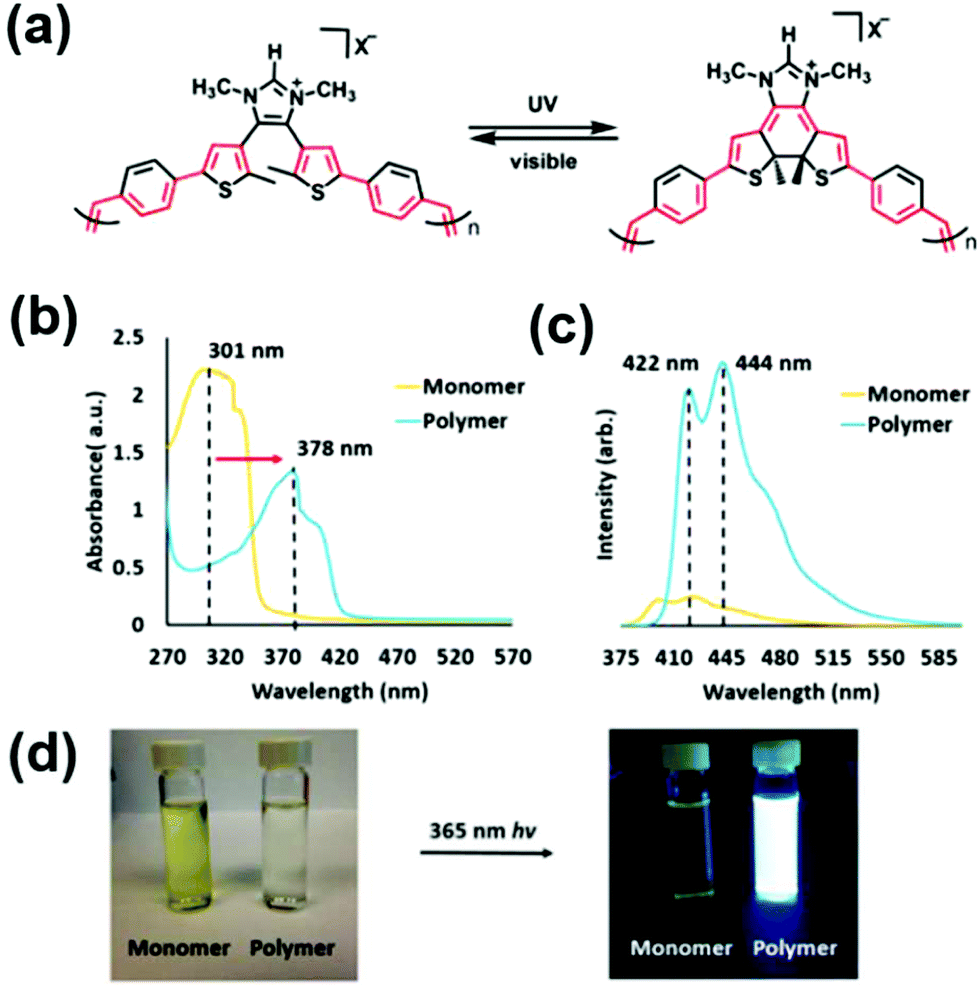 | ||
| Fig. 37 (a) Structure of photoswitchable CP upon irradiation with UV or visible light, (b) comparison of UV-Vis spectra for CP monomer and polymer of structure shown in (a), (c) emission spectra for monomer and polymer after excitation at 365 nm, and (d) photographs of vials containing solutions of monomer or polymer before and after excitation at 365 nm. (Adapted with permission from ref. 94. Copyright©2019 Royal Society of Chemistry.) | ||
Biologically inspired materials
Polymer chemists often attempt to emulate the sophisticated structural and functional characteristics of natural proteins. Many advances have been made in this area, especially in terms of sequence-controlled polymers (mimicking protein primary structure) and foldamers or single-chain nanoparticles (mimicking protein secondary and tertiary structure). Still, no synthetic polymer achieves anything close to natural protein structure or function. An excellent in-depth discussion of these topics can be found elsewhere.95 ADMET polymerization offers the opportunity to incorporate biologically-inspired motifs into synthetic polymers with precision spacing. Incorporation of biological motifs is generally done with the intention of leveraging their strong intramolecular interactions to affect the arrangement of synthetic polymer chains both in the solid state and in solution, and then extracting structure/property relationships therefrom.ADMET polymers with varying amino acids incorporated at every 19th carbon in the main chain were investigated by Freudenberg et al.96 The effect of chiral, achiral, polar, and non-polar amino acids on the thermal properties was examined (see Fig. 38 for a schematic of materials tested). It was found that polymers bearing non-polar amino acid side chains formed only amorphous materials. Polar amino acid side chains in the glutamic acid and aspartic acid residues resulted in semi-crystalline polymers with sharp melting endotherms observed via DSC. WAXS analysis suggested a lamellar structure for both glutamic acid and aspartic acid systems, but the authors could not make a more specific assignment of secondary structure based on the data presented.
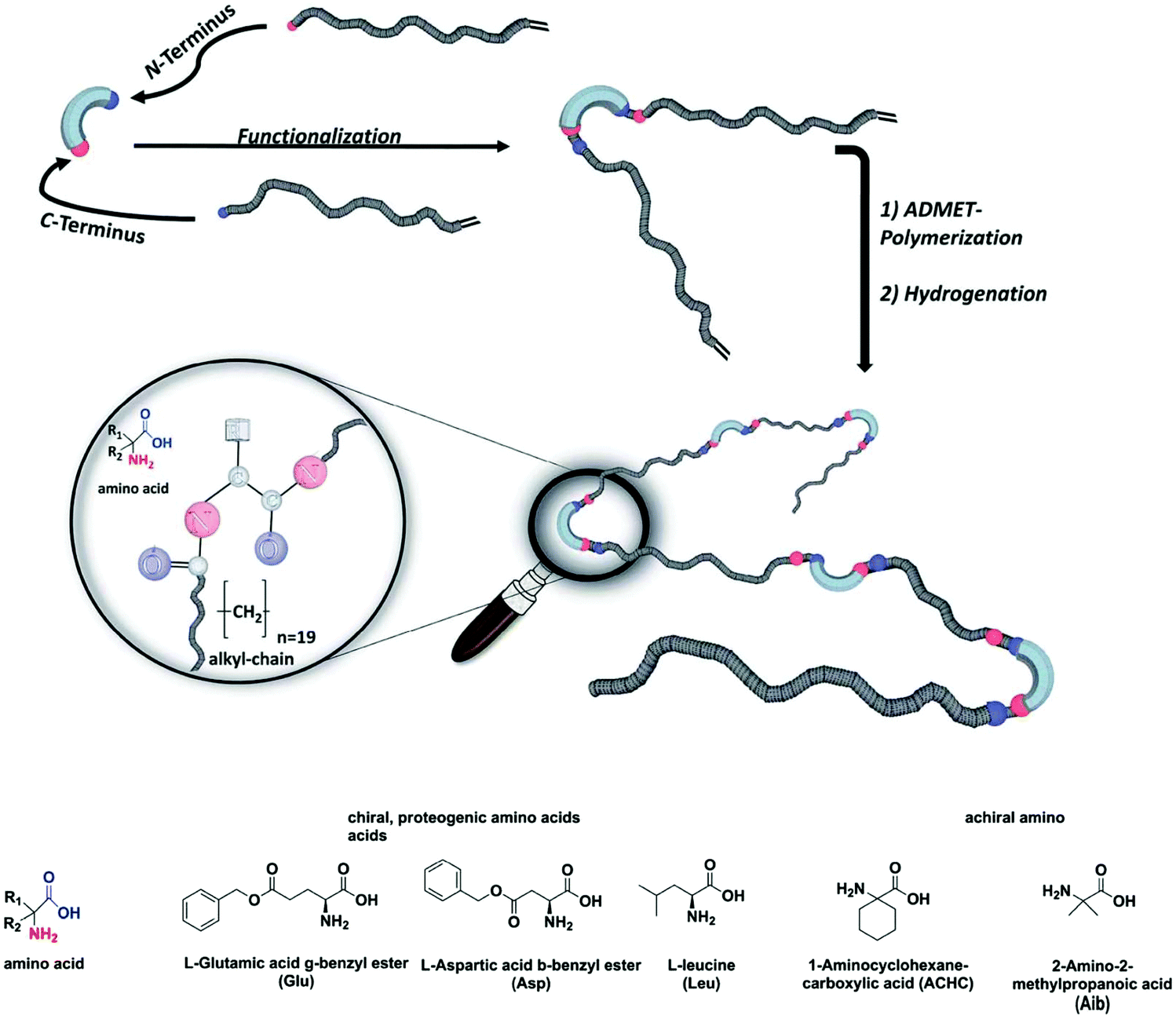 | ||
| Fig. 38 Schematic of ADMET polymers containing amino acid residues in the main chain. (Reprinted from ref. 96. Published by The Royal Society of Chemistry.) | ||
Glycolipids, generally found decorating the outside of cell membranes, are important for maintaining membrane stability and for cell recognition. A clever enzymatic approach to prepare α,ω-unsaturated trehalose diesters was described by Hibert et al.97 When crystallized from the bulk, the resulting ADMET polymer of the pure trehalose-derived glycolipid exhibited an EC ordered morphology driven by the association of carbohydrate and lipidic regions, respectively. Nanoscale self-assembly into micelles was observed in water due to the amphiphilic nature of the synthetic glycolipid (Fig. 39a and b). In another study, oligodeoxynucleosides linked with olefin bridges via head-to-tail ADMET polymerization were shown to form remarkably long fibrils via self-assembly similar to that which drives the formation of the double helix found in deoxyribonucleic acid (DNA, Fig. 39c and d).98
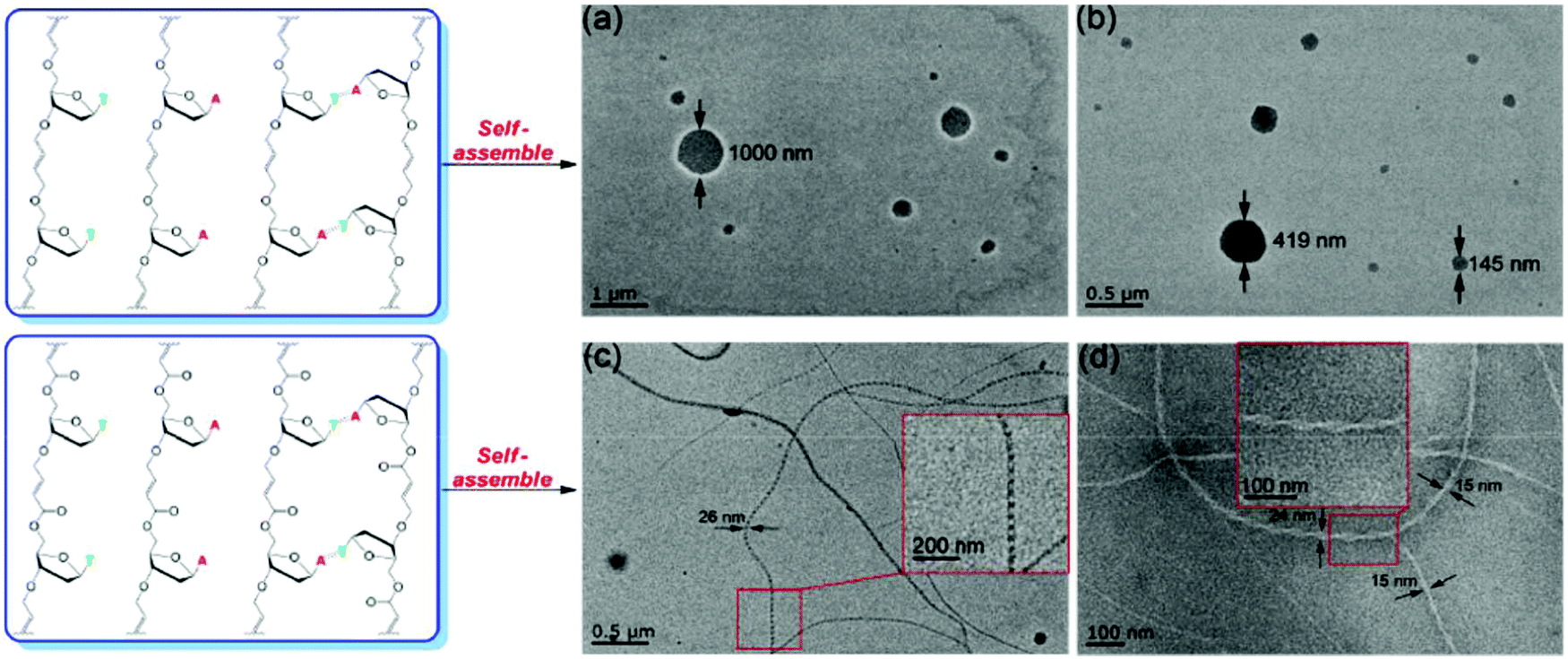 | ||
| Fig. 39 Schematic of synthesized oligodeoxynucleosides from regio-irregular ADMET polymerization (top) or head-to-tail ADMET polymerization (bottom), and TEM images of corresponding self-assembled nanostructures. (Reprinted with permission from ref. 98. Copyright©2019, American Chemical Society.) | ||
Sustainably derived materials
A blossoming sub-field in the polymer chemistry community is the synthesis of polymers derived from renewable feedstocks. The phrase “renewable feedstocks” has become a catch-all term for any polymer precursor obtained from a plant or animal source, often from what would normally be a waste byproduct in manufacturing. Specifically, carbohydrates, vegetable oils, lignocellulose, chitin, etc., have all been used to create polymeric building blocks.99–101 The interest in synthesizing polymers from renewable feedstocks is reaching a fever pitch to help address the societal problems of plastic pollution and heavy reliance on fossil fuel feedstocks. Sustainably derived polymers are generally carbon sinks and are often biodegradable, but overall there are still challenges associated with their poor processability and recyclability. A benefit of ADMET polymerization in this field is that the featured biomass-derived moieties are incorporated into the polymer backbone in a precise manner, giving rise to properties different from those of sustainable polymers prepared by controlled radical polymerization.For aliphatic polyesters and polyamides, precision spacing of the carbonyl moieties is essential, because dipole–dipole or hydrogen bonding interactions are responsible for the famously high melting points and good mechanical properties of these materials. Sustainable polyesters and amides are not generally aliphatic, so structure/property relationships are the focus of the cutting-edge research in this area. Sustainably derived α,ω-diene ester monomers were synthesized from glucose derivatives isosorbide and glucarodilactone and 10-undecenoyl chloride (a castor oil derivative).102 The two monomers (glucarodilactone undecenoate (GDLU) and isosorbide undecenoate (IU)) were homopolymerized and copolymerized, and Mn values of 50–60 kDa were achieved (see monomer and polymer structures in Fig. 40). The two homopolymers and two copolymers with very similar GDLU/IU compositions were analyzed for their thermal and mechanical characteristics. Poly(IU) exhibited the highest thermal stability (Td = 369 °C) and all of the polymers exhibited melting points between 22 and 59 °C. Tensile testing of homopolymers poly(GDLU) and poly(IU) revealed brittle behavior, while both copolymers exhibited rubbery behavior and shape-memory properties. Further work by the same group found that the percentage of GDLU incorporated was positively correlated with desirable degradation properties, and rheological measurements on GDLU-containing polymers suggested that a transient network was responsible for the observed elastic and shape-memory properties.103
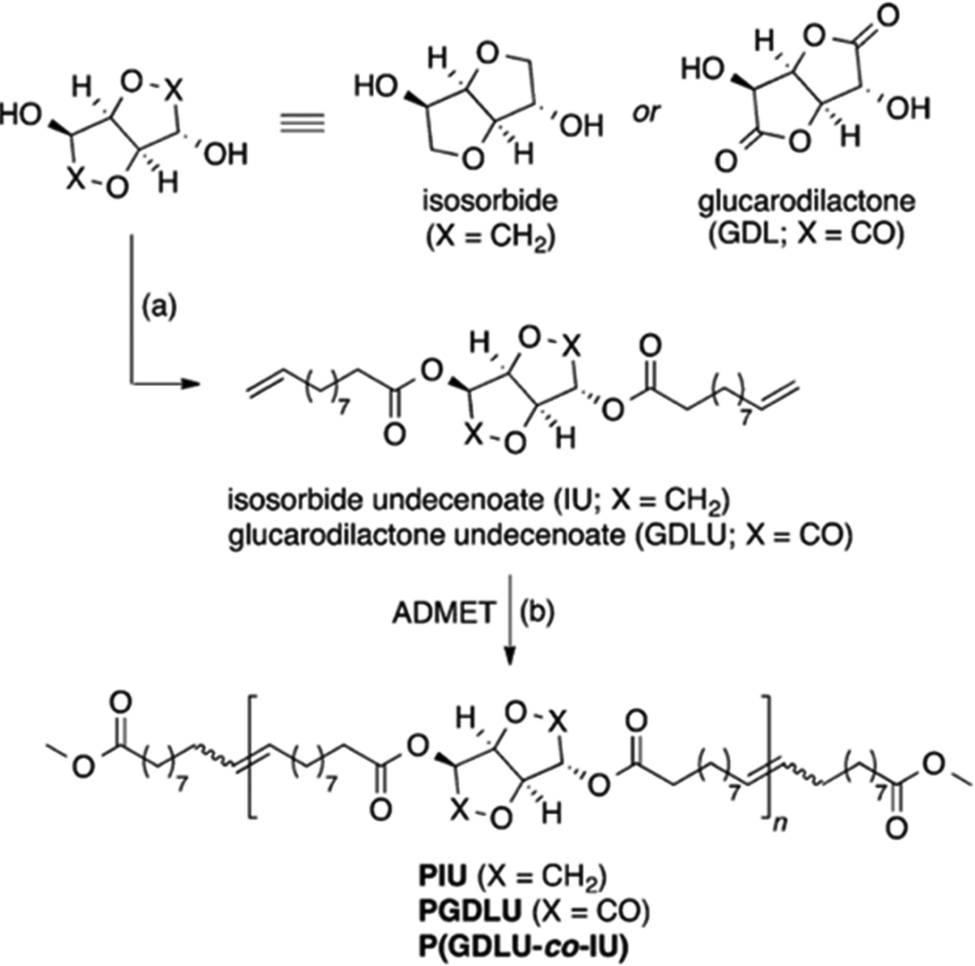 | ||
| Fig. 40 Synthesis of GDLU and IU and subsequent ADMET polymerization to form glucose and castor oil-derived polyesters. Conditions: (a) 4-(dimethylamino) pyridine, 10-undecenoyl chloride, tetrahydrofuran/chloroform, 0 °C to RT, 16 h; (b) 1.0 mol% Grubbs’ 2nd generation catalyst, 1.0 mol% methyl 10-undecenoate, toluene (0.33 M), 80 °C, 16 h, vacuum. (Reprinted with permission from ref. 102. Copyright©2015, American Chemical Society.) | ||
Barbara et al., reported the synthesis of poly(ester-alkenamers) prepared by ADMET polymerization of ferulic acid (from lignocellulosic biomass), biosourced diols (isosorbide and butanediol), and bromo-alkenes.104 Thermal stability up to ∼350 °C was reported and the Tgs were tunable by adjusting the nature of the internal diester and the alkene length of the α,ω-dienes. A structurally complex bisphenol polyester derived from syringaresinol which followed similar structure/property trends was prepared by Hollande and co-workers.105 Mutulu and Meier reported an ADMET polymerization approach to prepare unsaturated precision polyamide-X,20 (X corresponds to the number of carbons in the aliphatic diamine) from methyl-10-undecenoate (derived from castor oil).106 The symmetric amide-containing α,ω-diene was synthesized by treating 2.5 equivalents of methyl-10-undecenoate with the diamine of interest (1 equivalent) with 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as a catalyst. ADMET polymerization of the prepared monomers yielded unsaturated polyamides with modest Mn values.
A large library of phenolic polyethers were prepared by Vlaminck and co-workers from lignin-derived aromatic diols.107 A summary of the α,ω-dienes prepared is presented in Fig. 41. Diols were created by either a Dakin reaction (n = 0) or reduction (n = 1) of the aldehyde, and the dienes were formed by a Williamson ether synthesis with allyl bromide (A, m = 1), butenyl bromide (B, m = 2), or pentenyl bromide (P, m = 3). Several structure/property relationships in the ADMET polymers were observed due to the large structural variety including phenyl substitution pattern, alkyl spacer length, and varying methoxy substituents (0, 1, or 2). As might be expected, ortho-substitution on the aromatic ring resulted in lower Tg values compared to para- and meta-substituted rings. Chain flexibility afforded by longer alkyl spacers between aromatic groups also resulted in lower Tgs. Added methoxy substituents on the aromatic ring generally increased the Tg values. More structurally complex polyethers and polyesters were constructed from vanillin and eugenol derivatives as well as polyethers from L-ascorbic acid.108,109
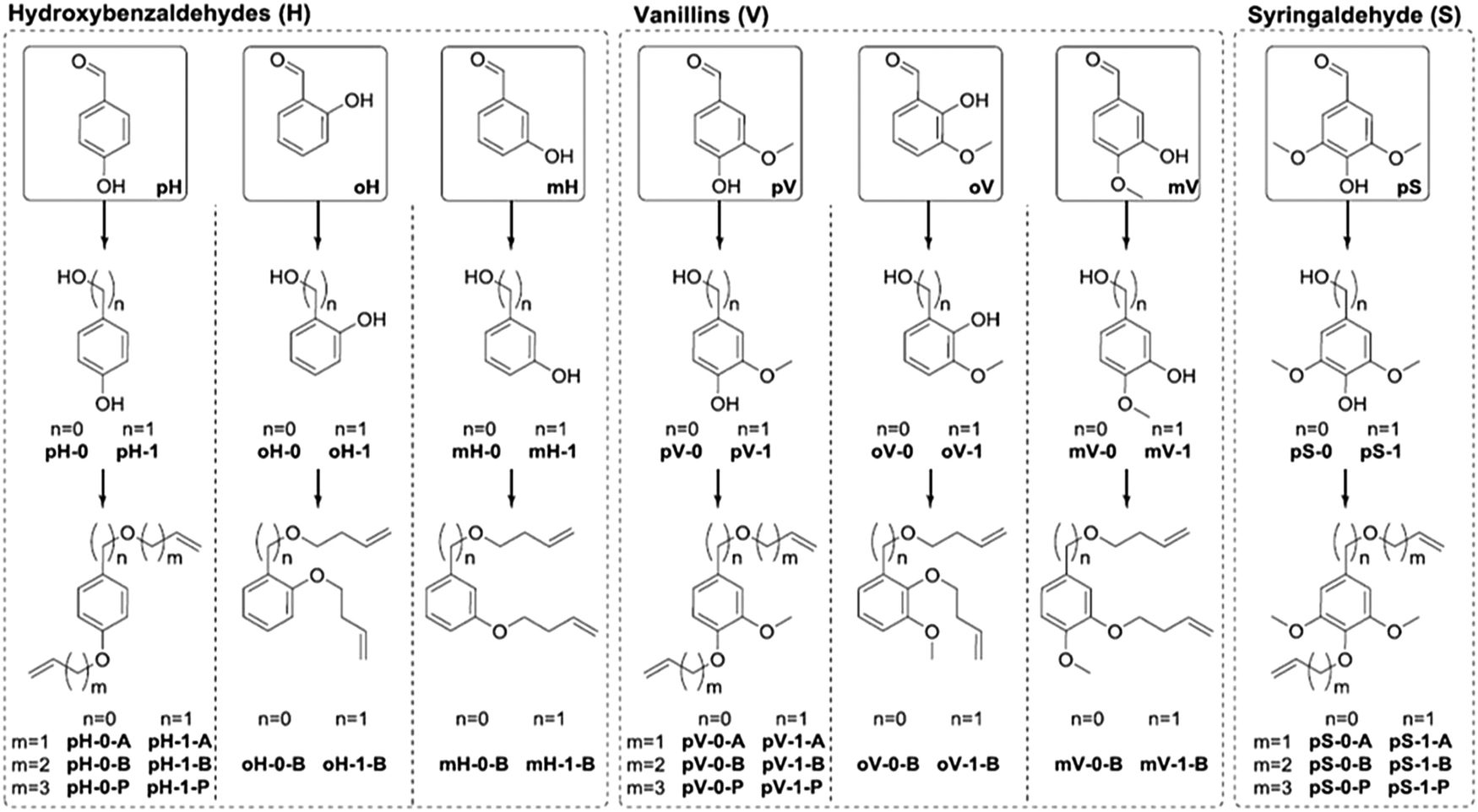 | ||
| Fig. 41 Overview of synthesized diene monomers starting from hydroxybenzaldehydes (H), vanillins (V) and syringaldehyde (S), with different substitution patterns (ortho (o), meta (m), para (p)). (Reprinted with permission from ref. 107. Copyright©2017 Elsevier Ltd.) | ||
5. Conclusion
During the past two-and-a-half decades, the trajectory of ADMET polymer research closely mirrors that of the metathesis catalysts that make them possible. Early synthetic work focused on preparing all aliphatic symmetric α,ω-dienes which were useful in carefully modeling ethylene/alkylene copolymers with unprecedented control of functional group spacing. Creativity in this area resulted in universal syntheses to prepare precision ethylene/alkylene copolymers with nearly any branch size or methylene run length. Later work focused on the incorporation of other functional groups pendant to the polyethylene backbone, such as halides and functional groups containing oxygen, phosphorus, sulfur, and boron. Of particular note are the ADMET polymers prepared with heteroatoms in the backbone; chiefly silanes, siloxanes, sulfites, and sulfones. The lack of tacticity in these polymers gave rise to unique properties compared to ADMET polymers with pendant functional groups. A key trend within this work is that incorporation of polar functionality correlated with the development of well-defined metathesis catalysts which were sufficiently robust to tolerate the presence of these functional groups. Because of its functional group tolerance, as well as a well-documented resistance to 1,2-olefin isomerization, Grubbs’ first-generation catalyst holds a privileged position among catalysts used for ADMET polymerization.It is clear from this body of work that the precision afforded by ADMET polymerization has led to structure/property understanding that would be impossible to study in imprecise systems. Effect of functional group frequency and size on precision polymer secondary structure is perhaps the most well-studied vignette in this area. Major advances in both identifying and leveraging highly ordered secondary structures in ADMET polymers has occurred in only the past few years. Many functional polymer systems that make use of the olefin bond left behind after ADMET polymerization have been explored and are discussed in this work, among other functional polymers. Still, much of the future research potential in ADMET polymers lies in taking advantage of well-controlled nanoscale secondary structures. The design rules to achieve each possible semi-crystalline structure are becoming clearer, and this understanding will lead to the design of precision polymers which may contribute greatly to the devices of the future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is dedicated with thanks to Prof. Bob Grubbs, a long-time friend. Much of the science reviewed herein was done with long term support from the Army Research Laboratory, Chemical Sciences Division and the National Science Foundation, Division of Materials Research. The Wagener Group has benefited from collaborations with many organizations and extend our gratitude to the Army Research Laboratory, ExxonMobil, LyondellBasell, Teijin Aramid, Sumitomo Chemical Company, Materia, Inc, and the Max Planck Institute for Polymer Research. The authors would like to thank Dr Kathryn R. Williams for manuscript editing and Dr Paula Delgado for her contribution of the abstract artwork.References
- C. J. Schaverien, J. C. Dewan and R. R. Schrock, A Well-Characterized, Highly Active, Lewis Acid Free Olefin Metathesis Catalyst1, J. Am. Chem. Soc., 1986, 108, 2771–2773 CrossRef CAS.
- R. Toreki and R. R. Schrock, A well-defined rhenium(VII) olefin metathesis catalyst, J. Am. Chem. Soc., 1990, 112, 2448–2449 CrossRef CAS.
- T. M. Trnka and R. H. Grubbs, The development of L2X2Ru CHR olefin metathesis catalysts: an organometallic success story, Acc. Chem. Res., 2001, 34, 18–29 CrossRef CAS.
- K. B. Wagener, J. M. Boncella and J. G. Nel, Acyclic diene metathesis (ADMET) polymerization, Macromolecules, 1991, 24, 2649–2657 CrossRef CAS.
- H. Li, L. Caire da Silva, M. D. Schulz, G. Rojas and K. B. Wagener, A review of how to do an acyclic diene metathesis reaction, Polym. Int., 2017, 66, 7–12 CrossRef CAS.
- M. Reisch, Potter Plans Vibrant Enterprise of Monsanto Chemical Spin-Off, Chem. Eng. News, 1997, 75, 14–15 Search PubMed.
- J. A. Smith, K. R. Brzezinska, D. J. Valenti and K. B. Wagener, Precisely controlled methyl branching in polyethylene via acyclic diene metathesis (ADMET) polymerization, Macromolecules, 2000, 33, 3781–3794 CrossRef CAS.
- T. W. Baughman, E. van der Aa, S. E. Lehman and K. B. Wagener, Circumventing the Reactivity Ratio Dilemma: Synthesis of Ethylene-co-Methyl Vinyl Ether Copolymer, Macromolecules, 2005, 38, 2550–2551 CrossRef CAS.
- J. C. Sworen, J. A. Smith, J. M. Berg and K. B. Wagener, Modeling Branched Polyethylene: Copolymers Possessing Precisely Placed Ethyl Branches, J. Am. Chem. Soc., 2004, 126, 11238–11246 CrossRef CAS.
- J. A. Smith, K. R. Brzezinska, D. J. Valenti and K. B. Wagener, Precisely Controlled Methyl Branching in Polyethylene via Acyclic Diene Metathesis (ADMET) Polymerization, Macromolecules, 2000, 33, 3781–3794 CrossRef CAS.
- J. C. Sworen, J. A. Smith, K. B. Wagener, L. S. Baugh and S. P. Rucker, Modeling Random Methyl Branching in Ethylene/Propylene Copolymers Using Metathesis Chemistry: Synthesis and Thermal Behavior, J. Am. Chem. Soc., 2003, 125, 2228–2240 CrossRef CAS.
- J. C. Sworen and K. B. Wagener, Linear low-density polyethylene containing precisely placed hexyl branches, Macromolecules, 2007, 40, 4414–4423 CrossRef CAS.
- G. Rojas, B. Inci, Y. Wei and K. B. Wagener, Precision polyethylene: Changes in morphology as a function of alkyl branch size, J. Am. Chem. Soc., 2009, 131, 17376–17386 CrossRef CAS.
- G. Rojas and K. B. Wagener, Avoiding olefin isomerization during decyanation of alkylcyano alpha,omega-dienes: A deuterium labeling and structural study of mechanism, J. Org. Chem., 2008, 73, 4962–4970 CrossRef CAS.
- L. C. da Silva, G. Rojas, M. D. Schulz and K. B. Wagener, Acyclic diene metathesis polymerization: History, methods and applications, Prog. Polym. Sci., 2017, 69, 79–107 CrossRef.
- B. Inci, I. Lieberwirth, W. Steffen, M. Mezger, R. Graf, K. Landfester and K. B. Wagener, Decreasing the alkyl branch frequency in precision polyethylene: effect of alkyl branch size on nanoscale morphology, Macromolecules, 2012, 45, 3367–3376 CrossRef CAS.
- H. Li, G. Rojas and K. B. Wagener, Long-chain branched random polyethylene via acyclic diene metathesis (ADMET) copolymerization, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 1705–1710 CrossRef CAS.
- D. Mäder, J. Heinemann, P. Walter and R. Mülhaupt, Influence ofn-Alkyl Branches on Glass-Transition Temperatures of Branched Polyethylenes Prepared by Means of Metallocene- and Palladium-Based Catalysts, Macromolecules, 2000, 33, 1254–1261 CrossRef.
- T. W. Baughman, J. C. Sworen and K. B. Wagener, Sequenced ethylene− propylene copolymers: Effects of short ethylene run lengths, Macromolecules, 2006, 39, 5028–5036 CrossRef CAS.
- T. W. Baughman, J. C. Sworen and K. B. Wagener, The facile preparation of alkenyl metathesis synthons, Tetrahedron, 2004, 60, 10943–10948 CrossRef CAS.
- N. F. Sauty, H. Li, L. C. da Silva and K. B. Wagener, Large-Scale Preparation of Long-Chain ADMET Synthons, Synth. Commun., 2014, 44, 2409–2415 CrossRef CAS.
- E. Boz, K. B. Wagener, A. Ghosal, R. Fu and R. G. Alamo, Synthesis and crystallization of precision ADMET polyolefins containing halogens, Macromolecules, 2006, 39, 4437–4447 CrossRef CAS.
- E. Boz, A. J. Nemeth, I. Ghiviriga, K. Jeon, R. G. Alamo and K. B. Wagener, Precision ethylene/vinyl chloride polymers via condensation polymerization, Macromolecules, 2007, 40, 6545–6551 CrossRef CAS.
- E. Boz, A. J. Nemeth, R. G. Alamo and K. B. Wagener, Precision ethylene/vinyl bromide polymers, Adv. Synth. Catal., 2007, 349, 137–141 CrossRef CAS.
- T. W. Baughman, C. D. Chan, K. I. Winey and K. B. Wagener, Synthesis and morphology of well-defined poly(ethylene-co-acrylic acid) copolymers, Macromolecules, 2007, 40, 6564–6571 CrossRef CAS.
- L. M. Hall, M. E. Seitz, K. I. Winey, K. L. Opper, K. B. Wagener, M. J. Stevens and A. L. Frischknecht, Ionic aggregate structure in ionomer melts: effect of molecular architecture on aggregates and the ionomer peak, J. Am. Chem. Soc., 2012, 134, 574–587 CrossRef CAS.
- M. E. Seitz, C. D. Chan, K. L. Opper, T. W. Baughman, K. B. Wagener and K. I. Winey, Nanoscale morphology in precisely sequenced poly(ethylene- co -acrylic acid) zinc ionomers, J. Am. Chem. Soc., 2010, 132, 8165–8174 CrossRef CAS.
- K. L. Opper, D. Markova, M. Klapper, K. Müllen and K. B. Wagener, Precision phosphonic acid functionalized polyolefin architectures, Macromolecules, 2010, 43, 3690–3698 CrossRef CAS.
- K. L. Opper, B. Fassbender, G. Brunklaus, H. W. Spiess and K. B. Wagener, Polyethylene functionalized with precisely spaced phosphonic acid groups, Macromolecules, 2009, 42, 4407–4409 CrossRef CAS.
- K. A. Mauritz and R. B. Moore, State of understanding of Nafion, Chem. Rev., 2004, 104, 4535–4586 CrossRef CAS.
- D. Fischer and H. H. Eysel, Analysis of polyethylene surface sulfonation, J. Appl. Polym. Sci., 1994, 52, 545–548 CrossRef CAS.
- F. Kučera and J. Jančář, Homogeneous and heterogeneous sulfonation of polymers: A review, Polym. Eng. Sci., 1998, 38, 783–792 CrossRef.
- T. W. Gaines, M. H. Bell, E. B. Trigg, K. I. Winey and K. B. Wagener, Precision Sulfonic Acid Polyolefins via Heterogenous to Homogenous Deprotection, Macromol. Chem. Phys., 2018, 219, 1700634 CrossRef.
- E. B. Trigg, T. W. Gaines, M. Maréchal, D. E. Moed, P. Rannou, K. B. Wagener, M. J. Stevens and K. I. Winey, Self-assembled highly ordered acid layers in precisely sulfonated polyethylene produce efficient proton transport, Nat. Mater., 2018, 17, 725–731 CrossRef CAS.
- T. W. Gaines, E. B. Trigg, K. I. Winey and K. B. Wagener, High Melting Precision Sulfone Polyethylenes Synthesized by ADMET Chemistry, Macromol. Chem. Phys., 2016, 217, 2351–2359 CrossRef CAS.
- V. M. Marx, A. H. Sullivan, M. Melaimi, S. C. Virgil, B. K. Keitz, D. S. Weinberger, G. Bertrand and R. H. Grubbs, Cyclic alkyl amino carbene (caac) ruthenium complexes as remarkably active catalysts for ethenolysis, Angew. Chem., Int. Ed., 2015, 54, 1919–1923 CrossRef CAS.
- M. Bell, H. G. Hester, A. N. Gallman, V. Gomez, J. Pribyl, G. Rojas, A. Riegger, T. Weil, H. Watanabe, Y. Chujo and K. B. Wagener, Bulk Acyclic Diene Metathesis Polycondensation, Macromol. Chem. Phys., 2019, 220, 1–6 CrossRef.
- J. N. Cambre and B. S. Sumerlin, Biomedical applications of boronic acid polymers, Polymer, 2011, 52, 4631–4643 CrossRef CAS.
- D. Roy and B. S. Sumerlin, Glucose-sensitivity of boronic acid block copolymers at physiological pH, ACS Macro Lett., 2012, 1, 529–532 CrossRef CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Triply-responsive boronic acid block copolymers: solution self-assembly induced by changes in temperature, pH, or sugar concentration, Chem. Commun., 2009, 2106–2108 RSC.
- J. N. Cambre, D. Roy, S. R. Gondi and B. S. Sumerlin, Facile strategy to well-defined water-soluble boronic acid (co) polymers, J. Am. Chem. Soc., 2007, 129, 10348–10349 CrossRef CAS.
- C. Simocko, T. C. Young and K. B. Wagener, ADMET polymers containing precisely spaced pendant boronic acids and esters, Macromolecules, 2015, 48, 5470–5473 CrossRef CAS.
- C. Simocko and K. B. Wagener, Effects of boron-containing Lewis acids on olefin metathesis, Organometallics, 2013, 32, 2513–2516 CrossRef CAS.
- A. C. Church, J. H. Pawlow and K. B. Wagener, Synthesis of Functionalized Polycarbosilanes via One-Pot ADMET Polymerization− Macromolecular Substitution, Macromolecules, 2002, 35, 5746–5751 CrossRef CAS.
- A. C. Church, J. H. Pawlow and K. B. Wagener, ADMET polymerization as a route to functionalized polycarbosilanes, Macromol. Chem. Phys., 2003, 204, 32–39 CrossRef CAS.
- P. P. Matloka and K. B. Wagener, The acyclic diene metathesis (ADMET) polymerization approach to silicon containing materials, J. Mol. Catal. A: Chem., 2006, 257, 89–98 CrossRef CAS.
- K. R. Brzezinska, K. B. Wagener and G. T. Burns, Silicon-terminated telechelic oligomers by ADMET chemistry: Synthesis and copolymerization, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 849–856 CrossRef CAS.
- T. E. Hopkins, J. H. Pawlow, D. L. Koren, K. S. Deters, S. M. Solivan, J. A. Davis, F. J. Gómez and K. B. Wagener, Chiral polyolefins bearing amino acids, Macromolecules, 2001, 34, 7920–7922 CrossRef CAS.
- T. E. Hopkins and K. B. Wagener, Amino acid and dipeptide functionalized polyolefins, Macromolecules, 2003, 36, 2206–2214 CrossRef CAS.
- T. E. Hopkins and K. B. Wagener, Chiral polyolefins, Adv. Mater., 2002, 14, 1703–1715 CrossRef CAS.
- T. E. Hopkins and K. B. Wagener, ADMET Synthesis of Polyolefins Targeted for Biological Applications, Macromolecules, 2004, 37, 1180–1189 CrossRef CAS.
- S. M. Bush and M. North, Synthesis of homochiral addition polymers derived from N-trityl-(S)-serine, Polymer, 1996, 37, 4649–4652 CrossRef CAS.
- E. B. Berda and K. B. Wagener, Probing the effects of hydrophilic branch size, distribution, and connectivity in amphiphilic polyethylene, Macromol. Chem. Phys., 2008, 209, 1601–1611 CrossRef CAS.
- E. B. Berda, R. E. Lande and K. B. Wagener, Precisely defined amphiphilic graft copolymers, Macromolecules, 2007, 40, 8547–8552 CrossRef CAS.
- E. B. Berda and K. B. Wagener, Inducing pendant group interactions in precision polyolefins: synthesis and thermal behavior, Macromolecules, 2008, 41, 5116–5122 CrossRef CAS.
- W. Qiu, J. Sworen, M. Pyda, E. Nowak-Pyda, A. Habenschuss, K. B. Wagener and B. Wunderlich, Effect of the precise branching of polyethylene at each 21st CH 2 group on its phase transitions, crystal structure, and morphology, Macromolecules, 2006, 39, 204–217 CrossRef CAS.
- D. L. VanderHart, R. G. Alamo, M. R. Nyden, M. H. Kim and L. Mandelkern, Observation of resonances associated with stereo and regio defects in the crystalline regions of isotactic polypropylene: toward a determination of morphological partitioning, Macromolecules, 2000, 33, 6078–6093 CrossRef CAS.
- R. G. Alamo, D. L. VanderHart, M. R. Nyden and L. Mandelkern, Morphological partitioning of ethylene defects in random propylene-ethylene copolymers, Macromolecules, 2000, 33, 6094–6105 CrossRef CAS.
- C. F. Buitrago, T. M. Alam, K. L. Opper, B. S. Aitken, K. B. Wagener and K. I. Winey, Morphological trends in precise acid-and ion-containing polyethylenes at elevated temperature, Macromolecules, 2013, 46, 8995–9002 CrossRef CAS.
- C. F. Buitrago, J. E. Jenkins, K. L. Opper, B. S. Aitken, K. B. Wagener, T. M. Alam and K. I. Winey, Room temperature morphologies of precise acid-and ion-containing polyethylenes, Macromolecules, 2013, 46, 9003–9012 CrossRef CAS.
- U. H. Choi, L. R. Middleton, M. Soccio, C. F. Buitrago, B. S. Aitken, H. Masser, K. B. Wagener, K. I. Winey and J. Runt, Dynamics of precise ethylene ionomers containing ionic liquid functionality, Macromolecules, 2015, 48, 410–420 CrossRef CAS.
- L. R. Middleton, J. D. Tarver, J. Cordaro, M. Tyagi, C. L. Soles, A. L. Frischknecht and K. I. Winey, Heterogeneous Chain Dynamics and Aggregate Lifetimes in Precise Acid-Containing Polyethylenes: Experiments and Simulations, Macromolecules, 2016, 49, 9176–9185 CrossRef.
- E. B. Trigg, M. J. Stevens and K. I. Winey, Chain Folding Produces a Multilayered Morphology in a Precise Polymer: Simulations and Experiments, J. Am. Chem. Soc., 2017, 139, 3747–3755 CrossRef CAS.
- S. Hosoda, Y. Nozue, Y. Kawashima, S. Utsumi, T. Nagamatsu, K. Wagener, E. Berda, G. Rojas, T. Baughman and J. Leonard, Perfectly controlled lamella thickness and thickness distribution: A morphological study on ADMET polyolefins, Macromol. Symp., 2009, 282, 50–64 CrossRef CAS.
- S. Hosoda, Y. Nozue, Y. Kawashima, K. Suita, S. Seno, T. Nagamatsu, K. B. Wagener, B. Inci, F. Zuluaga, G. Rojas and J. K. Leonard, Effect of the sequence length distribution on the lamellar crystal thickness and thickness distribution of polyethylene: Perfectly equisequential ADMET polyethylene vs ethylene/α-Olefin Copolymer, Macromolecules, 2011, 44, 313–319 CrossRef CAS.
- B. Inci and K. B. Wagener, Decreasing the alkyl branch frequency in precision polyethylene: Pushing the limits toward longer run lengths, J. Am. Chem. Soc., 2011, 133, 11872–11875 CrossRef CAS.
- E. Boz, A. J. Nemeth, K. B. Wagener, K. Jeon, R. Smith, F. Nazirov, M. R. Bockstaller and R. G. Alamo, Well-defined precision ethylene/vinyl fluoride polymers: Synthesis and crystalline properties, Macromolecules, 2008, 41, 1647–1653 CrossRef CAS.
- R. G. Alamo, K. Jeon, R. L. Smith, E. Boz, K. B. Wagener and M. R. Bockstaller, Crystallization of polyethylenes containing chlorines: Precise vs random placement, Macromolecules, 2008, 41, 7141–7151 CrossRef CAS.
- P. Kaner, C. Ruiz-Orta, E. Boz, K. B. Wagener, M. Tasaki, K. Tashiro and R. G. Alamo, Kinetic control of chlorine packing in crystals of a precisely substituted polyethylene. Toward advanced polyolefin materials, Macromolecules, 2014, 47, 236–245 CrossRef CAS.
- M. Tasaki, H. Yamamoto, M. Hanesaka, K. Tashiro, E. Boz, K. B. Wagener, C. Ruiz-Orta and R. G. Alamo, Polymorphism and phase transitions of precisely halogen-substituted polyethylene. (1) Crystal structures of various crystalline modifications of bromine-substituted polyethylene on every 21st backbone carbon, Macromolecules, 2014, 47, 4738–4749 CrossRef CAS.
- X. Zhang, L. Santonja-Blasco, K. B. Wagener, E. Boz, M. Tasaki, K. Tashiro and R. G. Alamo, Infrared Spectroscopy and X-ray Diffraction Characterization of Dimorphic Crystalline Structures of Polyethylenes with Halogens Placed at Equal Distance along the Backbone, J. Phys. Chem. B, 2017, 121, 10166–10179 CrossRef CAS.
- S. Chanda and S. Ramakrishnan, Controlling Interlamellar Spacing in Periodically Grafted Amphiphilic Copolymers, Macromolecules, 2016, 49, 3254–3263 CrossRef CAS.
- L. Yan, K. C. Bustillo, O. Panova, A. M. Minor and K. I. Winey, Solution-grown crystals of precise acid- and ion-containing polyethylenes, Polymer, 2018, 135, 111–119 CrossRef CAS.
- O. Diat and G. Gebel, Proton channels, Nat. Mater., 2008, 7, 13–14 CrossRef CAS.
- G. Lieser, G. Wegner, J. A. Smith and K. B. Wagener, Morphology and packing behavior of model ethylene/propylene copolymers with precise methyl branch placement, Colloid Polym. Sci., 2004, 282, 773–781 CrossRef CAS.
- T. W. Gaines, T. Nakano, Y. Chujo, E. B. Trigg, K. I. Winey and K. B. Wagener, Precise Sulfite Functionalization of Polyolefins via ADMET Polymerization, ACS Macro Lett., 2015, 4, 624–627 CrossRef CAS.
- A. Lv, Z. L. Li, Y. H. Wu, F. S. Du and Z. C. Li, Synthesis of precision polymers with regularly placed perfluoroalkyl segments and sulfonic acid groups via ADMET polymerization and internal alkene modification, Polymer, 2018, 153, 123–130 CrossRef CAS.
- J. K. Leonard, D. Turek, K. B. Sloan and K. B. Wagener, Polyethylene prodrugs using precisely placed pharmaceutical agents, Macromol. Chem. Phys., 2010, 211, 154–165 CrossRef CAS.
- K. A. Miller, E. G. Morado, S. R. Samanta, B. A. Walker, A. Z. Nelson, S. Sen, D. T. Tran, D. J. Whitaker, R. H. Ewoldt, P. V. Braun and S. C. Zimmerman, Acid-Triggered, Acid-Generating, and Self-Amplifying Degradable Polymers, J. Am. Chem. Soc., 2019, 141, 2838–2842 CrossRef CAS.
- D. Tao and K. B. Wagener, Conjugated Monomers in Acyclic Diene Metathesis (ADMET) Polymerization, Macromolecules, 1994, 27, 1281–1283 CrossRef CAS.
- K. Nomura, H. Morimoto, Y. Imanishi, Z. Ramhani and Y. Geerts, Synthesis of high molecular weight trans-poly(9,9-di-n-octylfluorene-2,7-vinylene) by the acyclic diene metathesis polymerization using molybdenum catalysts, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2463–2470 CrossRef CAS.
- T. Miyashita, M. Kunisawa, S. Sueki and K. Nomura, Synthesis of Poly(arylene vinylene)s with Different End Groups by Combining Acyclic Diene Metathesis Polymerization with Wittig-type Couplings, Angew. Chem., Int. Ed., 2017, 56, 5288–5293 CrossRef CAS.
- T. Yamada, K. Nomura and M. Fujiki, Noticeable Chiral Center Dependence of Signs and Magnitudes in Circular Dichroism (CD) and Circularly Polarized Luminescence (CPL) Spectra of all - Trans -Poly(9,9-dialkylfluorene-2,7-vinylene)s Bearing Chiral Alkyl Side Chains in Solution, Aggregates, an, Macromolecules, 2018, 51, 2377–2387 CrossRef CAS.
- Y. Fushimi, M. Koinuma, Y. Yasuda, K. Nomura and M. S. Asano, Effects of End-Groups on Photophysical Properties of Poly(9,9-di-n-octylfluorene-2,7-vinylene)s Linked with Metalloporphyrins: Synthesis and Time-Resolved Fluorescence Spectroscopy, Macromolecules, 2017, 50, 1803–1814 CrossRef CAS.
- K. Nomura, N. Yamamoto, R. Ito and Y. Geerts, Synthesis of all-trans high molecular weight poly(n-alkylcarbazole-2,7-vinylene)s and poly(9,9-dialkylfluorene-2,7-vinylene)s by acyclic diene metathesis (admet) polymerization using ruthenium-carbene complex catalysts, Macromolecules, 2009, 42, 5104–5111 CrossRef.
- K. Nomura, Y. Miyamoto, H. Morimoto and Y. Geerts, Acyclic diene metathesis polymerization of 2,5-dialkyl-1,4-divinylbenzene with molybdenum or ruthenium catalysts: Factors affecting the precise synthesis of defect-free, high-molecular-weight trans-poly(p-phenylene vinylene)s, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 6166–6177 CrossRef CAS.
- Y. Qin and M. A. Hillmyer, Poly(3-hexyl-2, 5-thienylene vinylene) by ADMET polymerization of a dipropenyl monomer, Macromolecules, 2009, 42, 6429–6432 CrossRef CAS.
- Z. Zhang and Y. Qin, Cross-conjugated poly(selenylene vinylene)s, Polym. Chem., 2019, 10, 1018–1025 RSC.
- H. Zhang, F. Liu, J. Cao, L. Ling and R. Sun, Ferrocene-containing polymers synthesized by acyclic diene metathesis (ADMET) polymerization, Chin. J. Polym. Sci., 2016, 34, 242–252 CrossRef CAS.
- R. H. Friend, R. W. Gymer, A. B. Holmes, J. H. Burroughes, R. N. Marks, C. Taliani, D. D. C. Bradley, D. A. Dos Santos, J. L. Bredas, M. Logdlund and W. R. Salaneck, Electroluminescence in Conjugated Polymers, Nature, 1995, 397, 121–128 CrossRef.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Conjugated polymer-based organic solar cells, Chem. Rev., 2007, 107, 1324–1338 CrossRef.
- Z. Zhang and Y. Qin, Synthesis and Characterization of Poly(selenylene vinylene) and Poly(selenylene vinylene)-co-Poly(thienylene vinylene) through Acyclic Diene Metathesis (ADMET) Polymerization, ACS Macro Lett., 2015, 4, 679–683 CrossRef CAS.
- J. C. Speros, B. D. Paulsen, B. S. Slowinski, C. D. Frisbie and M. A. Hillmyer, Band gap and HOMO level control in poly(thienylene vinylene)s prepared by ADMET polymerization, ACS Macro Lett., 2012, 1, 986–990 CrossRef CAS.
- W. Joo and C. W. Bielawski, Design, synthesis and study of a photochromic α,ω-diene: Toward new classes of photoswitchable polymers, Org. Biomol. Chem., 2019, 17, 2486–2491 RSC.
- J. P. Cole, A. M. Hanlon, K. J. Rodriguez and E. B. Berda, Protein-like structure and activity in synthetic polymers, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 191–206 CrossRef CAS.
- J. Freudenberg, S. Poppe and W. H. Binder, Precision polymers containing main-chain-amino acids: ADMET polymerization and crystallization, RSC Adv., 2017, 7, 47507–47519 RSC.
- G. Hibert, E. Grau, D. Pintori, S. Lecommandoux and H. Cramail, ADMET polymerization of α,ω-unsaturated glycolipids: synthesis and physico-chemical properties of the resulting polymers, Polym. Chem., 2017, 8, 3731–3739 RSC.
- L. Wang, M. Wang, L. X. Guo, Y. Sun, X. Q. Zhang, B. P. Lin and H. Yang, Oligodeoxynucleosides with Olefin Bridges, Macromolecules, 2019, 52, 649–659 CrossRef CAS.
- V. Zargar, M. Asghari and A. Dashti, A Review on Chitin and Chitosan Polymers: Structure, Chemistry, Solubility, Derivatives, and Applications, ChemBioEng Rev., 2015, 2, 204–226 CrossRef.
- A. Gandini, T. M. Lacerda, A. J. F. Carvalho and E. Trovatti, Progress of Polymers from Renewable Resources: Furans, Vegetable Oils, and Polysaccharides, Chem. Rev., 2016, 116, 1637–1669 CrossRef CAS.
- M. S. Ganewatta, H. N. Lokupitiya and C. Tang, Lignin biopolymers in the age of controlled polymerization, Polymers, 2019, 11, 1176 CrossRef.
- W. C. Shearouse, L. M. Lillie, T. M. Reineke and W. B. Tolman, Sustainable Polyesters Derived from Glucose and Castor Oil: Building Block Structure Impacts Properties, ACS Macro Lett., 2015, 4, 284–288 CrossRef CAS.
- L. M. Lillie, W. B. Tolman and T. M. Reineke, Structure/property relationships in copolymers comprising renewable isosorbide, glucarodilactone, and 2,5-bis(hydroxymethyl)furan subunits, Polym. Chem., 2017, 8, 3746–3754 RSC.
- I. Barbara, A. L. Flourat and F. Allais, Renewable polymers derived from ferulic acid and biobased diols via ADMET, Eur. Polym. J., 2015, 62, 236–243 CrossRef CAS.
- L. Hollande, A. S. Jaufurally, P. H. Ducrot and F. Allais, ADMET polymerization of biobased monomers deriving from syringaresinol, RSC Adv., 2016, 6, 44297–44304 RSC.
- H. Mutlu and M. A. R. Meier, Unsaturated PA X,20 from renewable resources via metathesis and catalytic amidation, Macromol. Chem. Phys., 2009, 210, 1019–1025 CrossRef CAS.
- L. Vlaminck, S. Lingier, A. Hufendiek and F. E. Du Prez, Lignin inspired phenolic polyethers synthesized via ADMET: Systematic structure-property investigation, Eur. Polym. J., 2017, 95, 503–513 CrossRef CAS.
- A. Llevot, E. Grau, S. Carlotti, S. Grelier and H. Cramail, ADMET polymerization of bio-based biphenyl compounds, Polym. Chem., 2015, 6, 7693–7700 RSC.
- S. Luleburgaz, M. Abuaf, U. Tunca, G. Hizal and H. Durmaz, Synthesis of Poly(vitamin C) through ADMET, Macromol. Rapid Commun., 2017, 38, 1–6 CrossRef.
| This journal is © the Partner Organisations 2021 |