
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSilicon phthalocyanines: synthesis and resurgent applications
Koushambi
Mitra
 *ab and
Matthew C. T.
Hartman
*ab
*ab and
Matthew C. T.
Hartman
*ab
aDepartment of Chemistry, Virginia Commonwealth University, 1001 W Main St, Richmond, 23284, VA, USA
bMassey Cancer Center, Virginia Commonwealth University, 401 College St, Richmond, 23219, Virginia, USA. E-mail: mchartman@vcu.edu; kmitrachem@gmail.com; Tel: +1-804-628-4095
First published on 8th January 2021
Abstract
The intense far-red absorption and emission features have made silicon phthalocyanines (SiPcs) distinct from the structurally related porphyrin analogues. Unlike most other phthalocyanines, SiPcs possess two additional axial bonds which reduce aggregation in solution and can be synthetically tailored, thereby creating further scope for modulation of optical, chemical and electronic properties. Multiple synthetic strategies have been employed for facile construction of symmetrical or unsymmetrical SiPc variants bearing desired substitutents at the axial and the aromatic ring positions. The overarching motive of this concise review article is to highlight and summarize the key synthetic routes and the fast-emerging applications of SiPcs in photouncaging techniques, photothermal and photoimmunotherapy, photovoltaics, optoelectronics and photocatalysis.
 Koushambi Mitra | Koushambi Mitra graduated from Presidency College, University of Calcutta, India in 2009 with a B.Sc. degree in chemistry. She obtained her integrated Ph.D. degree from Indian Institute of Science in 2016. Her doctoral thesis focused on development of visible light-activated platinum(II) complexes as dual-action photo-chemotherapeutic anticancer agents. She undertook postdoctoral studies in Virginia Commonwealth University where she explored synthesis of near-infrared photocages and their applications as cancer drug delivery systems. She then transitioned to The University of Texas at Dallas, where she developed activity-based fluorescent sensors for live cell imaging of the phase II metabolic enzymes, sulfotransferases. Currently, she is a postdoctoral researcher at The University of Chicago and her research involves development of quantitative imaging platforms for studying cations and anions in organelles of living systems. |
 Matthew C. T. Hartman | Matthew C. T. Hartman received his Ph.D. degree from the University of Michigan in 2002, studying under Professor James Coward. He was then a HHMI postdoctoral fellow with Professor Jack Szostak at Harvard University (2002–2006). Since 2007 he has been at Virginia Commonwealth University where he currently is an Associate Professor in the Department of Chemistry and the Massey Cancer Center. His research interests focus on the discovery of natural-product like peptide inhibitors of protein–protein interactions and the delivery and activation of chemotherapeutic agents with light. |
1. Introduction
The mid-twentieth century witnessed a wave of enthusiasm among researchers in developing new classes of macroheterocyclic compounds by inserting different elements in the core of phthalocyanines (Pcs).1–4 Like their closely related porphyrin (Por) analogues, Pcs are also 18 π-electronic systems. However, a Pc construct has imine bridges and isoindole rings as opposed to the methine bridges and pyrrole rings of a Por structure (Fig. 1A and B).5 Due to these structural variations, Pcs exhibit intense absorption at ∼700 nm (Q-band), and thus qualify as one of the elite classes of far-red light absorbing organic compounds. Further modification by replacing benzenes with naphthalenes as the peripheral rings results in the formation of naphthalocyanines (Ncs).6,7 Due to increase in π-delocalization, Q-bands of Ncs are red shifted by ∼100 nm compared to Pcs, and thus, these dyes classify as near-infrared (NIR) absorbing dyes. Despite their interesting optical properties in the visible, red and NIR region, the applications of these hydrophobic planar molecules were not fully realized because of poor solubility and tendency to form aggregates. Aggregate formation quenches excited state lifetimes and diminishes photophysical properties.8,9 Structural variations such as incorporation of appropriate aromatic substitutions or an element in the core structure were sought after to reduce the degree of planarity and hydrophobicity.10–12 These attempts have led to the development of facile synthetic routes for a new generation of Pcs with improved solubility, reduced aggregation, interesting photophysical and photochemical properties.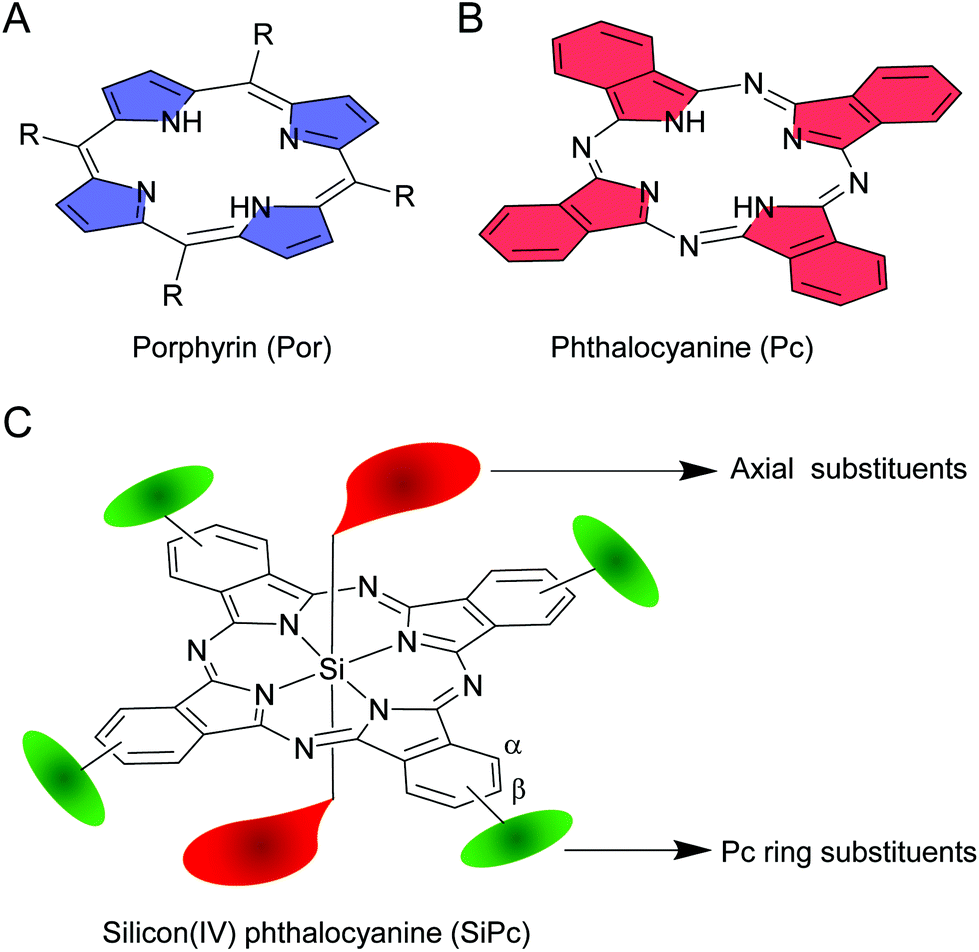 | ||
| Fig. 1 Chemical structures of (A) porphyrin (Por), (B) phthalocyanine (Pc) and (C) silicon(IV) phthalocyanine (SiPc). R denotes various alkyl or aryl groups. The axial substituents are highlighted as red teardrops. The phthalocyanine ring (α) and (β) substituents are highlighted as green ovals. | ||
Amidst all core-substituted Pcs, silicon phthalocyanines (SiPcs) immediately caught attention because of the unusual hexacoordinated silicon(IV) and marked chemical stability of the Si–N bonds.13 Moreover, the presence of two unique axial positions which can be readily functionalized with ligands of choice rendered it unparalleled in the plethora of Pcs (Fig. 1C). These axial ligands serve as pivotal handles for fine-tuning of the properties of SiPcs as desired. Furthermore, bulky or polar substitutions in the axial positions perturb planarity and are more effective than the α or β phthalocyanine ring substitutions in reducing aggregation and improving solubility.14,15 Extensive synthetic methodologies have been developed for preparation of symmetrical and unsymmetrical SiPcs, often containing both tetravalent (axial) and hexavalent (core) silicon in the same structure.16,17 Remarkably, axial Si–O and Si–C bonds exhibit NIR light induced dissociation rendering novel applications of SiPcs in medicinal biology, beyond the context of photodynamic therapy.18–22 These unique properties coupled with the inexpensive cost of synthesis and low toxicity have resulted in fascinating applications of SiPcs in major fields of research including cancer phototherapy, NIR imaging, organic photovoltaics, organic electronics and photocatalysis.
In this condensed review article, we present an overview of the broad synthetic methodologies adapted to improve the design of symmetrical and non-symmetrical SiPcs, since their first successful isolation in around 1960. Here, we also account for the interesting properties and the related applications of SiPcs over the past decade, emphasizing mostly on the emerging and novel applications. In the concluding section, we have included a brief perspective to provide guidance for rationally designing an envelope of new SiPc derivatives, to discern areas of impending developments, and to anticipate the future directions of this interesting field.
2. Syntheses of silicon phthalocyanines
Facile, short and high yielding synthetic strategies comprising of non-hazardous and easily accessible starting materials are an essential prerequisite to realize the complete potential of any material. Various synthetic approaches have been developed to increase the structural flexibility of SiPcs. These approaches offer easy modulation of the physico-chemical properties as desired and help to rapidly generate a vast library of SiPc derivatives with diverse functionalities and applications. For example, successful applications of SiPcs in biological fields can be achieved by incorporation of water soluble or biocompatible groups.11,23 On the other hand, insertion of electron donating and electron withdrawing moieties can alter the opto-electronic properties of SiPc molecules.5,12 Likewise, availability of electrophilic or nucleophilic reactive handles in the SiPc structure will allow to append desired functionalities for versatile applications.Such fine-tuning of SiPc structures is mainly obtained by reactions at two major sites: (i) the axial and (ii) the phthalocyanine ring (α) and (β) positions (Fig. 1C). While the axial handles are exploited for imparting variants without considerably affecting the absorption and emissive properties of SiPc, the α/β substitutions result in a prominent change of spectral features. Below, we have elaborated on the discovery of SiPc and the various synthetic methods developed henceforth resulting in the formation of several symmetrical and unsymmetrical SiPcs.
2.1 Synthesis of SiPc precursors
The synthesis of most SiPcs is based on two main precursors, the dichloro SiPc (SiPcCl2), 1 (Schemes 1 and 2) and the bis-hydroxy adduct (SiPc(OH)2, 2, (Scheme 3). In this section, we have described the synthetic strategies for these important building blocks.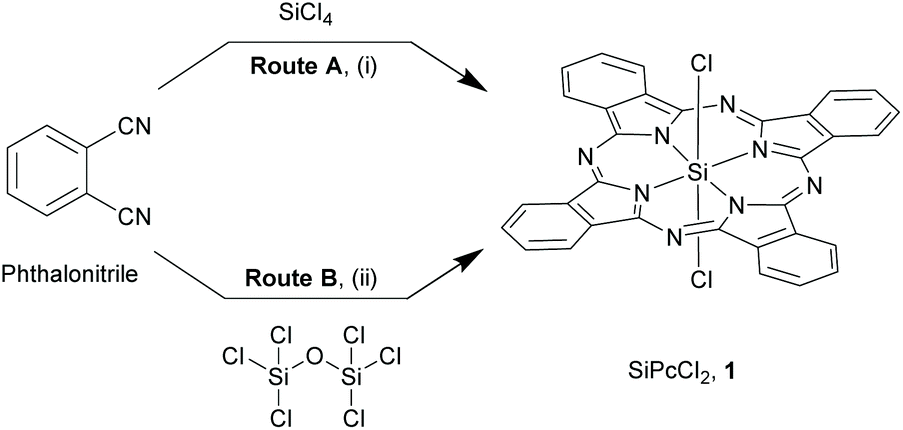 | ||
| Scheme 1 Early synthetic routes for silicon phthalocyanines, 1. Reagents and conditions: (i) SiCl4, dry quinoline, >200 °C, 2 h; (ii) hexachlorodisiloxane, dry quinoline, reflux, 4 h. | ||
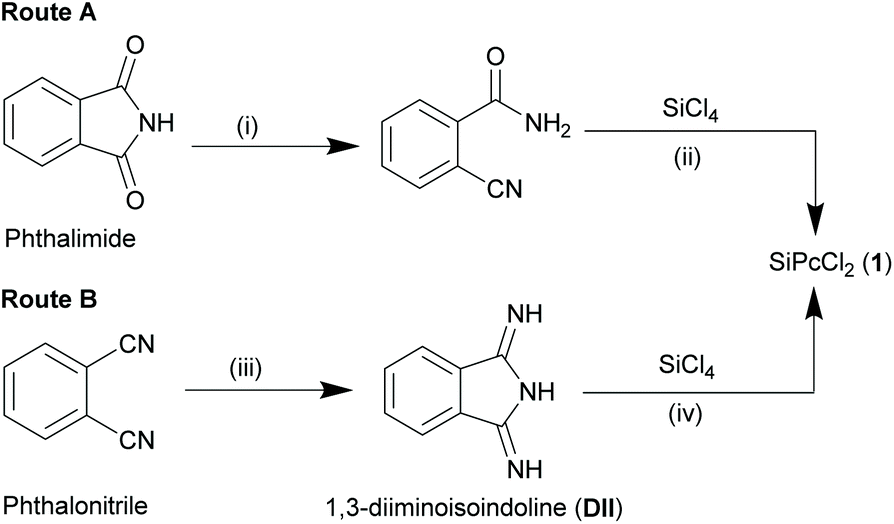 | ||
| Scheme 2 Synthetic routes for obtaining SiPcCl2 (1) starting from precursors phthalimide and phthalonitrile. Reagents and conditions: (i) conc. ammonium hydroxide, 24 h, r.t. (ii) SiCl4, dry quinoline, dry 1,2-dichlorobenzene, 205 °C, 5 min; (iii) (a) NH3 gas, NaOCH3, methanol, 40 min, r.t.; (b) reflux, 3.2 h; (iv) SiCl4, dry quinoline, reflux (219 °C), 30 min. | ||
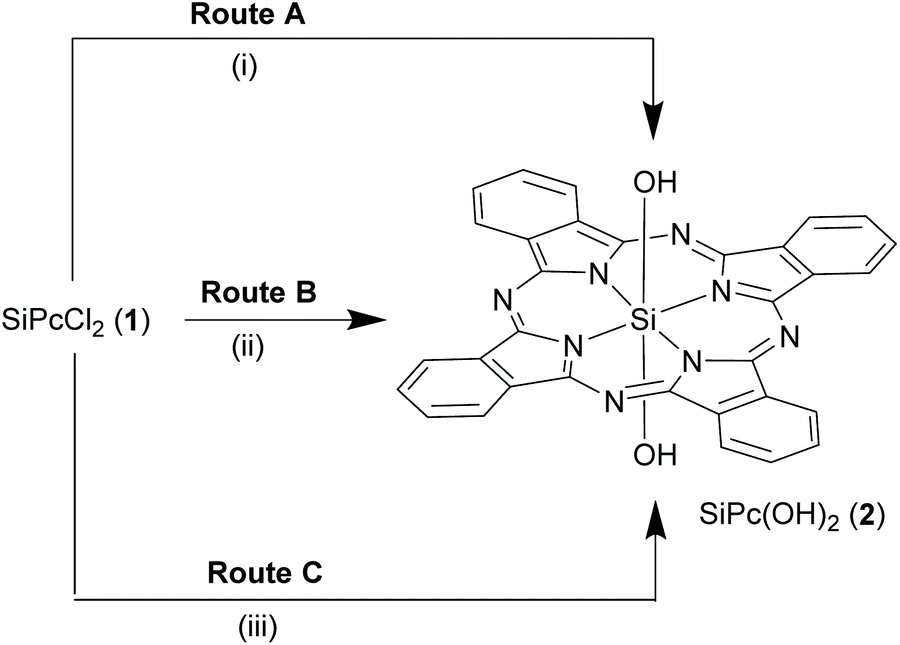 | ||
Scheme 3 Synthetic routes for hydrolysis of the axial chloro bonds to form bis-hydroxyl SiPc derivative, 2. Reagents and conditions: (i) 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) pyridine and conc. aq. NH3, 10 h, reflux; (ii) large excess of conc. H2SO4, r.t.; (iii) NaOCH3, 95% ethanol, reflux, 1 h. :1 (v/v) pyridine and conc. aq. NH3, 10 h, reflux; (ii) large excess of conc. H2SO4, r.t.; (iii) NaOCH3, 95% ethanol, reflux, 1 h. | ||
Though Kenney and co-workers successfully isolated this extraordinary class of compounds, the initial synthetic routes were experimentally inconvenient due to poor yields and laborious purification methods. This entailed exploration of refined strategies with alternate starting materials and/or inclusion of additional reaction steps. Reaction of 2-cyanobenzamide (obtained from hydrolysis of phthalimide with concentrated ammonium hydroxide) with SiCl4 in quinoline at elevated temperatures (∼205 °C) resulted in the formation of a blue solid (Route A, Scheme 2).25 The crude precipitate was washed with quinoline, benzene, pyridine, acetic acid, ethanol and ether to obtain the pure 1 (yield ∼35%). Another two-step synthetic route comprised of ammonolysis of phthalonitrile in methanol and sodium methoxide to form 1,3-diiminoisoindolline (DII) in the initial step, followed by reflux of DII with SiCl4 in quinoline to afford compound 1 (yield ∼71%) (Route B, Scheme 2).25
Brusso and co-workers recently proposed an alternate route to synthesize DII from phthalonitrile by using air-stable lithium bis(trimethylsilyl)amide etherate, LiN(TMS)2·Et2O in dry toluene at room temperature for 5 h, followed by exposure to gaseous hydrochloric acid at ice bath temperature.26 The dichloride salt, H2DIICl2, thus formed when refluxed with SiCl4 in quinoline afforded 1 (yield ∼51%). Among all these synthetic pathways, route B of Scheme 2 remains the most convenient and preferred choice for the construction of SiPcs.
The axial chloro groups of SiPc 1 can be replaced with other halogens. The chlorides are replaced by fluoride atoms on treatment with 49% hydrofluoric acid (HF) on a steam bath, and resulted in the formation of SiPcF2.27,28 Recently, Bender and co-workers synthesized SiPcF2 by refluxing 1 with either cesium fluoride (CsF) in dimethylformamide or with tetrabutylammonium fluoride in dimethyl sulfoxide.29 These alternate strategies involved relatively safer fluorinating agents compared to HF and resulted in high reaction yields of 100% and 73% respectively. In a similar approach, boron tribromide was used as a brominating reagent for the synthesis of SiPcBr2.27
:1 v/v mixture) for 10 h13,24 or treated with large excess of concentrated sulfuric acid at room temperature followed by dilution with water, hydrolysed to form SiPc(OH)2, 2 in ∼80% yield (Routes A and B, Scheme 3).13 Alternatively, sodium methoxide in 5% aqueous-ethanol hydrolysed 1 under reflux conditions to yield 2 (∼83%) and provided a comparatively safer method (Route C, Scheme 3).25 A recent report used 50% aqueous cesium hydroxide in DMF to obtain 2 from 1.30 Interestingly, unlike ordinary silanols, the axial hydroxyl groups of SiPc 2 are less acidic in nature and require harsh reaction conditions for deprotonation.13
2.2 Synthesis of SiPcs with axial substitutions
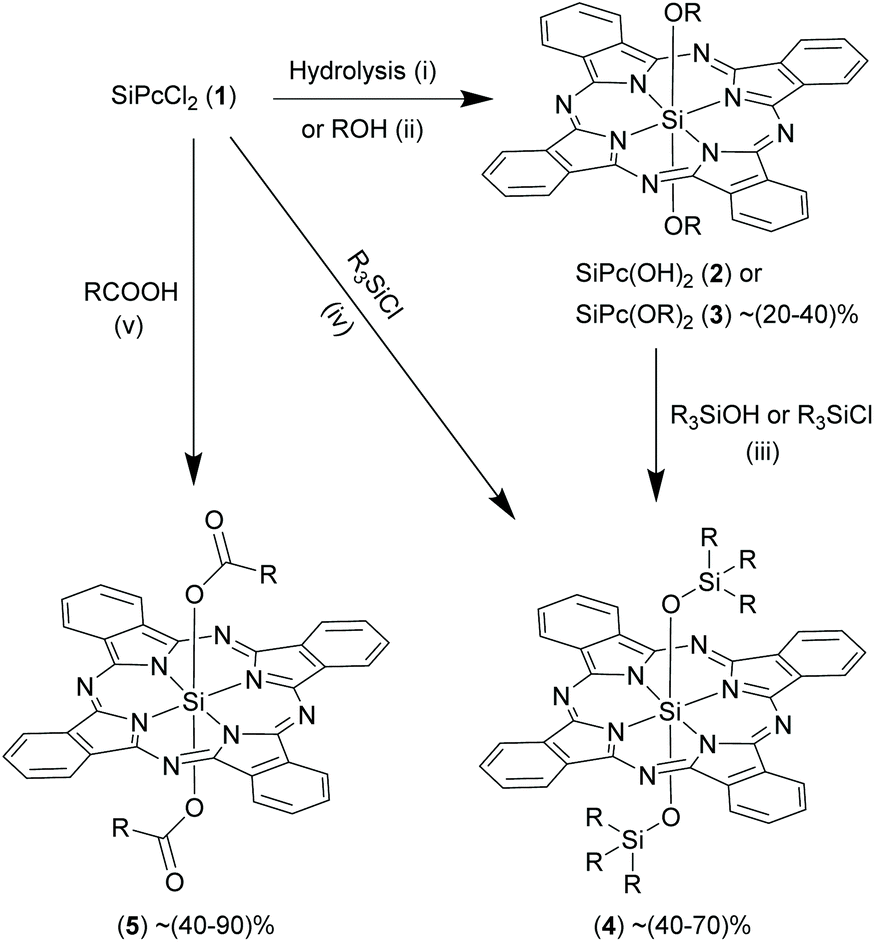 | ||
| Scheme 4 Reactions of SiPcCl2, (1) where axial chlorides are replaced by alcohols, phenols, silanes and carboxylic acids. Reagents and conditions: (i) NaOCH3, 95% CH3CH2OH, reflux, 1 h; (ii) ROH, toluene, pyridine or sodium hydride, reflux, 120 °C, 24–96 h; (iii) R3SiOH or R3SiCl, toluene, pyridine, reflux, 120 °C, 3–6 h; (iv) R3SiCl, NaOH, Aliquat HTA-1, chlorobenzene, reflux, 132 °C; (v) RCOOH, methoxyethyl ether, reflux, 160 °C, 0.5–6 h. R represent aryl or alkyl groups. | ||
In order to form a siloxane bond in the axial position, precursor 2 is refluxed with an excess of silanols or monochlorosilanes to obtain 4 (yields ∼40–70%).13,16,17,39–42 Reactions were usually carried out for short durations ranging from 3–6 h in non-polar solvents (e.g. dichlorobenzene and toluene) in presence of a mild base (e.g. pyridine) (Scheme 4).6,16,39 Interestingly, the axial Si–O-R linkages in certain bis-alkoxy SiPc derivatives, 3, can react with silanols under reflux conditions in dry pyridine to generate siloxane (Si–O–Si) bonds thereby forming SiPc derivatives, 4.13,43 Alternatively, 4 were obtained by directly refluxing 1 with trialkylchlorosilanes in sodium hydroxide and chlorobenzene in presence of Aliquat HTA-1, a high temperature phase transfer catalyst (Scheme 4).44
Oligomeric and polymeric SiPc derivatives having axial Si–O and Si–O–Si linkages have been reported as well. Such μ-oxo bridged SiPc polymers were formed when compound 1 was allowed to react with 2 and the resulting hydroxy-capped oligomers were reacted with a chlorosilane.6 The desired polymer was isolated by column chromatography. Another way to obtain oligomers is from reaction of 1 or 2 with dendritic hydroxylic compounds (e.g. polyethelyene glycols and triethanolamines) or silanols containing multiple hydroxyl moieties in the axial position. The properties of these polymers were investigated in both solid and solvent phases and some of these derivatives were even processed into thin polymeric films.45–50
Attempts to react 1 with alternate ligand functionalities such as carboxylic acids were successful and expanded the domain of axial functionalization of SiPcs (Scheme 4).27 Esterification reactions of 1 occurred under reflux conditions in polar solvents such as 2-methoxyethyl ether (∼160 °C), diglyme (∼160 °C) or 1,4-dioxane (∼100 °C), DMF (∼160 °C) to produce various SiPc esters, 5. Unlike etherification, esterification reactions proceeded easily in the absence of a base. The reaction completion times ranged from as short as of 30 min to as long as 48 h.20,51–56 The SiPc bis-esters, 5 were obtained as blue solids in moderate (∼30%) to high (∼90%) yields.
Kenney and co-workers noticed that only one of the two axial Si–O–Si linkages hydrolysed when 4a (R = [(CH3)2Si(CH2CH2CH2N(CH3)2)] in 4) was stirred with trichloroacetic acid in dichloromethane at room temperature for 5 h (Scheme 5A).22,57 This hydrolysed product, 6 was then reacted with methanol in dry pyridine to form unsymmetrical SiPc, 7 in high yields of ∼80% (Scheme 5A). Another synthetic route to obtain 6 is strategized on exploiting the photolabile nature of Si–C bonds which will be discussed in the proceeding sections of organo SiPcs.58
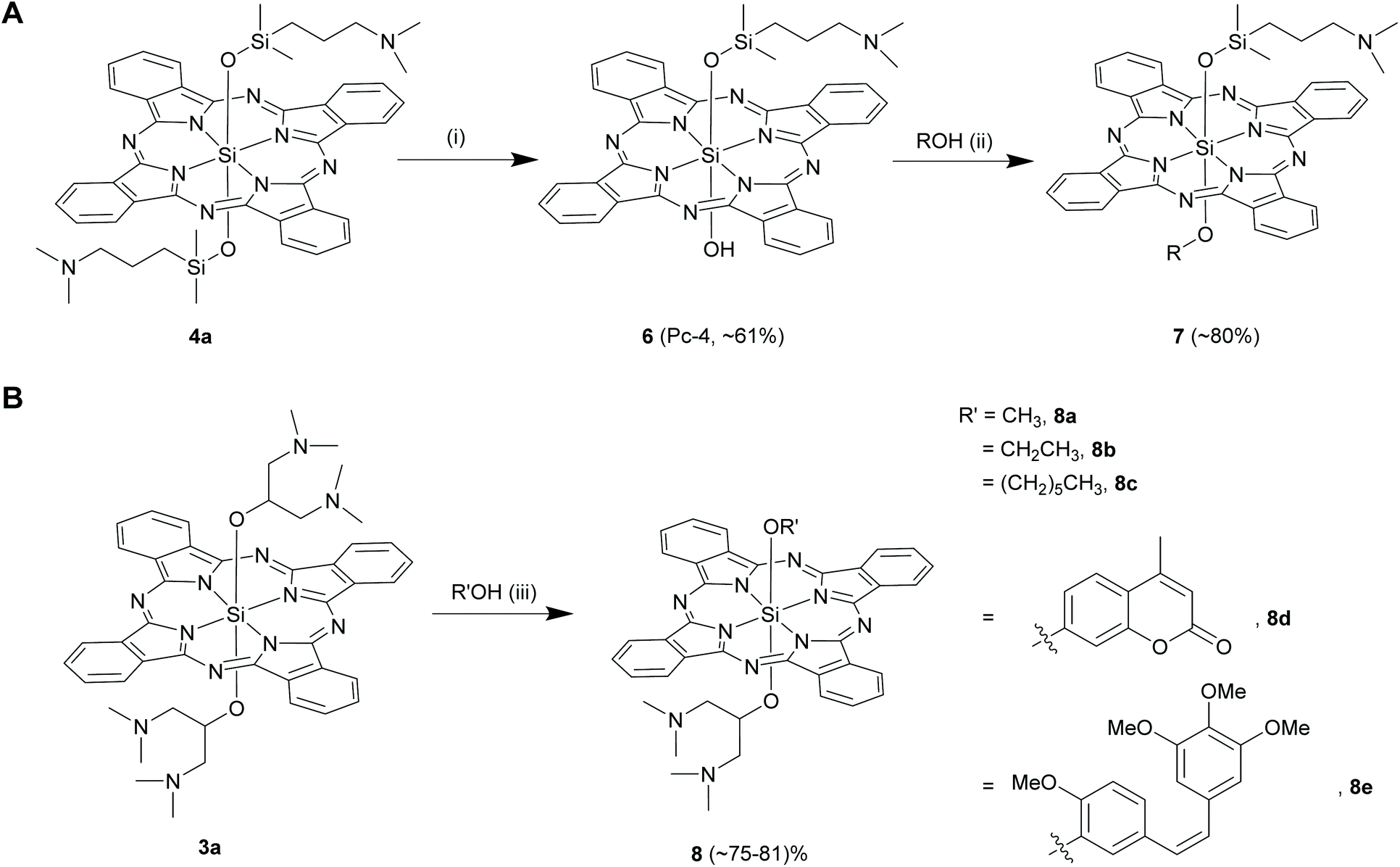 | ||
| Scheme 5 Synthesis of axially unsymmetrical Si–O bond containing SiPc derivatives starting from a symmetrical (A) bis-siloxy or (B) bis-ether SiPc compounds. Reagents and conditions: (i) (a) trichloroacetic acid, dichloromethane, r.t., 4 h (b) pyridine, water; (ii) CH3OH, pyridine, reflux, 30 min; (iii) R′OH, chloroform, r.t. or 70–95 °C, 18–48 h. R and R′ denotes aryl and alkyl groups as shown in the figure. Yield of 61% for Pc-4 is calculated based on initial starting material 2, SiPc(OH)2. | ||
In an endeavour to synthesize and explore amphiphilic SiPc derivatives as PDT agents, Ng and co-workers serendipitously discovered a facile route to prepare unsymmetrical SiPc derivatives having axial Si–O linkages (Scheme 5B).59 In an attempt to purify a bis-alkoxy symmetrical SiPc, 3a (R = 1,3-bis(dimethylamino)-2-propanol in 3) by crystallization from methanol-chloroform solvent at room temperature, they observed the formation of an unsymmetrical SiPc, 8a in high reaction yields (∼73%). Similar displacements reactions of 3a occurred with slight excess of ethanol and n-hexanol, but only under reflux conditions (∼70 °C) in chloroform for 48 h to form 8b and 8c (yields ∼29–37%). In contrast, no mono-substituted unsymmetrical SiPc could be isolated when 3a was refluxed with dodecanol for 48 h. All these observations led to the conclusion that the length of the alkyl group of the alcohols played a crucial role in determining the ratio of the mono and di-substituted SiPc products, however their different reactivities were unaccounted. Recently, Schnermann and co-workers utilized this method and displaced one of the bulky axial ligands from 3a with biologically relevant phenols such as coumarins and combretastatin to obtain SiPcs, 8d and 8e (Scheme 5B).19 The use of phenols and prior neutralization of chloroform over anhydrous potassium carbonate possibly led to improved reaction yields of ∼80%.
A recent report also utilized a similar displacement methodology to synthesize SiPc with unsymmetrical axial Si–OR bonds.60 Oligomeric ethylene glycols of various lengths were incorporated in one of the axial positions by refluxing a symmetrical amino-containing bis-phenoxy SiPcs with large excess (40×) of desired glycols in toluene in presence of sodium hydride. Moderate yields of 23–31% were achieved using this strategy.
In another report, Ng and co-workers used a slightly different strategy to synthesize SiPcs with unsymmetrical axial substituents, 9 (Scheme 6).61 Compound 1 was initially refluxed with various alkyl alcohols bearing sugar or diamino moieties in dry toluene and sodium hydride. After an hour of reaction, mono-6-hydroxy permethylated β-cyclodextrin and sodium hydride were added to the mixture and the reaction was continued for another 48 h. In these reactions, 1 was always taken in 1.5-folds molar excess of that of the alcohols to prevent exclusive formation of axially symmetrical SiPcs. However, the reaction yields of 9 were comparatively low (10–14%).
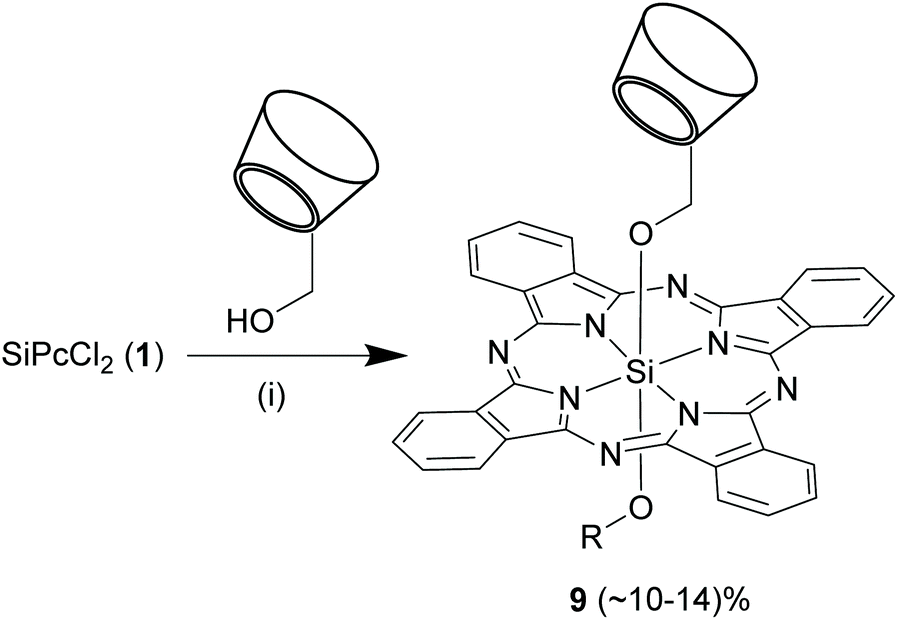 | ||
| Scheme 6 Synthesis of axially unsymmetrical Si–O bond containing SiPcs. Reagents and conditions: (i) (a) ROH, sodium hydride, toluene, r.t., 30 min, (b) 1, reflux, 1 h, (c) mono-6-hydroxy permethylated β-cyclodextrin (shown as cylindrical structure), sodium hydride, toluene, reflux, 48 h. R represents various alkyl moieties. | ||
SiPcs with unsymmetrical axial Si–O bonds have also been synthesized by selectively functionalizing one of the axial groups of a symmetrical SiPc. In a recent example, only one of the axial carboxylic acid groups in a symmetrical SiPc was subjected to a typical amide coupling reaction to form a series of unsymmetrical SiPc–peptide conjugates.62
Symmetrical and unsymmetrical SiPcs with axial Si–C bonds. The formation and isolation of SiPcs with direct Si–C axial bonds have been comparatively difficult because of two prime reasons: (i) cationic nature of hexa-coordinated silicon which instead prefers bonding with electronegative elements such as oxygen and chloride and (ii) photo-instability of Si–C bonds.49 Kenney and co-workers isolated monoorgano monochloro SiPc derivatives, (RSiPcCl, 10), such as methyl (10a), phenyl (10b) or 3-chloropropyl (10c), by refluxing an alkyl or aryl trichlorosilane (RSiCl3) with DII in solvents like quinoline, 2,6-lutidine or 2-picoline and using light-protected synthetic set-ups (Scheme 7).28 These reactions afforded crude or pure 10 as green solids with yields of ∼5–52%. Use of allyl and benzyl trichlorosilanes in these strategies, resulted in formation of only bis-siloxy ether SiPcs, 4 therefore, indicating the limited scope of axial Si–C ligands that can be installed in SiPcs.63 Symmetrical SiPcs, 11 containing trans-bis Si–C linkages were obtained in high yields (∼90%) by refluxing 1 with 2–3 fold excess of Grignard reagents of choice (RMgI) in dry THF for several hours under inert and dark conditions (Scheme 7).49,64 However, only primary alkyl, aryl or alkyne groups (such as methyl, ethyl and phenyl) were successfully inserted using the Grignard reactions. Kumada et al. aptly combined the above two strategies and successfully prepared unsymmetrical SiPcs, 12 with direct Si–C bonds in axial positions.65 They reacted 10 with 6-fold excess of the corresponding Grignard reagent (R′MgI) in dry diethyl ether and stirred at room temperature overnight (Scheme 7). Hydrolysis of the intermediate magnesium adducts was carefully accomplished using aqueous hydrochloric acid at 0 °C. The pure products, 12 were obtained as dark green solids in high yields of ∼85%. In contrast to the SiPc analogues with axial Si–O bonds, certain organo SiPcs exhibited better solubility in organic solvents such as chloroform and dichloromethane.
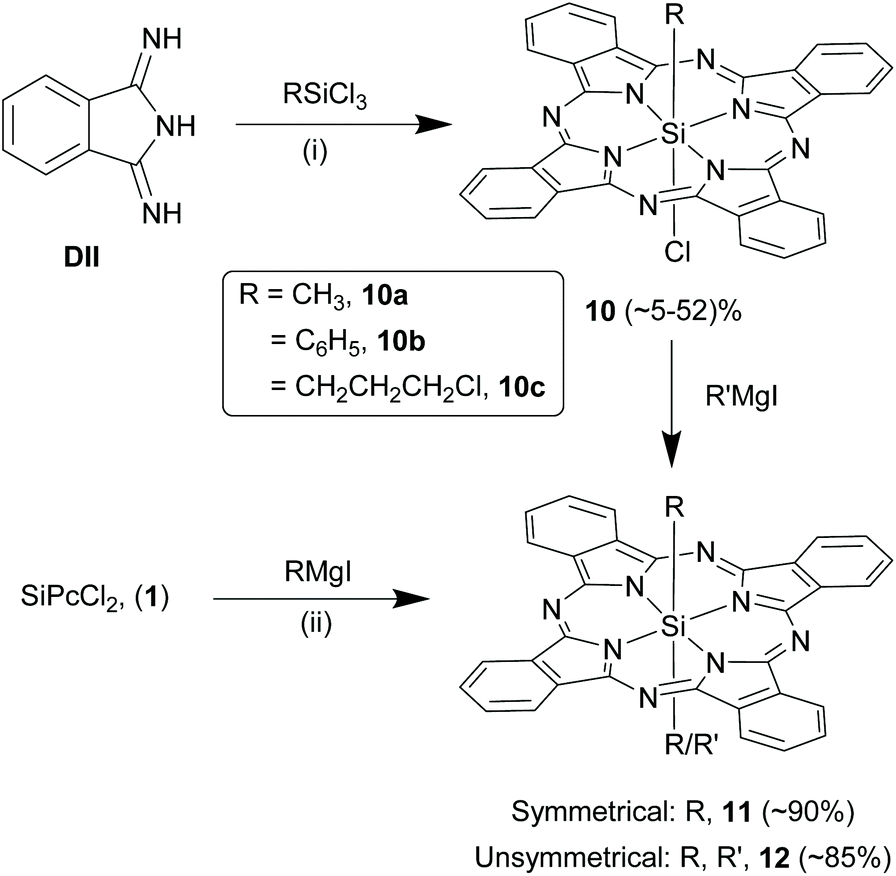 | ||
| Scheme 7 Synthesis of axially symmetrical and unsymmetrical Si–C bond containing SiPcs. Reagents and conditions: (i) RSiCl3, dry quinoline, reflux, 90 min; (ii) (a) RMgI, dry diethyl ether, N2, r.t., overnight, (b) 6 M HCl, 0 °C. R denotes alkyl groups. | ||
SiPcs with mixed axial Si–O and Si–C bonds. The axial chloride of 10 was hydrolysed with concentrated sulfuric acid, followed by addition of ice, to produce monoorgano monohydroxy SiPc derivatives, 13 [R′SiPc(OH)] (Scheme 8A).28 These hydroxyl derivatives were subsequently refluxed with phenols (e.g. phenol, 4-chlorophenol) in 1,2,4-trimethyl benzene for 30–90 min to yield SiPcs, [R′SiPc(OR)] with trans axial Si–C and Si–O bonds.28 In a similar fashion, compounds having an axial silyl group, 14 (where R′ = silyl) was obtained by directly exposing crude 10 to siloxy ethers (R3SiOCH3).22
 | ||
| Scheme 8 Synthesis of axially unsymmetrical SiPc derivatives containing Si–O and Si–C bonds. Reagents and conditions: (i) R′OH, 1,2,4-trimethyl benzene, 30 min, reflux; (ii) CH3OSiR3, chlorobenzene, reflux, 3 h; (iii) potassium thioacetate, dimethylformamide, 50 °C, 6 h; (iv) aqueous sodium hydroxide (2 N), ethanol, 0 °C, 75 min. | ||
Though organo SiPcs were long discovered, appendage of suitable reactive handles in the Si–C bonds was not achieved until recently. Kenney et al. synthesized and isolated crude organo SiPc derivative, 10c (R = ClCH2CH2CH2 in 10) where the axial Si–C ligand contained a reactive alkyl halide substituent (Scheme 7).22 Compound 10c was obtained by refluxing equimolar amounts of DII and 3-chloropropyltrichlorosilane in slightly polar solvents like 2,6-lutidine or 2-picoline for 6 h in argon atmosphere (Scheme 7). A similar bromo SiPc derivative was also obtained by using the corresponding bromosilane precursor. However, these compounds could not be purified substantially, presumably owing to their extremely low synthetic yields.22,66 The displacement the axial Si–Cl bonds of 10a by a siloxy ether, [(CH3)2Si(OCH3)(CH2CH2CH2N(CH3)2)] led to the formation of 14a which was isolated in yield of ∼5% (Scheme 8A). Next, using the chloride group, 14a was converted to a thiol bearing SiPc, 14b in two successive steps via formation of a thioacetate intermediate (Scheme 8B).22 The presence of a nucleophilic thiol moiety dangling at the end of the Si–C axial bond created the possibility of functionalization of the organo SiPcs.
The photolabile axial Si–C bonds triggered the development of interesting synthetic routes for the preparation of unsymmetrical SiPcs with axial bis Si–O bonds.58 For example, compounds 14 (R′ = silyl) on UV or visible light irradiation in trimethylamine saturated water and toluene solvent system at room temperature rendered the hydroxyl SiPc analogues, 6 in high yields which were further derivatized with hydroxyl groups to form SiPcs, 7.
2.3 Synthesis of SiPcs with phthalocyanine ring substitutions
Like axial substitutions, the peripheral rings of SiPcs can be modified at α and β positions to perturb planarity, reduce aggregation, and increase hydrophilicity or to insert reactive handles for attachment of desired functionalities (Fig. 1C). More importantly, α/β substitutions are exploited to modulate the spectral properties of SiPcs. Various synthetic strategies have been developed for incorporation of halides, alkyl, alkoxy, phenoxy, phenylthio, sulphonate, glycols and vinyl groups at the peripheral and non-peripheral positions and often result in the formation of tetra- or octa-substituted symmetrical SiPcs. A few routes have been also proposed for synthesis of SiPcs with unsymmetrical peripheral substitutions. However, the latter reactions are synthetically more challenging as they usually result in low synthetic yields and formation of mixtures of products which are difficult to separate.Direct reaction of the aromatic rings in SiPc. By means of a direct reaction, the aromatic rings of SiPc can be sulphonated,66,67 to obtain enhanced photostability as well as aqueous solubility. In a typical reaction, 1 was heated with excess chlorosulphonic acid (ClSO3H) at 130 °C for 5 h and then cooled to 85 °C. Thionyl chloride (30-fold excess) was slowly added to the solution and the reaction was continued for another 4 h. The resulting sulphonyl chloride SiPc adduct was isolated in pure form (yield ∼68%) and then hydrolysed in methanol–water (1
:5 v/v mixture) under reflux conditions for 3 h to obtain the tetrasulphonated SiPc, 15 in high reaction yields of ∼87% (Scheme 9).67 Importantly, the varying degrees of sulphonation depend on the SiPc:ClSO3H molar ratio and thus can be controlled. For example, use of SiPc and ClSO3H in 1:4 molar ratio resulted in isomeric mixtures of α or β substituted (Fig. 1) monosulphonated unsymmetrical SiPcs.66
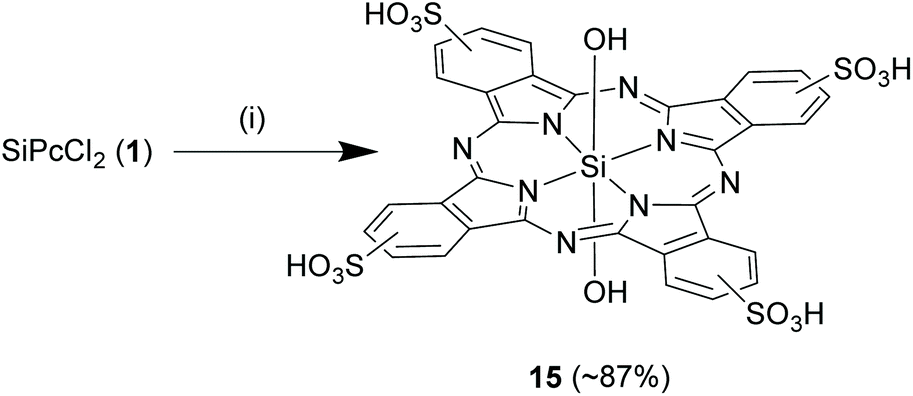 | ||
| Scheme 9 Sulphonation reaction of the aromatic rings of SiPc. Reagents and conditions: (a) chlorosulphonic acid, 130 °C, 5 h, (b) thionyl chloride, 4 h, (c) methanol–water (1:5 v/v), reflux, 3 h. | ||
Using substituted phthalonitriles as precursors. The choice of substituted phthalonitriles, 16 or 17 as precursors varied according to the nature and position of the desired substituents (Scheme 10). For synthesis of symmetrical β, β substituted SiPcs (22, where X = H), 4,5-disubstituted phthalonitriles (16, where X = H) were used as precursors. Depending on the nature of Y groups, compounds 16 were synthesized from either 4,5-dibromo catechol or 4,5-dichloro dicyanobenzene. The subsequent steps for formation of SiPcs, 22 were common in both the cases and proceeded via the formation of the corresponding DII derivatives, 18 (Scheme 10). In some cases, purification of the SiPc chloro analogues were difficult and required further transformation of the axial chloro group to a hydroxyl or siloxy group.
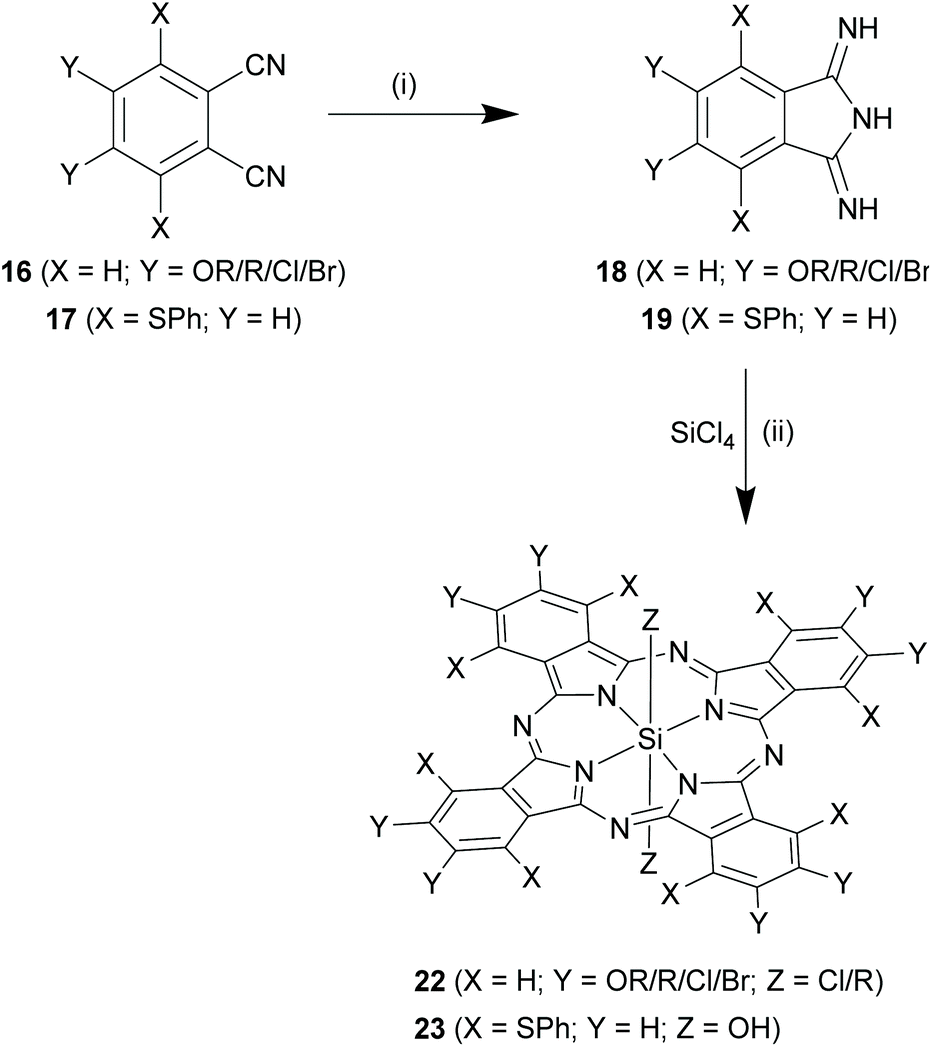 | ||
| Scheme 10 Synthesis of SiPc derivatives symmetrically substituted at the α or β positions by using DII derivatives. Reagents and conditions: (i) Na, liq NH3, digylme or (a) NH3 gas, NaOCH3, methanol, 40 min, r.t.; (b) reflux, 3.2 h; (ii) SiCl4, dry quinoline, reflux (180–219 °C), 0.5–24 h. X and Y denote different functional groups. | ||
Diether dicyano phthalonitriles (16a, where X = H and Y = OR) were obtained from 4,5-dibromo catechol by initial base mediated nucleophilic reaction of the hydroxyl groups and subsequent conversion of the bromides to cyanides via Rosemund-von Braun reaction. Lieberman et al. used this approach to obtain a crude product mixture containing octa-pentenyloxy SiPc derivatives, 22a (X = H; Y = pentenyloxy; Z = Cl in 22) via the DII derivative 18a.63 A similar approach was also adopted to obtain pentyloxy groups in the peripheral positions of SiPc (yield ∼45%).49
The other route involved 4,5-dichloro dicyanobenzene as the starting material. The two chloro groups were readily substituted with nucleophiles in presence of a base in polar solvents such as DMF and DMSO and afforded phthalonitriles, 16 (where X = H) in a single step reaction. This approach has been utilized for the synthesis of SiPc variants bearing phenoxy (∼yield ∼67%),15 and thioalkoxy as peripheral groups.68 In another report, 4,5-dihalo dicyanobenzenes (Y = Cl/Br), as such, was converted to phthalonitriles, 16b and further reacted to form DII18b and insert halogens in the peripheral positions of SiPcs, 22b (where X = H, Y = Cl/Br; Z = Cl in 22; yields ∼72–80%).31
In another example, eight peripheral pentyl groups were incorporated in a SiPc structure (22c, where X = H; Y = pentyl; Z = CH3 in 22) using 1,2-dichlorobenzene as the starting material.49 In the first step, Grignard coupling of 1,2-dichlorobenzene with pentylbromomagnesium was performed to obtain the 1,2-dipentyl benzene. This product was initially subjected to bromination and next, the aromatic bromides were converted to cyanides using CuCN. Finally, the resulting phthalonitrile 16c was converted into DII derivative, 18c and subsequently to 22c with a reaction yield of ∼42%.49 Similarly, use of naphthalonitrile (2,3-naphthalenedicarbonitrile) instead of a phthalonitrile, generated silicon naphthalocyanines (SiNcs) with a reaction yield of ∼82%.6 The SiNcs, a subclass of SiPcs, absorb and emit in the NIR region owing to the extended π-conjugation of the aromatic rings.
SiPcs bearing α,α substituents (23, where Y = H in Scheme 10) were generated through the use of 3,6-disubstituted phthalonitriles (17, where Y = H). One such example has been reported by Nakagawa and co-workers.69 The synthesis involved prior conversion of 3,6-hydroxyphthalonitrile to 3,6-(bisphenylthio)phthalonitrile via tosylation of the hydroxyl groups and base-mediated displacement by thiophenol. The phthalonitrile 17, thus obtained was first converted to corresponding DII derivative, 19 and eventually to α,α-octaphenylthio SiPc derivative, 23a (X = phenylthio; Y = H; Z = OH in 23; yield ∼84%).
Direct insertion of the core silicon to a pre-formed Pc. A more flexible synthetic methodology to form SiPc variants at α/β positions involved initial formation of core element-free Pcs, followed by insertion of core silicon atom (Scheme 11).70 In a typical reaction, phthalonitriles (16 or 17) were refluxed with lithium carbonate (∼4-fold excess) in 1-pentanol for 66 h to obtain Pcs (20 or 21) in ∼30% reaction yields.71 This conversion proceeded faster (∼48 h) and efficiently (high reaction yields ∼55–72%) in presence of non-nucleophilic strong bases like 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU).68 In the final step, these Pcs (20 and 21) were reacted with 10–30-folds excess of trichlorosilane (HSiCl3) to form the desired SiPcs with symmetrical α/β substituents and hydroxyl axial ligands, 23 and 24 (Scheme 11). Reactions were conventionally carried out in anhydrous and inert atmosphere using organic solvents such as DCM, tetrahydrofuran, acetonitrile or toluene at room temperature or under reflux conditions.70 The nature of the base (such as tripropylamine, tributylamine and DBU) employed affected both reaction completion times and yields (∼12–53%) of desired SiPcs (23 or 24).72,73
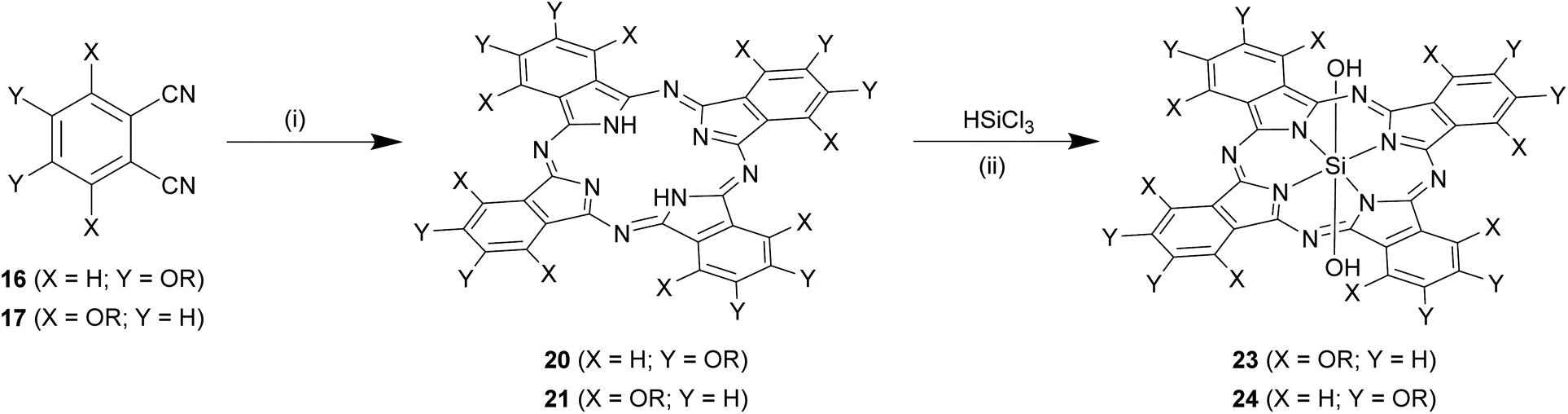 | ||
| Scheme 11 Synthesis of symmetrical SiPc derivatives by direct insertion. Reagents and conditions: (i) Lithium carbonate, 1-pentanol, reflux, 66 h or 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), reflux, 48 h; (ii) (a) HSiCl3, DBU, dichloromethane, argon, r.t., 80 min, (b) H2O, r.t. X and Y denote different functional groups. | ||
 | ||
| Fig. 2 Chemical structure of a SiPc polymeric derivative, La Jolla Blue with unsymmetrical peripheral (β) substitutions. | ||
In conclusion to this section, all the aforementioned strategies provided diverse reaction schemes to fabricate SiPcs with ligands of choice such as acidic, basic, neutral, hydrophilic, lipophilic, electron rich or electron withdrawing substituents. This led to exploration and exploitation of the NIR optical properties of SiPc in various contexts.
3. Applications of silicon phthalocyanines
SiPc derivatives display interesting optical, redox, electronic properties and high thermal stability. In addition, availability of multiple synthetic strategies have resulted in enormous flexibility of SiPc frameworks. Easy modulation of solubility, optical and redox properties, bio-compatibility, bioactivity or solid-state structural arrangements has been achieved through decorating both axial and peripheral positions. This led to a multitudinous array of SiPc derivatives that have been crafted according to the demands for particular applications. The past decade has experienced an upsurge of novel applications of SiPcs in medicinal chemistry, photovoltaics, optoelectronics and photocatalysis. Selected examples of these emerging applications are discussed below.3.1 Applications of SiPcs in medicinal chemistry
Silicon phthalocyanines have been extensively pursued in light-based medical applications, primarily as anticancer, antibacterial and antifungal agents.23,76–78 Owing to the strong NIR emission, SiPcs have been potentially applied for in vitro and in vivo bioimaging.32,52,79–81 Two fluorescent SiPc derivatives, namely La Jolla Blue® and IRD700DX®, are commercially available as labelling agents for biomolecules.82–84 Likewise, due to the intense far red and NIR Q-band absorptions, several photosensitive SiPc molecules have been deployed in photodynamic therapy (PDT), photouncaging techniques, photoimmunotherapy (PIT), and photothermal therapy (PTT). A few recent noteworthy examples are highlighted in the subsequent sections.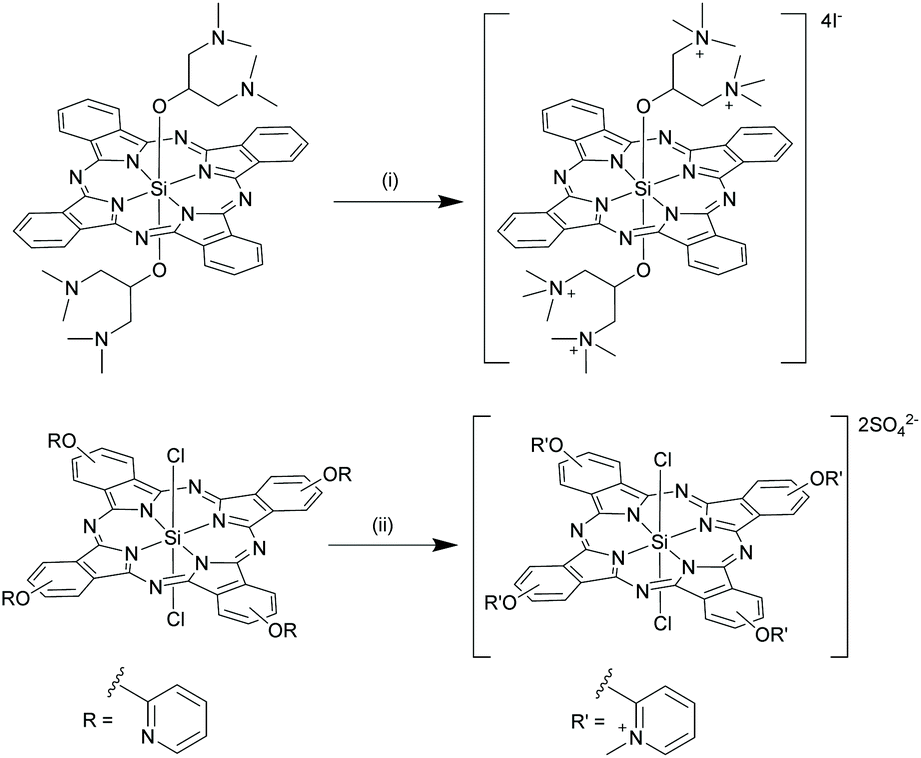 | ||
| Scheme 12 Synthesis of water soluble SiPc derivatives. Reagents and conditions: (i) Methyliodide, chloroform, reflux, 85 °C, 6 h; (ii) dimethyl sulphate, dimethylformamide, 120 °C; 24 h. | ||
Recently, You's research group developed SiPc pseudoprodrugs to execute multipurpose objectives such as (i) fluorescence imaging, (ii) far red light-mediated uncaging, and (iii) combined effects of singlet-oxygen induced PDT and local chemotherapy.100–103 They utilized singlet oxygen-labile aminoacrylate bonds to append clinical chemotherapeutic drugs such as combretastatin and paclitaxel in the axial positions of SiPcs (3b and 3c in Fig. 3A).101,103 The caged forms 3b and 3c exhibited significantly reduced dark cytotoxicity as evident from 19 and 190-folds lower IC50 values when compared to the respective free drugs. On light irradiation, SiPc produced singlet oxygen which led to an immediate PDT effect and simultaneously cleaved the aminoacrylate linker to uncage the intact chemotherapeutic drugs. This bystander or local chemotherapeutic effect after the illumination was demonstrated by exposing only one-half of each well containing cancer cells treated with vehicle only, 3b or a structurally equivalent non-photocleavable SiPc conjugate, NCL-3b (Fig. 3B). The PDT effect of NCL-3b was restricted only in the light exposed half of the well due to the short half-life (10–320 ns) and limited diffusion distance (20–200 nm) of singlet oxygen (panel (c) of Fig. 3B).88 However, the 3b-treated wells showed prominent bystander effects. The photoreleased combretastatin diffused throughout the well and caused similar toxicity in the light unexposed cells as compared to light-exposed cells (panel (b) of Fig. 3B).
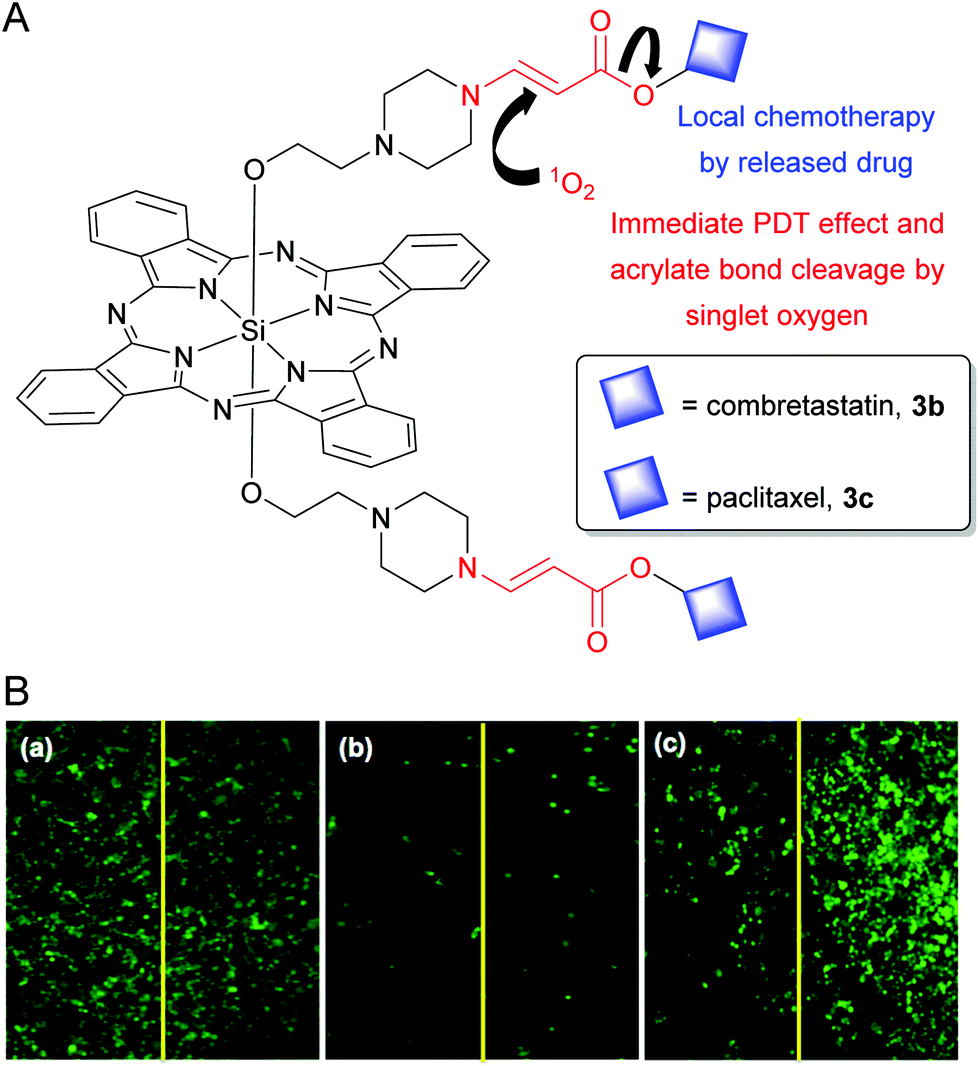 | ||
| Fig. 3 (A) Structure of SiPc 3b and 3c showing dual PDT and singlet oxygen mediated release of chemotherapeutic drug (shown as blue squares). (B) Fluorescence live cell images of the center of each well treated with (a) vehicle control, (b) 25 nM 3b, and (c) 25 nM NCL-3b. The left half of each well was illuminated with a 690 nm diode laser (11 mW cm−2 for 15 min). At these concentrations, 3b and NCL-3b did not produce any significant dark toxicity. Reproduced with permission from ref. 101. Further permissions related to the material excerpted should be directed to the ACS (https://pubs.acs.org/doi/10.1021/jm5000722). | ||
Compound 3b was further modified to incorporate a tumor-targeting folic acid at the second axial position via a hydrophilic PEG linker.102 It was observed that the longer length of the PEG spacer enhanced specific uptake and localization in folate receptor overexpressing colon 26 tumors. The SiPc conjugate having PEG linker length of ∼45 showed the most impressive light induced cytotoxicity giving a IC50 value of 1.65 × 10−8 M (690 nm, 10 J cm−2), while being non-toxic in the dark at <2 μM concentration.
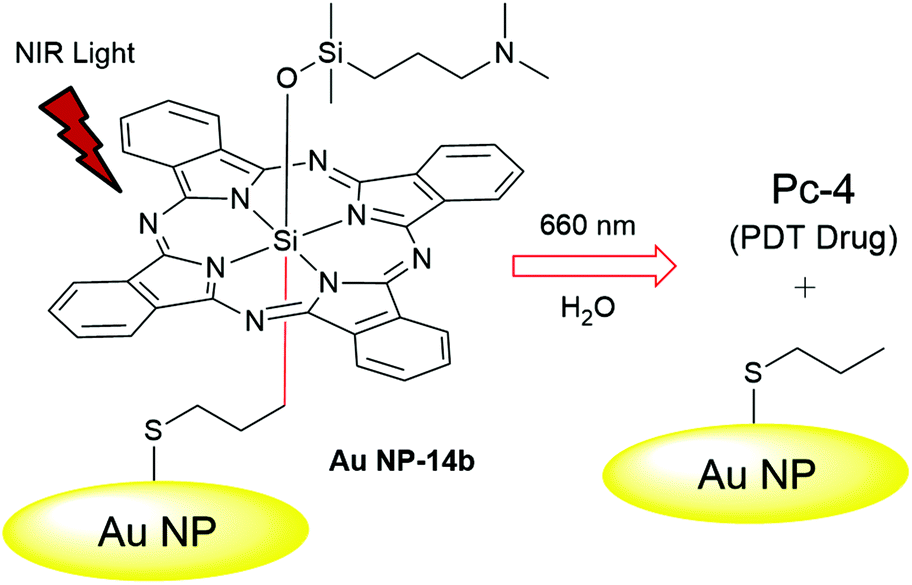 | ||
| Fig. 4 Schematic representation of phototriggered PDT drug Pc-4 release from Au NP-14b. The thiol group in 14b allowed covalent bonding of the drug precursor to the Au NP surface. Adapted from ref. 21 with permission. © 2014 WILEY–VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
The research groups of Kenney and Burda performed theoretical and experimental measurements to identify the preferred mechanistic pathway of the light triggered Si–C bond cleavage process.22,104,105 Characterization of the photoproducts using various techniques such as steady state spectroscopy, electrospray ionization mass spectrometry, liquid chromatography mass spectrometry, nuclear magnetic resonance and electron paramagnetic resonance hinted towards formation of a relatively long-lived SiPc radical as the key intermediate which consequently undergoes solvolysis.104 Computational studies additionally confirmed homolytic bond cleavage process involving radical intermediates. Bond-dissociation energy (BDE) calculations indicated that homolytic cleavage (∼50 kcal mol−1) is energetically more favoured than heterolytic cleavage (∼190 kcal mol−1).22 This is due to the fact that the unpaired electron is largely delocalized over the SiPc aromatic rings as implied by natural bond orbital (NBO) analysis and spin density calculations.
In another recent report, Schnermann et al. demonstrated the preferential photocleavage of Si–O bonds and release of phenols in axially unsymmetrical SiPcs molecules.19 On NIR photoexposure at the SiPc Q-band (690 nm), the axial Si–O bonds in 8d and 8e (Scheme 5, above) instantly dissociated in oxygen-deprived physiological conditions and in presence of reducing glutathione (GSH, 5 mM). Thus, photo uncaging of small phenolic molecules such as fluorescent 4-methyl umbelliferone or clinical anticancer drug combretastatin was achieved (Fig. 5A). In contrast, compounds 8d and 8e were fairly stable on light irradiation under normoxic conditions and produced singlet oxygen as the major reactive oxygen species (Fig. 5A). Thus, compound 8e resulted in NIR mediated cytotoxicity in cancer cells which was a manifestation of either PDT or chemotherapy depending on the availability of tissue oxygen. Compound 8e (200 nM) showed NIR (690 nm, 50 J cm−2) induced phototoxicity by a PDT mechanism in presence of oxygen leading to 48% cell growth inhibition. In hypoxic conditions, the cell growth inhibition increased to 80% which is comparable to that of free combretastatin suggesting photorelease of the chemotherapeutic agent from the SiPc molecule triggered by NIR light and cytosolic glutathione (Fig. 5A).
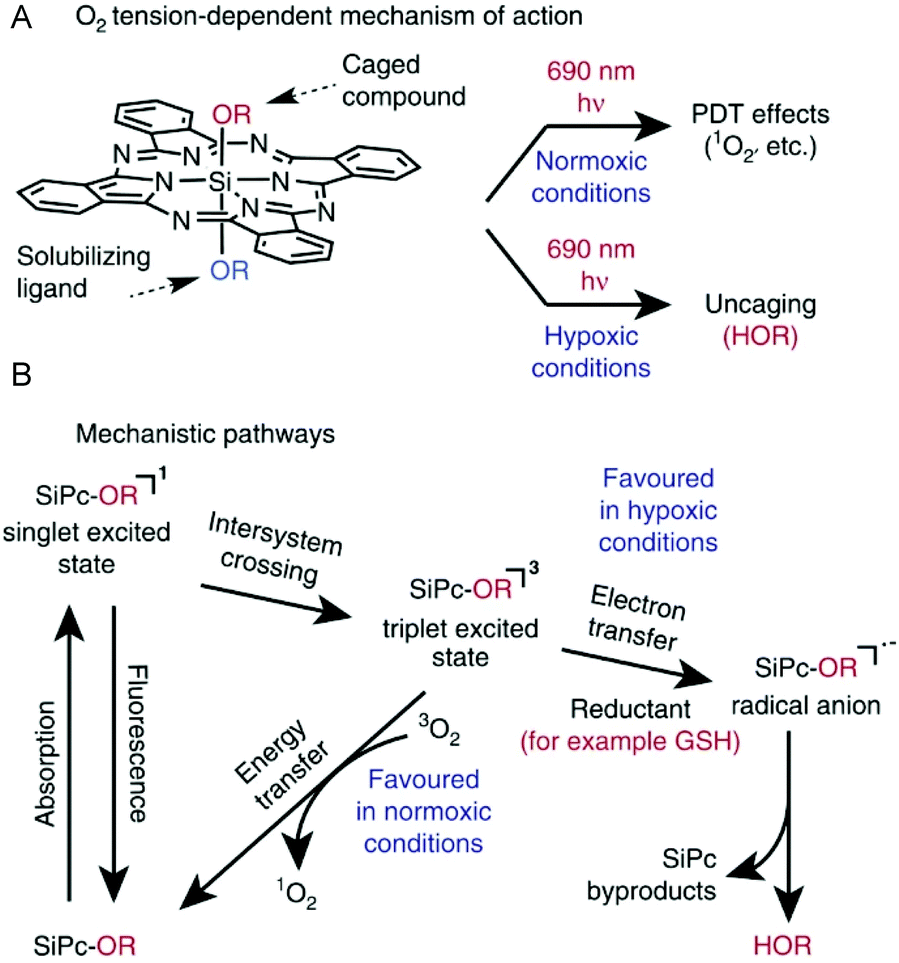 | ||
| Fig. 5 (A) O2 tension dependent mechanism of action of SiPc. (B) Mechanistic pathways leading to ROS generation or small molecule uncaging. R denotes aryl group. Reproduced from ref. 19. | ||
Detailed spectroscopic and computational studies were performed to understand the photoredox uncaging reactions.106 The proposed mechanistic pathway involved a key radical anion intermediate formed through photoinduced electron transfer from the SiPc triplet state (Fig. 5B). This explained the role of reducing agent, glutathione in accelerating the cleavage phenomenon. The transient radical anion was proposed to undergo solvolytic ligand exchange through a heptacoordinate silicon transition state.
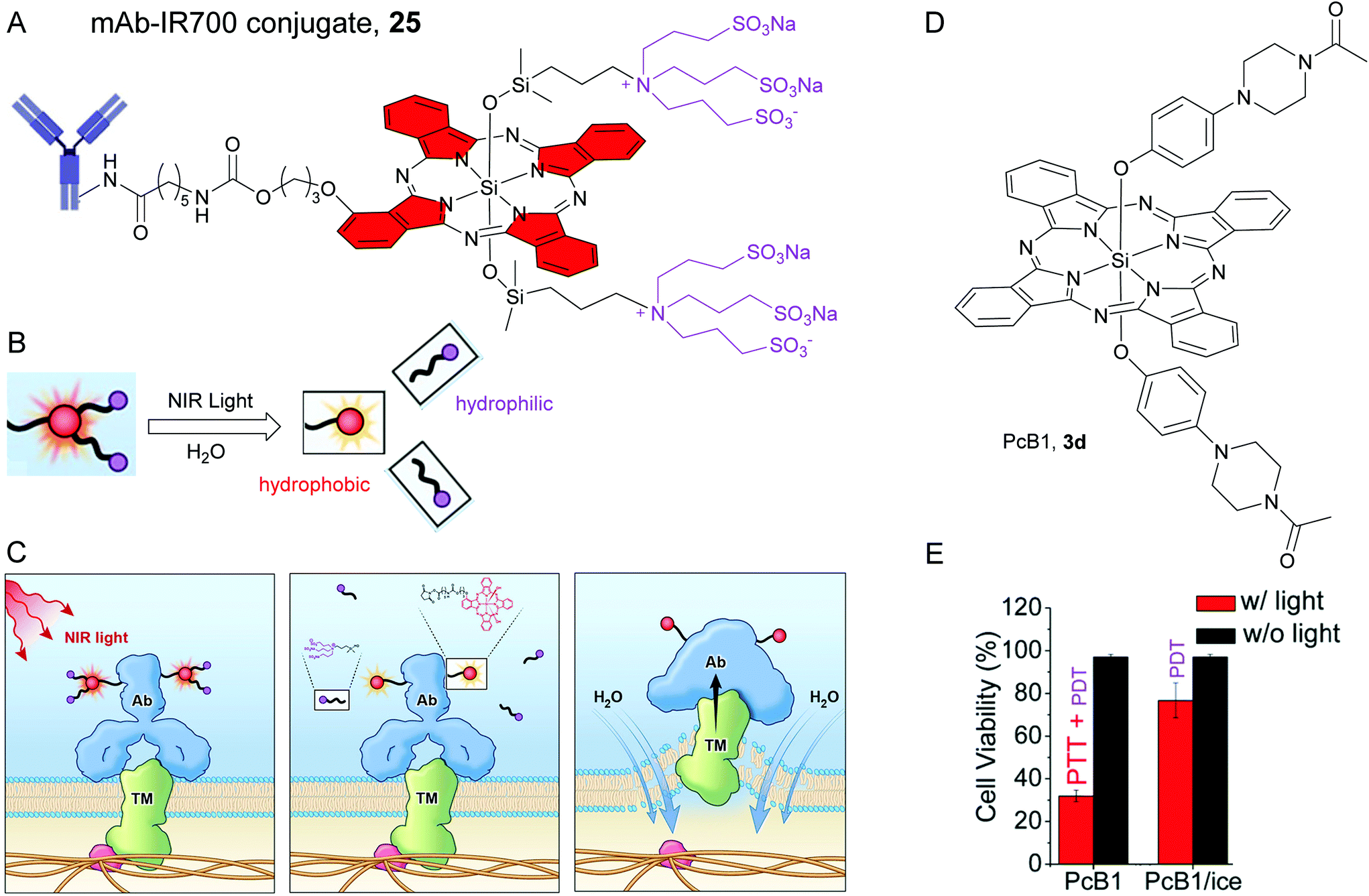 | ||
| Fig. 6 (A) Structure of mAB-IR700 conjugate, 25. The red colour is used to highlight hydrophobic SiPc core rings and the violet colour is used to highlight the hydrophilic axial ligands. (B) NIR light induced decomposition of the SiPc molecule attached to the antibody. NIR light irradiation causes mAb-IR700 to release the hydrophilic ligand (violet circles), which makes mAb-IR700 hydrophobic and causes aggregation. (C) Scheme indicates the proposed mechanism of NIR-PIT. An antibody-IR700-antigen complex is formed on the cell membrane. With NIR light irradiation, the ligands of IR700 were released form the antibody-IR700-antigen complex. The physical changes in aggregation and solubility of the antibody-IR700-antigen complex may produce physical stress on the membrane locally impairing cellular membrane function for maintaining membrane pressure, and then the water outside of the cell was flown into the cell to burst the cell. Reproduced with permission from ref. 18. Further permissions related to the material excerpted should be directed to the ACS (https://pubs.acs.org/doi/10.1021/acscentsci.8b00565). (D) Chemical structure of SiPc, 3d or PcB1. (E) Cytotoxic effect of 3d or PcB1 on HepG2 cells with (red bars) and without (black bars) laser irradiation. PcB1/ice mean controlling the temperature of cells at below 30 °C via an ice-bath during laser treatment. Reproduced from ref. 110 with permissions from Royal Society of Chemistry. | ||
Recently, Kobayashi and co-workers unravelled the distinct mechanism of PIT for SiPc–antibody conjugates. They observed NIR light induced dissociation of the axial Si–O–Si linkages in 25 in presence of an electron donor such as ascorbic acid sodium salt, NaAA (Fig. 6B).18,109 The NIR light triggered detachment of hydrophilic sulphonated axial ligands led to an increase in hydrophobicity of 25 and consequently resulted in aggregate formation and quenching of NIR fluorescence. Atomic force microscopy (AFM) revealed alteration in the protein conformation such as enlargement of the conjugates and loss of normal Y-shape of the antibody. Thus, the photoinduced degradation not only affected the physical shape and solubility of the covalently-conjugated antibody, but also influenced the antibody–antigen complexes. These profound morphological changes were speculated to enhance the transmembrane (TM) water flow which created physical stress within the cellular membrane and eventually led to cell bursting and necrotic/immunogenic cell death (ICD) (Fig. 6C).
3.2 Applications in organic photovoltaic solar cells
SiPcs have recently generated interest for multifunctional applications in dye-sensitized (DSSCs), bulk heterojunction (BHJ) and planar heterojunction (PHJ) solar cells as additives, acceptors, donors and crosslinking components.111 The intense absorption of chemically and thermally robust SiPcs and SiNcs in the red and far-red spectral region has been utilized to expand the light harvesting window and boost the overall photoconversion efficiency (PCE). The structural tunability has led to easy integration and multi-functional behaviour. Finally, the low toxicity levels and the elemental abundance of silicon in nature have particularly advocated for their candidacy in organic photovoltaics and optoelectronics. | ||
| Fig. 7 Chemical structures of P3HT, PCBM and SiPc derivatives 4a–4c. | ||
| Type of solar cell | Donor/acceptor pair | Additive dye(s) | Loading (wt%) | J SC/mA cm−2 | V OC/V | FF | PCE (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| C60 is fullerene. α-6 T is α-sexithiophene. PTB7 is an organic semiconducting polymer. All values are taken from the references as listed in the last column. | ||||||||
| BHJ | P3HT/PCBM | — | — | 8.48 | 0.57 | 0.56 | 2.74 | 30 |
| BHJ | P3HT/PCBM | 4b | 4.8 | 10.3 | 0.57 | 0.69 | 4.1 | 112 |
| BHJ | P3HT/PCBM | 4b | 3.7 | 9.84 | 0.57 | 0.59 | 3.29 | 30 |
| BHJ | P3HT/PCBM | 4c | 3.7 | 10.0 | 0.56 | 0.59 | 3.30 | 40 |
| BHJ | P3HT/PCBM | 4d | 3.7 | 10.1 | 0.57 | 0.60 | 3.40 | 40 |
| BHJ | P3HT/PCBM | 3e | 3.7 | 9.49 | 0.59 | 0.54 | 3.02 | 30 |
| BHJ | P3HT/PCBM | 5a | 20 | 10.02 | 0.65 | 0.63 | 4.14 | 115 |
| BHJ | P3HT/PCBM | 26 | 8 | 11.4 | 0.62 | 0.63 | 4.46 | 116 |
| BHJ | P3HT/PCBM | 4b/27 | 4.8/1.5 | 10.9 | 0.57 | 0.69 | 4.3 | 112 |
| BHJ | P3HT/PCBM | 5b/28 | 7.5/5 | 10.80 | 0.65 | 0.55 | 3.89 | 117 |
| BHJ | P3HT/PCBM | 28 | 10 | 9.72 | 0.65 | 0.58 | 3.68 | 117 |
| DSSC | — | 29 | — | 19.0 | 0.46 | 0.51 | 4.5 | 118 |
| BHJ | P3HT/4b | — | — | 0.99 | 0.70 | 0.37 | 0.25 | 40 |
| BHJ | P3HT/4c | — | — | 2.61 | 0.75 | 0.40 | 0.78 | 40 |
| BHJ | P3HT/4d | — | — | 3.0 | 0.84 | 0.43 | 1.07 | 40 |
| PHJ | 1/C60 | — | — | 0.47 | 0.24 | 0.32 | 0.04 | 36 |
| PHJ | 3f/C60 | — | — | 1.4 | 0.76 | 0.31 | 0.34 | 36 |
| PHJ | 3g/C60 | — | — | 4.6 | 0.87 | 0.44 | 1.8 | 119 |
| PHJ | α-6 T/3h | — | — | 3.5 | 0.66 | 0.43 | 1.0 | 119 |
| BHJ | PTB7/5c | — | — | 0.60 | 1.00 | 0.45 | 0.27 | 120 |
| BHJ | PTB7/5d | — | — | 6.18 | 1.03 | 0.42 | 2.67 | 120 |
Encouraged by these inferences, Bender and co-workers varied the alkyl spacer of the axial silyl groups of dye 4b and observed that use of smaller molecular fragments such as tri-n-butyl silyl (4c) and tri-isopropyl silyl (4d) in the axial positions led to ∼10% increase in JSC values when compared to 4b (Fig. 7, Table 1).40 These experimental outcomes led to the conclusion that the SiPc dyes with contrasting properties of high solubility and extreme tendency to crystallize favoured charge transfer and augmented the overall device efficiency. A more rigorous study using eight SiPc variants with axial alkylsilyl groups of different chain lengths has been recently reported.44 It was observed that SiPc variants displayed similar optical, electrochemical and hydrophobicity properties and frontier orbital energy levels. However, the alkyl chain length had a pronounced effect in molecular packing and solubility of the SiPc dyes and thus governed the overall device performance. Best PCE values were achieved in cases where solubility of the ternary additive SiPc dye matched closely to that of P3HT.
In parallel, researchers explored SiPc structural variants with axial functionalities other than siloxy ethers, such as alkyl/aryl ethers and esters, as alternate additives in BHJ solar cells. Bender and co-workers investigated the device performance of bis(3-pentadecylphenoxy)-SiPc, 3e (Fig. 8).30 Albeit having similar spectral features like 4b, the dye 3e failed to significantly increase the JSC and the efficiency (ηeff) of the polymer–fullerene based ternary cascade BHJ solar cell (Table 1). Compound 3e exhibited lower tendencies to crystallize and migrate to the P3HT/PCBM interface as compared to 4a. The authors speculated that these factors might be responsible for its inferior performance.
 | ||
| Fig. 8 Chemical structures of SiPc derivatives, 3e–3j. | ||
In another approach, Ameri et al. synthesized SiPc derivatives with bulky t-butyl groups in the periphery and flexible axial pyrene carboxylic acid groups to fabricate ternary BHJ P3HT/PCBM solar cell devices.115 The ternary devices with these variants displayed an increase of over 20% in JSC and over 50% in PCE values compared to the baseline binary device. Among the variants, best fill factor and charge carrier mobility were obtained for the device constructed with 5a (Fig. 9, Table 1).
 | ||
| Fig. 9 Chemical structures of bis-ester SiPc derivatives, 5a–5h. | ||
In an attempt to further increase the light harvesting window up to 800 nm, SiNcs, having appropriate energy levels, were deployed alone or with SiPcs (Fig. 10).112,116 The research group of Sellinger used a symmetrical 2,3-SiNc derivative, 26 with peripheral t-butyl groups and two axial trihexylsilyloxide groups as the only light harvesting additive in a ternary P3HT/PMBC solar cell (Fig. 10A).116 The presence of bulky t-butyl functional groups conferred higher solubility to 26 and the BHJ morphology of P3HT/PCBM was preserved even with excess dye incorporation. Thus, higher optimal dye loading contents up to 20% was achieved without causing a decrease in JSC. The blending of 26 resulted in ∼40% enhancement of JSC values and a PCE of up to 4.9% was reported (Table 1).
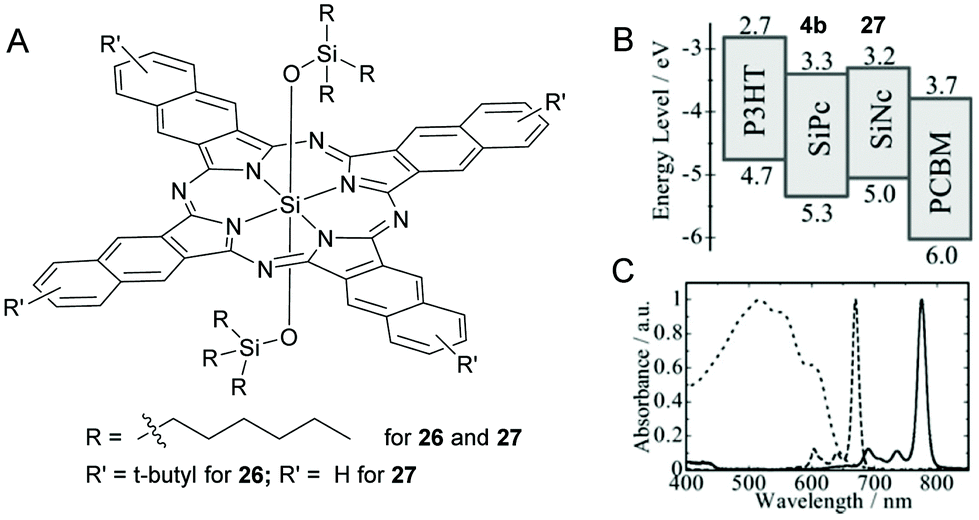 | ||
| Fig. 10 (A) Chemical structure of the SiPc dyes 26 and 27. (B) energy diagrams of P3HT, PCBM, SiPc 4b, and SiNc 27 (c) Absorption spectra of the P3HT/PCBM film (dotted line), SiPc 4b (broken line), and SiNc 27 (solid line) in solution. Adapted from ref. 112 with permissions from Royal Society of Chemistry. | ||
In another example, Honda et al. blended a SiNc dye, 27 along with 4b, as a fourth component in the polymer–fullerene-SiPc solar cell (Fig. 7, 10A, B and C).112 The quaternary solar cell P3HT/PCBM/SiPc(4a)/SiNc(27) exhibited a higher photoconversion efficiency (PCE) of 4.3% than the unblended (P3HT/PCBM) or ternary (P3HT/PCBM/SiPc(4b)) or (P3HT/PCBM/SiNc(27)) solar cells. The observed values for the quaternary device were JSC, VOC and FF were 10.9 mA cm−2, 0.57 V and 0.69 respectively at a loading content of 4.8 wt% for 4b and 1.5 wt% for 27 (Table 1). This improvement in device performance was attributed to the efficient photocurrent generation in the broad absorption profile in the visible region stretching from 400 to 800 nm.
Ameri and co-workers fabricated a quaternary solar cell using SiNc, 28 and SiPc, 5b containing axial pyrene units in order to investigate the effect of elongated π-systems on the device characteristics (Fig. 9 and 11A).117 They observed an improved PCE of 3.89 at a loading content of 7.5 and 5 wt% respectively for 5b and 28 as compared to the ternary blend solar cells with individual dyes (Table 1).
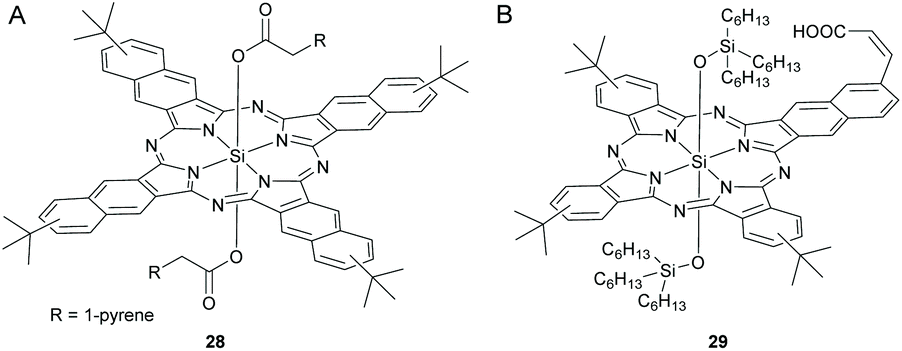 | ||
| Fig. 11 Chemical structures of dyes (A) 28 and (B) 29. R denotes 1-pyrene group. | ||
Instead of using two distinct SiPc and SiNc dyes, Sellinger et al. developed a hybrid SiNcPc, 29 bearing a mononaphthalo triphthalo cyanine core and investigated its photosensitizing properties in a DSSC (Fig. 11B).118 A vinyl group was introduced in the mononaphthalo ring to establish conjugation with an anchoring carboxylic acid unit. The hybrid dye, 29 exhibited high solubility, high extinction coefficients and desirable energy levels. The integration of 29 in solar devices resulted in high photocurrents of 19.0 A cm−2 and a total efficiency of 4.5% (Table 1).
In another report, the electron donating and accepting dual-nature functionalities of SiPcCl2, 1 and bis(pentafluorophenoxy)-SiPc, 3f (Fig. 8) in un-optimized planar heterojunction (PHJ) solar cell devices were demonstrated (Fig. 8, Table 1).36 Compound 3f displayed superior donor and acceptor properties which improved the device performance as compared to 1. These striking device characteristics of 3f were partially attributed to the solid-state arrangement and improved π-shielding as evident from its X-ray crystal structure. On the contrary, such π–π interactions were completely missing in the crystals of 1. It was further noted that the frequency and position of the fluorine atoms in the axial phenoxy group governed the dual-characteristics of the SiPc derivatives.119 The crude structure–activity analysis revealed SiPc analogues, 3g and 3h as potential donors and acceptors, respectively for future device configuration (Fig. 8, Table 1).
Samuel and co-workers investigated symmetrical SiPcs with axial bis-esters of eicosanoic acid (5c) and 3,5-di-tert-butylbenzoic acid (5d) as NIR absorbing dyes and electron acceptors in binary BHJ organic solar cells (Fig. 9, Table 1).120 The best device characteristics were obtained on using PTB7, a dithiophene based polymer and 5d as the donor acceptor pair at a 1:1.5 weight ratio and were attributed to the increased photocurrent and strong visible absorption of 5d.
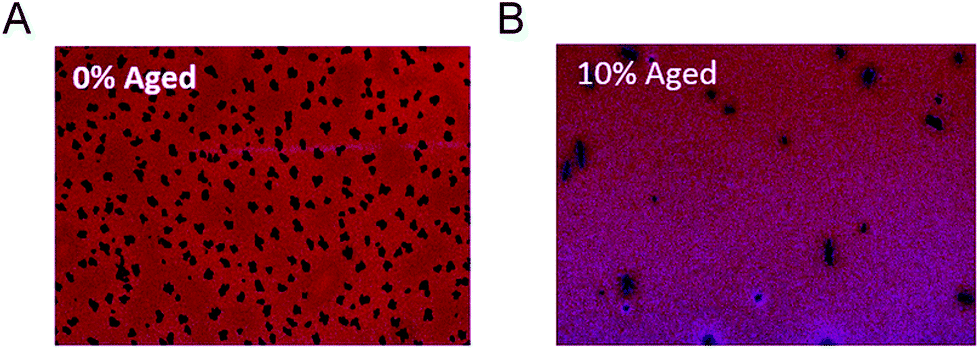 | ||
| Fig. 12 Microscopic images (20× magnification) of (A) after thermal ageing of baseline P3HT/PCBM devices with 0% 5e (B) after thermal ageing of P3HT/PCBM devices with 10% 5e. Adapted from ref. 121 with permission from The Royal Society of Chemistry. | ||
3.4 Applications of SiPc in OLEDs
SiPcs exhibit intense and narrow NIR luminescence spectra and thus find applications as emitters in organic light emitting diodes (OLEDs). Samuel and co-workers recently studied the optoelectronic properties and the electroluminescence of two SiPc derivatives, 5c and 5d (Fig. 9).120 Both derivatives displayed electroluminescence features in OLEDs similar to their photoluminescence profiles with a maxima around 698–709 nm and a narrow full width at half maxima (FWHM) of 21–27 nm. It was observed that doping of the emitting layer of the device with 10% 5c resulted in better quality of OLEDs with fewer pinholes and reduced roughness as compared to 5d indicating that structure of the axial ligands influence device characteristics (Fig. 13).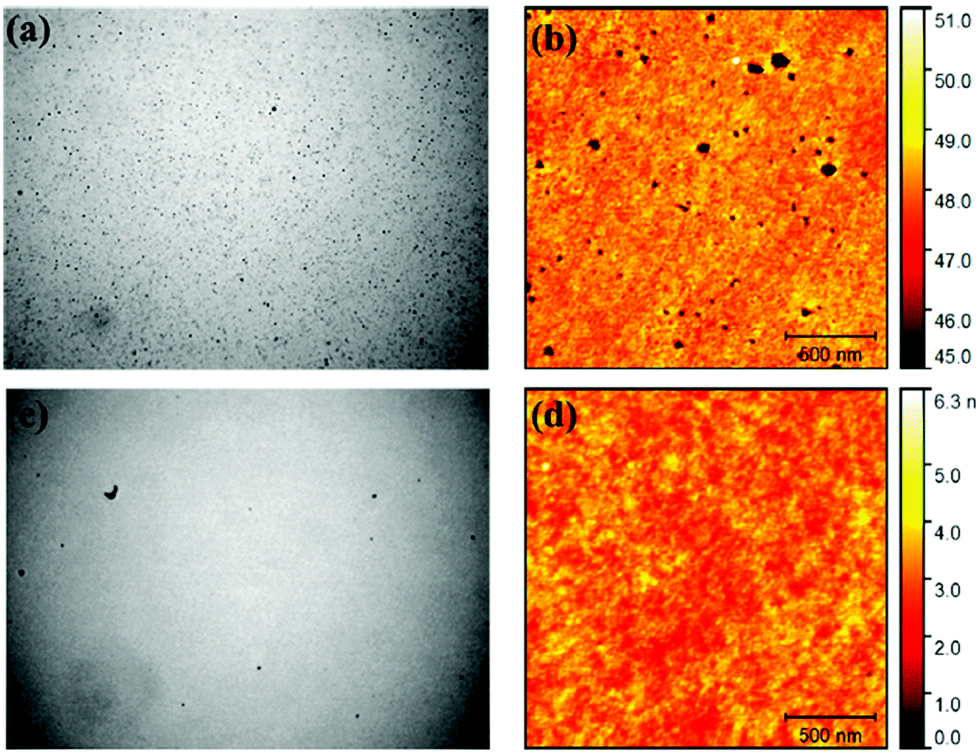 | ||
| Fig. 13 Optical microscopy [(a and c); 150 × 113 μm2 each] and atomic-force microscopy [(b and d); 2 × 2 μm2 each; 500 nm scale bar] of films ITO/PEDOT:PSS/PVK/CBP:PBD:SiPc (30:60:10; SiPc = 5c or 5d; 30 nm). The film doped with 5d [(a and b)] has more pinholes and higher RMS roughness Ra = 0.72 nm than the film doped with 5c [(c and d); Ra = 0.4 nm]. Adapted with permission from ref. 120. Further permissions related to the material excerpted should be directed to the ACS (https://pubs.acs.org/doi/10.1021/acsami.5b12408). | ||
Instead of using long chain carbons to reduce aggregation and confer solubility, Greenham and co-workers chose carbon-efficient and electron withdrawing pentafluorophenoxy moieties as the axial ligands.122 They deployed the SiPc derivative, 3f as dopant red emitters in both solution-processed and vapour-deposited OLEDs (Fig. 8). Compound 3f displayed a photoluminescence quantum yield value of 0.66 and a band gap of 1.77 eV. The OLED devices constructed with a polymeric host and 3f as a guest showed narrow electroluminescence spectra (FWHM of 20 nm) with a maximum external quantum efficiency (EQE) value of 2.5%. The vapour-deposited host:guest OLEDs incorporating 5 wt% doped 3f displayed similar emission profile with maximum around 715 nm, albeit, with a much lower EQE value of ∼0.09%. Though the energy transfer was clearly confirmed in this device, low EQE values indicated that a proper choice of host is crucial in order to achieve a significantly higher dopant emission over the host emission.
3.5 Applications of SiPc in OTFTs
Like other nitrogen heterocycle-based compounds,123,124 certain SiPcs also possess electronic properties of n-type semiconductors.26 Additionally, the readily tunable axial ligands, high field-effect mobility, robust environmental stability and ease of processability make them ideal to perform as organic thin film transistors (OTFTs).Lessard et al., first deployed SiPcs as n-type semiconductors in bottom-gate bottom-contact (BGBC) OTFE devices.125 SiPc bis-esters with 1-benzoic (5f), 1-naphthoic (5g) and 9-anthroic (5h) acid axial pendants were explored to establish a link between solid state arrangements of the semiconductors and the overall device performances (Fig. 9). The varying degree of π-bonding interactions of the Pc chromophores was expected to partly affect the 2D charge transfer pathways in the devices. The crystal structures revealed that 5f and 5g showed significant parallel stacking of the Pc rings. This probably led to higher electron field effect mobility (μe) and increased on-current (Ion) in devices with 5f and 5g. Although 5g revealed the closest π–π interactions, its device performance was lower than that of 5f. On the other hand, the bulky anthracene group hindered the overlap of the Pc rings and devices with 5h displayed lowest μe and Ion values. However, devices with 5h exhibited the lowest threshold voltage (VT). This is possibly because the lowest unoccupied molecular orbital (LUMO) of 5h was energetically comparable to the work function of gold. Overall, 5f outclassed the other SiPcs in the series in terms of device performance. Further increment of device performance of 5f was achieved with octyltrichlorosilane (OTS) dielectric modification and a substrate temperature of 200 °C maintained during deposition.
Motivated by these results, Lessard et al., investigated the behaviour of fluorophenoxy derivatives of SiPc as n-type transistors.126,127 It was noticed that the frequency and position of fluorine atoms affected electron mobility and the compounds 3f–3h exhibited some ambipolarity of charge transport (Fig. 8). In the series, devices built with 3f deposited on OTS-modified surface displayed superior performance. Taken together, these reports demonstrated the potential roles of axial substituents in governing the device performances and hinted towards a possibility of obtaining high performance SiPc based n-type semiconductors.
3.6 Applications in photocatalysis
The design of tetrapyrrole complexes and investigation of their photocatalytic behaviour stemmed from the key roles played by naturally occurring tetrapyrrole complexes (e.g. chlorophyll) in photosynthesis.128,129 In this context, SiPcs are recently being explored for potential applications in solar energy conversion and homogeneous photocatalysis. The fulfilling criteria of SiPcs to function as photocatalysts include: (i) robust photostability, (ii) intense absorption features in the solar spectrum which are tunable at the molecular level and (iii) ability to initiate photoinduced electron transfer reactions.130Knör and co-workers synthesized a cationic, water soluble SiPc, 3i and investigated its potential for construction of hydrogen evolving photocatalytic systems (Fig. 8, see above).131 Compound 3i reacted with aerial oxygen predominantly through a type II sensitization mechanism involving formation of singlet oxygen. Trace amounts of 1,4-benzoquinone and triethanolamine efficiently quenched the fluorescence signal of 3i following a Stern–Volmer kinetics of bimolecular quenching mechanism. Excess of triethanolamine led to rapid accumulation of permanent photoredox products of 3i. This confirmed the presence of a photoinduced electron transfer process possibly involving reduced Pc π-radical anion intermediates. Such radical species play important roles in mediating photochemical water splitting, thereby making SiPcs attractive candidates for construction of solar energy driven catalytic systems.
In another study, the catalytic behaviour of another SiPc derivative, 3j, was investigated using the 4-nitrophenol degradation probe reaction (Fig. 8, see above).132 Visible light induced degradation of 4-nitrophenol was observed with catalytic amounts of 3j and hydrogen peroxide as a source of oxygen. Hydroquinone and benzoquinone were determined as the major and minor photoproducts respectively. A substrate/oxidant/catalyst ratio of 2000/500/1 resulted in 88% conversion with a turnover number of 1758 and turnover frequency of 586 for 3j. Interestingly, photoconversion (37%) of 4-nitrophenol was observed without hydrogen peroxide, indicating the ability of 3j to act as a photocatalyst in absence of any oxygen source.
Recent reports exploited covalently functionalized SiPc-graphene based organic hybrid composites as improved solar catalysts.133,134 SiPc covalently functionalized with N-doped ultrasmall reduced graphene oxide (N-usRGO/SiPc) displayed strong absorption in the visible spectrum.133 The fluorescence quenching (90%) of SiPc and improved photocurrent responses for N-usRGO/SiPc demonstrated the efficient photoinduced electron transfer process from SiPc to N-usRGO through covalent bonds (Fig. 14A). These properties attributed to higher visible light induced catalytic activity of (N-usRGO/SiPc) in comparison to SiPc free N-usRGO. Platinum nanoparticles (Pt NP, 5 wt% Pt) deposited on N-heteroatoms of graphene acted as cocatalyst and further enhanced the catalytic performance of the composite (Fig. 14B). Pt NP decorated N-usRGO/SiPc construct produced a maximum of 4.5 μmol mg−1 of hydrogen from water on 6 h of UV-vis irradiation. The proposed mechanism suggests sequential shuttling of electrons from light harvesting SiPc to graphene layers and consequently to Pt NP where water molecules accept electrons and evolve hydrogen (Fig. 14B). Finally, triethanolamine (TEA) donates electron to excited SiPc and brings it back to the ground state (Fig. 14B).
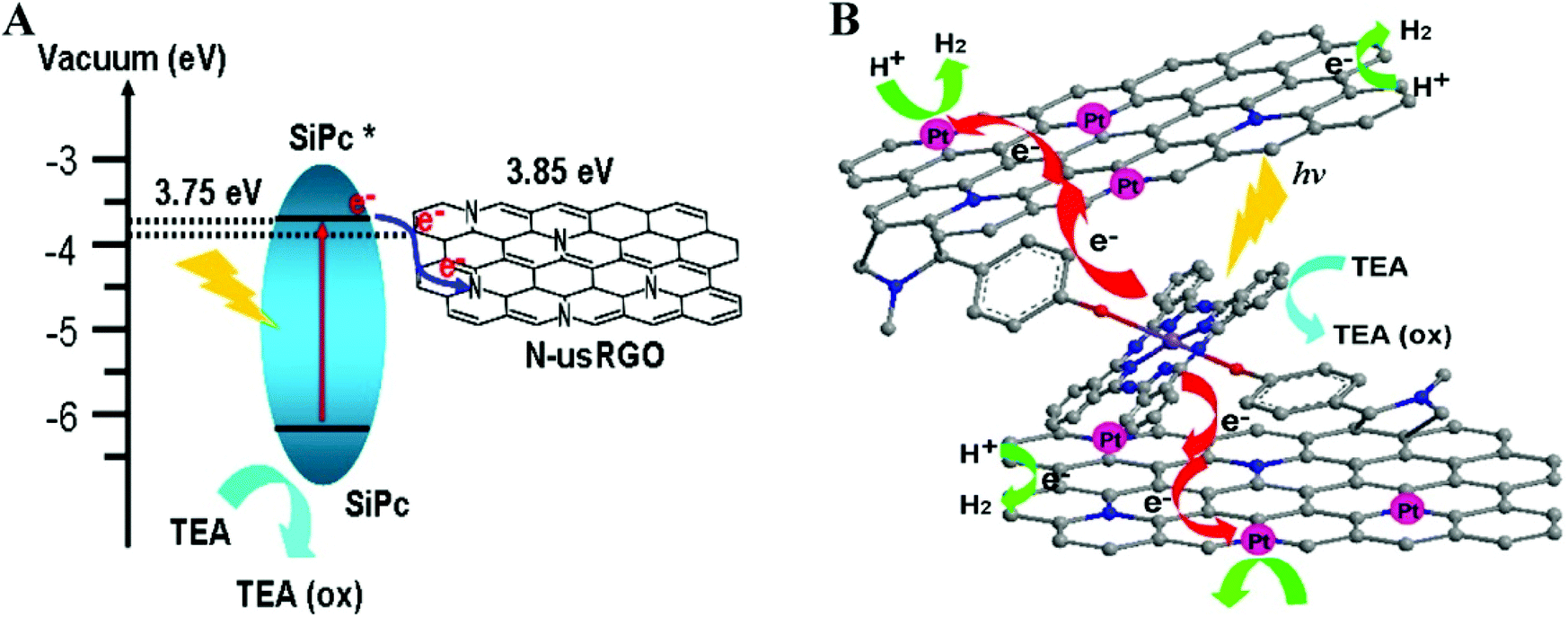 | ||
| Fig. 14 (A) Energy band structure diagram of heterostructure between N-usRGO and SiPc and (B) Electron transfer processes and evolution of hydrogen in N-usRGO/SiPc/Pt photocatalyst. Reprinted with permission from ref. 133. Copyright (2015) American Chemical Society. | ||
Lu et al. observed that addition of surfactants like cetyltrimethylammonium bromide (CTAB) prevented aggregation and boosted the photocatalytic behaviour of a SiPc(phenyl) covalently functionalized and Pt NP loaded graphene construct, SiPc(phenyl)2G2/Pt (Ref: IntJHydE2016).134 The integrated photocatalytic system, SiPc(phenyl)2G2/Pt/CTAB produced 18.2 μmol mg−1 of hydrogen on 10 h or UV-visible irradiation. These studies together established the potential of SiPcs in construction of water splitting photocatalytic systems.
4. Conclusions and perspectives
Despite their long existence, SiPcs continue to attract unabated attention due to their emerging properties and applications. Like most other phthalocyanines, SiPcs have gained profound recognition as far-red and NIR photodynamic therapeutic and fluorescence imaging agents. Recent findings corroborated the photodissociative nature of the axial ligands and added a new dimension to these old molecules. Novel mechanisms proposed to be involved in photouncaging and photoimmunotherapy have recommenced exploration of SiPcs in light-based therapeutic approaches, other than PDT. While the Si–O bonds exhibit additive and/or oxygen dependent photocleavage, the Si–C bonds can rapidly dissociate without any dependence on oxygen or redox reagents. Besides medicinal applications, SiPcs are rapidly acquiring a foothold in the emerging fields of photovoltaics, organic electronics and photocatalysis. However, thoughtful considerations of energy levels, bulk heterojunction morphology, solubility and crystallinity are essential while designing SiPc variants in order to observe enhanced efficiency and performances of the devices. Though few SiPc molecules seem propitious, further structure–activity studies are required to correlate the structural modifications with the device characteristics. Altogether, the promising results bode well for future explorations of SiPc in multifaceted research areas.However, these outstanding applications rest heavily on the structural modifications of the axial and phthalocyanine ring positions of SiPcs. The synthetic challenges to construct SiPc variants have been the bottleneck of this field and have impeded many applications. Oftentimes, the syntheses of SiPcs employ harsh reagents and drastic conditions. Furthermore, the reactions suffer from low synthetic yields and produce multiple undesired products. The purification techniques are complicated and laborious. Thus, the current scenario highlights the need for convenient and reliable synthetic methodology using non-hazardous and affordable reagents. Future efforts to define elegant synthetic approaches will be of paramount importance for complete realization of potential of SiPcs in phototherapy and energy harvesting applications.
Conflicts of interest
M. C. T. H. is a founder and shareholder of Light Switch Bio, LLC., a startup company that is developing light-based therapeutics for commercial applications.Acknowledgements
This work was supported by the Virginia Commonwealth Health Research Board (236-03-16).Notes and references
- P. A. Barrett, C. E. Dent and R. P. Linstead, Phthalocyanines. Part VII. Phthalocyanine as a co-ordinating group. A general investigation of the metallic derivatives, J. Chem. Soc., 1936, 382, 1719–1736 RSC.
- J. A. Elvidge and R. P. Linstead, Conjugated macrocycles. Part XXVII. The formation of tetrazaporphins from imidines. Tribenzotetrazaporphin, J. Chem. Soc., 1955, 3536–3544 RSC.
- R. P. Linstead and A. R. Lowe, Phthalocyanines. Part III. Preliminary experiments on the preparation of phthalocyanines from phthalonitrile, J. Chem. Soc., 1934, 214, 1022–1027 RSC.
- J. M. Robertson, An X-ray study of the structure of the phthalocyanines. Part I. The metal-free, nickel, copper, and platinum compounds, J. Chem. Soc., 1935, 136, 615–621 RSC.
- H. Lu and N. Kobayashi, Optically Active Porphyrin and Phthalocyanine Systems, Chem. Rev., 2016, 116, 6184–6261 CrossRef CAS.
- B. L. Wheeler, G. Nagasubramanian, A. J. Bard, L. A. Schechtman and M. E. Kenney, A silicon phthalocyanine and a silicon naphthalocyanine: synthesis, electrochemistry, and electrogenerated chemiluminescence, J. Am. Chem. Soc., 1984, 106, 7404–7410 CrossRef CAS.
- M. Matsuoka, Phthalocyanine and Naphthalocyanine Dyes, in Infrared Absorbing Dyes. Topics in Applied Chemistry, ed. M. Matsuoka, Springer, Boston, MA, 1990, pp. 45–55 Search PubMed.
- S. Kakade, R. Ghosh and D. K. Palit, Excited State Dynamics of Zinc–Phthalocyanine Nanoaggregates in Strong Hydrogen Bonding Solvents, J. Phys. Chem. C, 2012, 116, 15155–15166 CrossRef CAS.
- G. v. Bünau and J. B. Birks, Photophysics of Aromatic Molecules. Wiley-Interscience, London 1970. 704 Seiten. Preis: 210s, Ber. Bunsenges. Phys. Chem., 1970, 74, 1294–1295 Search PubMed.
- J. M. Dąbrowski, B. Pucelik, A. Regiel-Futyra, M. Brindell, O. Mazuryk, A. Kyzioł, G. Stochel, W. Macyk and L. G. Arnaut, Engineering of relevant photodynamic processes through structural modifications of metallotetrapyrrolic photosensitizers, Coord. Chem. Rev., 2016, 325, 67–101 CrossRef.
- X. Li, B.-D. Zheng, X.-H. Peng, S.-Z. Li, J.-W. Ying, Y. Zhao, J.-D. Huang and J. Yoon, Phthalocyanines as medicinal photosensitizers: Developments in the last five years, Coord. Chem. Rev., 2019, 379, 147–160 CrossRef CAS.
- C. G. Claessens, U. Hahn and T. Torres, Phthalocyanines: From outstanding electronic properties to emerging applications, Chem. Rec., 2008, 8, 75–97 CrossRef CAS.
- R. D. Joyner and M. E. Kenney, Phthalocyaninosilicon Compounds, Inorg. Chem., 1962, 1, 236–238 CrossRef CAS.
- J.-W. Hofman, F. van Zeeland, S. Turker, H. Talsma, S. A. G. Lambrechts, D. V. Sakharov, W. E. Hennink and C. F. van Nostrum, Peripheral and Axial Substitution of Phthalocyanines with Solketal Groups: Synthesis and In Vitro Evaluation for Photodynamic Therapy, J. Med. Chem., 2007, 50, 1485–1494 CrossRef CAS.
- H. Li, T. J. Jensen, F. R. Fronczek and M. G. H. Vicente, Syntheses and Properties of a Series of Cationic Water-Soluble Phthalocyanines, J. Med. Chem., 2008, 51, 502–511 CrossRef CAS.
- J. C. Berlin, Silicon phthalocyanines for photodynamic therapy, Phd Thesis, Case Western Reserve University, 2006 Search PubMed.
- E. van de Winckel, B. David, M. M. Simoni, J. A. González-Delgado, A. de la Escosura, Â. Cunha and T. Torres, Octacationic and axially di-substituted silicon(IV) phthalocyanines for photodynamic inactivation of bacteria, Dyes Pigm., 2017, 145, 239–245 CrossRef CAS.
- K. Sato, K. Ando, S. Okuyama, S. Moriguchi, T. Ogura, S. Totoki, H. Hanaoka, T. Nagaya, R. Kokawa, H. Takakura, M. Nishimura, Y. Hasegawa, P. L. Choyke, M. Ogawa and H. Kobayashi, Photoinduced Ligand Release from a Silicon Phthalocyanine Dye Conjugated with Monoclonal Antibodies: A Mechanism of Cancer Cell Cytotoxicity after Near-Infrared Photoimmunotherapy, ACS Cent. Sci., 2018, 4, 1559–1569 CrossRef CAS.
- E. D. Anderson, A. P. Gorka and M. J. Schnermann, Near-infrared uncaging or photosensitizing dictated by oxygen tension, Nat. Commun., 2016, 7, 13378 CrossRef.
- M. D. Maree, N. Kuznetsova and T. Nyokong, Silicon octaphenoxyphthalocyanines: photostability and singlet oxygen quantum yields, J. Photochem. Photobiol., A, 2001, 140, 117–125 CrossRef CAS.
- Y. Cheng, T. L. Doane, C.-H. Chuang, A. Ziady and C. Burda, Near infrared light-triggered drug generation and release from gold nanoparticle carriers for photodynamic therapy, Small, 2014, 10, 1799–1804 CrossRef CAS.
- J. Li, Y. Yang, P. Zhang, J. R. Sounik and M. E. Kenney, Synthesis, properties and drug potential of the photosensitive alkyl- and alkylsiloxy-ligated silicon phthalocyanine Pc 227, Photochem. Photobiol. Sci., 2014, 13, 1690–1698 RSC.
- P.-C. Lo, M. S. Rodríguez-Morgade, R. K. Pandey, D. K. P. Ng, T. Torres and F. Dumoulin, The unique features and promises of phthalocyanines as advanced photosensitisers for photodynamic therapy of cancer, Chem. Soc. Rev., 2020, 49, 1041–1056 RSC.
- R. D. Joyner, J. Cekada, R. G. Linck and M. E. Kenney, Diphenoxysilicon phthalocyanine, J. Inorg. Nucl. Chem., 1960, 15, 387–388 CrossRef CAS.
- M. K. Lowery, A. J. Starshak, J. N. Esposito, P. C. Krueger and M. E. Kenney, Dichloro(phthalocyanino)silicon, Inorg. Chem., 1965, 4, 128 CrossRef CAS.
- N. J. Yutronkie, T. M. Grant, O. A. Melville, B. H. Lessard and J. L. Brusso, Old Molecule, New Chemistry: Exploring Silicon Phthalocyanines as Emerging N-Type Materials in Organic Electronics, Materials, 2019, 12, 1334 CrossRef CAS.
- R. Rafaeloff, F. J. Kohl, P. C. Krueger and M. E. Kenney, New group IV phthalocyanines, J. Inorg. Nucl. Chem., 1966, 28, 899–902 CrossRef CAS.
- J. N. Esposito, J. E. Lloyd and M. E. Kenney, The Synthesis and Physical Properties of Some Organo- and Organosiloxysilicon Phthalocyanines, Inorg. Chem., 1966, 5, 1979–1984 CrossRef CAS.
- T. M. Grant, V. McIntyre, J. Vestfrid, H. Raboui, R. T. White, Z.-H. Lu, B. H. Lessard and T. P. Bender, Straightforward and Relatively Safe Process for the Fluoride Exchange of Trivalent and Tetravalent Group 13 and 14 Phthalocyanines, ACS Omega, 2019, 4, 5317–5326 CrossRef CAS.
- B. H. Lessard, J. D. Dang, T. M. Grant, D. Gao, D. S. Seferos and T. P. Bender, Bis(tri-n-hexylsilyl oxide) Silicon Phthalocyanine: A Unique Additive in Ternary Bulk Heterojunction Organic Photovoltaic Devices, ACS Appl. Mater. Interfaces, 2014, 6, 15040–15051 CrossRef CAS.
- J.-D. Huang, S. Wang, P.-C. Lo, W.-P. Fong, W.-H. Ko and D. K. P. Ng, Halogenated silicon(IV) phthalocyanines with axial poly(ethylene glycol) chains. Synthesis, spectroscopic properties, complexation with bovine serum albumin and in vitro photodynamic activities, New J. Chem., 2004, 28, 348–354 RSC.
- J. Mao, Y. Zhang, J. Zhu, C. Zhang and Z. Guo, Molecular combo of photodynamic therapeutic agent silicon(IV) phthalocyanine and anticancer drug cisplatin, Chem. Commun., 2009, 908–910 RSC.
- D. Demirkapı, A. Şirin, B. Turanlı-Yıldız, Z. P. Çakar and B. Ş. Sesalan, The synthesis of new silicon phthalocyanines and analysis of their photochemical and biological properties, Synth. Met., 2014, 187, 152–159 CrossRef.
- L. Luan, L. Ding, J. Shi, W. Fang, Y. Ni and W. Liu, Effect of Axial Ligands on the Molecular Configurations, Stability, Reactivity, and Photodynamic Activities of Silicon Phthalocyanines, Chem. – Asian J., 2014, 9, 3491–3497 CrossRef CAS.
- W. Harada, M. Hirahara, T. Togashi, M. Ishizaki, M. Kurihara, M. Haga and K. Kanaizuka, Wisely Designed Phthalocyanine Derivative for Convenient Molecular Fabrication on a Substrate, Langmuir, 2018, 34, 1321–1326 CrossRef CAS.
- B. H. Lessard, R. T. White, M. AL-Amar, T. Plint, J. S. Castrucci, D. S. Josey, Z.-H. Lu and T. P. Bender, Assessing the Potential Roles of Silicon and Germanium Phthalocyanines in Planar Heterojunction Organic Photovoltaic Devices and How Pentafluoro Phenoxylation Can Enhance π–π Interactions and Device Performance, ACS Appl. Mater. Interfaces, 2015, 7, 5076–5088 CrossRef CAS.
- X. Leng, C.-F. Choi, P.-C. Lo and D. K. P. Ng, Assembling a Mixed Phthalocyanine−Porphyrin Array in Aqueous Media through Host−Guest Interactions, Org. Lett., 2007, 9, 231–234 CrossRef CAS.
- J. T. F. Lau, P.-C. Lo, W.-P. Fong and D. K. P. Ng, Preparation and Photodynamic Activities of Silicon(IV) Phthalocyanines Substituted with Permethylated β-Cyclodextrins, Chem. – Eur. J., 2011, 17, 7569–7577 CrossRef CAS.
- H. M. Anula, J. C. Berlin, H. Wu, Y.-S. Li, X. Peng, M. E. Kenney and M. A. J. Rodgers, Synthesis and Photophysical Properties of Silicon Phthalocyanines with Axial Siloxy Ligands Bearing Alkylamine Termini, J. Phys. Chem. A, 2006, 110, 5215–5223 CrossRef CAS.
- M.-T. Dang, T. M. Grant, H. Yan, D. S. Seferos, B. H. Lessard and T. P. Bender, Bis(tri-n-alkylsilyl oxide) silicon phthalocyanines: a start to establishing a structure property relationship as both ternary additives and non-fullerene electron acceptors in bulk heterojunction organic photovoltaic devices, J. Mater. Chem. A, 2017, 5, 12168–12182 RSC.
- T. Gessner, R. Sens, W. Ahlers and C. Vamvakaris, Preparation of silicon phthalocyanines and germanium phthalocyanines and related substances, U.S. Patent, US20100113767A1, 2010 Search PubMed.
- Y.-S. Li, S. I. A. Zaidi, M. A. J. Rodgers, H. Mukhtar, M. E. Kenney, N. L. Oleinick, J. He, H. E. Larkin and B. D. Rihter, The Synthesis, Photophysical and Photobiological Properties and in vitro Structure-Activity Relationships of a Set of Silicon Phthalocyanine PDT Photosensitizers, Photochem. Photobiol., 1997, 65, 581–586 CrossRef.
- X. Peng, D. R. Draney and J. Chen, Phthalocyanine Dyes, U.S. Patent, US7005518B2, 2004 Search PubMed.
- M. C. Vebber, T. M. Grant, J. L. Brusso and B. H. Lessard, Bis(trialkylsilyl oxide) Silicon Phthalocyanines: Understanding the Role of Solubility in Device Performance as Ternary Additives in Organic Photovoltaics, Langmuir, 2020, 36, 2612–2621 CrossRef CAS.
- A. K. Sinha and B. K. Mandal, Thin Polyurethane Films of Polyhydroxysilicon Phthalocyanine and Bis-phthalocyanine Derivatives, Polym. J., 1995, 27, 1079–1084 CrossRef CAS.
- D. W. DeWulf, J. K. Leland, B. L. Wheeler, A. J. Bard, D. A. Batzel, D. R. Dininny and M. Kenney, Isolation, spectroscopic properties, and electrochemical properties of two oligomeric silicon phthalocyanines, Inorg. Chem., 1987, 26, 266–270 CrossRef CAS.
- Y. Yang, B. Samas, V. O. Kennedy, D. Macikenas, B. L. Chaloux, J. A. Miller, R. L. Speer, J. Protasiewicz, A. A. Pinkerton and M. E. Kenney, Long, Directional Interactions in Cofacial Silicon Phthalocyanine Oligomers, J. Phys. Chem. A, 2011, 115, 12474–12485 CrossRef CAS.
- K. Oniwa, S. Shimizu, Y. Shiina, T. Fukuda and N. Kobayashi, A μ-oxo hetero dimer of silicon phthalocyanine and naphthalocyanine, Chem. Commun., 2013, 49, 8341–8343 RSC.
- Z. Li and M. Lieberman, Axial Reactivity of Soluble Silicon(IV) Phthalocyanines, Inorg. Chem., 2001, 40, 932–939 CrossRef CAS.
- J. B. Davison and K. J. Wynne, Silicon Phthalocyanine-Siloxane Polymers: Synthesis and 1H Nuclear Magnetic Resonance Study, Macromolecules, 1978, 11, 186–191 CrossRef CAS.
- C. A. Barker, K. S. Findlay, S. Bettington, A. S. Batsanov, I. F. Perepichka, M. R. Bryce and A. Beeby, Synthesis of new axially-disubstituted silicon-phthalocyanine derivatives: optical and structural characterisation, Tetrahedron, 2006, 62, 9433–9439 CrossRef CAS.
- K. Mitra, M. Samsó, C. E. Lyons and M. C. T. Hartman, Hyaluronic acid grafted nanoparticles of a platinum(II)–silicon(IV) phthalocyanine conjugate for tumor and mitochondria-targeted photodynamic therapy in red light, J. Mater. Chem. B, 2018, 6, 7373–7377 RSC.
- A. K. Pal, S. Varghese, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, Near-Infrared Fluorescence of Silicon Phthalocyanine Carboxylate Esters, Sci. Rep., 2017, 7, 12282 CrossRef.
- C. Farren, S. FitzGerald, M. R. Bryce, A. Beeby and A. S. Batsanov, Synthesis, structure and optical characterisation of silicon phthalocyanine bis-esters, J. Chem. Soc., Perkin Trans. 2, 2002, 59–66 CAS.
- C. Farren, C. A. Christensen, S. FitzGerald, M. R. Bryce and A. Beeby, Synthesis of Novel Phthalocyanine−Tetrathiafulvalene Hybrids; Intramolecular Fluorescence Quenching Related to Molecular Geometry, J. Org. Chem., 2002, 67, 9130–9139 CrossRef CAS.
- W. Rodríguez-Córdoba, R. Noria, C. A. Guarín and J. Peon, Ultrafast Photosensitization of Phthalocyanines through Their Axial Ligands, J. Am. Chem. Soc., 2011, 133, 4698–4701 CrossRef.
- Y.-S. Li and M. E. Kenney, Methods of syntheses of phthalocyanine compounds, U.S. Patent, US5763602A, 1998 Search PubMed.
- N. L. Oleinick, A. R. Antunez, M. E. Clay, B. D. Rihter and M. E. Kenney, New phthalocyanine photosensitizers for photodynamic therapy, Photochem. Photobiol., 1993, 57, 242–247 CrossRef CAS.
- P.-C. Lo, J.-D. Huang, D. Y. Y. Cheng, E. Y. M. Chan, W.-P. Fong, W.-H. Ko and D. K. P. Ng, New Amphiphilic Silicon(IV) Phthalocyanines as Efficient Photosensitizers for Photodynamic Therapy: Synthesis, Photophysical Properties, and in vitro Photodynamic Activities, Chem. – Eur. J., 2004, 10, 4831–4838 CrossRef CAS.
- B.-Y. Zheng, L. Wang, Q.-Y. Hu, J. Shi, M.-R. Ke and J.-D. Huang, Novel unsymmetrical silicon(IV) phthalocyanines as highly potent anticancer photosensitizers. Synthesis, characterization, and in vitro photodynamic activities, Dyes Pigm., 2020, 177, 108286 CrossRef CAS.
- J. T. F. Lau, P.-C. Lo, Y.-M. Tsang, W.-P. Fong and D. K. P. Ng, Unsymmetrical β-cyclodextrin-conjugated silicon(IV) phthalocyanines as highly potent photosensitisers for photodynamic therapy, Chem. Commun., 2011, 47, 9657–9659 RSC.
- Q. Liu, M. Pang, S. Tan, J. Wang, Q. Chen, K. Wang, W. Wu and Z. Hong, Potent peptide-conjugated silicon phthalocyanines for tumor photodynamic therapy, J. Cancer, 2018, 9, 310–320 CrossRef.
- Z. Li and M. Lieberman, Synthesis and characterization of functionalized silicon phthalocyanines for fabrication of self-assembled monolayers, Supramol. Sci., 1998, 5, 485–489 CrossRef CAS.
- M. Hanack, K. Mitulla, G. Pawlowski and L. R. Subramanian, Synthesis and properties of a new kind of one-dimensional conductor VIII. Synthesis and characterization of trans-bis-1-alkynyl substituted silicon, germanium, tin phthalocyanines and germanium hemiporphyrazines, J. Organomet. Chem., 1981, 204, 315–325 CrossRef.
- K. Tamao, M. Akita, H. Kato and M. Kumada, Electrophilic cleavage reations of carbon-silicon bonds in neutral hexacoordinate silicon compounds: diorgano(phthalocyaninato)silicon, J. Organomet. Chem., 1988, 341, 165–179 CrossRef CAS.
- J. Li, Silicon phthalocyanines for photodynamic therapy of studies, PhD Thesis, Case Western Reserve University, 2008 Search PubMed.
- A. G. Gürek, G. Appel, R. P. Mikalo and D. Schmeißer, Synthesis of dihydroxy silicon phthalocyanine tetrasulfonic acid and poly-μ-oxo silicon phthalocyanine tetrasulfonic acid, J. Porphyrins Phthalocyanines, 2001, 5, 751–757 CrossRef.
- D. Woehrle, M. Eskes, K. Shigehara and A. Yamada, A Simple Synthesis of 4,5-Disubstituted 1,2-Dicyanobenzenes and 2,3,9,10,16,17,23,24-Octasubstituted Phthalocyanines, Synthesis, 1993, 1993, 194–196 CrossRef.
- T. Furuyama, T. Ishii, N. Ieda, H. Maeda, M. Segi, M. Uchiyama and H. Nakagawa, Cationic axial ligands on sulfur substituted silicon(IV) phthalocyanines: improved hydrophilicity and exceptionally red-shifted absorption into the NIR region, Chem. Commun., 2019, 55, 7311–7314 RSC.
- G. Cheng, G. Hao, H. Wu, Y.-S. Li and M. E. Kenney, Methods for inserting silicon into phthalocyanines and naphthalocyanines, U.S. Patent, US5872248A, 1999 Search PubMed.
- S. Foley, G. Jones, R. Liuzzi, D. J. McGarvey, M. H. Perry and T. G. Truscott, The synthesis and photophysical properties of polyether substituted phthalocyanines of potential use in photodynamic therapy, J. Chem. Soc., Perkin Trans. 2, 1997, 2, 1725–1730 RSC.
- S. I. Büyükekşi, S. Z. Topal and D. Atilla, Novel Silicon Phthalocyanines Bearing Triethylene Glycol Groups: Photophysical and Photochemical Properties as well as pH-Induced Spectral Behaviour, J. Fluoresc., 2017, 27, 1257–1266 CrossRef.
- J. J. Bergkamp, B. D. Sherman, E. Mariño-Ochoa, R. E. Palacios, G. Cosa, T. A. Moore, D. Gust and A. L. Moore, Synthesis and characterization of silicon phthalocyanines bearing axial phenoxyl groups for attachment to semiconducting metal oxides, J. Porphyrins Phthalocyanines, 2011, 15, 943–950 CrossRef CAS.
- P.-C. Lo, C. M. H. Chan, J.-Y. Liu, W.-P. Fong and D. K. P. Ng, Highly Photocytotoxic Glucosylated Silicon(IV) Phthalocyanines. Effects of Peripheral Chloro Substitution on the Photophysical and Photodynamic Properties, J. Med. Chem., 2007, 50, 2100–2107 CrossRef CAS.
- R. F. Devlin, W. B. Dandliker and P. O. G. Arrhenius, Fluorescence immunoassays using fluorescence dyes free of aggregation and serum binding, U.S. Patent, US6060598A, 2000 Search PubMed.
- M. Lam, M. L. Dimaano, P. Oyetakin-White, M. A. Retuerto, J. Chandra, P. K. Mukherjee, M. A. Ghannoum, K. D. Cooper and E. D. Baron, Silicon phthalocyanine 4 phototoxicity in Trichophyton rubrum, Antimicrob. Agents Chemother., 2014, 58, 3029–3034 CrossRef.
- A. Galstyan, R. Schiller and U. Dobrindt, Boronic Acid Functionalized Photosensitizers: A Strategy To Target the Surface of Bacteria and Implement Active Agents in Polymer Coatings, Angew. Chem., Int. Ed, 2017, 56, 10362–10366 Search PubMed.
- B. Aru, A. Günay, E. Şenkuytu, G. Yanıkkaya Demirel, A. G. Gürek and D. Atilla, A Translational Study of a Silicon Phthalocyanine Substituted with a Histone Deacetylase Inhibitor for Photodynamic Therapy, ACS Omega, 2020, 5, 25854–25867 CrossRef CAS.
- K. Li, W. Dong, Q. Liu, G. Lv, M. Xie, X. Sun, L. Qiu and J. Lin, A biotin receptor-targeted silicon(IV) phthalocyanine for in vivo tumor imaging and photodynamic therapy, J. Photochem. Photobiol., B, 2019, 190, 1–7 CrossRef CAS.
- M. Mitsunaga, M. Ogawa, N. Kosaka, L. T. Rosenblum, P. L. Choyke and H. Kobayashi, Cancer cell–selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules, Nat. Med., 2011, 17, 1685–1691 CrossRef CAS.
- J. T. F. Lau, P.-C. Lo, X.-J. Jiang, Q. Wang and D. K. P. Ng, A Dual Activatable Photosensitizer toward Targeted Photodynamic Therapy, J. Med. Chem., 2014, 57, 4088–4097 CrossRef CAS.
- R. Devlin, R. M. Studholme, W. B. Dandliker, E. Fahy, K. Blumeyer and S. S. Ghosh, Homogeneous detection of nucleic acids by transient-state polarized fluorescence., Clin. Chem., 1993, 39, 1939–1943 CrossRef CAS.
- G. T. Walker, J. G. Nadeau, C. P. Linn, R. F. Devlin and W. B. Dandliker, Strand displacement amplification (SDA) and transient-state fluorescence polarization detection of Mycobacterium tuberculosis DNA., Clin. Chem., 1996, 42, 9–13 CrossRef CAS.
- X. Peng, D. R. Draney, W. M. Volcheck, G. R. Bashford, D. T. Lamb, D. L. Grone, Y. Zhang and C. M. Johnson, Phthalocyanine dye as an extremely photostable and highly fluorescent near-infrared labeling reagent, Proc. SPIE-Int. Soc. Opt. Eng., 2006, 6097, 60970E CrossRef.
- N. Masilela and T. Nyokong, The synthesis and photophysical properties of novel cationic tetra pyridiloxy substituted aluminium, silicon and titanium phthalocyanines in water, J. Lumin., 2010, 130, 1787–1793 CrossRef CAS.
- M. Göksel, Z. Biyiklioglu and M. Durmuş, The water soluble axially disubstituted silicon phthalocyanines: photophysicochemical properties and in vitro studies, JBIC, 2017, 22, 953–967 CrossRef.
- X. Li, J. F. Lovell, J. Yoon and X. Chen, Clinical development and potential of photothermal and photodynamic therapies for cancer, Nat. Rev. Clin. Oncol., 2020, 17, 657–674 CrossRef.
- P. Agostinis, K. Berg, K. A. Cengel, T. H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, Photodynamic therapy of cancer: an update, Ca-Cancer J. Clin., 2011, 61, 250–281 CrossRef.
- E. D. Baron, C. L. Malbasa, D. Santo-Domingo, P. Fu, J. D. Miller, K. K. Hanneman, A. H. Hsia, N. L. Oleinick, V. C. Colussi and K. D. Cooper, Silicon phthalocyanine (pc 4) photodynamic therapy is a safe modality for cutaneous neoplasms: results of a phase 1 clinical trial, Lasers Surg. Med., 2010, 42, 888–895 CrossRef.
- M. A. Hutnick, S. Ahsanuddin, L. Guan, M. Lam, E. D. Baron and J. K. Pokorski, PEGylated Dendrimers as Drug Delivery Vehicles for the Photosensitizer Silicon Phthalocyanine Pc 4 for Candidal Infections, Biomacromolecules, 2017, 18, 379–385 CrossRef CAS.
- Y. Cheng, J. D. Meyers, A.-M. Broome, M. E. Kenney, J. P. Basilion and C. Burda, Deep Penetration of a PDT Drug into Tumors by Noncovalent Drug-Gold Nanoparticle Conjugates, J. Am. Chem. Soc., 2011, 133, 2583–2591 CrossRef CAS.
- A. M. Master, Y. Qi, N. L. Oleinick and A. Sen Gupta, EGFR-mediated intracellular delivery of Pc 4 nanoformulation for targeted photodynamic therapy of cancer: in vitro studies, Nanomedicine, 2012, 8, 655–664 CrossRef CAS.
- D. C. Soler, J. Ohtola, H. Sugiyama, M. E. Rodriguez, L. Han, N. L. Oleinick, M. Lam, E. D. Baron, K. D. Cooper and T. S. McCormick, Activated T cells exhibit increased uptake of silicon phthalocyanine Pc 4 and increased susceptibility to Pc 4-photodynamic therapy-mediated cell death, Photochem. Photobiol. Sci., 2016, 15, 822–831 RSC.
- A. M. Master, M. Livingston, N. L. Oleinick and A. Sen Gupta, Optimization of a Nanomedicine-Based Silicon Phthalocyanine 4 Photodynamic Therapy (Pc 4-PDT) Strategy for Targeted Treatment of EGFR-Overexpressing Cancers, Mol. Pharm., 2012, 9, 2331–2338 CrossRef CAS.
- X.-M. Shen, B.-Y. Zheng, X.-R. Huang, L. Wang and J.-D. Huang, The first silicon(IV) phthalocyanine–nucleoside conjugates with high photodynamic activity, Dalton Trans., 2013, 42, 10398–10403 RSC.
- M. K. Burdette, R. Jenkins, Y. Bandera, R. R. Powell, T. F. Bruce, X. Yang, Y. Wei and S. H. Foulger, Bovine serum albumin coated nanoparticles for in vitro activated fluorescence, Nanoscale, 2016, 8, 20066–20073 RSC.
- J.-J. Chen, Y.-Z. Huang, M.-R. Song, Z.-H. Zhang and J.-P. Xue, Silicon Phthalocyanines Axially Disubstituted with Erlotinib toward Small-Molecular-Target-Based Photodynamic Therapy, ChemMedChem, 2017, 12, 1504–1511 CrossRef CAS.
- K. Li, L. Qiu, Q. Liu, G. Lv, X. Zhao, S. Wang and J. Lin, Conjugate of biotin with silicon(IV) phthalocyanine for tumor-targeting photodynamic therapy, J. Photochem. Photobiol., B, 2017, 174, 243–250 CrossRef CAS.
- X.-H. Peng, S.-F. Chen, B.-Y. Zheng, B.-D. Zheng, Q.-F. Zheng, X.-S. Li, M.-R. Ke and J.-D. Huang, Comparison between amine-terminated phthalocyanines and their chlorambucil conjugates: Synthesis, spectroscopic properties, and in vitro anticancer activity, Tetrahedron, 2017, 73, 378–384 CrossRef CAS.
- A. M. L. Hossion, M. Bio, G. Nkepang, S. G. Awuah and Y. You, Visible Light Controlled Release of Anticancer Drug through Double Activation of Prodrug, ACS Med. Chem. Lett., 2013, 4, 124–127 CrossRef CAS.
- M. Bio, P. Rajaputra, G. Nkepang and Y. You, Far-Red Light Activatable, Multifunctional Prodrug for Fluorescence Optical Imaging and Combinational Treatment, J. Med. Chem., 2014, 57, 3401–3409 CrossRef CAS.
- G. Nkepang, M. Bio, P. Rajaputra, S. G. Awuah and Y. You, Folate receptor-mediated enhanced and specific delivery of far-red light-activatable prodrugs of combretastatin A-4 to FR-positive tumor, Bioconjugate Chem., 2014, 25, 2175–2188 CrossRef CAS.
- P. Thapa, M. Li, M. Bio, P. Rajaputra, G. Nkepang, Y. Sun, S. Woo and Y. You, Far-Red Light-Activatable Prodrug of Paclitaxel for the Combined Effects of Photodynamic Therapy and Site-Specific Paclitaxel Chemotherapy, J. Med. Chem., 2016, 59, 3204–3214 CrossRef CAS.
- T. Doane, Y. Cheng, N. Sodhi and C. Burda, NIR Photocleavage of the Si–C Bond in Axial Si-Phthalocyanines, J. Phys. Chem. A, 2014, 118, 10587–10595 CrossRef CAS.
- B. Maiti, A. K. Manna, C. McCleese, T. L. Doane, S. Chakrapani, C. Burda and B. D. Dunietz, Photoinduced Homolytic Bond Cleavage of the Central Si–C Bond in Porphyrin Macrocycles Is a Charge Polarization Driven Process, J. Phys. Chem. A, 2016, 120, 7634–7640 CrossRef CAS.
- E. D. Anderson, S. Sova, J. Ivanic, L. Kelly and M. J. Schnermann, Defining the conditional basis of silicon phthalocyanine near-IR ligand exchange, Phys. Chem. Chem. Phys., 2018, 20, 19030–19036 RSC.
- L. C. Gomes-da-Silva, O. Kepp and G. Kroemer, Regulatory approval of photoimmunotherapy: photodynamic therapy that induces immunogenic cell death, OncoImmunology, 2020, 9, 1841393 CrossRef.
- K. Ito, M. Mitsunaga, T. Nishimura, M. Saruta, T. Iwamoto, H. Kobayashi and H. Tajiri, Near-Infrared Photochemoimmunotherapy by Photoactivatable Bifunctional Antibody–Drug Conjugates Targeting Human Epidermal Growth Factor Receptor 2 Positive Cancer, Bioconjugate Chem., 2017, 28, 1458–1469 CrossRef CAS.
- M. Kobayashi, M. Harada, H. Takakura, K. Ando, Y. Goto, T. Tsuneda, M. Ogawa and T. Taketsugu, Theoretical and Experimental Studies on the Near-Infrared Photoreaction Mechanism of a Silicon Phthalocyanine Photoimmunotherapy Dye: Photoinduced Hydrolysis by Radical Anion Generation, ChemPlusChem, 2020, 85, 1959–1963 CrossRef CAS.
- X. Li, X.-H. Peng, B.-D. Zheng, J. Tang, Y. Zhao, B.-Y. Zheng, M.-R. Ke and J.-D. Huang, New application of phthalocyanine molecules: from photodynamic therapy to photothermal therapy by means of structural regulation rather than formation of aggregates, Chem. Sci., 2018, 9, 2098–2104 RSC.
- T. M. Grant, D. S. Josey, K. L. Sampson, T. Mudigonda, T. P. Bender and B. H. Lessard, Boron Subphthalocyanines and Silicon Phthalocyanines for Use as Active Materials in Organic Photovoltaics, Chem. Rec., 2019, 19, 1093–1112 CrossRef CAS.
- S. Honda, H. Ohkita, H. Benten and S. Ito, Multi-colored dye sensitization of polymer/fullerene bulk heterojunction solar cells, Chem. Commun., 2010, 46, 6596–6598 RSC.
- S. Honda, H. Ohkita, H. Benten and S. Ito, Selective Dye Loading at the Heterojunction in Polymer/Fullerene Solar Cells, Adv. Energy Mater., 2011, 1, 588–598 CrossRef CAS.
- S. Honda, S. Yokoya, H. Ohkita, H. Benten and S. Ito, Light-Harvesting Mechanism in Polymer/Fullerene/Dye Ternary Blends Studied by Transient Absorption Spectroscopy, J. Phys. Chem. C, 2011, 115, 11306–11317 CrossRef CAS.
- L. Ke, J. Min, M. Adam, N. Gasparini, Y. Hou, J. D. Perea, W. Chen, H. Zhang, S. Fladischer, A.-C. Sale, E. Spiecker, R. R. Tykwinski, C. J. Brabec and T. Ameri, A Series of Pyrene-Substituted Silicon Phthalocyanines as Near-IR Sensitizers in Organic Ternary Solar Cells, Adv. Energy Mater., 2016, 6, 1502355 CrossRef.
- B. Lim, J. T. Bloking, A. Ponec, M. D. McGehee and A. Sellinger, Ternary Bulk Heterojunction Solar Cells: Addition of Soluble NIR Dyes for Photocurrent Generation beyond 800 nm, ACS Appl. Mater. Interfaces, 2014, 6, 6905–6913 CrossRef CAS.
- L. Ke, N. Gasparini, J. Min, H. Zhang, M. Adam, S. Rechberger, K. Forberich, C. Zhang, E. Spiecker, R. R. Tykwinski, C. J. Brabec and T. Ameri, Panchromatic ternary/quaternary polymer/fullerene BHJ solar cells based on novel silicon naphthalocyanine and silicon phthalocyanine dye sensitizers, J. Mater. Chem. A, 2017, 5, 2550–2562 RSC.
- B. Lim, G. Y. Margulis, J.-H. Yum, E. L. Unger, B. E. Hardin, M. Grätzel, M. D. McGehee and A. Sellinger, Silicon-Naphthalo/Phthalocyanine-Hybrid Sensitizer for Efficient Red Response in Dye-Sensitized Solar Cells, Org. Lett., 2013, 15, 784–787 CrossRef CAS.
- B. H. Lessard, T. M. Grant, R. White, E. Thibau, Z.-H. Lu and T. P. Bender, The position and frequency of fluorine atoms changes the electron donor/acceptor properties of fluorophenoxy silicon phthalocyanines within organic photovoltaic devices, J. Mater. Chem. A, 2015, 3, 24512–24524 RSC.
- E. Zysman-Colman, S. S. Ghosh, G. Xie, S. Varghese, M. Chowdhury, N. Sharma, D. B. Cordes, A. M. Z. Slawin and I. D. W. Samuel, Solution-Processable Silicon Phthalocyanines in Electroluminescent and Photovoltaic Devices, ACS Appl. Mater. Interfaces, 2016, 8, 9247–9253 CrossRef CAS.
- T. M. Grant, T. Gorisse, O. Dautel, G. Wantz and B. H. Lessard, Multifunctional ternary additive in bulk heterojunction OPV: increased device performance and stability, J. Mater. Chem. A, 2017, 5, 1581–1587 RSC.
- A. J. Pearson, T. Plint, S. T. E. Jones, B. H. Lessard, D. Credgington, T. P. Bender and N. C. Greenham, Silicon phthalocyanines as dopant red emitters for efficient solution processed OLEDs, J. Mater. Chem. C, 2017, 5, 12688–12698 RSC.
- P. Gawrys, T. Marszalek, E. Bartnik, M. Kucinska, J. Ulanski and M. Zagorska, Novel, Low-Cost, Highly Soluble n-Type Semiconductors: Tetraazaanthracene Tetraesters, Org. Lett., 2011, 13, 6090–6093 CrossRef CAS.
- N. T. Boileau, O. A. Melville, B. Mirka, R. Cranston and B. H. Lessard, P and N type copper phthalocyanines as effective semiconductors in organic thin-film transistor based DNA biosensors at elevated temperatures, RSC Adv., 2019, 9, 2133–2142 RSC.
- O. A. Melville, T. M. Grant and B. H. Lessard, Silicon phthalocyanines as N-type semiconductors in organic thin film transistors, J. Mater. Chem. C, 2018, 6, 5482–5488 RSC.
- O. A. Melville, T. M. Grant, K. Lochhead, B. King, R. Ambrose, N. A. Rice, N. T. Boileau, A. J. Peltekoff, M. Tousignant, I. G. Hill and B. H. Lessard, Contact Engineering Using Manganese, Chromium, and Bathocuproine in Group 14 Phthalocyanine Organic Thin-Film Transistors, ACS Appl. Electron. Mater., 2020, 2, 1313–1322 CrossRef CAS.
- O. A. Melville, T. M. Grant, B. Mirka, N. T. Boileau, J. Park and B. H. Lessard, Ambipolarity and Air Stability of Silicon Phthalocyanine Organic Thin-Film Transistors, Adv. Electron. Mater., 2019, 5, 1900087 CrossRef.
- G. Knör and U. Monkowius, Photosensitization and photocatalysis in bioinorganic, bio-organometallic and biomimetic systems, Adv. Inorg. Chem., 2011, 63, 235–289 CrossRef.
- R. Menting, J. T. F. Lau, H. Xu, D. K. P. Ng, B. Röder and E. A. Ermilov, Formation and photoinduced processes of a self-assembled subphthalocyanine–porphyrin–phthalocyanine supramolecular complex, Chem. Commun., 2012, 48, 4597–4599 RSC.
- S. Zhu and D. Wang, Photocatalysis: Basic Principles, Diverse Forms of Implementations and Emerging Scientific Opportunities, Adv. Energy Mater., 2017, 7, 1700841 CrossRef.
- C. Uslan, K. T. Oppelt, L. M. Reith, B. Ş. Sesalan and G. Knör, Characterization of a non-aggregating silicon(IV) phthalocyanine in aqueous solution: toward red-light-driven photocatalysis based on earth-abundant materials, Chem. Commun., 2013, 49, 8108–8110 RSC.
- E. T. Saka, H. Yalazan, Z. Bıyıklıoglu, H. Kantekin and K. Tekintas, Synthesis, aggregation, photocatalytical and electrochemical properties of axially 1-benzylpiperidin-4-oxy units substituted silicon phthalocyanine, J. Mol. Struct., 2020, 1199, 126994 CrossRef CAS.
- J. Huang, Y. Wu, D. Wang, Y. Ma, Z. Yue, Y. Lu, M. Zhang, Z. Zhang and P. Yang, Silicon Phthalocyanine Covalently Functionalized N-Doped Ultrasmall Reduced Graphene Oxide Decorated with Pt Nanoparticles for Hydrogen Evolution from Water, ACS Appl. Mater. Interfaces, 2015, 7, 3732–3741 CrossRef CAS.
- B. Xiao, M. Zhu, X. Li, P. Yang, L. Qiu and C. Lu, A stable and efficient photocatalytic hydrogen evolution system based on covalently linked silicon-phthalocyanine-graphene with surfactant, Int. J. Hydrogen Energy, 2016, 41, 11537–11546 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |