
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The rich physics of A-site-ordered quadruple perovskite manganites AMn7O12
Alexei A.
Belik
 *a,
Roger D.
Johnson
b and
Dmitry D.
Khalyavin
c
*a,
Roger D.
Johnson
b and
Dmitry D.
Khalyavin
c
aInternational Center for Materials Nanoarchitectonics (WPI-MANA), National Institute for Materials Science (NIMS), Namiki 1-1, Tsukuba, Ibaraki 305-0044, Japan. E-mail: Alexei.Belik@nims.go.jp
bDepartment of Physics and Astronomy, University College London, Gower Street, London, WC1E 6BT, UK
cISIS Facility, Rutherford Appleton Laboratory, Chilton, Didcot OX11 0QX, UK
First published on 2nd October 2021
Abstract
Perovskite-structure AMnO3 manganites played an important role in the development of numerous physical concepts such as double exchange, small polarons, electron–phonon coupling, and Jahn–Teller effects, and they host a variety of important properties such as colossal magnetoresistance and spin-induced ferroelectric polarization (multiferroicity). A-site-ordered quadruple perovskite manganites AMn7O12 were discovered shortly after, but at that time their exploration was quite limited. Significant progress in their understanding has been reached in recent years after the wider use of high-pressure synthesis techniques needed to prepare such materials. Here we review this progress, and show that the AMn7O12 compounds host rich physics beyond the canonical AMnO3 materials.
 Alexei A. Belik | Alexei A. Belik is a principal investigator at the National Institute for Materials Science (NIMS), Japan, which he jointed as a permanent member in 2006. He holds a PhD in Inorganic Chemistry from Moscow State University, Russia. He was a postdoctoral researcher at NIMS (2000–2002 and 2004–2006) and Kyoto University, Japan (2002–2004). His current interests are in high-pressure synthesis of new materials (mainly oxide perovskites) and their characterization. |
 Roger D. Johnson | Roger Johnson is a lecturer in condensed matter physics at University College London and a Royal Society University Research Fellow. He works in the area of quantum materials and physical crystallography. Crystal and magnetic structure solutions are obtained by multiple diffraction-based methods, and combined with symmetry analysis by group and representation theory, Roger determines structure–property relationships in inorganic oxides, halides, and hybrid organic–inorganic materials. |
 Dmitry D. Khalyavin | Dmitry D. Khalyavin is an instrument scientist at the ISIS Facility of the Rutherford Appleton Laboratory. He runs the high-resolution cold-neutron diffractometer WISH primarily designed for powder and single crystal diffraction in magnetic and large unit cell systems. The main Dmitry's expertise is magnetic crystallography, structural and magnetic phase transitions as well as diffraction methods in solid state physics. |
1. Introduction
Perovskite-structure manganites have experienced waves of intensive study,1 starting in the late 1940s with the discovery of conductive and ferromagnetic (FM) properties and metal–insulator transitions in mixed-valence R1−xAxMnO3 (R = trivalent cations and A = divalent cations). A theoretical understanding of these properties was quickly developed, including the theory of double exchange2 and Goodenough's one-electron electronic structures.3 Then followed the discovery of colossal magnetoresistance (CMR),4 prompting intensive work on single crystals and thin films, and the proposal of magnetic and electronic phase separation scenarios.5 Perovskite manganites also played an important role in the revival of interest in multiferroic and magnetodielectric materials with RMnO3 (R = Tb and Gd) hosting some of the largest values of spin-induced ferroelectric polarization.6 They were also intensively studied as catalysts,7 solid mixed-conductive electrolytes, electrode materials, thermoelectric ceramics and sensors. This remarkable diversity in physical properties is rooted in considerable structural and chemical flexibility. As such, the perovskite manganites have become canonical systems for the study of structure–property relationships in strongly correlated electron systems.AMn7O12 manganites (with A = Na, divalent and trivalent cations) are a special subfamily of perovskite-type materials, namely the A-site-ordered quadruple perovskites8 with general chemical formula AA′3B4O12. Three quarters of large A cations are replaced by small Mn3+ cations at the A′ sites with an effective square-planar coordination (Fig. 1). By heterovalent substitution of the A cations, which have a large 12-fold coordination (Fig. 1), one can obtain mixed-valence states of Mn at the B sites, i.e. An+Mn3+3(Mn3+1+nMn4+3−n)O12, in close analogy to R1−xAxMnO3. The AMn7O12 compounds are stabilized by very large tilts of the a+a+a+ type in Glazer's notation,9 requiring high-pressure high-temperature treatments for their preparation (except for CaMn7O12, which can be synthesised at ambient pressure). Such an ‘exotic’ synthesis procedure could be the main reason why they did not attract much attention immediately after their discovery despite great interest in the classical mixed-valence R1−xAxMnO3 perovskites. Only with the widespread use of high-pressure facilities has research on AMn7O12 intensified, and significant progress in their understanding has been reached in recent years. In this work, we review the recent progress on AMn7O12 manganites.
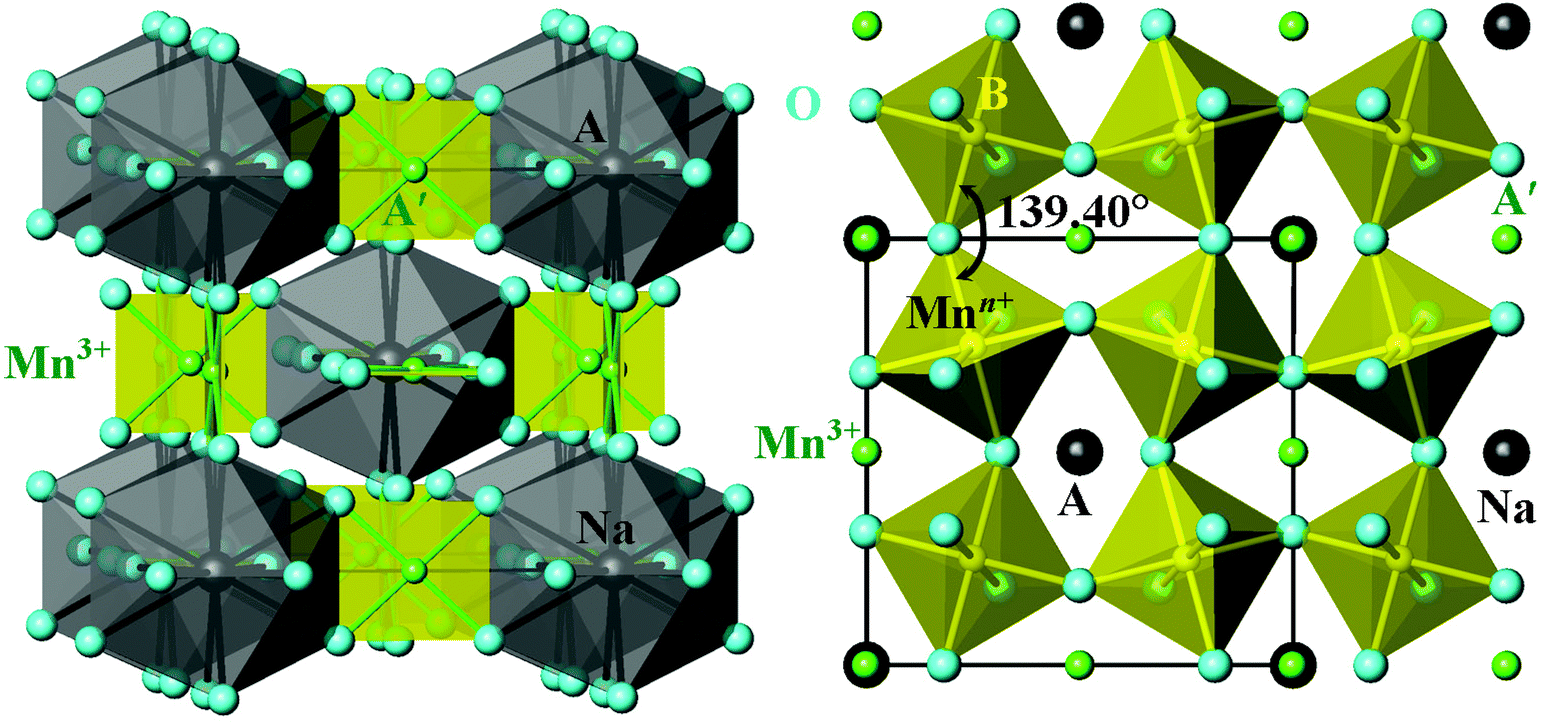 | ||
Fig. 1 Crystal structure of the parent cubic modification of A-site-ordered quadruple perovskites AMn7O12 and AA′3B4O12 in space group Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) exemplified by the structure of NaMn7O12 at RT.12 An Mn–O–Mn (B–O–B) angle is given. A-site polyhedra are shown on the left, and B-site octahedra – on the right. exemplified by the structure of NaMn7O12 at RT.12 An Mn–O–Mn (B–O–B) angle is given. A-site polyhedra are shown on the left, and B-site octahedra – on the right. | ||
Because of the space limitations we cannot cite all references, especially those devoted to CaMn7O12. Therefore, only foundational references are given that are widely cited in latter works.
2. NaMn7O12
NaMn7O12 was the first compound of the AMn7O12 series to be discovered (in 1973) and was found to crystallize in an Im cubic structure at room temperature (RT) (Fig. 1); now identified as the parent aristotype to all AMn7O12.10 A low-temperature (LT) structural phase transition near TCO = 176–190 K (CO stands for charge order) was soon found (Fig. 2), below which a monoclinic I2/m crystal symmetry with 1Mn3+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1Mn4+ B site charge order was suggested (here, the pattern of charge ordering is analogous to the pattern of orbital ordering associated with the I2/m structure of LaMn7O12 (see part 4)).11 Despite clear parallels with the widely studied half-doped R0.5A0.5MnO3 perovskite manganites also containing a 1Mn3+:1Mn4+ mixture at the B sites, NaMn7O12 attracted little attention for nearly 30 years.
:1Mn4+ B site charge order was suggested (here, the pattern of charge ordering is analogous to the pattern of orbital ordering associated with the I2/m structure of LaMn7O12 (see part 4)).11 Despite clear parallels with the widely studied half-doped R0.5A0.5MnO3 perovskite manganites also containing a 1Mn3+:1Mn4+ mixture at the B sites, NaMn7O12 attracted little attention for nearly 30 years.
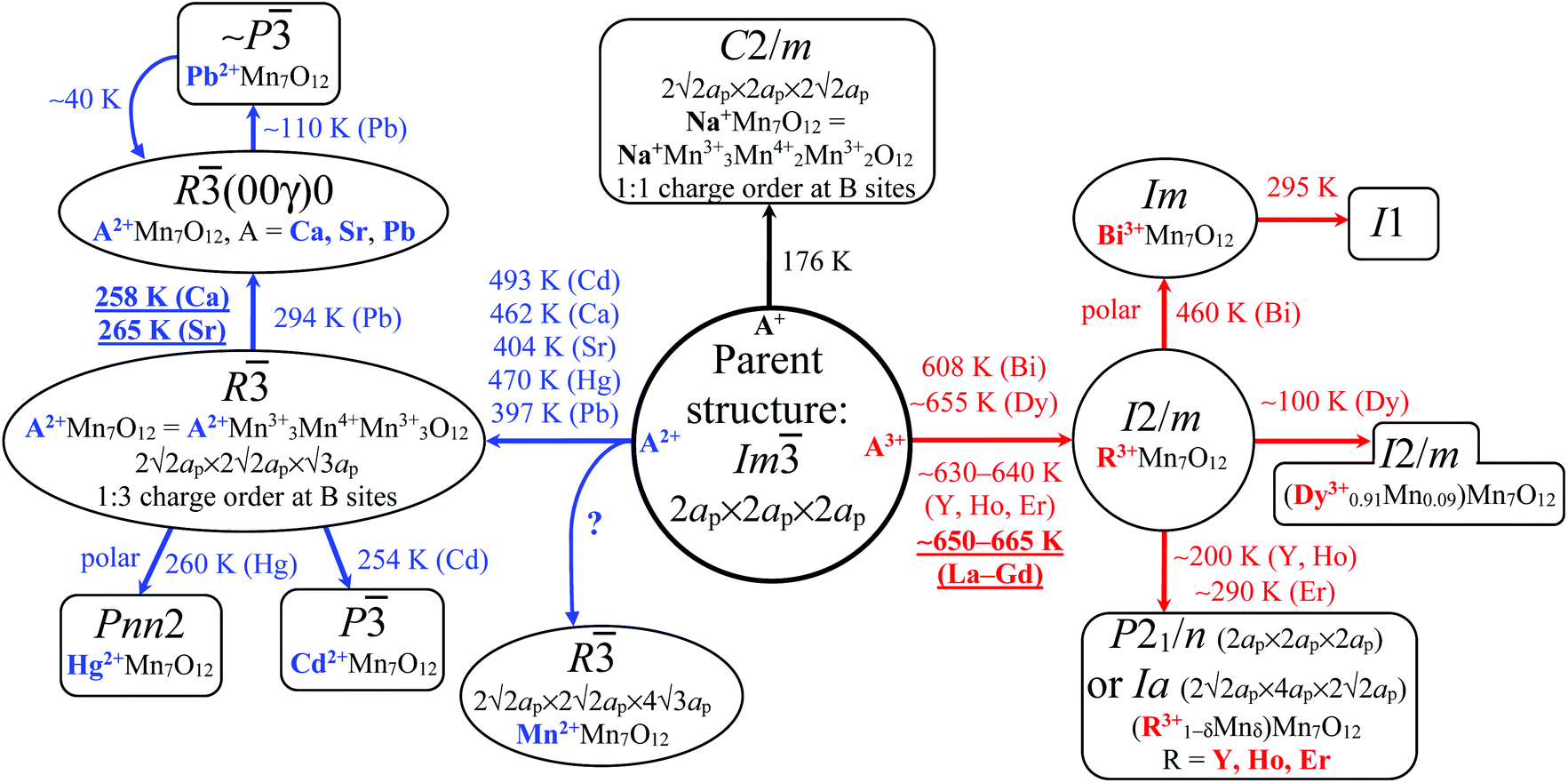 | ||
Fig. 2 A diagram of temperature-driven structural distortions in AMn7O12 manganites from the parent cubic structure. Transition temperatures are reported on the figure when available. All distortions keep the 2ap × 2ap × 2ap cell (with α ≈ β ≈ γ ≈ 90°) except trigonal systems (with α = β = 90° and γ = 120°), C2/m (ref. 13 and 14; with α = γ = 90° and β ≈ 90°) and Ia (ref. 68; with α = γ = 90° and β ≈ 90°). Note that for the R distortion with 2√2ap × 2√2ap × √3ap, the 2ap × 2ap × 2ap cell (with α = β = γ ≈ 90°) can also be constructed (using rhombohedral axes and non-standard setting). For MnMn7O12, a 4ap × 4ap × 4ap cell with F![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) symmetry was also reported at RT.37ap ≈ 3.8 Å is a lattice parameter of the cubic ABO3 perovskite (space group Pmm). For underlined compositions there are no further structural transitions. symmetry was also reported at RT.37ap ≈ 3.8 Å is a lattice parameter of the cubic ABO3 perovskite (space group Pmm). For underlined compositions there are no further structural transitions. | ||
Magnetic studies were performed in 2004, which showed two magnetic transitions at TN1 = 125 K and TN2 = 92 K.12 It was suggested that Mn magnetic moments at the B sites order first at 125 K with an AFM-CE-type structure (AFM stands for antiferromagnetic), which can be described by two propagation vectors; k = (1/2, 0, −1/2) and k = (0, 0, 0). Mn moments at the A′ sites were reported to order at 92 K with an anti-body-centered arrangements of spins with propagation vector k = (0, 1, 0).12 In this study the symmetry of the LT phase was taken to be I2/m (see above) with compressed Jahn–Teller (JT) distortions of the Mn3+ B sites. However, a theoretical study in 2014 pointed out inconsistencies between the reported experimental crystal and magnetic structures.13 The theoretical work13 proposed an alternative LT crystal structure, which was soon confirmed experimentally.14 This correct LT structure has C2/m symmetry, a 2√2ap × 2ap × 2√2ap superstructure with α = γ = 90° and β ≈ 90° (where ap is a lattice parameter of an ideal cubic ABO3 perovskite), full static Mn3+/Mn4+ charge ordering, and elongated JT distortions of the Mn3+ B sites. This LT crystal structure can be described in terms of a commensurate modulation of the parent cubic structure with propagation vector k = (1/2, 0, −1/2). Importantly, this structural modulation is consistent with the magnetic propagation vectors observed below 125 K.
We believe that the magnetic structures of NaMn7O12 need re-investigation considering recent results on the magnetic structures of all other AMn7O12 and RMn7O12 manganites (see parts 3 and 4), where simultaneous ordering of Mn at the B and A′ sites was observed. An additional magnetic anomaly near 34 K was recently reported in NaMn7O12,15 which, in our opinion likely originates in an impurity: MnCO3 has a magnetic transition with weak FM-like properties at this temperature, and an X-ray diffraction pattern reported in (ref. 16) gave evidence for the presence of an MnCO3 impurity (a 100% peak of MnCO3 near 31.5° was observed; note that (ref. 16) incorrectly assigned impurity peaks).
NaMn7O12 is a weak insulator with thermally activated charge transport, and resistivity increases by about one order of magnitude below TCO.12 NaMn7O12 ceramics also showed dielectric anomalies at TCO and additional relatively sharp step-like frequency-dependent dielectric anomalies from 50 K (at f = 1 kHz) to 80 K (at f = 1 MHz).15,17 These steps were originally left without explanation.17 Similar step-like dielectric behavior was observed in RMn7O12 (R = La, Ce, Sm, Eu and Gd) between 17 and 35 K, which was assigned to extrinsic effects (see part 4). No apparent dielectric anomalies were detected in NaMn7O12 at TN1 and TN2.17 Broad and symmetrical pyroelectric current anomalies were observed in NaMn7O12 from 20 K to 40 K, which were assigned to a (spin-induced) ferroelectric transition associated with the 34 K magnetic anomaly.15 Following the results of more recent detailed studies of other AMn7O12 compounds (see part 4), we expect that the dielectric and pyroelectric current anomalies in NaMn7O12 are most probably caused by extrinsic effects.
A number of works were devoted to the single-crystal growth of NaMn7O12.16,18 It was found that small amounts of water and Na excess facilitate the crystal growth. The formation of good samples was observed in the pressure range of 2–6 GPa and temperature range of 670–1120 K with the best conditions at 6 GPa and 1100 K. Single-crystal X-ray diffraction studies gave the real chemical composition of the studied crystal as (Na0.95Mn0.05)Mn7O12.16 No structural phase transitions were found up to 40 GPa (at RT), but some anomalies in resistivity near 18 GPa gave evidence for charge transfer from the B site to the A′ site.19
3. AMn7O12 (A = Mn, Cd, Ca, Sr, Hg and Pb)
Perovskites with A = Ca, Cd and Sr were first reported by Bochu et al.20 and synthesized under high-pressure and high-temperature (HT) conditions. Later, Horowitz and Longo21 prepared powder samples of CaMn7O12 at ambient pressure and so far this is the only member whose synthesis does not require extreme conditions. The authors also pointed out the mixed-valence character of this oxide. The charge degree of freedom has been found to be mainly localized on the B-site Mn ions, resulting in 3:1 charge order between Mn3+ and Mn4+ in the RT trigonal structure with R symmetry and apically compressed Mn3+O6 octahedra.22 The latter implies that, unlike the vast majority of known JT systems with two longer and four shorter Mn–O distances (JT elongation), the Mn3+O6 octahedra in CaMn7O12 are characterized by four longer and two shorter Mn–O distances (JT contraction, Fig. 3a). Hence, it was suggested that this perovskite represented a new type of JT distortion of Mn3+ cations in oxides, with x2–y2-type orbital ordering.22 This is a very unusual situation because the occupation of x2–y2 orbitals of Mn3+ with locally compressed octahedra has been shown by Khomskii and van den Brink to be unstable due to anharmonic effects which always stabilize elongated octahedra.23 The transition to the trigonal charge-ordered phase takes place well above RT and is associated with phase coexistence in the temperature range of 409–448 K where the LT trigonal phase coexists with the HT cubic one.24 The LT structural studies25–28 revealed another transition at TOO = 260 K (OO stands for orbital order), where the crystal structure of CaMn7O12 develops an incommensurate structural modulation with propagation vector kS = (0, 0, γ) (γ = 0.9215 at T = 113 K,29 in the following discussion we will use the equivalent propagation vector (0, 0, 3) − kS = (0, 0, 2.0785) to emphasize its relation with the modulation of the magnetic order). Initially, the modulation was assigned to charge ordering,25 however, subsequent quantitative structure refinement of synchrotron X-ray and neutron powder diffraction data in the R(0,0,γ)0 superspace group27 did not support this interpretation and instead revealed a complex modulation of Mn–O bond distances. The model was confirmed by Perks et al.30 using X-ray single-crystal data, and the modulation was interpreted as an incommensurate orbital ordering (orbital density wave) localised on the B-site Mn3+ cations. The density wave implies a periodic change of the 3x2–r2 and 3y2–r2 orbital occupancies passing through the x2–y2 state only at the nodal points (Fig. 3b). In other words, in the modulated structure most of the octahedra are locally elongated along one of two axes that alternate periodically within the crystal, giving rise to the apical compression of octahedra on average.
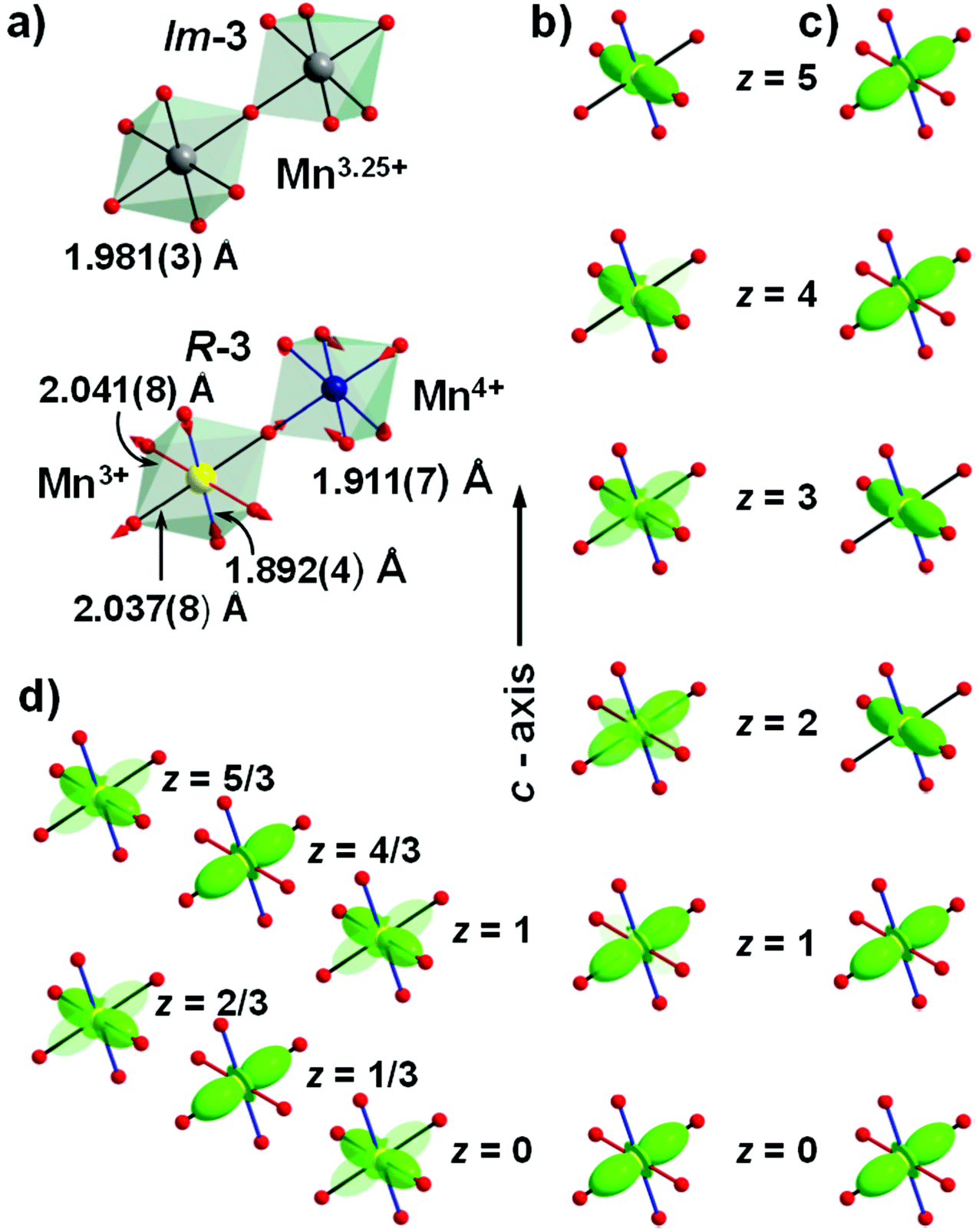 | ||
| Fig. 3 (a) Compression of Mn3+O6 octahedra in CaMn7O12 at the phase transition from the disordered cubic Im to the charge-ordered trigonal structure (Mn–O bond distances are taken from ref. 24 and 22 for the cubic and trigonal structures, respectively). Orbital density wave in CaMn7O12 (b), ξ-Mn2O3 (c) and CdMn7O12 (d), illustrating the change of orbital state of Mn3+ upon propagation along the trigonal axis. In the latter case, the commensurate orbital density wave splits the Mn position into two sites with fully and partially polarised states. | ||
The systematic structural study by Belik et al.29 revealed a similar modulation in other members of the family with A = Sr, Cd and Pb, taking place at TOO = 265 K, 254 K and 294 K, respectively (Fig. 2). The propagation vector for the Sr-perovskite is also incommensurate with the magnitude very close to the Ca-counterpart; kS = (0, 0, 2.0765) at T = 113 K. The modulation was found to be commensurate, kS = (0, 0, 2), in the case of CdMn7O12, giving rise to the primitive unit cell with P symmetry. The commensurate nature of the orbital density wave was interpreted as an activation of a third-power lock-in term allowed in the Landau free-energy decomposition when the γ-component of the propagation vector takes the commensurate value.31 The centrosymmetric space group fixes the global phase of the commensurate orbital density wave and imposes a stacking of fully and partially polarized orbital states (Fig. 3d).
Note that some samples of CdMn7O12 showed phase separation below about 60 K,31,32 characterised by a mixture of P and I2/m phases and large hysteresis in properties.33 This coexistence likely reflects small variations in the real chemical compositions of such samples, and the appearance of the I2/m phase needs detailed investigations as it usually appears in R3+Mn7O12 (Fig. 2).
The situation with PbMn7O12 has been found to be more complicated.34 Just below the transition at TOO = 294 K the modulation is incommensurate with γ ∼2.08. At TOO2 = 110 K another structural transition takes place where the propagation vector suddenly drops down to a quasicommensurate value kS = (0, 0, 2.0060(6)). The quasicommensurate phase is stable in the temperature range of 40–110 K, and below TOO3 = 40 K the propagation vector jumps back to the incommensurate value kS = (0, 0, 2.060(6)). Both the LT structural transitions are strongly first order with large thermal hysteresis. The orbital density wave in the quasicommensurate phase has been found to be substantially suppressed in comparison with the incommensurate phases, and this behaviour was attributed to a competition between the Pb2+ lone electron pair and Mn3+ JT instabilities. This makes this perovskite particularly attractive as a new playground for various charge doping strategies by analogy with the BiMn7O12 system (see section 5, below), where a light hole doping has been shown to stabilize exotic electric dipole and orbital textures due to a fine tuning of the relative strength of these two electronic instabilities.35 Similar to CaMn7O12, the perovskites with A = Sr, Cd and Pb exhibit a HT transition to the cubic Im structure at TCO = 404 K, 493 K and 397 K, respectively, as determined by differential scanning calorimetry.33 We note that in other studies, TCO for PbMn7O12 was reported to be 380 K,36 and for CaMn7O12 prepared at high pressure – to be 462 K,33 suggesting that TCO, and also TOO, are sensitive to the sample quality.
Another member of the A2+Mn7O12 family is the ξ-polymorph of Mn2O3. As has been shown by Ovsyannikov et al.,37 at high temperature (above 1000 K) and at high-pressure (above 18 GPa), Mn2O3 transforms to a perovskite modification which can be quenched into a metastable phase at ambient conditions. This metastable phase exhibits a unique charge disproportionation phenomenon stabilizing the quadruple perovskite structure Mn2+Mn3+3Mn3.25+4O12 with an additional 3Mn3+:1Mn4+ charge ordering and commensurate orbital density wave with kS = (0, 0, 9/4), localized in the B-site perovskite position. The charge ordered modulated structure possesses R symmetry with a 2√2ap × 2√2ap × 4√3ap supercell defined with respect to the pseudocubic simple perovskite structure which is prototype for all families of perovskite materials. Here, the orbital density wave represents a sequence of 3x2–r2 and 3y2–r2 orbital states ordered in a ++−− fashion (where + and − indicate distinct states) upon propagation along the trigonal axis (Fig. 3c). The commensurate nature of the orbital density wave and the associated structural modulation has been explained by a coupling of the orbital ordering to an independent structural distortion, which improves the poor bonding conditions of Mn2+ in the A-site perovskite position.38 The trigonal structure has been reported to be stable at RT,37 but the temperatures at which the B-site charge and orbital ordering onset are yet to be determined.
One more perovskite with a divalent A-site cation, HgMn7O12, has been recently reported by Chen et al.39 Similar to other members of the family, it exhibits a cubic to trigonal structural transition due to the 3Mn3+:1Mn4+ charge ordering in the B-site manganese position. The transition is first order and takes place via phase coexistence in the temperature range of 470–490 K. However, instead of the modulated trigonal ground state, the LT structure of HgMn7O12 to a good approximation is commensurate orthorhombic with the polar Pnn2 symmetry. The transition to the orthorhombic phase takes place at 240–260 K and is associated with a further charge disproportionation where 1/3 of Mn3+ in the A′ position become Mn2+, giving rise to 1Mn2+:2Mn3+ charge ordering in the A′ square planar position as well as 1Mn3+:1Mn4+ charge and orbital ordering in the B-site position (Fig. 4). The polar symmetry is a result of the common action of two irreducible order parameters. One of them is the charge/orbital order and another serves to improve the coordination environment of the A′-site Mn2+ cations by making the environment more regular through shortening the second-nearest-neighbour distances. The charge transfer between the A′ and B sites has been suggested as an alternative mechanism to the orbital density wave formation to release the instability associated with the compressed octahedra in the 3:1 charge ordered trigonal phase. The B-site Mn3+ in this phase was assumed to exhibit orbital disorder between the 3x2–r2/3y2–r2 states. Whether this is the case, or the x2–y2 orbital state is imposed by the bilinear coupling to the atomic displacement mode accompanying the 3:1 charge order40 is an interesting open question.
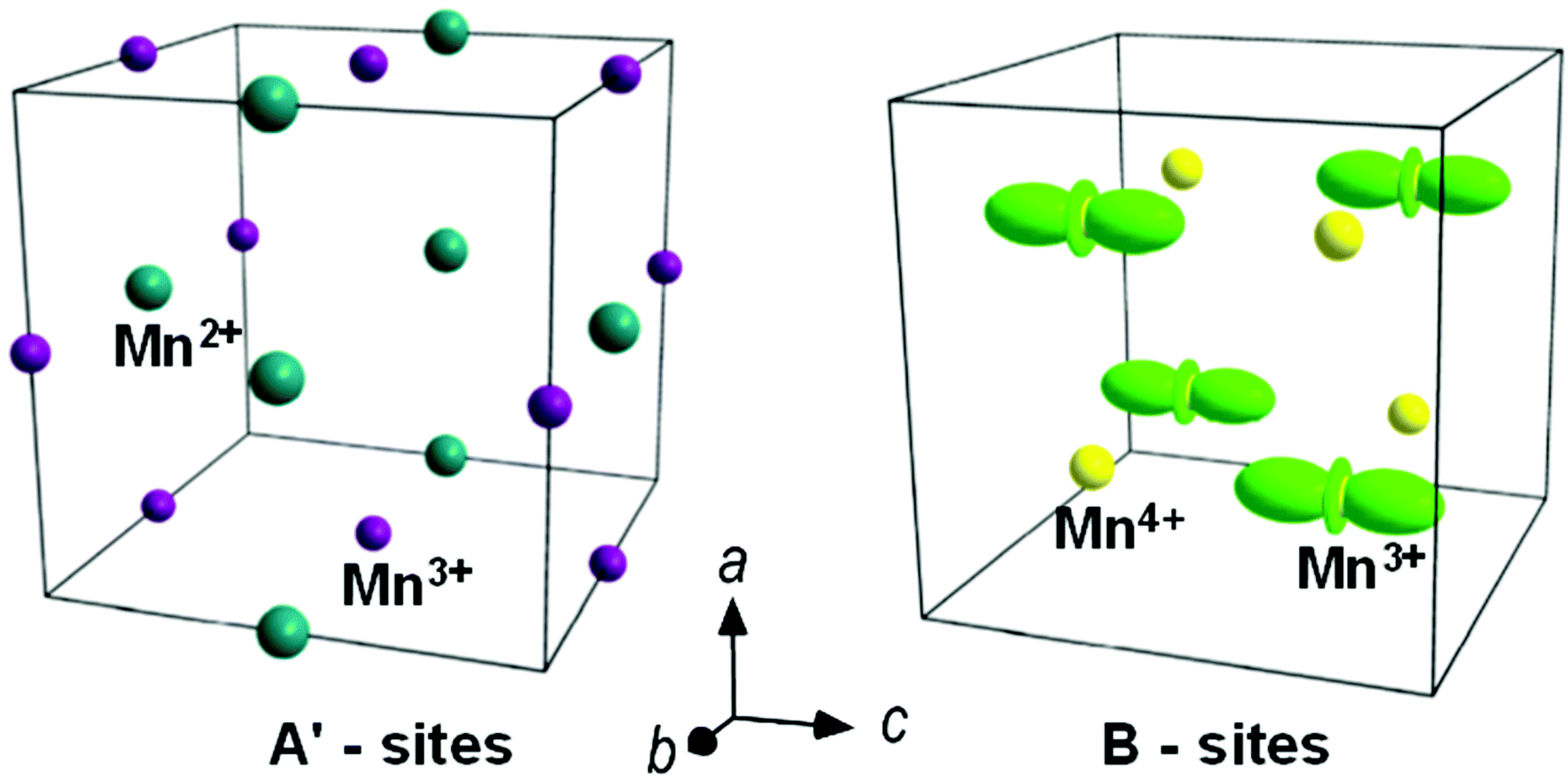 | ||
| Fig. 4 Charge (left) and orbital (right) ordering in the orthorhombic Pnn2 phase of HgMn7O12 perovskite.39 | ||
The perovskites of the A2+Mn7O12 family attracted great attention after the discovery of prominent multiferroic properties in CaMn7O12.41,42 Long-range magnetic ordering in this material takes place at TN1 ∼90 K with the incommensurate propagation vector, k0, locked to the structural modulation such that k0 = kS/2.26–28 Below TN2 ∼48 K, the magnetic subsystem delocks from the structural modulation giving rise to a complex multi-k magnetic ground state.43 The initially reported giant spin-driven polarization was found to onset at TN1. Later studies44 confirmed the multiferroic properties of CaMn7O12 but the electrical polarization was found only below the second magnetic transition at TN2 with substantially lower magnitude. The corresponding ground state magnetic structure was quantitatively determined using high-resolution neutron powder and single crystal diffraction data43 and was found to represent an unprecedented example of helical order with modulated spin chirality (Fig. 5). Unlike usual helical structures with a constant magnetic phase between spins related by a lattice translation along the propagation vector, the magnetic phase of the spins in the ground state of CaMn7O12 is incommensurately modulated with the periodicity of the orbital density wave. The magnetic structure decomposes into a set of magnetic order parameters with distinct propagation vectors, kn±, along the trigonal axis. A symmetry based phenomenological approach43 revealed that this exotic ground state is a result of coupling between the primary magnetic order, stabilized by competing exchange interactions and referred to as the fundamental component k0 = (0, 0, 1.12354(8)), and the orbital density wave kS = (0, 0, 2.0775(1)) at T = 1.5 K (magneto-orbital coupling). By symmetry, the fundamental helical order alone cannot lock to the structural modulation, and hence the coupling introduces additional magnetic components with kn± = k0 ± nkS propagation vectors. Experimentally, the components up to n = 2 have been confirmed.43 Microscopically, the magneto-orbital coupling implies that the orbital density wave modulates the competing exchange interactions and local anisotropies resulting in faster and slower rotations of spins upon propagation along the trigonal axis.
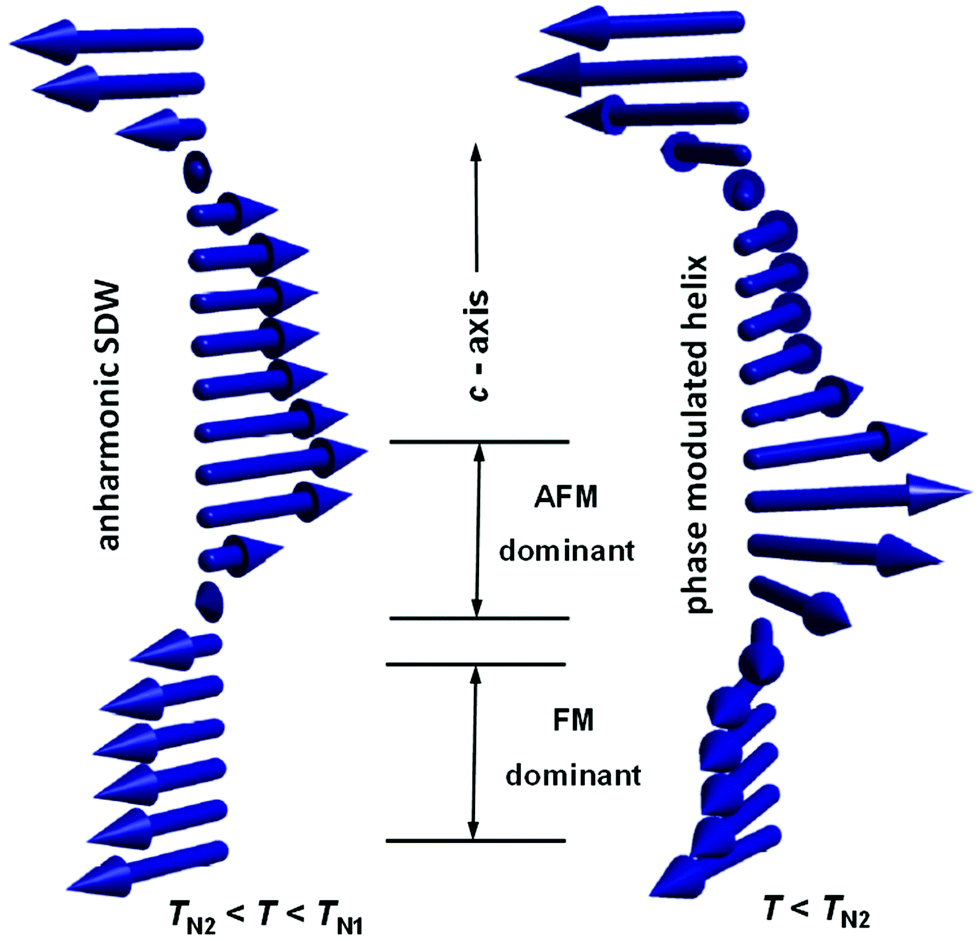 | ||
| Fig. 5 Schematic representation of the anharmonic spin density wave (SDW) and the phase modulated helix, corresponding to the high-temperature lock-in phase and the ground state of A2+Mn7O12 manganites.43 | ||
The spin-driven electric polarization in CaMn7O12 has been explained by the ferroaxial mechanism41 first proposed to explain multiferroic properties of Cu3Nb2O8.45 The mechanism describes a bi-linear coupling between spin chirality and polarization in crystals with axial distortions, known as ferroaxiality. The latter is a ferroic property like ferroelectricity and ferromagnetism, characterized by the presence of a well-defined rotation between different elements of the crystal structure (octahedral tilting in the given case). The trigonal crystal structure of A2+Mn7O12 with point group belongs to the ferroaxial class. The relevant microscopic interaction giving rise to the macroscopic polarization is antisymmetric Dzyaloshinskii–Moriya (DM) exchange. This exchange can gain energy by distorting the crystal structure and/or the electronic density in the presence of non collinear spins.46 A discussion of the ferroaxiality and the polar distortions optimizing the antisymmetric exchange in CaMn7O12 can be found in the work by Perks et al.30
Unlike the helical order, a spin density wave can be locked to the structural modulation associated with the orbital density wave.43 This type of ordering therefore is the prime candidate for the magnetic structure of CaMn7O12 in the temperature range of TN2 < T < TN1, where the magnetic and structural modulations are commensurate with each other, holding the k0 = kS/2 ratio (Fig. 5). The magneto-orbital coupling is expected to yield additional magnetic components making the spin density wave anharmonic, which was confirmed experimentally through the observation of higher order satellites (in particular the 3k0 propagation vector).27,43 It has to be pointed out that experimentally it is challenging to distinguish the spin-density wave with the spins confined within the (ab)-plane of the trigonal structure and the helical order. However, the commensurate relation between the magnetic and structural modulations and the absence of electrical polarization in the lock-in phase44 strongly support the existence of a spin density wave above TN2. Another argument in favour of this scenario comes from the precise magnetic structure determination in the ξ-polymorph of Mn2O3.38,47 Similar to CaMn7O12, this perovskite also exhibits two magnetic transitions at TN1 = 100 K and TN2 = 50 K, and mutiferroic properties below TN2. The HT magnetic phase has been found to be commensurate and locked to the structural modulation. The spins in this phase are polarized along the c-axis forming an anharmonic longitudinal spin density wave. The magnetic structure is only partially ordered and contains 1/4 of the A-site Mn2+ cations with zero ordered moment. Below TN2, the magnetic propagation vector becomes incommensurate (k0 = (0, 0, 1.2439(3)) at T = 1.5 K) and the magnetic structure turns into a phase modulated constant moment cycloid with only a small admixture of a helical component. Thus, the magnetic behaviour of CaMn7O12 and ξ-Mn2O3 is very similar apart from the different magnetic anisotropy which is an easy-plane type in the former case and easy axis (along the trigonal axis) in the latter. The longitudinal spin density wave can be unambiguously distinguished from any other solution and therefore the sequence paramagnetic – spin density wave – constant moment magnetic structure with rotating spins is a well-established experimental observation for this perovskite.
Magnetic structures of the perovskites with A = Sr and Cd are very similar to the Ca-counterparts.31 For these materials TN1 is at 87 K and 88 K, respectively and the LT TN2 transition occurs at 63 K for A = Sr and 33 K for A = Cd. The magnetic propagation vector k0 for the former is (0, 0, 1.15427(6)) and for the latter (0, 0, 1.0682(1)) at T = 1.5 K, indicating that the periodicity of the fundamental helical component depends on the radius of the A-site cation. The PbMn7O12 perovskite exhibits an additional intermediate magnetic phase31,48 related to the fact that the magnetic ordering onsets within the quasicommensurate structural phase with a substantially suppressed orbital density wave. Like in other manganites of this series, the magnetic propagation vector of PbMn7O12 is locked to the structural modulation just below TN1 = 83 K. The lock-in phase is stable only in a narrow temperature range and at TN2 = 77 K the magnetic propagation vector delocks from the structural modulation giving rise to the incommensurate helical order with only weakly modulated spin chirality. In an early study,31 this weakly modulated intermediate magnetic phase was interpreted as an unmodulated single-k helix, where the magneto-orbital coupling is fully suppressed. However, the latest work34 confirmed the presence of very weak magnetic components kn± indicating a finite magneto-orbital coupling. At TOO3 = 40 K, the re-entrant transition to the incommensurate structural phase takes place, re-establishing the orbital polarization which in turn triggers a strong magneto-orbital coupling in the ground state helical structure of PbMn7O12 with the periodicity of the fundamental magnetic component being k0 = (0, 0, 1.18410(8)) at 1.5 K.
Thus, the magnetic properties of the divalent A-site quadruple perovskites reveal a universal interplay between magnetic and orbital degrees of freedom, leading to the concept of magneto-orbital coupling. In the HT ordered state the periodicities of the magnetic and orbital subsystems lock together via commensurate magneto-orbital coupling. On cooling, these periodicities delock, allowing the system to evolve towards a multi-k magnetic ground state in which incommensurate magneto-orbital coupling gives rise to a modulation of the spin helicity. The stability of the locked and delocked magnetic phases is controlled by the balance between the entropy term (promoting a partially ordered state at higher temperatures), lock-in term, and magnetic exchange interaction requiring a propagation vector different from the orbital modulation.31,43 While TN1 remains approximately constant across the series (apart from ξ-Mn2O3 which has the additional A-site magnetic sublattice and therefore a noticeably higher TN1), the periodicity of the ground state magnetic structure and the LT transition TN2 both monotonically increase with increasing A2+ ionic radius. The only member of the family whose magnetic properties might differ significantly from that described above is HgMn7O12. Although, the magnetic structure is yet to be reported for this perovskite, there is no reason to believe that it should obey the common scenario where the lock-in phase is followed by the delocked phase-modulated ground state, imposed by the magneto-orbital coupling. Its crystal structure does not support the orbital density wave, adopting instead the alternative mechanism associated with the inter-site charge transfer that results in distinct charge and orbital patterns. Interplay between these electronic degrees of freedom with magnetism is therefore a very exciting topic for future studies.
Finally, let us mention the very recent X-ray resonant elastic scattering study of CaMn7O12,49 where using polarization analysis it was found that the satellites with k0 propagation vector are not purely magnetic. The authors conclude that there is one more phase transition around 30 K, where the periodicity of the magnetic ground state locks to the second harmonic of the orbital modulation (a second lock-in phase). It is quite hard to understand this conclusion based on symmetry grounds because this would require a free-energy term linear in the magnetic order parameter components and quadratic in the orbital density wave which is forbidden by time reversal symmetry. As discussed above, the ground state of CaMn7O12 is the multi-k modulated helix which consists of a set of magnetic order parameters creating additional structural modulations through magneto-elastic coupling. The periodicities of these structural modulations are twice bigger than the periodicities of the corresponding magnetic components and they have distinct symmetries from the orbital density wave. For instance, if one takes the second harmonic of the k1+ = k0 + kS (k0 and kS are found to be (0, 0, 1.12) and (0, 0, 0.94), respectively for the studied crystal), it coincides with k0 and can be the origin of the observed charge-like contribution to the corresponding satellites. Since k1+ is not an independent order parameter, its second harmonic can also be expressed through a higher order coupling term using the k0 and kS order parameters. In particular, the relevant term will be linear in respect of the magneto-elastic structural modulation and quadratic in respect of both k0 and kS, whose explicit form can be worked out using the information provided in (ref. 43). The presence of this structural modulation is a natural consequence of the magneto-elastic coupling and does not require a phase transition. Thus, the interesting results reported by Gautam et al.49 need a further detailed study, with a particular attention to the observed splitting of the orbital modulation below TN2.
As CaMn7O12 is often prepared at ambient pressure in air it may contain different impurities. Magnetic anomalies from impurities are sometimes erroneously attributed to additional magnetic transitions of the main CaMn7O12 phase, for example, a FM-like anomaly near 43 K from an Mn3O4 impurity.
4. RMn7O12 (R = Y, La, Ce, Pr, Nd and Sm–Er)
Quadruple perovskite manganites with trivalent rare earth cations at the A site (R3+Mn7O12) with R3+ = La,7,20,50–61 Ce,59,62 Pr,63 Nd,20,59 Sm,59,64 Eu,59,64 Gd,64 Tb,64 Dy,65 and Y66–68 (in order of decreasing ionic radii) have been reported in the literature. The authors have also synthesised compounds with R3+ = Ho and Er, but the results are yet to be published. Compounds with R3+ = Tm–Lu appear to lie beyond the stability range of the AMn7O12 structural framework (when prepared at 6–8 GPa) owing to their small ionic radii. Indeed, emergent dipolar-glass-like states have been observed in DyMn7O12 and YMn7O12,65,67 understood to arise from the underbonding of R3+ cations as the boundary of structural stability is approached (discussed in detail below).Given the trivalency of the rare-earth cations, all manganese ions adopt a formal 3+ oxidation state in the stoichiometric charge balanced system. RMn7O12 can be prepared in the stoichiometric composition for R3+ = La–Gd. However, they take (R1−xMnx)Mn7O12 compositions for R3+ = Tb–Er, determined through (1) the synthesis of single-phase samples of corresponding compositions, (2) the neutron powder diffraction structural analysis of (Dy0.91Mn0.09)Mn7O12 (ref. 65) and (3) the single-crystal X-ray diffraction structural analysis of (Y1−xMnx)Mn7O12 (N.B. in this case, the structural analysis cannot distinguish between the Y1−yMn7O12 and (Y1−xMnx)Mn7O12 models).68 Therefore, manganese at the A sites could then take the 2+ oxidation state in (R1−xMnx)Mn7O12. For simplicity, we will often use the RMn7O12 chemical formula.
B site ions are predominantly Mn3+ and hence JT active. LaMnO3-type69 B site orbital order has been observed at RT in all RMn7O12 compounds, which imposes I2/m space group symmetry (N.B. PrMn7O12 was found to adopt both I2/m (two variations with different degrees of monoclinic distortions) and R structures at RT, with the phase fractions controllable by synthesis conditions).63 The I2/m orbital order can be understood in terms of a long-range checkerboard alternation of JT axes in the monoclinic ac plane, which are coaligned on stacking along the b axis (Fig. 6). Melting of the orbital order has been reported for all RMn7O12 except R = Pr, Nd, and Dy, as evidenced by a first order phase transition to the Im aristotype occurring in the range 630–665 K at ambient pressure,51,62,64,67 and under hydrostatic pressures greater than 34 GPa at RT (in LaMn7O12).61 This Im phase appears to be ubiquitous to the AMn7O12 materials (Fig. 2), and we anticipate its discovery in the R = Pr, Nd, and Dy compounds should HT diffraction measurements be made. The orbitally ordered I2/m crystal structure has been found to persist down to the lowest measured temperatures with the exception of YMn7O12, in which a P21/n ground state was found to accompany the disordered yttrium dipolar glass.67 Ferroelectric ground states have recently been proposed for compounds with R = La and Y, which would imply non-centrosymmetric structures at LT.60,68
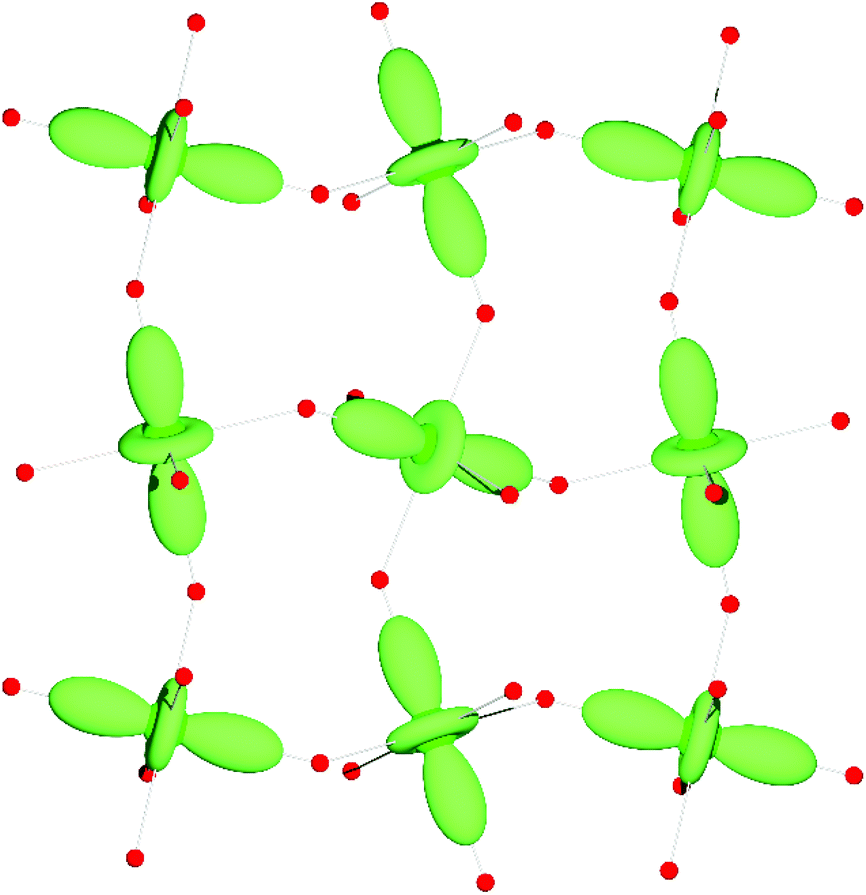 | ||
| Fig. 6 LaMnO3-type orbital order spanning the B site Mn3+ ions in any given ac-plane in monoclinic R3+Mn7O12. The orbital order is co-aligned on moving along b (perpendicular to this figure) from one layer to the next. | ||
The RMn7O12 family of quadruple perovskite manganites have been considered in the context of functional material properties such as catalysis,7,56,57 however most studies have focused on the microscopic cross-coupling of dipolar, orbital and magnetic degrees of freedom. Hence, it is this latter topic that we will focus on in the remainder of this section.
CeMn7O12, SmMn7O12 and EuMn7O12 undergo a single magnetic phase transition at TN1 = 80, 87 and 87 K, respectively.59,62,64 Both A′ and B site Mn3+ magnetic moments adopt long range order below TN1 with propagation vector k = (0, 0, 0). The A′-sites form a collinear ferrimagnetic structure, with magnetic moments lying in the ac plane and rotated away from the c-axis towards −a by 38.7(3)°, 34.7(4)°, and 32.1(5)° for R = Ce, Sm and Eu, respectively.59 One in every three A′ site moments is uncompensated, giving a net ferrimagnetic moment as observed in magnetometry measurements. The B site Mn3+ magnetic moments form a C-type AFM structure (following the notation of Bertaut),70 in which nearest neighbour moments are antiferromagnetically aligned in the ac plane, and coupled ferromagnetically along the b axis. B site spin canting is allowed by symmetry but could not be detected in neutron powder diffraction experiments. Hence, a minimal model with B site moments collinear with the A′ site moments was proposed (Fig. 7).59 Importantly, the magnetic structures on both sublattices transform by the same irreducible representation (Γ2+, defined w.r.t. the I2/m paramagnetic parent), hence the two are directly coupled and can develop together below a single phase transition. Based upon the Goodenough–Kanamori–Anderson rules3 taken in the limit of 180° Mn–O–Mn bonds, the LaMnO3 type orbital order should stabilise an A-type magnetic structure on the B sites (FM layers stacked AFM). This magnetic structure was indeed observed in LaMnO3,71 but it is the exact opposite of what is found in the above RMn7O12 compounds. The octahedral tilting in RMn7O12 is large compared to that of the RMnO3 perovskites, leading to a more significant departure from 180° Mn–O–Mn bonds that may weaken or even change the sign of the superexchange interaction. In addition, the RMn7O12 compounds host strong d–d superexchange between A′ and B sublattices. Such A′–B exchange may compete with B–B exchange and play a key role in stabilising the C-type structure. We note that a B site A-type structure transforms by a different k = (0, 1, 0) irreducible representation to the observed k = (0, 0, 0) C-type structure, hence the energy gain through A′–B interactions would average to zero in this case.
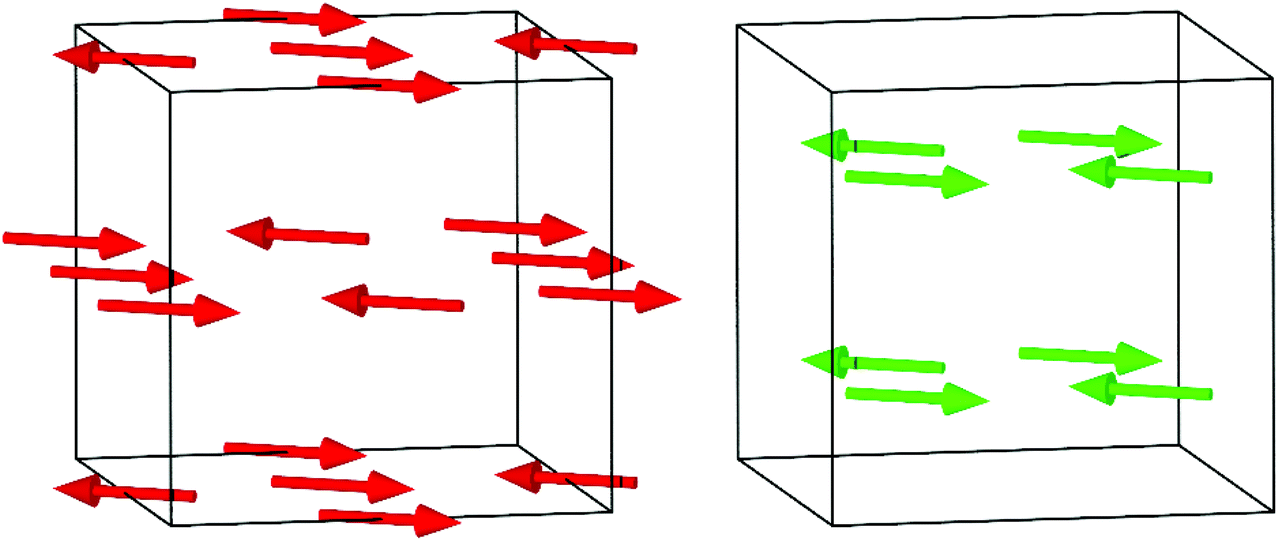 | ||
| Fig. 7 The A′ site ferrimagnetic structure (red, left) and B site C-type antiferromagnetic structure (green, right) observed for R = La, Ce, Nd, Sm, Eu, Dy and Y, and likely common to all R3+Mn7O12 compounds below TN1.59,65,67 The cubes represent the unit cell. The monoclinic b-axis points up the page, and the c-axis to the right. | ||
Magnetic susceptibility and heat capacity measurements of GdMn7O12 and TbMn7O12 show anomalous behaviour at TN1 = 86 and 82 K, respectively, qualitatively similar to the behaviour of the R = Ce, Sm and Eu samples; hence consistent with a phase transition to the same ferrimagnetic ground state.64 However, the microscopic nature of the long range magnetic order is yet to be confirmed, for example, by neutron powder diffraction experiments. Measurements of the isothermal magnetisation at 5 K indicate a sizeable uncompensated ferrimagnetic moment in both GdMn7O12 and TbMn7O12, which is enhanced compared to the R = Ce, Sm and Eu compounds likely due to the large moments carried by Gd and Tb. For all five samples discussed above, no direct evidence has been provided for long-range magnetic order of the rare-earth ions. However, a Schottky-like anomaly was observed in the specific heat of GdMn7O12 below ∼10 K that was largely suppressed by a 9 T magnetic field.64 This result may point towards Gd ions playing a significant role in the ground state. In addition, magnetic and specific heat measurements gave evidence that a second LT magnetic transition could emerge under magnetic fields in R = Sm and Eu.64
The dielectric constant of the R = Ce, Sm, Eu, Gd and Tb compounds has been reported,62,64 measured as a function of both temperature and excitation frequency. In all cases a small kink-like anomaly was observed at TN1, with the LT behaviour (frequency-dependent sharp steps between 18 and 35 K) otherwise dominated by extrinsic relaxation processes typical of these and related material systems. As such, there exists no evidence for dipolar or ferroelectric properties in these five compounds. N.B. Sharp steps in dielectric constant near 30 K were also reported in LaMn7O12, discussed below.60
PrMn7O12 is the only member of the RMn7O12 family that has two polymorphs at RT.63 The I2/m phase supports the same orbital order and ferrimagnetic magnetization below a single phase transition (TN1 = 70 K) as is characteristic of the compounds with R = Ce, Sm, Eu, Gd and Tb. Again, one might therefore speculate that the I2/m polymorph of PrMn7O12 adopts the same ferrimagnetic structure below TN1, but this is yet to be confirmed. The R polymorph was reported to have the same crystal structure as CaMn7O12, implying a 3:1 splitting of B sites. In CaMn7O12 this splitting can be understood in terms of a 3:1 charge ordering of Mn3+ and Mn4+ ions from an average valence of Mn3.25+ imposed by the divalent Ca cation. It is therefore somewhat surprising to find this polymorph in the RMn7O12 family (Fig. 2). Nonetheless, this R phase displays altogether different magnetic behaviour with TN1 = 44 K,63 and it has been proposed that in the R symmetry the B-site Mn3+ ions split into three high spin and one low spin per formula unit, with the low spin ion at the undistorted octahedral site occupied by Mn4+ in CaMn7O12.
LaMn7O12 is arguably the most studied of all RMn7O12 compounds. Unlike the above, LaMn7O12 displays two magnetic phase transitions at TN1 = 79.5 and TN2 = 22.5 K.52 The HT transition was first assigned to the onset of the same B site C-type AFM structure described above, but independent of A′ site Mn order which was instead assigned to the LT transition – an observation akin to the behaviour of NaMn7O12.12 The proposed LT A′ site order had propagation vector k = (0, 1, 0), with FM layers in the ac plane, stacked AFM along b. Owing to the different symmetries, the A′ sites could naturally order independently of the B sites. Both A′ and B sublattice magnetic structures refined against neutron powder diffraction experiments52 did not have a net ferrimagnetic moment – a result inconsistent with magnetometry measurements showing a net magnetisation similar to that measured for R = Ce, Pr, Sm, Eu, Gd and Tb. It was therefore proposed that the net moment in LaMn7O12 arose through an exceptionally large canting of the B site magnetic structure induced by the Dzyaloshinskii–Moriya interaction.52 The application of the same model to YMn7O12 led to the suggestion that the A′ Mn sublattice remains disordered as only one magnetic transition was found.66 More recently, analysis of high resolution neutron powder diffraction data has shown that both A′ and B Mn moments order together at the HT transition with the same magnetic structure found for R = Ce, Sm and Eu, hence providing a more natural explanation for the sizeable ferrimagnetic moment.59 We note that these two different scenarios manifest in remarkably subtle changes in the neutron powder diffraction pattern.59 The latter scenario requires a more complex explanation for the LT transition, and it was proposed that LaMn7O12 enters into a ground state in which B site moments adopt a canted magnetic structure characterised by an admixture of commensurate C-type and A-type modes.59 Intriguingly, such a ground state points towards an underlying instability towards the A-type structure expected in the presence of LaMnO3-type orbital order – perhaps acting in competition A′–B superexchange.
NdMn7O12 was found to undergo three magnetic phase transitions, one at TN1 = 85 K, and a further two at low temperature (TN2 = 12 and TN3 = 8.5 K). Both A′ and B sublattices were found to order below TN1 with ferrimagnetic and C-type magnetic structures, respectively – in common with all of the above RMn7O12 compounds.59 It was reported that below the second transition there developed an incommensurate modulation of the B-site magnetic structure with k = (0.248(2), 1, 0.064(3)), which corresponds to an admixture of commensurate C-type and incommensurate A-type modes. In comparison with LaMn7O12, the incommensurate modulation of NdMn7O12 can be thought of as a spatially varying unidirectional canting (addition of an orthogonal spin density wave) or as a conical rotation of moments (addition of an orthogonal cycloid). These two scenarios could not be differentiated by neutron powder diffraction, but both support the argument for competing magnetic instabilities in both R = La and Nd compounds (it is well established that incommensurate structures can be stabilised by competing interactions). Below the third transition the incommensurate propagation vector was found to jump to k = (0.3231(7), 1, 0.0069(7)), accompanied by a finite ordered moment appearing on the Nd ions.
It is apparent that the additional LT magnetic transitions occur for compounds with rare earth ions of largest ionic radii, with the exceptions of CeMn7O12 and PrMn7O12. It was suggested that this discrepancy may arise due to Ce1−xMn7+xO12 or PrMn7O12+d off-stoichiometry of the measured samples. Indeed, the second transition was found to be suppressed in non-stoichiometric La0.9Mn7.1O12.59,62 The experimental results published to date indicate complex behaviour at low temperatures, and we suggest future studies, both experimental and theoretical, are required to establish the full microscopic details of the ground state magnetic structures of LaMn7O12 and NdMn7O12, and the underlying competing interactions in the RMn7O12 family.
Unpublished dielectric measurements of LaMn7O12 and NdMn7O12 indicate qualitatively similar behaviour to the non-polar compounds with R = Ce, Sm, Eu, Gd and Tb. Recently, however, a large electric polarisation of up to 0.56 μC cm−2 has been suggested for LaMn7O12 developing below TN1,60 based on the observation of very strong, broad and symmetrical pyroelectric-current peaks. Following the reported giant ferroelectric polarisation in CaMn7O12 (ref. 41) some debate ensued regarding the intrinsic nature of the polarisation in these systems. Indeed, other works have assigned such pyroelectric-current peaks to extrinsic effects.44,48,62,64 In light of this, we suggest that it is important for extrinsic effects, for example those due to thermally stimulated currents, to be ruled out in the case of LaMn7O12.
Specific heat and magnetometry measurements showed that YMn7O12 supports a single ferrimagnetic phase transition at TN1 = 108 K;66 approximately 20–30 K higher than the RMn7O12 compounds discussed above. Unlike the other RMn7O12 compounds, YMn7O12 was found to undergo a structural phase transition at Ts = 200 K that was clearly marked by anomalous behaviour in the temperature dependence of the specific heat and spontaneous negative thermal expansion of the lattice.66 The microscopic details of both phase transitions were established in a later publication, in which the same ferrimagnetic structure discussed above was reported to occur below TN1, apparently independent of the structural transition at Ts.67 The ground state crystal structure was characterised by an ordered pattern of atomic displacements that lowered the crystal symmetry to P21/n, accompanied by disordered displacements of yttrium ions forming a so-called dipolar glass that naturally explained the observed negative thermal expansion.67 A recent publication based on single crystal diffraction measurements showed that the yttrium ions can form partial antiferroelectric order out of the dipolar glass state,68 the extent of which is likely sample dependent. The structural modifications below Ts could in principle occur independent of one another, but they all improve the bonding conditions of the yttrium ions that were shown to be somewhat underbonded above Ts.67 Hence, YMn7O12 has a pseudo-JT instability associated with A site cations – phenomena usually associated with B site cations in perovskite-derived materials – that can drive unusual LT structural phase transitions. We note that, compared to the RMnO3 systems, the quadruple perovskites may be prone to underbonding of R cations as the octahedral tilts primarily generate square planar coordinations for the majority A′ site Mn3+ ions, perhaps at the expense of the minority A site R ions. Pyroelectric current measurements similar to those reported for LaMn7O12 have also led to the suggestion of a large ferroelectric polarisation developing in YMn7O12 at low temperature.68 As before, we stress a similar level of caution when interpreting the results of these measurements.
Finally, the reported phenomenology of DyMn7O12 is similar to that of YMn7O12 but with one key difference; the structural and magnetic transitions come together (TN1 and Ts are merged) to create a single, first order magneto-structural phase transition at TN1 ∼95 K.65 Below TN1 the Dy dipolar glass and ferrimagnetic order parameters were found to be cross-coupled (note that in DyMn7O12 ordered atomic displacements were not observed below TN1),65 as demonstrated by temperature and field dependent dielectric constant measurements (Fig. 8) (N.B. when TN1 and Ts are separated as in YMn7O12 no dielectric anomalies were found at the both temperatures).65 In addition, long-range FM ordering of Dy magnetic moments was found below 5.5 K.65
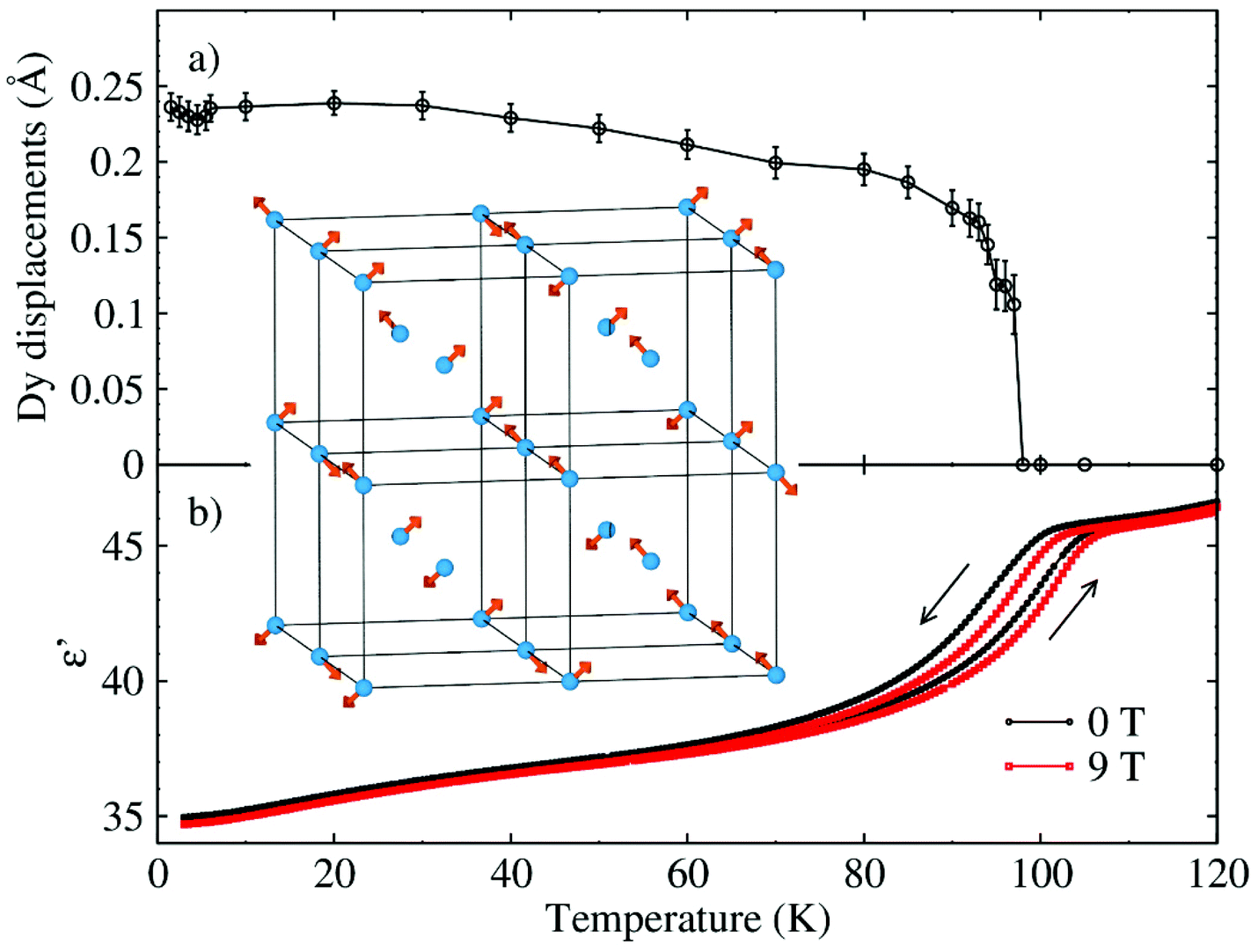 | ||
| Fig. 8 The magnetostructural dipolar glass found in (Dy0.91Mn0.09)Mn7O12: (a) The temperature dependence of the disordered Dy atomic displacement amplitudes established through structural refinements against neutron powder diffraction data,65 and (b) the temperature dependence of the real dielectric constant measured in 0 T (black) and 9 T (red) applied magnetic fields.65 The inset illustrates the disordered Dy dipolar glass (circles are ideal positions of Dy atoms at the (0,0,0) site; arrows show displacements). | ||
In summary, all RMn7O12 compounds host a common magnetic structure composed of a ferrimagnetic A′ site Mn3+ sublattice with one uncompensated Mn3+ moment per unit cell, and a C-type AFM B site sublattice (Fig. 7). In addition, LaMn7O12 and NdMn7O12 show LT magnetic phase transitions likely associated with the competition between A′–B and B–B exchange. For compounds with small R ionic radius (R = Dy and Y), the R cations adopt a pseudo-JT instability that leads to exotic LT phases characterised by cross-coupled magnetic and electric degrees of freedom.
5. BiMn7O12
Compared with RMn7O12, BiMn7O12 has an electronic instability originating from the presence of the lone-electron pair of Bi3+ cations. As a result, it exhibits distinct structural, magnetic and dielectric properties. BiMn7O12 was first reported in 2008,72 and its RT crystal structure was often described using a centrosymmetric I2/m model derived from powder diffraction experiments.53,58,73,74 On the other hand, more sensitive single-crystal structural investigations gave evidence for polar Im symmetry at RT.58,75 A number of additional structural phase transitions were later found,58,76 and the sequence of phase transitions is now well established (on cooling) as Im ⇒ I2/m ⇒ [R(αβγ)0] ⇒ Im ⇒ I1 with transitions at about 608 K, 460 K, and 295 K, respectively, where [R(αβγ)0] denotes an incipient phase, which appears only as diffuse scattering in undoped BiMn7O12 between the I2/m and Im phases.58 The proper stabilisation of the R(αβγ)0 phase can be triggered by small Cu2+ or Fe3+ doping.35 The Im ⇒ I2/m transition is related to orbital ordering of Mn3+ similar to RMn7O12, and the I2/m ⇒ Im transition – to the development of a ferroelectric polarization associated with the Bi3+ lone pair electrons. However, no direct confirmation of ferroelectricity (such as P–E loops) has been obtained to date.
Two magnetic transitions were widely reported in BiMn7O12 at about TN1 = 59 K and TN2 = 21 K in analogy with LaMn7O12,72,74,75,77 despite the first paper (ref. 72) reporting evidence for three magnetic transitions from specific heat measurements. In comparison with RMn7O12, frequency-independent peaks in the dielectric constant of BiMn7O12 were observed at all (three) magnetic transition temperatures without any extrinsic steps,72,75,78 but no pyroelectric current anomalies have been detected. Therefore, the origin of dielectric peaks remains unclear and needs further investigation.
The only neutron diffraction study of the magnetic structures assumed that Mn at the B sites are ordered at TN1, and Mn at the A′ sites are ordered at TN2, in analogy with LaMn7O12.74 In addition, the total zero moment was assumed for both A′ and B sites despite the observation of large uncompensated moments on M versus H curves (e.g., about 1μB at 2 K and 0.8μB at 40 K).72,77 Furthermore, in the analysis of the magnetic structures the (crystal) symmetry was considered to be Im,74 while the real ground-state symmetry is I1. Hence, we suggest that the magnetic structures of BiMn7O12 need careful re-investigation.
Similar to PrMn7O12, several modifications of BiMn7O12 were reported at RT (monoclinic, rhombohedral and cubic).73,77–79 However, in this case, the polymorphism was assigned to cation non-stoichiometry (Bi1−x/3(Mn3+3)(Mn3+4−xMn4+x)O12 and Bi0.94Mn6.91O12) and the appearance of Mn4+ at the B sites. We believe the same should be true for PrMn7O12. Magnetic properties of non-stoichiometric BiMn7O12 were noticeably different from stoichiometric BiMn7O12.
6. Conclusions
We have reviewed the current state of research on AMn7O12 quadruple perovskite manganites and identified some future directions, such as the re-investigation of magnetic structures of NaMn7O12 and BiMn7O12 and understanding dielectric behavior of all members, in particular, BiMn7O12. Investigations of different solid solutions35,80–82 may also yield fascinating results, as it allows continuous variations of Mn oxidation states at the B sites, while keeping nearly the same B-site Mn–O–Mn bond angles,82 as was extensively studied in the mixed-valence R1−xAxMnO3 perovskites.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by JSPS KAKENHI Grant Number JP20H05276 and Innovative Science and Technology Initiative for Security from Acquisition, Technology, and Logistics Agency (ATLA), Japan. R. D. J. acknowledges support from a Royal Society University Research Fellowship.References
- J. M. D. Coey, M. Viret and S. von Molnar, Mixed-valence manganites, Adv. Phys., 2009, 58, 571–697 CrossRef CAS.
- C. Zener, Interaction between the d-shells in the transition metals. II. Ferromagnetic compounds of manganese with perovskite structure, Phys. Rev., 1951, 82, 403–405 CrossRef CAS.
- J. B. Goodenough, Theory of the role of covalence in the perovskite-type manganites [La,M(II)]MnO3, Phys. Rev., 1955, 100, 564–573 CrossRef CAS.
- S. Jin, T. H. Tiefel, M. McCormack, R. A. Fastnacht, R. Ramesh and L. H. Chen, Thousandfold change in resistivity in magnetoresistive La-Ca-Mn-O films, Science, 1994, 264, 413–415 CrossRef CAS PubMed.
- E. Dagotto, T. Hotta and A. Moreo, Colossal magnetoresistant materials: the key role of phase separation, Phys. Rep., 2001, 344, 1–153 CrossRef CAS.
- T. Kimura, T. Goto, H. Shintani, K. Ishizaka, T. Arima and Y. Tokura, Magnetic control of ferroelectric polarization, Nature, 2003, 426, 55–58 CrossRef CAS PubMed.
- I. Yamada, H. Fujii, A. Takamatsu, H. Ikeno, K. Wada, H. Tsukasaki, S. Kawaguchi, S. Mori and S. Yagi, Bifunctional oxygen reaction catalysis of quadruple manganese perovskites, Adv. Mater., 2017, 29, 1603004 CrossRef PubMed.
- A. N. Vasil'ev and O. S. Volkova, New functional materials AC3B4O12 (review), Low Temp. Phys., 2007, 33, 895–914 CrossRef.
- A. M. Glazer, The classification of tilted octahedra in perovskites, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 3384–3392 CrossRef CAS.
- M. Marezio, P. D. Dernier, J. Chenavas and J. C. Joubert, High pressure synthesis and crystal structure of NaMn7O12, J. Solid State Chem., 1973, 6, 16–20 CrossRef CAS.
- J. Chenavas, F. Sayetat, A. Collomb, J. C. Joubert and M. Marezio, X-ray study of the low-temperature phase of [NaMn3+3](Mn3+2Mn4+2)O12, a perovskite-like compound, Solid State Commun., 1975, 16, 1129–1132 CrossRef CAS.
- A. Prodi, E. Gilioli, A. Gauzzi, F. Licci, M. Marezio, F. Bolzoni, Q. Huang, A. Santoro and J. W. Lynn, Charge orbital and spin ordering phenomena in the mixed valence manganite (NaMn3+3)(Mn3+2Mn4+2)O12, Nat. Mater., 2004, 3, 48–52 CrossRef CAS PubMed.
- S. V. Streltsov and D. I. Khomskii, Jahn-Teller distortion and charge, orbital, and magnetic order in NaMn7O12, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 201115 CrossRef.
- A. Prodi, A. Daoud-Aladine, F. Gozzo, B. Schmitt, O. Lebedev, G. van Tendeloo, E. Gilioli, F. Bolzoni, H. Aruga-Katori, H. Takagi, M. Marezio and A. Gauzzi, Commensurate structural modulation in the charge- and orbitally ordered phase of the quadruple perovskite (NaMn3)Mn4O12, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 180101 CrossRef.
- V. P. Gastaldo, Y. Klein, B. Baptiste, R. Cabassi, E. Gilioli, A. Gauzzi and A. J. A. de Oliveira, Unconventional magnetic ferroelectricity in the quadruple perovskite NaMn7O12, Phys. Rev. B: Condens. Matter Mater. Phys., 2020, 102, 161120 CrossRef.
- E. Gilioli, G. Calestani, F. Licci, A. Gauzzi, F. Bolzoni, A. Prodi and M. Marezio, P–T phase diagram and single crystal structural refinement of NaMn7O12, Solid State Sci., 2005, 7, 746–752 CrossRef CAS.
- R. Cabassi, F. Bolzoni, A. Gauzzi, E. Gilioli, A. Prodi and F. Licci, Dielectric properties of doping-free NaMn7O12: Origin of the observed colossal dielectric constant, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 045212 CrossRef.
- E. Gilioli, G. Calestani, F. Licci, C. Paorici, A. Gauzzi, F. Bolzoni and A. Prodi, High-pressure growth of NaMn7O12 crystals, J. Solid State Chem., 2006, 179, 3839–3848 CrossRef CAS.
- D. Delmonte, F. Mezzadri, F. Orlandi, G. Calestani, Y. Amiel and E. Gilioli, High pressure induced insulator-to-semimetal transition through intersite charge transfer in NaMn7O12, Crystals, 2018, 8, 81 CrossRef.
- B. Bochu, J. Chenavas, J. C. Joubert and M. Marezio, High pressure synthesis and crystal structure of a new series of perovskite-like compounds CMn7O12 (C = Na, Ca, Cd, Sr, La, Nd), J. Solid State Chem., 1974, 11, 88–93 CrossRef CAS.
- H. S. Horowitz and J. M. Longo, Phase relations in the Ca-Mn-O system, Mater. Res. Bull., 1978, 13, 1559–1369 CrossRef.
- B. Bochu, J. L. Buevoz, J. Chenavas, A. Collomb, J. C. Joubert and M. Marezio, Bond lengths in [CaMn3](Mn4)O12: A new Jahn–Teller distortion of Mn3+ octahedral, Solid State Commun., 1980, 36, 133–138 CrossRef CAS.
- D. Khomskii and J. van den Brink, Anharmonic effects on charge and orbital order, Phys. Rev. Lett., 2000, 85, 3329 CrossRef CAS PubMed.
- R. Przeniosło, I. Sosnowska, E. Suard, A. Hewat and A. N. Fitch, Phase coexistence in the charge ordering transition in CaMn7O12, J. Phys.: Condens. Matter, 2002, 14, 5747–5753 CrossRef.
- W. Sławiński, R. Przeniosło, I. Sosnowska, M. Bieringer, I. Margiolaki, A. N. Fitch and E. Suard, Charge ordering in CaCuxMn7−xO12 (x = 0.0 and 0.1) compounds, J. Phys.: Condens. Matter, 2008, 20, 104239 CrossRef.
- W. Sławiński, R. Przeniosło, I. Sosnowska and M. Bieringer, Structural and magnetic modulations in CaCuxMn7−xO12, J. Phys.: Condens. Matter, 2010, 22, 186001 CrossRef PubMed.
- W. Sławiński, R. Przeniosło, I. Sosnowska, M. Bieringer, I. Margiolakic and E. Suard, Modulation of atomic positions in CaCuxMn7−xO12 (x ≤0.1), Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 535–542 CrossRef PubMed.
- W. Sławiński, R. Przeniosło, I. Sosnowska and V. Petříček, Helical screw type magnetic structure of the multiferroic CaMn7O12 with low Cu-doping, Acta Crystallogr., Sect. B: Struct. Sci., 2012, 68, 240–249 CrossRef PubMed.
- A. A. Belik, Y. S. Glazkova, Y. Katsuya, M. Tanaka, A. V. Sobolev and I. A. Presniakov, Low-temperature structural modulations in CdMn7O12, CaMn7O12, SrMn7O12, and PbMn7O12 perovskites studied by synchrotron X-ray powder diffraction and Mössbauer spectroscopy, J. Phys. Chem. C, 2016, 120, 8278–8288 CrossRef CAS.
- N. J. Perks, R. D. Johnson, C. Martin, L. C. Chapon and P. G. Radaelli, Magneto-orbital helices as a route to coupling magnetism and ferroelectricity in multiferroic CaMn7O12, Nat. Commun., 2012, 3, 1277 CrossRef CAS PubMed.
- R. D. Johnson, D. D. Khalyavin, P. Manuel, P. G. Radaelli, I. S. Glazkova, N. Terada and A. A. Belik, Magneto-orbital ordering in the divalent A-site quadruple perovskite manganites AMn7O12 (A = Sr, Cd, and Pb), Phys. Rev. B: Condens. Matter Mater. Phys., 2017, 96, 054448 CrossRef.
- H. Guo, M. T. Fernández-Daz, L. Zhou, Y. Yin, Y. Long and A. C. Komarek, Non-collinear magnetic structure of manganese quadruple perovskite CdMn7O12, Sci. Rep., 2017, 7, 45939 CrossRef CAS PubMed.
- Y. S. Glazkova, N. Terada, Y. Matsushita, Y. Katsuya, M. Tanaka, A. V. Sobolev, I. A. Presniakov and A. A. Belik, High-pressure synthesis, crystal structures, and properties of CdMn7O12 and SrMn7O12 perovskites, Inorg. Chem., 2015, 54, 9081–9091 CrossRef CAS PubMed.
- R. D. Johnson, D. D. Khalyavin, P. Manuel and A. A. Belik, Competing electronic instabilities in the quadruple perovskite manganite PbMn7O12, Phys. Rev. B: Condens. Matter Mater. Phys., 2021, 103, 134115 CrossRef CAS.
- D. D. Khalyavin, R. D. Johnson, F. Orlandi, P. G. Radaelli, P. Manuel and A. A. Belik, Emergent helical texture of electric dipoles, Science, 2020, 369, 680–684 CrossRef CAS PubMed.
- T. Locherer, R. Dinnebier, R. K. Kremer, M. Greenblatt and M. Jansen, Synthesis and properties of a new quadruple perovskite: A-site-ordered PbMn3Mn4O12, J. Solid State Chem., 2012, 190, 277–284 CrossRef CAS.
- S. V. Ovsyannikov, A. M. Abakumov, A. A. Tsirlin, W. Schnelle, R. Egoavil, J. Verbeeck, G. Van Tendeloo, K. V. Glazyrin, M. Hanfland and L. Dubrovinsky, Perovskite-like Mn2O3: a path to new manganites, Angew. Chem., Int. Ed., 2013, 52, 1494–1498 CrossRef CAS PubMed.
- D. D. Khalyavin, R. D. Johnson, P. Manuel, A. A. Tsirlin, A. M. Abakumov, D. P. Kozlenko, Y. Sun, L. Dubrovinsky and S. V. Ovsyannikov, Magneto-orbital texture in the perovskite modification of Mn2O3, Phys. Rev. B: Condens. Matter Mater. Phys., 2018, 98, 014426 CrossRef CAS.
- W. T. Chen, C. W. Wang, H. C. Wu, F. C. Chou, H. D. Yang, A. Simonov and M. S. Senn, Improper ferroelectric polarization in a perovskite driven by intersite charge transfer and ordering, Phys. Rev. B: Condens. Matter Mater. Phys., 2018, 97, 144102 CrossRef CAS.
- A. A. Belik, Y. Matsushita and D. D. Khalyavin, Reentrant structural transitions and collapse of charge and orbital orders in quadruple perovskites, Angew. Chem., Int. Ed., 2017, 56, 10423–10427 CrossRef CAS PubMed.
- R. D. Johnson, L. C. Chapon, D. D. Khalyavin, P. Manuel, P. G. Radaelli and C. Martin, Giant improper ferroelectricity in the ferroaxial magnet CaMn7O12, Phys. Rev. Lett., 2012, 108, 067201 CrossRef CAS PubMed.
- G. Zhang, S. Dong, Z. Yan, Y. Guo, Q. Zhang, S. Yunoki, E. Dagotto and J.-M. Liu, Multiferroic properties of CaMn7O12, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 174413 CrossRef.
- R. D. Johnson, D. D. Khalyavin, P. Manuel, A. Bombardi, C. Martin, L. C. Chapon and P. G. Radaelli, Modulated spin helicity stabilized by incommensurate orbital density waves in a quadruple perovskite manganite, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 93, 180403(R) CrossRef.
- N. Terada, Y. S. Glazkova and A. A. Belik, Differentiation between ferroelectricity and thermally stimulated current in pyrocurrent measurements of multiferroic MMn7O12 (M = Ca, Sr, Cd, Pb), Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 93, 155127 CrossRef.
- R. D. Johnson, S. Nair, L. C. Chapon, A. Bombardi, C. Vecchini, D. Prabhakaran, A. T. Boothroyd and P. G. Radaelli, Cu3Nb2O8: a multiferroic with chiral coupling to the crystal structure, Phys. Rev. Lett., 2011, 107, 137205 CrossRef CAS PubMed.
- S. W. Cheong and M. Mostovoy, Multiferroics: a magnetic twist for ferroelectricity, Nat. Mater., 2007, 6, 13–20 CrossRef CAS PubMed.
- J. Cong, K. Zhai, Y. Chai, D. Shang, D. D. Khalyavin, R. D. Johnson, D. P. Kozlenko, S. E. Kichanov, A. M. Abakumov, A. A. Tsirlin, L. Dubrovinsky, X. Xu, Z. Sheng, S. V. Ovsyannikov and Y. Sun, Spin-induced multiferroicity in the binary perovskite manganite Mn2O3, Nat. Commun., 2018, 9, 2996 CrossRef PubMed.
- A. A. Belik, Y. S. Glazkova, N. Terada, Y. Matsushita, A. V. Sobolev, I. A. Presniakov, N. Tsujii, S. Nimori, K. Takehana and Y. Imanaka, Spin-driven multiferroic properties of PbMn7O12 perovskite, Inorg. Chem., 2016, 55, 6169–6177 CrossRef CAS PubMed.
- K. Gautam, S. S. Majid, S. Francoual, A. Ahad, K. Dey, M. C. Rahn, R. Sankar, F. C. Chou and D. K. Shukla, Magnetic and orbital correlations in multiferroic CaMn7O12 probed by X-ray resonant elastic scattering, Phys. Rev. B: Condens. Matter Mater. Phys., 2020, 101, 224430 CrossRef CAS.
- A. Prodi, G. Allodi, E. Gilioli, F. Licci, M. Marezio, F. Bolzoni, A. Gauzzi and R. De Renzi, μSR study of AA′3Mn4O12 double perovskites, Phys. B, 2006, 374–375, 55–58 CrossRef CAS.
- H. Okamoto, M. Karppinen, H. Yamauchi and H. Fjellvag, High-temperature synchrotron X-ray diffraction study of LaMn7O12, Solid State Sci., 2009, 11, 1211–1215 CrossRef CAS.
- A. Prodi, E. Gilioli, R. Cabassi, F. Bolzoni, F. Licci, Q. Huang, J. W. Lynn, M. Affronte, A. Gauzzi and M. Marezio, Magnetic structure of the high-density single-valent eg Jahn-Teller system LaMn7O12, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 085105 CrossRef.
- H. Okamoto, N. Imamura, M. Karppinen, H. Yamauchi and H. Fjellvag, Square coordinated MnO2-units in BiMn7O12, Inorg. Chem., 2010, 49, 8709–8712 CrossRef CAS PubMed.
- X. J. Liu, S. H. Lv, E. Pan, J. Meng and J. D. Albrecht, First-principles study of crystal structural stability and electronic and magnetic properties in LaMn7O12, J. Phys.: Condens. Matter, 2010, 22, 246001 CrossRef CAS PubMed.
- R. Cabassi, F. Bolzoni, E. Gilioli, F. Bissoli, A. Prodi and A. Gauzzi, Jahn-Teller-induced crossover of the paramagnetic response in the singly valent eg system LaMn7O12, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 214412 CrossRef.
- I. Yamada, Novel catalytic properties of quadruple perovskites, Sci. Technol. Adv. Mater., 2017, 18, 541–548 CrossRef CAS PubMed.
- A. Takamatsu, I. Yamada, S. Yagi and H. Ikeno, Oxygen evolution via the bridging inequivalent dual-site reaction: first-principles study of a quadruple-perovskite oxide catalyst, J. Phys. Chem. C, 2017, 121, 28403–28411 CrossRef CAS.
- A. A. Belik, Y. Matsushita, Y. Kumagai, Y. Katsuya, M. Tanaka, S. Yu. Stefanovich, B. I. Lazoryak, F. Oba and K. Yamaura, Complex structural behavior of BiMn7O12 quadruple perovskite, Inorg. Chem., 2017, 56, 12272–12281 CrossRef CAS PubMed.
- R. D. Johnson, D. D. Khalyavin, P. Manuel, L. Zhang, K. Yamaura and A. A. Belik, Magnetic structures of the rare-earth quadruple perovskite manganites RMn7O12, Phys. Rev. B: Condens. Matter Mater. Phys., 2018, 98, 104423 CrossRef CAS.
- A. Gauzzi, F. P. Milton, V. Pascotto Gastaldo, M. Verseils, A. J. Gualdi, D. von Dreifus, Y. Klein, D. Garcia, A. J. A. de Oliveira, P. Bordet and E. Gilioli, Ferroelectricity in the 1 μC cm−2 range induced by canted antiferromagnetism in (LaMn3)Mn4O12, Appl. Phys. Lett., 2019, 115, 152902 CrossRef.
- V. S. Bhadram, B. Joseph, D. Delmonte, E. Gilioli, B. Baptiste, Y. Le-Godec, R. P. S. M. Lobo and A. Gauzzi, Reentrant phase transition and suppression of Jahn-Teller distortion in the quadruple perovskite structure under high pressure, arXiv:2102.09998, 2021.
- A. A. Belik, L. Zhang, N. Terada, Y. Katsuya, M. Tanaka, Y. Matsushita and K. Yamaura, A-site-ordered quadruple perovskite manganite CeMn7O12 with trivalent cations, J. Solid State Chem., 2020, 283, 121161 CrossRef CAS.
- F. Mezzadri, M. Calicchio, E. Gilioli, R. Cabassi, F. Bolzoni, G. Calestani and F. Bissoli, High-pressure synthesis and characterization of PrMn7O12 polymorphs, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 014420 CrossRef.
- L. Zhang, N. Terada, R. D. Johnson, D. D. Khalyavin, P. Manuel, Y. Katsuya, M. Tanaka, Y. Matsushita, K. Yamaura and A. A. Belik, High-pressure synthesis, structures, and properties of trivalent A-site-ordered quadruple perovskites RMn7O12 (R = Sm, Eu, Gd, and Tb), Inorg. Chem., 2018, 57, 5987–5998 CrossRef CAS PubMed.
- R. D. Johnson, D. D. Khalyavin, P. Manuel, L. Zhang, K. Yamaura and A. A. Belik, Emergence of a magnetostructural dipolar glass in the quadruple perovskite Dy1−δMn7+δO12, Phys. Rev. Lett., 2020, 125, 097601 CrossRef CAS PubMed.
- M. Verseils, F. Mezzadri, D. Delmonte, B. Baptiste, Y. Klein, S. Shcheka, L. C. Chapon, T. Hansen, E. Gilioli and A. Gauzzi, Effect of chemical pressure induced by La3+/Y3+ substitution on the magnetic ordering of (AMn3)Mn4O12 quadruple perovskites, Phys. Rev. Mater., 2017, 1, 064407 CrossRef.
- R. D. Johnson, D. D. Khalyavin, P. Manuel, Y. Katsuya, M. Tanaka, Y. Matsushita, L. Zhang, K. Yamaura and A. A. Belik, Displacive structural phase transitions and the magnetic ground state of quadruple perovskite YMn7O12, Phys. Rev. B: Condens. Matter Mater. Phys., 2019, 99, 024107 CrossRef CAS.
- M. Verseils, F. Mezzadri, D. Delmonte, R. Cabassi, B. Baptiste, Y. Klein, G. Calestani, F. Bolzoni, E. Gilioli and A. Gauzzi, Centrosymmetry breaking and ferroelectricity driven by short-range magnetic order in the quadruple perovskite (YMn3)Mn4O12, Inorg. Chem., 2019, 58, 14204–14211 CrossRef CAS PubMed.
- J. Rodríguez-Carvajal, M. Hennion, F. Moussa, A. H. Moudden, L. Pinsard and A. Revcolevschi, Neutron-diffraction study of the Jahn-Teller transition in stoichiometric LaMnO3, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, R3189–R3192 CrossRef.
- E. F. Bertaut, Magnetism, ed. G. T. Rado and H. Suhl, Academic, New York, 1963, vol. 3, p. 149 Search PubMed.
- E. O. Wollan and W. C. Koehler, Neutron diffraction study of the magnetic properties of the series of perovskite-type compounds [(1−x)La,xCa]MnO3, Phys. Rev., 1955, 100, 545–563 CrossRef CAS.
- N. Imamura, M. Karppinen, T. Motohashi, D. Fu, M. Itoh and H. Yamauchi, Positive and negative magnetodielectric effects in A-site ordered (BiMn3)Mn4O12 perovskite, J. Am. Chem. Soc., 2008, 130, 14948–14949 CrossRef CAS PubMed.
- H. Okamoto, N. Imamura, M. Karppinen, H. Yamauchi and H. Fjellvag, Crystal structure of the monoclinic and cubic polymorphs of BiMn7O12, J. Solid State Chem., 2010, 183, 186–191 CrossRef CAS.
- A. Gauzzi, G. Rousse, F. Mezzadri, G. L. Calestani, G. Andre, F. Bouree, M. Calicchio, E. Gilioli, R. Cabassi, F. Bolzoni, A. Prodi, P. Bordet and M. Marezio, Magnetoelectric coupling driven by inverse magnetostriction in multiferroic BiMn3Mn4O12, J. Appl. Phys., 2013, 113, 043920 CrossRef.
- F. Mezzadri, G. Calestani, M. Calicchio, E. Gilioli, F. Bolzoni, R. Cabassi, M. Marezio and A. Migliori, Synthesis and characterization of multiferroic BiMn7O12, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 100106 CrossRef.
- W. A. Slawinski, H. Okamoto and H. Fjellvag, Triclinic crystal structure distortion of multiferroic BiMn7O12, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2017, 73, 313–320 CrossRef CAS PubMed.
- N. Imamura, M. Karppinen and H. Yamauchi, Synthesis and properties of monoclinic and cubic forms of the A-site-ordered (BiMn3)Mn4O12 perovskite, Chem. Mater., 2009, 21, 2179–2183 CrossRef CAS.
- F. Mezzadri, M. Buzzi, C. Pernechele, G. Calestani, M. Solzi, A. Migliori and E. Gilioli, Polymorphism and multiferroicity in Bi1−x/3(MnIII3)(MnIII4−xMnIVx)O12, Chem. Mater., 2011, 23, 3628–3635 CrossRef CAS.
- N. Imamura, K. Singh, D. Pelloquin, C. Simon, T. Sasagawa, M. Karppinen, H. Yamauchi and A. Maignan, Magnetodielectric response of square-coordinated MnO2 unit in cubic BiMn7O12, Appl. Phys. Lett., 2011, 98, 072903 CrossRef.
- A. A. Belik, Y. Matsushita, M. Tanaka, R. D. Johnson and D. D. Khalyavin, A plethora of structural transitions, distortions and modulations in Cu-doped BiMn7O12 quadruple perovskites, J. Mater. Chem. C, 2021, 9, 10232–10242 RSC.
- R. D. Johnson, F. Mezzadri, P. Manuel, D. D. Khalyavin, E. Gilioli and P. G. Radaelli, Evolution of magneto-orbital order upon B-site electron doping in Na1−xCaxMn7O12 quadruple perovskite manganites, Phys. Rev. Lett., 2018, 120, 257202 CrossRef CAS PubMed.
- W.-T. Chen, C.-W. Wang, C.-C. Cheng, Y.-C. Chuang, A. Simonov, N. C. Bristowe and M. S. Senn, Striping of orbital-order with charge-disorder in optimally doped manganites, arXiv:2104.14274, 2021.
| This journal is © The Royal Society of Chemistry 2021 |