
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCancer molecular biology and strategies for the design of cytotoxic gold(I) and gold(III) complexes: a tutorial review
Danielle
van der Westhuizen
 a,
Daniela I.
Bezuidenhout
*b and
Orde Q.
Munro
*a
a,
Daniela I.
Bezuidenhout
*b and
Orde Q.
Munro
*a
aMolecular Sciences Institute, School of Chemistry, University of the Witwatersrand, Johannesburg 2050, South Africa. E-mail: orde.munro@wits.ac.za
bLaboratory of Inorganic Chemistry, Environmental and Chemical Engineering, University of Oulu, P. O. Box 3000, 90014 Oulu, Finland. E-mail: daniela.bezuidenhout@oulu.fi
First published on 18th October 2021
Abstract
This tutorial review highlights key principles underpinning the design of selected metallodrugs to target specific biological macromolecules (DNA and proteins). The review commences with a descriptive overview of the eukaryotic cell cycle and the molecular biology of cancer, particularly apoptosis, which is provided as a necessary foundation for the discovery, design, and targeting of metal-based anticancer agents. Drugs which target DNA have been highlighted and clinically approved metallodrugs discussed. A brief history of the development of mainly gold-based metallodrugs is presented prior to addressing ligand systems for stabilizing and adding functionality to bio-active gold(I) and gold(III) complexes, particularly in the burgeoning field of anticancer metallodrugs. Concepts such as multi-modal and selective cytotoxic agents are covered where necessary for selected compounds. The emerging role of carbenes as the ligand system of choice to achieve these goals for gold-based metallodrug candidates is highlighted prior to closing the review with comments on some future directions that this research field might follow. The latter section ultimately emphasizes the importance of understanding the fate of metal complexes in cells to garner key mechanistic insights.
 Danielle van der Westhuizen | Danielle van der Westhuizen completed her PhD in 2021 from the University of the Witwatersrand in South Africa. Her PhD was focused on the syntheses of pincer carbene gold(III) complexes as potential cytotoxic agents and their ability to interact with macromolecular targets. In 2021, she joins the Chemical Synthesis and Analysis group at UiT the Arctic University of Norway, Tromsø as a postdoctoral fellow in organic chemistry and homogeneous catalysis. |
 Daniela I. Bezuidenhout | Daniela I. Bezuidenhout obtained her PhD from the University of Pretoria, South Africa in 2010. After a Fulbright postdoctoral fellowship at the University of California, San Diego 2011–2012, she returned to the University of Pretoria. From 2016–2019 she was appointed as an Associate Professor at the University of the Witwatersrand (WITS), South Africa. In 2019 she joined the University of Oulu, Finland as an Associate Professor of Inorganic Chemistry. |
 Orde Q. Munro | Orde Q. Munro completed his PhD at the University of the Witwatersrand in 1995. After a post-doctoral fellowship with Bob Scheidt at the University of Notre Dame (USA), he joined the Department of Chemistry at the University of Natal (1997) as a Lecturer in Inorganic Chemistry and was promoted to Full Professor in 2008. In 2014, he was awarded the SASOL Innovator of the Year Medal for his work on anticancer metallodrugs prior to being appointed as the DSI/NRF Chair in Bioinorganic Chemistry at WITS University (2015), where he is currently Professor and Chair of Bioinorganic and Inorganic Chemistry. |
Background
Since cancer is predicted to become the leading cause of mortality in every region of the world,1,2 the quest for effective, yet selective anticancer therapy remains paramount. In this tutorial review, we aim to provide a brief description of the general molecular mechanism of cancer and its fundamental origin in genomic instability. Cancer cells are not only associated with an increased rate of proliferation but are further endorsed by their ability to evade cell death pathways.3,4 Disrupting mitochondrial function, which often (but not exclusively5,6) initiates apoptosis,7 has emerged as a key chemotherapeutic strategy to elicit cytotoxicity in cancer cells.8,9 Currently, many anticancer drugs in clinical use (mitomycin C,10 dactinomycin,11 bleomycin,12 chlorambucil,13 doxorubicin,14etc.) target DNA15,16 and/or the enzymes which regulate DNA function.17 One such drug, cisplatin—a metallodrug,18 has paved the way for the potential use of metal complexes as anticancer agents. The active design and development of metallodrugs reflects their structural diversity and electronic tunability.19 To the drug developer, these advantages provide access to multiple macromolecular targets. This versatility is exemplified by the widespread exploration of Au(I) and Au(III) complexes as potential new chemotherapeutic agents.20 Specifically, Au(I) complexes preferentially bind to cysteine (Cys) and selenocysteine (Sec) residues in proteins, with thioredoxin reductase (TrxR) being an especially suitable target for drug development as TrxR inhibition induces the mitochondrial apoptotic pathway.21 Gold(III) complexes are often hampered by their instability under physiological conditions.22–24 However, the use of highly basic C- and N-donor ligands to stabilize higher metal ion oxidation states,25,26 allows metallodrug designers to routinely target crucial enzymes and DNA with Au(III) complexes. This review will follow the general rubric outlined in this paragraph, culminating in a critical analysis of the way forward for carbene complexes of Au(I) and Au(III).Cancer biology: an introduction
Cancer is associated with irregularities in cell replication
The proliferation of healthy cells is controlled by their response to extracellular signals that influence the internal circuitry of the cell, i.e., the cell cycle. For the duration of the cycle (Fig. 1), the cell grows (G1 and G2-phase), replicates DNA (S-phase) and divides into two genetically identical daughter cells (M-phase). Cells can withdraw from this cycle to enter a non-dividing state (G0) as a response to intra- or extracellular signals to either temporarily halt proliferation, to differentiate and mature, or undergo cell death. The progression from one stage to the next within the cell cycle is tightly regulated by a cascade of protein kinases (cyclin-dependent kinases, CDKs) and a checkpoint control system.27 The regulation of the cell cycle is crucial to ensure cell number homeostasis and maintenance of normal tissue function.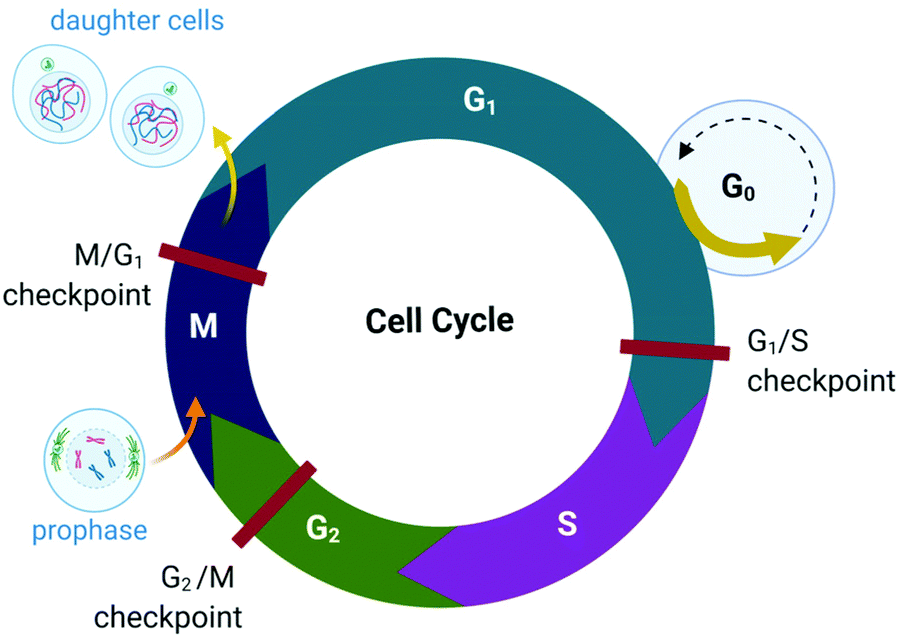 | ||
| Fig. 1 Phases of the eukaryotic somatic cell cycle. The cell cycle describes the life of a cell from where it is initially formed from a dividing parent cell until the cell itself divides into two daughter cells. The cycle is divided into sequential phases namely G1 (first gap), S (synthesis), G2 (second gap), and M (mitotic). During the G1 and G2 phases, the cell grows and carries out functions such as forming new organelles and protein synthesis. Between these two phases, the cell undergoes DNA replication (S-phase) to prepare for cell division and cytokinesis (M-phase). If too few growth factors are available when the cell reaches the G1/S checkpoint, the cell enters a quiescent stage, G0. The illustration (and those presented in Fig. 2–4, 9 and Fig. 16) was created with BioRender.com. | ||
Protein kinases (such as CDKs) are enzymes that activate or inactivate other proteins and play significant roles in signal transduction pathways within cells. In the case of CDKs, these kinases themselves are only activated by proteins named cyclins (hence the term “cyclin-dependent kinase”). Cyclins are present at different concentrations at different phases of the cell cycle and therefore orchestrate the events of the cell cycle depending on the type, activity and concentration of specific CDK-cyclin complexes within the cell.28,29 CDKs are themselves regulated by phosphorylation involving other kinases.29
The checkpoint control system evaluates if critical events such as replication of DNA or chromosome segregation occurred correctly and signals the cell cycle machinery to either stop or proceed with the cycle. Critical checkpoints are found in the G1, G2 and M phase, but the G1/S checkpoint (Fig. 1) is considered to be the most important as cells that proceed past this checkpoint usually transit through the S, G2 and M phase.30 The cell responds to extracellular signals only in the G1 phase, after which the progression to division is autonomous. Cells that receive a stop signal at the G1/S checkpoint withdraw from the cycle and transition to G0.30
The lack of cell cycle regulation can result in limitless replicative potential and is therefore associated with the development of cancer.28 Indeed, cancer cells can proceed through the G1/S checkpoint independent of extracellular signals (both growth and antigrowth signals) and remain in the cycle to avoid terminal differentiation or cell death.30 However, the progression from healthy cells to cancerous cells is a multistep process and is associated with many more acquired capabilities that are common to most types of cancer cells. Cancer cells can evade the immune system, generate neovascular support by the process of angiogenesis and can invade tissues and metastasize. Recent studies suggest that cancer cells can also reprogram cellular energy metabolism to sustain uncontrolled proliferation.31,32
The origin of cancer: genomic instability
Aside from the intrinsic genomic predisposition of certain cell types to become cancerous,33,34 cancer cells typically achieve their characteristic hallmarks because of changes in their genomes. More specifically, the viability of a cancer cell relies on multiple mutations in the genes where the gene products are key players in maintaining the genomic stability in healthy cells.31,32 Mutations in two types of genes have been identified in the pathogenesis of cancer, namely oncogenes and tumour-suppressor genes.Proto-oncogenes, which are the nonmutated form of oncogenes, code for proteins that function in signal transduction pathways that stimulate cell growth and division. Mutations in proto-oncogenes (e.g., the ras gene that codes for the Ras protein) usually leads to increased activity. The products of tumour suppressor genes, on the other hand, play key roles in signalling pathways that result in inhibited cell division. Mutations in tumour suppressor genes (e.g., the TP53 gene that codes for the p53 protein) lead to a loss in function. Both scenarios can result in increased cellular proliferation and destabilize the genome.27,28,31,32,35 For instance, in about 40% of human melanomas and 25% of human tumours, the MAPK (mitogen-activated protein kinase) signalling pathway is deregulated due to mutated proteins that play key roles in the pathway. The MAPK pathway is a signal transduction pathway that stimulates cellular proliferation and consists of a cascade of proteins, which are products of proto-oncogenes (Fig. 2). Ras proteins initiate the cascade by relaying signals that are received from an extracellular growth factor (e.g., epidermal growth factor, EGF). In some cancers, mutated Ras can initiate the signalling cascade without the extracellular growth factor. Moreover, this activation can affect downstream transcription factors, which regulate a repertoire of cellular functions such as DNA replication, cell cycle progression, apoptosis, differentiation and proliferation.36–38 Cancer cells can potentially acquire multiple hallmark capabilities such as sustained proliferative signalling, evasion of apoptosis, angiogenesis, modified metabolism and invasion by the mutations of these oncogenic genes.32,36
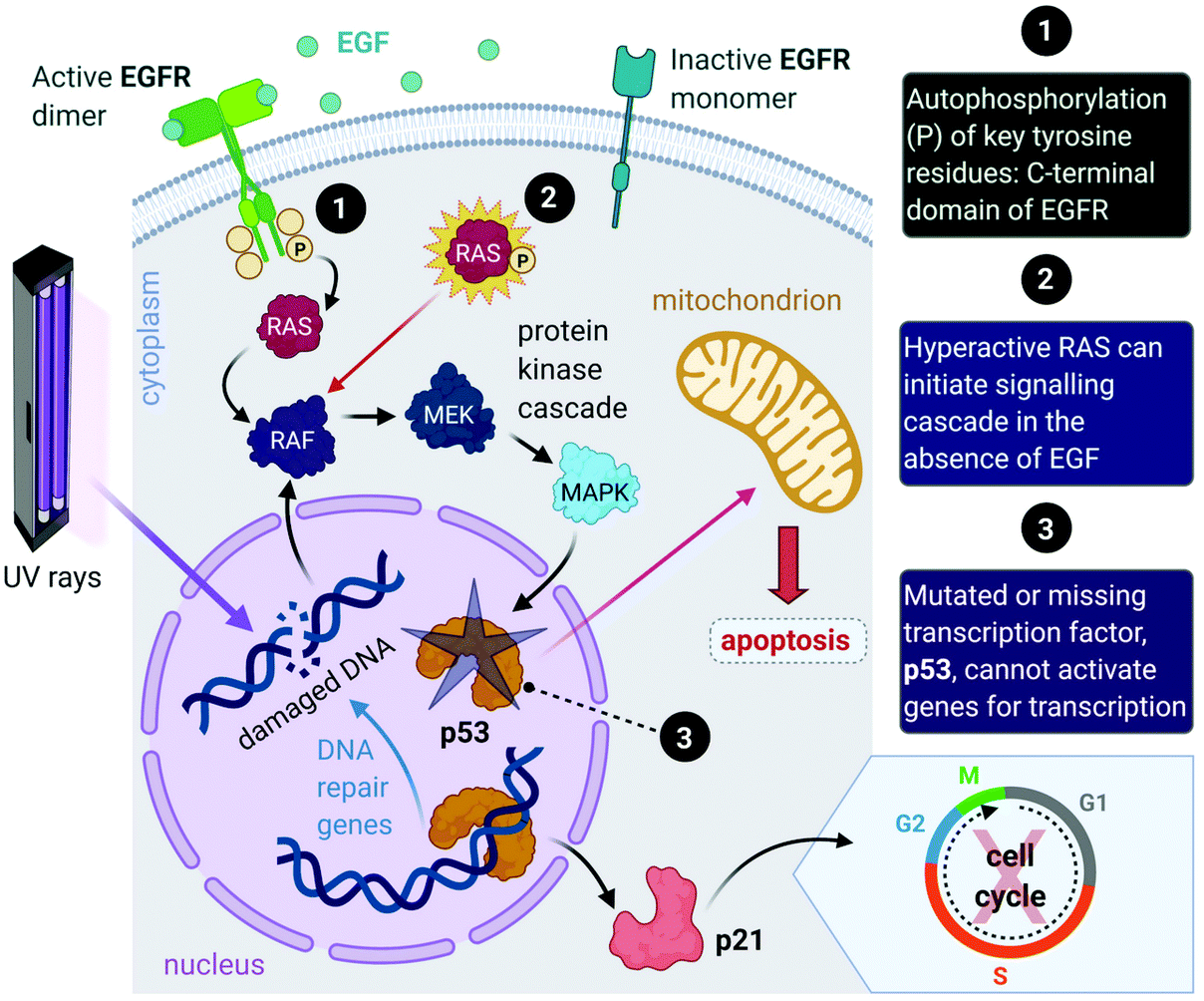 | ||
| Fig. 2 A simplified representation of the MAPK cell signalling pathway. This pathway is triggered by the binding of external growth factor (e.g., epidermal growth factor, EGF) to the EGF receptor (EGFR) on the membrane of the cell. Ras proteins (small GTPase signalling proteins) are activated by the receptor and initiate the signalling cascade involving a series of protein kinases. The last kinase (MAPK) activates a transcription factor (p53) that allows the synthesis of a protein (p21) that stimulates the cell cycle. (Note: p21 transcription can be initiated independently of p53.) Excessive cell replication can occur because of a mutated Ras protein, as the pathway can be initiated without the binding of EGF. In the case of UV radiation induced DNA damage, p53 is activated and promotes either the transcription of genes directly involved in DNA repair, the transcription of p21 that can halt the cell cycle to prevent damaged DNA from being copied or induce apoptosis if the DNA is damaged beyond repair. If p53 is missing or defective due to a mutation in TP53, the response system fails to prevent the cell cycle from copying faulty genetic material and fails to induce apoptosis. Acronyms: RAF, proto-oncogene serine/threonine protein kinase; MEK; tyrosine/threonine protein kinase; MAPK, mitogen-activated protein kinase; P, inorganic phosphate. | ||
Furthermore, mutations in tumour suppressor genes (e.g., TP53) and the subsequent inactivation of their products (e.g., p53 protein) enable cancer cells to be insensitive to growth inhibitory signals, evade differentiation and avoid programmed cell death (apoptosis).39 Tumour suppressors control cellular proliferation and inhibit tumour development. In more than half of human cancers, TP53 is mutated and leads to a loss of function in p53; this protein is an important transcription factor that prevents, as a response to damaged DNA, mutated DNA from being replicated by activating several genes (Fig. 2). The protein product (p21) of one such gene, p21, can halt the cell cycle by binding to CDKs to allow time for DNA repair to take place.40 (Transcription of p21 can also occur via a p53-independent pathway.41) Moreover, p53 activates DNA repair genes, however when the damage is irreparable, p53 initiates apoptosis by activating pro-apoptotic genes that induce cell death. Cells with damaged DNA or intracellular regulatory circuits have an increased frequency of mutations and loss of genomic stability.27,31,32
Triggering apoptosis: the key objective
The ability of cancer cells to develop into a tumour is not only attributed to a rise of uncontrolled proliferation, but also to the reduction in the rate of cell death. Although cancer cells can successfully evade cell death,31 they are not fully immortal. The goal of cytotoxic anticancer drugs is to kill tumour cells directly, thereby decreasing the tumour mass,42 while clinically, chemotherapy aims to reduce the tumour burden for the patient.Apoptosis is a highly conserved mechanism of cell death that regulates the number of cells in the body. There are two general cell signalling pathways that induce apoptosis, namely the extrinsic and intrinsic pathway. The extrinsic pathway is initiated by extracellular death receptors and has a significant role in immune system functions. The intrinsic pathway, which is also known as the mitochondrial pathway, is of interest as this pathway is initiated intracellularly as a response to DNA damage, anticancer therapy, depleted growth factors and oxidative stress.43,44
These diverse apoptotic signals all converge on the mitochondria and stimulate changes in the mitochondria that result in mitochondrial outer membrane permeabilization (MOMP),45 an event that induces apoptosis by either the release of pro-apoptotic proteins such as cytochrome c from the mitochondria or leads to the loss of vital mitochondrial functions (e.g., respiration).46 MOMP involves the opening of permeability transition (PT) pores located on the inner mitochondrial membrane (IMM) and is accompanied by a sudden increase in IMM permeability allowing ions and water to enter the matrix, leading to a loss in mitochondrial transmembrane potential and swelling of the matrix.47,48 The outer mitochondrial membrane then ruptures49,50 and releases pro-apoptotic proteins into the cytoplasm of the cell. The respiratory chain and production of ATP cease in response to MOMP as vital proteins leak out of the mitochondria, also leading to cell death.51–53
The release of the pro-apoptotic protein, cytochrome c,54,55 initiates a signalling cascade of the major protein group involved in apoptosis namely caspases (Fig. 3). Cytochrome c binds to Apaf-1 (apoptosis protease-activating factor-1) facilitating the binding of procaspase-9, forming a star-shaped apoptosome (recently structurally elucidated by cryo-electron microscopy).56 Procaspase-9 is cleaved to form active caspase-9, which subsequently activates procaspase-3, yielding the effector caspase (caspase-3). Effector caspases initiate apoptosis by activating proteins that are responsible for the cellular changes associated with apoptosis and are characterized by cell membrane blebbing, cell shrinkage, chromatin condensation and DNA fragmentation. Thereafter, the contents are packaged into apoptotic bodies and ingested by neighbouring or immune cells through phagocytosis.35,51,53,57
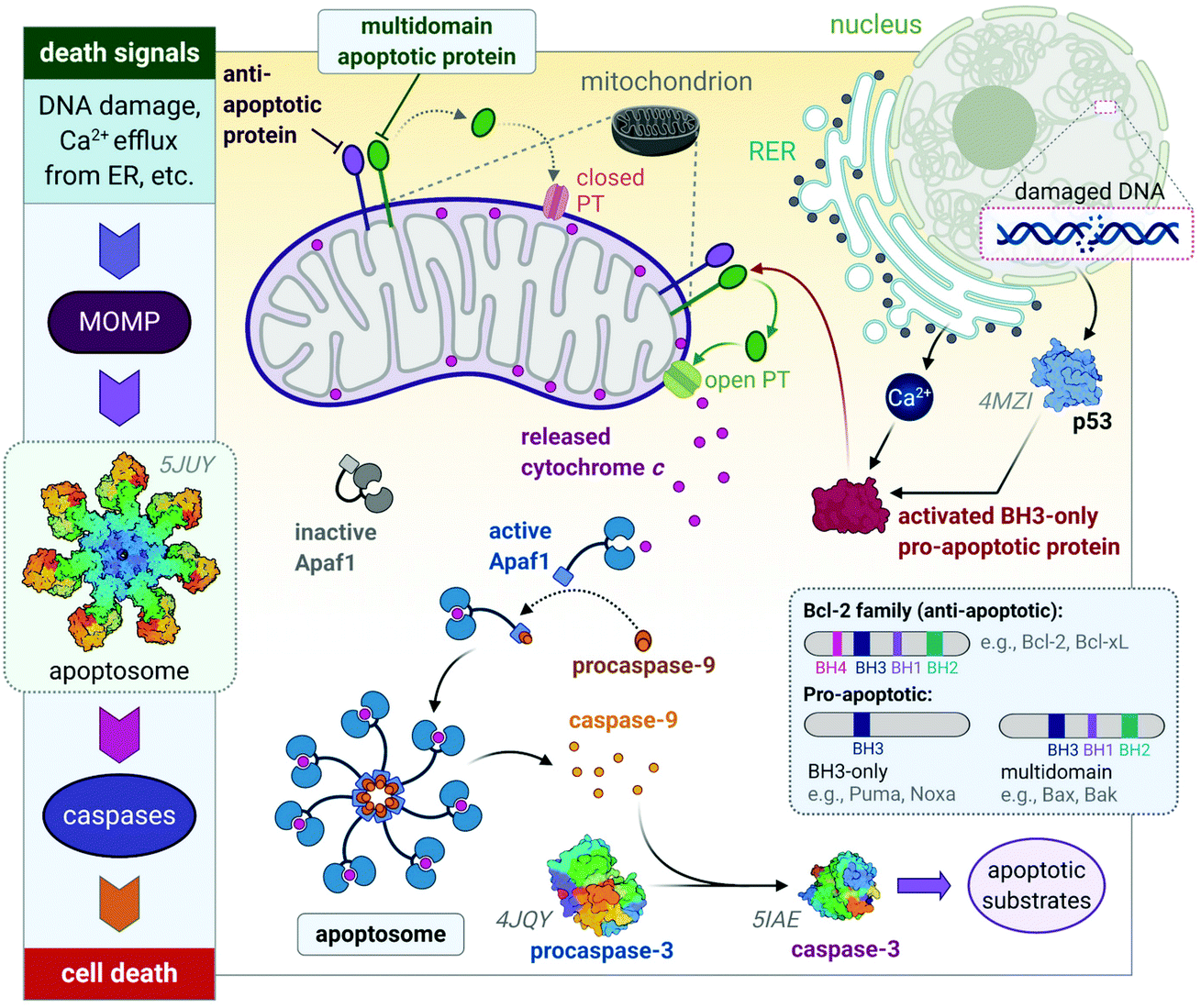 | ||
| Fig. 3 A basic representation of the intrinsic or mitochondrial apoptotic signalling pathway. Internal signals from the cell converge on the mitochondria, resulting in MOMP mediated by pro-apoptotic proteins of the Bcl-2 family. MOMP results in the release of cytochrome c, which binds to Apaf-1 to form an apoptosome with procaspase-9, which is subsequently activated to caspase 9. The effector caspase, procaspase-3, is activated by caspase-9 and activates proteins that execute apoptosis. The insert illustrates how the members of the Bcl-2 family are group based on their functions (i.e., anti- and pro-apoptotic) and number of BH domains. Acronyms: Apaf-1, apoptosis protease-activating factor-1; MOMP, mitochondrial outer membrane permeabilization; PT, permeability transition pore. Protein structures are rendered from their protein data bank (PDB) codes: apoptosome (5JUY), p53 (4MZI), procaspase-3 (4JQY), and caspase-3 (5IAE; the monomeric V266F mutant has been shown for simplicity). | ||
The Bcl-2 protein family are considered as the regulators of apoptosis but more specifically, they mediate and control the events leading to MOMP.45 The Bcl-2 protein family consists of a group of proteins that all have at least one of the four relatively conserved Bcl-2 homology (BH) domains (Fig. 3 inset). Members of this protein family are either initiators (pro-apoptotic) or inhibitors (anti-apoptotic) of apoptosis. The pro-apoptotic members are further divided into BH3-only (e.g., Noxa, Puma) and multi-domain members (e.g., Bax, Bak), the latter sequestered or neutralized by anti-apoptotic members (e.g., Bcl-2, Bcl-xL). The relative ratio of anti- and proapoptotic members work together to control cell death.35,51–53
Apoptosis is triggered once BH3-only proteins receive a death signal. For instance, in response to DNA damage,58 p53 activates the BH3-only proteins, Noxa and Puma, to induce activation of downstream multi-domain proapoptotic proteins.59,60 BH3-only proteins can release multi-domain pro-apoptotic proteins such as Bax and Bak from anti-apoptotic proteins (e.g., Bcl-2). Unbound Bax and Bak subsequently permeabilize the OMM for the release of cytochrome c by either forming pores in the OMM themselves or opening the PT pores to induce MOMP (Fig. 3).45,46 The anti-apoptotic proteins such as Bcl-2 maintain the mitochondrial membrane status to prevent the release of cytochrome c, balance the interactions between the members of the Bcl-2 family and counterbalance the damage done to the mitochondria, nucleus and endoplasmic reticulum (ER) by reactive oxygen species (ROS).51,52
Stress to the ER can also initiate apoptosis. The main function of the ER involves protein processing and sorting. The ER also plays a key role in the homeostasis of calcium,61 as it is the major store of intracellular calcium.35,48 When the ER experiences stress from exposure to chemical toxins, oxidative stress, etc., proteins are misfolded and the ER initiates repair mechanisms to either remedy or degrade the faulty proteins. When the response fails to resolve the problem, the ER releases calcium into the cytoplasm.47,61 The mitochondria respond by releasing a small amount of cytochrome c, where it interacts with ER receptors. The ER releases more calcium, resulting in a mass release of cytochrome c from the mitochondria, thereby initiating stress-induced apoptosis.62,63
Cancer cells have various mechanisms to avoid apoptosis. In B-cell lymphoma, the anti-apoptotic protein, Bcl-2 is overexpressed, thereby tipping the scales in favour of survival. The loss of p53 is one of the most successful ways cancer cells avoid cell death, as p53 initiates the apoptotic caspase cascade not only in response to DNA damage but also under abnormal conditions such as hypoxia and overexpression of oncogenes (e.g., Bcl-2).64–66
Other mechanisms of cell death include necrosis and autophagy.67,68 Cell death by necrosis is often associated with cells that suffer excessive damage. Necrotic conditions are favoured when normal physiological function (e.g., ion transport, pH balance and cellular respiration) is inhibited due to either pathological events (e.g., microbial or viral infection) or injury. Necrosis is characterized by the release of cellular contents into surrounding tissues due to the swelling of the cytoplasm and the breakdown of the cell membrane, thereby eliciting an inflammatory response by the immune system.51,53,68 Cell death can also occur through autophagy,67,69,70 an intracellular lysosome-mediated catabolic mechanism. Autophagy recovers nutrients such as amino acids during periods of nutrient scarcity by degrading proteins within the cell and digesting damaged or dysfunctional cellular components to ensure cytoplasmic homeostasis. At first glance, autophagy promotes cell survival, but cell death can occur due to extensive vacuolization of the cytoplasm, which engenders cell destruction.51,53,71
The relationship between cancer and cell death is unfortunately not straightforward, with apoptosis being able to promote tumourigenesis.72 Furthermore, cancer cells can initiate autophagy under the same stressful conditions (starvation, oxidative stress, etc.) as healthy cells, to the extent that this state becomes protective and reversable, especially when exposed to chemotherapy.73 In the event of necrotic death, pro-inflammatory signals are elicited, alerting immune inflammatory cells to the site of damage. Bioactive molecules such as growth factors, survival factors and enzymes that promote angiogenesis, invasion, and metastasis are thereby supplied to the tumour microenvironment as a result. In early tumour development, the sacrificial death of a pre-malignant cell possibly allows the development of more aggressive- and apoptosis-resistant cancer cells.32,42
This evidence aside, cancer cells avoid death and apoptosis is still the core intended outcome of chemotherapy. However, considering the tumour environment, drugs that elicit inflammatory responses and unnecessary tissue damage are best avoided. Drugs that are highly effective at killing all tumour cells are ideal as death induced by chemotherapeutic agents imposes a selective pressure on cancer cells. The chance of a treatment-resistant cancer arising increases markedly when low efficacy anticancer agents are used,42 which is one reason why multi-drug chemotherapy regimens are favoured for certain malignancies.74
Cancer is the result of defects in cellular regulatory mechanisms and the examples discussed above do not do the complexity around the disease justice. The list of possible faults in the internal circuitry and potential checkpoint fails is virtually infinite. The advances made in elucidating the molecular biology of distinct cancer types allow for the development of target-specific treatments.75,76 Unfortunately, many cancer cell targets match those operating in healthy cells; achieving selective chemotherapy thus remains elusive. Ideally, chemotherapeutic agents should have a multi-faceted approach77,78 to successfully induce apoptosis in cancer cells, a demise such cells dutifully attempt to avoid.
DNA as a cancer drug target
The chemotherapeutic effects of mustard gas, ironically discovered during the First World War, is attributed to its ability to alkylate DNA. The use of mustard gas and nitrogen mustard, an easier-to-manage alternative in clinical trials, represents one of the first examples of DNA-targeted chemotherapy.17,79 The appropriateness of DNA as an anticancer drug target is validated by the dominant role it plays in mitosis (cell division), the transmission of genetic material, the control of cellular processes (gene transcription), and how closely these functions relate to the development of cancer.80 However, DNA can be a risky target for anticancer drugs. Topoisomerase II mediated DNA cleavage in the presence of etoposide, for instance, causes chromosomal translocations and acute myeloid leukaemias (AMLs) in 2–3% of patients.81 Problematically, strategies used to develop compounds that selectively target tumour cell DNA82 have not been entirely effective.83 Furthermore, because of their inherent lack of selectivity (vide infra), many clinically relevant DNA-targeting drugs extensively damage normal cells such as those constituting bone marrow,84,85 causing Grade 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 86 (i.e., life threatening) adverse effects.
86 (i.e., life threatening) adverse effects.
The structure of DNA
DNA is a polynucleotide, where each nucleotide consists of a D-2-deoxyribose sugar, a phosphate group, and an aromatic nitrogenous base, of which there are four types: adenine (A), guanine (G), thymine (T) and cytosine (C). The individual nucleotide residues are joined by a phosphodiester bond between the sugar of one nucleotide and the phosphate group of another, forming one DNA strand. The direction of the DNA strand depends on the direction of the phosphodiester bond i.e., a 3′–5′ phosphodiester bond is between carbon-3 of the sugar of one nucleotide and carbon-5 of the sugar of the following nucleotide. The structure of duplex DNA, first presented in 1953 by Watson and Crick,87 takes on the form of a double helix, whereby two DNA strands are aligned antiparallel (5′–3′ and 3′–5′) and are held together by hydrogen bonds between complementary base pairs (A–T, 2 H-bonds; G–C, 3 H-bonds), as illustrated in Fig. 4a. The base pairs (bp) are stacked in the centre of the helix and the sugar-phosphate backbone strands frame the outside of the helix.88 Within the eukaryotic cell nucleus, the DNA is wrapped around histone proteins prior to further packaging to form chromatin.89,90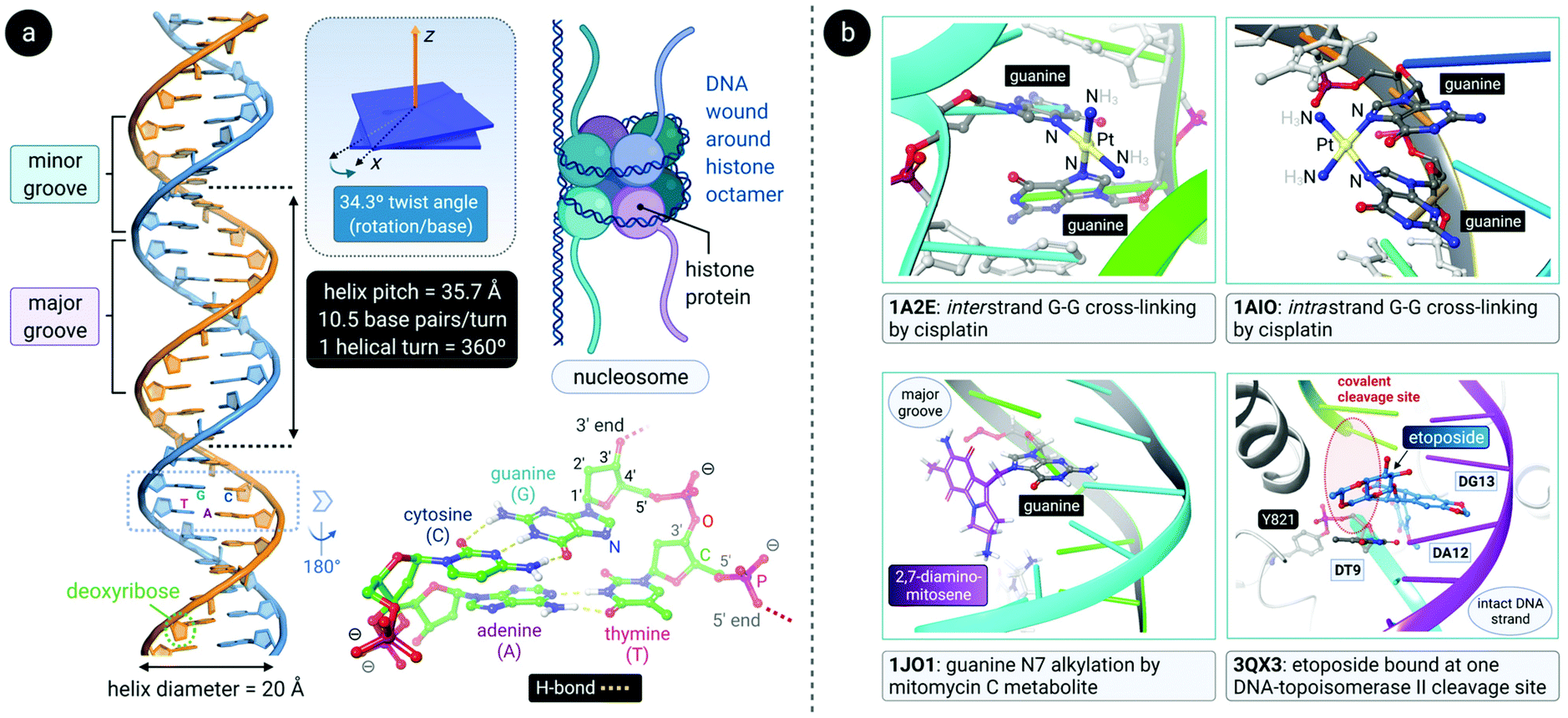 | ||
| Fig. 4 (a) Double-helical structure of the most common conformer of DNA, B-form DNA, with distinct major and minor grooves. The structural parameters (helical pitch of 35.7 Å, 10.5 residues per turn, 34.3° twist angle between residues and a diameter of 20 Å) associated with B-DNA are indicated. The DNA is wound around histone protein octamers prior to further packaging into chromatin in eukaryote cell nuclei (upper right). The structure of the residues in a 3′-GT-5′ tetramer motif within the left DNA duplex is shown (lower right) to highlight the complementary H-bonding between the nucleobase pairs (polar H atoms are shown along with D-2-deoxyribose C-atom numbering). (b) Illustration of some key DNA interactions by clinically relevant anticancer drugs. Top: cisplatin forms both inter- and intrastrand cross-linkages (via covalent bonds to N7 of guanine residues), thereby bending the DNA toward the major groove. Bottom: mitomycin C's main metabolite covalently attached to guanine N7 (left) and etoposide intercalated at one of two covalent cleavage sites (3′-TA-5′) created by topoisomerase II (Top2) during its catalytic cycle with a suitable DNA substrate (right). PDB codes are indicated in bold font for each structure. | ||
The helical structure of B-DNA, the most common conformer of DNA (the others being A- and Z-form), is presented in Fig. 4a. The helix is right-handed and is defined by a helix pitch of 35.7 Å and 10.5 base pairs per turn. Since each helical turn is 360°, each base is rotated 34.3° from the subsequent base. These structural parameters of B-DNA change dramatically (twice the curvature) when DNA is wrapped around the histone protein octamer within the nucleosome core particle.91 As revealed by an analysis of numerous X-ray structures of DNA and DNA–protein complexes, small local variations in the ideal geometry of B-form DNA induce a wide range of global geometries that accommodate DNA-binding protein motifs.92 In short, B-form DNA is conformationally flexible. A-form DNA deviates from the B-DNA parameters because of sugar puckering that results in a smaller helix pitch (28.15 Å), 11 base pairs per turn and a significant base tilt of 20° from the helical axis.93 Another variant, Z-DNA, is a left-handed helix with a zig-zag backbone.80,88 Of the conformations, the helical structure of B-DNA offers the best distinction between the major and the minor grooves. These grooves vary quite significantly due to the exposure of different nucleobase functional groups as potential binding sites for proteins, small molecules and drugs.80,88,94
Interactions of clinically approved anticancer drugs with DNA
Alkylating agents such as nitrogen mustard (N(CH3)(CH2CH2Cl)2) are among the most effective anticancer drugs due to their ability to crosslink DNA strands.95,96 Two nucleotide residues on the same strand (intrastrand) or opposite strands (interstrand) can be covalently connected by dialkylating agents. In the case of nitrogen mustard, interstrand crosslinks are formed, subsequently preventing the separation of DNA strands during critical cellular processes such as replication and gene transcription. Therefore, nitrogen mustard and its derivatives are most effective against highly proliferating cancers such as leukaemias and lymphomas. Unfortunately, cells in rapidly proliferating healthy tissues are also targeted and this results in side effects such as nausea, vomiting, hair loss and bone marrow toxicity (myelosuppression).17,79,97Other clinically-approved drugs, including cisplatin,98 mitomycin C99 and its primary metabolite 2,7-DAM (2,7-diaminomitosene),100 as well as the natural product trabectedin (Et-743),101 also covalently bind to DNA (Fig. 4b).102 Each drug elicits a markedly different cellular response and therefore has a distinctive chemotherapeutic mechanism. Often, drugs that bind to DNA influence the interaction of proteins with DNA since DNA is the substrate to which proteins responsible for DNA replication, gene transcription, packaging, recombination, repair, etc., bind.17
After aquation,103 cisplatin preferentially binds to N7 of two guanine residues on the same (intrastrand)102 or adjacent98 strands (Fig. 4b), forming cross-linked covalent G–Pt–G DNA lesions. These crosslinks significantly bend the DNA towards the major groove (Fig. 5),102 exposing a minor groove binding pocket to DNA-binding proteins, e.g., high-motility group box proteins (HMGB),104 repair proteins, transcription factors and other damage recognition proteins. Trapped proteins are prevented from carrying out their intended functions or mask the distortion of DNA from the nucleotide excision repair (NER) mechanism,105 a process that repairs DNA lesions caused by environmental or chemical damage by recognizing, removing, and resynthesizing the damaged region (these activities form part of the DNA damage response, DDR106,107). Precise details regarding the progression from initial damage to cell death remain unclear, but several DNA-damage transduction signal pathways (e.g., AKT, c-ABL, MAPK, p53, etc.) are thought to be influenced in response to cisplatin-induced damage.17,108–117
 | ||
| Fig. 5 (a) Illustration of the crystal structure of cisplatin covalently bound to two N7 atoms of adjacent guanine residues. The dsDNA target adopts a distorted helix conformation with a marked kink close to the DNA lesion (PDB 1AIO). Cisplatin itself undergoes aquation to form the mono(aqua) adduct prior to coordination to guanine N7, as depicted in the equilibrium ligand exchange (left side). PDB codes are indicated in italics font for each structure. (b) X-ray structure of the complex formed between a linear Au(I) mesoionic carbene complex and a G-quadruplex DNA target stabilized by centrally located K+ ions (PDB 5CCW). The planar Au(I) bis(carbene) complexes π-stack atop guanine bases in the central region of the G-quadruplex above and below the cylindrical structure's core. | ||
While cisplatin kinks the DNA toward the major groove, the naturally occurring anticancer drug trabectedin covalently binds within the minor groove of DNA and unveils the major groove pocket for protein binding. Thus, a range of different proteins are targeted including key players in transcription-coupled NER (the repair of genes that are coded into proteins).117 Et-743, which targets the DNA sequences 5′-TCCA-3′ and 5′-TGCC-3′,117 and mitomycin C,118,119 also a minor groove binder, have far greater DNA binding selectivity than normal DNA alkylators due to their sequence specificity.17
Small molecule compounds that influence DNA–protein complexes are not limited to covalent binders. Doxorubicin120 and etoposide,121 the former a non-specific DNA intercalator122 and the latter a non-intercalating compound,123 are both notably cytotoxic and operate by inducing DNA double strand breaks where the enzyme topoisomerase II (topo II) is covalently bound to its DNA substrate via a pair of tyrosine phosphodiester links positioned four bases apart (Fig. 4b).124 The two drugs act as interfacial poisons of the enzyme by binding noncovalently at the pair of DNA strand nick sites, thereby blocking the enzyme from resealing the transient DNA double strand break to complete its catalytic cycle.125 Topoisomerases are a family of enzymes which are responsible for maintaining the correct DNA topology during critical cellular functions such as replication and transcription. They introduce temporary single- (topo I) or double-stranded (topo II) DNA breaks.126 Cancer cells are thought to be more susceptible to topo II poisons than non-tumorigenic cells due to the elevated levels of topoisomerases found in most cancer cells.17 Interestingly, an investigational cytotoxic Au(III) isoquinoline-amide complex has been shown to act as a dual-mode inhibitor of topo II, poisoning the enzyme at low concentrations (KD ∼240 nM) but catalytically inhibiting it at higher concentrations (KD ∼9 μM).127 Since the ancillary cis-chloride ligands were substitution inert, this class of compounds likely does not covalently bind to DNA (unlike cisplatin), suggesting that future clinically approved topo II-targeting drugs might include metal chelates with unique behaviour.
Although not yet clinically relevant, metallodrug candidates that target DNA secondary structures such as G-quadruplexes are also being investigated.128 G-Quadruplexes form readily under physiological conditions and consist of intermittent runs of guanine residues. These quadruplex structures mainly form in the promoter (DNA sequences to which the enzyme RNA polymerase binds to initiate protein synthesis) and telomeric regions of the DNA sequence.129 Telomeres protect the ends of chromosomes during DNA replication and shorten progressively during each cell division until cell death ensues. Cancer cells have highly expressed telomerase activity, the enzyme responsible for the maintenance of telomeres, amounting to another mechanism of acquired immortality. G-quadruplex interacting compounds, which span a range of transition metal complexes,130 including Au(I) carbenes131,132 and a polypyridine Ru(II)133 complex, and which have recently been crystallized with different binding site preferences to G-quadruplex DNA targets (e.g., Fig. 5b), can inhibit telomerase by blocking enzyme-DNA binding or by preventing the formation of G-quadruplexes (i.e., telomeres and promoters) altogether.17,31,32,35,134 Other DNA secondary structures such as DNA three-way junctions (3WJs) and DNA four-way junctions (4WJs) have also emerged as suitable targets for several investigational organic compounds and metal chelates,135 as discussed by Zell et al. in their excellent review.136 Notably, metallopeptides137 and metallocages138 feature among the more active compounds for targeting 3WJs, benefitting from the ability of the metal ions to assemble the ligands and create the required triangular shape complementarity for the DNA target.139 Regarding 4WJs, the first metal complex (square planar Pt2+ chelated by a tetradentate polypyridyl ligand) that acts as an effective template for the assembly of a 4WJ-like DNA superstructure has been described.140
Metal complexes, due to their own high tolerance for structural and electronic modification, can clearly exploit various DNA binding sites with great success.80,94,134,141 Moreover, metal complexes may interact with a diverse range of molecular targets, not limited to DNA, which might allow the development of metallodrugs with multi-faceted cellular targets. A noteworthy caveat is that such compounds should ideally be designed to accumulate selectively in neoplastic tissue to overcome the limitations of cisplatin142 and to be realistic future contenders for clinical use. The current surge of investigations into the anticancer potential of metal complexes ultimately stems from the initial discovery of cisplatin's anticancer properties,143 the delineation of its mechanism of action,102 and the intrigue that such a simple complex (and structures related to it) has held for chemists, biologists, and pharmaceutical researchers over many decades. The field, however, has a firm foundation and is now poised for significant new discoveries of novel biomolecular targets for metal complexes, as evidenced by the recent report that metallo-supramolecular cylinders which target DNA 3WJs can also target bulge structures in the 5′ untranslated regions of the RNA genome of SARS-CoV-2 and inhibit viral replication.144 The emergent picture is that “DNA (and RNA145) folds threaten genetic stability and can be leveraged for chemotherapy”,136 whether the target is cancer or pathogenic viruses. The question is will pharmaceutical firms take promising preclinical metallodrug candidates seriously enough to offer them the same interest, ADMET scrutiny, and development opportunities as seemingly preferred “mono-target” organic compounds? Binding to multiple macromolecular targets, which is often a hallmark (but can also be a serious limitation) of many metallodrug candidates,146 might be a useful strategy for overcoming drug resistance since multiple simultaneous mutations in unrelated genes would be required by the organism in question to develop resistance to such a drug. This idea is now in vogue147,148 in the face of the current SARS-CoV-2 pandemic and could become a feasible approach to limit drug resistance in cancer chemotherapy.
Cytotoxic gold complexes
Historic use of medicinal gold
The earliest known application of medicinal gold was in China dating back to 2500 BC in pursuit of longevity; arguably so, as gold itself is resistant to corrosion and rust. In early times, gold powder was rubbed on open wounds and said to remove the toxins associated with smallpox and skin ulcers as well as mercury from the body. Heated gold relieved toothache and golden amulets were worn to repel evil spirits.149The medicinal use of gold was limited to topical ailments as there was no way to dissolve gold. In medieval times, potable gold (i.e., drinkable gold) emerged as elixirs of life to restore youth. These elixirs contained very low concentrations of dissolved gold, as cited in one of the many recipes for potable gold: “to quench a heated gold plate in wine four to five times”. Not surprisingly, scepticism arose around the medicinal properties of gold as some potable gold recipes cured ailments (in these recipes aqua regia was used to generate the soluble salt, AuCl3) and others did not (e.g., heated gold in wine).149,150
In the 17th century, several preparations for medicinal gold were known. One of the preparations required the distillation of dissolved gold and aqua regia, resulting in the soluble AuCl3 salt. A second preparation, the fulminate of gold, was made by adding ammonia to aqua regia and gold. Not unexpectedly, gold chloride was deemed too corrosive and gold fulminate too dangerous (explosive) for any medicinal use. In the 19th century, after the use of medicinal gold had declined dramatically, a French physician used a less caustic mixture of gold and sodium chloride, a so-called “muriate of gold and soda”, to treat syphilis. Although the reproducibility of his findings was ambiguous, by the mid-19th century, this double chloride of gold was prescribed as a secondary drug, only when the primary would fail, to treat morphine addiction, nephritis, anaemia, premature senility, neurasthenia, lupus and alcoholism.150
The first rational use of a gold metal complex, K[Au(CN)2], in medicine is ascribed to Koch in ∼1890.151 The many attributes of metal complexes, such as their variable coordination numbers, geometries, and redox states, in combination with appropriate organic ligands, are particularly useful for drug discovery and design. Metal ions generally target diverse biological molecules depending on the element, type of coordinated ligands, coordination geometry, kinetic lability (ligand exchange), and redox potential. Indeed, the same metal ion in different oxidation states can bind to different drug targets.152–156
Gold(I) compounds, for instance, are classified as soft electrophiles in Pearson's HSAB theory157,158 and interact preferentially with soft sulfur-donor and phosphane ligands (nucleophiles). Because of the ubiquity of cysteine in proteins, Au(I) can bind to solvent-exposed cysteine thiolate groups and subsequently inactivate enzymes, particularly those with catalytic cysteine residues such as the cysteine proteases.159,160 Hard Au(III) ions favour harder nitrogen and oxygen donor ligands, which is one reason why Au(III) salts were initially used to stain nucleic acids for imaging by electron microscopy.151 Moreover, the redox chemistry and associated change in coordination number of gold complexes can be an advantage as the reduction of the square-planar Au(III) ion to the linear Au(I) ion is often associated with the release of ligands, whereby these ligands can have inherent medicinal properties.24 In principle, Au(III) complexes can be used to deliver both a clinically approved drug and Au(I) to a tumour if appropriately designed, akin to Pt(IV) systems.161,162
Koch found that K[Au(CN)2] was bacteriostatic against “tubercle bacillus”, a strain of bacteria now known as Mycobacterium tuberculosis and the causative agent of tuberculosis.163 During clinical trials, however, K[Au(CN)2] (“gold cyanide”) proved ineffective in treating tuberculosis and had serious toxic side effects,151 stemming any further use of the compound. Gold(I)-thiols, such as sodium aurothiomalate (Fig. 6 and 7) were much less toxic and were administered to tuberculosis patients during the period 1925–1935 (known as the Gold Decade) but the usage declined as reports on toxicity increased along with very little evidence of treatment efficacy. Later on, these gold thiolates were investigated against rheumatoid arthritis after the idea that tubercle bacillus caused rheumatoid arthritis.150,163,164
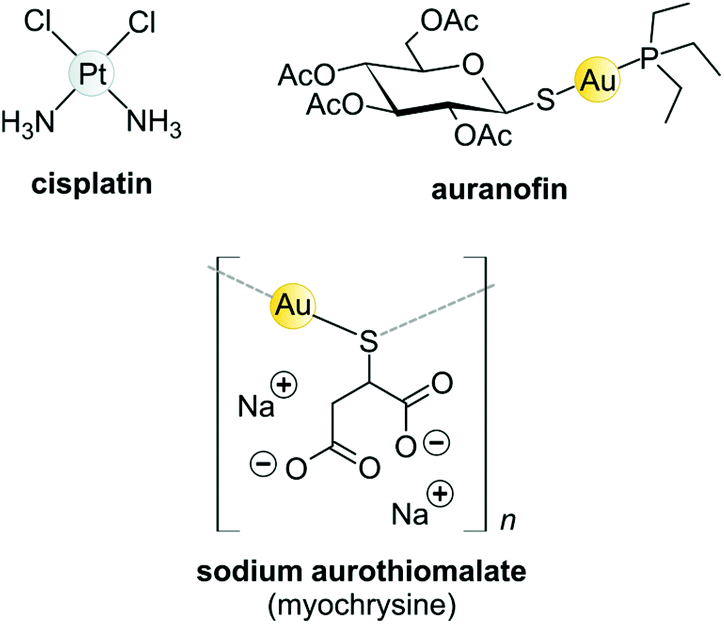 | ||
| Fig. 6 Structures of selected metal-based chemotherapeutic drugs in clinical use (Pt2+ and Au+ complexes). | ||
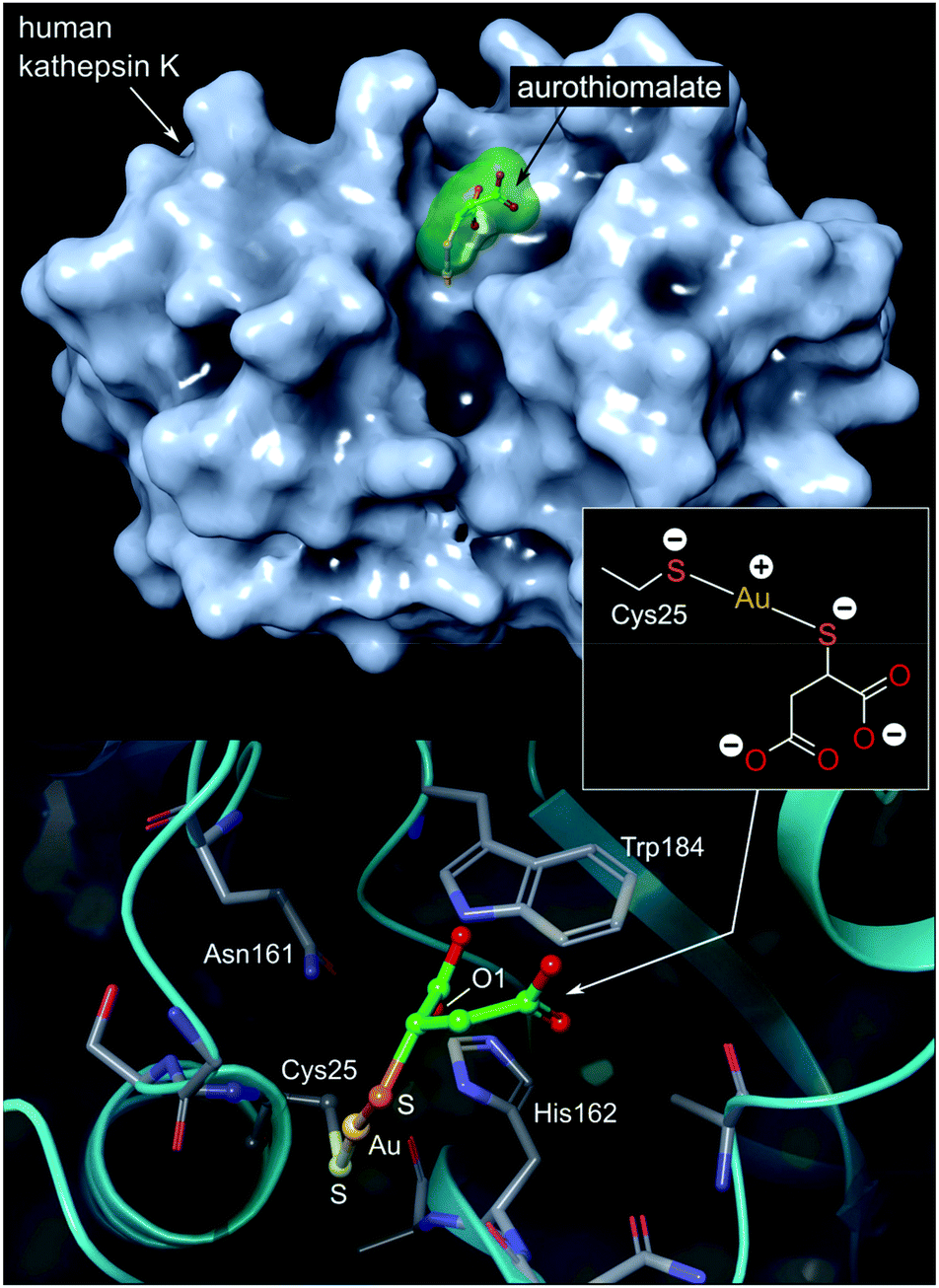 | ||
| Fig. 7 Top: View of the X-ray structure (PDB 2ATO) of human cathepsin K (a cysteine protease) covalently bound to myochrysine (aurothiomalate). The linear Au(I) thiolate targets Cys25 within the enzyme's active site cleft. Bottom: close-up view of the coordination of the Au(I) ion to the thiolate ligand of Cys25. The S–Au–S bond angle measures 173.1°; the Au–S distances are equivalent at 2.5 Å. The O1 atom of thiomalate is an acceptor for three NH⋯O hydrogen bonds with Gln19, His162, Trp184 (not shown). | ||
Today, after years of debate and results obtained from a large, well-controlled double-blind trial concluded that gold compounds have a beneficial effect on rheumatoid arthritis (RA),165 injectable Au(I)-thiolates have been included in a group of FDA-approved disease-modifying antirheumatic drugs (DMARDs).164,166,167 These drugs do have notable efficacy limitations as they are slow acting and accumulate in the kidneys, giving rise to nephrotoxicity.163 In the 1970s, an orally administered gold(I)-phosphane drug, auranofin (tetraacetyl-β-D-thioglucose gold(I) triethylphosphine, Fig. 6)168,169 was introduced and although only mild side-effects were reported, auranofin was less effective than the injectable gold drugs in treating RA.134,141 However, auranofin is still used clinically today for psoriatic arthritis and juvenile rheumatoid arthritis when other front-line DMARDs like methotrexate170 fail to treat refractory RA.164,166,167
The Au(I) complexes, auranofin and myochrysine (Fig. 6), another approved antiarthritic drug, are currently undergoing clinical trials as anticancer drugs.171,172 Auranofin is an inhibitor of the enzyme thioredoxin reductase, increases cellular ROS, and induces the mitochondrial apoptotic pathway.166,167,173 Myochrysine, an aurothiomalate complex, specifically binds to a cysteine residue in the binding pocket of protein kinase C iota (PKCί), preventing the downstream protein, par-6, from binding to the kinase. The PKCί⋯par-6 complex is a signalling intermediate within a cellular pathway that stimulates tumour growth and invasion.174,175 Myochrysine covalently bound to the cysteine residue (Cys25) in the active site of human cathepsin K has been structurally elucidated by X-ray crystallography (Fig. 7), representing one of the first gold–protein adducts to be studied at near-atomic resolution. The thiomalate ligand does not dissociate from the Au(I) ion and participates in hydrogen bonding via its carboxylate groups with several nearby residues.176
Biological targets and antitumour activity of Au(I) compounds
The use of gold(I) compounds as potential anticancer agents was sparked by the in vitro cytotoxicity of auranofin against HeLa (human cervical epithelioid carcinoma) cells177 and further studies that revealed auranofin's similar, or in some cases superior, cytotoxicity compared to cisplatin.171,178,179 The in vivo activity of auranofin, however was found to be limited to leukaemia P388 mouse models only.180–182 Once injected, auranofin undergoes rapid ligand exchange and binds covalently to the cysteine residue (Cys34) of human serum albumin, the most abundant blood protein,183 an event which subsequently decreases the concentration of the active gold species and the rate of cellular uptake.184 The first structural evidence of Cys34 being a gold-binding site was recently reported (Fig. 8). Messori and co-workers co-crystallized BSA (bovine serum albumin) with a dithiocarbamate Au(III) complex. Upon binding to the protein, the Au(III) ion was reduced to Au(I) with concomitant loss of the ligands (2 Br− and C6H10NO2S2−), resulting in a “naked” Au(I) ion bound to Cys34 of BSA.185 Because of the low resolution of the structure, the identity of the second ligand (probably water) was not established.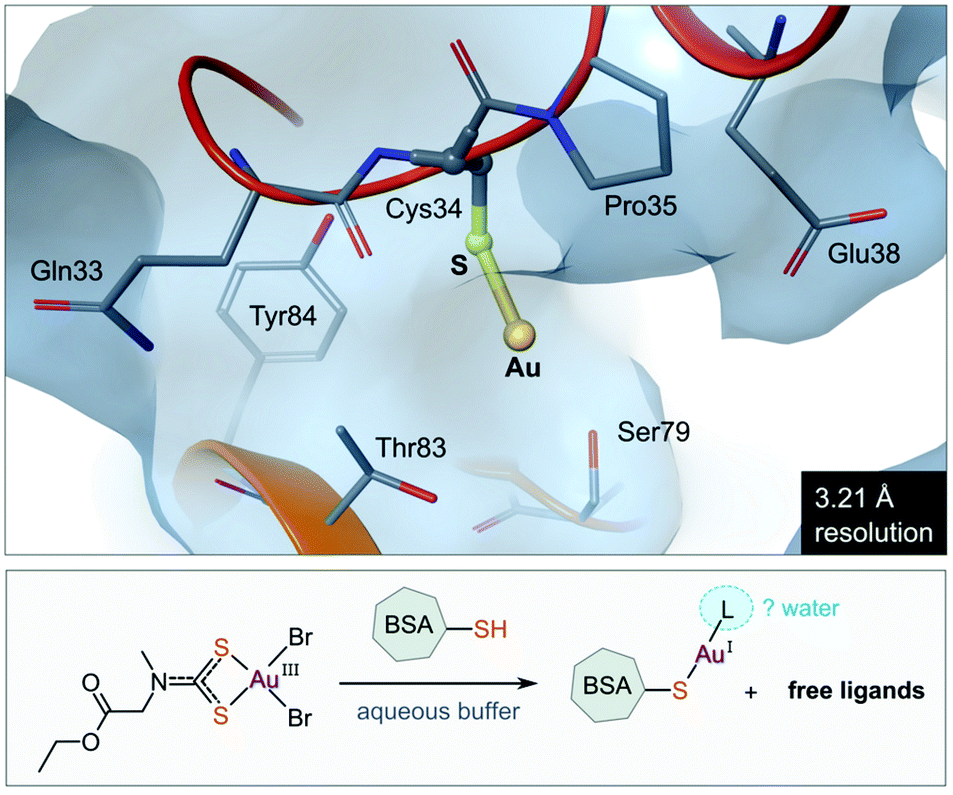 | ||
| Fig. 8 View of the X-ray structure of the Au(I) adduct of bovine serum albumin (BSA) formed after incubation of the protein with the Au(III) complex of (2-ethoxy-2-oxoethyl)methylcarbamodithioate (PDB 6RJV). Reduction of the Au(III) complex occurs with concomitant release of the ligands to form the linear Au(I) adduct with Cys34 thiolate (Au–S, 2.14 Å). Water molecules were not located in the low-resolution X-ray structure, but it is possible that a second ligand (water) completes the coordination sphere of the linear Au(I) ion. | ||
Despite limited in vivo activity, auranofin is a potent inhibitor of mitochondrial thioredoxin reductase (TrxR2), elevating this enzyme's status to that of a promising new drug target for investigational Au(I) complexes,173,186 but perhaps realistically only if compounds can be designed to selectively target tumour cells (since TrxR2, though not overexpressed as in tumour cells,187 is present in the mitochondria of all human cells which utilize this organelle188). Thioredoxin reductases (TrxR) are a class of enzymes that catalyse the reduction of thioredoxins (Trx), proteins which themselves are responsible for maintaining other cellular proteins in their reduced state. TrxR, Trx and the redox co-enzyme, NADP(H) collectively form the Trx system, a ubiquitous arrangement that is crucial in maintaining intracellular redox conditions (Fig. 9). Reduced Trx is responsible for the reduction of cellular proteins that play critical roles in cell division, transcription of genes, apoptosis and proteins that protect the cell against ROS species. Specifically, Trx reduces proteins such as ribonucleotide reductase, a critical enzyme in DNA synthesis, and thioredoxin peroxidase which catalyses the reduction of hydrogen peroxide to water.189–191
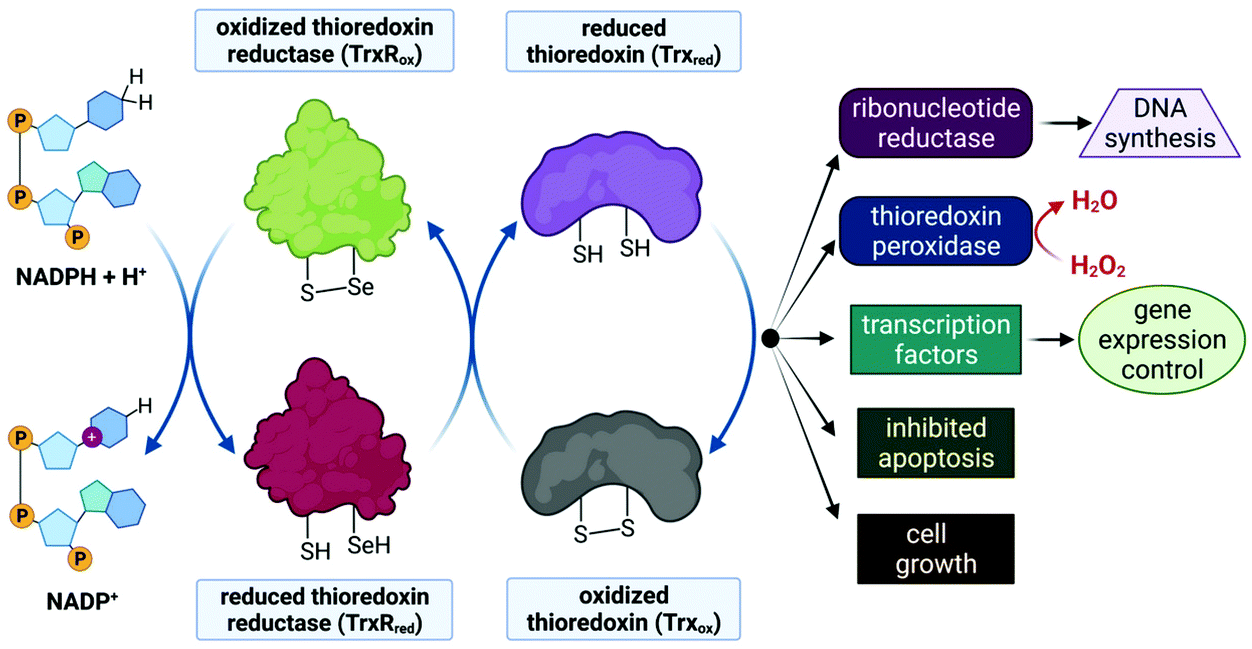 | ||
| Fig. 9 Schematic illustration of thiol-disulphide exchange in the Trx system. Left: the co-enzyme NADPH is oxidized to NADP+ to reduce TrxRox (–S–Se–) to TrxRred (–SH, –SeH), which subsequently reduces Trxox (–S–S–) to Trxred (–SH, –SH), as depicted in the centre two cycles. Trxred is then available to reduce substrate proteins or to carry out required cellular functions (right side). Thus Trx reduces ribonucleotide reductase, an enzyme that reduces ribose to deoxyribose (the sugar of DNA nucleotides required for DNA synthesis). Trx also reduces other proteins such as thioredoxin peroxidase, which is responsible for converting toxic H2O2 to water as well as transcription factors that regulate gene expression. In addition, Trx functions as a cell growth factor and can inhibit apoptosis. | ||
The Au(I) ion of auranofin binds covalently to the selenocysteine (Sec, U) residue in the active site of TrxR with impressive selectivity since the drug binds to the mechanically and structurally similar, but selenium-free enzyme glutathione reductase (GR) at a 1000-fold higher concentration.173,192 Au(I) binding prevents the substrate, Trx, from binding and undergoing subsequent reduction by TrxR. The mitochondrial PT pores (Fig. 3) sense the imbalance of thiol proteins, open and induce MOMP, which ultimately leads to cell death due to the release of cytochrome c. MOMP is also induced by the accumulation of H2O2, as auranofin prevents the removal of hydrogen peroxide by inhibiting TrxR. Furthermore, MOMP leads to the leaking of important respiratory chain components into the cytosol, inevitably causing the respiration chain to uncouple and the production of ATP to cease. Indeed, a decrease in basal oxygen consumption and ATP output of cells has been observed in auranofin-treated cells.52,173,187
The mitochondrion is, in principle, a suitable drug target for chemotherapeutics that can be selectively taken up by tumour cells as this organelle is a crucial regulator of cell death, an endpoint most cancer cells are resistant to. These resistance pathways, due to either defective pro-apoptotic or overexpressed antiapoptotic members, are potentially avoided with compounds that induce MOMP directly.193,194 The challenge, however is to target the mitochondria of cancer cells selectively since mitochondria are present in most human cells. The selective inhibition of mitochondrial TrxR is a strategy which exploits the fact that cancer cells have elevated levels of these proteins to protect themselves from the high levels of ROS generated due to increased proliferation and respiration.187 When these proteins are inhibited, cancer cells are much more vulnerable to these elevated intracellular ROS levels than healthy cells.189,191 Unfortunately, most known TrxR inhibitors cannot distinguish between mitochondrial TrxR and the cytosolic TrxR, and MOMP is only associated with the effective inhibition of mitochondrial TrxR.173,187
Another approach to targeting the mitochondria of cancer cells is the development of a diverse group of compounds called delocalized lipophilic cations (DLCs).195 These compounds, which include metal chelates,196 are characterized by elevated partition coefficient values to readily pass lipid membranes and a delocalized positive charge,197 which allows them to be driven towards negatively charged species. In fact, the mitochondria of cancer cells have elevated mitochondrial membrane potential (Δψm) due to an increased rate of ATP production. Hyperpolarized mitochondria are linked to more aggressive and treatment-resistant cancers. DLCs selectively accumulate in the mitochondria of cancer cells due to the difference of Δψm between healthy and cancer cells.173,187
The DLCs reported by the group of Berners-Price such as the tetrahedral Au(I) phosphane complex 3 (Fig. 10)198,199 and the cationic bis-N-heterocyclic (NHC) carbene Au(I) complex 4 (Fig. 10)200,201 showed high selectivity towards breast cancer cell lines. These findings were not due to chance but a result of carefully considered lipophilic characteristics that are required for the selective accumulation of DLCs in cancer cells.202,203
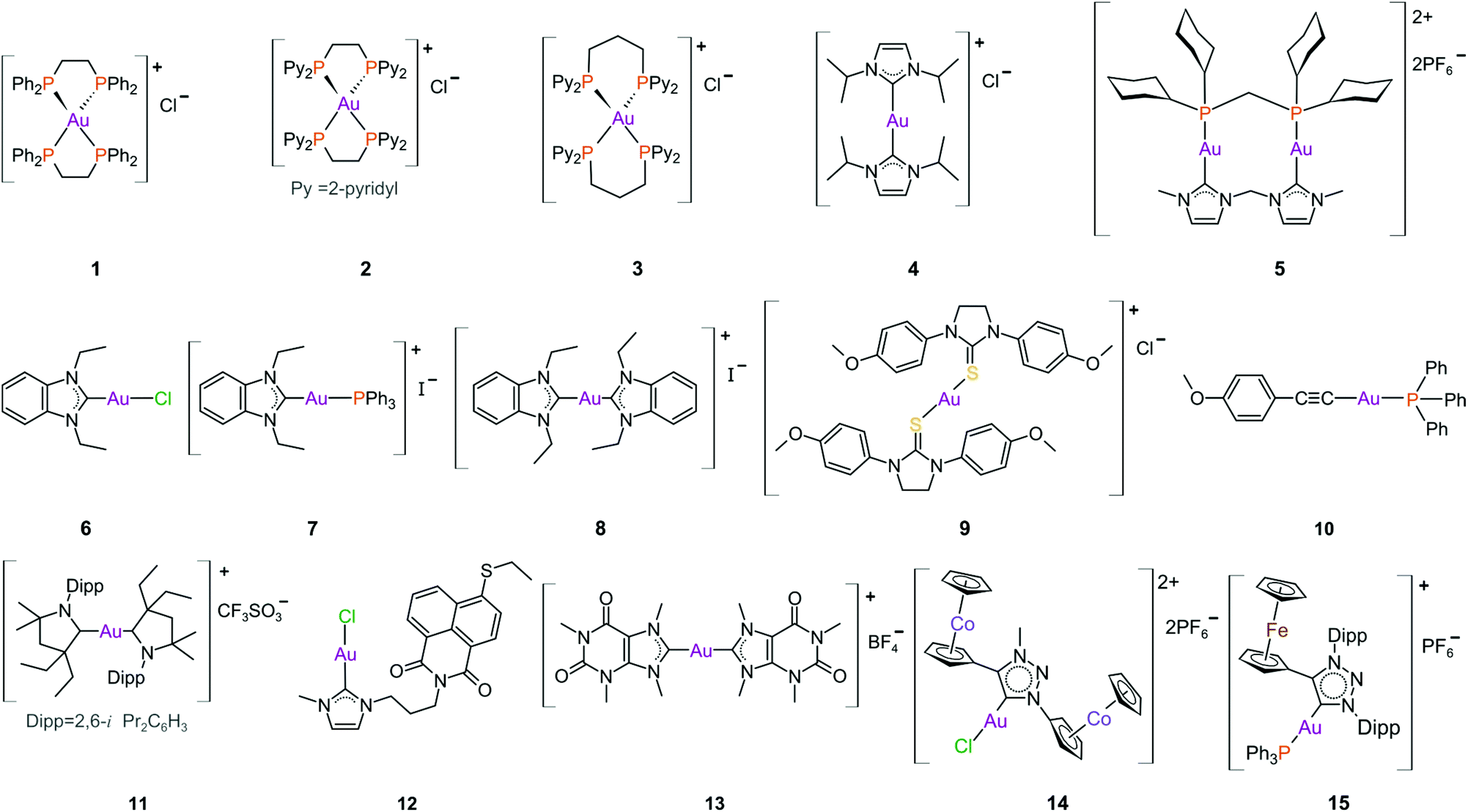 | ||
| Fig. 10 Structures of selected cationic and neutral gold(I) complexes with promising anticancer activity. | ||
The parent compound of 3, the tetrahedral Au(I) phosphane complex 1 (Fig. 10), was developed as an attempt to decrease the known tendency of linear two-coordinate Au(I) complexes to rapidly undergo ligand exchange reactions under physiological conditions. They are very reactive towards thiol proteins in blood serum, limiting their in vivo cytotoxicity, such as the case with auranofin.181,204 Complex 1 is stable against GSH (reduced glutathione), a thiol-containing tripeptide used as a model protein to assess the reactivity of Au(I) complexes towards either blood thiol proteins such albumin or the potential of inhibiting a thiol-containing protein target such as TrxR. Although complex 1 exhibited excellent in vitro and in vivo activity,205 it was found to be very toxic during preclinical trials, likely due to the high lipophilicity of the complex as non-selective accumulation in the mitochondria leads to severe mitochondrial dysfunction. Moreover, the extreme stability of 1 towards GSH suggests that TrxR would be an unlikely intracellular target.167,173
The first-generation tetrahedral phosphane complexes (type 1, Fig. 10) were redeveloped to assess the influence of the lipophilic/hydrophilic ratio on in vitro and in vivo anticancer activity. By substituting the phenyl substituents with more hydrophilic 2-, 3- or 4-pyridyl moieties (salts of type 2), a range of complexes could be assessed with varying degrees of lipophilic and hydrophilic character. The complexes with higher lipophilic character were more cytotoxic, but also less selective. The more hydrophilic complexes were significantly less cytotoxic, due to low cellular uptake, high rates of excretion, and low protein binding affinity. From these results, the complex with intermediate lipophilicity, 2, was the best performing complex, in both in vitro and in vivo studies.167,173,206
The hydrophilic/lipophilic balance was further fine-tuned for the second-generation complexes, where the ethyl linker of 2 was replaced with a propyl bridge (3, Fig. 10). By increasing the chelate ring size, ligand exchange reactions with thiol/selenol proteins are more favourable. Complex 3 therefore possesses lipophilic properties resembling those of the first-generation complex 2, but potentially has auranofin's protein inhibiting properties due to improved ligand exchange.167 Complex 3 is highly selective towards breast cancer cells and mechanistic studies reveal that 3 selectively accumulates in the mitochondria and inhibits TxrR.199
From the same research group, the cationic bis(NHC) Au(I) complexes of type 4 (Fig. 10) were also reported to be selective towards breast cancer cells.201 Imidazol-2-ylidene-based NHCs are a very attractive class of ligands mostly because a wide range of metal complexes can be synthesized from their precursor imidazolium salts.186,207–215 NHCs have captured significant attention over the last few decades, predominantly as highly successful ancillary ligands to transition metals in catalytic processes.216–220 This popularity is warranted as NHCs are easy to synthesize, exceptionally stable, and are amenable to both structural and electronic modulation. Further, the metal–carbene bond is strong, which underpins the traction that NHC metal complexes have in chemical biology as promising anticancer and antimicrobial agents.221–226 Gold(I) bis(NHC) complexes of type 4 also accumulate in the mitochondria and trigger mitochondrial swelling.200 Kinetic studies on these complexes show that the substitution of the NHC ligands is faster for selenium-containing residues (Sec) than for thiols (Cys) thereby offering an explanation for the high, selective inhibition of TrxR over GR. Bulkier NHC N-substituents (e.g., isopropyl in complex 4) induce a slower reactivity towards the protein, most likely due to the added steric protection preventing the substitution with Cys/Sec residues.201
Combining the properties of both phosphanes and NHCs, Che's group reported on the anticancer properties of a dinuclear Au(I) complex with a bridging diphosphane and a bridging bis(NHC) ligand (5, Fig. 10).227 The complex displayed sufficient stability against serum proteins and adequate reactivity towards thiol proteins, which culminated in a potent TrxR inhibitor (0.038 μM, entry 1, Table 1). This complex is the first reported Au(I) derivative to inhibit cancer stem cell activity and inhibit angiogenesis in vivo.24,184
| Entry | Complex | IC50a (μM): cancer cells (cell line, incubation time) | IC50a (μM): non-cancer cells (cell line, incubation time) | IC50a (μM): isolated TrxR |
|---|---|---|---|---|
| Cell viability data were reported as [a] the half maximal inhibitory concentration (μM) required to inhibit the growth of 50% of cells in the culture or 50% of the enzyme activity (IC50 values) and were determined by either [b] the MTT assay (MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide), [c] crystal violet assay, or [d] the cell titre assay. | ||||
| 1 | 5 | 1.8 ± 0.6 (HeLa, 72 h)b | 0.038 (TxrR1, rat) | |
| 2 | 2.5 ± 0.4 (MCF-7, 72 h)b | |||
| 3 | 6 | 4.6 ± 0.03 (MCF-7, 96 h)c | 10.3 ± 1.1 (HEK293, 72 h)c | 0.4 ± 0.04 (TxrR, rat) |
| 4 | 7 | 0.9 ± 0.4 (MCF-7, 96 h)c | 0.4 ± 0.2 (HEK293, 72 h)c | 0.7 ± 0.02 (TxrR, rat) |
| 5 | 8 | 0.8 ± 0.1 (MCF-7, 96 h)c | 3.1 ± 0.5 (HEK293, 72 h)c | 4.9 ± 1.15 (TxrR, rat) |
| 6 | 9 | 14.6 ± 0.7 (HeLa, 72 h)b | K i = 18–36 pM (TxrR, rat) | |
| 7 | 10 | 1.0 ± 0.2 (MCF-7, 96 h)c | 0.05 ± 0.03 (TxrR, rat) | |
| 8 | 11 | 0.3 ± 0.2 (HeLa, 96 h)d | >1 (TxrR, rat) | |
| 9 | 12 | 2.1 ± 0.5 (MCF-7, 96 h)c | 0.4 ± 0.1 (TxrR, rat) | |
| 10 | 13 | 16.2 ± 2.1 (A2780, 72 h)b | >100 (HEK293T, 72 h)b | |
| 11 | 14 | 49.6 ± 4.2 (MDA-MB-231, 72 h)c | 34.3 ± 6.7 (RC-124, 72 h)c | |
| 12 | 15 | 0.2 ± 0.1 (H1975, 48 h)b | 5.4 ± 0.3 (HEK293, 48 h)b | |
| 13 | Cisplatin | 4.7 ± 0.3 (HeLa, 72 h)b | 11.0 ± 2.9 (HEK293T, 72 h)b | |
| 14 | 5.2 ± 1.9 (A2780, 72 h)b | |||
| 15 | 33.7 ± 7.8 (MCF-7, 72 h)b | |||
| 16 | 7.8 ± 0.8 (MDA-MB-231, 72 h)c | |||
| 17 | Auranofin | 1.8 ± 0.1 (HeLa, 72 h)b | 1.7 ± 0.3 (HEK293T, 72 h)b | 0.02 (TxrR, human) |
| 18 | 1.2 ± 0.5 (A2780, 72 h)b | K i = 4 nM | ||
| 19 | 1.0 ± 0.3 (MCF-7, 72 h)b | 0.009 ± 0.000 (TxrR, rat) | ||
The benzimidazole NHC complexes reported by Ott et al.,228 although not very selective in terms of cytotoxicity, contributed valuable information regarding the anticancer properties for this class of NHCs. For instance, the cationic complexes (7 and 8, Fig. 10) were more cytotoxic than the neutral complex 6, with higher cellular uptake and increased accumulation in the mitochondria. However, the neutral complex, due to the more labile trans chloride ligand, was the best and most selective inhibitor of TrxR (24-fold over GR) in this series.228–230
Gold(I) adducts of other ligands such as thiourea (9, Fig. 10) and alkynes, 10, are notably potent TrxR inhibitors. In fact, complex 9 is the most potent complex ever reported with a Ki (inhibitory dissociation constant) value ∼18 to 36 pM (entry 6, Table 1).231 Auranofin by comparison has a Ki value of 4 nM (entry 18, Table 1).192 However, the cytotoxicity of both cisplatin (entry 13, Table 1) and auranofin (entry 17, Table 1) against HeLa cells surpass that of 9. Alkynes like 10 are also potent TrxR inhibitors (entry 7, Table 1) with very promising anticancer metallodrug potential.232 In contrast, the CAAC (cyclic (alkyl)(amino)carbene) gold(I) DLCs of type 11 (Fig. 10) did not inhibit TxrR and were unreactive towards to BSA. This was attributed to very strong metal–carbene bonds involving the CAAC ligands. Interestingly, the most cytotoxic complex (11, Fig. 10), which transcended that of auranofin (entry 8, Table 1), was the least lipophilic of the series examined and exhibited the highest cellular uptake. However, no mitochondrial localization occurred in the presence of 11. Therefore, the cytotoxicity of 11 is likely cell-based, but independent of the mitochondria or the inhibition of related proteins.233
To enhance the cytotoxicity of Au(I) complexes, some Au(I) NHC derivatives have been synthesized with macromolecule-targeting substituents, such as the DNA intercalator naphthalimide (12, Fig. 10), to garner a multi-faceted cytotoxic response. Indeed, complex 12 functions as both a TxrR inhibitor and a DNA intercalator.234
Taking advantage of the imidazole ring of caffeine, Casini et al. developed a series of symmetric bis(carbene) complexes of type 13 (Fig. 10).235 Even though the cytotoxicity of 13 against A2780 cancer cells (entry 10, Table 1) was somewhat poorer than that of both cisplatin (entry 14, Table 1) and auranofin (entry 18, Table 1), its cytotoxicity towards human embryonic kidney HEK-293T cells (non-tumorigenic) was practically negligible (IC50 >100 μM), signalling great specificity towards the investigated tumour cell lines.235 Impressively, complex 13 selectively interacts with DNA G-quadruplex structures in a non-covalent manner by π-stacking atop guanine bases between stacks of telomeric sequences (Fig. 5b).131,235 Following extensive cell-based approaches and shotgun proteomic studies, it was revealed that 13 can enter the nuclei of A2780 cells and bind to DNA. The mechanism of action is considered multi-modal, not only inducing stress responses, but also interfering with the expression of nuclear and telomeric proteins.236
After the first report of Ru(II) and Os(II) NHC complexes based on 1,2,3-triazolylidene ligands (trz) detailing potential anticancer properties and high selectivity towards cancerous cells,237 the cytotoxicity of bimetallic gold(I) trz complexes 14 and 15 (Fig. 10) was investigated. Complex 14238 was moderately cytotoxic towards several breast and colon cancer cell lines (entry 11, Table 1). The complex was, unfortunately, more cytotoxic towards the non-tumorigenic RC-124 kidney cell line (entry 11, Table 1). The monocationic Au(I) complex 15 showed antiproliferative activity against the lung cancer cell lines A549 and H1975 in the sub micromolar range (IC50, 0.2–0.9 μM, entry 12, Table 1) with a rather limited selectivity ratio relative to the non-tumorigenic human embryonic kidney cell line HEK-293 (IC50 ∼5 μM).239
In comparison to the salts of conventional NHCs (i.e., imidazol-2-ylidenes) trz salts are more σ-donating as the removal of one σ-withdrawing nitrogen adjacent to the carbene increases the basicity of the coordinated carbene.240–243 Further fine-tuning of the required properties can be accomplished by altering the wingtip- and C5-substituents of the heterocycle. Synthetic modifications are inherently tractable thanks to the high functional group tolerance of the CuAAC reaction (CuAAC = CuI-catalysed azide–alkyne cycloaddition) employed for the synthesis of the precursors. Since the first report, trz complexes have been successfully incorporated as suitable pre-catalysts for a number of organic transformations and explored for their photophysical properties.244–247 In the realm of bioinorganic chemistry, however, the use of trz derivatives as ligands remains underexplored.
Many more Au(I) complexes bearing structurally diverse ligands have been reported that show promising in vitro and in vivo cytotoxicity. The most probable biomolecular target(s) for binding Au(I) complexes are proteins with readily deprotonated cysteine (–SH) and selenocysteine (–SeH) residues. Covalent adduct formation involving these residues results in outright inhibition or structural changes in the protein which, in turn, may elicit mitochondrial changes and thence apoptosis. Gold(I) complexes do undergo ligand exchange and careful selection of the ligands248 can dictate the reactivity of Au(I) species with thiol proteins, which can subsequently lead to either their selective targeting of cellular proteins or inactivation by serum proteins.249–253 The other biologically relevant oxidation state of gold, namely Au(III), has recently re-emerged as a class of compounds with potent cytotoxicity against cancer cells.254–258
Biological targets and antitumour activity of Au(III) compounds
Gold(III) complexes were among the first complexes investigated as alternatives to platinum drugs and justifiably so, as Au(III) and Pt(II) are both isoelectronic (d8), square planar complexes259–261 with a high binding affinity for DNA nucleotides.151,262 However, the kinetic lability, light sensitivity and the tendency for the Au(III) ion to reduce to Au(I) or Au(0) under physiological conditions hampered investigations considerably until some Au(III) complexes, years later, were reported with improved stability and antitumour activity.253,263Regarding the earlier literature, of the most noteworthy were the complexes reported by Buckley et al. where 16 (Fig. 11) was stable against reduction in the presence of GSH and underwent hydrolysis in water to form the bis(aqua) species [Au(damp)(H2O)2] (damp = 2-[(dimethylamino)methyl]phenyl), similar to cisplatin's reactivity in biological medium. Both the in vitro (entry 1, Table 2) and in vivo cytotoxicity showed promising but modest results compared to cisplatin.23,264–266 During the same period, the monodentate (17, Fig. 11) and bidentate (18) pyridine-based Au(III) complexes were reported by Messori et al.; however, the complexes underwent rapid ligand exchange in water. Despite showing promising in vitro cytotoxicity, especially against cisplatin resistant cell lines (entries 2 and 3, Table 2), these complexes were deemed too unstable for medicinal purposes and were not pursued further.267 Polydentate N-donor ligands considerably improved the stability of Au(III) complexes in solution and soon numerous complexes bearing these ligands were reported. However, reduction to Au(I) in the presence of ascorbic acid or GSH occurs more readily with N-donor functionalities followed by the release of coordinated ligands.24,184,255,268,269 For instance, the cytotoxicity of complexes 19 and 20 (Fig. 12), although very promising (entries 4 and 5, Table 2), was close to the cytotoxicity reported for the gold-free phenanthroline and terpyridine (terpy) ligands, respectively.22 It has been suggested that complex 21 (Fig. 12) also undergoes reduction to Au(I) intracellularly, associates with and causes oxidative damage to proteins and DNA.256,257
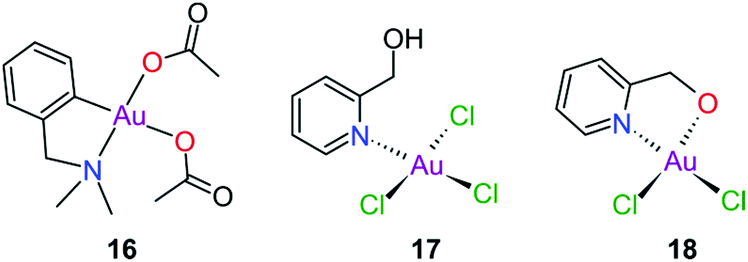 | ||
| Fig. 11 Early examples of Au(III) complexes evaluated for their antitumour activity. | ||
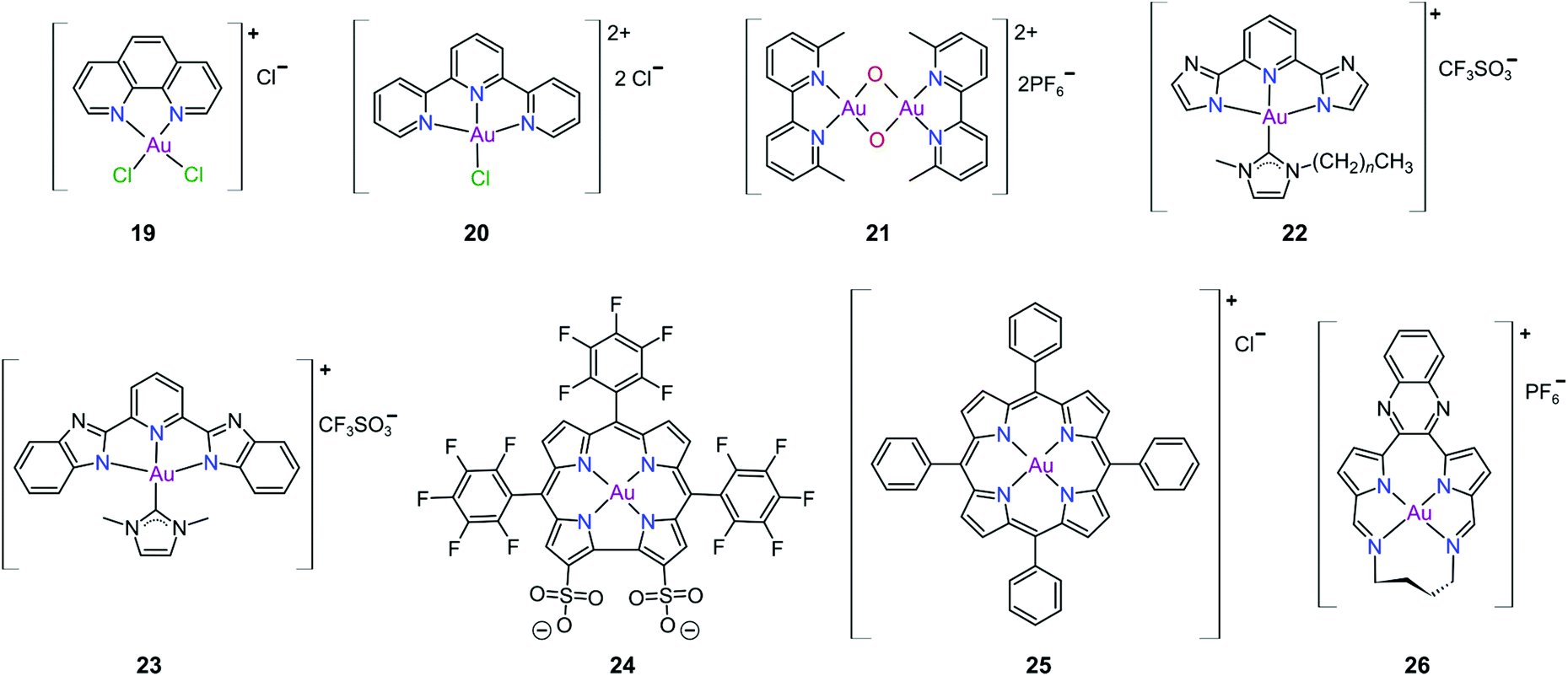 | ||
| Fig. 12 Representative Au(III) chelates of polydentate N-donor ligands (19–23), a corrole derivative (24), a tetra(aryl) porphyrin derivative (25), and a bis(pyrrolide-imine) macrocycle (26). Ligands 24–26 effectively stabilize the Au(III) ion against reduction under physiological conditions. | ||
| Entry | Complex | Cytotoxicity (IC50/μM)a,b A2780c cell line | Cytotoxicity (IC50/μM)a,b A2780Rd cell line |
|---|---|---|---|
| Cell viability data were reported as [a] the half maximal inhibitory concentration (μM) required to inhibit the growth of 50% of cells in the culture (IC50 values) during [b] indicated time of drug exposure and were determined by the sulforhodamine assay. [c] Human ovarian carcinoma cell line. [d] Cisplatin-resistant human ovarian carcinoma cell line. | |||
| 1 | 16 | 3.5, 96 h (1.2) | 35, 96 h (10) |
| 2 | 17 | 10.0 (5.28) | 21.0 (57.0) |
| 3 | 18 | 8.4 (5.28) | 13.8 (57.0) |
| 4 | 19 | 3.8 ± 1.1, 72 h (1.2 ± 0.4) | 3.5 ± 0.9, 72 h (14.2 ± 2.7) |
| 5 | 20 | 0.1, 72 h (1.2 ± 0.4) | 0.4, 72 h (14.2 ± 2.7) |
| 6 | 21 | 1.8 ± 0.2, 72 h (2.1 ± 0.1) | 4.8 ± 0.5, 72 h (24.4 ± 0.1) |
| 7 | 27 | 3.3 ± 1.4, 72 h | 8.2 ± 1.5, 72 h |
Che's group used the redox chemistry of Au(III) chelated by tridentate N-donor pincer ligands to design a range of complexes (cf.22 and 23, Fig. 12) that act dually as fluorescent thiol probes and exhibit cytotoxicity. These complexes bear the highly emissive 2,6-bis(imidazol-2-yl)pyridine (22) and 2,6-bis(benzimidazol-2-yl)pyridine (23) ligands which act as fluorescent probes when the Au(III) ion is reduced to Au(I) by thiol proteins or GSH. Reduction switches on emission (upon excitation) from the metal-free pincer ligand. The released Au(I) ion remains coordinated to the NHC ligand and subsequently inhibits TrxR, therefore being cytotoxic to cancer cells.268
The water soluble corrole-based Au(III) complex 24 (Fig. 12) effectively protects the Au(III) ion from reduction and demetallation. Complex 24 exhibited greater cytotoxicity (entry 3, Table 3) compared with the Ga(III) analogue and outperformed cisplatin on most cell lines. The activity is likely attributed to the low binding affinity of 24 towards HSA.270
| Entry | Complex | IC50a (μM): cancer cells (cell line, timeb) | IC50a (μM): non-cancer cells (cell line, timeb) | Selectivity |
|---|---|---|---|---|
| Cell viability data were reported as [a] the half maximal inhibitory concentration required to inhibit the growth of 50% of cells in the culture (IC50 values, μM) during [b] indicated time of drug exposure and were determined by either [c] the MTT assay (MTT = 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide), or [d] the MTS assay (MTS = 5-(3-carboxymethoxyphenyl)-2-(4,5-dimethylthiazolyl)-3-(4-sulfophenyl) tetrazolium, or [e] the crystal violet assay. [f] IC50 of auranofin measured under the same conditions. | ||||
| 1 | 22 | 1.4 ± 0.2 (HeLa, 72 h)c, (1.8 ± 0.1)f | ||
| 2 | 23 | 14.4 ± 2.2 (HeLa, 72 h)c, (1.8 ± 0.1)f | ||
| 3 | 24 | 19.7 (MDA-MB-231, 72 h), (44.8) | ||
| 4 | 25 | 0.3 ± 0.02 (HeLa, 48 h)c, (11.2 ± 1.0) | 0.91 (CCD-19Lu, 48 h)c | 3.3 |
| 5 | 28 | 7.7 ± 0.1 (HeLa, 72 h)c, (5.2 ± 0.1) | 32.8 ± 2.6 (CCD-19Lu, 72 h)c, (36.2 ± 0.4) | 4.1 |
| 6 | 29a | 3.4 ± 0.6 (HeLa, 72 h)c, (11 ± 1.0) | ||
| 7 | 29b | 7.2 ± 0.6 (HeLa, 72 h)c | 18 ± 0.6 (CCD-19Lu, 72 h)c | 2.5 |
| 8 | 29c | 8.0 ± 0.5 (HeLa, 72 h)c, (11 ± 1.0) | ||
| 9 | 29d | 8.2 ± 0.5 (HeLa, 72 h)c, (11 ± 1.0) | ||
| 10 | 30 | 0.04 ± 0.006 (HeLa, 72 h)c, (11 ± 1.0) | 1.6 ± 0.1 (CCD-19Lu, 72 h)c | 37 |
| 11 | 31a | 0.08 ± 0.007 (HeLa, 72 h)c, (11 ± 1.0) | 1.9 ± 0.1 (CCD-19Lu, 72 h)c | 23 |
| 12 | 31b | 0.2 ± 0.05 (NCI-H460, 72 h)c, (3.5 ± 1.0) | 25 ± 3.8 (CCD-19Lu, 72 h)c | 147 |
| 13 | 32 | 0.3 ± 0.2 (HL60, 72 h)d, (3.7 ± 0.3) | 1.4 ± 0.4 (MRC-5, 72 h)d, (10.7 ± 3.0) | 4.5 |
| 14 | 33 | 2.3 ± 0.8 (MDA-MB-231, 48 h)e | 8.6 ± 2.1 (EA.hy926, 48 h) | 3.8 |
Au(III) porphyrin complexes of type 25 (Fig. 12) are characterized by high redox stability and cannot be reduced by GSH. These complexes are reportedly very successful cytotoxic agents as 25 is significantly more potent in vitro than cisplatin (entry 4, Table 3), especially in cisplatin- and multidrug resistant cell lines. Moreover, 25 can inhibit the self-renewal ability of cancer stem-like cells. The molecular target of 25 remains elusive but extensive mechanistic studies suggest that 25 reduces the mitochondrial membrane potential and targets the mitochondrial chaperone Hsp60 (heat shock protein 60), a protein that is involved in the transport and folding of mitochondrial proteins.24,184,271
Munro's group272 reported the effective inhibition of human topoisomerase IB (topo I) with the use of predominantly planar cationic pyrrole-based Au(III) macrocycles (26, Fig. 12) and proved that these complexes’ mode of action involves highly specific unconventional catalytic inhibition of topo I. Cation 26 intercalates DNA directly at the sequence targeted by the enzyme and therefore blocks binding of topo I to its DNA substrate. Molecular simulations demonstrated the pivotal role of the Au(III) ion in both the intercalation to DNA and enzyme inhibition. The in vitro cytotoxicity of 26 was investigated against the NCI's (National Cancer Institute) panel of 60 human cancer cell lines and the average IC50 value was 16 ± 7 μM; the most sensitive cancer cell line was non-small lung cancer, for which an IC50 of 280 nM was obtained.
The complexes reported by Buckley et al. in the 1990s (e.g., 16, Fig. 11) are stable against reducing agents such as ascorbic acid and GSH, whereas the similarly structured pyridine-based complexes (17 and 18, Fig. 11) were unstable in solution. These early organogold complexes, however, prompted widespread studies to develop multidentate ligand scaffolds for Au(III), which ultimately revealed that at least one C-deprotonated (C−) ligand coordinated to the metal is sufficient to stabilize the Au(III) ion against reduction.24,184,255 Representative examples of cytotoxic organogold(III) complexes chelated by tridentate (CNN)-(27) and bidentate (CN)-(28) ligands are shown in Fig. 13. Compared to cisplatin and several other (NN)-, (NNC)- and (NC)-Au(III) complexes in the series reported by Messori et al.,27327 was among the most cytotoxic (entry 7, Table 2). Interaction with nucleic acids was found to be weak, but 27 does inhibit TrxR2 (0.28 μM) and interacts strongly with other proteins such as lysozyme and cytochrome c.273 Proteomic studies were conducted to identify the effect of 27 on protein expression in whole cells and found that only a few proteins had modified expressions. Of those modified proteins, most of them were involved in the stress response of the cell and proteins associated with glucose metabolism were downregulated, identifying mitochondria as a key target in the cytotoxicity of this complex.255 Compound 28 is a stable, water-soluble salt that is able to form AuIII–GSH adducts and induce ER stress. The in vitro cytotoxicity of 28 was evaluated against several cancer cell lines and although not as potent as the porphyrin complex 25, this simple complex had marginally better selectivity than 25 (entries 4 and 5, Table 3).274
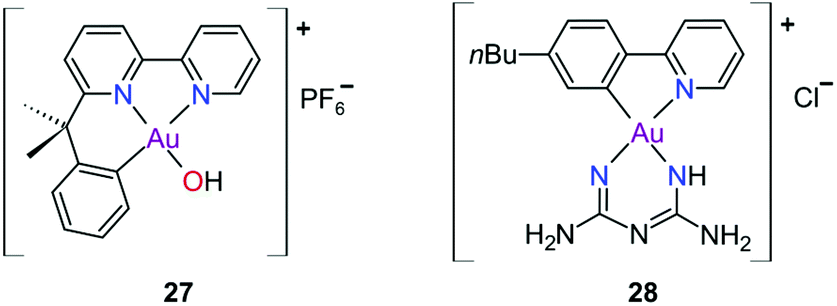 | ||
| Fig. 13 Representative examples of Au(III) chelated by tridentate CNN-donor (27) and bidentate CN-donor pyridine-based ligands (28). | ||
In pursuit of suitable Au(III) DNA-metallointercalators (to potentially mimic the mode of action of [Pt(terpy)L]+), Che and co-workers synthesized structurally varied CNC Au(III) complexes based on 2,6-diphenylpyridine (29–31, Fig. 14). Conceptually, not only would the dianionic [CNC]2− ligand scaffold provide sufficient stabilization for the Au(III) ion, but it also lends itself to diverse structural modifications. Although these modifications included varying the scaffold using 2,4,6-triphenylpyridine and 2,6-dinaphthylpyridine, structural adjustments of the ancillary ligand, L, in [Au(CNC)L]+ ultimately had the largest impact on cytotoxicity (complexes 29a–29d in Fig. 14 and entries 6–9, Table 3).184,275,276 The mechanism of action was also influenced by structural variations of L. For instance, the N-methylimidazole (N-MeIm) complex, 29c, intercalates double-stranded DNA and arrests the cell cycle in S-phase, the phase in which DNA replication takes place. Therefore, it has been suggested that 29c induces apoptosis by inhibiting DNA replication. Conversely, the triphenylphosphine complex (29b) interacts only weakly with DNA.184,275,276 The cytotoxicity of both 29b and 29c were drastically improved by modifying the mononuclear 29b to a dinuclear Au(III) derivative (30, Fig. 14) by utilizing a bridging bis(diphenylphosphino)propane (μ-dppp) ligand277 and substituting the N-MeIm ligand of 29c with NHCs, resulting in complexes 31a and 31b (Fig. 14).231 Complex 30 showed impressive cytotoxicity in the submicromolar range against several cancer cell lines (entry 10, Table 3) with a 37-fold selectivity ratio. Complex 30 is a potent TrxR inhibitor (2.7 nM, rat TxrR1) and induces ER stress.277
 | ||
| Fig. 14 Representative examples of cytotoxic Au(III) CNC pincer complexes. | ||
Complex 31a is 100-fold more active against HeLa cancer cells (entry 11, Table 3) compared with 29c (entry 8, Table 3). The dimethylated NHC Au(III) complex 31b showed remarkable selectivity for non-small cell lung cancer cells (NCI-H460, entry 12, Table 3) relative to normal fibroblast cells (CCD-19Lu, entry 12, Table 3). Complex 31b is able to intercalate into DNA and subsequently prevent topo I mediated relaxation of supercoiled DNA.24,184,231 Moreover, multiple targets for Au(III) CNC NHC complexes of type 31 were identified by Che and co-workers using a clickable photoaffinity probe.278 This study indicated that multiple proteins bind to type 31 cations, such as hsp60, vimentin, nucleoside disphosphatase kinase A, nucleophosmin, nuclease-sensitive element binding protein, and peroxiredoxin 1. Notably, all of these proteins have been identified as potential anticancer drug targets.184,255,276
The pyrazine-based CNC complex 32 also had high in vitro cytotoxicity with a 4.5-fold selectivity ratio (measured against healthy foetal lung fibroblast cells, MRC-5: entry 13, Table 3).279 DNA binding studies revealed that 32 is capable of stabilizing G-quadruplex DNA secondary structures. Surprisingly, the caffeine analogue of 32 was 10 times less cytotoxic and this was attributed to lower cellular uptake.279 The more polar caffeine-based ligand possibly hindered cellular uptake of the complex, which potentially occurs through passive diffusion.
The Au(III) ion of the sterically encumbered CNC pincer complex 33 is sufficiently stabilized by the bis-1,2,3-triazol-5-ylidenes and the anionic carbazolide ligand scaffold against reduction by GSH.280 Furthermore, 33 showed promising selectivity during in vitro cytotoxicity studies (entry 14, Table 3). Further mechanistic studies revealed that 33 binds to dsDNA via partial intercalation, while in silico experiments suggested that 33 might play a role in the stabilization of DNA 3WJs due to its charge (+1) and shape complementarity.280
Since the discovery that Au(III) can be stabilized for use in biological media and protected against reduction under physiological conditions by judicious choice of appropriate ligand scaffolds, many more examples have been reported and mechanistically explored for their antitumour potential. In particular, porphyrin- and CNC pincer-stabilized Au(III) complexes show exceptional promise, although selectivity towards cancer over healthy cells remains a challenge calling for future solutions. Using non-toxic ligands such as NHCs generally improves selectivity. However, too few Au(III) complexes, including Au(III)-NHC derivatives, have been evaluated against healthy cell lines to currently draw any firm conclusions. NHCs, however, are strong σ-donors and have been shown to effectively stabilize both Au(I) and Au(III) under physiological conditions.254–258 Indeed, the first crystal structure of an Au(I)-NHC protein adduct has recently been reported (Fig. 15).281 The adduct involves the plant protein thaumatin (from Thaumatococcus daniellii), which was used as a generic globular protein model. In contrast to most other Au(I/III) complexes, where ligands are often lost upon binding to proteins,282 the NHC moiety in this case was retained by half of the bound Au(I) ions. Since thaumatin lacks His and free Cys residues (Cys in thaumatin is involved exclusively in S–S bridges), the Au(I) complex interacts with several solvent-exposed lysine and arginine residues. We have depicted the likely coordination283 and acid–base chemistry for NHCs284 leading to the two different types of protein-bound Au(I) ions in this system (Fig. 15), rectifying this omission from the original literature.
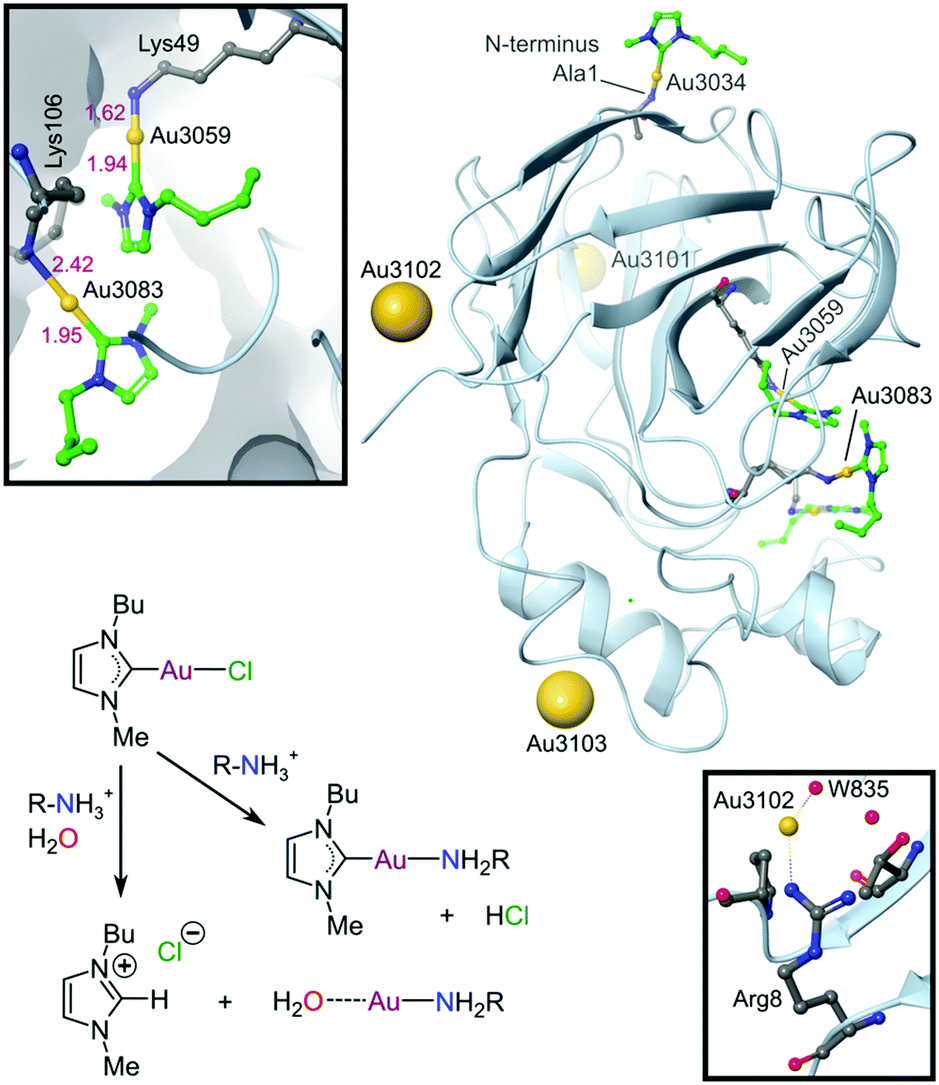 | ||
| Fig. 15 X-ray structure (1.70 Å resolution) of the Au(I)-NHC adduct with thaumatin (PDB 5JVX). The three Au(I) ions bound to surface residues (Arg8, Lys97, Arg175) are indicated by large spheres. These Au(I) ions have water weakly coordinated trans to the protein N-donor ligand and the NHC has dissociated (presumably as the protonated imidazolium ion). Coordination of Au(I) to Arg8 and water 835 is indicated (lower inset) to illustrate a surface-bound NHC-free Au(I) ion. Three other amino groups (Ala1, Lys49, and Lys106) are coordinated to AuI–NHC moieties (green carbon atoms). The upper inset displays the coordination environments for the AuI–NHC moieties bound to Lys49 and Lys106. The reaction scheme summarizes the most likely coordination chemistry for the system. | ||
Outlook
The ability of cytotoxic gold-based anticancer agents to elicit apoptosis has been amply illustrated by both Au(I) and Au(III) complexes over several decades. Judicious choice of the ligand scaffold for the metal ion is critical to achieving this goal and many suitable ligand classes now exist, including conventional N-heterocyclic and triazolylidene-based mesoionic carbenes. The attractive features of carbene ligands, namely their ease of preparation, good stability under physiological conditions, structural diversity, and functional group tunability, foreshadow further exploitation of this class of C-donor ligands in the design of ever more specific and better-targeted metallodrugs.Given the need to develop anticancer drugs with fewer side-effects,110 the use of specific macromolecule-, organelle- or cancer cell-targeting groups (e.g., glucose, folic acid, biotin, oestrogen, peptides, proteins, polymers, micelles, nanoparticles, etc.),285–287 perhaps as part of an ancillary ligand such as a thiolate coordinated trans to a compact, exchange-inert carbene in a two-coordinate Au(I) complex,288 might produce metallodrug candidates better suited to preclinical development. The idea of adding targeting groups or other functional moieties, such as fluorophores, to Au(I)–carbene derivatives has indeed been recently explored. Specifically, the cytotoxic acridine derivatives 34–37 (Fig. 16) were found to localize and fluoresce predominantly in tumour cell lysosomes.289 Some localization within the nucleoli was also observed, consistent with nuclear uptake of the complexes. Complexes 36 and 37 had good IC50 values against MiaPaca2 cells (human pancreatic cancer) of 2.8 ± 0.8 and 3.4 ± 0.8 μM, respectively, and did not disrupt mitochondria,289 as found with several other NHC–Au(I) complexes.222,290 The advantage of fluorescently labelled compounds is that their biodistribution can be imaged at high resolution by confocal microscopy and important mechanistic information regarding their cellular fate straightforwardly delineated.
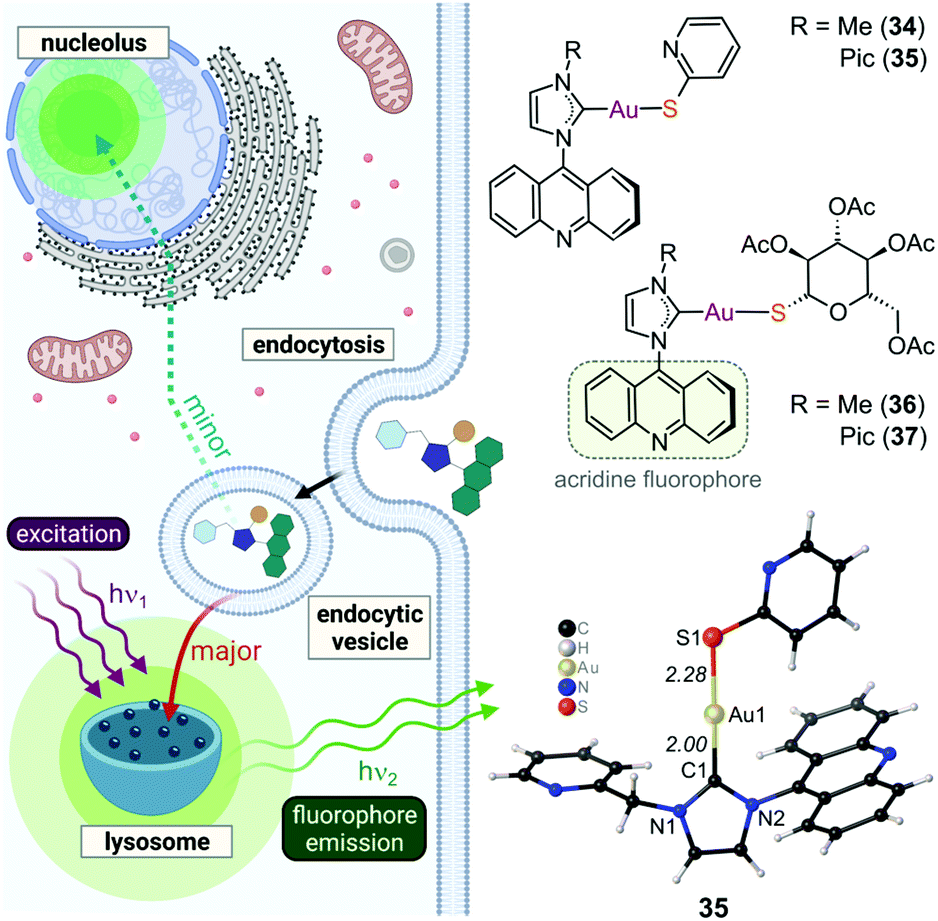 | ||
| Fig. 16 Illustration of the structures of selected cytotoxic NHC–Au(I) thiolate complexes with an acridine fluorophore tag that localize mainly in tumour cell lysosomes and, to a lesser extent, in the nucleolus. The complexes are readily detected by confocal microscopy (405 nm excitation). Localization within lysosomes indicates a probable endocytosis internalization pathway for the compounds. The X-ray structure of 35 is shown at the lower right (CSD code AVAJIS, ref. 289). The bulky substituted NHC ligand probably prevents 34–37 from targeting thioredoxin reductase. | ||
For metallodrug design more broadly, the challenge going forward will be to use in silico design methods and conventional discovery techniques with suitable metal complexes to target specific proteins, for instance TrxR2 in mitochondria188—exclusively in cancer cells. To do this will require selectively targeting: [1] tumour cells (thereby reducing general toxicity), [2] a specific organelle within the cytoplasm (the mitochondria), and [3] the active site Cys-Sec residue pair291 of TrxR2. In short, a molecular recognition trifactor on top of achieving optimal ADMET (absorption, distribution, metabolism, excretion, and toxicity) parameters in vivo.292 The task is clearly formidable, but careful analysis of existing strategies alongside innovative new ideas, coupled with artificial intelligence algorithms293,294 to process large data sets, might yield some truly successful Au(I/III) metallodrug candidates in the next two decades.
Author contributions
DvdW prepared the initial manuscript draft and figures. OQM created the final versions of all figures, some de novo, and others by reconceptualizing and artistically extending notions present in DvdW's draft figures. All three authors co-wrote the final draft of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors (DvdW and DIB) gratefully acknowledge financial support from the National Research Foundation (NRF), South Africa (grant numbers: NRF 108521, NRF 1057404, and NRF 105529). OQM thanks the South African Research Chairs Initiative of the Department of Science and Innovation and NRF of South Africa (SARChI grant number 64799) for generous financial support and the University of the Witwatersrand. DIB thanks the University of Oulu for current financial support. We thank Dr Leonie Harmse (Pharmacology, WITS Medical School) for her insightful comments on the paper.References
- L. A. Torre, R. L. Siegel, E. M. Ward and A. Jemal, Cancer Epidemiol. Biomarkers Prev., 2016, 25, 16–27 CrossRef PubMed.
- F. Bray, A. Jemal, N. Grey, J. Ferlay and D. Forman, Lancet Oncol., 2012, 13, 790–801 CrossRef PubMed.
- S. Bedoui, M. J. Herold and A. Strasser, Nat. Rev. Mol. Cell Biol., 2020, 21, 678–695 CrossRef CAS PubMed.
- J. S. Long and K. M. Ryan, Oncogene, 2012, 31, 5045–5060 CrossRef CAS PubMed.
- N. Zamzami, T. Hirsch, B. Dallaporta, P. X. Petit and G. Kroemer, J. Bioenerg. Biomembr., 1997, 29, 185–193 CrossRef CAS PubMed.
- S. M. Ogbourne, A. Suhrbier, B. Jones, S.-J. Cozzi, G. M. Boyle, M. Morris, D. McAlpine, J. Johns, T. M. Scott, K. P. Sutherland, J. M. Gardner, T. T. T. Le, A. Lenarczyk, J. H. Aylward and P. G. Parsons, Cancer Res., 2004, 64, 2833–2839 CrossRef CAS PubMed.
- J. Zeng, Y. Sun, K. Wu, L. Li, G. Zhang, Z. Yang, Z. Wang, D. Zhang, Y. Xue, Y. Chen, G. Zhu, X. Wang and D. He, Mol. Cancer Ther., 2011, 10, 104–116 CrossRef CAS PubMed.
- A. Rovini, A. Savry, D. Braguer and M. Carré, Biochim. Biophys. Acta, Bioenerg., 2011, 1807, 679–688 CrossRef CAS PubMed.
- D. Decaudin, I. Marzo, C. Brenner and G. Kroemer, Int. J. Oncol., 1998, 12, 141–193 CAS.
- M. Vijayaraj Reddy and K. Randerath, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1987, 179, 75–88 CrossRef.
- A. Rabbani-Chadegani, S. Keyvani-Ghamsari and N. Zarkar, Spectrochim. Acta, Part A, 2011, 84, 62–67 CrossRef CAS PubMed.
- S. M. Hecht, J. Nat. Prod., 2000, 63, 158–168 CrossRef CAS PubMed.
- D. Mohamed, S. Mowaka, J. Thomale and M. W. Linscheid, Chem. Res. Toxicol., 2009, 22, 1435–1446 Search PubMed.
- S. M. Zeman, D. R. Phillips and D. M. Crothers, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 11561–11565 CrossRef CAS PubMed.
- P. D. Lawley and D. H. Phillips, Mutat. Res., Fundam. Mol. Mech. Mutagen., 1996, 355, 13–40 CrossRef PubMed.
- S. R. Rajski and R. M. Williams, Chem. Rev., 1998, 98, 2723–2796 CrossRef CAS PubMed.
- L. H. Hurley, Nat. Rev. Cancer, 2002, 2, 188–200 CrossRef CAS PubMed.
- G. Chu, J. Biol. Chem., 1994, 269, 787–790 CrossRef CAS.
- J. J. Soldevila-Barreda and P. J. Sadler, Curr. Opin. Chem. Biol., 2015, 25, 172–183 CrossRef CAS PubMed.
- A. Casini, R. W.-Y. Sun and I. Ott, in Metal Ions in Life Sciences, ed. A. Sigel, H. Sigel, E. Freisinger and R. K. O. Sigel, De Gruyter, Berlin, Boston, 2018, vol. 18, pp. 199–217 Search PubMed.
- C. Gabbiani, G. Mastrobuoni, F. Sorrentino, B. Dani, M. P. Rigobello, A. Bindoli, M. A. Cinellu, G. Pieraccini, L. Messori and A. Casini, MedChemComm, 2011, 2, 50–54 RSC.
- L. Messori, F. Abbate, G. Marcon, P. Orioli, M. Fontani, E. Mini, T. Mazzei, S. Carotti, T. O'Connell and P. Zanello, J. Med. Chem., 2000, 43, 3541–3548 CrossRef CAS PubMed.
- R. V. Parish, R. G. Buckley, S. P. Fricker, J. Mack, L. Hargreaves, J. P. Wright, A. M. Elsome and B. R. C. Theobald, J. Chem. Soc., Dalton Trans., 1996, 69–74 RSC.
- T. Zou, C. Tung Lum, C.-N. Lok, J.-J. Zhang and C.-M. Che, Chem. Soc. Rev., 2015, 44, 8786–8801 RSC.
- K.-C. Tong, D. Hu, P.-K. Wan, C.-N. Lok and C.-M. Che, in Advances in Inorganic Chemistry, ed. P. J. Sadler and R. van Eldik, Academic Press, 2020, vol. 75, pp. 87–119 Search PubMed.
- W. Henderson, in Advances in Organometallic Chemistry, ed. R. West and A. F. Hill, Academic Press, 2006, vol. 54, pp. 207–265 Search PubMed.
- N. A. Campbell, J. B. Reece, L. A. Urry, M. L. Cain, S. A. Wasserman, P. V. Minorsky and R. B. Jackson, Biology, 8th Edition, Pearson Benjamin Cummings, 8th edn, 2008 Search PubMed.
- K. Collins, T. Jacks and N. P. Pavletich, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 2776–2778 CrossRef CAS PubMed.
- M. J. Solomon and P. Kaldis, in Cell Cycle Control, ed. M. Pagano, Springer, Berlin, Heidelberg, 1998, pp. 79–109 Search PubMed.
- C. J. Sherr, Science, 1996, 274, 1672–1674 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2000, 100, 57–70 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- D. X. Nguyen and J. Massagué, Nat. Rev. Genet., 2007, 8, 341–352 CrossRef CAS PubMed.
- S. A. Frank, Nat. Rev. Genet., 2004, 5, 764–772 CrossRef CAS PubMed.
- G. M. Cooper and R. E. Hausman, The cell: A Molecular Approach, ASM Press, Washington, 4th edn, 2007 Search PubMed.
- E. Stefan and K. Bister, in Viruses, Genes, and Cancer, ed. E. Hunter and K. Bister, Springer, Cambridge, 2017, vol. 407, pp. 117–151 Search PubMed.
- C. Braicu, M. Buse, C. Busuioc, R. Drula, D. Gulei, L. Raduly, A. Rusu, A. Irimie, A. G. Atanasov, O. Slaby, C. Ionescu and I. Berindan-Neagoe, Cancers, 2019, 11, 1–25 Search PubMed.
- P. Bachireddy, P. K. Bendapudi and D. W. Felsher, Clin. Cancer Res., 2005, 11, 4278–4281 CrossRef CAS PubMed.
- P. W. Hinds and R. A. Weinberg, Curr. Opin. Genet. Dev., 1994, 4, 135–141 CrossRef CAS PubMed.
- A. L. Gartel and S. K. Radhakrishnan, Cancer Res., 2005, 65, 3980–3985 CrossRef CAS PubMed.
- A. L. Gartel and A. L. Tyner, Exp. Cell Res., 1999, 246, 280–289 CrossRef CAS PubMed.
- V. Labi and M. Erlacher, Cell Death Dis., 2015, 6, e1675–e1675 CrossRef CAS PubMed.
- K. Murphy, Janeway's Immunobiology, Garland Science, 8th edn, 2011 Search PubMed.
- Z. Jin and W. S. El-Deiry, Cancer Biol. Ther., 2005, 4, 147–171 CrossRef PubMed.
- S. W. G. Tait and D. R. Green, Nat. Rev. Mol. Cell Biol., 2010, 11, 621–632 CrossRef CAS PubMed.
- S. H. Suhaili, H. Karimian, M. Stellato, T.-H. Lee and M.-I. Aguilar, Biophys. Rev., 2017, 9, 443–457 CrossRef CAS PubMed.
- V. Giorgio, L. Guo, C. Bassot, V. Petronilli and P. Bernardi, Cell Calcium, 2018, 70, 56–63 CrossRef CAS PubMed.
- B. Wacquier, L. Combettes and G. Dupont, Sci. Rep., 2020, 10, 3924 CrossRef CAS PubMed.
- A. Sesso, J. E. Belizário, M. M. Marques, M. L. Higuchi, R. I. Schumacher, A. Colquhoun, E. Ito and J. Kawakami, Anat. Rec., 2012, 295, 1647–1659 CrossRef CAS PubMed.
- H. Zischka, N. Larochette, F. Hoffmann, D. Hamöller, N. Jägemann, J. Lichtmannegger, L. Jennen, J. Müller-Höcker, F. Roggel, M. Göttlicher, A. M. Vollmar and G. Kroemer, Anal. Chem., 2008, 80, 5051–5058 CrossRef CAS PubMed.
- Z. Jin and W. S. El-Deiry, Cancer Biol. Ther., 2005, 4, 147–171 CrossRef PubMed.
- K. Kannan and S. K. Jain, Pathophysiology, 2000, 7, 153–163 CrossRef CAS PubMed.
- S. Elmore, Toxicol. Pathol., 2007, 35, 495–516 CrossRef CAS PubMed.
- S. J. Korsmeyer, M. C. Wei, M. Saito, S. Weiler, K. J. Oh and P. H. Schlesinger, Cell Death Differ., 2000, 7, 1166–1173 CrossRef CAS PubMed.
- M. Zhang, J. Zheng, R. Nussinov and B. Ma, Sci. Rep., 2017, 7, 2635 CrossRef PubMed.
- T. C. Cheng, C. Hong, I. V. Akey, S. Yuan and C. W. Akey, eLife, 2016, 5, e17755 CrossRef PubMed.
- K. Murphy, Janeway's Immunobiology, Garland Science, 8th edn, 2011 Search PubMed.
- H.-L. Ou and B. Schumacher, Blood, 2018, 131, 488–495 CrossRef CAS PubMed.
- B. J. Aubrey, G. L. Kelly, A. Janic, M. J. Herold and A. Strasser, Cell Death Differ., 2018, 25, 104–113 CrossRef CAS PubMed.
- A. Villunger, E. M. Michalak, L. Coultas, F. Müllauer, G. Böck, M. J. Ausserlechner, J. M. Adams and A. Strasser, Science, 2003, 302, 1036–1038 CrossRef CAS PubMed.
- S. Marchi, S. Patergnani, S. Missiroli, G. Morciano, A. Rimessi, M. R. Wieckowski, C. Giorgi and P. Pinton, Cell Calcium, 2018, 69, 62–72 CrossRef CAS PubMed.
- E. Szegezdi, S. E. Logue, A. M. Gorman and A. Samali, EMBO Rep., 2006, 7, 880–885 CrossRef CAS PubMed.
- A. M. Gorman, S. J. M. Healy, R. Jäger and A. Samali, Pharmacol. Ther., 2012, 134, 306–316 CrossRef CAS PubMed.
- D. D. Bufalo, A. Biroccio, C. Leonetti and G. Zupi, FASEB J., 1997, 11, 947–953 CrossRef PubMed.
- T. Knight, D. Luedtke, H. Edwards, J. W. Taub and Y. Ge, Biochem. Pharmacol., 2019, 162, 250–261 CrossRef CAS PubMed.
- I. Kapoor, J. Bodo, B. T. Hill, E. D. Hsi and A. Almasan, Cell Death Dis., 2020, 11, 1–11 CrossRef PubMed.
- M. S. D'Arcy, Cell Biol. Int., 2019, 43, 582–592 CrossRef PubMed.
- P. Golstein and G. Kroemer, Trends Biochem. Sci., 2007, 32, 37–43 CrossRef CAS PubMed.
- J. Doherty and E. H. Baehrecke, Nat. Cell Biol., 2018, 20, 1110–1117 CrossRef CAS PubMed.
- D. Verzella, A. Pescatore, D. Capece, D. Vecchiotti, M. V. Ursini, G. Franzoso, E. Alesse and F. Zazzeroni, Cell Death Dis., 2020, 11, 1–14 CrossRef PubMed.
- G. Kroemer, L. Galluzzi, P. Vandenabeele, J. Abrams, E. S. Alnemri, E. H. Baehrecke, M. V. Blagosklonny, W. S. El-Deiry, P. Golstein, D. R. Green, M. Hengartner, R. A. Knight, S. Kumar, S. A. Lipton, W. Malorni, G. Nuñez, M. E. Peter, J. Tschopp, J. Yuan, M. Piacentini, B. Zhivotovsky and G. Melino, Cell Death Differ., 2009, 16, 3–11 CrossRef CAS PubMed.
- J. A. Hickman, Curr. Opin. Genet. Dev., 2002, 12, 67–72 CrossRef CAS PubMed.
- S. Fulda, Front. Oncol., 2017, 7, 128 CrossRef PubMed.
- X. Wang, H. Zheng, T. Shou, C. Tang, K. Miao and P. Wang, J. Orthop. Surg., 2017, 12, 52 CrossRef PubMed.
- M. A. Knowles and C. D. Hurst, Nat. Rev. Cancer, 2015, 15, 25–41 CrossRef CAS PubMed.
- R. M. Neve, K. Chin, J. Fridlyand, J. Yeh, F. L. Baehner, T. Fevr, L. Clark, N. Bayani, J.-P. Coppe, F. Tong, T. Speed, P. T. Spellman, S. DeVries, A. Lapuk, N. J. Wang, W.-L. Kuo, J. L. Stilwell, D. Pinkel, D. G. Albertson, F. M. Waldman, F. McCormick, R. B. Dickson, M. D. Johnson, M. Lippman, S. Ethier, A. Gazdar and J. W. Gray, Cancer Cell, 2006, 10, 515–527 CrossRef CAS PubMed.
- M. A. Pierotti, F. Berrino, M. Gariboldi, C. Melani, A. Mogavero, T. Negri, P. Pasanisi and S. Pilotti, Oncogene, 2013, 32, 1475–1487 CrossRef CAS PubMed.
- M. Olivo, R. Bhuvaneswari, S. S. Lucky, N. Dendukuri and P. S.-P. Thong, Pharmaceuticals, 2010, 3, 1507–1529 CrossRef PubMed.
- K. W. Kohn, Cancer Res., 1996, 56, 5533–5546 CAS.
- N. Farrell, in Advances in Inorganic Chemistry, 1989, vol. 75, pp. 8–45 Search PubMed.
- J. E. Deweese and N. Osheroff, Nucleic Acids Res., 2009, 37, 738–748 CrossRef CAS PubMed.
- X. Wang and Z. Guo, Chem. Soc. Rev., 2013, 42, 202–224 RSC.
- C. Wu, D. Han, T. Chen, L. Peng, G. Zhu, M. You, L. Qiu, K. Sefah, X. Zhang and W. Tan, J. Am. Chem. Soc., 2013, 135, 18644–18650 CrossRef CAS PubMed.
- N. Aydemir and R. Bilaloğlu, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2003, 537, 43–51 CrossRef CAS.
- T. Tamaki, Y. Naomoto, S. Kimura, R. Kawashima, Y. Shirakawa, K. Shigemitsu, T. Yamatsuji, M. Haisa, M. Gunduz and N. Tanaka, J. Int. Med. Res., 2003, 31, 6–16 CrossRef CAS PubMed.
- E. Basch, A. Iasonos, T. McDonough, A. Barz, A. Culkin, M. G. Kris, H. I. Scher and D. Schrag, Lancet Oncol., 2006, 7, 903–909 CrossRef PubMed.
- J. D. Watson and F. H. C. Crick, Cold Spring Harbor Symp. Quant. Biol., 1953, 18, 123–131 CrossRef CAS PubMed.
- R. R. Sinden, DNA Structure and Function, Elsevier, 1994, pp. 1–57 Search PubMed.
- P. Cheung, C. D. Allis and P. Sassone-Corsi, Cell, 2000, 103, 263–271 CrossRef CAS PubMed.
- A. J. Bannister and T. Kouzarides, Cell Res., 2011, 21, 381–395 CrossRef CAS PubMed.
- T. J. Richmond and C. A. Davey, Nature, 2003, 423, 145–150 CrossRef CAS PubMed.
- A. Marathe, D. Karandur and M. Bansal, BMC Struct. Biol., 2009, 9, 24 CrossRef PubMed.
- S. B. Zimmerman, Annu. Rev. Biochem., 1982, 51, 395–427 CrossRef CAS PubMed.
- B. J. Pages, D. L. Ang, E. P. Wright and J. R. Aldrich-Wright, Dalton Trans., 2015, 44, 3505–3526 RSC.
- M. I. Nejad, K. M. Johnson, N. E. Price and K. S. Gates, Biochemistry, 2016, 55, 7033–7041 CrossRef CAS PubMed.
- S. M. Rink and P. B. Hopkins, Bioorg. Med. Chem. Lett., 1995, 5, 2845–2850 CrossRef CAS.
- Y. Huang and L. Li, Transl. Cancer Res., 2013, 2, 144–154 CAS.
- F. Coste, J.-M. Malinge, L. Serre, M. Leng, C. Zelwer, W. Shepard and M. Roth, Nucleic Acids Res., 1999, 27, 1837–1846 CrossRef CAS PubMed.
- M. Sastry, R. Fiala, R. Lipman, M. Tomasz and D. J. Patel, J. Mol. Biol., 1995, 247, 338–359 CrossRef CAS PubMed.
- G. Subramaniam, M. M. Paz, G. Suresh Kumar, A. Das, Y. Palom, C. C. Clement, D. J. Patel and M. Tomasz, Biochemistry, 2001, 40, 10473–10484 CrossRef CAS PubMed.
- Y. Pommier, G. Kohlhagen, C. Bailly, M. Waring, A. Mazumder and K. W. Kohn, Biochemistry, 1996, 35, 13303–13309 CrossRef CAS PubMed.
- P. M. Takahara, A. C. Rosenzweig, C. A. Frederick and S. J. Lippard, Nature, 1995, 377, 649–652 CrossRef CAS PubMed.
- M. S. Davies, S. J. Berners-Price and T. W. Hambley, Inorg. Chem., 2000, 39, 5603–5613 CrossRef CAS PubMed.
- D. Tang, R. Kang, H. J. Zeh and M. T. Lotze, Biochim. Biophys. Acta, Gene Regul. Mech., 2010, 1799, 131–140 CrossRef CAS PubMed.
- J. T. Reardon and A. Sancar, Progress in Nucleic Acid Research and Molecular Biology, Academic Press, 2005, vol. 79, pp. 183–235 Search PubMed.
- A. Ciccia and S. J. Elledge, Mol. Cell, 2010, 40, 179–204 CrossRef CAS PubMed.
- J. W. Harper and S. J. Elledge, Mol. Cell, 2007, 28, 739–745 CrossRef CAS PubMed.
- J. C. Dabrowiak, Metals in Medicine, John Wiley & Sons Ltd, 2017, pp. 91–156 Search PubMed.
- S. J. Berners-Price, L. Ronconi and P. J. Sadler, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 49, 65–98 CrossRef CAS.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- T. W. Hambley, J. Chem. Soc., Dalton Trans., 2001, 2711–2718 RSC.
- A. Eastman, in Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug, ed. B. Lippert, Wiley, 1999, pp. 111–134 Search PubMed.
- C. A. Rabik and M. E. Dolan, Cancer Treat. Rev., 2007, 33, 9–23 CrossRef CAS PubMed.
- Z. H. Siddik, Oncogene, 2003, 22, 7265–7279 CrossRef CAS PubMed.
- T. Makovec, Radiol. Oncol., 2019, 53, 148–158 CrossRef CAS PubMed.
- E. R. Jamieson and S. J. Lippard, Chem. Rev., 1999, 99, 2467–2498 CrossRef CAS PubMed.
- M. D'Incalci and C. M. Galmarini, Mol. Cancer Ther., 2010, 9, 2157–2163 CrossRef PubMed.
- V. N. Iyer and W. Szybalski, Proc. Natl. Acad. Sci. U. S. A., 1963, 50, 355–362 CrossRef CAS PubMed.
- M. Tomasz, R. Lipman, D. Chowdary, J. Pawlak, G. Verdine and K. Nakanishi, Science, 1987, 235, 1204–1208 CrossRef CAS PubMed.
- M. Binaschi, M. Bigioni, A. Cipollone, C. Rossi, C. Goso, C. A. Maggi, G. Capranico and F. Animati, Curr. Med. Chem.: Anti-Cancer Agents, 2001, 1, 113–130 CrossRef CAS PubMed.
- P. Meresse, E. Dechaux, C. Monneret and E. Bertounesque, Curr. Med. Chem., 2004, 11, 2443–2466 CrossRef CAS PubMed.
- O. Hovorka, V. Šubr, D. Větvička, L. Kovář, J. Strohalm, M. Strohalm, A. Benda, M. Hof, K. Ulbrich and B. Říhová, Eur. J. Pharm. Biopharm., 2010, 76, 514–524 CrossRef CAS PubMed.
- W. Ross, T. Rowe, B. Glisson, J. Yalowich and L. Liu, Cancer Res., 1984, 44, 5857–5860 CAS.
- C.-C. Wu, T.-K. Li, L. Farh, L.-Y. Lin, T.-S. Lin, Y.-J. Yu, T.-J. Yen, C.-W. Chiang and N.-L. Chan, Science, 2011, 333, 459–462 CrossRef CAS PubMed.
- Y. Pommier, E. Leo, H. Zhang and C. Marchand, Chem. Biol., 2010, 17, 421–433 CrossRef CAS PubMed.
- J. J. Champoux, Annu. Rev. Biochem., 2001, 70, 369–413 CrossRef CAS PubMed.
- C. R. Wilson, A. M. Fagenson, W. Ruangpradit, M. T. Muller and O. Q. Munro, Inorg. Chem., 2013, 52, 7889–7906 CrossRef CAS PubMed.
- G. W. Collie and G. N. Parkinson, Chem. Soc. Rev., 2011, 40, 5867–5892 RSC.
- A. T. Phan, FEBS J., 2010, 277, 1107–1117 CrossRef CAS PubMed.
- S. N. Georgiades, N. H. Abd Karim, K. Suntharalingam and R. Vilar, Angew. Chem., Int. Ed., 2010, 49, 4020–4034 CrossRef CAS PubMed.
- C. Bazzicalupi, M. Ferraroni, F. Papi, L. Massai, B. Bertrand, L. Messori, P. Gratteri and A. Casini, Angew. Chem., Int. Ed., 2016, 55, 4256–4259 CrossRef CAS PubMed.
- F. Guarra, T. Marzo, M. Ferraroni, F. Papi, C. Bazzicalupi, P. Gratteri, G. Pescitelli, L. Messori, T. Biver and C. Gabbiani, Dalton Trans., 2018, 47, 16132–16138 RSC.
- K. McQuaid, J. P. Hall, L. Baumgaertner, D. J. Cardin and C. J. Cardin, Chem. Commun., 2019, 55, 9116–9119 RSC.
- Ö. Karaca, S. M. Meier-Menches, A. Casini and F. E. Kühn, Chem. Commun., 2017, 53, 8249–8260 RSC.
- K. Duskova, J. Lamarche, S. Amor, C. Caron, N. Queyriaux, M. Gaschard, M.-J. Penouilh, G. de Robillard, D. Delmas, C. H. Devillers, A. Granzhan, M.-P. Teulade-Fichou, M. Chavarot-Kerlidou, B. Therrien, S. Britton and D. Monchaud, J. Med. Chem., 2019, 62, 4456–4466 CrossRef CAS PubMed.
- J. Zell, F. R. Sperti, S. Britton and D. Monchaud, RSC Chem. Biol., 2021, 2, 47–76 RSC.
- I. Gamba, G. Rama, E. Ortega-Carrasco, J.-D. Maréchal, J. Martínez-Costas, M. E. Vázquez and M. Vázquez López, Chem. Commun., 2014, 50, 11097–11100 RSC.
- A. Oleksi, A. G. Blanco, R. Boer, I. Usón, J. Aymamí, A. Rodger, M. J. Hannon and M. Coll, Angew. Chem., 2006, 118, 1249–1253 CrossRef.
- L. Cerasino, M. J. Hannon and E. Sletten, Inorg. Chem., 2007, 46, 6245–6251 CrossRef PubMed.
- V. H. S. van Rixel, A. Busemann, M. F. Wissingh, S. L. Hopkins, B. Siewert, C. van de Griend, M. A. Siegler, T. Marzo, F. Papi, M. Ferraroni, P. Gratteri, C. Bazzicalupi, L. Messori and S. Bonnet, Angew. Chem., 2019, 131, 9478–9482 CrossRef.
- A. L. c. eczkowska and R. Vilar, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2013, 109, 299 RSC.
- A. Gelasco and S. J. Lippard, in Metallopharmaceuticals I: DNA Interactions, ed. M. J. Clarke and P. J. Sadler, Springer, Berlin, Heidelberg, 1999, pp. 1–43 Search PubMed.
- B. Rosenberg, L. Van Camp and T. Krigas, Nature, 1965, 205, 698–699 CrossRef CAS PubMed.
- L. Melidis, H. J. Hill, N. J. Coltman, S. P. Davies, K. Winczura, T. Chauhan, J. S. Craig, A. Garai, C. A. J. Hooper, R. T. Egan, J. A. McKeating, N. J. Hodges, Z. Stamataki, P. Grzechnik and M. J. Hannon, Angew. Chem., Int. Ed., 2021, 60, 18144–18151 CrossRef CAS PubMed.
- F. Arjmand, Z. Afsan, S. Sharma, S. Parveen, I. Yousuf, S. Sartaj, H. R. Siddique and S. Tabassum, Coord. Chem. Rev., 2019, 387, 47–59 CrossRef CAS.
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F.-X. Wang, W.-Y. Zhang and Z. Zhang, Chem. Sci., 2020, 11, 12888–12917 RSC.
- K. Sargsyan, C.-C. Lin, T. Chen, C. Grauffel, Y.-P. Chen, W.-Z. Yang, H. S. Yuan and C. Lim, Chem. Sci., 2020, 11, 9904–9909 RSC.
- L. Thurakkal, S. Singh, R. Roy, P. Kar, S. Sadhukhan and M. Porel, Chem. Phys. Lett., 2021, 763, 138193 CrossRef CAS PubMed.
- H. Zhao and Y. Ning, Gold Bull., 2001, 34, 24–29 CrossRef CAS.
- G. J. Higby, Gold Bull., 1982, 15, 130–140 CrossRef CAS PubMed.
- P. J. Sadler, Gold Bull., 1976, 9, 110–118 CrossRef CAS.
- P. V. Simpson, N. M. Desai, I. Casari, M. Massi and M. Falasca, Future Med. Chem., 2019, 11, 119–135 CrossRef CAS PubMed.
- C. S. Allardyce and P. J. Dyson, Dalton Trans., 2016, 45, 3201–3209 RSC.
- T. Gianferrara, I. Bratsos and E. Alessio, Dalton Trans., 2009, 7588–7598 RSC.
- S. P. Fricker, Dalton Trans., 2007, 4903–4917 RSC.
- U. Ndagi, N. Mhlongo and M. E. Soliman, Drug Des., Dev. Ther., 2017, 11, 599–616 CrossRef CAS PubMed.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- R. G. Pearson, Coord. Chem. Rev., 1990, 100, 403–425 CrossRef CAS.
- H.-H. Otto and T. Schirmeister, Chem. Rev., 1997, 97, 133–172 CrossRef CAS PubMed.
- L. Massai, L. Messori, N. Micale, T. Schirmeister, L. Maes, D. Fregona, M. A. Cinellu and C. Gabbiani, BioMetals, 2017, 30, 313–320 CrossRef CAS PubMed.
- D. Gibson, J. Inorg. Biochem., 2019, 191, 77–84 CrossRef CAS PubMed.
- X. Han, J. Sun, Y. Wang and Z. He, Med. Res. Rev., 2015, 35, 1268–1299 CrossRef CAS PubMed.
- S. P. Pricker, Gold Bull., 1996, 29, 53–60 CrossRef.
- P. J. Sadler and R. E. Sue, Met.-Based Drugs, 1994, 1, 107–144 CrossRef CAS PubMed.
- J. R. Ward, Am. J. Med., 1988, 85, 39–44 CrossRef CAS PubMed.
- I. Ott, Coord. Chem. Rev., 2009, 253, 1670–1681 CrossRef CAS.
- S. J. Berners-Price and A. Filipovska, Metallomics, 2011, 3, 863–873 CrossRef CAS PubMed.
- B. M. Sutton, Gold Bull., 1986, 19, 15–16 CrossRef CAS.
- B. M. Sutton, E. McGusty, D. T. Walz and M. J. DiMartino, J. Med. Chem., 1972, 15, 1095–1098 CrossRef CAS PubMed.
- K. Albrecht, K. Krüger, J. Wollenhaupt, R. Alten, M. Backhaus, C. Baerwald, W. Bolten, J. Braun, H. Burkhardt, G. R. Burmester, M. Gaubitz, A. Gause, E. Gromnica-Ihle, H. Kellner, J. Kuipers, A. Krause, H.-M. Lorenz, B. Manger, H. Nüßlein, H.-G. Pott, A. Rubbert-Roth, M. Schneider, C. Specker, H. Schulze-Koops, H.-P. Tony, S. Wassenberg and U. Müller-Ladner, Rheumatol. Int., 2014, 34, 1–9 CrossRef CAS PubMed.
- C. Roder and M. J. Thomson, Drugs RD, 2015, 15, 13–20 CrossRef CAS PubMed.
- X. Zhang, K. Selvaraju, A. A. Saei, P. D'Arcy, R. A. Zubarev, E. SJ. Arnér and S. Linder, Biochimie, 2019, 162, 46–54 CrossRef CAS PubMed.
- P. J. Barnard and S. J. Berners-Price, Coord. Chem. Rev., 2007, 251, 1889–1902 CrossRef CAS.
- R. Garg, L. G. Benedetti, M. B. Abera, H. Wang, M. Abba and M. G. Kazanietz, Oncogene, 2014, 33, 5225–5237 CrossRef CAS PubMed.
- A. P. Fields and R. P. Regala, Pharmacol. Res., 2007, 55, 487–497 CrossRef CAS PubMed.
- E. Weidauer, Y. Yasuda, B. K. Biswal, M. Cherny, M. N. G. James and D. Brömme, Biol. Chem., 2007, 388, 331–336 CAS.
- T. M. Simon, D. H. Kunishima, G. J. Vibert and A. Lorber, Cancer, 1979, 44, 1965–1975 CrossRef CAS PubMed.
- C. Marzano, V. Gandin, A. Folda, G. Scutari, A. Bindoli and M. P. Rigobello, Free Radicals Biol. Med., 2007, 42, 872–881 CrossRef CAS PubMed.
- S. T. Crooke and C. K. Mirabelli, Am. J. Med., 1983, 75, 109–113 CrossRef CAS PubMed.
- T. M. Simon, D. H. Kunishima, G. J. Vibert and A. Lorber, Cancer Res., 1981, 41, 94–97 CAS.
- C. K. Mirabelli, R. K. Johnson, C. M. Sung, L. Faucette, K. Muirhead and S. T. Crooke, Cancer Res., 1985, 45, 32–39 CAS.
- D. T. Felson, J. J. Anderson and R. F. Meenan, Arthritis Rheum., 1990, 33, 1449–1461 CrossRef CAS PubMed.
- G. Fanali, A. Di Masi, V. Trezza, M. Marino, M. Fasano and P. Ascenzi, Mol. Aspects Med., 2012, 33, 209–290 CrossRef CAS PubMed.
- D. Hu, C.-N. Lok and C.-M. Che, Metal-based Anticancer Agents, 2019, pp. 120–142 Search PubMed.
- A. Pratesi, D. Cirri, D. Fregona, G. Ferraro, A. Giorgio, A. Merlino and L. Messori, Inorg. Chem., 2019, 58, 10616–10619 CrossRef CAS PubMed.
- I. Ott, in Inorganic and Organometallic Transition Metal Complexes with Biological Molecules and Living Cells, Elsevier, 2017, pp. 147–179 Search PubMed.
- V. Scalcon, A. Bindoli and M. P. Rigobello, Free Radicals Biol. Med., 2018, 127, 62–79 CrossRef CAS PubMed.
- A. Miranda-Vizuete, A. E. Damdimopoulos, J. R. Pedrajas, J.-Å. Gustafsson and G. Spyrou, Eur. J. Biochem., 1999, 261, 405–412 CrossRef CAS PubMed.
- J. Zhang, X. Li, X. Han, R. Liu and J. Fang, Trends Pharmacol. Sci., 2017, 38, 794–808 CrossRef CAS PubMed.
- J. Huang and L. Zhong, in Selenoproteins and Mimics, ed. J. Liu, G. Luo and Y. Mu, Springer, Berlin, Heidelberg, 2012, pp. 41–64 Search PubMed.
- P. Nguyen, R. T. Awwad, D. D. K. Smart, D. R. Spitz and D. Gius, Cancer Lett., 2006, 236, 164–174 CrossRef CAS PubMed.
- S. Gromer, L. D. Arscott, C. H. Williams, R. H. Schirmeri and K. Becker, J. Biol. Chem., 1998, 273, 20096–20101 CrossRef CAS PubMed.
- K. M. Debatin, D. Poncet and G. Kroemer, Oncogene, 2002, 21, 8786–8803 CrossRef CAS PubMed.
- G. Ferrin, C. I. Linares and J. Muntane, Curr. Pharm. Des., 2011, 17, 2002–2016 CrossRef CAS PubMed.
- J. S. Modica-Napolitano and J. R. Aprille, Adv. Drug Delivery Rev., 2001, 49, 63–70 CrossRef CAS PubMed.
- G. Filomeni, S. Piccirillo, I. Graziani, S. Cardaci, A. M. Da Costa Ferreira, G. Rotilio and M. R. Ciriolo, Carcinogenesis, 2009, 30, 1115–1124 CrossRef CAS PubMed.
- K. Qian, H. Chen, C. Qu, J. Qi, B. Du, T. Ko, Z. Xiang, M. Kandawa-Schulz, Y. Wang and Z. Cheng, Nanomedicine, 2020, 23, 102087 CrossRef CAS PubMed.
- A. S. Humphreys, A. Filipovska, S. J. Berners-Price, G. A. Koutsantonis, B. W. Skelton and A. H. White, Dalton Trans., 2007, 4943–4950 RSC.
- O. Rackham, S. J. Nichols, P. J. Leedman, S. J. Berners-Price and A. Filipovska, Biochem. Pharmacol., 2007, 74, 992–1002 CrossRef CAS PubMed.
- M. V. Baker, P. J. Barnard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, Dalton Trans., 2006, 6, 3708–3715 RSC.
- J. L. Hickey, R. A. Ruhayel, P. J. Barnard, M. V. Baker, S. J. Berners-Price and A. Filipovska, J. Am. Chem. Soc., 2008, 130, 12570–12571 CrossRef CAS PubMed.
- S. Trapp and R. W. Horobin, Eur. Biophys. J., 2005, 34, 959–966 CrossRef CAS PubMed.
- I. Kandela, W. Lee and G. L. Indig, Biotech. Histochem., 2003, 78, 157–169 CrossRef CAS PubMed.
- S. J. Berners-Price and P. J. Sadler, in Bioinorganic Chemistry. Strcuture and Bonding, Springer, Berlin Heidelberg, 1988, vol. 70, pp. 27–102 Search PubMed.
- S. J. Berners-Price, C. K. Mirabelli, R. K. Johnson, M. R. Mattern, F. L. McCabe, L. F. Faucette, C. M. Sung, S. M. Mong, P. J. Sadler and S. T. Crooke, Cancer Res., 1986, 46, 5486–5493 CAS.
- M. J. McKeage, S. J. Berners-Price, P. Galettis, R. J. Bowen, W. Brouwer, L. Ding, L. Zhuang and B. C. Baguley, Cancer Chemother. Pharmacol., 2000, 46, 343–350 CrossRef CAS PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- S. B. Aher, P. N. Muskawar, K. Thenmozhi and P. R. Bhagat, Eur. J. Med. Chem., 2014, 81, 408–419 CrossRef CAS PubMed.
- W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755–773 RSC.
- S. Jürgens and A. Casini, Chim. Int. J. Chem., 2017, 71, 92–101 CrossRef PubMed.
- A. Gautier and F. Cisnetti, Metallomics, 2012, 4, 23–32 CrossRef CAS PubMed.
- P. O. Wagers, M. J. Panzner, M. R. Southerland, M. A. DeBord, M. C. Deblock, C. A. Tessier, C. L. Cannon and W. J. Youngs, in N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools 2nd Edition, ed. S. Díez-González, Royal Society of Chemistry, Cambridge, 2nd edn, 2016, pp. 567–595 Search PubMed.
- T. Zou, C.-N. Lok, P.-K. Wan, Z.-F. Zhang, S.-K. Fung and C.-M. Che, Curr. Opin. Chem. Biol., 2018, 43, 30–36 CrossRef CAS PubMed.
- L. Oehninger, R. Rubbiani and I. Ott, Dalton Trans., 2013, 42, 3269–3284 RSC.
- S. Y. Hussaini, R. A. Haque and M. R. Razali, J. Organomet. Chem., 2019, 882, 96–111 CrossRef CAS.
- H. V. Huynh, The Organometallic Chemistry of N-heterocyclic Carbenes, John Wiley & Sons, Ltd, Chichester, UK, 1st edn, 2017 Search PubMed.
- N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools: Edition 2, ed. S. Diez-Gonzalez, Royal Society of Chemistry, Cambridge, 2016 Search PubMed.
- N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, ed. S. Diez-Gonzalez, Royal Society of Chemistry, Cambridge, 2010 Search PubMed.
- N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, ed. C. S. J. Cazin, Springer, Netherlands, Dordrecht, 2011, vol. 32 Search PubMed.
- A. T. Biju, N-Heterocyclic Carbenes in Organocatalysis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2018 Search PubMed.
- M. Mora, M. C. Gimeno and R. Visbal, Chem. Soc. Rev., 2019, 48, 447–462 RSC.
- K. M. Hindi, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Chem. Rev., 2009, 109, 3859–3884 CrossRef CAS PubMed.
- M. C. Deblock, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, in N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, ed. S. Díez-González, Royal Society of Chemistry, Cambridge, 2011, pp. 119–133 Search PubMed.
- W. Liu and R. Gust, Coord. Chem. Rev., 2016, 329, 191–213 CrossRef CAS.
- L. Mercs and M. Albrecht, Chem. Soc. Rev., 2010, 39, 1903–1912 RSC.
- M.-L. Teyssot, A.-S. Jarrousse, M. Manin, A. Chevry, S. Roche, F. Norre, C. Beaudoin, L. Morel, D. Boyer, R. Mahiou and A. Gautier, Dalton Trans., 2009, 6894–6902 RSC.
- T. Zou, C. T. Lum, C.-N. N. Lok, W.-P. P. To, K.-H. H. Low and C.-M. M. Che, Angew. Chem., Int. Ed., 2014, 53, 5810–5814 CrossRef CAS PubMed.
- R. Rubbiani, I. Kitanovic, H. Alborzinia, S. Can, A. Kitanovic, L. A. Onambele, M. Stefanopoulou, Y. Geldmacher, W. S. Sheldrick, G. Wolber, A. Prokop, S. Wölfl and I. Ott, J. Med. Chem., 2010, 53, 8608–8618 CrossRef CAS PubMed.
- A. Pratesi, C. Gabbiani, E. Michelucci, M. Ginanneschi, A. M. Papini, R. Rubbiani, I. Ott and L. Messori, J. Inorg. Biochem., 2014, 136, 161–169 CrossRef CAS PubMed.
- R. Rubbiani, S. Can, I. Kitanovic, H. Alborzinia, M. Stefanopoulou, M. Kokoschka, S. Mönchgesang, W. S. Sheldrick, S. Wölfl and I. Ott, J. Med. Chem., 2011, 54, 8646–8657 CrossRef CAS PubMed.
- K. Yan, C. N. Lok, K. Bierla and C. M. Che, Chem. Commun., 2010, 46, 7691–7693 RSC.
- A. Meyer, C. P. Bagowski, M. Kokoschka, M. Stefanopoulou, H. Alborzinia, S. Can, D. H. Vlecken, W. S. Sheldrick, S. Wölfl and I. Ott, Angew. Chem., Int. Ed., 2012, 51, 8895–8899 CrossRef CAS PubMed.
- M. T. Proetto, K. Alexander, M. Melaimi, G. Bertrand and N. C. Gianneschi, Chem. – Eur. J., 2021, 27, 3772–3778 CrossRef CAS PubMed.
- A. Meyer, L. Oehninger, Y. Geldmacher, H. Alborzinia, S. Wölfl, W. S. Sheldrick and I. Ott, ChemMedChem, 2014, 9, 1794–1800 CAS.
- B. Bertrand, L. Stefan, M. Pirrotta, D. Monchaud, E. Bodio, P. Richard, P. Le Gendre, E. Warmerdam, M. H. De Jager, G. M. M. Groothuis, M. Picquet and A. Casini, Inorg. Chem., 2014, 53, 2296–2303 CrossRef CAS PubMed.
- S. M. Meier-Menches, B. Neuditschko, K. Zappe, M. Schaier, M. C. Gerner, K. G. Schmetterer, G. D. Favero, R. Bonsignore, M. Cichna-Markl, G. Koellensperger, A. Casini and C. Gerner, Chem. – Eur. J., 2020, 26, 15528–15537 CrossRef CAS PubMed.
- K. J. Kilpin, S. Crot, T. Riedel, J. A. Kitchen and P. J. Dyson, Dalton Trans., 2014, 43, 1443–1448 RSC.
- S. Vanicek, M. Podewitz, J. Stubbe, D. Schulze, H. Kopacka, K. Wurst, T. Müller, P. Lippmann, S. Haslinger, H. Schottenberger, K. R. Liedl, I. Ott, B. Sarkar and B. Bildstein, Chem. – Eur. J., 2018, 24, 3742–3753 CrossRef CAS PubMed.
- D. Aucamp, S. V. Kumar, D. C. Liles, M. A. Fernandes, L. Harmse and D. I. Bezuidenhout, Dalton Trans., 2018, 47, 16072–16081 RSC.
- M. Albrecht, in Advances in Organometallic Chemistry, Elsevier Inc., 1st edn, 2014, vol. 62, pp. 111–158 Search PubMed.
- M. Albrecht, Chem. Commun., 2008, 3601 RSC.
- A. Krüger and M. Albrecht, in N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, ed. S. Díez-González, Royal Society of Chemistry, Cambridge, 2011, pp. 134–165 Search PubMed.
- Á. Vivancos, C. Segarra and M. Albrecht, Chem. Rev., 2018, 118, 9493–9586 CrossRef PubMed.
- D. Schweinfurth, L. Hettmanczyk, L. Suntrup and B. Sarkar, Metal Complexes of Click-Derived Triazoles and Mesoionic Carbenes: Electron Transfer, Photochemistry, Magnetic Bistability, and Catalysis, 2017, vol. 643 Search PubMed.
- G. Guisado-Barrios, M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2018, 51, 3236–3244 CrossRef CAS PubMed.
- K. O. Marichev, S. A. Patil and A. Bugarin, Tetrahedron, 2018, 74, 2523–2546 CrossRef CAS.
- K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145–1159 RSC.
- D. I. Bezuidenhout, G. Kleinhans, G. Guisado-Barrios, D. C. Liles, G. Ung and G. Bertrand, Chem. Commun., 2014, 50, 2431–2433 RSC.
- A. Casini and L. Messori, Curr. Top. Med. Chem., 2011, 11, 2647–2660 CrossRef CAS PubMed.
- S. Nobili, E. Mini, I. Landini, C. Gabbiani, A. Casini and L. Messori, Med. Res. Rev., 2010, 30, 550–580 CrossRef CAS PubMed.
- V. Milacic, D. Fregona and Q. P. Dou, Histol. Histopathol., 2008, 23, 101–108 CAS.
- E. R. T. Tiekink, Gold Bull., 2003, 36, 117–124 CrossRef CAS.
- C. F. Shaw, Chem. Rev., 1999, 99, 2589–2600 CrossRef CAS PubMed.
- N. Cutillas, G. S. Yellol, C. de Haro, C. Vicente, V. Rodríguez and J. Ruiz, Coord. Chem. Rev., 2013, 257, 2784–2797 CrossRef CAS.
- B. Bertrand, M. R. M. Williams and M. Bochmann, Chem. – Eur. J., 2018, 24, 11840–11851 CrossRef CAS PubMed.
- A. Casini, C. Hartinger, C. Gabbiani, E. Mini, P. J. Dyson, B. K. Keppler and L. Messori, J. Inorg. Biochem., 2008, 102, 564–575 CrossRef CAS PubMed.
- C. Gabbiani, A. Casini and L. Messori, Gold Bull., 2007, 40, 73–81 CrossRef CAS.
- X. Wang and Z. Guo, Dalton Trans., 2008, 1521–1532 RSC.
- E. R. T. Tiekink, Inflammopharmacology, 2008, 16, 138–142 CrossRef CAS PubMed.
- P. J. Sadler, M. Nasr and V. L. Narayanan, Platinum Coordination Complexes in Cancer Chemotherapy: Proceedings of the Fourth International Symposium on Platinum Coordination Complexes in Cancer Chemotherapy convened in Burlington, Vermont by the Vermont Regional Cancer Center and the Norris Cotton Cancer Center, June 22–24, 1983, ed. M. P. Hacker, E. B. Douple and I. H. Krakoff, Springer US, Boston, MA, 1984, pp. 290–304.
- E. R. T. Tiekink, Crit. Rev. Oncol. Hematol., 2002, 42, 225–248 CrossRef PubMed.
- C. K. Mirabelli, C. M. Sung, J. P. Zimmerman, D. T. Hill, S. Mong and S. T. Crooke, Biochem. Pharmacol., 1986, 35, 1427–1433 CrossRef CAS PubMed.
- C. F. Shaw, in Metal Compounds in Cancer Therapy, ed. S. P. Fricker, Springer, Netherlands, Dordrecht, 1994, pp. 46–64 Search PubMed.
- J. Vicente, M. T. Chicote and M. D. Bermúdez, J. Organomet. Chem., 1984, 268, 191–195 CrossRef CAS.
- R. G. Buckley, A. M. Elsome, S. P. Fricker, G. R. Henderson, B. R. C. Theobald, R. V. Parish, B. P. Howe and L. R. Kelland, J. Med. Chem., 1996, 39, 5208–5214 CrossRef CAS PubMed.
- R. V. Parish, B. P. Howe, J. P. Wright, J. Mack, R. G. Pritchard, R. G. Buckley, A. M. Elsome and S. P. Fricker, Inorg. Chem., 1996, 35, 1659–1666 CrossRef CAS PubMed.
- P. Calamai, S. Carotti, A. Guerri, L. Messori, E. Mini, P. Orioli and G. P. Speroni, J. Inorg. Biochem., 1997, 66, 103–109 CrossRef CAS PubMed.
- T. Zou, C. T. Lum, S. S. Y. Chui and C. M. Che, Angew. Chem., Int. Ed., 2013, 52, 2930–2933 CrossRef CAS PubMed.
- A. N. Wein, A. T. Stockhausen, K. I. Hardcastle, M. R. Saadein, S. (Bruce) Peng, D. Wang, D. M. Shin, Z. (Georgia) Chen and J. F. Eichler, J. Inorg. Biochem., 2011, 105, 663–668 CrossRef CAS PubMed.
- R. D. Teo, H. B. Gray, P. Lim, J. Termini, E. Domeshek and Z. Gross, Chem. Commun., 2014, 50, 13789–13792 RSC.
- C.-M. Che, R. W.-Y. Sun, W.-Y. Yu, C.-B. Ko, N. Zhu and H. Sun, Chem. Commun., 2003, 1718–1719 RSC.
- K. J. Akerman, A. M. Fagenson, V. Cyril, M. Taylor, M. T. Muller, M. P. Akerman and O. Q. Munro, J. Am. Chem. Soc., 2014, 136, 5670–5682 CrossRef CAS PubMed.
- M. Coronnello, E. Mini, B. Caciagli, M. A. Cinellu, A. Bindoli, C. Gabbiani and L. Messori, J. Med. Chem., 2005, 48, 6761–6765 CrossRef CAS PubMed.
- J.-J. Zhang, R. W.-Y. Sun and C.-M. Che, Chem. Commun., 2012, 48, 3388–3390 RSC.
- C. K.-L. Li, R. W.-Y. Sun, S. C.-F. Kui, N. Zhu and C.-M. Che, Chem. – Eur. J., 2006, 12, 5253–5266 CrossRef CAS PubMed.
- B. Bertrand, M. Bochmann, J. Fernandez-Cestau and L. Rocchigiani, in Pincer Compounds: Chemistry and Applications, 2018, pp. 673–699 Search PubMed.
- R. W.-Y. Sun, C.-N. Lok, T. T.-H. Fong, C. K.-L. Li, Z. F. Yang, T. Zou, A. F.-M. Siu and C.-M. Che, Chem. Sci., 2013, 4, 1979–1988 RSC.
- S. K. Fung, T. Zou, B. Cao, P.-Y. Lee, Y. M. E. Fung, D. Hu, C.-N. Lok and C. M. Che, Angew. Chem., Int. Ed., 2017, 56, 3892–3896 CrossRef CAS PubMed.
- B. Bertrand, J. Fernandez-Cestau, J. Angulo, M. M. D. Cominetti, Z. A. E. Waller, M. Searcey, M. A. O'Connell and M. Bochmann, Inorg. Chem., 2017, 56, 5728–5740 CrossRef CAS PubMed.
- D. van der Westhuizen, C. A. Slabber, M. A. Fernandes, D. F. Joubert, G. Kleinhans, C. J. van der Westhuizen, A. Stander, O. Q. Munro and D. I. Bezuidenhout, Chem. – Eur. J., 2021, 27, 8295–8307 CrossRef CAS PubMed.
- G. Ferraro, C. Gabbiani and A. Merlino, Bioconjugate Chem., 2016, 27, 1584–1587 CrossRef CAS PubMed.
- A. Giorgio and A. Merlino, Coord. Chem. Rev., 2020, 407, 213175 CrossRef CAS.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- M. C. Jahnke and F. E. Hahn, in N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, Royal Society of Chemistry, 2016, vol. 2, pp. 1–45 Search PubMed.
- C. Sanchez-Cano and M. J. Hannon, Dalton Trans., 2009, 10702–10711 RSC.
- S. Yashveer, P. Matthew and J. S. Patrick, Curr. Med. Chem., 2008, 15, 1802–1826 CrossRef PubMed.
- M. Soler, L. Feliu, M. Planas, X. Ribas and M. Costas, Dalton Trans., 2016, 45, 12970–12982 RSC.
- E. Schuh, C. Pflüger, A. Citta, A. Folda, M. P. Rigobello, A. Bindoli, A. Casini and F. Mohr, J. Med. Chem., 2012, 55, 5518–5528 CrossRef CAS PubMed.
- R. Visbal, V. Fernández-Moreira, I. Marzo, A. Laguna and M. C. Gimeno, Dalton Trans., 2016, 45, 15026–15033 RSC.
- M. M. Jellicoe, S. J. Nichols, B. A. Callus, M. V. Baker, P. J. Barnard, S. J. Berners-Price, J. Whelan, G. C. Yeoh and A. Filipovska, Carcinogenesis, 2008, 29, 1124–1133 CrossRef CAS PubMed.
- K. Fritz-Wolf, S. Urig and K. Becker, J. Mol. Biol., 2007, 370, 116–127 CrossRef CAS PubMed.
- M. P. Gleeson, A. Hersey, D. Montanari and J. Overington, Nat. Rev. Drug Discovery, 2011, 10, 197–208 CrossRef CAS PubMed.
- K.-K. Mak and M. R. Pichika, Drug Discovery Today, 2019, 24, 773–780 CrossRef PubMed.
- A. Zhavoronkov, Mol. Pharm., 2018, 15, 4311–4313 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |